

Abstract
Given the endemic seroprevalence of herpes simplex viruses (HSV), its associated human diseases, and the emergence of acyclovir-resistant strains, there is a continuous need for better antiviral therapies. Towards this aim, identifying mechanistic details of how HSV-1 manipulates infected cells, how it modulates the immune responses, and how it causes diseases are essential. Measuring titers and growth kinetics of clinical isolates and viral mutants are important for a thorough characterization of viral phenotypes in vitro and in vivo. We provide protocols for the preparation as well as titration of HSV-1 stocks, and explain how to perform single-step growth curves to characterize the functions of viral proteins or host factors during infection. In particular, we describe methods to prepare and characterize high-titer HSV-1 stocks with low genome to titer ratios that are required for infection studies in cell culture and animal experiments.
Keywords: Herpes simplex virus type 1, Virus propagation, Plaque assay, Single-step growth curve
Background
Herpes simplex virus type 1 (HSV-1) is the prototypic genus of the human pathogenic subfamily Alphaherpesvirinae. Worldwide, it has a seroprevalence of approximately 67% that varies from 30% to more than 90% in different human groups, depending on the age, the socio-economic status, and the geographical region ( Nahmias et al., 1990 ; reviewed in Smith and Robinson, 2002; Looker et al., 2015 ). HSV-1 shares several features with all herpesviruses: (i) a conserved virion architecture, (ii) productive and lytic infection of cells and (iii) establishment of life-long-latent infection in the host (Whitley and Griffiths, 2002; Schleiss, 2009).
During acute infection, HSV-1 causes cutaneous and mucosal herpetic lesions, and in very young or immune-compromised individuals more severe sequelae, such as potentially blinding keratitis and life-threatening disseminated disease or encephalitis (Thompson and Whitley, 2011; Kennedy and Steiner, 2013). Despite a potent antiviral immune response inside the host, the immune system fails to clear the virus. This is achieved by HSV-1-mediated immune evasion mechanisms ( Theodoridis et al., 2011 ; Su et al., 2016 ; Zheng, 2018; Tognarelli et al., 2019 ), and the establishment of life-long latent infection in neurons of peripheral ganglia which are reached via retrograde axonal transport (Smith, 2012; Roizman and Whitley, 2013; Koyuncu et al., 2018 ). During latency, viral antigen expression in the neurons is restricted, but the virus sporadically reactivates upon lowered immune surveillance and systemic stress ( Padgett et al., 1998 ; Huang et al., 2011 ). Recurrent productive infections after reactivation comprise anterograde axonal spread, virus release from the neurons back to mucocutaneous sites, and reinfection of epithelial cells ( Enquist et al., 1998 ; Whitley and Roizman, 2001; Diefenbach et al., 2008 ; Smith, 2012; Koyuncu et al., 2018 ).
Here, we describe protocols to prepare HSV-1 stocks. We generate virus stocks by harvesting post-nuclear supernatants from repeatedly freeze-thawed infected cells, or by pelleting cell-free virions released from infected cells into the conditioned medium. For cell entry and animal experiments, we recommend purifying such preparations further on sedimentation density gradients ( Sathananthan et al., 1997 ; Sodeik et al., 1997 ; Döhner et al., 2006 ; Dai and Zhou, 2014).
Viral stocks will maintain and acquire genomic mutations, if they provide an evolutionary advantage and are not deleterious during virus amplification. To achieve a high genomic homogeneity, a passage 1 preparation from a single viral plaque or a single clone generated by mutagenesis should be generated. Subsequent passage numbers should be kept to a minimum (Harland and Brown, 1998). We characterize virus stocks by measuring the number of plaque-forming units (pfu) by plaque-titration (see Procedure B) and the number of viral genomes by real-time detection PCR ( Döhner et al., 2006 ; Engelmann et al., 2008 ). In addition, the particle to pfu ratio can be determined by electron microscopy (Harland and Brown, 1998; Döhner et al., 2006 ). To this end, virus stocks are mixed with latex beads of known concentration and appropriate heavy metal salts for negative contrast, and analyzed by electron microscopy. Viral particles and latex beads are counted, and using the known concentration of the latex beads as a reference, the number of viral particles can be determined. Low ratios of genome to pfu, total protein to pfu, or particle to pfu are indicative of a high-quality virus stock.
Since HSV-1 causes strong cytopathic effects, we use plaque assays to determine the number of infectious units within a given sample. Due to the high HSV-1 prevalence, it suffices to add a pooled fraction of human IgGs to the cell culture medium to prevent virus spread via the culture medium. Alternatively, an overlay with agarose or methylcellulose could also neutralize extracellular HSV-1 virions; however, such assays require a bit advanced experimental skills. The direct cell-to-cell spread of HSV-1 leads to the formation of macroscopic plaques, which indicates the occurrence of an infectious particle present in this sample.
Titration of virus stocks is not only crucial for subsequent in vitro or in vivo infection experiments, i.e., to use a defined amount of plaque forming units (pfu), but also during the analysis of viral loads in different tissues of infected animals, when comparing phenotypes of different viral mutants, or during the characterization of clinical isolates. Moreover, the impact of perturbing a host function by pharmacological inhibitors, gene silencing, genetic knock-out, or overexpression is determined by performing a time course of virus production.
The growth kinetics of viral mutants lacking essential protein functions are compared in non-complementing versus complementing cells, the latter expressing the ablated protein in trans ( Schipke et al., 2012 ; Sandbaumhüter et al., 2013 ). When analyzing the impact of particular host factors on virus propagation, viral yields on deficient cells obtained by RNAi or CRISPR/Cas9 technology or from knock-out animals are compared to yields on the corresponding wildtype cells ( Döhner et al., 2018 ).
Single-step or multi-step growth curves in susceptible cell lines provide a standardized method to compare different clinical isolates or viral mutants with the respective wildtype strain, or to determine the impact of a particular host factor during infection. In single-step growth curves, a high multiplicity of infection (MOI) of 5 to 20 pfu/cell ensures complete and simultaneous infection. We recommend monitoring virus propagation up to 24 hpi during one replication cycle (Harland and Brown, 1998). In contrast, multi-step growth curves amplify the effect of more subtle phenotypes during several cycles of replication (Harland and Brown, 1998). Thus, cells are infected using a low MOI of 0.01 to 0.1 pfu/cell for 3 days. While both methods involve the same procedures, we focus here on the experimental setup for a synchronous single-step infection.
Materials and Reagents
Aluminum foil
Pipette
JA-17 or JA-19 centrifuge tubes (Beckman Coulter, catalog number: 357000-357007)
JA-17 or JA-19 rotor (Beckman Coulter, catalog numbers: 369691, 325632)
SW28 ultra-clear tubes (25 x 89 mm, Beckman Coulter, catalog number: 344058)
SW40 ultra-clear tubes (14 x 95 mm, Beckman Coulter, catalog number: 344060)
Cell culture flasks 175 cm2, 75 cm2 (from any qualified supplier)
6- and 12-well cell culture plates (from any qualified supplier)
Cell scraper (SARSTEDT, catalog number: 83.1830)
-
Examples of permissive cells
BHK-21 cell line (baby hamster kidney, ATCC® CCL-10TM)
Vero cell line (African green monkey kidney, ATCC® CCL-81TM)
HeLa (human cervical cancer, ATCC® CCL-2TM)
Note: Not all of these exemplary cell lines propagated further in the laboratory will turn out to be permissive for HSV-1; e.g., in our hands, certain HeLa cell lines are permissive while others are not. Thus, it is beneficial to obtain a proven permissive cell line from a laboratory working already on HSV-1, or to do some pilot experiments, before scaling up the virus preparation.
-
HSV-1(17+)Lox ( Snijder et al., 2012 ) or HSV-1 strain 17+CMV-GFP/UL43 (BioVex; Coffin et al., 1996 and 1998)
In these reporter strains, a GFP expression cassette controlled by the CMV promoter has been inserted between the loci UL55 and UL56 or into the UL43 locus. HSV-1 belongs to biosafety class L2 for clinical isolates or S2 for genetically engineered strains, and thus any work has to be done in certified and governmentally registered L2 or S2 laboratories.
Fetal calf serum (FCS, optimal batch for the cell line to be used for virus production; e.g., Sigma-Aldrich Chemie GmbH, catalog number: F7524)
Bovine serum albumin fraction V (BSA, Sigma-Aldrich Chemie GmbH, catalog number: 10735094001)
Bovine serum albumin, fraction V, pH 7.0 (Capricorn Scientific, catalog number: BSA-1S)
Pooled human immunoglobulin G (IgG, Sigma-Aldrich, Chemie GmbH, catalog number: #I4506)
α-HSV-gD antibody (mouse monoclonal, Santa Cruz Biotechnology, catalog number: sc-21719)
Goat α-mouse IgG-alkaline phosphatase secondary antibody (catalog number: 115-055-166; Jackson ImmunoResearch Laboratories, Inc.)
DMEM cell culture medium (Lonza, catalog number: AMF4300)
RPMI 1640 cell culture medium (Lonza, catalog number: 12-167F)
PBS (Lonza, catalog number: 17-512F)
CO2-independent medium (Thermo Fisher Scientific, catalog number: 18045-054)
HEPES (from any qualified supplier)
Penicillin-Streptomycin mixture (from any qualified supplier)
L-Glutamine (from any qualified supplier)
Trypsin-EDTA (from any qualified supplier)
MEM non-essential amino acids (from any qualified supplier)
Nycodenz (Axis-Shield PoC, catalog number: 1002424)
Methanol (from any qualified supplier)
Ethanol (from any qualified supplier)
CaCl2 (from any qualified supplier)
NaOH (from any qualified supplier)
Tris (from any qualified supplier)
Tris-HCl (pH 9.5)
NaCl (from any qualified supplier)
2-(N-morpholino) ethane sulfonic acid (MES, Sigma-Aldrich Chemie GmbH, catalog number: M5287)
Trypan Blue solution 0.4% (from any qualified supplier)
Liquid N2
Aqua bidest
DAPI (4′,6-diamidino-2-phenylindole, from any qualified supplier)
NP-40 (from any qualified supplier)
DMSO (dimethyl sulfoxide, from any qualified supplier)
Nitroblue tetrazolium chloride (from any qualified supplier)
5-bromo-4-chloro-3-indolyl phosphate (from any qualified supplier)
MgCl2 (from any qualified supplier)
Cell culture medium (see Recipes)
Formalin (see Recipes)
TSM (see Recipes)
Inoculation medium (1) (see Recipes)
Inoculation medium (2) (see Recipes)
MNT buffer (see Recipes)
Crystal violet (from any qualified supplier, see Recipes)
Paraformaldehyde (PFA; from any qualified supplier, see Recipes)
Nuclei stain stock solution (see Recipes)
Equipment
Beckman Optima L-90K ultracentrifuge (Beckman Coulter, catalog number: COL11F05)
Megafuge 2.0 RS (Heraeus, catalog number: 75015505)
Rocking Platform wt 15 (Biometra, catalog number: 042-590)
Cell Imaging System (e.g., EVOS FL AMG/Life Technologies, catalog number: AMF4300)
Perfect Spin 24R refrigerated microcentrifuge (Peqlab, catalog number: C2500R-PL-230)
HERAcell 150i CO2 Incubator (Thermo Fisher Scientific, catalog number: 51026282)
Neubauer chamber (from any qualified supplier)
Ultrasound bath Sonorex RK 100 (Bandelin Elektronik GmbH & Co., catalog number: 66253)
Cup sonifier Sonopuls UW 3200 (Bandelin Elektronik GmbH & Co., catalog number: 364000064495003)
Biocomp gradient master Model 106 (BioComp Instruments, catalog number: 613; http://biocompinstruments.com/our-approach/gradient-forming)
Fume hood
Procedure
-
Virus propagation
Virus propagation can be scaled up to 40 T175 cell culture flasks. For virus propagation, we recommend using BHK-21 cells, although several other permissive cell lines such as Vero or HeLa cells can be used as well. HSV-1 mutants lacking the expression of essential proteins are propagated on appropriate complementing cell lines.
Use a passage 1 virus isolate, clone or mutant to generate a medium-scale passage 2 virus stock that provides the inoculum for a large-scale preparation of passage 3 to 4 for infection experiments. To produce virus stocks, cells should be infected at a low multiplicity of infection (MOI). This fosters amplification and packaging of complete viral genomes, and prevents an enrichment of viral particles harboring defective genomes (Harland and Brown, 1998).
Three days prior to inoculation, seed BHK-21 cells into five cell culture flasks (T175) in 30 ml cell culture medium (10% FCS; Recipe 1) for confluency two days after (= one day prior to infection).
-
One day prior to inoculation, split BHK-21 cells approximately 1:3 into 16 cell culture flasks (T175) in 30 ml of fresh cell culture medium (10% FCS) to achieve approximately 90 to 95% confluence on the next day. Alternatively, seed cells at lower cell density (1:10 to 1:20) three days prior to inoculation to achieve approximately 90 to 95% confluence on the day of inoculation. However, to facilitate achieving an appropriate cell density, we recommend seeding cells at higher density one day prior to inoculation.
Note: Use 15 cell culture flasks for virus inoculation and one cell culture flask for determining the cell density to calculate the amount of the HSV-1 inoculum. The latter flask can be used for further cell culture.
One day prior to inoculation, aspirate the cell culture medium and wash cells with 10 ml PBS/cell culture flask.
Aspirate PBS, immediately add 3 ml of trypsin/EDTA, ensure complete distribution on the cell layer and incubate at 37 °C for 3-4 min.
Meanwhile, prepare 16 T175 cell culture flasks each containing 27 ml pre-warmed cell culture medium.
Microscopically verify trypsinization of the cells. When cells are detached from the bottom of the cell culture flask, add 7 ml cell culture medium/flask and resuspend cells by pipetting up and down.
Pool 10 ml cell suspensions from each of the five cell culture flasks into one 50 ml tube.
Gently add 3 ml cell suspension to the 16 cell culture flasks to reach a final volume of 30 ml/flask.
Mix cell suspension by gently moving the cell culture flasks vertically, horizontally and diagonally.
-
Place cell culture flasks into a humidified incubator at 37 °C and 5% CO2.
Note: If your incubator has uneven shelves, place cell culture flasks for 10 min on a planar bench to achieve a more uniform cell seeding. Do not leave cells too long outside the incubator, since the pH of the DMEM medium will drop in the absence of an additional buffer substance or CO2.
-
Grow cells for one day until they reach approximately 90 to 95% confluency (Figure 1).
Note: The density of optimally cultured cells influences virus yields, as subconfluent cells are more susceptible to HSV-1 than confluent ones. If seeded at low density, some cell lines tend to grow to small confluent cell islets. Cells in the periphery of such islets are more susceptible to HSV-1 infection than confluent cells in a monolayer, since the major HSV-1 receptor nectin-1 is sequestered into cell-cell adhesion contacts and not accessible for virus binding (Schelhaas et al., 2003; Marozin et al., 2004; Snijder et al., 2012).
-
At the day of virus propagation, inoculate BHK-21 cells with the respective HSV-1 strain or mutant.
Wash one T175 cell culture flask with 10 ml PBS, aspirate, trypsinize with 3 ml trypsin/EDTA as described in Step A2b.
-
Resuspend cells in 25 ml of medium and count the number of cells using a Neubauer chamber or an alternative method.
Note: Typically, the cell amount per T175 cell culture flask is approximately 1.5 x 107 to 3 x 107 cells. Use these cells for further cell culture.
-
Calculate the volume of a passage 2 virus stock required to infect 15 T175 cell culture flasks with a multiplicity of infection (MOI) of 0.01 pfu/cell using the formula below:
Formula:
Example calculation: Titer of stock preparation HSV-1 p2: 6 x 108 pfu/ml = 6 x 105 pfu/µl
MOI of 0.01 pfu/cell; 40 flasks with 2 x 107 cells/flask
-
Prepare one inoculum for all flasks by diluting the calculated virus volume into RPMI 1640 supplemented with 20 mM HEPES (use 5 ml/flask). Alternatively, dilute calculated virus in CO2-independent medium supplemented with 0.1% (w/v) cell culture grade BSA (use 5 ml/flask). Both media warrant a stable pH during subsequent inoculation of the cells outside a CO2 incubator.
Note: As CO2 is a gas, it evaporates quickly from the medium outside a CO2 incubator, which will therefore turn more basic. While cells recover quickly from a short-term incubation in a more alkaline medium, HSV-1 is very sensitive to higher pH. Already at pH 7.6, the titer of a given virus suspension drops significantly.
-
Harvest cell culture medium from each cell culture flask.
Note: To reduce the costs for virus preparation, the cell culture medium removed from the flasks can be pooled in a sterile flask, placed in a 37 °C incubator and added to the BHK-21 cells after inoculation.
-
Prior to infection, wash cells once with 10 ml/cell culture flask PBS, aspirate, and immediately add 5 ml inoculum.
Note: Wash and infect up to five cell culture flasks simultaneously.
Inoculate cells on a gently moving rocking platform at room temperature (RT) for 1 h. Ensure that the cells are completely covered by the inoculum.
After 1 h, add 25 ml/flask (preserved) cell culture medium.
Incubate HSV-1-infected BHK-21 cells at 37 °C and 5% CO2 for three to four days.
-
Harvest extracellular HSV-1 virions.
Note: Microscopically monitor the infected cells daily (Figure 2). In our hands, the time point of harvesting is crucial and can vary from three to four dpi. For good yields of high-quality HSV-1 extracellular virions, harvest the cell culture supernatants when 80 to 90% of the cells display cytopathic effects, i.e., cells are rounded up and detach from the bottom of the flask. When harvesting HSV-1 too early, the yields for extracellular virus will be lower. When harvesting HSV-1 too late, the virus stocks will be of lower quality due to cell debris. In particular, released DNA/RNA will crosslink many viral particles and thus hampers sufficient resuspension of aggregated virions (see Step A4d).
At three or four dpi, detach cells by tapping the cell culture flask, rinse remaining cells from the bottom and collect the cell-virus suspension in 50 ml tubes.
To pellet the cells centrifuge at 1,900-2,850 × g and 4 °C for 10 min.
The sedimented infected cells can be subsequently used to isolate nuclear capsids ( Wolfstein et al., 2006 ; Bucks et al., 2007 ; Radtke et al., 2010 ), or infectious virions from the cytoplasm by freeze-thaw cycles. For this, resuspend the cell pellets in an appropriate small volume of MNT buffer (Recipe 8), freeze in liquid N2, and store at -80 °C.
-
Transfer the supernatant containing HSV-1 virions to precooled centrifuge tubes. Centrifuge in a JA-17 rotor at 4 °C and 17,000 rpm (maximal 39,813 × g) for 2 h, or in a type 19 rotor at 4 °C and 12,000 rpm (maximal 21,500 × g) for 90 min. While the first centrifugation setting warrants a higher yield of pelleted virions, the latter one warrants easier and more homogenous resuspension of the virus pellet to single virions.
Note: After centrifugation, label the positions of the pellets on the outside of the tubes.
-
Place centrifuge tubes on ice, aspirate supernatants completely, but carefully, and add 200 to 500 μl/tube MNT buffer to resuspend the pellets.
Note: We recommend using MNT buffer instead of PBS for storage of HSV-1 stocks since sodium phosphate salts precipitate upon cooling causing dramatic pH changes. MES and Tris-HCl maintain a more stable pH during the freezing procedure (Gómez et al., 2001).
Tilt tubes for complete coverage of the virus pellet with MNT buffer and allow virus pellets to swell at 4 °C over-night. Alternatively, resuspend the pellets in MNT buffer by pipetting, but avoid the formation of foam or bubbles. Transfer the resuspended pellets, for sonication on the next day, to a fresh glass vial coated with BSA (fraction V or cell culture grade) to prevent particle loss, due to unspecific binding to the glass surface. Close the vial tightly with parafilm and allow viral particles to swell at 4 °C for 24 to 36 h. This glass vial will be used for sonication on the next day (see Step A5b).
-
Resuspend virus suspension.
On the next day, pool and/or further resuspend the medium pellet. Work on ice and avoid generating foam and bubbles.
We recommend further homogenizing the medium pellet in an ultrasonic bath filled with ice water (pulse three times for 30 s with 30 s pauses in between). If necessary, repeat Step A5b two to three times until the virus suspension appears homogenous.
-
Aliquot the resuspended medium pellet virus for storage into small volumes (25-100 μl, depending on the experiments planned) into sterile screw cap vials, snap-freeze in liquid N2, and store at -80 °C. To prepare gradient-purified virus stocks, skip this step and directly proceed with the purification protocol (see Step A6).
Note: Avoid repeated freeze-thaw cycles of virus stocks. Always store stocks at -80 °C, since they continuously lose infectivity at -20 °C. Aliquoted vials should be snap-frozen rapidly in liquid N2, thawed quickly and kept at 0 to 4 °C until use.
-
Gradient purify HSV-1 stocks.
Virus stocks for virus entry or animal experiments should be gradient-purified to remove residual cell debris and cytokines that might have been synthesized during infection. HSV-1 virions can be purified using sucrose, Ficoll, nycodenz or iodixanol (Opti-prep) gradients ( Sathananthan et al., 1997 ; Sodeik et al., 1997 ; Döhner et al., 2006 ; Marconi and Manservigi, 2014). At the same density, Ficoll, nycodenz and iodixanol solutions have a lower osmotic pressure than sucrose solutions. When HSV-1 virions enter a sucrose sedimentation gradient, they lose water due to an increasing osmotic pressure at higher sucrose concentration, and more importantly swell abruptly upon dilution from their sucrose fraction into MNT or PBS buffers. Such changes in osmotic pressure impair the physical integrity of enveloped virions. Therefore, we developed protocols using nycodenz for gradient purification of HSV-1 stocks ( Döhner et al., 2006 ).
Prepare a linear nycodenz gradient. Pipette MNT buffer supplemented with 10% nycodenz into ultraclear tubes up to half height and label fill level. Carefully underlay with a 40% nycodenz in MNT using a syringe attached to a long needle until the interphase is at the half-full line (Video 1).
-
If using a Biocomp gradient master: Close tubes with corresponding caps, level the gradient master (see manual) and prepare a linear gradient using the parameters provided by the vendor. Place the tubes with the step gradient into the tube holder, place the tube holder onto the magnetic platform of the gradient master and start the program. The platform will tilt and then rotate. The tilting angle (°), the speed (rpm), and duration of rotation (min:s) are specified by the user as indicated in the table below (Table 1). For better visualization, we refer to https://www.youtube.com/watch?v=wizxYB5brz8.
*SW28 for larger or SW40 for medium volumes.
Alternative procedure: Prepare a two-step gradient as described in Step A6a and seal tightly. To receive a linear gradient via diffusion, gently place the tube horizontally, stabilize and keep at RT for 90 min.
-
If necessary, discard the respective sample volume from the top of the gradient. Subsequently, overlay the gradient with 1 ml and 2-3 ml of resuspended medium pellet for SW40 and SW28 gradient, respectively.
Note: Avoid overloading of the gradients.
-
Centrifuge in a pre-cooled ultracentrifuge (e.g., Beckman Optima L-90K) at 4 °C. When using the SW28 rotor, centrifuge at 20,000 rpm (maximal 72,128 × g) for 105 min, when using the SW40 rotor, centrifuge at 20,000 rpm (maximal 71,142 × g) for 120 min.
Note: If using other settings or rotors, recalculate speed and time. Choose the appropriate rotor depending on the volume of virus suspension to be gradient-purified.
Label the light-scattering virus band slightly beneath the middle of the gradient (Figure 3). Remove the upper gradient solution and harvest the light-scattering band with a yellow tip pipette without disturbing the gradient. We harvest 1 to 4 ml gradient-purified virus from one tube of an SW28 bucket. Transfer the harvested band into a vial, mix gently by pipetting, prepare single-use aliquots (10 to 100 µl, depending on purpose) in autoclaved 0.5 ml screw caps with O-rings, snap-freeze in liquid nitrogen, and store at -80 °C. The given volume depends on the volume that had been layered onto the gradient, and whether one aims for a highly concentrated stock for cell entry experiments, or complete yield for experiments performed at a lower MOI.
-
Plaque Assay
Note: We recommend technical duplicates for each virus stock. When comparing different viruses in single-step growth kinetics (e.g., parental vs. mutant or clinical isolates), perform plaque assays in parallel including three biological and two technical replicates for each sample.
-
Prepare one T75 cell culture flask of confluent Vero cells for 6-well or 12-well plate plaque assay.
Note: One T75 cell culture flask with confluent Vero cells is sufficient for seeding two 6-well or three 12-well plates.
-
One day prior to inoculation, seed Vero cells from one confluent T75 cell culture flask into one 6-well plate at 3 x 105 to 5 x 105 cells/well or 12-well plate at 1.5 x 105 to 2.5 x 105 cells/well to achieve just confluence on the following day.
Add 5 ml PBS, gently wash cell layer and aspirate PBS.
Immediately add 1.5 ml of trypsin/EDTA, ensure even distribution on the entire cell layer and incubate at 37 °C for 5 min.
Microscopically monitor cell rounding. When cells are completely detached, add 8.5 ml of prewarmed medium and resuspend cells by pipetting up and down.
Count the cells using a Neubauer chamber or an alternative method.
Prepare an appropriate volume of medium to achieve a final concentration of 0.5 x 106 cells/ml, and transfer 2 ml/6-well or 1 ml/12-well into each well.
Incubate Vero cells at 37 °C and 5% CO2 for 16 to 20 h to reach just-confluence.
-
At the day of inoculation, prepare serial 10-fold dilutions of the virus stocks to be titrated.
-
Prepare 30 ml of inoculation medium (Recipes 6 and 7): RPMI 1640 supplemented with 20 mM HEPES and 0.1% (w/v) BSA or CO2-independent medium supplemented with 0.1% (w/v) cell culture grade BSA. Both media maintain a stable pH while inoculating the cells outside a CO2 incubator on a rocking platform.
Note: For an uncharacterized virus stock, use dilutions ranging from 10-2 to 10-9 (see Figure 4). For an uncharacterized sample of a growth curve, use dilutions ranging from 10-2 to 10-6. If prior experiments permit an estimation of the titer, it suffices to plate fewer dilutions. Do not forget an uninfected mock control.
Thaw a virus stock and prepare a 10-2 dilution by adding 10 μl of the stock to 990 μl of inoculation medium and vortex thoroughly.
Prepare the 10-3 dilution by transferring 130 μl of the 10-2 dilution with a fresh pipette tip to 1,170 μl inoculation medium. Vortex.
Complete this serial dilution until you reach the desired final dilution (see Figure 4).
Gently wash Vero cells once with 1.5 ml/well inoculation medium.
Add 500 μl/6-well or 400 μl/12-well of the 10-5 to 10-9 dilutions to titer a virus stock, or the 10-2 to 10-6 dilutions for samples of a growth curve to the designated well. Perform in single-sample or in duplicates and incubate cells on a gently rocking platform at RT for 1 h.
-
At 1 hpi, aspirate the inoculum and add 2 ml/6-well or 800 μl/12-well of medium freshly supplemented with pooled human IgG.
Caution: As the titers of neutralizing antibodies vary among the human population, we titrate every batch by pretreating a standard HSV-1 stock with 2-fold serial dilutions of a pooled human IgG stock solution. An IgG concentration of about 10 to 20 μg/ml usually suffices to neutralize a virus stock diluted for plaque assays. It may require a higher concentration of human IgGs to neutralize a synchronous infection at a higher MOI.
Incubate cells at 37 °C and 5% CO2 for three days.
-
At 2 dpi, microscopically analyze plaque formation (Figure 5). A fluorescent microscope could be used to monitor cells infected with a strain expressing a fluorescent protein.
-
Variant 1: At 3 dpi, stain the cells with crystal violet.
-
Aspirate the medium from each well, and fix the cells using 1 ml/6-well or 400 μl/12-well of 9% formalin (Recipe 3) at RT for 10 min. Alternatively, fix cells using pre-cooled (-20 °C) 100% methanol for 3 min.
Note: Carefully add the fixative to the wall of the wells to avoid cell detachment.
Aspirate the fixative, and, in the case of methanol fixation, air-dry the wells for at least 5-10 min. Add 1 ml/6-well or 400 μl/12-well of 0.1% (w/v) crystal violet (Recipe 4) staining solution, and incubate at RT for 10 min.
Aspirate staining solution, wash twice with 1 ml/well Aqua bidest and let dry (Figure 6).
-
-
Variant 2, for small plaques in a 6-well plate: At 2 dpi, label infected cells with an antibody directed against the HSV-1 envelope glycoprotein D (gD; Döhner et al., 2002 ).
-
Aspirate the medium from each well and fix cells with -20 °C pre-cooled methanol at RT for 3 min.
Note: Carefully add the fixative to the wall of the wells to avoid cell detachment.
Aspirate the fixative, air-dry the cells and incubate on a gently rocking platform at RT for 30 min with 0.5 ml/well PBS supplemented with 10% FCS (PBS/FCS) to block unspecific protein binding.
Replace PBS/FCS with 0.5 ml/well primary α-gD antibody diluted in PBS/FCS and incubate on a rocking platform at RT for 30 min. We use our present batch of the anti-gD antibody (mAb DL6) at a dilution of 1:2000. Since antibody concentrations vary among suppliers and batches, we recommend titrating each batch using dilutions ranging from 1:50 to 1:5,000.
Remove the primary antibody and wash cells three times with 2 ml/well PBS for 10 min.
Replace PBS with 0.5 ml/well of secondary goat α-mouse IgG coupled to alkaline phosphatase in PBS/FCS and incubate on a rocking platform at RT for 30 min. We use our present batch of the alkaline phosphatase-coupled anti-mouse IgG antibody at a dilution of 1:2000. Since antibody concentrations vary among suppliers and batches, we recommend titrating each batch using dilutions ranging from 1:500 to 1:10,000.
Remove the secondary antibody and wash three times with 2 ml/well PBS for 10 min, and two times with 2 ml/well TSM (Recipe 5) for 5 min each.
Replace with 0.5 ml/well of 0.2 mM nitroblue tetrazolium chloride and 0.8 mM 5-bromo-4-chloro-3-indolyl phosphate in TSM buffer. Protect from light with aluminum foil, and incubate on a rocking platform at RT until dark plaques appear.
Replace staining solution, and rinse the wells twice with Aqua bidest to stop alkaline phosphatase reaction by lowering the pH and removing the substrate.
Aspirate water and air-dry the plates (Figure 7).
-
-
Count the plaques in wells containing approximately 20 to 50 plaques in a 6-well or 10 to 40 in a 12-well.
Note: In wells with too many plaques the titer is underestimated as fused plaques will be recorded as only one plaque. In contrast, in wells with few plaques the titer is overestimated as they may contain irregularities in the cell monolayer that may be interpreted falsely as plaques.
-
Calculate the titer in pfu/ml using these formulas:
6-well plate:
12-well plate:
Example calculation: Titration in 6-well plates in duplicates, using 0.5 ml of the inoculum.
Assumption: Wells treated with the 10-8 dilution of the inoculum show plaques in the range of 20 to 50, namely 28 and 32.
-
-
Single-step growth kinetics
Note: Measuring samples from at least three biological independent experiments once is better than measuring samples from a single experiment in triplicates. We usually measure samples from at least three independent experiments in duplicates.
-
One day prior to infection, seed susceptible cells, e.g., Vero, into 6-well plates at 0.25 x 106 cells/well in 2 ml medium, and incubate at 37 °C and 5% CO2 for 16 to 20 h.
Note: The cells should be 90 to 95% confluent at the time of inoculation for single-step growth kinetics. For each virus isolate, virus mutant, cell line, and time point, one well is required to monitor virus propagation, and another one to estimate the cell density.
On the next day, remove the medium and wash the cells three times with 1 ml/well PBS.
-
Assess the cell density in one well of 6-well plate by performing DAPI staining.
Fix cells with 1 ml/well 3% paraformaldehyde (PFA) in PBS at RT for 20 min and wash three times with 1 ml/well PBS.
Prepare a 1:200 dilution of nuclei stain stock solution in PBS supplemented with 0.1% (v/v) Triton X-100, and stain the cells with 1 ml/well at RT for 10 min.
Remove staining solution and carefully wash the cells three times with 1 ml/well PBS. Measure the cell density by automated fluorescence microscopy. There are several automated fluorescence microscopes available with proprietary software to obtain cell number per are. At present, we use a Cytation 3 imaging reader (BioTek, Agilent, Winnoski, VT, US).
-
Prepare virus inoculum.
Calculate the volume of the inoculum for all wells, and dilute the virus stock to yield approximately 5 to 10 pfu/cell with CO2-independent medium supplemented with 0.1% (w/v) cell culture grade BSA. Prepare one sample CO2-independent medium lacking virus as mock-treated control.
Inoculate or mock-treat cells with 1 ml/well, and incubate cells on a rocking platform at RT for 1 h.
At 1 hpi, wash cells three times with 1 ml/well PBS, and incubate with 2 ml/well medium supplemented with 1% FCS at 37 °C and 5% CO2. This is 0 h of the time scale.
-
Determine the amount of extracellular and cell-associated intracellular virus at various time points, e.g., in 3 h intervals from 3 to 24 hpi.
To determine extracellular viral yields, transfer supernatants from a 6-well plate into 2 ml tubes kept on ice. Centrifuge at 4 °C and 150 × g for 5 min to pellet cells.
Meanwhile, add 600 μl/well MNT buffer to prevent cells from drying out.
After centrifugation, harvest supernatants, aliquot for plaque assay (6 x 300 μl), snap-freeze in liquid N2, and store at -80 °C. Keep the resulting cell pellets on ice.
To determine intracellular viral yields, harvest residual Vero cells of each well by scraping, and combine with the cells harvested from the medium (Step C5c).
Transfer 600 μl of this cell suspension to fresh 2 ml tubes.
Add 400 μl MNT buffer to each well, to wash the well and scraper, and transfer this also to the respective 2 ml tube (Step C5e) reaching a final volume of 1 ml.
Vortex cell suspensions, snap-freeze in liquid N2 and store at -80 °C.
To release the cell-associated virus, the cells are subjected to three cycles of freeze-thaw, or sonicated in an ultrasound bath until the solution appears clear.
Centrifuge these suspensions at 4 °C and 150 × g for 3 min, aliquot the supernatants in 4x 200 μl, snap-freeze in liquid N2, and store at -80 °C. Furthermore, store the 2 ml vials with residual 200 μl containing any potential pellets.
-
Determine the intra- and extracellular viral yields by plaque assay (see Procedure B).
Note: In addition, the number of intra- and extracellular viral genomes can be determined by real-time detection PCR using a standard diagnostic probe (e.g., Engelmann et al., 2008).
-
Figure 1. Subconfluent BHK-21 cells for HSV-1 inoculation.

Scale bar = 0.4 mm.
Figure 2. BHK-21 cells infected with HSV-1 strain 17+CMV-GFP/UL43 at 1 dpi (upper panel) and 4 dpi (lower panel).

Scale bar= 0.4 mm.
Video 1. Visualization of the interphase formation during preparation of a step gradient.
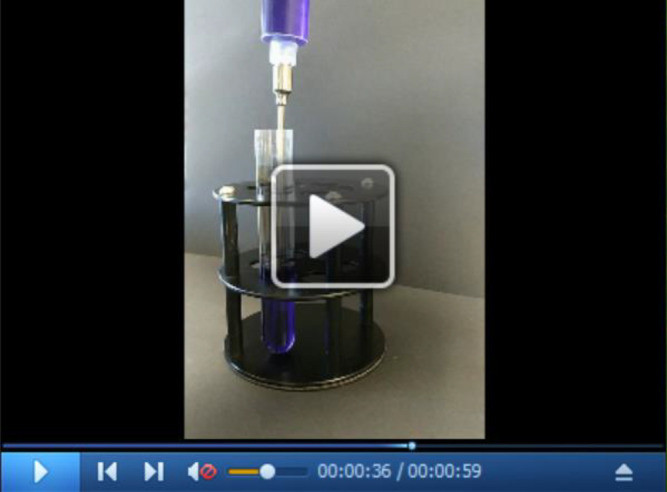
The tube contains 10% sucrose (transparent). We underlay with the 40% sucrose (blue) using a syringe. To better visualize the interphase formation, we stained the 40% sucrose solution here with a blue dye. If preparing gradients for virus preparations we do not add any dye. It is crucial to add the heavy solution smoothly without pressing the needle to the bottom of the tube and without pushing the plunger too abruptly. An interphase between the 10% and 40% solution forms and migrates up.
Table 1. Centrifugation settings for the Gradient master.
| Rotor* | Step | Time [min:s] | Angle | Speed | Temperature |
|---|---|---|---|---|---|
| SW28 | 1 | 7:00 | 50° | 25 rpm | RT |
| 2 | 0:15 | 80° | 15 rpm | RT | |
| SW40 | 1 | 1:11 | 80° | 25 rpm | RT |
Figure 3. Gradient purification of HSV-1 virions.

Exemplary result of a linear nycodenz sedimentation gradient with a light-scattering virus band (arrow) slightly beneath the middle of the tube (red line).
Figure 4. Pipetting scheme of a serial dilution for plaque assays.

Figure 5. HSV-1 plaques in a Vero cell monolayer at 2 dpi (left) or 3 dpi (right).

Scale bar = 0.4 mm.
Figure 6. Crystal violet stained HSV-1 plaque assay.

Vero cells cultured in a 12-well plate were inoculated with 10-fold serial dilutions of a virus suspension, fixed at 3 dpi and stained with crystal violet.
Figure 7. α-gD stained HSV-1 plaque assay.

Vero cells were inoculated with serial dilutions of HSV-1 suspensions and infected for 3 days. Cells were fixed and subsequently labeled with an α-gD antibody followed by labeling with a secondary alkaline phosphatase antibody.
Data analysis
When comparing different viruses in single-step growth kinetics (e.g., parental vs. mutant or clinical isolates), perform plaque assays in parallel including three biological and two technical replicates for each sample. For some statistical considerations on virological experiments, please refer to Richardson and Overbaugh (2005) and Wang and Bushman (2006).
Notes
Primary risks for people working with HSV are due to droplet exposure of mucous membranes of the eyes, nose or mouth, inhalation of concentrated aerosols, or accidental injection. Lab workers thus require a health check, and regular intensive training in biosafety L2/S2 work. Working with HSV presupposes an approved biosafety level S2 laboratory. Handling of virus preparations needs to be performed in a biosafety L2/S2 cabinet, while wearing gloves and goggles. We avoid the use of needles, syringes, or any other sharp devices to prepare high-titer virus stock solutions. All waste that contains or has encountered infectious virus needs to be inactivated either chemically (bleach or aldehyde-containing disinfections), or by autoclaving for at least 20 min at 121°C, according to the biosafety L2/S2 regulation in the respective country and the L2/S2 guidelines of the respective institution. We also recommend to UV-irradiate all waste as well as the biosafety L2/S2 cabinet after working with virus-containing samples.
Recipes
-
Cell culture medium (store at 4 °C, use within 3 months after opening and before expiry date)
DMEM supplemented with:
100 mg/ml Streptomycin (optional)
100 U/ml Penicillin (optional)
2 mM L-glutamine
1x of MEM nonessential amino acids solution (100x)
1% or 7.5-10% FCS (optimized for the cell line to be used for propagation)
-
3% paraformaldehyde in PBS, pH 7.4 (store at -20 °C, stable for at least 6 months)
Mix 12 g paraformaldehyde with 40 ml 10x PBS, fill up to 300 ml with Aqua bidest
To dissolve the PFA, heat up to maximal 60 °C (fume hood) while stirring continuously
Add 40 µl of 1 M CaCl2 and 40 µl of 1 M MgCl2
After the PFA is dissolved, cool the solution down to RT, adjust the pH to 7.4 using 1 M NaOH and fill up to 400 ml with Aqua bidest
-
Formalin (store at RT, stable for at least 1 month)
Dilute a 36.5% stock solution 1:4 in PBS to reach a final concentration of 9% formaldehyde
-
Crystal violet (store at RT, stable for at least 1 month)
Prepare a 5% stock solution by dissolving 0.25 g crystal violet in 5 ml 100% ethanol (store at RT, stable for at least 6 months)
Prepare a working solution by 1:50 dilution in Aqua bidest to reach a final concentration of 0.1% crystal violet in 2% ethanol
-
TSM buffer (store at RT, stable for at least 1 month)
100 mM Tris-HCl, pH 9.5
100 mM NaCl
5 mM MgCl2
-
Inoculation medium (1) (store at 4 °C, use within 3 months after opening and before expiry date)
RPMI 1640
20 mM HEPES
0.1% (w/v) cell-culture grade BSA
-
Inoculation medium (2) (store at 4 °C, use within 3 months after opening and before expiry date)
CO2-independent medium
20 mM HEPES
0.1% (w/v) cell-culture grade BSA
-
MNT buffer (filter sterilize and store at RT, stable for at least 12 months)
30 mM MES
100 mM NaCl
20 mM Tris
-
Nuclei stain stock solution (store at 4°C, protect from light, stable for at least 12 months)
10 mg/ml DAPI
10% (v/v) DMSO
0.1% (v/v) NP-40
5% (w/v) BSA fraction V
10 mM Tris-HCl, pH 7.4
146 mM NaCl
22 mM CaCl2
22 mM MgCl2
Dilute 1:200 for a working solution
Acknowledgments
This work was supported by the ELAN Program (Faculty of Medicine, Friedrich-Alexander-Universität Erlangen-Nürnberg; project 18-12-21-1, awarded to LG), and by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG; project STE 432/11-1 awarded to AS; the Germany’s Excellence Strategy, EXC 2155, project number 390874280, awarded to BS; the Collaborative Research Center SFB 900, Project C2, project number 158989968, awarded to BS). We thank Manutea Serrero (Institute of Virology, Hannover Medical School) for providing Figure 3 and Timmy Riccardo (Institute of Virology, Hannover Medical School) for providing Figure 7.
Competing interests
The authors declare no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Bucks M. A., O'Regan K. J., Murphy M. A., Wills J. W. and Courtney R. J.(2007). Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361(2): 316-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coffin R. S., MacLean A. R., Latchman D. S. and Brown S. M.(1996). Gene delivery to the central and peripheral nervous systems of mice using HSV1 ICP34.5 deletion mutant vectors. Gene Ther 3(10): 886-891. [PubMed] [Google Scholar]
- 3. Coffin R. S., Thomas S. K., Thomas N. S., Lilley C. E., Pizzey A. R., Griffiths C. H., Gibb B. J., Wagstaff M. J., Inges S. J., Binks M. H., Chain B. M., Thrasher A. J., Rutault K. and Latchman D. S.(1998). Pure populations of transduced primary human cells can be produced using GFP expressing herpes virus vectors and flow cytometry. Gene Ther 5(5): 718-722. [DOI] [PubMed] [Google Scholar]
- 4. Dai X. and Zhou Z. H.(2014). Purification of herpesvirus virions and capsids. Bio Protoc 4(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diefenbach R. J., Miranda-Saksena M., Douglas M. W. and Cunningham A. L.(2008). Transport and egress of herpes simplex virus in neurons. Rev Med Virol 18(1): 35-51. [DOI] [PubMed] [Google Scholar]
- 6. Döhner K., Radtke K., Schmidt S. and Sodeik B.(2006). Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26. J Virol 80(16): 8211-8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Döhner K., Ramos-Nascimento A., Bialy D., Anderson F., Hickford-Martinez A., Rother F., Koithan T., Rudolph K., Buch A., Prank U., Binz A., Hügel S., Lebbink R. J., Hoeben R. C., Hartmann E., Bader M., Bauerfeind R. and Sodeik B.(2018). Importin α1 is required for nuclear import of herpes simplex virus proteins and capsid assembly in fibroblasts and neurons. PLoS Pathog 14(1): e1006823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Döhner K., Wolfstein A., Prank U., Echeverri C., Dujardin D., Vallee R. and Sodeik B.(2002). Function of dynein and dynactin in herpes simplex virus capsid transport. Mol Biol Cell 13(8): 2795-2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Engelmann I., Petzold D. R., Kosinska A., Hepkema B. G., Schulz T. F. and Heim A.(2008). Rapid quantitative PCR assays for the simultaneous detection of herpes simplex virus, varicella zoster virus, cytomegalovirus, Epstein-Barr virus, and human herpesvirus 6 DNA in blood and other clinical specimens. J Med Virol 80(3): 467-477. [DOI] [PubMed] [Google Scholar]
- 10. Enquist L. W., Husak P. J., Banfield B. W. and Smith G. A.(1998). Infection and spread of alphaherpesviruses in the nervous system. Adv Virus Res 51: 237-347. [DOI] [PubMed] [Google Scholar]
- 11. Gómez G., Pikal M. J. and Rodríguez-Hornedo N.(2001). Effect of initial buffer composition on pH changes during far-from-equilibrium freezing of sodium phosphate buffer solutions. Pharm Res 18(1): 90-97. [DOI] [PubMed] [Google Scholar]
- 12. Harland J. and Brown S. M.(1998). HSV growth, preparation, and assay. Methods Mol Med 10: 1-8. [DOI] [PubMed] [Google Scholar]
- 13. Huang J., Lazear H. M. and Friedman H. M.(2011). Completely assembled virus particles detected by transmission electron microscopy in proximal and mid-axons of neurons infected with herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology 409(1): 12-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kennedy P. G. and Steiner I.(2013). Recent issues in herpes simplex encephalitis. J Neurovirol 19(4): 346-350. [DOI] [PubMed] [Google Scholar]
- 15. Koyuncu O. O., MacGibeny M. A. and Enquist L. W.(2018). Latent versus productive infection: the alpha herpesvirus switch. Future Virol 13(6): 431-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Looker K. J., Magaret A. S., May M. T., Turner K. M., Vickerman P., Gottlieb S. L. and Newman L. M.(2015). Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One 10(10): e0140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marconi P. and Manservigi R.(2014). Herpes simplex virus growth, preparation, and assay. Methods Mol Biol 1144: 19-29. [DOI] [PubMed] [Google Scholar]
- 18. Marozin S., Prank U. and Sodeik B.(2004). Herpes simplex virus type 1 infection of polarized epithelial cells requires microtubules and access to receptors present at cell-cell contact sites. J Gen Virol 4): 775-786. [DOI] [PubMed] [Google Scholar]
- 19. Nahmias A. J., Lee F. K. and Beckman-Nahmias S.(1990). Sero-epidemiological and-sociological patterns of herpes simplex virus infection in the world. Scand J Infect Dis Suppl 69: 19-36. [PubMed] [Google Scholar]
- 20. Padgett D. A., Sheridan J. F., Dorne J., Berntson G. G., Candelora J. and Glaser R.(1998). Social stress and the reactivation of latent herpes simplex virus type 1. Proc Natl Acad Sci U S A 95(12): 7231-7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Radtke K., Kieneke D., Wolfstein A., Michael K., Steffen W., Scholz T., Karger A. and Sodeik B.(2010). Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6(7): e1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richardson B. A. and Overbaugh J.(2005). Basic statistical considerations in virological experiments. J Virol 79: 669-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roizman B. and Whitley R. J.(2013). An inquiry into the molecular basis of HSV latency and reactivation. Annu Rev Microbiol 67: 355-374. [DOI] [PubMed] [Google Scholar]
- 24. Sandbaumhüter M., Döhner K., Schipke J., Binz A., Pohlmann A., Sodeik B. and Bauerfeind R.(2013). Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell Microbiol 15(2): 248-269. [DOI] [PubMed] [Google Scholar]
- 25. Sathananthan B., Rødahl E., Flatmark T., Langeland N. and Haarr L.(1997). Purification of herpes simplex virus type 1 by density gradient centrifugation and estimation of the sedimentation coefficient of the virion. APMIS 105(3): 238-246. [DOI] [PubMed] [Google Scholar]
- 26. Schelhaas M., Jansen M., Haase I. and Knebel-Mörsdorf D.(2003). Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells. J Gen Virol 9): 2473-2484. [DOI] [PubMed] [Google Scholar]
- 27. Schipke J., Pohlmann A., Diestel R., Binz A., Rudolph K., Nagel C. H., Bauerfeind R. and Sodeik B.(2012). The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J Virol 86(7): 3682-3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schleiss M. R.(2009). Persistent and recurring viral infections: the human herpesviruses. Curr Probl Pediatr Adolesc Health Care 39(1): 7-23. [DOI] [PubMed] [Google Scholar]
- 29. Smith G.(2012). Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66: 153-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith J. S. and Robinson N. J.(2002). Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis 186 Suppl 1: S3-28. [DOI] [PubMed] [Google Scholar]
- 31. Snijder B., Sacher R., Rämö P., Liberali P., Mench K., Wolfrum N., Burleigh L., Scott C. C., Verheije M. H., Mercer J., Moese S., Heger T., Theusner K., Jurgeit A., Lamparter D., Balistreri G., Schelhaas M., De Haan C. A., Marjomäki V., Hyypiä T., Rottier P. J., Sodeik B., Marsh M., Gruenberg J., Amara A., Greber U., Helenius A. and Pelkmans L.(2012). Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol 8: 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sodeik B., Ebersold M. W. and Helenius A.(1997). Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J Cell Biol 136(5): 1007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Su C., Zhan G. and Zheng C.(2016). Evasion of host antiviral innate immunity by HSV-1, an update. Virol J 13: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Theodoridis A. A., Eich C., Figdor C. G. and Steinkasserer A.(2011). Infection of dendritic cells with herpes simplex virus type 1 induces rapid degradation of CYTIP, thereby modulating adhesion and migration. Blood 118(1): 107-115. [DOI] [PubMed] [Google Scholar]
- 35. Thompson C. and Whitley R.(2011). Neonatal herpes simplex virus infections: where are we now? Adv Exp Med Biol 697: 221-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tognarelli E. I., Palomino T. F., Corrales N., Bueno S. M., Kalergis A. M. and González P. A.(2019). Herpes simplex virus evasion of early host antiviral responses. Front Cell Infect Microbiol 9: 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang G. P. and Bushman F. D.(2006). A statistical method for comparing viral growth curves. J Virol Methods 135(1): 118-123. [DOI] [PubMed] [Google Scholar]
- 38. Whitley R. J. and Griffiths P. D.(2002). Chap. 6: Herpesviruses: an introduction with a focus of herpes simplex virus. In: Boucher, C. A. B., Galasso, G. A., Katzenstein, D. A. and Copper D. A.(Eds.). Practical Guidelines in Antiviral Therapy. 1st edition. Amsterdam: Elsevier Science, 127-149. [Google Scholar]
- 39. Whitley R. J. and Roizman B.(2001). Herpes simplex virus infections. Lancet 357(9267): 1513-1518. [DOI] [PubMed] [Google Scholar]
- 40. Wolfstein A., Nagel C. H., Radtke K., Döhner K., Allan V. J. and Sodeik B.(2006). The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro . Traffic 7(2): 227-237. [DOI] [PubMed] [Google Scholar]
- 41. Zheng C.(2018). Evasion of cytosolic DNA-stimulated innate immune responses by herpes simplex virus 1. J Virol 92(6). [DOI] [PMC free article] [PubMed] [Google Scholar]