

Abstract
Recently, we published a large and versatile set of plasmids, the chromatic bacteria toolbox, to deliver eight different fluorescent protein genes and four combinations of antibiotic resistance genes to Gram-negative bacteria. Fluorescent tags are important tools for single-cell microbiology, synthetic community studies, biofilm, and host-microbe interaction studies. Using conjugation helper strain E. coli S17-1 as a donor, we show how plasmid conjugation can be used to deliver broad host range plasmids, Tn5 transposons delivery plasmids, and Tn7 transposon delivery plasmids into species belonging to the Proteobacteria. To that end, donor and recipient bacteria are grown under standard growth conditions before they are mixed and incubated under non-selective conditions. Then, transconjugants or exconjugant recipients are selected on selective media. Mutant colonies are screened using a combination of tools to ensure that the desired plasmids or transposons are present and that the colonies are not containing any surviving donors. Through conjugation, a wide range of Gram-negative bacteria can be modified without prior, often time-consuming, establishment of competent cell and electroporation procedures that need to be adjusted for every individual strain. The here presented protocol is not exclusive for the delivery of Chromatic bacteria plasmids and transposons, but can also be used to deliver other mobilizable plasmids to bacterial recipients.
Keywords: Fluorescent proteins, GFP, Plasmid, Tn5 transposon , Tn7 transposon , Conjugation, Mobilisable plasmids
Background
Fluorescent proteins have become an important tool for the study of bacteria, e.g., in biofilms, gene expression studies, or host-microbe interactions (Tomlin et al ., 2004; Monier and Lindow, 2005; Ma and Bryers, 2010; Remus-Emsermann et al., 2011; Schada von Borzyskowski et al., 2015; Remus-Emsermann et al., 2016a; Remus-Emsermann and Schlechter, 2018; Schlechter et al., 2018).
In a recently published study, we have described the construction of 96 plasmids, each different in fluorescent protein gene, antibiotic resistance marker combination, or plasmid backbone. The complete collection offers eight different fluorescent protein genes, each with its own unique excitation and emission spectrum, four different antibiotic resistance marker combinations, a broad host range plasmid, or delivery plasmids based on two different transposon systems, Tn5 and Tn7 (Schlechter et al., 2018).
The broad-host plasmid backbone and the two transposon systems have different advantages and disadvantages: 1) The plasmid usually exists in several copies in the recipient cell, which results in multiple gene copies and thereby often higher fluorescent signals. However, plasmids may be lost more easily, leading to subpopulations of cells that might lose their ability to express fluorescent proteins (Bahl et al., 2004). 2) The Tn5 transposon inserts into Proteobacteria with high efficiency and, as it integrates into the bacterial genome, the chance of losing the fluorescent protein tag is minimal. However, the Tn5 transposon randomly inserts DNA into the genome, thereby potentially disrupting genes that are important for bacterial fitness (de Lorenzo et al., 1990). 3) The Tn7 transposon integrates into the genome of the recipient bacterium in a site-specific manner, where no genes are disrupted and thereby should have minor impact on the bacterial fitness. However, the Tn7 transposon only inserts in a narrow range of bacteria, e.g., most γ-Proteobacteria, some α-Proteobacteria, and β-Proteobacteria. Outside of the γ-Proteobacteria, its insertion frequency may be low (McKenzie and Craig, 2006; Schlechter et al., 2018).
In this protocol, we describe the procedure to equip a bacterial strain with fluorescent labels using Pantoea eucalypti 299R (Remus- Emsermann et al., 2013 ; Tecon and Leveau, 2016) as an example recipient strain. P. eucalypti 299R is a competent recipient of all three different kinds of delivery systems, the broad host range plasmid and the two transposon systems. We could show that the broad-host plasmid as well as Tn5 and Tn7 transposon-based delivery of fluorescent proteins is feasible in this recipient strain (Schlechter et al., 2018). Other Enterobacteriaceae, such as E. coli, Erwinia amylovora and Pantoea vagans were also successfully tested with all three systems as well as Pseudomonas strains such as Pseudomonas citronellolis P3B5 (Remus-Emsermann et al., 2016b; Schlechter et al., 2018). Additionally, bacteria from other taxa, such as the α-Proteobacterial genera Sphingomonas and Methylobacterium were modified at least with the Tn5 transposon delivery system and in some cases with the broad-host-range plasmids and Tn7 transposons.
The here presented protocol is based on bacterial mating using the E. coli S17-1 helper strain, which is able to transfer mobilizable plasmids to recipients. Conjugation is a promiscuous process that crosses strain, genus, and phylum level and even works across domains of life (Heinemann and Sprague, 1989; Waters, 2001) . Transformation methods such as heat-shock and electroporation are often used when transferring genetic material into laboratory model hosts, however, newly isolated strains are usually recalcitrant to these methods and the establishment of transformation protocols is often tedious and time-consuming. By relying on conjugation, transformation can be avoided and can be as simple as scraping recipient bacterial colonies from an agar plate and mixing them with the donor bacterium.
Materials and Reagents
Autoclaved, untreated, wooden toothpicks (local supermarket of choice)
Pipette tips, 100-1,000 μl
Pipette tips, 1-200 μl
Pipette tips, 0.1-10 μl
1.5 ml Eppendorf tubes (Corning, Axygen®, catalog number: MCT-150-C)
0.2 ml PCR tubes (Merck, Corning®, catalog number: CLS6531-960EA)
Parafilm (Bemis, catalog number: PM996)
Petri dishes 90 mm diameter (Merck, catalog number: P5731-500EA)
Disposable cuvettes (Merck, catalog number: Z330418-1PAK)
Disposable loops (Citotest Labware Manufacturing Co., catalog number: 2121-0001)
Disposable spreaders (VWR, catalog number: 612-1560)
15 ml centrifuge tubes (Electron Microscopy Sciences, catalog number: 64760-01)
0.22 µm membrane filter (Merck, catalog number: GSTF02500)
Donor strain E. coli S17-1 equipped with plasmids listed in Table 1 (strains can be acquired from Addgene, https://www.addgene.org/browse/article/28196767/)
Recipient strain (e.g., Pantoea eucalypti 299R)
-
Plasmids (Table 1) encoding for fluorescent proteins (Table 2)
Source of all plasmids: Schlechter et al., 2018 , plasmids can be obtained at Addgene plasmid number 118484 to 118579. X is a placeholder for either 0, 1, 2, 3, 4, 5, 6, 7 representing mTagBFP2, mTurquoise2, sGFP2, sYFP2, mOrange2, mScarlet-I, mCardinal, or mClover3 encoding versions of the plasmids, respectively. KanR = Kanamycin resistance; CamR = Chloramphenicol resistance; GentR = Gentamicin resistance; TetR = Tetracycline resistance; AmpR = Ampicillin resistance.
Table 2. Fluorescent protein properties*.
Name Excitation peak Emission peak Emission color mTagBFP2 402 nm 457 nm blue mTurquoise2 435 nm 475 nm cyan sGFP2 495 nm 512 nm green mClover3 506 nm 518 nm green sYFP2 515 nm 527 nm yellow mOrange2 549 nm 565 nm orange mScarlet-I 569 nm 593 nm red mCardinal 604 nm 659 nm near infrared *A more detailed spectrum figure can be found in Schlechter et al., 2018
Lysogeny broth (Merck, catalog number: 110285)
Agar No. 1 (Oxoid, catalog number: LP0011)
Tris free base (Sigma, catalog number: T6791-500G)
Glacial acetic acid (Sigma, catalog number: 695092-500ML)
Glycerol (Sigma, catalog number: G6279-500ML)
Succinate (Sigma, catalog number: S9637-500G)
DMSO (Sigma, catalog number: D8418-100ML)
dNTPs (Thermo Fisher, catalog number: N8080260)
Multi-purpose HyAgaroseTM LE Agarose (HydraGene, catalog number: R9012LE-500g)
Phusion polymerase (Thermo Fisher, catalog number: F530S)
Oligonucleotides, synthesis amount 0.025 µmol, purified by desalting (see Table 3, Macrogen)
Nucleic acid stain RedSafe (Intron, catalog number: 21141)
Ampicillin (Duchefa Biochemie, catalog number: A0104)
Gentamicin (Duchefa Biochemie, catalog number: G0124)
Kanamycin (Duchefa Biochemie, catalog number: K0126)
Chloramphenicol (Duchefa Biochemie, catalog number: C0113)
Tetracycline (Duchefa Biochemie, catalog number: T0150)
Ethanol (Carl Roth, catalog number: T171.5)
K2HPO4 (Sigma, catalog number: 795496-100G)
NaH2PO4·2H2O (Sigma, catalog number: 71505-250G)
NH4Cl (Sigma, catalog number: A9434-500G)
MgSO4·7H2O (Sigma, catalog number: 63138-250G)
Na2EDTA·2H2O (Sigma, catalog number: E6635-500G)
ZnSO4·7H2O (Sigma, catalog number: Z0251-100G)
CoCl2·6H2O (Sigma, catalog number: C8661-25G)
MnCl2 (Sigma, catalog number: 244589-10G)
H3BO3 (Sigma, catalog number: 202878-10G)
CaCl2 (Sigma, catalog number: C5670-500G)
Na2MoO4·2H2O (Sigma, catalog number: 480967-25G)
FeSO4·7H2O (Sigma, catalog number: F8263-500G)
CuSO4·5H2O (Sigma, catalog number: 469130-50G)
NaCl (Sigma, catalog number: S7653-1KG)
KCl (Sigma, catalog number: P9333-500G)
Na2HPO4 (Sigma, catalog number: S7907-500G)
KH2PO4 (Sigma, catalog number: P5655-500G)
NaOH (Sigma, catalog number: S8045-500G)
Demineralized water
Lysogeny broth (LB) (see Recipes)
Lysogeny broth agar (LB agar) (see Recipes)
-
Minimal medium (see Recipes)
10x ammonium solution
10x phosphate buffer solution
10x sulfate solution
1,000x trace elements solution
20% succinate solution
3% agar
1x Phosphate buffered saline (1x PBS) (see Recipes)
-
Antibiotic stock solutions and working concentrations (see Recipes)
Ampicillin 100 mg ml-1 stock solution
Gentamicin 15 mg ml-1 stock solution
Kanamycin 50 mg ml-1 stock solution
Chloramphenicol 15 mg ml-1 stock solution
Tetracycline 5 mg ml-1 stock solution
-
TAE buffer (see Recipes)
0.5 M EDTA stock solution
50x TAE stock solution
1x TAE working solution
Table 1. Plasmids.
| Plasmid Name | Notable features |
| pMRE13X | KanR, CamR |
| pMRE14X | KanR, CamR, GentR |
| pMRE15X | KanR, CamR, KanR |
| pMRE16X | KanR, CamR, TetR |
| pMRE-Tn7-13X | Tn7 transposon, AmpR, CamR |
| pMRE-Tn7-14X | Tn7 transposon, AmpR, CamR, GentR |
| pMRE-Tn7-15X | Tn7 transposon, AmpR, CamR, KanR |
| pMRE-Tn7-16X | Tn7 transposon, AmpR, CamR, TetR |
| pMRE-Tn5-13X | Tn5 transposon, AmpR, CamR |
| pMRE-Tn5-14X | Tn5 transposon, AmpR, CamR, GentR |
| pMRE-Tn5-15X | Tn5 transposon, AmpR, CamR, KanR |
| pMRE-Tn5-16X | Tn5 transposon, AmpR, CamR, TetR |
Table 3. Primers.
| Name | Target | Sequence 5' to 3' * | tm (°C)# |
| FWD Tn5/7_gt | Tn5 and Tn7 mutant genotyping | ATGGTGAGCAAGGGCGAG | 58 |
| REV_Tn5/7_gt | Tn5 and Tn7 mutant genotyping | CAACAGGAGTCCAAGCTCAG | 56 |
| FWD_Tn5_gt | Tn5 mutant genotyping | CTGAGTAGGACAAATCCGCCG | 58 |
| REV_Tn5_gt | Tn5 mutant genotyping | GCCTCGGCAGAAACGTTGG | 60 |
| Tn7_gt1 | Tn7 mutant genotyping (backbone) | GAATTACAACAGTACTGCGATGAG | 55 |
| Tn7_gt2 | Tn7 mutant genotyping (backbone) | GATCAACTCTATTTCTCGCGGG | 56 |
| Tn7_gt3 | Tn7 mutant genotyping (backbone) | TACATAACGGACTAAGAAAAACACTACAC | 64 |
| FWD_uidA | uidA gene | AACAGGTGGTTGCAACTGGA | 60 |
| REV_uidA | uidA gene | TTGCTGAGTTTCCCCGTTGA | 60 |
Equipment
Forceps
Pipettes (Eppendorf, P1000, catalog number: 3120000062; P200, catalog number: 3120000054; P10, catalog number: 3120000020)
Erlenmeyer flasks 125 ml (e.g., Merck, catalog number: CLS4980125-12EA)
Automatic micropipettes (Eppendorf or Gilson)
Metal tweezers (VWR, catalog number: 232-0092)
Bunsen burner (Eisco, catalog number: CH0087A)
Vortex (Scientific Industries, catalog number: SI-0236)
Autoclave (Heidolph, catalog number: 023210745CA)
-20 °C Freezer (Fisher and Paykel, catalog number: E388LXFD1)
Sterile bench (Labconco, catalog number: 302389100)
Microcentrifuge for 1.5 ml centrifuge tubes with max speed > 6,000 × g (Eppendorf, catalog number: 5418000017)
Centrifuge for 15 ml centrifuge tubes with max speed > 6,000 × g (Eppendorf, catalog number: 5805000327)
Shaking incubators 30 °C and 37 °C (Infors, catalog number: Multitron Pro)
Incubators 30 °C and 37 °C (Binder, catalog number: BD 56)
Spectrophotometer (Biochrom WPA CO8000 Cell Density meter, catalog number: 80-3000-45)
PCR thermocycler (Eppendorf, catalog number: 6311000010)
Horizontal agarose gel electrophoresis setup including running chamber and power supply (VWR International, catalog number: 76196-448)
Blue light or UV transilluminator (optional: camera for documentation) (Fisher Scientific, catalog number: UV95042001)
Fluorescent microscope with appropriate filter sets for the excitation and emission spectra of the fluorescent protein tags and equipped with a 5x objective with a long working distance (Zeiss Axio Imager.M1 equipped with Objective A-Plan 5x/0.12 M27 [Zeiss, catalog number: 421030-9900-000])
Procedure
An overview of the workflow is shown in Figure 1.
Figure 1. Flowsheet of protocol.

Prepare or have ready media and stock solutions for the conjugation
Prepare 2 lysogeny broth (LB) agar plates without antibiotics.
Prepare 1 LB plate containing kanamycin when working with a pMRE1XX plasmid or 1 LB plate containing ampicillin and chloramphenicol when working with transposon bearing plasmids (Ampicillin will select for the plasmid and chloramphenicol for the transposon).
Prepare 3 ml and 50 ml LB broth without antibiotics.
Prepare 3 ml and 50 ml LB broth containing kanamycin when working with a pMRE1XX plasmid or 3 ml and 50 ml LB broth containing ampicillin and chloramphenicol when working with transposon bearing plasmids.
-
Prepare ~10 minimal media plates containing succinate as the sole carbon source and antibiotics selecting for the plasmids or transposons as explained in Table 1. In case you are selecting for transposon insertions, do not include ampicillin, since it selects for the plasmid, not the transposon. Store at 4 °C and in the dark if the light-sensitive tetracycline is used in the plates.
Note: Select a carbon source for minimal media plates that can be metabolized/utilized by the recipient strain.
Prepare 100 ml sterile 1x PBS.
Day 1
-
Streak donor strain E. coli S17-1 containing pMRE1XX, pMRE-Tn5-1XX, or pMRE-Tn7-1XX on LB agar plates containing appropriate antibiotics from a glycerol stock using a disposable loop and incubate overnight at 37 °C or 30 °C when working with pMRE-Tn7-1XX plasmids.
Note: pMRE-Tn7-1XX plasmids do not replicate above 32 °C. Thus, to maintain it in the bacterial culture, it must be grown at lower temperatures.
Streak recipient strain P. eucalypti 299R on an LB agar plate from a glycerol stock using a disposable loop and incubate overnight at 30 °C.
Day 2
Inoculate 3 ml LB broth containing the same antibiotics as before with an individual colony of donor E. coli S17-1 containing pMRE1XX, pMRE-Tn5-1XX, or pMRE-Tn7-1XX using a sterile toothpick and ethanol flamed forceps. Incubate overnight at 200 rounds per minute (rpm) and 37 °C or 30 °C when working with pMRE-Tn7-1XX plasmids.
Inoculate 3 ml LB broth without antibiotics with an individual colony of P. eucalypti 299R using a sterile toothpick and ethanol flamed forceps. Incubate overnight at 30 °C and 200 rpm.
Day 3
-
Inoculate 50 ml LB broth in a 125 ml Erlenmeyer flask containing the same antibiotics as before with 500 µl overnight culture E. coli S17-1 containing pMRE1XX, pMRE-Tn5-1XX, or pMRE-Tn7-1XX using a sterile pipette tip and a P1000 pipette. Incubate at 200 rpm and 37 °C or 30 °C when working with pMRE-Tn7-1XX plasmids. Grow to an optical density at 600 nm (OD600nm) of ~0.5, which will take between 1.5 h and 3 h depending on the incubation temperature.
Note: Measure OD600nm regularly using disposable cuvettes and a spectrophotometer and note down the final OD600nm.
-
Inoculate 50 ml LB broth in a 125 ml Erlenmeyer flask containing no antibiotics with 500 µl overnight culture P. eucalypti 299R using a sterile pipette tip and a P1000 pipette. Incubate at 200 rpm and 30 °C. Grow until P. eucalypti 299R has reached late exponential/early stationary phase, which will take ~3-4 h (OD600nm of 0.7 or above). Measure OD600nm using disposable cuvettes and a spectrophotometer. Alternatively, recipient strain P. eucalypti 299R can be scraped off directly from the plate using a sterile disposable loop and resuspended in a 1.5 ml microcentrifuge tube containing 1 ml sterile 1x PBS by gentle vortexing.
Note: P. eucalypti 299R is a fast growing strain with a similar growth rate to E. coli S17-1. For others, such as slow-growing strains, the inoculation volume or time of inoculation may vary accordingly. For other strains, growth rate, exponential growth phase and early/late stationary phase will have to be determined.
-
Harvest 10 ml of both cultures by centrifugation at 2,000 × g for 5 min in a centrifuge. Discard the supernatants, resuspend each culture in 5 ml sterile 1x PBS by gentle vortexing and measure OD600nm.
Note: Vortex gently for less than 5 s to avoid shearing of the donor which could impact conjugation efficiency. Mixing by inversion is also recommended.
-
Mix the cultures to reach similar amounts of donor and recipient cells.
Example: If the 10 ml E. coli S17-1 culture had an OD600nm of 0.5, it should be concentrated to OD600nm of 1. If the 10 ml P. eucalypti 299R culture had an OD600nm of 0.4, it should be concentrated to OD600nm of 0.8. The P. eucalypti 299R culture, therefore, contains only 80% of the cells that the E. coli S17-1 culture contains. Therefore, mix the 5 ml P. eucalypti 299R culture with 4 ml (80% of 5 ml) E. coli S17-1 culture.
Note: For other strains, the number of CFU per OD600nm will have to be determined beforehand.
-
Harvest the combined cultures by centrifugation at 2,000 × g for 5 min in a centrifuge, discard the supernatant and resuspend in 100 µl sterile 1x PBS using a vortex.
Note: Vortex gently for less than 5 s to avoid shearing of donor pili which could impact conjugation efficiency. Mixing by inversion is also recommended.
Dry an LB agar plate containing no antibiotics in a sterile bench for 15 min. In the sterile bench, dropspot the resuspended culture mix onto the agar using a sterile pipette tip and a 200 µl pipette. Keep the plate open and wait until all the liquid evaporates. Afterwards, close the plate and incubate overnight at 30 °C.
Day 4
Harvest the culture drop using a wire loop and transfer to a 1.5 ml microcentrifuge tube containing 1 ml sterile 1x PBS. Resuspend the bacteria using a vortex. Plate 10 µl, 100 µl and the rest of the bacterial suspensions on the minimal media plates containing antibiotics selecting for the used plasmid or transposon using a disposable or flame-sterilized spreader. Minimal media will select against the auxotrophic E. coli donor.
Incubate at 30 °C until visible colonies appear on the plates.
Days 5-8
Note: Slow growing strains may appear weeks after the conjugation.
-
Check morphology and fluorescence emission in emerging colonies. Fluorescence can often be seen using a transilluminator (e.g., UV or Blue light) that is also used for agarose gel electrophoresis (e.g., for most green, yellow, orange, and red emitting proteins see Figure 2). Otherwise, and additionally, it is useful to investigate emerging colonies with a fluorescent microscope equipped with a very low magnification and long working distance objectives. Use filters that have appropriate excitation and emission filters. Often colonies are made up of both, the donor and the recipient strain, for an example see Figure 3A. Select colonies that appear homogeneously fluorescent under the fluorescent microscope and streak them on minimal medium (as above) to individual colonies (Figure 3B).
Note: A fun and beautiful example of a forced consortium. The recipient strain provides nutrients for the otherwise auxotroph E. coli and E. coli is providing antibiotic resistance to the recipient strain.
Restreak the colonies at least three times using a disposable loop in the subsequent days on minimal media plates.
Figure 2. Bacterial colonies expressing fluorescent proteins.

A and B. Bacteria were streaked as crude streaks on agar plates. Expressed fluorescent proteins were visualized on a blue light gel reader. Green, yellow, orange, red and far-red fluorescent colonies were easily detectable on gel readers, blue and cyan fluorescent colonies were difficult to distinguish from non-fluorescent wild type colonies. C. Expressed fluorescent proteins were visualized using a UV light tray with attached gel documentation system. Fluorescent protein expressing colonies appear brighter than the wild type.
Figure 3. Overlay of fluorescent and brightfield microscope channels.

A. Example of unsuccessful conjugation that yielded colonies. The colonies are mixes of a Sphingomonas species and red fluorescent protein expressing E. coli S17-1. B. Clonal colony of P. eucalypti 299R::MRE-Tn7-145. Scale bars = 500 µm.
Day 9, or after subsequent subcultivations
At this stage, you will have to follow different procedures, depending on the delivery systems you choose:
pMRE1XX plasmids
As final check, perform PCRs to test for E. coli contamination. For that, perform a crude DNA extraction from a freshly grown individual colony by picking a part of the colony with an autoclaved toothpick and dislodging the cells in a 0.2 ml PCR tube filled with 100 µl 0.05 M NaOH.
-
Boil the sample for 15 min at 95 °C in a PCR machine. Use 1 µl of crude DNA extract as template for a PCR using Phusion polymerase and primers FWD_uidA and REV_uidA following the recommendations of the manufacturer (for an example refer to Figure 4).
Note: Presence of E. coli will yield a ~500 bp PCR product. To check for the presence of uidA in a bacterial strain, the sequence of the E. coli gene (https://www.ncbi.nlm.nih.gov/gene/946149) can be blasted against the genome of the target strain.
Figure 4. Colony PCR of uidA for detection of Escherichia coli.
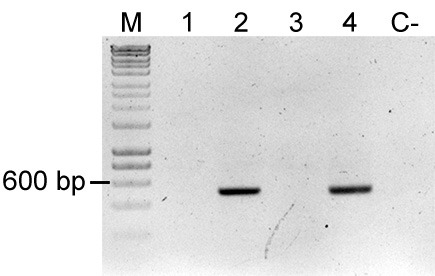
uidA is an E. coli and Shigella sp. specific gene, thereby amplification of this gene fragment indicates mixed colony or false positive colony after restreak and colony isolation. Primers used were FWD_uidA and REV_uidA (Table 3) and yield a 500 bp product. Lane M: Molecular marker (HyperLadderTM, Bioline); Sample 1: P. eucalypti 299R::MRE-Tn5-145; Sample 2: E. coli (pMRE-Tn5-145); Sample 3: P. eucalypti 299R::MRE-Tn7-145; Sample 4: E. coli (pMRE-Tn7-145); C-: water control. In this example, samples #1 and #3 showed no detection of E. coli, while sample #2 and #4 showed presence of uidA.
pMRE-Tn5-1XX–Tn5 transposon delivery plasmids
Perform the E. coli contamination check as described above for pMRE1XX plasmids.
Additionally, use primers FWD_Tn5_gt, REV_Tn5_gt, FWD_Tn5/7_gt, and REV_Tn5/7_gt in a multiplex PCR. The primer combination tests of the presence of the delivery plasmid and for the presence of the fluorescent protein coding sequence in the sample (for an example refer to Figure 5). Samples containing the plasmid will yield two bands (~700 bp and ~400 bp), which suggests E. coli contamination. Samples containing only the fluorescent protein coding sequence yield only the larger band (~700 bp).
Figure 5. Colony PCR to confirm Tn5 insertions.
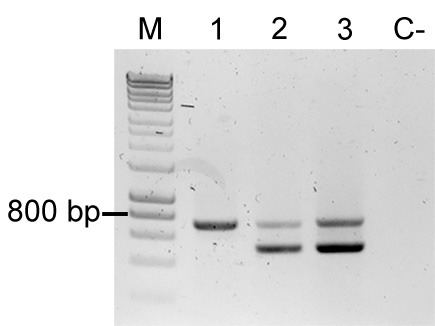
Multiplex PCR result using primers FWD_Tn5_gt, REV_Tn5_gt, FWD_Tn5/7_gt, and REV_Tn5/7_gt. M: Molecular marker (HyperLadder, Bioline); Sample 1: P. eucalypti 299R::MRE-Tn5-145; Sample 2: E. coli (pMRE-Tn5-145); Sample 3: pMRE-Tn5-145; C-: water control.
pMRE-Tn7-1XX–Tn7 transposon delivery plasmids
-
Curing of plasmid (depending on the recipient strain)
pMRE-Tn7-1XX plasmid series are conditional suicide plasmids in many, but not all, Enterobacteriaceae. P. eucalypti belongs to the Enterobacteriaceae, i.e., the plasmid is able to replicate in P. eucalypti since the plasmids origin of replication pSC101 is functional in the strain. pSC101 is however heat instable and does not replicate at temperatures above 32 °C (McKenzie and Craig, 2006). Therefore, to cure the strain from the plasmid, it has to be grown at high temperatures.
Inoculate a 5 ml LB broth culture using an individual fluorescent colony and grow it overnight at 37 °C.
Streak out the culture on an LB agar plate using an inoculation loop and incubate overnight at 37 °C.
Select 10 individual colonies and streak them each onto a new LB agar plate containing Amp and on an LB agar plate containing no antibiotic. Colonies that still contain the plasmid will be able to grow on ampicillin, while colonies where the transposition occurred will only grow on the LB agar plate without antibiotics.
Perform the E. coli contamination check as described above for pMRE1XX plasmids.
Additionally, perform a PCR to identify the presence of Tn7 insertion and/or Tn7-derived plasmid. Use FWD_Tn5/7_gt and REV_Tn5/7_g for the presence of the fluorescent protein coding sequence in the sample (~700 bp) and primers Tn7_gt1, Tn7_gt2, and Tn7_gt3 for the presence of the delivery plasmid (~1,000 bp). Tn7_gt1, Tn7_gt2, and Tn7_gt3 are needed because pMRE-Tn7-1XX plasmids were constructed by blunt-end cloning, so the insert orientation may vary between plasmids (Tn7_gt2, and Tn7_gt3 anneal to a flanking sequence of the insert in the plasmid backbone). For an example refer to Figure 6.
Figure 6. Colony PCRs to confirm Tn7 insertions.
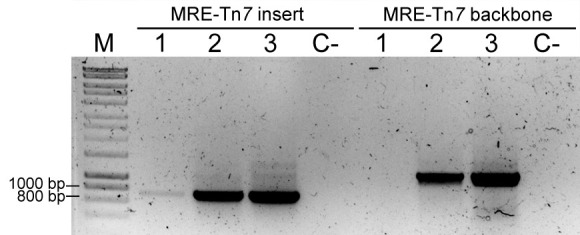
PCR result using primers FWD_Tn5/7_gt and REV_Tn5/7_gt for the amplification of Tn7-delivered insert (MRE-Tn7 insert), and PCR result using primers Tn7_gt1, Tn7_gt2, and Tn7_gt3 for amplification of a fragment of plasmid pMRE-Tn7 backbone (MRE-Tn7 backbone). M: Molecular marker. Sample 1: P. eucalypti 299R::MRE-Tn7-145; Lane 2: E. coli (pMRE-Tn7-145); Lane 3: pMRE-Tn7-145; C-: water control.
Notes
Depending on the recipient strain, addition of 100 µl drop spotted 1 M CaCl2 onto the agar plate used to drop spot the bacterial mixture onto the CaCl2 might increase conjugation efficiencies (Wang and Jin, 2014; McCully et al., 2018).
Conjugation efficiency can be optimized by mixing donor and recipient strains in different ratios (e.g., perform three conjugations by using a 1:3, 1:1, and 3:1 ratio of donor and recipient strain, respectively).
Recipes
-
Lysogeny broth (LB)
Add 25 g of lysogeny broth medium to 1 L of demineralized water
Autoclave
-
Lysogeny broth agar (LB agar)
Add 25 g of lysogeny broth medium and 15 g agar no. 1 to 1 L of demineralized water
Autoclave
-
Minimal medium (Attwood and Harder, 1972)
-
Prepare the following stock solutions:
-
10x ammonium solution
Dissolve 16.2 g of NH4Cl in 1 L of demineralized water
Autoclave
-
10x phosphate buffer solution
Dissolve 15.9 g of K2HPO4 and 18 g of NaH2PO4·2H2O in 1 L of demineralized water (pH should be 6.7 without adjustment)
Autoclave
-
10x sulfate solution
Dissolve 2 g of MgSO4 in 1 L of demineralized water
Filter sterilize using a 0.22 µm filter to avoid precipitation
-
1,000x trace elements solution (Harder and Veldkamp, 1968)
Dissolve 15 g Na2EDTA2·2H2O, 4.5 g ZnSO4·7H2O, 3 g CoCl2·6H2O, 0.6 g MnCl2, 1 g H3BO3, 3 g CaCl2, 0.4 g Na2MoO4·2H2O, 3 g FeSO4·7H2O, and 0.3 g CuSO4·5H2O in 1 L of demineralized water
Filter sterilize using a 0.22 µm filter
-
20% succinate solution
Dissolve 20 g succinate in 100 ml of demineralized water
Filter sterilize using a 0.22 µm filter
Note: P. eucalypti is able to grow on succinate as the sole carbon source, other strains may require other carbon source supplements to grow on minimal medium.
-
3% agar
Add 15 g Agar No. 1 in 500 ml of demineralized water
Autoclave
Note: 3% agar should be at a temperature below 60 °C, since higher temperatures might lead to salts precipitation. This also prevents boiling retardation. Pour quickly after thoroughly mixing the agar medium.
-
To prepare 1 L of minimal medium agar supplemented with 0.5% succinate as sole carbon source, combine 100 ml of 10x ammonium solution with 100 ml 10x phosphate buffer solution, 100 ml 10x sulphate solution, 1 ml of 1,000x trace element solution and 25 ml 20% succinate solution into 500 ml of molten 3% agar. Top up with sterile demineralized water to a final volume of 1 L
-
-
1x Phosphate buffered saline (1x PBS)
Dissolve 8 g NaCl, 0.24 g KCl, 1.42 g Na2HPO4, 0.24 g KH2PO4 in 1 L of demineralized water and autoclave
The buffer can alternatively be prepared as a 10x concentrated stock which is diluted to a 1x concentration with sterile water prior use
-
Antibiotic stock solutions and working concentrations
Antibiotic Stock concentration Final concentration Ampicillin 100 mg ml-1 100 µg ml-1 Gentamicin 15 mg ml-1 15 µg ml-1 Kanamycin 50 mg ml-1 50 µg ml-1 Chloramphenicol 15 mg ml-1 15 µg ml-1 Tetracycline 5 mg ml-1 15 µg ml-1 -
Ampicillin stock solution
Dissolve ampicillin in water to a concentration of 100 mg ml-1 and filter sterilized using a 0.22 µm filter
-
Gentamicin stock solution
Dissolve gentamicin in water to a concentration of 15 mg ml-1 and filter sterilized using a 0.22 µm filter
-
Kanamycin stock solution
Dissolve kanamycin in water to a concentration of 50 mg ml-1 and filter sterilized using a 0.22 µm filter
-
Chloramphenicol stock solution
Dissolve chloramphenicol in ethanol to a concentration of 15 mg ml-1
-
Tetracycline stock solution
Dissolve tetracycline in ethanol to a concentration of 5 mg L-1
Note: Store all antibiotic stock solutions in 1.5 ml aliquots at -20 °C.
-
-
TAE buffer (40 mM Tris, 20 mM acetic acid, 1 mM EDTA)
-
0.5 M EDTA stock solution
Dissolve 93.05 g Na2EDTA·2H2O in 400 ml of demineralized water and adjust the pH to 8.0 with NaOH
Top up the solution to a final volume of 500 ml
Note: EDTA will start dissolving when pH gets close to 8, use 10 M NaOH to adjust the pH.
-
50x TAE stock solution
Dissolve 242 g Tris base in 750 ml of demineralized water
Add 57.1 ml of 100% glacial acetic acid and 100 ml of 0.5 M EDTA solution
Adjust the volume to 1 L with demineralized water
This solution can be stored at room temperature
-
1x TAE working solution
Dilute 20 ml of 50x TAE stock solution in 980 ml of demineralized water
-
Acknowledgments
The protocol presented here was adapted from two previously published studies (Remus- Emsermann et al., 2016a ; Schlechter et al., 2018 ) and majorly influenced by a study by McKenzie and Craig (2006). The authors thank Michał Bernach, Hyunwoo Jun and the other authors of Schlechter et al., 2018 for their help during the original study.
This work was funded by a seed grant of the Biomolecular Interaction Centre of the University of Canterbury to MR-E and the Royal Society of New Zealand Marsden Fast Start grant (UOC1704) to MR-E. RS was supported by an NZIDRS doctoral scholarship.
Competing interests
The authors declare no competing interests.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1. Bahl M. I., Sorensen S. J. and Hestbjerg Hansen L.(2004). Quantification of plasmid loss in Escherichia coli cells by use of flow cytometry. FEMS Microbiol Lett 232(1): 45-49. [DOI] [PubMed] [Google Scholar]
- 2. de Lorenzo V., Herrero M., Jakubzik U. and Timmis K. N.(1990). Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J Bacteriol 172(11): 6568-6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Attwood M. M. and Harder W., (1972). A rapid and specific enrichment procedure for Hyphomicrobium spp. Antonie van Leeuwenhoek 38: 369–377.. [DOI] [PubMed] [Google Scholar]
- 4. Harder W. and Veldkamp H.(1968). Physiology of an obligately psychrophilic marine Pseudomonas species. J Appl Microbiol 31: 12-23. [Google Scholar]
- 5. Heinemann J. A. and Sprague G. F. Jr, (1989). Bacterial conjugative plasmids mobilize DNA transfer between bacteria and yeast. Nature 340(6230): 205-209. [DOI] [PubMed] [Google Scholar]
- 6. Ma H. and Bryers J. D.(2010). Non-invasive method to quantify local bacterial concentrations in a mixed culture biofilm. J Ind Microbiol Biotechnol 37(10): 1081-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCully L. M., Bitzer A. S., Seaton S. C., Smith L. M. and Silby M. W.(2018). Social Motility: Interaction between two sessile soil bacteria leads to emergence of surface motility. bioRxiv: 296814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McKenzie G. J. and Craig N. L.(2006). Fast, easy and efficient: site-specific insertion of transgenes into enterobacterial chromosomes using Tn7 without need for selection of the insertion event. BMC Microbiol 6: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Monier J. M. and Lindow S. E.(2005). Spatial organization of dual-species bacterial aggregates on leaf surfaces. Appl Environ Microbiol 71(9): 5484-5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Remus-Emsermann M. N. P., de Oliveira S., Schreiber L. and Leveau J. H.(2011). Quantification of lateral heterogeneity in carbohydrate permeability of isolated plant leaf cuticles. Front Microbiol 2: 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Remus-Emsermann M. N. P., Gisler P. and Drissner D.(2016). MiniTn7-transposon delivery vectors for inducible or constitutive fluorescent protein expression in Enterobacteriaceae. FEMS Microbiol Lett 363(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Remus-Emsermann M. N. P., Kim E. B., Marco M. L., Tecon R. and Leveau J. H.(2013). Draft genome sequence of the phyllosphere model bacterium Pantoea agglomerans 299R. Genome Announc 1(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Remus-Emsermann M. N. P., Schmid M., Gekenidis M. T., Pelludat C., Frey J. E., Ahrens C. H. and Drissner D.(2016). Complete genome sequence of Pseudomonas citronellolis P3B5, a candidate for microbial phyllo-remediation of hydrocarbon-contaminated sites. Stand Genomic Sci 11: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Remus-Emsermann M. N. P. and Schlechter R. O.(2018). Phyllosphere microbiology: at the interface between microbial individuals and the plant host. New Phytol 218(4): 1327-1333. [DOI] [PubMed] [Google Scholar]
- 15. Schada von Borzyskowski L., Remus-Emsermann M., Weishaupt R., Vorholt J. A. and Erb T. J.(2015). A set of versatile brick vectors and promoters for the assembly, expression, and integration of synthetic operons in Methylobacterium extorquens AM1 and other alphaproteobacteria. ACS Synth Biol 4(4): 430-443. [DOI] [PubMed] [Google Scholar]
- 16. Schlechter R. O., Jun H., Bernach M., Oso S., Boyd E. F., Muñoz-Lintz D. A., Dobson R. C., Remus D. M. and Remus-Emsermann M.(2018). Chromatic Bacteria- A broad host-range plasmid and chromosomal insertion toolbox for fluorescent protein expression in bacteria. bioRxiv: 402172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tecon R. and Leveau J. H.(2016). Symplasmata are a clonal, conditional, and reversible type of bacterial multicellularity. Sci Rep 6: 31914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tomlin K. L., Clark S. R. and Ceri H.(2004). Green and red fluorescent protein vectors for use in biofilm studies of the intrinsically resistant Burkholderia cepacia complex. J Microbiol Methods 57(1): 95-106. [DOI] [PubMed] [Google Scholar]
- 19. Wang X. K. and Jin J. L.(2014). Crucial factor for increasing the conjugation frequency in Streptomyces netropsis SD-07 and other strains. FEMS Microbiol Lett 357(1): 99-103. [DOI] [PubMed] [Google Scholar]
- 20. Waters V. L.(2001). Conjugation between bacterial and mammalian cells. Nat Genet 29(4):375-6. [DOI] [PubMed] [Google Scholar]