

Abstract
We evaluated the administration of ARI-0001 cells (chimeric antigen receptor T cells targeting CD19) in adult and pediatric patients with relapsed/refractory CD19+ malignancies. Patients received cyclophosphamide and fludarabine followed by ARI-0001 cells at a dose of 0.4–5 × 106 ARI-0001 cells/kg, initially as a single dose and later split into 3 fractions (10%, 30%, and 60%) with full administration depending on the absence of cytokine release syndrome (CRS). 58 patients were included, of which 47 received therapy: 38 with acute lymphoblastic leukemia (ALL), 8 with non-Hodgkin’s lymphoma, and 1 with chronic lymphocytic leukemia. In patients with ALL, grade ≥3 CRS was observed in 13.2% (26.7% before versus 4.3% after the amendment), grade ≥3 neurotoxicity was observed in 2.6%, and the procedure-related mortality was 7.9% at day +100, with no procedure-related deaths after the amendment. The measurable residual disease-negative complete response rate was 71.1% at day +100. Progression-free survival was 47% (95% IC 27%–67%) at 1 year: 51.3% before versus 39.5% after the amendment. Overall survival was 68.6% (95% IC 49.2%–88%) at 1 year. In conclusion, the administration of ARI-0001 cells provided safety and efficacy results that are comparable with other academic or commercially available products. This trial was registered as ClinicalTrials.gov: NCT03144583.
Keywords: CART-cells, ARI-0001, CD19, A3B1, ALL, NHL
Graphical Abstract
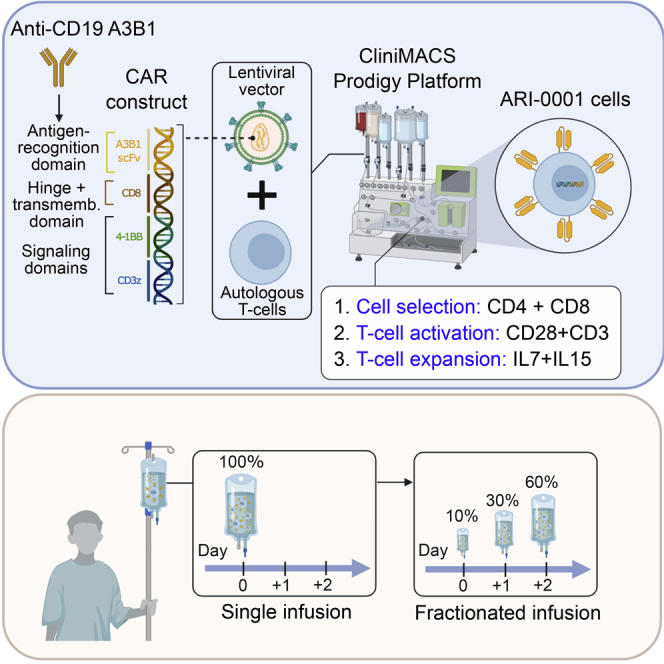
Ortiz-Maldonado and colleagues evaluated the administration of ARI-0001 cells (chimeric antigen receptor T cells incorporating A3B1, a novel anti-CD19), in 47 adult and pediatric patients with relapsed/refractory CD19+ malignancies. The efficacy was comparable with other academic or commercially available products, while safety was improved with the fractionated administration of cells.
Introduction
Despite currently available therapies, most patients with relapsed/refractory (R/R) B cell malignancies such as acute lymphoblastic leukemia (ALL) and diffuse large B cell lymphoma (DLBCL) remain incurable. R/R ALL is associated with a complete response rate (CRR) around 30%–45% and a median overall survival (OS) around 4–8 months, depending on age, type of salvage therapy, and response to it.1, 2, 3, 4 The prognosis is particularly poor for patients relapsing after allogeneic hematopoietic cell transplantation (alloHCT).5,6 Novel agents such as inotuzumab ozogamicin (IO) or blinatumomab have increased the CR rate in this patient population, but responses are generally not durable.7,8 Outcomes are also poor in patients with R/R DLBCL, defined as no response to the last line of chemotherapy or relapse within 1 year of autologous hematopoietic cell transplant (autoHCT). A meta-analysis of patients with refractory DLBCL found that these patients achieve an overall response rate of 26% and median OS of 6.6 months.9
Adoptive cell therapy with T cells genetically engineered to express chimeric antigen receptors (CAR) targeting CD19 is a well-established approach for the treatment of B cell malignancies.10 These CAR constructs usually comprise an extracellular single-chain variable fragment (scFv) of a monoclonal antibody (mAb) attached to a transmembrane domain and two or more signaling domains. Two of these cell products (tisagenlecleucel and axicabtagene ciloleucel) were recently approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the treatment of R/R ALL and DLBCL. Both products share the same scFv (derived from the FMC63 mAb) but have a different composition, including a different co-stimulatory domain and transduction vector, and yet both have remarkable clinical efficacy and a comparable safety profile.11, 12, 13
In 2013, we started developing our own CAR19 construct. We identified the scFv sequence from our own CD19-A3B1 hybridoma, incorporated a transmembrane CD8 domain, a 4-1BB costimulatory domain, and a CD3z signaling domain next to it, cloned the sequence into a 3rd generation lentiviral vector, and transduced healthy-donor T cells.14 Once the preclinical tests were completed, we scaled up both the lentiviral and cell production, this leading to the Spanish Agency for Medicines and Health Products (AEMPS) approval of our investigational new drug, called ARI-0001 cells (IMP nº 16-187), and also this clinical trial (CART19-BE-01) in May 2017. Here we present the results of this clinical trial.
Results
Baseline Characteristics
58 patients were enrolled in the study, but 4 of them never made it through the screening phase: 2 patients did not comply with inclusion/exclusion criteria and 2 were eventually referred for therapy with commercial products that became available at the time (Figure 1). Out of the remaining 54 patients, 47 received therapy with ARI-0001 cells, 19 in cohorts 1 and 2 (single dose infusion) and 28 in cohort 3 (fractionated infusion). These 47 patients (modified full analysis set [mFAS]) were diagnosed with ALL (38), DLBCL (4, one of them a Richter’s transformation from chronic lymphocytic leukemia [CLL]), primary mediastinal B cell lymphoma,2 follicular lymphoma (FL),2 and CLL1. Baseline characteristics of patients included in the mFAS are shown in Table 1. Median age was 26 years (range, 3–67), and 17 patients (36%) were female. The full details of the only CLL patient were published elsewhere.15 The data cutoff date was November 5, 2019, when all infused patients had a minimum follow-up of 100 days or had experienced disease relapse or death. At that time, the median follow-up for survivors was 5.5 months (range, 1.9–23.6) from ARI-0001 cell infusion.
Figure 1.

Consort Diagram of Patients Included in the CART19-BE-01 Trial
Table 1.
Patients’ Demographics and Baseline Characteristics
| Characteristic | Cohorts 1 and 2 (Single Infusion) | Cohort 3 (Fractionated Infusion) | All Patients |
|---|---|---|---|
|
Acute lymphoblastic leukemia |
n = 15 | n = 23 | n = 38 |
| Age (y), median (range) | 20 (3–35) | 29 (4–67) | 24.5 (3–67) |
| Female sex, n (%) | 4 (27) | 10 (44) | 14 (37) |
| ECOG performance status ≥ 1, n (%) | 4 (31) | 5 (22) | 9 (25) |
| Prior regimens, median (range) | 4 (3-10) | 4 (2–8) | 4 (2–10) |
| Prior inotuzumab, n (%) | 4 (27) | 9 (39) | 13 (34) |
| Prior blinatumomab, n (%) | 3 (20) | 6 (26) | 9 (24) |
| Prior allogeneic HCT, n (%) | 11 (73) | 22 (96) | 33 (87) |
| Bone marrow blast cell count ≥ 5%, n (%) | 10 (67) | 7 (30) | 17 (45) |
|
Non-Hodgkin’s lymphoma |
n = 3 | n = 5 | n = 8 |
| Histology: | |||
| • Diffuse large B cell lymphoma | 1 (33.3) | 3 (60) | 4 (50) |
| • Primary mediastinal B cell lymphoma | 2 (66.6) | 0 | 2 (25) |
| • Follicular lymphoma | 0 (0) | 2 (40) | 2 (25) |
| Age (y), median (range) | 25 (19–51) | 50 (22–62) | 47.5 (19–62) |
| Female sex, n (%) | 1 (33) | 2 (40) | 3 (37.5) |
| ECOG performance status ≥ 1, n (%) | 1 (33) | 3 (60) | 4 (50) |
| Prior regimens, median (range) | 6 (5–7) | 6 (4–9) | 6 (4–9) |
| Prior allogeneic/autologous HCT, n (%) | 3 (100) | 1 (20) | 4 (50) |
HCT, hematopoietic cell transplantation; NA, not applicable/available.
Out of 54 patients who proceeded to apheresis, 47 (87%) and 7 (13%) required one and two procedures, respectively. Reasons for a second procedure were bacterial contamination of the cell product5 and insufficient viral transduction.2 All infused patients received fludarabine + cyclophosphamide lymphodepletion and received ARI-0001 cells a median of 54 days (range, 34–215) after study inclusion. The median vein-to-vein time was 42 days (range, 25–190) for the entire population. The original target dose ranged from 0.5 to 5 × 106 ARI-0001 cells/kg, with the condition imposed by the AEMPS that the first patient had to receive the minimum dose (0.5 × 106 ARI-0001 cells/kg). In cohort 3, one patient received 0.4 × 106 ARI-0001 cells/kg (i.e., the last fraction was omitted) due to cytokine release syndrome (CRS).
Toxicity
All adverse events (AEs) occurring from study inclusion, even before ARI-0001 cell infusion, were graded and reported (Tables S1 and S2). Grade ≥ 3 AEs were documented in 68.4% of patients with ALL and 75% of patients with non-Hodgkin’s lymphoma (NHL) at day +100, whereas serious AEs (SAEs) were observed in 44.7% and 50% of patients with ALL and NHL, respectively (Table 2). Procedure-related mortality (PRM) at day +100 was 7.9% (95% confidence interval [CI] 1.7%–21.4%) for patients with ALL and 0% for patients with NHL. Suspected unexpected serious adverse reactions (SUSARs) were observed in 4 patients: 2 patients (aged 11 and 19, respectively) developed lethal CRS and a 35-year-old patient died of pseudomembranous colitis while recovering from grade 4 CRS. These 3 patients, belonging to cohorts 1 and 2, motivated the second major amendment of the study as previously described. The fourth SUSAR was reported in a patient with FL from cohort 3 who developed grade 4 toxic epidermal necrolysis while recovering from grade 2 CRS. In the last case, causality was investigated but could not be demonstrated (i.e., ARI-0001 cells could not be identified in the skin).
Table 2.
Adverse Events of Special Interest
| Cohorts 1 and 2 (Single Infusion) | Cohort 3 (Fractionated Infusion) | All Patients | |
|---|---|---|---|
| Acute lymphoblastic leukemia | n = 15 | n = 23 | n = 38 |
| Grade ≥ 3 adverse events, n (%) | 11 (73.3) | 15 (65.2) | 26 (68.4) |
| Severe adverse events, n (%) | 8 (53.3) | 9 (39.1) | 17 (44.7) |
| Grade ≥ 3 CRS, n (%) | 4 (26.7) | 1 (4.3) | 5 (13.2) |
| Grade ≥ 3 ICANS, n (%) | 1 (6.7) | 0 (0.0) | 1 (2.6) |
| Grade ≥ 3 s malignancies, n (%) | 1 (6.7) | 0 (0.0) | 1 (2.6) |
| Non-Hodgkin’s lymphoma | n = 3 | n = 5 | n = 8 |
| Grade ≥ 3 adverse events, n (%) | 3 (100) | 3 (60) | 6 (75) |
| Severe adverse events, n (%) | 3 (100) | 1 (20) | 4 (50) |
| Grade ≥ 3 CRS, n (%) | 1 (33.3) | 1 (20) | 2 (25) |
| Grade ≥ 3 ICANS, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Grade ≥ 3 s malignancies, n (%) | 0 (0) | 0 (0) | 0 (0) |
CRS, cytokine release syndrome; ICANS, immune effector cell-associated neurotoxicity syndrome.
Regarding AEs of special interest, CRS was reported in 55.3% (13.2% grade ≥ 3) and 87.5% (25% grade ≥ 3) of patients with ALL and NHL, respectively. In patients with ALL, we observed a reduction in the rate of grade ≥3 CRS after the second amendment, dropping from 26.7% (cohorts 1 and 2) to 4.3% (cohort 3; Table 2), this leading to a reduction in tocilizumab administration from 26.7% (cohorts 1 and 2) to 8.7% (cohort 3), and a reduction in corticosteroid use from 20% (cohorts 1 and 2) to 0% (cohort 3). Due to CRS, 1/23 (4%) patients from cohort 3 received only two ARI-0001 cell fractions, while no patient received only one fraction. The occurrence of CRS (any grade) was also reduced and delayed in cohort 3 (Figure S1). Moreover, grade ≥ 3 immune effector-cell associated neurotoxicity syndrome (ICANS) was only observed in 1 (2.6%) patient with ALL. The only grade ≥ 3 s malignancy observed in the study was myelodysplasia in a 7-year-old girl diagnosed with ALL who had already received 6 lines of therapy, including IO and alloHCT.
Globally speaking, the most common AEs in patients with ALL were neutropenia (97.4%), anemia (84.2%), hypogammaglobulinemia (78.9%), thrombocytopenia (76.3%), and lymphopenia (73.7%). Liver toxicity was also frequent, including increased AST (50%), increased ALT (47.4%), increased GGT (39.5%) and increased alkaline phosphatase (36.8%), mostly in patients with prior alloHCT (Table S1). Similar figures were observed in patients with NHL (Table S2). Two patients with ALL (2/38, 5%) with prior history of alloHCT and IO therapy developed severe hepatic sinusoidal obstruction syndrome (SOS) that resolved with conventional supportive care.
Efficacy
In patients with ALL, the measurable residual disease (MRD)-negative CRR was 84.2% (95% CI 69%–94%) at day +28 and 71.1% (95% CI 54%–85%) at day +100. All evaluable patients (i.e., excluding those who died prematurely) developed absolute B cell aplasia that lasted for a median of 100 days (95% CI 56–100 days). Progression-free survival (PFS) at 1 year was 47% (27%–67%) for the whole ALL cohort, while the 1-year OS was 68.6% (49%–88%; 78% for children, 65% for adults; Figure 2). The median duration of response (DOR), considering only patients who responded to therapy by day +100, was 14.8 months. Out of 15 patients with progressive disease after ARI-0001 cell infusion, tumor cells expressed CD19 in 13 (87%), while 2 (13%) were CD19-negative. When we considered all patients with ALL recruited into the study, including those who did not receive therapy (n = 43), the 1-year PFS and OS were 43.5% (24%–63%) and 77.7% (62%–93%), respectively.
Figure 2.

Clinical Outcome of Patients with Acute Lymphoblastic Leukemia
(A–D) Progression-free survival (A), overall survival (B), in vivo survival of ARI-0001 cells, as measured by persistence of B cell aplasia (C), and procedure-related mortality (D) of patients with acute lymphoblastic leukemia belonging to the modified full analysis set (n = 38) according to type of administration (cohorts 1 and 2 versus cohort 3).
Subgroup analyses according to type of administration (cohorts 1 and 2 versus cohort 3) and age are depicted in Table 3. The apparent lower response rate observed in the pediatric population at day +100 is due to the early administration of a second ARI-0001 cell dose before that day in 2 patients. Both patients were in MRD-negative CR on day +100 but received the second infusion shortly before this time point due to early B cell recovery. If we count them both as responders, the CRR for pediatric patients would be 72% instead of 55%, and the CRR of the entire population would be 76% instead of 71%. On the other hand, the B cell aplasia lasted for longer in the pediatric population compared to adult patients (48% versus 10.5% at 1 year).
Table 3.
Efficacy of ARI-0001 Cells in Patients with Acute Lymphoblastic Leukemia
| Population | n | MRD-CRR at day +28, rate (95% CI) | MRD-CRR at day +100, rate (95% CI) | PFS Median (95% CI) 1-year rate (95% CI) | DOR Median (95% CI) 1-year rate (95% CI) | OS Median (95% CI) 1-year rate (95% CI) |
|---|---|---|---|---|---|---|
| Total | 38 | 84.2 (69–94) | 71.1 (54–85) | 12.0 mo (4.2–20.2) 47% (27-67%) | 14.8 mo (6.0–NA) 59% (34%–83%) | 20.2 mo (10.4–NA) 69% (49%–88%) |
| Cohorts 1 and 2 (single dose) | 15 | 73.3 (45–92) | 66.7 (38–88) | 17.6 mo (0.3–NA) 51% (25%–77%) | 15.7 (3.6–NA) 70% (42%–98%) | 20.2 mo (0.3–NA) 65.5% (41%–90%) |
| Cohort 3 (fractionated) | 23 | 91.3 (72–99) | 73.9 (52–90) | 12.0 mo (4.2–14.5) 39.5% (4%–75%) | 8.7 mo (2.0–NA) NA (NA–NA) | 14.5 mo (6.8–14.5) 62% (24%–100%) |
| Age groups | ||||||
| ≤18 years | 11 | 81.8 (48–98) | 54.5 (23–83) | 18.1 mo (14.5–NA) 82% (59%–100%) | NA mo (14.8–NA) 100% (100%–100%) | NR (7.1–NA) 78% (50%–100%) |
| >18 years | 27 | 85.2 (66–96) | 77.8 (58–91) | 9.4 mo (3.3–20.2) 34% (12%–57%) | 8.7 mo (3.9–16.6) 46% (18%–75%) | 20.2 mo (12.8–NA) 64.5% (40%–89%) |
| ≤25 years | 19 | 78.9 (54–94) | 63.2 (38–84) | 17.6 mo (4.2–20.2) 64% (40%–89%) | 14.8 mo (6.0–NR) 79% (51%–100%) | 20.2 mo (14.5–NA) 82% (63%–100%) |
| >25 years | 19 | 89.4 (67–99) | 78.9 (54–94) | 7.2 mo (3.2–NA) 25% (0%–52%) | 8.7 mo (2.0–NR) 34% (0%–70%) | NR (6.9–NA) 50.5% (18%–84%) |
MRD-CRR, complete response rate with negative measurable residual disease; PFS, progression-free survival; DOR, duration of response; OS, overall survival; mo, months; NA, not available.
In patients with NHL, the overall response rate at day +100 was 75% (35%–97%), while the CRR was 50% (16%–84%). Full details, including the patient with CLL, are depicted in Figure 3.
Figure 3.

Swimmer Plot of Patients with Non-Hodgkin’s Lymphoma and Chronic Lymphocytic Leukemia Belonging to the Modified Full Analysis Set (n = 9)
Green denotes complete remission, blue denotes partial remission, and red denotes refractory disease or disease progression. Sharp edges denote “alive at last follow-up,” round edges denote “dead at last follow-up.”
Cytokines
Cytokine levels were available from 34 patients, 19 diagnosed with ALL, 5 diagnosed with NHL, and 1 diagnosed with CLL. The most informative cytokine levels were those determined at day +7, but peak values are also displayed for reference. The median absolute and relative values (to the pre-administration level) are depicted in Table S3. Figure S4 shows the association between cytokine levels on day +7 and the development of CRS for patients with ALL. The only cytokine significantly associated with the occurrence of CRS was IL-6. Figure S5 shows the non-significant impact of fractionated administration of ARI-0001 cells on cytokine levels on day +7, also for patients with ALL.
ARI-0001 Expansion
ARI-0001 cells expanded with a median area under the curve (AUC) of 129.1 copies per cell per day (range: 15.6–4271.3). Peak expansion occurred at a median of 14.5 days after infusion (range: 2–236 days). There was no correlation between the AUC of ARI-0001 expansion and patient’s age (younger or older than 18 years), type of administration (cohorts 1 and 2 [single dose] versus cohort 3 [fractionated]), development of CRS, diagnosis (ALL versus NHL), bone marrow blast percentage at study inclusion or response rate. Representative plots are displayed in Figures S6 and S7.
Human Anti-murine Antibodies
In patients with ALL, human anti-murine antibodies (HAMAs) were clearly detected (>20% positive cells by flow cytometry) in 9 patients, but we observed borderline results (10%–20% positive cells) in 5 further patients. Of note, 3 patients already had HAMAs before ARI-0001 cell infusion, which is quite intriguing because 2 of them were pediatric patients who had not received any monoclonal antibodies or an alloHCT. 2 of these patients experienced B cell recovery 1.8 and 3.2 months after ARI-0001 cell infusion but have not experienced disease relapse with current follow-up (one of them received a second infusion and is depicted in Figure S2D). The third patient who had detectable HAMAs before the cell infusion died of severe CRS. Out of the remaining 6 patients with HAMAs, 2 of them developed HAMAs after the second infusion of ARI-0001 cells (Figures S2A and S2E) and 1 before the second infusion of ARI-0001 cells (Figure S2F).
There was no significant association between the presence of HAMAs and B cell recovery or disease relapse (Fisher’s exact test). Furthermore, there was no association between persistence of B cell aplasia and PFS (Mantel-Byar test, Figure S3).
Second ARI-0001 Cell Infusion
In cohorts 2 and 3, a second infusion was allowed as per protocol, and this was performed in 6 patients, 2 due to CD19+ relapse and 4 due early B cell recovery. Unfortunately, it only resulted in brief responses and/or B cell aplasia, and also led to the appearance of HAMAs in 3 patients (Figure S2).
Discussion
To our knowledge, this is one of the first European academic clinical trials of CART19-cell therapy in patients with B cell malignancies, and certainly the first with the new construct A3B1:CD8:4-1BB:CD3z. The preliminary results observed in the first 10 patients led to the extension of the trial to include 54 patients in total, most of them diagnosed with ALL. An important caveat is that our study included both pediatric and adult patients with ALL, which are known to have a different prognosis,16 and this complicates the comparison with other studies. Other important differences are that (1) 87% of our patients with ALL had already received an alloHCT, compared to 33%–62% in other studies;11,17, 18, 19, 20, 21 and (2) response evaluation was performed at day +100, 4–10 weeks later than in other studies.11,17, 18, 19, 20, 21 This late response evaluation was necessary to capture ARI-0001 activity in patients with both ALL and NHL.
The preparation of ARI-0001 cells could be accomplished with one leukocytapheresis procedure in 87% of patients, and the cell production time ranged from 7 to 10 days. Still, the median vein-to-vein time was 42 days (range, 25–190) mostly due to 7 patients who required 2 aphereses and also numerous intervening medical complications that forced us to delay lymphodepleting chemotherapy and cell infusion.
We have observed that the safety profile of ARI-0001 cells is comparable to that of commercially available or other academic products.11,17, 18, 19, 20, 21 Of note, the excessive severity/lethality of CRS observed in the first 19 patients led to a major amendment of the protocol mandating the fractionated administration of ARI-0001 cells (cohorts 1 and 2 versus cohort 3). Consequently, the grade ≥3 CRS rate dropped from 27% to 4% and the PRM was reduced from 20% to 0%. These results almost replicate the experience with CTL019 (later known as tisagenlecleucel) in adult patients with ALL, in which the grade ≥3 CRS rate was reduced from 50% to 4%, and the PRM from 50% to 0%, after the fractionated administration of this drug was introduced into the protocol.17 Still, the global CRS rate of 55% (13% grade ≥ 3) observed in our trial, considering all patients together, is quite comparable to similar trials of CART19 cells in ALL.11,18, 19, 20, 21 Regarding neurotoxicity, there were no apparent differences before and after the amendment since only one (3%) patient developed grade ≥ 3 ICANS. These results are also comparable (if not better) to those obtained in similar trials of CART19 cells in ALL.11,17, 18, 19, 20, 21 We have no explanation for the reduced incidence of ICANS observed in our trial. Since the cell dose and patient management were comparable with other trials, we hypothesize that this could be a genuine feature of our A3B1-based construct. Moreover, there were no differences in safety according to age: grade ≥ 3 CRS rate was 18% for patients ≤18 years and 11% for those >18 years; and grade ≥3 ICANS was 0% and 4% for the same patient groups. Of note, two patients with ALL developed severe hepatic SOS, which, to the best of our knowledge, has not been reported before in patients receiving CART19 cells. Both patients had in common their relatively advanced age (54 and 67 years), prior alloHCT and prior administration of IO, which are all known risk factors for SOS. As a result, we currently perform a comprehensive liver evaluation in this patient population before the administration of ARI-0001 cells. From these results we can conclude that the fractionated administration of 1 × 106 ARI-0001 cells/kg can be considered safe and merits further evaluation in phase 2 trials.
In this study, ARI-0001 cells achieved a 71% MRD-negative CRR at day +100 with a median PFS of 12 months, which is comparable to results obtained with similar products in pediatric patients,11,18 and perhaps superior to studies performed exclusively in adults, in which the median PFS ranged from 6 to 8 months.17,21 Equally comparable are the median DOR of patients who were in remission by day +100 (14.8 months) and the median OS (20.2 months). If we focus on the survival of ARI-0001 cells, as measured by the persistence of B cell aplasia, it is also comparable to other clinical trials evaluating other CART19 constructs incorporating the 4-1BB costimulatory domain,18,19 but significantly shorter to what was obtained in the ELIANA trial.11 Probably, T cell exhaustion accounts for this phenomenon, which also mirrors the tendency toward a longer survival of CART19-cells in pediatric compared to adult ALL populations.11,19,21
Then, we evaluated the results obtained in cohorts 1 and 2 (single-dose) versus cohort 3 (fractionated administration) and observed that the CRR (67% versus 74%), 1-year PFS (51% versus 39.5%), and 1-year OS (66% versus 62%) were not remarkably different. These comparisons should, however, be handled with care since the follow-up is significantly shorter for patients in cohort 3. On the other hand, we observed some important differences between pediatric and adult patients. The CRR was apparently lower for pediatric patients (55% versus 78%), clearly influenced by the second administration of ARI-0001 cells in two pediatric patients before day +100. In contrast, the 1-year PFS of pediatric patients was significantly higher compared to adults (82% versus 34%), and also the duration of B cell aplasia (48% versus 10.5%), all in keeping with prior experience in pediatric versus adult populations.11,17, 18, 19, 20, 21 Remarkably, almost all (96%) adult patients recruited into this trial had already failed an alloHCT and none underwent a second procedure as consolidation therapy. As such, the effect of ARI-0001 on PFS or OS was not confounded by the effect of a subsequent allograft. On the other hand, most (87%) relapses were CD19+ in keeping to other studies performed in adult ALL,19,21 but almost the opposite to what was observed with tisagenlecleucel in pediatric patients.11 Once again, shorter T cell persistence may explain this phenomenon.11,19,21 Unfortunately, the low number of patients and the trial design prevented us from identifying any relationship between persistence of B cell aplasia and PFS. Finally, second ARI-0001 infusions had little efficacy in reverting early B cell recovery and/or disease relapse as seen by other investigators with similar products.22
In conclusion, the administration of ARI-0001 cells, produced in a European academic setting, provided safety and efficacy results that are comparable with other academic or even commercial products. In February 2020, a marketing authorization application under the Hospital Exemption Rule was submitted to the AEMPS. This exemplifies how initiatives from Academia and Pharmaceutical companies can be complementary and synergistic in the best interest of patients.
Materials and Methods
Patient Population
The CART19-BE-01 study is a single-arm, multicenter, open-label pilot study evaluating the safety and efficacy of ARI-0001 cells in patients with R/R B cell malignancies. Eligible patients had to have all of the following: (1) CD19-positive B cell malignancy, including ALL, DLBCL, CLL, FL, or mantle-cell lymphoma (MCL); (2) age from 2 to 80 years; (3) ECOG performance status 0–2; (4) estimated life expectancy from 3 months to 2 years; and (5) adequate venous access. Patients were eligible if they suffered from (1) ALL in ≥2nd relapse, either ineligible for or with relapsed disease after alloHCT; (2) DLBCL or MCL in ≥2nd relapse, ineligible for or with relapsed disease after autoHCT; and (3) CLL or FL who had received a minimum of 2 lines of therapy (including rituximab) and experienced disease progression within 2 years of last therapy. Patients with CLL/FL could be ineligible for or with relapsed disease after alloHCT/autoHCT. Key exclusion criteria included history of other malignancy unless it had been in remission for more than 3 years; severe renal, hepatic, pulmonary, or cardiac impairment; active immunosuppressive therapy; HIV infection; active HBV or HCV infection; and active infection requiring systemic therapy. Of note, neither central nervous system involvement nor prior alloHCT were exclusion criteria for this trial. All patients provided written, informed consent. The AEMPS and Institutional Review Boards/Ethics Committees of each study site approved the trial, which was conducted in accordance with the principles of the Declaration of Helsinki (last updated version, Fortaleza, Brazil, 2013).
Study Design, Endpoints, and Sample Size
The primary endpoint was safety as determined by PRM and grade 3–4 toxicity at day +100 and 1 year. AEs of special interest were CRS, neurotoxicity (currently known as ICANS) and second neoplasia. AEs were graded according to common terminology criteria (CTC), version 4.0. SAEs were defined as those AEs that were fatal, life-threatening, leading to (or prolonging) a stay in hospital or a transfer to the intensive care unit, or resulting in severe disability. For CRS, we used the grading system by Lee et al.23 Secondary endpoints included objective response rate (ORR) as per conventional criteria;24, 25, 26 PFS, OS, DOR, B cell aplasia duration, and impact of therapy on quality of life. The ORR was assessed at day +100 for all patients (prespecified endpoint) and at day +28 for patients with ALL (exploratory endpoint).
The original sample size was 10 patients (cohort 1). 5 months after study initiation, a major amendment increased the sample size to 39 patients and allowed patients with either normal B cell recovery within 3 months (early B cell recovery), CD19+ disease relapse, or CD19+ refractory disease to receive a second dose of ARI-0001 cells (cohort 2). 12 months after study initiation, with 19 patients already recruited, a second major amendment increased the sample size to a total of 54 patients (cohort 3), mandated the fractionated administration of ARI-0001 cells (10%, 30%, and 60% of the total dose) contingent on the lack of CRS after each fraction, and allowed the early administration of tocilizumab in patients with grade 2 CRS (it was initially reserved to those with grade ≥ 3). This second amendment was motivated by 3 toxic deaths.
ARI-0001 Cells Production and Treatment
Full details of ARI-0001 cell production, including a comprehensive phenotypic characterization of these cells can be found elsewhere.27 Patients were enrolled following screening and confirmation of eligibility, and underwent leukocytapheresis to obtain peripheral blood mononuclear cells (PBMCs). ARI-0001 cells were manufactured using the CliniMACS Prodigy system (Miltenyi). Approximately 100 × 106 T cells were stimulated to expand with anti-CD3 and anti-CD28 antibodies together with IL-7 and IL-15. 24 h after T cell activation, cells were transduced with a lentiviral vector containing the CAR gene construct at a multiplicity of infection of 10. Following expansion, that lasted from 7 to 10 days, the final ARI-0001 cell product was washed, cryopreserved, and tested for identity, potency, sterility, and adventitious agents.
Before ARI-0001 cell infusion, patients received fludarabine at 30 mg/m2/day plus cyclophosphamide at 300 mg/m2/day on days −6, −5, and −4 followed by ARI-0001 cells. In cohorts 1 and 2, patients received a single intravenous infusion of ARI-0001 cells, at a dose of 0.5–5 × 106 cells/kg, on day 0. Adult patients with ALL received 0.5–1 × 106 cells/kg, while pediatric patients with ALL and adult patients with NHL/CLL received 5 × 106 cells/kg (if available). In cohort 3, patients received the first fraction (10%) of ARI-0001 cells on day 0, followed by the second (30%) and third (60%) fraction. The second fraction was administered 24–48 h after the first, and the third 24–48 h after the second only if the patient had no signs or symptoms of CRS. Moreover, in cohort 3 all patients with ALL received a maximum ARI-0001 cell dose of 1 × 106 cells/kg regardless of age. All patients remained hospitalized to recovery through day +21 or until all procedure-related non-hematological toxicity returned to grade ≤ 1 or baseline. Patients were followed in the post-treatment assessment period and returned to the clinic at weeks 4, 5, 6, 8, 10, 12, and 14 (day +100), and monthly afterward for the first year. All patients completing the first year of follow-up were followed up for survival and disease status every 3 months in years 2 and 3. In case of a second ARI-0001 cell infusion, the entire procedure was repeated, including lymphodepleting chemotherapy, ARI-0001 cell dose (if available), hospital admission, and close follow-up.
The methods used to measure cytokines, ARI-0001 cells and HAMAs are available in the Supplemental Information.
Statistical Analysis
Adverse events and response rates are presented with 95% exact Clopper-Pearson confidence intervals. PRM was calculated as a cumulative incidence considering disease relapse as a competing event. OS, PFS, DOR, and persistence of B cell aplasia, were plotted using the Kaplan-Meier method. Due to the open-label non-randomized nature of the study, the statistical analysis was descriptive and no formal comparisons between cohorts are provided. However, for illustrative reasons, some p values are provided, which should be considered informative but not conclusive. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA) and R (R Foundation for Statistical Computing, Vienna, Austria). The trial (EUDRA nº 2016-2972-29) was registered at https://clinicaltrials.gov/ (ClinicalTrials.gov: NCT03144583).
Author Contributions
V.O.-M., S.R., M.J., and J.D. designed the clinical trial and wrote the paper; J.E., G.C., and A.U.-I. provided significant contribution to the study planning and analysis; V.O.-M., T.B., J.E., E.G., M.D.-B., L.G., M.M., P.C., S.F., and J.D. looked after adult patients; S.R., A.A.-S., C.L., A.C., A.F., L.A., F.R., and I.J. looked after pediatric patients; D.B.-R., M.C.-B., E.A.G., M.E.-R., N.K.-G., E.T., and M.J. manufactured ARI-0001 cells and monitored ARI-0001-cell expansion and cytokine production; M.C., G.S., J.M.C., U.P., and M.J. designed the CAR construct and produced the lentiviral vector; N.V. and M.T. performed MRD tests; J.C., E.G.-R., and M.L. performed leukocytaphereses; F.T. and J.D. performed the statistical analysis; J.S.-P. was responsible for pharmacovigilance; S.V. was the study manager; A.S. drafted the regulatory dossier for the submission to the AEMPS; G.C. is the coordinator of Advanced Therapies at Hospital Clínic. All authors reviewed the study data, read, and approved the manuscript.
Conflicts of Interest
V.O.-M.: consultant or advisory role (Kite Gilead, Celgene, Novartis), travel grants (Kite Gilead, Celgene, Novartis, Roche, Takeda, Janssen), honoraria (Kite Gilead). S.R.: consultant or advisory role (Novartis, Jazz, Shire/Servier, Amgen), travel grants (Novartis, Jazz, Shire/Servier, Amgen), honoraria (Novartis, Jazz, Shire/Servier, Amgen). A.A.-S.: consultant or advisory role (Novartis), travel grants (Novartis), honoraria (Novartis). T.B.: consultant of advisory role (Janssen), travel grants (Kite Gilead, Abbvie, Janssen), honoraria (Janssen, Abbvie, Roche, Kite Gilead). E.G.-R.: honoraria (Novartis). C.L.: travel grants (Novartis), honoraria (Novartis). M.T.: travel grants (Novartis, Jazz Pharma, Shire/Servier, Amgen). E.G.: consultant or advisory role (Kite Gilead, Janssen, Roche), research funding (Kite Gilead, Janssen, Roche). M.D.-B.: consultant or advisory role (Celgene, Novartis, Jazz, Astellas). A.C.: consultant or advisory role (Novartis, Celgene), travel grants (Novartis, Celgene), honoraria (Novartis, Celgene). A.F.: consultant or advisory role (Novartis), travel grants (Novartis), honoraria (Novartis). P.C.: consultant or advisory role (Kite Gilead, Celgene, Janssen), travel grants (Kite Gilead, MSD, Janssen). F.R.: consultant or advisory role (GW), travel grants (GW), honoraria (GW). M.L.: Honoraria (Terumo BCT, Cerus, Grifols), research funding (Terumo BCT, Sanofi-Genzyme, Maco). A.S.: current employment (Bayer). J.S.-P.: consultant or advisory role (Bayer). G.C.: consultant or advisory role (Celgene, Novartis). J.E.: consultant or advisory role (Abbvie, Novartis, Celgene, Astellas, Jazz, Daiichi Dankyo, Roche, Amgen, Pfizer), travel grants (Celgene, Roche, Astellas, Daiichi Dankyo), research funding (Novartis, Celgene). A.U.-I.: consultant or advisory role (Kite Gilead, Celgene, Miltenyi), travel grants (Kite Gilead, Celgene). M.J.: consultant or advisory role (Kite Gilead, Grifols), honoraria (Kite Gilead, Grifols). M.C., D.B.-R., M.C.-B., J.C., N.V., L.G., M.M., E.A.G., M.E.-R., N.K.-G., L.A., I.J., S.F., G.S., U.P., J.M.P., E.T., S.V., F.T., and J.D. have no competing interests to disclose.
Acknowledgments
This study was funded by CatSalut, Projecte ARI, and grants co-financed by the Instituto de Salud Carlos III –Subdirección General de Evaluación y Fomento de la Investigación Sanitaria and Fondo Europeo de Desarrollo Regional (FEDER) PICI14/122, PI13/676, PIE13/33, and PI18/775. V.O.-M. is a recipient of a research grant from FEHH and J.D. is a recipient of a research grant from the Generalitat de Catalunya (PERIS IPFE SLT006/17/301). We thank the patients who participated in the study and their families, friends, caregivers, and referring physicians. We are very grateful to Drs. Miguel Ángel Perales (Memorial Sloan Kettering Cancer Center, New York), Shannon Maude (Children’s Hospital of Philadelphia), Juan José Lasarte (Centro de Investigación Médica Aplicada, Navarra), and Michael Schmitt (Universitäts Klinikum Heidelberg), members of our data safety monitoring board, who helped us immensely during the trial, particularly with the management of patients with fatal toxicity. We are also indebted to the study staff and health care providers at Hospitals Clínic and Sant Joan de Déu, specifically to Anna Boronat and Raquel Martín-Ibáñez, who participated in the initial development of the construct; Judit Pich, Gabriela Recalde, Leticia Pereira, and Joan Albert Arnáiz, from the Clinical Trials Unit; Nuria Coderch and Christopher Mann from Asphalion; and Josep Maria Campistol, Antoni Castells, Aurea Mira, Manel del Castillo, Miquel Pons, and Marc Roda as the main representatives from both hospitals.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.09.027.
Contributor Information
Manel Juan, Email: mjuan@clinic.cat.
Julio Delgado, Email: jdelgado@clinic.cat.
Supplemental Information
References
- 1.Tavernier E., Boiron J.-M., Huguet F., Bradstock K., Vey N., Kovacsovics T., Delannoy A., Fegueux N., Fenaux P., Stamatoullas A., GET-LALA Group. Swiss Group for Clinical Cancer Research SAKK. Australasian Leukaemia and Lymphoma Group Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia. 2007;21:1907–1914. doi: 10.1038/sj.leu.2404824. [DOI] [PubMed] [Google Scholar]
- 2.Oriol A., Vives S., Hernández-Rivas J.M., Tormo M., Heras I., Rivas C., Bethencourt C., Moscardó F., Bueno J., Grande C., Programa Español de Tratamiento en Hematologia Group Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica. 2010;95:589–596. doi: 10.3324/haematol.2009.014274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kantarjian H.M., Thomas D., Ravandi F., Faderl S., Jabbour E., Garcia-Manero G., Pierce S., Shan J., Cortes J., O’Brien S. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer. 2010;116:5568–5574. doi: 10.1002/cncr.25354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gökbuget N., Stanze D., Beck J., Diedrich H., Horst H.-A., Hüttmann A., Kobbe G., Kreuzer K.A., Leimer L., Reichle A., German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032–2041. doi: 10.1182/blood-2011-12-399287. [DOI] [PubMed] [Google Scholar]
- 5.Spyridonidis A., Labopin M., Schmid C., Volin L., Yakoub-Agha I., Stadler M., Milpied N., Socie G., Browne P., Lenhoff S., Immunotherapy Subcommittee of Acute Leukemia Working Party Outcomes and prognostic factors of adults with acute lymphoblastic leukemia who relapse after allogeneic hematopoietic cell transplantation. An analysis on behalf of the Acute Leukemia Working Party of EBMT. Leukemia. 2012;26:1211–1217. doi: 10.1038/leu.2011.351. [DOI] [PubMed] [Google Scholar]
- 6.Poon L.M., Hamdi A., Saliba R., Rondon G., Ledesma C., Kendrick M., Qazilbash M., Hosing C., Jones R.B., Popat U.R. Outcomes of adults with acute lymphoblastic leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2013;19:1059–1064. doi: 10.1016/j.bbmt.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kantarjian H.M., DeAngelo D.J., Stelljes M., Martinelli G., Liedtke M., Stock W., Gökbuget N., O’Brien S., Wang K., Wang T. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016;375:740–753. doi: 10.1056/NEJMoa1509277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kantarjian H., Stein A., Gökbuget N., Fielding A.K., Schuh A.C., Ribera J.-M., Wei A., Dombret H., Foà R., Bassan R. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017;376:836–847. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crump M., Neelapu S.S., Farooq U., Van Den Neste E., Kuruvilla J., Westin J., Link B.K., Hay A., Cerhan J.R., Zhu L. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800–1808. doi: 10.1182/blood-2017-03-769620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.June C.H., Sadelain M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak Ö., Brogdon J.L., Pruteanu-Malinici I., Bhoj V., Landsburg D. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castella M., Boronat A., Martín-Ibáñez R., Rodríguez V., Suñé G., Caballero M., Marzal B., Pérez-Amill L., Martín-Antonio B., Castaño J. Development of a Novel Anti-CD19 Chimeric Antigen Receptor: A Paradigm for an Affordable CAR T Cell Production at Academic Institutions. Mol. Ther. Methods Clin. Dev. 2018;12:134–144. doi: 10.1016/j.omtm.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delgado J., Caballero-Baños M., Ortiz-Maldonado V., Castellà M., Magnano L., Juan M., Urbano-Ispizua Á. Chimeric Antigen Receptor T Cells Targeting CD19 and Ibrutinib for Chronic Lymphocytic Leukemia. HemaSphere. 2019;3:e174. doi: 10.1097/HS9.0000000000000174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rafei H., Kantarjian H.M., Jabbour E.J. Targeted therapy paves the way for the cure of acute lymphoblastic leukaemia. Br. J. Haematol. 2020;188:207–223. doi: 10.1111/bjh.16207. [DOI] [PubMed] [Google Scholar]
- 17.Frey N.V., Shaw P.A., Hexner E.O., Pequignot E., Gill S., Luger S.M., Mangan J.K., Loren A.W., Perl A.E., Maude S.L. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults With Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020;38:415–422. doi: 10.1200/JCO.19.01892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gardner R.A., Finney O., Annesley C., Brakke H., Summers C., Leger K., Bleakley M., Brown C., Mgebroff S., Kelly-Spratt K.S. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hay K.A., Gauthier J., Hirayama A.V., Voutsinas J.M., Wu Q., Li D., Gooley T.A., Cherian S., Chen X., Pender B.S. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood. 2019;133:1652–1663. doi: 10.1182/blood-2018-11-883710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee D.W., Kochenderfer J.N., Stetler-Stevenson M., Cui Y.K., Delbrook C., Feldman S.A., Fry T.J., Orentas R., Sabatino M., Shah N.N. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bezerra E.D., Gauthier J., Hirayama A.V., Pender B.S., Hawkins R.M., Vakil A. Factors associated with response, CAR-T cell in vivo expansion, and progression-free survival after repeated infusiones of CD19 CAR-T cells. Blood. 2019;134:201. [Google Scholar]
- 23.Lee D.W., Gardner R., Porter D.L., Louis C.U., Ahmed N., Jensen M., Grupp S.A., Mackall C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarnas J.C., Brown P.A., Aoun P., Ballen K.K., Barta S.K., Borate U., Boyer M.W., Burke P.W., Cassaday R., Castro J.E. Acute Lymphoblastic Leukemia, Version 2.2015. J. Natl. Compr. Canc. Netw. 2015;13:1240–1279. doi: 10.6004/jnccn.2015.0153. [DOI] [PubMed] [Google Scholar]
- 25.Cheson B.D., Fisher R.I., Barrington S.F., Cavalli F., Schwartz L.H., Zucca E., Lister T.A., Alliance, Australasian Leukaemia and Lymphoma Group. Eastern Cooperative Oncology Group. European Mantle Cell Lymphoma Consortium. Italian Lymphoma Foundation. European Organisation for Research. Treatment of Cancer/Dutch Hemato-Oncology Group. Grupo Español de Médula Ósea. German High-Grade Lymphoma Study Group. German Hodgkin’s Study Group. Japanese Lymphorra Study Group. Lymphoma Study Association. NCIC Clinical Trials Group. Nordic Lymphoma Study Group. Southwest Oncology Group. United Kingdom National Cancer Research Institute Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J. Clin. Oncol. 2014;32:3059–3068. doi: 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hallek M., Cheson B.D., Catovsky D., Caligaris-Cappio F., Dighiero G., Döhner H., Hillmen P., Keating M.J., Montserrat E., Rai K.R., Kipps T.J., International Workshop on Chronic Lymphocytic Leukemia Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castella M., Caballero-Baños M., Ortiz-Maldonado V., González-Navarro E.A., Suñé G., Antoñana-Vidósola A., Boronat A., Marzal B., Millán L., Martín-Antonio B. Point-of-care CAR T-cell production (ARI-0001) using a closed semi-automatic bioreactor: experience from an academic phase I clinical trial. Front. Immunol. 2020;11:482. doi: 10.3389/fimmu.2020.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.