

Abstract
Immunotherapies designed to treat neurodegenerative tauopathies that primarily engage extracellular tau may have limited efficacy as tau is primarily intracellular. We generated tau-targeting single-chain variable fragments (scFvs) and intrabodies (iBs) from the phosphorylated tau-specific antibodies CP13 and PHF1 and the pan-tau antibody Tau5. Recombinant adeno-associated virus (rAAV) was utilized to express these antibody fragments in homozygous JNPL3 P301L tau mice. Two iBs (CP13i, PHF1i) and one scFv (PHF1s) abrogated tau pathology and delayed time to severe hindlimb paralysis. In a second tauopathy model (rTg4510), CP13i and PHF1i reduced tau pathology, but cognate scFvs did not. These data demonstrate that (1) disease-modifying efficacy does not require antibody effector functions, (2) the intracellular targeting of tau with phosphorylated tau-specific iBs is more effective than extracellular targeting with the scFvs, and (3) robust effects on tau pathology before neurodegeneration only resulted in modest disease modification as assessed by delay of severe motor phenotype.
Keywords: tauopathy, phosphorylation, single-chain variable fragments, scFv, immunotherapy, adeno-associated virus, intrabody, iB, neurodegeneration, Alzheimer's disease
Graphical Abstract
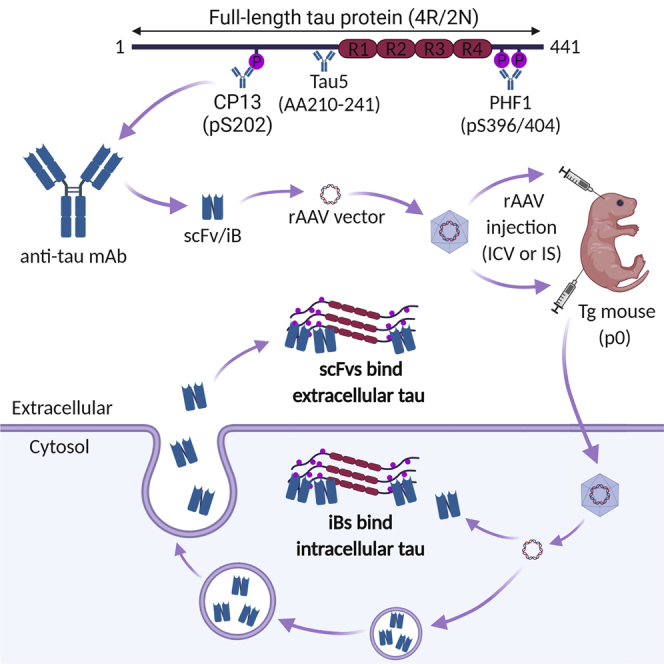
Tau-specific antibodies have been shown to reduce tau pathology in mice by binding extracellular tau. Goodwin et al. developed phosphorylated tau-specific single-chain variable fragments expressed in the cytoplasm and targeted to bind intracellular tau. These intrabodies reduced pathology and slowed the progression of tau-induced neurodegeneration in transgenic mice.
Introduction
Alzheimer’s disease (AD) is a complex neurodegenerative proteinopathy with both amyloid β (Aβ) protein accumulation in extracellular plaques and tau aggregation to form intracellular neurofibrillary tangles (NFTs).1, 2, 3 Tau pathology is also found in many other neurodegenerative disorders, termed tauopathies, and mutations in tau cause frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17).4,5 Modeling studies have linked tau to neurodegeneration both in primary culture and transgenic mouse models, making tau an attractive therapeutic target for disease modification.6,7 Tau forms intracellular aggregates in AD and other tauopathies, yet it is also present in extracellular fluids at very low picomolar levels.8,9 Extracellular tau levels in cerebrospinal fluid (CSF) and blood increase in AD, but these changes appear to be associated with Aβ deposition and not tau pathology, as increased extracellular tau has not been observed in other tauopathies.10 Most extracellular tau species, in both control and AD CSF, are truncated before the microtubule domains and are not capable of seeding tau pathology.8,9,11 In contrast, numerous studies have shown that exogenous aggregated tau species that are either full length or contain the carboxyl-terminal microtubule binding domains can, via prion-like propagation, seed intracellular tau pathology in cell culture and animal models.12, 13, 14, 15, 16, 17, 18, 19, 20, 21 Thus, as there is limited evidence in humans for extracellular tau capable of seeding tau aggregation, it is only a hypothesis that prion-like spread underlies the progressive accumulation of tau pathology across neuroanatomically connected routes observed in the brains of AD patients.22
In mice, both active and passive immunotherapies have been shown to be capable of reducing the formation of intracellular tau pathology.9,23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34 Active and passive immunotherapies that were modestly efficacious with respect to reducing tau pathology in these models have advanced to clinical trials. Several mechanisms have been proposed to contribute to the efficacy of tau-targeted immunotherapy. Peripherally injected anti-tau antibodies have been shown to cross the blood-brain barrier (BBB) and bind tau within the CNS.32 Mechanistically, tau-targeting antibodies are thought to engage extracellular tau “seeds” and inhibit the prion-like propagation of tau pathology.21 Alternatively, it has been proposed that antibodies might target tau inside the cell following internalization by Fc receptors or endocytosis of antibody-tau complexes.35, 36, 37, 38 Once inside cells, antibodies have been shown to promote the disassembly of tau aggregates, allowing for increased degradation via the lysosomal or proteasomal pathways, although these mechanisms remain controversial.31,34,39
Regardless of the mechanisms that contribute to efficacy, tau immunotherapies must first reach the brain and may be limited by the finding that antibody levels in the CNS only reach ∼0.1% of peripheral levels.40 Whether peripheral immunization will result in sufficient target engagement to modulate tau pathology and improve patient functional outcomes remains to be shown. Indeed, lack of clinical efficacy observed in multiple clinical trials assessing anti-tau and anti-Aβ immunotherapies highlights the importance of exploring alternative therapeutic strategies. Recombinant adeno-associated virus (rAAV) vectors encoding tau-targeted antibodies or antibody fragments have been used to more directly target tau within the mouse brain.41, 42, 43 One study expressed a full-length monoclonal antibody (mAb) (PHF1) specific for phosphorylated (p-)tau (serine 396 and 404) in adult P301S transgenic mice,43 and it showed robust (>70%) reductions of insoluble p-tau species in the hippocampus and cortex. Another study utilized neonatal injection of AAV to express scFvs specific for the N terminus of tau in the brains of P301S transgenic mice42 and found that scFv expression reduced soluble tau species by ∼30% in the hippocampus and pathological tau staining in specific hippocampal subregions by ∼80%. A third study expressed a scFv derived from the conformation-specific MC1 antibody in adult JNPL3 tau transgenic mice.41 Hippocampal expression of this scFv via an astrocyte-specific promoter significantly reduced pathological tau staining by ∼60%–70% in the hippocampus, as well as soluble and insoluble tau by ∼30% across several brain regions. Another study used rAAV vectors to express chimeric intrabodies (iBs) specific for the N terminus of tau and showed that fusion of iBs to mutant ubiquitin enhanced proteasomal targeting and resulted in robust reductions (∼75%–90%) of pathological tau staining in specific hippocampal subregions of P301S transgenic mice.44
Taken together, these studies demonstrate the potential of rAAV-delivered antibodies or antibody fragments to reduce tau aggregation and accumulation. However, most reports of efficacy in preclinical tauopathy models simply provide a readout of tau pathology, and results on modification of neurodegenerative phenotypes have been lacking. Furthermore, none of these studies directly compared multiple recombinant antibodies targeting different tau epitopes, nor directly compared the effects of a scFv with its corresponding iB. In this study, we compared the disease-modifying potential of three different tau-targeting scFvs and their cognate iBs. These recombinant antibodies were derived from three well-characterized tau mAbs, that is, CP13 (epitope pS202), PHF1 (epitope pS396/404), and Tau5 (epitope AA210–241).6,45, 46, 47, 48 Notably, both CP13 and PHF1 mAbs have been shown to modify tau pathology in passive immunotherapy paradigms,23,30 as well as recombinant PHF1 mAb showing efficacy in the previously mentioned rAAV study. We show that these recombinant antibodies retain tau-binding specificity and engage tau in a HEK293 cell tau aggregation assay. In homozygous JNPL3 tau P301L mice, CP13i, PHF1i, and PHF1s robustly suppressed tau pathology and modestly delayed hindlimb paralysis. When expressed in a second tau mouse model, rTg4510 mice, CP13i and PHF1i reduced tau pathology, but their cognate scFvs did not. These data extend our knowledge about the factors that predict efficacy of gene therapies using tau-targeted scFvs and highlight both the potential and current limitations of delivering such antibody fragments.
Results
Generation and Functional Assessment of Recombinant Tau scFvs and iBs
To directly compare the efficacy of targeting intracellular versus extracellular tau, we first generated single-chain variable fragments (scFvs) from the CP13, PHF1, and Tau5 mAbs (Figures 1A and 1B). iBs were engineered from each scFv by making specific mutations known to promote intracellular antibody stability49 and removing the secretion signal sequence (Table 1). To confirm expression and tau binding, HEK293T cells were transiently transfected with expression vectors encoding scFvs and iBs. Immunoblotting detected all three secreted scFvs in both cell lysates and conditioned media, whereas all iBs were detected only in cell lysates (Figure 1C). iBs were visualized as a single band at ∼25 kDa, while scFvs appeared as multiple bands ranging from ∼25 to 28 kDa. These additional bands are likely due to glycosylation or other post-translational modifications that occur in the secretory pathway. We did not attempt to further characterize these modifications, as all scFvs retained the ability to bind to tau. We confirmed this by using cell lysates or conditioned HEK293T media following transient transfection of scFvs or iBs to stain brain cortical tissue from 10-month-old rTg4510 mice with extensive tau pathology. Patterns of staining by scFvs and iBs were indistinguishable from the pattern observed when staining with the parental antibodies (Figure 1D). Finally, we confirmed that all iBs and scFvs retained epitope specificity by measuring antigen-binding reactivity of cell lysates and media with ELISAs (Figure 1E).
Figure 1.

Anti-tau iBs and scFvs Are Expressed in Mammalian Cells and Bind Tau In Vitro
(A) Schematic of tau (4R/2N) protein and three antibodies from which scFvs and iBs were derived. (B) Schematic of delivery of scFvs and iBs and hypothesized localization. (C) CP13, PHF1, and Tau5 iBs and scFvs were cloned into rAAV vectors in frame with a c-myc tag and expressed in HEK cells using calcium phosphate transfection. SDS-PAGE and immunoblotting for c-myc were used to detect expression in the conditioned media, the 1% Triton X-100 soluble fraction, and the Triton X-100 insoluble/2% SDS solubilized fraction. (D) Cell lysates and media transfected with iBs, scFvs, or EGFP as a control were applied to paraffin-embedded sections of 10-month-old rTg4510 tau transgenic mice. Cortical staining of neurofibrillary tangles was visualized with a biotinylated anti-c-myc secondary antibody. Scale bar, 50 μm. (E) Cell lysates and media containing scFvs and iBs were subjected to direct ELISA on plates coated with recombinant full-length tau, p-tau, or amyloid β as a control. Anti-c-myc HRP secondary antibody served as a detection. Data represent mean ± SD of three independent transfections and are normalized to non-transfected cell lysates and media.
Table 1.
Sequences of scFvs and iBs
| Parent mAb | Amino Acid Sequence | |
|---|---|---|
| Variable heavy chain (VH) | CP13 parent | EVQLVETGGGLVQPKGSLKLSCAASGFTFNTSAMNWVRQAPGKGLEWVARIKSKSNKYTTSYADSVKD RFTISRDDSQSMLYLQMYNLKTEDTAMYYCMNSAMDYWGQGTSVTVSS |
| CP13 iB | EVQLVESGGGLVQPGGSLRLSCAASGFTFNTSAMNWVRQAPGKGLEWVSRIKSSSNKYTTSYADSVKG RFTISRDNSKNTLYLQMNSLRAEDTAVYYCMNSAMDYWGQGTLVTVSS |
|
| PHF1 parent | EVQLQQSGPELVKPGASVKISCKTSGYTFTEYTIHWVKQSHGESLEWIGGINPNDGGTIYNQKFKGKA TLTVDKSSKTAYMELRSLTSEDSAVFYCARGPSARFPYWGQGTLVTVSA |
|
| PHF1 iB | EVQLVQSGPGLVQPGGSLRLSCATSGYTFTEYTIHWVRQAPGESLEWVSGINPSDGGTIYNQSVKGRF TLSVDNSKNTAYLQMNSLRAEDTAVYYCARGPSARFPYWGQGTLVTVSS |
|
| Tau5 parent | EVQLQQSGAEIVRSGASVKLSCAASGFNIKDYYMHWVKQRPEQGLEWIGWIDPENGDIAYAPKFQGKA TMTADTSSNTAYLQLSRLTSEDTAVYFCNGRGGMITTDFFDYWGQGTTLTVSS |
|
| Tau5 iB | EVQLVQSGAGLVQPGGSLRLSCAASGFNIKDYYMHWVRQAPGQGLEWVSWIDPSNGDIAYAPSVQGRF TMSADNSKNTAYLQMNSLRAEDTAVYFCNGRGGMITTDFFDYWGQGTLVTVSS |
|
| Variable light chain (VL) | CP13 parent | EIVMTQAVSSVPVTPGESVSISCRSSKSLLHSNGNTYLYWFLQRPGQSPHLLIYRMSNLASGVPDRFSGSGS GTAFTLRISRVEAEDVGVYYCMQHLEYPLTFGAGTKLELKR |
| CP13 iB | EIVMTQSPATLSLSPGERATLSCRSSQSLLHSNGNTYLYWFQQRPGQSPRLLIYRMSNRASGVPARFSGS GSGTAFTLTISSLEPEDFAVYYCMQHLEYPLTFGQGTKVEIKR |
|
| PHF1 parent | DVVMTQTPLTLSVTIGQPASISCKSSQSLLDSDGKTYLNWLLQRPGQSPKRLIYLVSKLDSGVPDRFTGS GSGTDFTLKISRVEAEDLGVYYCWQGTHFPRTFGGGTKLEIKR |
|
| PHF1 iB | DVVMTQSPATLSLSPGERATLSCRSSQSLLDSDGKTYLNWLQQRPGQSPRRLIYLVSKRDSGVPARFTG SGSGTDFTLTISSLEPEDFAVYYCWQGTHFPRTFGQGTKVEIKR |
|
| Tau5 parent | DVLMTQTP–LSLPVSLGDQASISCRSSQSIVHSNGNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGS GSGTDFTLKISRVEAEDLGVYYCFQGSHVPWTFGGGTKLEIKR |
|
| Tau5 iB | DVVMTQSPATLSL–SPGERATLSCRSSQSIVHSNGNTYLEWYQQKPGQSPRLLIYKVSNRFSGVPARFSGSG SGTDFTLTISSLEPEDFAVYYCFQGSHVPWTFGQGTKVEIKR |
Original sequences of variable regions from PHF1, CP13. and tau5 mAbs and modified sequences of iBs. Mutations in the VH and VL chains, which were made to promote stability of intracellularly expressed scFvs,49 are shown in bold.
We then assessed the ability of iBs and scFvs to prevent intrinsic tau aggregation in a recently developed cell culture assay. HEK293T cells were co-transfected with plasmids encoding either iBs or scFvs, along with full-length (4R/0N) tau protein harboring the FTDP-17-associated mutation S320F, which has been shown to promote intrinsic tau aggregation without the addition of exogenous fibrils.19 Biochemical fractionation followed by immunoblotting revealed that all three iBs and scFvs significantly reduced phosphorylated and total tau in the insoluble fraction when compared to controls co-transfected with enhanced yellow fluorescent protein (EYFP) (Figure 2). Expression of all three scFvs reduced soluble tau, while the effects of iB expression on soluble tau species varied. Expression of CP13i did not significantly alter soluble total or p-tau, while Tau5i reduced soluble total tau but not p-tau. Interestingly, expression of PHF1i increased levels of soluble p-tau. These results indicate that anti-tau iBs and scFvs successfully expressed from plasmid DNA maintained specificity and demonstrated target engagement in vitro.
Figure 2.

Anti-tau iBs and scFvs Prevent Tau Aggregation In Vitro
HEK293T cells were co-transfected with rAAV plasmids expressing S320F mutant human tau and CP13, PHF1, and Tau5 iBs or scFvs. Co-transfection with enhanced yellow fluorescent protein (EYFP) served as the control. Forty-eight hours after transfection, cells were harvested to obtain Triton X-100 soluble and Triton X-100 insoluble/2% SDS solubilized fractions. (A) Representative western blots of soluble and insoluble fractions stained with anti-p-tau mAb AT8, anti-total tau rabbit polyclonal mAb T44-3029, and anti-actin mAb shows alterations in tau levels as a result of iB and scFv expression. (B) Densitometric analysis of p-tau and total tau in the soluble and insoluble fractions normalized to actin. n = 7–14. Data represent mean ± SEM. A one-way ANOVA with Tukey’s multiple comparison test was used. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Expression of p-Tau-Specific iBs and scFvs Prevents Tau Pathology and Delays Hindlimb Paralysis in Homozygous JNPL3 Mice
The effects of tau-targeted iBs and scFvs on tau pathology and disease modification were assessed in JNPL3 transgenic mice that express human tau (4R/0N) containing the FTD-associated P301L mutation.50 In our colony, homozygous females develop significant tau pathology in the spinal cord and brainstem by 6 months of age, followed by hindlimb paralysis around 10 months of age. To express recombinant antibodies in the spinal cord and brainstem of these mice, we intraspinally injected rAAV vectors into neonatal mice as described previously.4 Expression of iBs and scFvs was assessed 6 months after injection by staining for the c-myc epitope tag present in each. Staining was detected throughout the spinal cord, with highest levels in the gray matter, indicating successful and widespread transgene delivery (Figure 3A). Expression of iBs and scFvs in the brain was probably below the threshold of sensitivity of anti-c-myc antibody for immunohistochemical detection (data not shown). We noted stronger intracellular staining in the spinal cords of mice expressing iBs than those expressing scFvs, although levels were not significantly different when we performed ELISA of whole spinal cord homogenates (Figure 3B). We also examined expression in brains and plasma from the injected mice by ELISA and found that all iBs and scFvs were present at low levels in the brain, whereas only scFvs were detected at significant levels above background in the plasma (Figure 3B).
Figure 3.

Expression of Anti-tau scFvs and iBs in the Midbrain and Spinal Cord Following Neonatal Intraspinal AAV Injection
JNPL3 mice were injected intraspinally at P0 with pAAV2/8 (2 × 1010 genomes) expressing PHF1, CP13, and Tau5 iBs and scFvs. Control mice were injected with rAAV-EGFP. (A) Widespread iB and scFv expression was visualized with an anti-c-myc antibody in the spinal cord on representative paraffin sections. Scale bar, 100 μm. (B) Relative iB and scFv levels in brain homogenates, spinal cord homogenates, and plasma of injected mice detected via sandwich ELISA. n = 4. Data represent mean ± SEM. ∗∗∗p < 0.001, by unpaired two-tailed Student’s t test.
We then evaluated whether iB or scFv expression altered tau pathology in the brainstem and spinal cord pathology of 6-month-old female JNPL3 mice. Immunohistochemical analysis and quantification revealed that CP13i expression significantly reduced p-tau staining in the brainstem and spinal cord compared to control mice expressing rAAV-EGFP (61% reduction of pS202(CP13) staining in the brainstem, p < 0.001, and 46% reduction in the spinal cord, p < 0.01) (Figures 4A and 4B). Expression of PHF1i also significantly reduced p-tau staining in both regions (45% reduction in brainstem, p < 0.001, and 30% reduction in spinal cord, p < 0.05), whereas expression of PHF1s reduced p-tau staining only in the brainstem (25% reduction, p < 0.05) but not the spinal cord. Expression of CP13s, Tau5i, and Tau5s did not significantly alter p-tau pathology at 6 months of age. To ensure that reduction in p-tau staining is not a result of epitope masking, we also assessed effects of scFv/iB overexpression on additional pathological tau epitopes by staining for misfolded tau (ALZ50) and neurofibrillary tangle pathology (thioflavin-S [Thio-S]). Immunohistochemical analysis and quantification revealed that ALZ50 staining is reduced in brainstems and spinal cords of mice overexpressing CP13i compared to control mice (38% reduction in the brainstem, p < 0.01, and 40% reduction in the spinal cord, p < 0.05) (Figures S1A and S1B). Expression of PHF1i also significantly reduced misfolded tau in the brainstem (32% reduction in brainstem, p < 0.05), whereas only a trend to reduction was observed in the spinal cord. Similar results were obtained by Thio-S staining in the spinal cords of scFv/iB-expressing mice (CPi3i overexpression resulted in a 45% reduction of Thio-S positivity in the spinal cord, p < 0.05) (Figures S2A and S2B).
Figure 4.

p-Tau-Specific iBs and scFvs Significantly Reduce p-Tau Pathology in Brainstems and Spinal Cords of Homozygous JNPL3 Mice at 6 Months
Homozygous JNPL3 mice were intraspinally injected at P0 with pAAV2/8 (2 × 1010 genomes) expressing CP13, PHF1, and Tau5 iBs or scFvs. Control mice were injected with rAAV-EGFP. Mice were harvested at 6 months, and pathology was assessed by staining with pS202 (CP13) mAb. (A) Representative paraffin sections of brainstem (Br St) and spinal cord (SC) stained with pS202 (CP13) mAb. Scale bars, 500 μm (top row), 50 μm (Br St), and 100 μm (SpC). (B) Immunoreactivity analysis shows reduction in CP13 staining in brainstem area of mice expressing CP13i, PHF1i, and PHF1s, and in the spinal cord area of mice expressing CP13i and PHF1i. n = 4. Data represent mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
In order to assess effects on disease modification, we evaluated whether expression of iBs or scFvs altered the development of hindlimb paralysis in aged JNPL3 mice. Previous characterization of the JNPL3 line found that homozygous females develop hindlimb paralysis around 10 months of age.51 Mice overexpressing iBs and scFvs were aged until hindlimb paralysis developed and were euthanized at a humane endpoint. Analysis of survival curves revealed that only CP13i, PHF1i, and PHF1s significantly delayed the onset of paralysis (24.1%, 18.7%, and 12.2% delay, respectively), with CP13i expression resulting in the largest lifespan extension (Figure 5). Notably, expression levels of CP13s or Tau5s that did not reduce pathology at 6 months were ineffective in delaying hindlimb paralysis (3.7% and 4.8% delay). Tau5i did not significantly delay hindlimb paralysis, but there was a trend toward significance (8% delay). We also compared tau pathology in mice harvested due to paralysis and found no significant differences in pathological tau staining between groups, indicating that all mice had similar tau burden at the final stage of disease (Figure S3). Collectively, these data show that in a prevention paradigm, rAAV-mediated expression of selected anti-tau iBs and scFvs substantially decreased accumulation of tau pathology and modestly delayed the onset of hindlimb paralysis.
Figure 5.

p-Tau-Specific iBs and scFvs Delay the Onset of Hindlimb Paralysis of Homozygous JNPL3 Mice
JNPL3 mice for the survival cohort were intraspinally injected at P0 with pAAV2/8 (2 × 1010 genomes) expressing PHF1, CP13, and Tau5 iBs or scFvs. Control mice were injected with rAAV-EGFP. All injected mice reached a terminal state (paralysis) within 12 months post-injection (n = 10–12). (A) Kaplan-Meier survival plots show an increased time to terminal state (killed due to paralysis) for mice expressing PHF1i, CP13i, and PHF1s. ∗∗p < 0.002, ∗∗∗∗p < 0.000002. (B) Median age of terminal state and relative increase of lifespan. (C) Mantel-Cox analysis of lifespan of mice overexpressing iBs and scFvs, all cohorts compared with each other, when multiple comparisons were taken into account. ∗p ≤ 0.008, ∗∗p ≤ 0.0002, ∗∗∗p ≤ 0.00002, ∗∗∗∗p ≤ 0.000002. ns, not significant (p > 0.008).
Expression of p-Tau-Specific iBs Reduces Accumulation of Tau Pathology in rTg4510 Mice
To confirm our findings in a second tauopathy model, we injected rAAV vectors encoding the PHF1 and CP13 iBs and scFvs into the brains of neonatal rTg4510 mice. These mice express a repressible form of human tau (4R/0N) containing the FTD-associated P301L mutation and rapidly develop tau pathology in the cortex and hippocampus as early as 2.5 months when tau expression is not repressed. For this study, we excluded Tau5i and Tau5s, as they were not effective (scFv) or minimally effective (Tau5i) in the JNPL3 study, and we only assessed CP13 and PHF1 iBs and scFvs. Control mice were injected with rAAV-EGFP. We first evaluated expression of each antibody fragment in non-transgenic littermates at 3 weeks of age and found that both iBs and scFvs were expressed throughout the cortex, hippocampus, midbrain, and choroid plexus (Figure 6A). Expression of scFvs was overall lower than that of iBs, with qualitative ELISA of brain homogenates indicating that CP13s was expressed at ∼76% of CP13i levels while PHF1s levels reach only ∼56% of PHF1i (Figure 6B). We were unable to detect iBs and scFvs in the plasma (data not shown).
Figure 6.

Anti-tau iBs and scFvs Are Expressed in the Brain Following Neonatal AAV Injection
rTg4510 mice were bilaterally injected i.c.v. at P0 with pAAV2/1 (4 × 1010 genomes) expressing CP13 and PHF1 iBs or scFvs. Control mice were injected with rAAV-EGFP. Non-transgenic littermates were used to evaluate expression. (A) Representative paraffin sections of anti-c-myc staining showing widespread iB and scFv expression in the cortex, hippocampus, midbrain, and choroid plexus. Scale bar, 100 μm. (B) Relative iB and scFv levels in the brain homogenates as detected with sandwich ELISA (n = 3–4). Data represent mean ± SEM.
Next, we compared the cortical and hippocampal tau pathology in 2.5-month-old rTg4510 mice expressing scFvs or iBs to controls expressing EGFP (Figure 7). Immunohistochemical analysis and quantification of tau pathology using CP13, ALZ50, and PHF1 mAbs revealed that CP13i expression significantly reduced both phosphorylated and misfolded tau in the frontal cortex and hippocampus (15%–25% reduction in CP13 staining, p < 0.01, 36%–38% reduction in PHF1 staining, p < 0.01, and 35%–38% reduction in ALZ50 staining, p < 0.01). PHF1i also reduced p-tau in the cortex (31% reduction in CP13 staining, p < 0.05, and 36% reduction in PHF1 staining, p < 0.05) and exhibited a trend toward reduction in the hippocampus. However, PHF1i did not significantly alter levels of misfolded tau in either brain region (Figures 7A and 7B). Additionally, expression of CP13s or PHF1s did not significantly reduce pathological tau staining. Cohorts were male/female mixed, but post hoc analysis did not reveal any significant differences between the sexes. Taken together, these findings confirm our results in the JNPL3 model that iBs were more effective at reducing tau pathology than were scFvs targeting the same p-tau epitopes.
Figure 7.

CP13 and PHF1 iBs Prevent Accumulation of Phosphorylated and Misfolded Tau in rTg4510 Mice
rTg4510 mice were bilaterally injected i.c.v. at P0 with pAAV2/1 (4 × 1010 genomes) expressing PHF1 and CP13 iBs and scFvs. Control mice were injected with rAAV-EGFP. At 2.5 months after injection, brains were harvested and tau pathology was visualized on paraffin sections with CP13 (pS202), ALZ50 (misfolded tau), and PHF1 (pS396/404) mAbs. (A) Representative staining for p-tau and misfolded tau in the cortex and hippocampus. Scale bars, 500 μm (cortex), 100 μm (hippocampus). Arrows point to neurons with accumulated tau. (B) Quantification of cortical and hippocampal staining. n = 3–4. ∗p < 0.05, ∗∗p < 0.01, by unpaired two-tailed Student’s t test. Data represent mean ± SEM.
Discussion
Direct comparison of the disease-modifying potential of three tau-targeting scFvs and iBs in mouse models of tauopathy reveals that rAAV-mediated delivery of selected recombinant antibodies can significantly reduce pathology prior to the onset of neurodegeneration and delay the neurodegenerative phenotype. To our knowledge, this is the first example of a preclinical gene therapy approach or any immunotherapy targeting tau, where efficacy is observed as both reduction of tau pathology and significant delays in the degenerative phenotype induced by tau. It is also the first example of a direct comparison of the efficacy of tau-targeting scFvs and iBs derived from the same parent antibody. Both CP13i and PHFi are more effective than their cognate scFvs with respect to lowering tau pathology and delaying time to hindlimb paralysis. Indeed, although PHF1s shows some efficacy, CP13s is not effective. Such data suggest that in order to maximize the disease-modifying potential of tau-targeting immunotherapies, it may be necessary to engage tau in the intracellular compartment, or perhaps even in both extracellular and intracellular compartments. These data also reinforce previous observations that effector functions of an antibody are not needed for an anti-tau immunotherapy to show some degree of efficacy, although additional functionalization of the recombinant antibody fragments may be necessary to further enhance efficacy.
In general, there is a lack of published preclinical data showing that any form of tau immunotherapy is sufficiently optimized. Most studies in preclinical models show modest effects on tau pathology, and only a few provide evidence for efficacy on other endpoints (behavioral or neurodegenerative).21,23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34,52 Furthermore, almost all studies of immunotherapies in tau models report on prevention paradigms where the treatment is initiated before pathology onset or when pathology is limited. However, previous studies have not examined long-term endpoints such as induction of a robust neurodegenerative phenotype as we have in the present study. Given that our data showing ∼30%–50% reductions in pathology at a time point before neurodegeneration translates into ∼10%–20% prolongation of time to paralysis, we would caution that a modest reduction in pathology may not translate into sufficient disease modification upon translation to observe a robust effect in a human trial. Indeed, numerous preclinical studies in amyotrophic lateral sclerosis (ALS) models have reported modest, but significant (10%–20%) survival benefits, and such therapeutic approaches have almost invariably failed when tested in humans (reviewed in Petrov et al.53). Some may argue that most tauopathy models have such robust and rapid progression of pathology (over months) relative to humans with tauopathies, where the disease progresses over years or decades, that therapeutics with modest disease modification in mouse models are worth advancing. However, given that prevention or early intervention trials in human tauopathy will be difficult to initiate and conduct, we would suggest that further optimization and evidence for disease modification at time points when significant pathology and neurodegeneration are present is needed before advancing any tau immunotherapy to the clinic. Especially when using a gene therapy-based approach to deliver the recombinant antibodies, it would seem that further preclinical optimization is warranted. Any gene therapy trial in humans with a degenerative tauopathy will have some inherent risk and will be initially tested on a relatively small number of patients. Such trials will likely produce ambiguous clinical readouts unless therapeutics are sufficiently optimized to achieve high degrees of target engagement and significant impacts on disease course.
Tau is a complex therapeutic target for immunotherapies. It is presumed that the pathological effects of tau are largely mediated by intracellular aggregation/accumulation and that intracellular tau pathology may be induced by extracellular tau seeds. Whether cell-to-cell spread of tau via extracellular seeds truly occurs in the human brain is not known; however, a number of observations indicate that this prion-like spread contributes in part to the development of brain and spinal cord pathology in some tau models.10,12, 13, 14, 15, 16, 17, 18, 19, 20,54 Indeed, one of the key findings to support this latter assertion is that antibodies that target extracellular tau can block pathology spread in tauopathy models.21,52 As the pathological seeds and forms of tau that are most tightly linked to neurodegeneration are not completely characterized, our finding that targeting tau with iBs is more effective than scFvs needs to be interpreted with some degree of caution. Certainly, these data suggest that intracellular targeting might be more effective, but it is unclear whether the epitopes targeted in this study are present in endogenous extracellular tau seeds. Furthermore, given that a large portion of extracellular tau is truncated before the microtubule binding domain and thought to be incapable of seeding,8,9,55 it is possible that engagement of this form of extracellular tau could actually reduce target engagement of seeding-competent forms. Such a phenomenon could explain the lack of efficacy observed with CP13s, which could bind these truncated extracellular tau species.
There are many possible epitopes within tau as well as assemblies or complexes that might theoretically be targeted by immunologic approaches. Indeed, tau has an extremely large number of (1) epitopes, both phosphorylated and not,56, 57, 58 (2) truncations,59 (3) other post-translational modifications,60,61 (4) aggregation states,62,63 and (5) numerous associations with other proteins.64,65 The data generated in the present study do support the concept that targeting phosphorylated epitopes associated with pathology results in a higher degree of efficacy. However, there are many other factors that might influence the efficacy of individual recombinant antibody fragments. In order to draw more general conclusions, a much larger panel of recombinant antibodies targeting many different epitopes will have to be evaluated in comparable preclinical studies. Alternatively, the field must identify the most pathogenic or most prion-like forms of tau, and then develop recombinant antibodies that are highly selective for those forms.66, 67, 68 The identification of such species may be possible, but it is not clear that there is really a single toxic or prion-like species. More likely, there are a number of forms of tau with various toxic properties. Even within our own data, there is some suggestion that targeting total tau is having a very modest impact. Although not significant after correction for multiple testing, Tau5i did show a slight benefit in survival compared to control or the Tau5s. Furthermore, the contrast between the effect of CP13i (highly effective) and CP13s (no efficacy) is striking. As noted above, besides intracellular versus extracellular targeting, one can only speculate about other possible reasons for this difference.
We note that all tau-targeting scFvs and iBs in this study show target engagement and the ability to reduce insoluble tau in HEK293 cell culture models. Expression of secreted scFvs also significantly reduced soluble tau species in vitro, while iB expression did not. These results were somewhat surprising, as scFvs are directed to the secretory pathway and would therefore be less likely to interact with cytoplasmic tau. However, it is possible that partially misfolded scFvs could interact with intracellular tau after being exported from the endoplasmic reticulum (ER) during ER-associated protein degradation (ERAD), as a fraction of secreted proteins are known to misfold and undergo intracellular degradation. Another potential explanation is that scFvs may re-enter cells after being secreted into the media, as a previous study has found anti-tau scFvs to co-localize with intracellular tau in vivo following peripheral injection.69 Despite reducing multiple tau species in vitro, CP13 scFv and Tau5 scFv did not significantly alter tau pathology or neurodegenerative phenotypes in vivo. Our data indicate that the mechanisms through which scFvs alter tau levels in vitro are likely not reflective of those that occur in vivo. It is unfortunate that the HEK293T cell culture model did not directly predict in vivo efficacy, as in vivo studies in tau models are time and resource intensive. Nevertheless, such culture studies are valuable, as they enable a rapid assessment of expression and binding of recombinant tau scFvs or iBs. For iBs, such studies are requisite, as iBs are often unstable, even when derived from a scFv that shows excellent stability.
If tau immunotherapy is not yet optimized, then it is important to think about the steps that might be required to improve efficacy. First, as scFvs and iBs should be targeting different pools of tau, one simple concept is that an efficacious scFv and iB should show additive or synergistic effects. Second, these data and those generated by others suggest the effector functions are not needed, but these data do not mean that functionalizing scFvs or iBs will not improve efficacy. Given the small size of these antibody fragments, there are many functionalizations that might be considered, although they would likely be different for iBs and scFvs. For scFvs, one might want to target to microglial receptors or the BBB to enhance clearance. For iBs, one might want to add functional motifs that increase protein clearance. Multimerization of scFvs or iBs might also increase avidity and specificity for toxic aggregated forms of tau.
In addition, we also need to more thoroughly explore whether these recombinant antibodies can alter disease in studies where the therapeutic is delivered long after onset of disease in the model. This preclinical therapeutic paradigm will be greatly facilitated by novel rAAV capsids that enhance CNS transduction upon peripheral delivery.70 A final concern about any gene therapy-based human tau therapy is whether one can achieve enough transgene expression to sufficiently engage the target. Sufficient transgene expression might be easier to achieve with scFvs that are predicted to function in a non-cell-autonomous fashion. In contrast, there will likely need to be improvements in widespread delivery to neurons in the adult brain in order for an iB-based gene therapy approach to have potential disease-modifying impacts.
In summary, our data demonstrate the therapeutic potential of a gene therapy-based approach to targeting tau with recombinant antibodies. These data also highlight the need for continued optimization of this approach. Evidence for successful optimization might include robust disease modification in a preclinical model defined as suppression of the neurodegenerative phenotype by >50% even when delivery of the therapy is delayed until the model has robust pre-existing pathology. Given the unmet medical need and the lack of alternative therapies, we think that additional studies to optimize both the tau-targeting recombinant antibody fragments and delivery vectors are warranted.
Materials and Methods
Mice
All animal husbandry procedures performed were approved by the Institutional Animal Care and Use Committee. Homozygous JNPL3 mice (on a Swiss Webster background) were generated by breeding non-sibling males and females of hemizygous JNPL3 mice. Only female JNPL3 mice were used in this study. Parent rTg4510 mice were obtained from Dr. J. Lewis (University of Florida). The rTg4510 mice were generated by crossing the Tg4510 responder line expressing human P301L mutant microtubule-associated protein tau (MAPT) with the CaMKIIα-tTA activator line.71 Mice were maintained under a 12-h light/12-h dark cycle, group housed, and had free access to food and water.
Production of rAAV
Purified rAAVs for injection were produced by plasmid transfection with helper plasmids in HEK293T cells as described previously.4 Forty-eight hours after transfection, cells were harvested and lysed by freeze thawing. Viral particles were then isolated using a discontinuous iodixanol gradient and affinity purified on a HiTrap HQ column (Amersham). The genomic titer of each virus was determined by quantitative PCR.
Neonatal rAAV Injections
rTg4510 was injected with 2 μL of rAAV intracerebroventricularly (i.c.v.) into both hemispheres using a 10-μL Hamilton syringe with a 30G needle on postnatal day 0 (P0) as described before72 and aged until 2.5 months. Neonatal JNPL3 pups were intraspinally injected on P0 as described before73 and aged until 6 months (pathology cohort) or development of hindlimb paralysis (survival cohort).
mRNA Isolation, cDNA Synthesis, Amplification of cDNAs Encoding VH and VL Regions, and Construction of scFvs
mRNA was isolated from hybridoma cell lines using a mRNA isolation kit (QIAGEN). cDNA was synthesized using Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega) and random hexamers. The cDNA was then poly(G)-tailed with terminal transferase (NE Biolabs). cDNAs encoding the variable heavy (VH) and variable light (VL) chains were amplified using anchor PCR with a forward poly(C) anchor primer and a reverse primer specific for the constant region sequence of immunoglobulin (Ig)G1, as previously described.74 PCR products were then sequenced using the same primers, and the consensus sequences of both variable regions were determined. cDNAs encoding scFvs of three anti-tau mAbs were constructed by ligating the VH and VL cDNAs in the VH-linker-VL orientation separated by a 15-aa glycine/serine linker (GGGGS)3 (Table 1). Optimized iBs were synthesized by GenScript by making mutations in the scFv sequence previously shown to promote stability of intracellularly expressed scFvs.49 All scFvs and iBs were ligated into rAAV vectors under the control of the hybrid cytomegalovirus enhancer/chicken β-actin (hCBA) promoter and in frame with a c-myc tag, polyhistidine tag, woodchuck hepatitis virus posttranscriptional regulatory element (WPRE), and the bovine growth hormone (BGH) poly(A).
Target Engagement by Immunohistochemistry and ELISA
Cell lysates and media containing scFvs and iBs were diluted 1:10 with PBS and applied to paraffin-fixed brain tissue slides from 10-month-old rTg4510 mice. Immunohistochemical staining was done with a biotinylated anti-c-myc secondary antibody. For direct ELISA, MaxiSorp plates (Thermo Scientific, Waltham, MA, USA) were coated with 1 μg/mL recombinant tau, or the same tau protein phosphorylated in vitro (p-tau), prepared as described previously75 in PBS and blocked with 5% fetal bovine serum (FBS)/PBS. Cell lysates and media were applied to plates, chicken anti-c-myc secondary antibody was conjugated to horseradish peroxidase (HRP; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) was used as detection, and absorbance was measured at 450 nm.
Cell Culture, Transfection, and Induction of Intracellular S320F Tau Aggregates
HEK293T cells, derived from human embryonic kidney cells, were maintained using Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% FBS and 100 U/mL penicillin/100 μg/mL streptomycin. A rAAV expression plasmid encoding full-length human tau (4R/0N) containing the FTDP-17-associated S320F mutation76 was provided by Dr. Benoit Giasson (CTRND, UF). Cells were transiently transfected in 12-well plates at ∼30% confluence with a mixture of 0.4 μg/mL S320F tau plasmid and 0.4 μg/mL of plasmids encoding anti-tau iBs and scFvs using calcium phosphate as described previously.50 Cells were harvested for analysis 48 h after transfection.
Biochemical Cellular Fractionation
For biochemical fractionation, HEK293T cells were washed with PBS and lysed in CSK buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 20 mM NaF, 2 mM EDTA, 1% Triton X-100) with protease inhibitors and incubated on ice for 10 min. Lysates were then centrifuged at 100,000 × g for 30 min at 4°C. Supernatants were kept as the Triton X-100-soluble fraction. Pellets were then washed with CSK buffer, re-sedimented at 100,000 × g for 30 min, and sonicated in 2% SDS to produce the Triton X-100-insoluble fractions.
SDS-PAGE and Immunoblotting
Protein concentrations were determined by a bicinchoninic acid (BCA) assay (Thermo Scientific) using bovine serum albumin (BSA) as the standard. SDS sample buffer was added and 5 μg of protein from the Triton X-100-insoluble fraction and 15 μg of protein from Triton X-100-soluble supernatant were separated on 4%–12% SDS-PAGE gels (Bio-Rad) and electrophoretically transferred to polyvinylidene fluoride membranes, as described previously.77 Membranes were blocked in 0.5% casein for 1 h and incubated overnight at 4°C submerged in 0.5% casein containing primary antibodies against c-myc, tau phosphorylated at S202/T205 (AT8; 1:1,000), total tau (T44-3029 from Dr. Benoit Giasson; 1:5,00075), or β-actin (AC15 Sigma-Aldrich; 1:5,000). Membranes were then washed three times with Tris-buffered saline (TBS) and incubated for 1 h with fluorophore-conjugated secondary antibodies Alexa Fluor 680 anti-mouse IgG (Thermo Fisher Scientific; 1:10,000) and IRDye 800 goat anti-rabbit IgG (LI-COR Biosciences; 1:10,000). Membranes were then washed three times with TBS and protein bands were detected using the multiplex LI-COR Odyssey Infrared Imaging system (LI-COR Biosciences).
Measurement of scFv/iB Expression in the Brain, Spinal Cord, and Plasma
Frozen hemi-brains and spinal cords from non-transgenic mice injected with rAAV scFv/iB were homogenized in radioimmunoprecipitation assay (RIPA) buffer (Thermo Fisher Scientific), centrifuged at 43,000 × g for 1 h, and supernatants were subjected to direct ELISA with anti-polyhistidine antibody as capture and anti-c-myc-HRP as detection. The same procedure was performed on plasma from injected mice. All ELISA results were analyzed using SoftMax Pro software (Molecular Devices).
Immunohistochemical Imaging and Image Processing
Brains were sagittally dissected and the right brain was fixed in 4% paraformaldehyde. Whole spinal columns were immersion fixed in 4% paraformaldehyde. Immunohistochemical staining was done on paraffin-embedded brain or spinal sections using mAbs specific for p-tau (PHF1, pS396/404, CP13, pS202) or a misfolded conformation of tau (ALZ50, AA2-10, and AA312-342, 1:500; P. Davies).6,45, 46, 47, 48 Color development was done using Vector ImmPRESS reagents (Vector Laboratories). Images were captured using the ScanScope XT image scanner (Aperio) and analyzed using the ImageScope program. Brightness and contrast alterations were applied identically on captured images using Adobe Photoshop CS3. Thio-S (Sigma-Aldrich) staining was performed on paraffin-embedded spinal cord sections using established protocols. Fluorescently stained sections were captured using the Zeiss slide scanner and analyzed using ZEN imaging software. For Thio-S quantification, one section per sample was used by a blinded observer to calculate the number of positive cells per spinal cord in the lumbar area using ImageJ.78
Statistical Analysis
Data were analyzed using Prism 6 (GraphPad) and presented as mean ± SEM. Specific tests used are noted in the figure legends. Overall data were tested for normality and, after being deemed to have a normal distribution, were analyzed via one-way ANOVA followed by Dunnett’s multiple comparison test, two-way ANOVA, or Dunn’s test. Survival curves were compared two at a time via Mantel-Cox analysis, and p values were adjusted. Sex differences in rTg4510 were assessed by a post hoc analysis of the cohorts. Final images were created using Photoshop CS3 (Adobe).
Author Contributions
Y.L. and T.E.G. planned experiments; Y.L., O.S., F.B., and V.O. performed animal studies; Y.L. and M.S.G. performed cell culture experiments; P.E.C., C.C.-D., and M.S.G. did molecular cloning; B.I.G. and P.D. provided valuable hybridoma lines and antibodies; J.L. provided mice strains; and Y.L., M.S.G., and T.E.G. wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was partially supported by funding from the National Institutes of Health (AG018454 to T.E.G.), an Alzheimer’s Association New Investigator Grant (NIRG-08-91673 to Y.L.), the BrightFocus Foundation (A2014105S to Y.L.), the Stop Alzheimer’s Now Fund (to T.E.G.), the Ed and Ethel Moore AD Research Program from the Florida Department of Health (7AZ19 to Y.L.), and by the Brooks Hogard Fellowship (M.S.G.). We would like to thank the Center for Translational Research in Neurodegenerative Disease and the McKnight Brain Institute.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.10.007.
Contributor Information
Todd E. Golde, Email: tgolde@ufl.edu.
Yona Levites, Email: levites.yona@ufl.edu.
Supplemental Information
References
- 1.Terry R.D., Katzman R. Senile dementia of the Alzheimer type. Ann. Neurol. 1983;14:497–506. doi: 10.1002/ana.410140502. [DOI] [PubMed] [Google Scholar]
- 2.Terry R.D. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J. Neuropathol. Exp. Neurol. 1996;55:1023–1025. [PubMed] [Google Scholar]
- 3.Alzheimer A., Stelzmann R.A., Schnitzlein H.N., Murtagh F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 4.Hutton M. Molecular genetics of chromosome 17 tauopathies. Ann. N Y Acad. Sci. 2000;920:63–73. doi: 10.1111/j.1749-6632.2000.tb06906.x. [DOI] [PubMed] [Google Scholar]
- 5.Hutton M., Lendon C.L., Rizzu P., Baker M., Froelich S., Houlden H., Pickering-Brown S., Chakraverty S., Isaacs A., Grover A. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 6.Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 7.Ramsden M., Kotilinek L., Forster C., Paulson J., McGowan E., SantaCruz K., Guimaraes A., Yue M., Lewis J., Carlson G. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J. Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cicognola C., Brinkmalm G., Wahlgren J., Portelius E., Gobom J., Cullen N.C., Hansson O., Parnetti L., Constantinescu R., Wildsmith K. Novel tau fragments in cerebrospinal fluid: relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 2019;137:279–296. doi: 10.1007/s00401-018-1948-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson G.V., Seubert P., Cox T.M., Motter R., Brown J.P., Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J. Neurochem. 1997;68:430–433. doi: 10.1046/j.1471-4159.1997.68010430.x. [DOI] [PubMed] [Google Scholar]
- 10.Brier M.R., Gordon B., Friedrichsen K., McCarthy J., Stern A., Christensen J., Owen C., Aldea P., Su Y., Hassenstab J. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci. Transl. Med. 2016;8:338ra66. doi: 10.1126/scitranslmed.aaf2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanmert D., Cantlon A., Muratore C.R., Jin M., O’Malley T.T., Lee G., Young-Pearse T.L., Selkoe D.J., Walsh D.M. C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J. Neurosci. 2015;35:10851–10865. doi: 10.1523/JNEUROSCI.0387-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson S.J., Kerridge C., Cooper J., Cavallini A., Falcon B., Cella C.V., Landi A., Szekeres P.G., Murray T.K., Ahmed Z. Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S tau. J. Neurosci. 2016;36:762–772. doi: 10.1523/JNEUROSCI.3542-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iba M., McBride J.D., Guo J.L., Zhang B., Trojanowski J.Q., Lee V.M. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015;130:349–362. doi: 10.1007/s00401-015-1458-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo J.L., Narasimhan S., Changolkar L., He Z., Stieber A., Zhang B., Gathagan R.J., Iba M., McBride J.D., Trojanowski J.Q., Lee V.M. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J. Exp. Med. 2016;213:2635–2654. doi: 10.1084/jem.20160833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmed Z., Cooper J., Murray T.K., Garn K., McNaughton E., Clarke H., Parhizkar S., Ward M.A., Cavallini A., Jackson S. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667–683. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frost B., Jacks R.L., Diamond M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaufman S.K., Sanders D.W., Thomas T.L., Ruchinskas A.J., Vaquer-Alicea J., Sharma A.M., Miller T.M., Diamond M.I. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. 2016;92:796–812. doi: 10.1016/j.neuron.2016.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato C., Barthélemy N.R., Mawuenyega K.G., Patterson B.W., Gordon B.A., Jockel-Balsarotti J., Sullivan M., Crisp M.J., Kasten T., Kirmess K.M. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;98:861–864. doi: 10.1016/j.neuron.2018.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strang K.H., Croft C.L., Sorrentino Z.A., Chakrabarty P., Golde T.E., Giasson B.I. Distinct differences in prion-like seeding and aggregation between tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018;293:2408–2421. doi: 10.1074/jbc.M117.815357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wegmann S., Bennett R.E., Delorme L., Robbins A.B., Hu M., McKenzie D., Kirk M.J., Schiantarelli J., Tunio N., Amaral A.C. Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 2019;5:eaaw6404. doi: 10.1126/sciadv.aaw6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yanamandra K., Kfoury N., Jiang H., Mahan T.E., Ma S., Maloney S.E., Wozniak D.F., Diamond M.I., Holtzman D.M. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braak H., Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging. 1995;16:271–278, discussion 278–284. doi: 10.1016/0197-4580(95)00021-6. [DOI] [PubMed] [Google Scholar]
- 23.Boutajangout A., Ingadottir J., Davies P., Sigurdsson E.M. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J. Neurochem. 2011;118:658–667. doi: 10.1111/j.1471-4159.2011.07337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Troquier L., Caillierez R., Burnouf S., Fernandez-Gomez F.J., Grosjean M.E., Zommer N., Sergeant N., Schraen-Maschke S., Blum D., Buee L. Targeting phospho-Ser422 by active tau immunotherapy in the THYTau22 mouse model: a suitable therapeutic approach. Curr. Alzheimer Res. 2012;9:397–405. doi: 10.2174/156720512800492503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sankaranarayanan S., Barten D.M., Vana L., Devidze N., Yang L., Cadelina G., Hoque N., DeCarr L., Keenan S., Lin A. Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS ONE. 2015;10:e0125614. doi: 10.1371/journal.pone.0125614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Theunis C., Crespo-Biel N., Gafner V., Pihlgren M., López-Deber M.P., Reis P., Hickman D.T., Adolfsson O., Chuard N., Ndao D.M. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS ONE. 2013;8:e72301. doi: 10.1371/journal.pone.0072301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.d’Abramo C., Acker C.M., Jimenez H.T., Davies P. Tau passive immunotherapy in mutant P301L mice: antibody affinity versus specificity. PLoS ONE. 2013;8:e62402. doi: 10.1371/journal.pone.0062402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chai X., Wu S., Murray T.K., Kinley R., Cella C.V., Sims H., Buckner N., Hanmer J., Davies P., O’Neill M.J. Passive immunization with anti-tau antibodies in two transgenic models: reduction of tau pathology and delay of disease progression. J. Biol. Chem. 2011;286:34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boutajangout A., Quartermain D., Sigurdsson E.M. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J. Neurosci. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.d’Abramo C., Acker C.M., Jimenez H., Davies P. Passive Immunization in JNPL3 transgenic mice using an array of phospho-tau specific antibodies. PLoS ONE. 2015;10:e0135774. doi: 10.1371/journal.pone.0135774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krishnamurthy P.K., Deng Y., Sigurdsson E.M. Mechanistic studies of antibody-mediated clearance of tau aggregates using an ex vivo brain slice model. Front. Psychiatry. 2011;2:59. doi: 10.3389/fpsyt.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Asuni A.A., Boutajangout A., Quartermain D., Sigurdsson E.M. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collin L., Bohrmann B., Göpfert U., Oroszlan-Szovik K., Ozmen L., Grüninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain. 2014;137:2834–2846. doi: 10.1093/brain/awu213. [DOI] [PubMed] [Google Scholar]
- 34.Gu J., Congdon E.E., Sigurdsson E.M. Two novel tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J. Biol. Chem. 2013;288:33081–33095. doi: 10.1074/jbc.M113.494922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Congdon E.E., Gu J., Sait H.B., Sigurdsson E.M. Antibody uptake into neurons occurs primarily via clathrin-dependent Fcγ receptor endocytosis and is a prerequisite for acute tau protein clearance. J. Biol. Chem. 2013;288:35452–35465. doi: 10.1074/jbc.M113.491001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura K., Hirai H., Torashima T., Miyazaki T., Tsurui H., Xiu Y., Ohtsuji M., Lin Q.S., Tsukamoto K., Nishimura H. CD3 and immunoglobulin G Fc receptor regulate cerebellar functions. Mol. Cell. Biol. 2007;27:5128–5134. doi: 10.1128/MCB.01072-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kam T.I., Song S., Gwon Y., Park H., Yan J.J., Im I., Choi J.W., Choi T.Y., Kim J., Song D.K. FcγRIIb mediates amyloid-β neurotoxicity and memory impairment in Alzheimer’s disease. J. Clin. Invest. 2013;123:2791–2802. doi: 10.1172/JCI66827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Kleij H., Charles N., Karimi K., Mao Y.K., Foster J., Janssen L., Chang Yang P., Kunze W., Rivera J., Bienenstock J. Evidence for neuronal expression of functional Fc (epsilon and gamma) receptors. J. Allergy Clin. Immunol. 2010;125:757–760. doi: 10.1016/j.jaci.2009.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McEwan W.A., Falcon B., Vaysburd M., Clift D., Oblak A.L., Ghetti B., Goedert M., James L.C. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. USA. 2017;114:574–579. doi: 10.1073/pnas.1607215114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levites Y., Smithson L.A., Price R.W., Dakin R.S., Yuan B., Sierks M.R., Kim J., McGowan E., Reed D.K., Rosenberry T.L. Insights into the mechanisms of action of anti-Aβ antibodies in Alzheimer’s disease mouse models. FASEB J. 2006;20:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- 41.Vitale F., Giliberto L., Ruiz S., Steslow K., Marambaud P., d’Abramo C. Anti-tau conformational scFv MC1 antibody efficiently reduces pathological tau species in adult JNPL3 mice. Acta Neuropathol. Commun. 2018;6:82. doi: 10.1186/s40478-018-0585-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ising C., Gallardo G., Leyns C.E.G., Wong C.H., Jiang H., Stewart F., Koscal L.J., Roh J., Robinson G.O., Remolina Serrano J., Holtzman D.M. AAV-mediated expression of anti-tau scFvs decreases tau accumulation in a mouse model of tauopathy. J. Exp. Med. 2017;214:1227–1238. doi: 10.1084/jem.20162125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu W., Zhao L., Blackman B., Parmar M., Wong M.Y., Woo T., Yu F., Chiuchiolo M.J., Sondhi D., Kaminsky S.M. Vectored intracerebral immunization with the anti-tau monoclonal antibody PHF1 markedly reduces tau pathology in mutant tau transgenic mice. J. Neurosci. 2016;36:12425–12435. doi: 10.1523/JNEUROSCI.2016-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gallardo G., Wong C.H., Ricardez S.M., Mann C.N., Lin K.H., Leyns C.E.G., Jiang H., Holtzman D.M. Targeting tauopathy with engineered tau-degrading intrabodies. Mol. Neurodegener. 2019;14:38. doi: 10.1186/s13024-019-0340-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duff K., Knight H., Refolo L.M., Sanders S., Yu X., Picciano M., Malester B., Hutton M., Adamson J., Goedert M. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol. Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- 46.Greenberg S.G., Davies P., Schein J.D., Binder L.I. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J. Biol. Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- 47.Otvos L., Jr., Feiner L., Lang E., Szendrei G.I., Goedert M., Lee V.M. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J. Neurosci. Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 48.Porzig R., Singer D., Hoffmann R. Epitope mapping of mAbs AT8 and Tau5 directed against hyperphosphorylated regions of the human tau protein. Biochem. Biophys. Res. Commun. 2007;358:644–649. doi: 10.1016/j.bbrc.2007.04.187. [DOI] [PubMed] [Google Scholar]
- 49.Ewert S., Honegger A., Plückthun A. Stability improvement of antibodies for extracellular and intracellular applications: CDR grafting to stable frameworks and structure-based framework engineering. Methods. 2004;34:184–199. doi: 10.1016/j.ymeth.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 50.Sobrido M.J., Miller B.L., Havlioglu N., Zhukareva V., Jiang Z., Nasreddine Z.S., Lee V.M., Chow T.W., Wilhelmsen K.C., Cummings J.L. Novel tau polymorphisms, tau haplotypes, and splicing in familial and sporadic frontotemporal dementia. Arch. Neurol. 2003;60:698–702. doi: 10.1001/archneur.60.5.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin W.L., Zehr C., Lewis J., Hutton M., Yen S.H., Dickson D.W. Progressive white matter pathology in the spinal cord of transgenic mice expressing mutant (P301L) human tau. J. Neurocytol. 2005;34:397–410. doi: 10.1007/s11068-006-8726-0. [DOI] [PubMed] [Google Scholar]
- 52.Yanamandra K., Jiang H., Mahan T.E., Maloney S.E., Wozniak D.F., Diamond M.I., Holtzman D.M. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann. Clin. Transl. Neurol. 2015;2:278–288. doi: 10.1002/acn3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petrov D., Mansfield C., Moussy A., Hermine O. ALS clinical trials review: 20 Years of failure. Are we any closer to registering a new treatment? Front. Aging Neurosci. 2017;9:68. doi: 10.3389/fnagi.2017.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo J.L., Lee V.M. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sato C., Barthélemy N.R., Mawuenyega K.G., Patterson B.W., Gordon B.A., Jockel-Balsarotti J., Sullivan M., Crisp M.J., Kasten T., Kirmess K.M. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97:1284–1298.e7. doi: 10.1016/j.neuron.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spillantini M.G., Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–622. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- 57.Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 58.Hanger D.P., Anderton B.H., Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 59.Novak M., Kabat J., Wischik C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993;12:365–370. doi: 10.1002/j.1460-2075.1993.tb05665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martin L., Latypova X., Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem. Int. 2011;58:458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 61.Gong C.X., Liu F., Grundke-Iqbal I., Iqbal K. Post-translational modifications of tau protein in Alzheimer’s disease. J. Neural Transm. (Vienna) 2005;112:813–838. doi: 10.1007/s00702-004-0221-0. [DOI] [PubMed] [Google Scholar]
- 62.Ayers J.I., Giasson B.I., Borchelt D.R. Prion-like spreading in tauopathies. Biol. Psychiatry. 2018;83:337–346. doi: 10.1016/j.biopsych.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Avila J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006;580:2922–2927. doi: 10.1016/j.febslet.2006.02.067. [DOI] [PubMed] [Google Scholar]
- 64.Strang K.H., Golde T.E., Giasson B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Invest. 2019;99:912–928. doi: 10.1038/s41374-019-0197-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brunden K.R., Trojanowski J.Q., Lee V.M. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat. Rev. Drug Discov. 2009;8:783–793. doi: 10.1038/nrd2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dujardin S., Commins C., Lathuiliere A., Beerepoot P., Fernandes A.R., Kamath T.V., De Los Santos M.B., Klickstein N., Corjuc D.L., Corjuc B.T. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020;26:1256–1263. doi: 10.1038/s41591-020-0938-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Falcon B., Zhang W., Murzin A.G., Murshudov G., Garringer H.J., Vidal R., Crowther R.A., Ghetti B., Scheres S.H.W., Goedert M. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561:137–140. doi: 10.1038/s41586-018-0454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janelidze S., Stomrud E., Smith R., Palmqvist S., Mattsson N., Airey D.C., Proctor N.K., Chai X., Shcherbinin S., Sims J.R. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020;11:1683. doi: 10.1038/s41467-020-15436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Krishnaswamy S., Lin Y., Rajamohamedsait W.J., Rajamohamedsait H.B., Krishnamurthy P., Sigurdsson E.M. Antibody-derived in vivo imaging of tau pathology. J. Neurosci. 2014;34:16835–16850. doi: 10.1523/JNEUROSCI.2755-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chakrabarty P., Rosario A., Cruz P., Siemienski Z., Ceballos-Diaz C., Crosby K., Jansen K., Borchelt D.R., Kim J.Y., Jankowsky J.L. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS ONE. 2013;8:e67680. doi: 10.1371/journal.pone.0067680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ayers J.I., Fromholt S., Sinyavskaya O., Siemienski Z., Rosario A.M., Li A., Crosby K.W., Cruz P.E., DiNunno N.M., Janus C. Widespread and efficient transduction of spinal cord and brain following neonatal AAV injection and potential disease modifying effect in ALS mice. Mol. Ther. 2015;23:53–62. doi: 10.1038/mt.2014.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gilliland L.K., Norris N.A., Marquardt H., Tsu T.T., Hayden M.S., Neubauer M.G., Yelton D.E., Mittler R.S., Ledbetter J.A. Rapid and reliable cloning of antibody variable regions and generation of recombinant single chain antibody fragments. Tissue Antigens. 1996;47:1–20. doi: 10.1111/j.1399-0039.1996.tb02509.x. [DOI] [PubMed] [Google Scholar]
- 75.Strang K.H., Goodwin M.S., Riffe C., Moore B.D., Chakrabarty P., Levites Y., Golde T.E., Giasson B.I. Generation and characterization of new monoclonal antibodies targeting the PHF1 and AT8 epitopes on human tau. Acta Neuropathol. Commun. 2017;5:58. doi: 10.1186/s40478-017-0458-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rosso S.M., van Herpen E., Deelen W., Kamphorst W., Severijnen L.A., Willemsen R., Ravid R., Niermeijer M.F., Dooijes D., Smith M.J. A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick’s disease. Ann. Neurol. 2002;51:373–376. doi: 10.1002/ana.10140. [DOI] [PubMed] [Google Scholar]
- 77.Li A., Ceballos-Diaz C., DiNunno N., Levites Y., Cruz P.E., Lewis J., Golde T.E., Chakrabarty P. IFN-γ promotes τ phosphorylation without affecting mature tangles. FASEB J. 2015;29:4384–4398. doi: 10.1096/fj.15-275834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.