

Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal dominant disorder characterized by a progressive, asymmetric weakening of muscles, starting with those in the upper body. It is caused by aberrant expression of the double homeobox protein 4 gene (DUX4) in skeletal muscle. FSHD is currently incurable. We propose to develop a therapy for FSHD using antisense 2′-O-methoxyethyl (2′-MOE) gapmers, to knock down DUX4 mRNA expression. Using immortalized patient-derived muscle cells and local intramuscular injections in the FLExDUX4 FSHD mouse model, we showed that our designed 2′-MOE gapmers significantly reduced DUX4 transcript levels in vitro and in vivo, respectively. Furthermore, in vitro, we observed significantly reduced expression of DUX4-activated downstream targets, restoration of FSHD signature genes by RNA sequencing, significant improvements in myotube morphology, and minimal off-target activity. This work facilitates the development of a promising candidate therapy for FSHD and lays down the foundation for in vivo systemic treatment studies.
Keywords: facioscapulohumeral muscular dystrophy, FSHD, DUX4, 2’-MOE gapmers, mRNA knockdown, antisense therapy, immortalized FSHD patient-derived cell line, FLExDUX4 mice, skeletal muscle, genetic disorder
Graphical Abstract
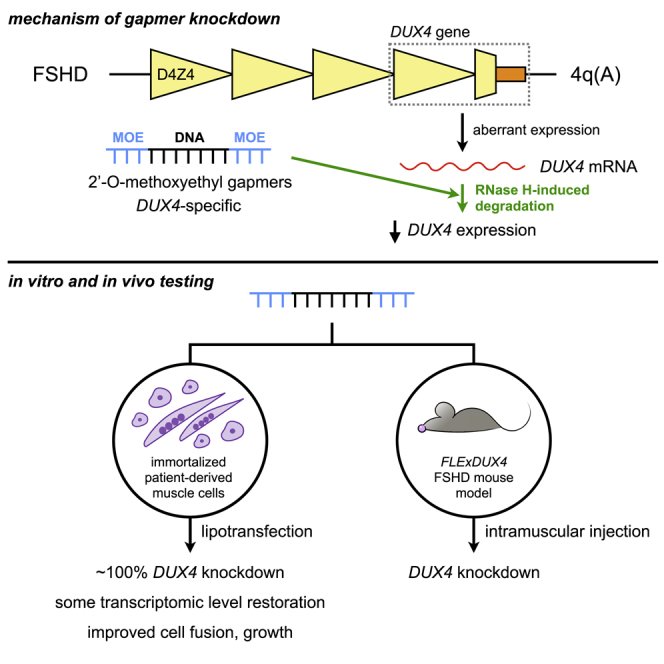
Facioscapulohumeral muscular dystrophy is a disabling, incurable genetic disease caused by aberrant DUX4 expression in skeletal muscle. Using 2′-O-methoxyethyl antisense gapmers, Lim and colleagues reduced DUX4 expression in patient-derived muscle cells with nearly 100% efficacy, improving cellular phenotypes. They also demonstrate DUX4 knockdown in a mouse model of the disease.
Introduction
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common form of muscular dystrophy in the world, with 1:8,000–22,000 people affected globally.1,2 FSHD is a disabling, autosomal dominant disorder primarily characterized by progressive muscle weakness that begins in the face, shoulders, and upper limbs, followed by the lower extremities.1 Muscle involvement in FSHD is distinctly asymmetric, with disease severity varying across individuals. Around 15%–20% of patients are wheelchair-bound.3 In certain cases, often in early-onset FSHD, patients present with additional extra-muscular features, e.g., hearing loss, retinal vasculopathy, and cognitive impairment.4, 5, 6 Surgery and physical therapy, among others, are available to help manage symptoms and improve patient quality of life.7 However, these approaches do not treat FSHD itself, and the disease remains incurable at present.
FSHD is caused by mutations promoting aberrant expression of the double homeobox protein 4 gene (DUX4) in skeletal muscle. On chromosome 4q35, there is a macrosatellite array of 3.3-kb D4Z4 repeats that is typically 11–100 repeats long in healthy individuals. Each D4Z4 unit contains the first two exons of the DUX4 gene.8 Its third and final exon is found immediately after the most distal unit of the array and possesses a functional polyadenylation signal (PAS) only in the 4qA haplotype.9 The presence of the PAS is required for successful DUX4 transcription. In most tissues, including skeletal muscle, the D4Z4 array is normally hypermethylated after early embryonic development, silencing DUX4 expression.10,11 However, in FSHD, D4Z4 methylation is reduced either through contraction of the array, mutations in genes coding for epigenetic regulators, or a combination of both.1,9,11, 12, 13 It is now known that strict cutoffs of 4q35 D4Z4 array length (i.e., ≤10 repeats) do not satisfactorily explain penetrance of the FSHD phenotype. For instance, individuals with less than 10 repeat units in this array can be asymptomatic.12 Although further study of the underlying genetics in this disease is warranted, it remains that these culminate in the de-repression of DUX4, which produces the DUX4 transcription factor whose downstream activities are thought to cause FSHD.14
Given its central role in FSHD, reducing DUX4 expression has been the focus of a number of therapies being developed for the disease. This was mostly achieved through the use of antisense oligonucleotides (AOs), such as those of the phosphorodiamidate morpholino oligomer (PMO) and 2′-O-methyl RNA (2′-OMe) chemistries. PMOs and 2′-OMe AOs were previously designed to target the DUX4 PAS15,16 or its splice sites,17,18 respectively. Both resulted in up to 50% reduced DUX4 transcript levels in vitro, as well as considerable decreases in DUX4 expression in vivo in xenograft and DUX4-transduced mouse models. More effective DUX4 knockdown is desirable, however, because it is known that low levels of DUX4 expression are sufficient to drive pathological changes in skeletal muscle.19 Compared with PMOs and 2′-OMe AOs, which passively knock down gene expression by interfering with DUX4 transcript maturation, use of antisense gapmers may prove more effective owing to their ability to actively induce the degradation of target mRNA transcripts via RNase H.
In the present study, we therefore aimed to explore the efficacy of antisense gapmers for DUX4 knockdown, specifically those with the 2′-O-methoxyethyl (2′-MOE) modification. The 2′-MOE chemistry has proved to be favorable for therapeutic AO development, with its enhanced resistance to nucleases, increased target binding affinity and specificity, as well as with four 2′-MOE-based AOs already having received approval in the United States and/or the European Union.20, 21, 22, 23 We designed 2′-MOE gapmers against mRNA in the coding region of DUX4 and evaluated their efficacy and specificity using immortalized FSHD patient-derived muscle cells as an in vitro model. We then test these gapmers in vivo via local intramuscular (i.m.) treatment in FLExDUX4 FSHD model mice.24 Overall, we show that our 2′-MOE gapmers are potent agents of DUX4 transcript knockdown and serve as promising clinical trial candidates for FSHD therapy.
Results
Designed 2′-MOE Gapmers Effectively Knock Down DUX4 Transcript Expression
Three 2′-MOE gapmers were designed and tested for their potential to knock down DUX4 transcript levels (Figure 1A; Table 1). The 2′-MOE gapmers were transfected at 100 nM into immortalized FSHD patient-derived muscle cells at 13 days post-differentiation, and cells were collected the following day for analysis. Quantitative real-time RT-PCR expression analysis revealed that all three 2′-MOE gapmers significantly reduced DUX4 transcript levels almost completely (n = 3; p < 0.005) (Figure 1B). No significant differences in knockdown efficacy were observed between the gapmers. Significant knockdown of the expression of DUX4 downstream transcriptional targets ZSCAN4, TRIM43, and MBD3L2 was observed for MOE2 and MOE3 (n = 3; p < 0.05) (Figure 1C). Transfection of lower doses of the 2′-MOE gapmers led to significant DUX4 transcript knockdown at the 10 nM, but not the 1 nM, dose (n = 3; p < 0.005) (Figure 1D). Up to ∼70% reduction in DUX4 levels on average was observed upon treatment with 10 nM of the 2′-MOE gapmers. ZSCAN4, TRIM43, and MBD3L2 expression levels were not affected at the 10 and 1 nM transfected doses of any of the 2′-MOE gapmers, however. Interestingly, treatment with lower doses of some of the 2′-MOE gapmers led to variably increased expression of DUX4 and its downstream target genes compared with the non-treated control, particularly at the 1 nM transfected concentration (Figure 1D). Overall, these results show that the designed 2′-MOE gapmers could knock down DUX4 transcript levels with high efficacy in vitro, even at reduced doses.
Figure 1.

DUX4 Knockdown Efficacy Evaluation of Designed 2′-MOE Gapmers
(A) Scheme showing the approximate locations targeted by our 2′-MOE gapmers on DUX4 exon 3. Transcript levels of (B) DUX4 and (C) ZSCAN4, TRIM43, and MBD3L2 were evaluated by quantitative real-time RT-PCR after overnight treatment of immortalized FSHD patient-derived myotubes with 100 nM of the 2′-MOE gapmers at 13 days post-differentiation. ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005 versus mock (M), one-way ANOVA with Dunnett’s test. δδδp < 0.0005, unpaired, two-tailed t test. (D) Transcript levels of these same four genes after treatment with 100, 10, or 1 nM of the 2′-MOE gapmers, following similar culture conditions. Error bars: SD; n = 3 independent experiments. ∗∗p < 0.005, ∗∗∗p < 0.0005 versus NT, one-way ANOVA with Dunnett’s test. δp < 0.05, δδp < 0.005, one-way ANOVA with Tukey’s test. NT, non-treated; PAS, polyadenylation signal; U, FSHD-unaffected/healthy.
Table 1.
Characteristics of Designed 2′-MOE Gapmers against the DUX4 Transcript
| ID | Sequencea (5′–3′) | Length (nt) | Target DUX4 Exon | GC Content (%) |
|---|---|---|---|---|
| MOE1 | TAGACAGCGTCGGAAGGTGG | 20 | 3 | 60.0 |
| MOE2 | CTAGACAGCGTCGGAAGGTG | 20 | 3 | 60.0 |
| MOE3 | CCTAGACAGCGTCGGAAGGT | 20 | 3 | 60.0 |
Fully phosphorothioated. Bold indicates 2′-MOE nucleotides, and nonbold indicates DNA.
A Subset of Transcriptome-Level Alterations Was Restored with 2′-MOE Gapmer Treatment
To obtain a better idea of the restorative effects of 2′-MOE gapmer treatment at the transcriptomic level, we performed RNA sequencing analysis on total RNA extracts from immortalized healthy control myotubes and patient-derived myotubes that were either treated or not with MOE3. MOE3 was chosen because it induced the greatest reduction in DUX4 transcript expression in our initial screen (Figure 1B). For the treatment, cells were transfected with 100 nM MOE3 at 13 days post-differentiation and harvested the following day, similar to what we did in the initial 2′-MOE gapmer screen. To obtain a list of transcripts linked to DUX4 expression, we compared our dataset with that of Rickard et al.,25 who performed RNA sequencing on extracts from flow cytometry-sorted DUX4 reporter-positive primary FSHD patient myotubes and who used similar cell culture conditions as we did in this study. We initially obtained 96 overlapping transcripts with that of the Rickard et al.25 dataset but excluded 2 because parameters for these were not present in all pairwise comparisons across our groups (Figure 2A). This led us to an FSHD signature of 94 transcripts whose expression levels were significantly affected by DUX4 expression (n = 3; p < 0.05) that consisted of 69 upregulated and 25 downregulated transcripts, representing 55 and 18 genes, respectively. A comparison of the expression levels obtained for these transcripts between our study and that of Rickard et al.25 revealed that the majority of genes had a similar direction of dysregulation (up/down) in both datasets (Figure 2B). We found that MOE3 treatment restored the expression of some FSHD signature transcripts to healthy levels (Figures 2C and 2D). Specifically, MOE3 significantly restored the expression of 8/69 (12%) upregulated FSHD transcripts and 1/25 (4%) downregulated transcripts (n = 3; p < 0.05), all corresponding to unique genes (Table S1). Of the significantly restored upregulated transcripts, two were validated by quantitative real-time RT-PCR (ZSCAN4 and TRIM43; Figure 1C); an additional four upregulated transcripts (MBD3L2, TRIM48, TRIM64B, PRAMEF4/5/9/11) that showed non-significant restoration in RNA sequencing were demonstrated to have significantly reduced expression post-treatment by quantitative real-time RT-PCR (Figure 1C; Figure S1; Table S2).
Figure 2.

RNA Sequencing Analysis of 2′-MOE Gapmer-Treated Muscle Cells
(A) FSHD signature transcripts from comparison with the RNA sequencing results of Rickard et al.,25 who used DUX4 reporter-positive primary FSHD patient myotubes. (B) Plot of log2(fold change) values (cutoff ± 2) for unique genes in the current study versus values obtained by Rickard et al.25 For genes with multiple transcripts in our dataset, the transcript with the least adjusted p value was used to represent the gene. (C) Volcano plot visualizations of RNA sequencing results from our dataset. Comparisons are indicated in the upper right, with the second listed sample as the reference. FSHD signature transcripts are shown as colored dots: upregulated (red) and downregulated (blue). The horizontal line represents the cutoff adjusted p value of 0.05, and the vertical lines represent log2(fold change) values of ±2. (D) Heatmap visualization of the expression levels of the 94 FSHD signature transcripts before and after treatment with 100 nM MOE3. Expression levels are colored from high to low with purple to yellow shades, respectively. Asterisks indicate transcripts significantly restored (p < 0.05) to healthy levels. n = 3 independent experiments. See also Figure S1.
Improvements in Cellular Phenotypes upon 2′-MOE Gapmer Treatment
Myotube fusion and size are two phenotypes negatively affected by aberrant DUX4 expression and signaling in skeletal muscle.14,26,27 We sought to determine whether 2′-MOE gapmer treatment could promote increased muscle fusion and decreased hypotrophic characteristics in vitro. Qualitatively, immunocytochemistry showed that immortalized FSHD patient-derived muscle cells treated with 10 nM of the 2′-MOE gapmers had larger, extensive myotubes with more nuclei than the non-treated or mock gapmer-treated controls (Figure 3A). All 2′-MOE gapmer-treated muscle cells had significantly increased myogenic fusion indices (MFIs), reaching up to 55% higher MFIs on average than the mock control (n = 3; p < 0.05) (Figure 3B). No significant MFI differences were observed across the gapmer-treated groups. Myotube diameters were also significantly increased by the treatment (n = 3; p < 0.0005), shifting the frequency distribution peak from 15–20 to 20–25 μm, similar to that of the healthy control (Figures 3C and 3D). Once again, no significant differences in myotube diameters were observed between gapmer-treated groups. Moreover, western blot analysis revealed that myosin heavy chain protein levels were observably but non-significantly increased by treatment with 100 nM MOE3 compared with mock-treated controls (n = 3; p = 0.0828) (Figure S2). We saw no effect of 10 nM 2′-MOE gapmer treatment on muscle cell apoptosis, another in vitro phenotype that characterizes FSHD (Figure 3E).14,28
Figure 3.

In Vitro Muscle Cell Phenotypes after 2′-MOE Gapmer Treatment
(A) Representative immunocytochemistry images of healthy immortalized control myotubes and NT, mock 2′-MOE gapmer-treated (M), and DUX4-specific MOE gapmer-treated (MOE1, MOE2, MOE3) immortalized FSHD patient-derived myotubes stained for nuclei (blue) and desmin (green). In this case, patient-derived myotubes were transfected with 10 nM of the various 2′-MOE gapmers at 4 days post-differentiation and then stained 3 days later. Scale bar: 100 μm. (B) Myogenic fusion index quantification for the various treatment groups. (C) Frequency distribution of myotube diameters across the different treatment groups. n = 3 independent experiments; 382 nuclei and 26 myotubes on average for each replicate, per condition, were counted for quantification of MFI and muscle cell diameters, respectively. (D) Individual myotube diameters from (C) were plotted. (E) Early and late apoptotic cell populations in immortalized FSHD patient-derived myotubes treated with 10 nM of the various 2′-MOE gapmers at 13 days post-differentiation were quantified by Annexin V/propidium iodide-based flow cytometry 1 day later. (B and E) Error bars: SD; n = 3 independent experiments. (D) The box represents the P25–P75 with the central line marking the median, and the whiskers represent the range; n = 3. ∗p < 0.05, ∗∗∗p < 0.0005 versus M, one-way ANOVA with Dunnett’s test. δp < 0.05, δδp < 0.005, δδδp < 0.0005, unpaired, two-tailed t test. See also Figure S2.
Off-Target Effect Analysis of 2′-MOE Gapmer Treatment
Using GGGenome, we compiled a list of sequences from other genes sharing the highest degree of similarity possible to the DUX4 target sequence of our 2′-MOE gapmers (Table 2). In part due to the length of the 2′-MOE gapmers, the closest sequences we could find were those having at least a 3-bp mismatch to the targeted DUX4 sequence. We examined whether the expression levels of these genes were knocked down upon 100 nM 2′-MOE gapmer treatment in immortalized FSHD patient-derived cells. Upon further testing, only BANF1, SSR4, FARP1, and ZBTB7B had detectable expression in FSHD patient-derived myotubes. Treatment with the 2′-MOE gapmers did not significantly reduce the transcript levels of these genes, except for BANF1, which was significantly knocked down by MOE1 and MOE2 (n = 3; p < 0.05 and p < 0.005, respectively) (Figures 4A–4D). MOE1 treatment also significantly increased the expression of ZBTB7B (n = 3; p < 0.005). However, by the nature of the change, this is not considered a direct off-target effect resulting from gapmer-mediated knockdown. Importantly, we note that the expression levels of all four potential off-target genes were not affected by MOE3.
Table 2.
Potential Off-Target Transcripts of the 2′-MOE Gapmers, as Determined by GGGenome
| Gene | Transcript Variant(s) | Sequence Showing Mismatch (5′-3′)a | No. of Mismatches to 2′-MOE Gapmer Target Sequence |
||
|---|---|---|---|---|---|
| MOE1 | MOE2 | MOE3 | |||
| DUX4 | N/A | CCACCTTCCGACGCTGTCTAGG | 0 | 0 | 0 |
| GALNT14 | 4 | CCACCTTCGGACGCTGACT-GG | 3 | 3 | 3 |
| FOXH1 | N/A | CC-CCTGCCCACGCTGTCTACC | 3 | 4 | 5 |
| SSR4 | 1–4 | CCA-CTTCTGACGCTGTC-ATT | 3 | 4 | 5 |
| ZBTB7B | 1–5 | ACACCTTCCGCCTCTCTCTAGC | 4 | 3 | 4 |
| TSPEAR-AS1 | N/A | CCACCTGCCGA-GCTGTC-AGC | 3 | 3 | 4 |
| FARP1 | 1,3 | TCACCTTTCCGA-GCTGTCT-GT | 4 | 3 | 4 |
| BANF1 | 1,2 | GTA-CTTCCGGCGCTGTCTCGG | 5 | 4 | 3 |
Bold indicates mispairing, and underline indicates indels versus the DUX4 target sequence.
Figure 4.

Off-Target Effect Evaluation upon 2′-MOE Gapmer Treatment
Immortalized patient-derived myotubes transfected with 100 nM MOE1, MOE2, or MOE3 (numbered) at 13 days post-differentiation were harvested a day later and used for quantitative real-time RT-PCR expression analysis of (A) BANF1, (B) SSR4, (C) FARP1, and (D) ZBTB7B. Error bars: SD; n = 3 independent experiments. ∗p < 0.05, ∗∗p < 0.005 versus NT, one-way ANOVA with Dunnett’s test. δp < 0.05, unpaired, two-tailed t test.
2′-MOE Gapmer Treatment Reduces DUX4 Expression in an FSHD Mouse Model
FLExDUX4 mice carry a floxed human full-length DUX4 transgene and express very low levels of DUX4 transcript even without Cre-mediated induction.24 To determine the in vivo efficacy of local 2′-MOE gapmer treatment, we treated adult hemizygous FLExDUX4 mice with 20 μg i.m. injections of MOE3 to the tibialis anterior (TA) every other day for 6 days. MOE3 was chosen given its increased capacity to reduce DUX4 transcript expression as previously mentioned and also from its favorable performance in the off-target effect analysis. For each mouse, MOE3 was injected in one of the legs, while vehicle (phosphate-buffered saline [PBS]) was injected in the other. Quantitative real-time RT-PCR was performed to determine DUX4 expression levels a day after the third injection; because the low level of DUX4 expression in FLExDUX4 mice is not sufficient to induce DUX4 downstream genes,24 we were not able to evaluate for their expression in this experiment. Our results showed that injection of MOE3 into the TA of FLExDUX4 mice significantly reduced DUX4 mRNA expression compared with the contralateral limb that received only a PBS injection (n = 5; p < 0.05) (Figure 5). Similar injection of a scrambled 2′-MOE gapmer control did not have an effect on DUX4 mRNA expression in FLExDUX4 mice; MOE3 significantly knocked down DUX4 transcript levels compared with the scrambled control (n = 5 MOE3-treated mice, n = 3 scrambled gapmer-treated mice; p < 0.05).
Figure 5.

In Vivo Efficacy of MOE3 Gapmer Treatment in FLExDUX4 Mice
Intramuscular injections of 20 μg MOE3 to the TA muscles (one leg with MOE3 and the contralateral leg with PBS) every other day for a total of three injections showed knockdown of DUX4 mRNA by quantitative real-time RT-PCR 1 day after the last injection. No knockdown was observed when a 2′-MOE gapmer control with a scrambled sequence was injected instead. Bars with similar patterns (block or hashed) indicate leg pairs. Error bars: SEM; n = 5 for MOE3/PBS mice and n = 3 for scrambled 2′-MOE/PBS mice. ∗p < 0.05, paired, two-tailed t test. δp < 0.05, unpaired, two-tailed t test.
Discussion
Since initial demonstrations of their effective knockdown abilities in vitro,29,30 2′-MOE gapmers have proceeded to become one of the most successful AO chemistries in clinical development. Two AO gapmers have been given US Food and Drug Administration (FDA) approval thus far: mipomersen (Kynamro; Ionis) for familial hypercholesterolemia and inotersen (Tegsedi; Akcea) for hereditary transthyretin amyloidosis, both of which are of the 2′-MOE chemistry.21,31,32 A third, volanesorsen (Waylivra; Akcea), has received conditional marketing authorization at the European Union for the treatment of familial chylomicronemia syndrome.22 Volanesorsen is currently under review for its second attempt at obtaining FDA approval. Although not a gapmer, there is the FDA-approved nusinersen (Spinraza, Biogen) for spinal muscular atrophy treatment, an AO composed entirely of 2′-MOE nucleotides.33,34 There are also many 2′-MOE gapmers under clinical and pre-clinical development, e.g., for Huntington’s disease, amyotrophic lateral sclerosis, and Alzheimer’s disease, among others.20 Furthermore, 2′-MOE gapmers have favorable safety and pharmacokinetic profiles in vivo. Integrated assessments of 2′-MOE gapmer toxicity in non-human primates and human subjects found no safety concerns for liver and kidney function. Cases of thrombocytopenia and complement activation were observed; however, these were limited to animal models and not humans.35,36 These AOs are stable in vivo, display broad tissue distribution, and have an elimination half-life of 2–4 weeks across tissues and species,36 indicating the possibility of reduced patient administrations. With such a proven track record, in this study we sought to adapt the use of these 2′-MOE gapmers for the treatment of FSHD by targeting DUX4 transcript knockdown.
We successfully demonstrated that our designed 2′-MOE gapmers could significantly reduce DUX4 expression in immortalized patient-derived differentiated muscle fibers (Figure 1B) and in the FLExDUX4 FSHD mouse model (Figure 5). At the highest in vitro tested dose of 100 nM, we observed corresponding reductions in the expression of DUX4 downstream target genes ZSCAN4, TRIM43, and MBD3L2 (Figure 1C). MOE3 was particularly effective among the three DUX4-targeting 2′-MOE gapmers. Notably, the extent of DUX4 knockdown achieved here was comparably better than what was previously observed with PMO or 2′-OMe AOs, supporting the notion that a direct, RNase H-mediated approach to transcript knockdown is more effective at reducing gene expression than an indirect approach that relies on steric blocking AOs. We also showed that 2′-MOE treatment did not significantly reduce the expression of three out of four potential off-target genes (Figures 4B–4D). However, we did observe significantly decreased BANF1 expression upon treatment with MOE1 and MOE2 (Figure 4A). BANF1 codes for a DNA-binding protein with roles in cell-cycle progression, chromatin organization, and early development.37,38 It has been reported that BANF1 is important for mouse and human embryonic stem cell (ESC) self-renewal, with BANF1 knockdown leading to their decreased survival and cloning efficiency.38 In mouse ESCs, this also led to decreased pluripotent gene expression and increased differentiation. Because DUX4 is known to upregulate genes associated with stem cells, as well as in generating an overall less-differentiated gene expression signature in skeletal muscle,39,40 there is the possibility that BANF1 is a downstream DUX4 target. As such, its decreased expression may have been an indirect result of DUX4 knockdown, but this remains to be proved.
We further confirmed the potential restorative effects of 100 nM MOE3 treatment at the transcriptomic level by RNA sequencing analysis (Figure 2). Aside from confirming the significant reduction of ZSCAN4 and TRIM43 expression (p < 0.05), we found that MOE3 treatment significantly decreased the expression of TRIM43B, TRIM51, TRIM64B, PRAMEF1, and PRAMEF8 (Figure 2D; Figure S1; Tables S1 and S2), which belong to gene families known to be upregulated by DUX4.14,41 We also observed a significant reduction in SLC34A2 and RFPL4B expression, which have both been associated with FSHD;42,43 SLC34A2 in particular has been recently reported as an FSHD protein biomarker.43 The roles of all these genes in FSHD pathogenesis are yet to be determined, however. On another note, although DUX4 downstream target expression was reduced at the 100 nM gapmer dose, this was not the case at the 10 nM dose despite significant knockdown of DUX4 transcript expression at this condition (Figure 1D). Increased 2′-MOE gapmer doses are recommended, because the level of DUX4 knockdown at 10 nM may not have been sufficient to generate observable downstream effects. This once again stresses the need to achieve complete DUX4 knockdown as much as possible, because any remaining low levels of DUX4 expression19 may be enough to maintain the dysregulated transcriptomic landscape seen in FSHD muscle. This scale-up of dose should be achievable in vivo, because 2′-MOE gapmers are non-toxic even at higher doses in humans, up to 475 mg in one study by subcutaneous or intravenous administration,35 and a repeated dosing regimen can be easily established. We also interestingly observed increased expression of DUX4 and its downstream target genes upon treatment with some 2′-MOE gapmers at lower doses. We are not certain why this occurs; however, this phenomenon has also been observed in a previous report that treated primary FSHD patient-derived muscle fibers with DUX4-targeting PMOs.16 In that study, expression levels of the downstream target genes ZSCAN4, TRIM43, and MBD3L2 were evaluated as well, and were found to be increased after treatment with 10 μM of certain PMOs. Because there are numerous differences between this work and ours, e.g., cell culture schedule, antisense chemistry employed, and transfection dose used, a direct comparison to identify potential reasons for this increased expression post-treatment is not possible. However, these observations provide valuable insight into considerations for future antisense therapy development, particularly concerning treatment dose, to minimize or prevent the occurrence of such off-target effects.
DUX4 orchestrates a large number of abnormal signaling events in skeletal muscle, whose cumulative effects give rise to FSHD.14 One critically affected pathway is muscle development, with DUX4 downregulating genes for Pax7, MyoD, and myogenin, among others.44 As a result, FSHD myoblasts exhibit defects in fusion and differentiation, giving rise to abnormal or deformed myotube morphologies.26,27 Treatment with DUX4-targeting 2′-MOE gapmers led to significant improvements in FSHD patient-derived muscle cell fusion, differentiation, and growth (Figures 3A–3D; Figure S2). In particular, treatment brought a large proportion of muscle fibers to over 20 μm in diameter, a size threshold below which characterized hypotrophic-type FSHD patient myotubes in a previous study.26 It is interesting to note that at a 10 nM transfected 2′-MOE dose, phenotypic improvements in muscle cell morphology were observed, but not apoptosis (Figure 3E). This may be partly explained by the knowledge that DUX4 dysregulates far more pathways contributing to apoptosis than those involved in muscle development.14 As we previously observed for low 2′-MOE gapmer doses on in vitro DUX4 downstream target gene expression (Figure 1D), even higher doses than those tested in this study may be required to achieve observable effects on apoptosis.
Finally, it is encouraging that we observed significant DUX4 transcript reduction with three 20-μg i.m. injections of MOE3 in FLExDUX4 mice (Figure 5), supporting the knockdown efficacy of our 2′-MOE gapmers not only in vitro but also in vivo. Practically, this supports the potential of i.m. injections for the muscle-specific treatment of FSHD, given how the disease exhibits asymmetric involvement of muscle groups.1 Furthermore, this promising proof-of-concept result sets the stage for future experiments, particularly evaluation of the therapeutic efficacy of our 2′-MOE gapmers upon systemic treatment. It would be beneficial to test these gapmers in Cre-induced FLExDUX4 mice,24 to examine whether 2′-MOE gapmer treatment can restore DUX4-mediated downstream gene expression to normal levels and rescue FSHD phenotypes. For instance, a recent study presented that FLExDUX4 mice crossed to ACTA1-Mer-cre-Mer (ACTA1-MCM; with a skeletal muscle-specific and tamoxifen-inducible promoter) mice produce bi-transgenic animals showing FSHD-like phenotypes, e.g., with significantly decreased skeletal muscle function.45 Because disease severity can be modulated in these mice, this would be an interesting potential model for further testing of our 2′-MOE gapmers. In summary, we were able to design DUX4-targeting 2′-MOE gapmers that could effectively reduce DUX4 transcript expression in vitro and in vivo, with improvements in some cellular phenotypes. Future work will evaluate the therapeutic efficacy and safety of these gapmers when administered systemically in a more severe FSHD animal model, to further assess their potential as candidate FSHD therapeutics.
Materials and Methods
AOs and Cell Culture
Experiments involving human cells were approved by the Health Research Ethics Board at University of Alberta. Our group designed three 2′-MOE gapmers (MOE1–3) with target sequences on DUX4 exon 3, before the PAS (Figure 1A). Target sites were chosen based on GC content and the mRNA secondary structure at the region. All gapmers were 20 bp long, fully phosphorothioated, consisted of a central 10-bp DNA segment flanked by 5 bp of 2′-MOE-modified nucleotides on each side, and synthesized by Eurogentec (Belgium). Gapmer sequences and characteristics are summarized in Table 1.
Cell culture was performed using immortalized WS229 FSHD patient-derived and WS234 healthy control myoblasts obtained in kind from the University of Massachusetts Medical School (MA, USA) Wellstone Program. Cells were derived from biceps biopsies of siblings and immortalized via stable CDK4/hTERT cassette integration46 (Table S3). For growing cells, a growth medium was prepared with 15% fetal bovine serum (FBS) (Sigma, St. Louis, MO, USA), 0.055 μg/mL dexamethasone, 2.5 ng/mL recombinant human hepatocyte growth factor (EMD Millipore, Burlington, MA, USA), and 10 ng/mL recombinant human fibroblast growth factor (BioPioneer, San Diego, CA, USA) in basal medium (BM; 20% Medium 199 [Life Technologies, Carlsbad, CA, USA], 0.03 μg/mL ZnSO4, 1.4 μg/mL vitamin B12, and 2.5% penicillin-streptomycin in DMEM [Life Technologies]). For differentiation, the following was prepared: 15% KnockOut Serum Replacement (KOSR; Life Technologies),47 10 μg/mL insulin (Sigma), and 100 μg/mL human apo-transferrin (R&D Systems, Minneapolis, MN, USA) in BM. All cells were cultured at 37°C and 5% CO2.
Transfection for Gapmer Screen
For AO transfection, 4 × 105 WS229 or WS234 cells/well were seeded onto six-well plates and differentiated the following day. Gapmers against DUX4 or a mock 2′-MOE gapmer were prepared at 100, 10, and 1 nM doses in 2% Lipofectamine RNAiMAX (Life Technologies) in OptiMEM (Life Technologies) following the manufacturer’s instructions. The transfection mixture was then diluted with differentiation medium at a 1:5 ratio, after which the final mixture was given to WS229 cells at 13 days post-differentiation. WS229 cells were also subjected to transfection but without any gapmer as non-treated control. Cells were transfected overnight and harvested the following day.
RNA Extraction and Quantitative Real-Time RT-PCR
Total cell RNA extracts were obtained using the RNeasy Mini Kit (QIAGEN, Germany) with on-column DNase treatment, following the manufacturer’s instructions. For cDNA synthesis, SuperScript IV Reverse Transcriptase (Life Technologies) was used following the manufacturer’s instructions with 1,400 ng RNA extract and 0.5 μg oligo(dT)12–18 primer (Life Technologies). Using this cDNA, we then performed quantitative real-time RT-PCR with the QuantStudio 3 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) to evaluate transcript expression of the following genes: DUX4, GAPDH, ZSCAN4, TRIM43, MBD3L2, TRIM48, TRIM64B, PRAMEF4/5/9/11, BANF1, SSR4, FARP1, and ZBTB7B. Probe-based TaqMan Gene Expression assays (Thermo Fisher, Waltham, MA, USA; reference numbers in parentheses) with the TaqMan Fast Advanced Master Mix (Thermo Fisher) were used for ZSCAN4 (Hs00537549_m1), TRIM43 (Hs00299174_m1), MBD3L2 (Hs00544743_m1), and TRIM64B (Hs04194067_mH) expression analysis. For the other genes, a SYBR-based system was used, with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). Primers were designed for these genes and are listed in Table S4. The default Fast cycling program corresponding to the detection system was used for quantitative real-time RT-PCR: (1) 95°C, 20 s; and (2) 40 cycles of 95°C, 1 s and then 60°C, 20 s. Expression levels were normalized to those of GAPDH and determined using the ΔΔCt method.
RNA Sequencing and Bioinformatics
Total RNA was extracted from WS234, non-treated WS229 myotubes, and MOE3-treated WS229 myotubes as described in RNA Extraction and Quantitative Real-Time RT-PCR. WS229 myotubes were treated with the MOE3 gapmer as in Transfection for Gapmer Screen. RNA quality was determined using an Agilent 2000 bioanalyzer instrument (Agilent, Santa Clara, CA, USA) and a high-sensitivity RNA chip (Agilent). Total RNA was quantified using Qubit 2.0 and an RNA high-sensitivity Assay Kit (Invitrogen, Carlsbad, CA, USA). RNA sequencing (RNA-seq) libraries were constructed from 100 ng total RNA with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA), according to the manufacturer’s instructions. In brief, polyadenylated mRNA was enriched with dT oligos conjugated to paramagnetic beads. Enriched mRNA was chemically fragmented, end repaired, A tailed, and ligated to sequencing linkers. Linker sequences were used as binding sites to incorporate indexed sequencing adapters by 12 cycles of PCR. Libraries were inspected using a High-Sensitivity DNA bioanalyzer chip (Agilent), and the DNA was quantified using a High-Sensitivity DNA Qubit assay (Invitrogen). Indexed libraries were pooled to a final concentration of 4 nM and were finally sequenced at a 10 pM concentration using a MiSeq instrument (Illumina, San Diego, CA, USA), with a 75-cycle paired-end protocol that included on-instrument de-multiplexing.
Sequences were inspected with fastqc, and bases with quality (Q) scores lower than 30 were trimmed with fastq-mcf. Pseudo-alignment of sequences against the GRCH38 version of the human cDNA database from the Ensembl database was conducted with Kallisto, using 100 bootstraps and bias correction.48 Transcripts that accumulated an average of at least 10 reads were subjected to statistical analysis. Differential expression analysis of RNA-seq data was conducted using negative binomial generalized linear models with the DESeq2 R package.49 Plots were generated with in-house R scripts. RNA sequencing datasets are available at the NCBI SRA: PRJNA629563.
Cellular Phenotype Analysis
For immunocytochemistry, WS229 cells in 24-well plates seeded at 2.5 × 104 cells/well were transfected as indicated in Transfection for Gapmer Screen with 10 nM 2′-MOE gapmers at 4 days post-differentiation (overnight transfection). Cells at 7 days post-differentiation or 3 days post-transfection were fixed for 5 min with 4% paraformaldehyde, incubated for 5 min with 0.5% Triton X-100 in PBS, and blocked for 1 h with 20% FBS in PBS. Cells were then stained for desmin (1:200 rabbit polyclonal antibody [Abcam, UK] for 1 h, followed by three 5-min PBS washes, and then 1:100 Alexa 594 goat anti-rabbit IgG [H+L] [Life Technologies]), given three final 5-min PBS washes, and mounted with SlowFade Gold Antifade Mountant with DAPI (Life Technologies). The Nikon Eclipse TE 2000-U fluorescence microscope was used for visualization. MFI determination and cell diameter measurements were done blinded using ImageJ (NIH), from three randomly chosen fields of view for each replicate, per condition. For MFI calculation, the number of nuclei in myotubes was divided by the total number of nuclei in a given field of view and then multiplied by 100 to get a percentage value. Approximately 382 nuclei on average were counted per replicate, per condition (range: 69–541). Myotube diameters were taken as the average of the three widest measurements across the length of a myotube, avoiding locations near branch points or overlaps between myotubes. Approximately 26 myotubes on average were counted per replicate, per condition (range: 15–36). For purposes of quantification, we considered a myotube as a cell with at least two nuclei having the same cytoplasm. For apoptosis analysis, WS229 cells in 24-well plates seeded at 5 × 104 cells/well were transfected as above with 10 nM 2′-MOE gapmers at 13 days post-differentiation. Apoptosis was then assessed by flow cytometry with the eBioscience Annexin V Apoptosis Detection Kit FITC (Thermo Fisher). WS234 cells were grown alongside WS229 cells and subjected to similar procedures to serve as a control.
Western Blotting
WS229 and WS234 cells were seeded and differentiated in six-well plates as described in AOs and Cell Culture. Transfection with 100 nM of either MOE1, MOE2, MOE3, or mock 2′-MOE gapmers to WS229 myotubes was done as in Transfection for Gapmer Screen, with transfection performed at 4 days post-differentiation (overnight). Total protein was extracted from cells at 7 days post-differentiation or 3 days post-transfection using radioimmunoprecipitation assay (RIPA) buffer (Sigma) with cOmplete, Mini, EDTA-free protease inhibitor cocktail (Sigma); protein was quantified using the Pierce BCA Protein Assay kit (Thermo Fisher). For SDS-PAGE, 12 μg of protein extracts was loaded and run on a NuPAGE 4%–12% Bis-Tris gel (Thermo Fisher) at 150 V for 70 min. Semi-dry transfer onto a polyvinylidene fluoride (PVDF) membrane (Millipore) was then performed at 20 V for 30 min. Blocking was done overnight at 4°C using 5% skim milk in PBS with 0.05% Tween 20 (PBST). The next day, the membrane was cut and incubated in primary antibodies against myosin heavy chain (MF20, 1:800 in blocking solution; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) or β-tubulin (ab6046, 1:5,000 in PBST; Abcam) for 1 h at room temperature, followed by three 10-min PBST washes. Membranes were then incubated in 1:10,000 of the corresponding HRP-conjugated secondary antibody (anti-mouse IgG H+L for the myosin heavy chain antibody, and anti-rabbit IgG H+L for the β-tubulin antibody; Invitrogen) in PBST for 1 h at room temperature, followed again by three 10-min PBST washes. Bands were visualized using the Amersham ECL Select detection kit (GE Healthcare, Chicago, IL, USA). Myosin heavy chain protein levels were determined based on band intensities normalized to those of β-tubulin and calculated relative to the normalized band intensity of one of the three WS234 replicates.
Searching for Potential Off-Target Genes
The GGGenome search engine (https://gggenome.dbcls.jp/) was used to find targets with highly similar sequences to those recognized by the various DUX4-targeting 2′-MOE gapmers.50 The RefSeq human RNA release 80 (Jan 2017) database was used for the search. The top hits are shown in Table S4. Only BANF1, SSR4, FARP1, and ZBTB7B were found to have detectable expression by quantitative real-time RT-PCR in our in vitro system, and so we focused on these for off-target analysis.
In Vivo Delivery of 2′-MOE Gapmers
Experiments involving animals were approved by the Institutional Animal Care and Use Committee at the Children’s National Health System, Washington, DC, USA. The animal experiment was also approved by the University of Alberta. To examine the effect of 2′-MOE gapmer treatment on DUX4 expression, five adult hemizygous FLExDUX4 mice (three male, two female; 2–3 months old) received three i.m. injections of MOE3 (20 μg dissolved in PBS, for a final 20 μL volume) to the TA muscle over 6 days, one injection every other day. The contralateral TA of each mouse was injected with PBS (20 μL) as a control. In addition, three adult male hemizygous FLExDUX4 mice were similarly injected i.m. (TA) with a scrambled 2′-MOE gapmer control following the same dose and treatment schedule; contralateral TAs were injected with PBS as before. Twenty-four hours after the final injection, mice were euthanized by CO2 asphyxiation with cervical dislocation, and TAs were dissected, cleaned of connective tissue, snap frozen in dry ice-cooled isopentane, and stored at −80°C for further analysis. For DUX4 expression analysis, quantitative real-time RT-PCR was performed as previously described.47,51 In brief, RNA was extracted using TRIzol (Ambion, Austin, TX, USA) and cleaned up using the RNeasy Plus Mini Kit (QIAGEN). cDNA was synthesized from 2 μg of total RNA using SuperScript IV Reverse Transcriptase (Life Technologies) and oligo(dT)12–18 primers. Gapdh was used as an internal control, and relative gene expression was analyzed using the ΔΔCt method.
Statistical Analysis
Statistical analyses for all data besides those from RNA sequencing were done with GraphPad Prism 7 (GraphPad Software). One-way ANOVAs with post hoc Tukey’s or Dunnett’s test, or unpaired two-tailed t tests were used as needed.
Author Contributions
K.R.Q.L., Y.-W.C., and T.Y. conceived and designed the study. Y.-W.C. and T.Y. supervised the work. K.R.Q.L., R.M., and Y.E. designed the gapmers. K.R.Q.L., A.B., Q.N., Y.H., K.D., and A.Z. performed the experiments. K.R.Q.L., A.B., Y.-W.C., and T.Y. wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We would like to thank Dr. Juan Jovel and The Applied Genomics Core (University of Alberta), as well as Dr. Aja Rieger and the Flow Cytometry Facility (University of Alberta), for their excellent advice and technical assistance toward this study. This work was supported by the FSH Society (grants 82018-2 and 82016-4), Muscular Dystrophy Canada (grant 82016-4), University of Alberta, Women and Children’s Health Research Institute Graduate Studentship award and Innovation Grant (2874), Alberta Innovates Graduate Studentship Award (201810476), Friends of FSH Research (grant 20170528), FSHD Global Research Foundation (34), FSHD Canada Foundation, The Friends of Garrett Cumming Research Fund, HM Toupin Neurological Science Research Fund, Canadian Institutes of Health Research (grant 143251), Canada Foundation for Innovation (grant 30819), Alberta Enterprise and Advanced Education, University Hospital Foundation, National Institutes of Health, Muscular Dystrophy Association—Strength, Science, and Stories of Inspiration Fellowship, and the FSHD Society New Investigator Grant Award.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.10.010.
Contributor Information
Yi-Wen Chen, Email: ychen@childrensnational.org.
Toshifumi Yokota, Email: toshifumi.yokota@ualberta.ca.
Supplemental Information
References
- 1.Wang L.H., Tawil R. Facioscapulohumeral Dystrophy. Curr. Neurol. Neurosci. Rep. 2016;16:66. doi: 10.1007/s11910-016-0667-0. [DOI] [PubMed] [Google Scholar]
- 2.Deenen J.C.W., Arnts H., van der Maarel S.M., Padberg G.W., Verschuuren J.J.G.M., Bakker E., Weinreich S.S., Verbeek A.L., van Engelen B.G. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–1059. doi: 10.1212/WNL.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richards M., Coppée F., Thomas N., Belayew A., Upadhyaya M. Facioscapulohumeral muscular dystrophy (FSHD): an enigma unravelled? Hum. Genet. 2012;131:325–340. doi: 10.1007/s00439-011-1100-z. [DOI] [PubMed] [Google Scholar]
- 4.Klinge L., Eagle M., Haggerty I.D., Roberts C.E., Straub V., Bushby K.M. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2006;16:553–558. doi: 10.1016/j.nmd.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Brouwer O.F., Padberg G.W., Wijmenga C., Frants R.R. Facioscapulohumeral muscular dystrophy in early childhood. Arch. Neurol. 1994;51:387–394. doi: 10.1001/archneur.1994.00540160085011. [DOI] [PubMed] [Google Scholar]
- 6.Goselink R.J.M., Voermans N.C., Okkersen K., Brouwer O.F., Padberg G.W., Nikolic A., Tupler R., Dorobek M., Mah J.K., van Engelen B.G.M. Early onset facioscapulohumeral dystrophy—a systematic review using individual patient data. Neuromuscul. Disord. 2017;27:1077–1083. doi: 10.1016/j.nmd.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Lu J., Yao Z., Yang Y., Zhang C., Zhang J., Zhang Y. Management strategies in facioscapulohumeral muscular dystrophy. Intractable Rare Dis. Res. 2019;8:9–13. doi: 10.5582/irdr.2019.01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabriëls J., Beckers M.C., Ding H., De Vriese A., Plaisance S., van der Maarel S.M., Padberg G.W., Frants R.R., Hewitt J.E., Collen D., Belayew A. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/s0378-1119(99)00267-x. [DOI] [PubMed] [Google Scholar]
- 9.Lemmers R.J.L.F., van der Vliet P.J., Klooster R., Sacconi S., Camaño P., Dauwerse J.G., Snider L., Straasheijm K.R., van Ommen G.J., Padberg G.W. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hewitt J.E., Lyle R., Clark L.N., Valleley E.M., Wright T.J., Wijmenga C., van Deutekom J.C., Francis F., Sharpe P.T., Hofker M. Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 1994;3:1287–1295. doi: 10.1093/hmg/3.8.1287. [DOI] [PubMed] [Google Scholar]
- 11.Hewitt J.E. Loss of epigenetic silencing of the DUX4 transcription factor gene in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2015;24(R1):R17–R23. doi: 10.1093/hmg/ddv237. [DOI] [PubMed] [Google Scholar]
- 12.Lemmers R.J.L.F., Goeman J.J., van der Vliet P.J., van Nieuwenhuizen M.P., Balog J., Vos-Versteeg M., Camano P., Ramos Arroyo M.A., Jerico I., Rogers M.T. Inter-individual differences in CpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum. Mol. Genet. 2015;24:659–669. doi: 10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Himeda C.L., Jones P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Annu. Rev. Genomics Hum. Genet. 2019;20:265–291. doi: 10.1146/annurev-genom-083118-014933. [DOI] [PubMed] [Google Scholar]
- 14.Lim K.R.Q., Nguyen Q., Yokota T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020;21:729. doi: 10.3390/ijms21030729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marsollier A.-C., Ciszewski L., Mariot V., Popplewell L., Voit T., Dickson G., Dumonceaux J. Antisense targeting of 3′ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: a new gene-silencing approach. Hum. Mol. Genet. 2016;25:1468–1478. doi: 10.1093/hmg/ddw015. [DOI] [PubMed] [Google Scholar]
- 16.Chen J.C., King O.D., Zhang Y., Clayton N.P., Spencer C., Wentworth B.M., Emerson C.P., Jr., Wagner K.R. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol. Ther. 2016;24:1405–1411. doi: 10.1038/mt.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ansseau E., Vanderplanck C., Wauters A., Harper S.Q., Coppée F., Belayew A. Antisense Oligonucleotides Used to Target the DUX4 mRNA as Therapeutic Approaches in FaciosScapuloHumeral Muscular Dystrophy (FSHD) Genes (Basel) 2017;8:93. doi: 10.3390/genes8030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanderplanck C., Ansseau E., Charron S., Stricwant N., Tassin A., Laoudj-Chenivesse D., Wilton S.D., Coppée F., Belayew A. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS One. 2011;6:e26820. doi: 10.1371/journal.pone.0026820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tassin A., Laoudj-Chenivesse D., Vanderplanck C., Barro M., Charron S., Ansseau E., Chen Y.W., Mercier J., Coppée F., Belayew A. DUX4 expression in FSHD muscle cells: how could such a rare protein cause a myopathy? J. Cell. Mol. Med. 2013;17:76–89. doi: 10.1111/j.1582-4934.2012.01647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scoles D.R., Minikel E.V., Pulst S.M. Antisense oligonucleotides: A primer. Neurol. Genet. 2019;5:e323. doi: 10.1212/NXG.0000000000000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shen X., Corey D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018;46:1584–1600. doi: 10.1093/nar/gkx1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paik J., Duggan S. Volanesorsen: First Global Approval. Drugs. 2019;79:1349–1354. doi: 10.1007/s40265-019-01168-z. [DOI] [PubMed] [Google Scholar]
- 23.Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones T., Jones P.L. A cre-inducible DUX4 transgenic mouse model for investigating facioscapulohumeral muscular dystrophy. PLoS One. 2018;13:e0192657. doi: 10.1371/journal.pone.0192657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rickard A.M., Petek L.M., Miller D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015;24:5901–5914. doi: 10.1093/hmg/ddv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barro M., Carnac G., Flavier S., Mercier J., Vassetzky Y., Laoudj-Chenivesse D. Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J. Cell. Mol. Med. 2010;14:275–289. doi: 10.1111/j.1582-4934.2008.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banerji C.R.S., Panamarova M., Pruller J., Figeac N., Hebaishi H., Fidanis E., Saxena A., Contet J., Sacconi S., Severini S., Zammit P.S. Dynamic transcriptomic analysis reveals suppression of PGC1α/ERRα drives perturbed myogenesis in facioscapulohumeral muscular dystrophy. Hum. Mol. Genet. 2019;28:1244–1259. doi: 10.1093/hmg/ddy405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kowaljow V., Marcowycz A., Ansseau E., Conde C.B., Sauvage S., Mattéotti C., Arias C., Corona E.D., Nuñez N.G., Leo O. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Garay M., Gaarde W., Monia B.P., Nero P., Cioffi C.L. Inhibition of hypoxia/reoxygenation-induced apoptosis by an antisense oligonucleotide targeted to JNK1 in human kidney cells. Biochem. Pharmacol. 2000;59:1033–1043. doi: 10.1016/s0006-2952(99)00412-8. [DOI] [PubMed] [Google Scholar]
- 30.Levesque L., Dean N.M., Sasmor H., Crooke S.T. Antisense oligonucleotides targeting human protein kinase C-alpha inhibit phorbol ester-induced reduction of bradykinin-evoked calcium mobilization in A549 cells. Mol. Pharmacol. 1997;51:209–216. doi: 10.1124/mol.51.2.209. [DOI] [PubMed] [Google Scholar]
- 31.Geary R.S., Baker B.F., Crooke S.T. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (kynamro(®)): a second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin. Pharmacokinet. 2015;54:133–146. doi: 10.1007/s40262-014-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keam S.J. Inotersen: First Global Approval. Drugs. 2018;78:1371–1376. doi: 10.1007/s40265-018-0968-5. [DOI] [PubMed] [Google Scholar]
- 33.Meijboom K.E., Wood M.J.A., McClorey G. Splice-Switching Therapy for Spinal Muscular Atrophy. Genes (Basel) 2017;8:161. doi: 10.3390/genes8060161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodkey K., Aslesh T., Maruyama R., Yokota T. Nusinersen in the treatment of spinal muscular atrophy. In: Yokota T., Maruyama R., editors. Exon Skipping and Inclusion Therapies. Vol. 1828. Humana Press; NY: 2018. pp. 69–76. [DOI] [PubMed] [Google Scholar]
- 35.Crooke S.T., Baker B.F., Kwoh T.J., Cheng W., Schulz D.J., Xia S., Salgado N., Bui H.H., Hart C.E., Burel S.A. Integrated Safety Assessment of 2′-O-Methoxyethyl Chimeric Antisense Oligonucleotides in NonHuman Primates and Healthy Human Volunteers. Mol. Ther. 2016;24:1771–1782. doi: 10.1038/mt.2016.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crooke S.T., Baker B.F., Xia S., Yu R.Z., Viney N.J., Wang Y., Tsimikas S., Geary R.S. Integrated Assessment of the Clinical Performance of GalNAc3-Conjugated 2′-O-Methoxyethyl Chimeric Antisense Oligonucleotides: I. Human Volunteer Experience. Nucleic Acid Ther. 2019;29:16–32. doi: 10.1089/nat.2018.0753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margalit A., Brachner A., Gotzmann J., Foisner R., Gruenbaum Y. Barrier-to-autointegration factor—a BAFfling little protein. Trends Cell Biol. 2007;17:202–208. doi: 10.1016/j.tcb.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 38.Cox J.L., Mallanna S.K., Ormsbee B.D., Desler M., Wiebe M.S., Rizzino A. Banf1 is required to maintain the self-renewal of both mouse and human embryonic stem cells. J. Cell Sci. 2011;124:2654–2665. doi: 10.1242/jcs.083238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knopp P., Krom Y.D., Banerji C.R.S., Panamarova M., Moyle L.A., den Hamer B., van der Maarel S.M., Zammit P.S. DUX4 induces a transcriptome more characteristic of a less-differentiated cell state and inhibits myogenesis. J. Cell Sci. 2016;129:3816–3831. doi: 10.1242/jcs.180372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Geng L.N., Yao Z., Snider L., Fong A.P., Cech J.N., Young J.M., van der Maarel S.M., Ruzzo W.L., Gentleman R.C., Tawil R., Tapscott S.J. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev. Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao Z., Snider L., Balog J., Lemmers R.J.L.F., Van Der Maarel S.M., Tawil R., Tapscott S.J. DUX4-induced gene expression is the major molecular signature in FSHD skeletal muscle. Hum. Mol. Genet. 2014;23:5342–5352. doi: 10.1093/hmg/ddu251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banerji C.R.S., Panamarova M., Hebaishi H., White R.B., Relaix F., Severini S., Zammit P.S. PAX7 target genes are globally repressed in facioscapulohumeral muscular dystrophy skeletal muscle. Nat. Commun. 2017;8:2152. doi: 10.1038/s41467-017-01200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mueller A.L., O’Neill A., Jones T.I., Llach A., Rojas L.A., Sakellariou P., Stadler G., Wright W.E., Eyerman D., Jones P.L., Bloch R.J. Muscle xenografts reproduce key molecular features of facioscapulohumeral muscular dystrophy. Exp. Neurol. 2019;320:113011. doi: 10.1016/j.expneurol.2019.113011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosnakovski D., Xu Z., Gang E.J., Galindo C.L., Liu M., Simsek T., Garner H.R., Agha-Mohammadi S., Tassin A., Coppée F. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones T.I., Chew G.-L., Barraza-Flores P., Schreier S., Ramirez M., Wuebbles R.D., Burkin D.J., Bradley R.K., Jones P.L. Transgenic mice expressing tunable levels of DUX4 develop characteristic facioscapulohumeral muscular dystrophy-like pathophysiology ranging in severity. Skelet. Muscle. 2020;10:8. doi: 10.1186/s13395-020-00227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stadler G., Chen J.C.J., Wagner K., Robin J.D., Shay J.W., Emerson C.P., Jr., Wright W.E. Establishment of clonal myogenic cell lines from severely affected dystrophic muscles - CDK4 maintains the myogenic population. Skelet. Muscle. 2011;1:12. doi: 10.1186/2044-5040-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pandey S.N., Khawaja H., Chen Y.-W. Culture Conditions Affect Expression of DUX4 in FSHD Myoblasts. Molecules. 2015;20:8304–8315. doi: 10.3390/molecules20058304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 49.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshida T., Naito Y., Sasaki K., Uchida E., Sato Y., Naito M., Kawanishi T., Obika S., Inoue T. Estimated number of off-target candidate sites for antisense oligonucleotides in human mRNA sequences. Genes Cells. 2018;23:448–455. doi: 10.1111/gtc.12587. [DOI] [PubMed] [Google Scholar]
- 51.Pandey S.N., Cabotage J., Shi R., Dixit M., Sutherland M., Liu J., Muger S., Harper S.Q., Nagaraju K., Chen Y.W. Conditional over-expression of PITX1 causes skeletal muscle dystrophy in mice. Biol. Open. 2012;1:629–639. doi: 10.1242/bio.20121305. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.