

Abstract
Oncolytic herpes simplex virus type 1 (HSV-1) has been investigated to expand its application to various malignancies. Because hematopoietic cells are resistant to HSV-1, its application to hematological malignancies has been rare. Here, we show that the third generation oncolytic HSV-1, T-01, infected and killed 18 of 26 human cell lines and 8 of 15 primary cells derived from various lineages of hematological malignancies. T-01 replicated at low levels in the cell lines. Viral entry and the oncolytic effect were positively correlated with the expression level of nectin-1 and to a lesser extent 3-O-sulfated heparan sulfate, receptors for glycoprotein D of HSV-1, on tumor cells. Transfection of nectin-1 into nectin-1-negative tumor cells made them susceptible to T-01. The oncolytic effects did not appear to correlate with the expression or phosphorylation of antiviral molecules in the cyclic GMP-AMP (cGAS)-stimulator of interferon genes (STING) and PKR-eIF2α pathways. In an immunocompetent mouse model, intratumoral injection of T-01 into lymphoma induced regression of injected, as well as non-injected, contralateral tumors accompanied by abundant infiltration of antigen-specific CD8+ T cells. These data suggest that intratumoral injection of oncolytic HSV-1 may be applicable to systemic hematological malignancies. Nectin-1 expression may be the most useful biomarker for optimal efficacy.
Keywords: oncolytic virus, HSV-1, hematological malignancies, nectin-1, CD8+ T cell, 3-O-sulfated heparan sulfate, glycoprotein D, STING, PKR
Graphical Abstract
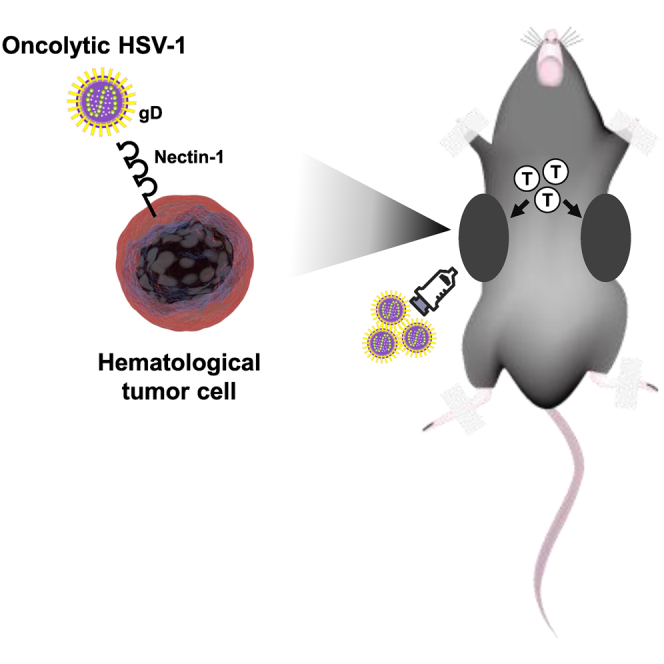
Oncolytic HSV-1 has been considered to be unsuitable to treat hematological malignancies because hematopoietic cells are resistant to HSV-1. Kadowaki and colleagues show that many hematological tumor cell lines and clinical samples are in fact susceptible to oncolytic HSV-1 in which nectin-1 expression plays a key role.
Introduction
Oncolytic virus therapy has been emerging as an important modality of cancer immunotherapy.1 Notably, oncolytic viruses provoke innate immune responses in the tumor site, thereby converting the immunosuppressive tumor microenvironment to an immune-potentiating one. This leads to activation of intratumor dendritic cells, which engulf the killed tumor cells and present an array of tumor antigens, presumably including neoantigens, in draining lymph nodes. Subsequently, antigen-specific T cells migrate to the inflamed tumor site. Thus, oncolytic virus therapy generates a so-called cancer-immunity cycle,2 thereby representing a rational tumor immunotherapy.
Herpes simplex virus type 1 (HSV-1) has been leading the field of oncolytic virus therapy.3 Mineta et al.4 generated a second generation oncolytic HSV-1, G207, which had deletion of the γ34.5 gene and inactivation of ICP6 with a LacZ gene insertion and could replicate only in cancer cells that can complement these mutations. The third generation G47Δ has further deletion of the overlapping region of the α47 gene and the promoter of the US11 gene, resulting in increasing the presentation of the peptide-major histocompatibility complex (MHC) class I complex by tumor cells and also in replicating more efficiently by placing the late US11 gene under control of the immediate-early α47 promoter.5
Many clinical trials of oncolytic HSV-1 have been conducted.3 T-VEC (talimogene laherparepvec), an oncolytic HSV-1 that has deletion of the γ34.5 and α47 genes and has the gene encoding human granulocyte macrophage colony-stimulating factor, has been approved for advanced melanoma in the United States, Europe, and Australia.3 We have been conducting clinical trials using G47Δ for brain tumors (University hospital Medical Information Network Clinical Trials Registry [UMIN-CTR]: UMIN000015995 and UMIN000011636) and prostate cancer (UMIN-CTR: UMIN000010463), and applications to other types of cancers are expected.
Originally, oncolytic viruses had been invented based on many anecdotal reports that hematological malignancies, such as leukemia and lymphoma, resolved after viral infection.6, 7, 8 Therefore, oncolytic virus therapy may be particularly effective for hematological malignancies. Several oncolytic viruses have been applied to hematological malignancies in preclinical and clinical trials.9 However, application of oncolytic HSV-1 to hematological malignancies has been rare10,11 because human hematopoietic cells are resistant to HSV-1 replication,12 and thus oncolytic HSV-1 has been considered unsuitable to treat hematological malignancies.
In this study, we used T-01, an HSV-1 containing modification of the same genes as G47Δ,13 and examined whether T-01 can infect and kill human hematological tumor cells in vitro. We showed that T-01 does so and that the expression of an HSV-1 receptor, nectin-1, is a determining factor in the oncolytic effect. Furthermore, using immunocompetent mice, we show that intratumoral injection of T-01 into lymphoma induces tumor-specific immune responses systemically in vivo. This study presents the rationale of applying G47Δ for the treatment of hematological malignancies.
Results
T-01 Kills Human Cell Lines Derived from Various Lineages of Hematological Malignancies
We first examined whether T-01 kills human cell lines derived from hematological malignancies. T-01 exhibited significant cytotoxicity at multiplicity of infection (MOI) 1.0 or less against 18 out of 26 cell lines, including myeloid-, B cell-, and T cell-derived tumors (Table S1; Figure S1). Figure 1A shows representative cell lines that were susceptible or resistant to T-01. Jurkat and GRANTA-519 cells were significantly killed by T-01 at MOI as low as 0.01, whereas Ramos cells were not killed even at MOI 1.0. Ultraviolet (UV)-inactivated T-01 lost cytotoxic activity in Jurkat cells even at a high concentration (MOI 10) (Figure 1B), indicating that intact T-01 is necessary for the cytotoxicity. These data indicate that T-01 kills a substantial fraction of cell lines from human hematological malignancies derived from various lineages.
Figure 1.

T-01 Induces Immunogenic Cell Death of Human Hematological Tumor Cell Lines
(A and B) Jurkat, GRANTA-519, and Ramos cells were treated with mock or T-01 (MOI 0.01, 0.1, and 1.0) (A). Jurkat cells were treated with mock, intact T-01, or UV-inactivated T-01 (MOI 0.1, 1.0, and 10) (B). After 3 days, the number of viable cells was counted. The data are shown as the mean ± SE of three independent experiments. Statistical analysis was conducted using non-repeated-measures ANOVA followed by Dunnett’s test. ∗∗p < 0.01. (C and D) Jurkat and GRANTA-519 cells were treated with mock or the indicated MOI of T-01. After 24 h, (C) ATP in the supernatants was measured using a CellTiter-Glo 2.0 Assay. The data are shown as the mean ± SE of three independent experiments. Statistical analysis was conducted using a two-tailed unpaired t test. ∗p < 0.05, ∗∗p < 0.01. (D) HMGB1 in the supernatants was detected using western blotting. The data are representative of three independent experiments. See also Figure S1.
In addition, Jurkat and GRANTA-519 cells treated with T-01 released ATP (Figure 1C) and HMGB1 (Figure 1D)14 more than mock-treated cells. These data suggest that hematological tumor cells treated with T-01 undergo immunogenic cell death.
A Low Level of Replication of T-01 in Hematological Tumor Cells
We examined whether replication of T-01 accompanies the cytotoxic activity. We quantified viral replication by real-time PCR of the glycoprotein B (gB) gene of HSV-1 in the T-01-susceptible cell lines. When T-01 was added at MOI 0.1, the copy number of the gB gene increased more than 1,000 times in 24 h in Vero cells as expected (Figure 2A). In contrast, only low levels of increase or no increase were detected in the T-01-susceptible cell lines (Figure 2A). We added a viral DNA polymerase inhibitor acyclovir to examine whether the low levels of replication contribute to the cytotoxic activity of T-01. Acyclovir at 10 μM suppressed the viral replication in Vero cells (Figure 2A), as well as in Jurkat and GRANTA-519 cells (Figure 2B), and attenuated the cytotoxic activity of T-01 in these cell lines (Figure 2C). When T-01 was added at MOI 0.1 or 1.0 to Jurkat cells or at MOI 1.0 to GRANTA-519 cells, acyclovir attenuated the cytotoxic effect of T-01 only partially (MOI 0.1 for Jurkat and MOI 1.0 for GRANTA-519) or not at all (MOI 1.0 for Jurkat) (Figure 1C). Such residual cytotoxicity observed at the higher concentrations (MOI 0.1 or 1.0) of T-01 in the presence of acyclovir may be caused by efficient entry of T-01 into these cell lines as shown below (Figures S2A and 2B). These data suggest that cytotoxic activity accompanying a low level of viral replication or even efficient viral entry alone contributes to the killing of the hematological tumor cell lines by T-01.
Figure 2.

A Low Level of Replication of T-01 in Hematological Tumor Cells
(A) Vero cells were treated with T-01 (MOI 0.1) for 24 h in the presence of DMSO or 10 μM acyclovir. Jurkat, GRANTA-519, ED-40515(+), MT-2, and RPMI 8226 cells were treated with T-01 (MOI 1.0) for the indicated periods. The amounts of the gB gene of HSV-1 were measured by real-time PCR and were expressed as the ratios to the amounts at the time of culture start. The data are shown as the mean ± SE of 2 (Vero cells) or 3 (the other cells) independent experiments. (B) Jurkat and GRANTA-519 cells were treated with T-01 (MOI 1.0) for the indicated periods in the presence of DMSO (control) or 10 μM acyclovir. The amounts of the gB gene were measured and expressed as in (A). The data are shown as the mean ± SE of 3 independent experiments. (C) Jurkat and GRANTA-519 cells were treated with mock or T-01 (MOI 0.01, 0.1, and 1.0) in the presence of DMSO (control) or 10 μM acyclovir. After 3 days, the number of viable cells was counted. The data are shown as the mean ± SE of 3 independent experiments. Statistical analysis was conducted using non-repeated-measures ANOVA followed by SNK test. ∗p < 0.05, ∗∗p < 0.01.
The Expression Levels of Nectin-1 on the Cell Lines Most Correlate with Viral Entry and Cytotoxicity
We next examined the differences between T-01-susceptible and -resistant cell lines. Viral entry was quantified by culturing cells with EGFP-expressing T-01, T-GFP, for a short period (4 h) to avoid contribution of viral replication. The efficacy of viral entry into cells was variable among the 26 cell lines (Figures S2A and S2B). Although viral entry to several T-01-susceptible cell lines (SET-2, ATL-43T, Raji, and KMS-12-BM) was low at 4 h, EGFP-positive cells increased in 24 h probably because of progression of viral entry and replication (Figure S2Ci). In contrast, almost no increase in EGFP-positive cells was observed in T-01-resistant cell lines (CHRF-288-11, HL-60, Ramos, SR-786, KIS-1, SU-DHL-1, and FL-318) after 24-h culture, except for HEL cells, which were killed with a higher concentration of T-01 (MOI 10; data not shown) (Figure S2Cii). The degrees of viral entry at 4 h were positively correlated with those of cytotoxicity (Figure 3A). Thus, we examined whether the expression levels of HSV-1 glycoprotein D (gD) receptors (nectin-1,15 herpes viral entry mediator [HVEM],16 and 3-O-sulfated heparan sulfate [3-OS HS]17) and gB receptors (paired immunoglobulin-like type 2 receptor α [PILRα],18 non-muscle myosin heavy chain [NMHC]-IIA,19 and NMHC-IIB20) correlated with the cytotoxicity. Lymphocytes in peripheral blood expressed HVEM, but not nectin-1, 3-OS HS, or PILRα (Figure S3). Monocytes expressed HVEM, PILRα, and a marginal level of nectin-1, but not 3-OS HS. Nectin-1 (Figure S4A), HVEM (Figure S4B), and 3-OS HS (Figure S4C) were expressed on many of the 26 cell lines at various levels, whereas PILRα was not expressed on 11 cell lines we examined (data not shown). Notably, the expression levels of nectin-1 positively correlated with degrees of viral entry and cytotoxicity (Figure 3B). The only exception was HL-60 (labeled with asterisk [∗] in Figure 3B); although it expressed a high level of nectin-1 (Figure S4A), T-01 did not enter (Figure S2) or kill (Figure S1) HL-60. In contrast with nectin-1, the expression levels of HVEM did not correlate with degrees of viral entry or cytotoxicity (Figure 3C). In addition, three nectin-1+ HVEM− cell lines (SKM-1, MEG-01, and Jurkat) underwent efficient viral entry, whereas two nectin-1− HVEM+ cell lines (CHRF-288-11 and Ramos) did not undergo viral entry (Figure S2). The expression levels of 3-OS HS positively correlated with degrees of viral entry and cytotoxicity (Figure 3D), although to the lesser extent than those of nectin-1. Two nectin-1+ cell lines (HuT 102 and RPMI 8226) underwent efficient viral entry (Figures S2A and S2B) despite the absence of 3-OS HS (Figure S4C). There was no T-01-susceptible cell line that is nectin-1− 3-OS HS+. These data suggest that nectin-1 plays a dominant role in viral entry compared with 3-OS HS. NMHC-IIA was expressed in all 14 cell lines examined (Figure S5). NMHC-IIB was expressed in all of them except for THP-1 and RPMI 8226 (Figure S5). Thus, the expression of NMHC-IIA and -IIB did not correlate with cytotoxicity.
Figure 3.

The Expression Levels of Nectin-1 and 3-OS HS on the Cell Lines Correlate with Viral Entry and Cytotoxicity
(A–D) Viral entry versus cytotoxicity (A), nectin-1 expression (B), HVEM expression (C), and 3-OS HS expression (D) versus viral entry and cytotoxicity. Viral entry is indicated as percentages of T-GFP-positive cells at MOI 1.0 after 4-h culture. Cytotoxicity was calculated as 1 − (viable cell number with T-01/viable cell number with mock) × 100 (%) after treatment with T-01 (MOI 1.0) for 3 days. The expression of nectin-1, HVEM, and 3-OS HS is indicated as the mean fluorescence intensity (MFI) ratios compared with cells stained with isotype-matched control mAbs (nectin-1 and HVEM) or those stained by omitting anti-heparan sulfate antibody HS4C3 (3-OS HS). Each dot indicates each cell line. Asterisks in (B) indicate HL-60. Statistical analysis was conducted using Spearman’s correlation. See also Figures S2–S4.
These data suggest that nectin-1 on hematological tumor cells plays a key role in viral entry and subsequent cytotoxicity, and that nectin-1 expression predicts the cytotoxicity of T-01 in hematological tumor cells. Although 3-OS HS may also play a role in viral entry and cytotoxicity, the contribution appears to be less than that of nectin-1.
The Expression Levels of Nectin-1 in Clinical Samples Correlate with Viral Entry and Cytotoxicity
We next examined primary hematological tumor cells and found that viable cells decreased after treatment with T-01 in 8 of 15 samples (Table S2; Figure S6). Although we added high concentrations (up to MOI 10) of T-01, UV inactivation abrogated cytotoxic activity of T-01 even at MOI 10 (Figure 1B), indicating that it is unlikely that contaminants of viral preparations were responsible for the cytotoxicity at MOI 10 for the clinical samples. All of the eight T-01-susceptible samples expressed nectin-1 (Figure S6). Of note, all of them were from relapsed patients, whereas none of the five samples from untreated patients expressed nectin-1 and were resistant to T-01. Seventeen of 22 additional samples from untreated patients failed to express nectin-1 (data not shown). The expression levels of nectin-1, but not of HVEM, were positively correlated with the degrees of cytotoxicity in clinical samples (Figure 4), as observed in the cell lines. These data suggest that T-01 may be more effective for relapsed hematological malignancies, which frequently express nectin-1, than untreated ones.
Figure 4.

The Expression Levels of Nectin-1 on Clinical Samples Correlate with Entry and Cytotoxicity by T-01
Nectin-1 expression (left) and HVEM expression (right) versus cytotoxicity. Nectin-1 and HVEM expression is indicated as the MFI ratio compared with cells stained with isotype-matched control mAbs. Cytotoxicity was calculated as 1 − (viable cell percentage with T-01/viable cell percentage with mock) × 100 (%) after treatment with T-01 (MOI 10) for 3 days. Each dot indicates each cell line. Red and black dots indicate samples from relapsed and untreated patients, respectively. Statistical analysis was conducted using Spearman’s correlation. See also Figure S6.
Viral Entry via Nectin-1 Is Functionally Important for Cytotoxicity
To examine the functional involvement of nectin-1 in viral entry and cytotoxicity, we knocked down the expression of nectin-1 on Jurkat and THP-1 cells. The knockdown significantly decreased the expression levels of nectin-1 and tended to decrease viral entry, although statistically not significant (Figure 5A). The residual entry may be because of incomplete reduction of nectin-1 expression by shRNA or by the low levels of expression of HVEM (THP-1) (Figure S4B) or 3-OS HS (Jurkat and THP-1) (Figure S4C). Conversely, human nectin-1 was transduced into Ramos cells and a mouse T cell lymphoma cell line E.G7-ovalbumin (OVA). The overexpression of nectin-1 induced viral entry and cytotoxicity in both cell lines (Figure 5B). These data indicate that the expression of nectin-1 is functionally important for viral entry and the cytotoxic effect of HSV-1 against human and mouse hematological tumor cells.
Figure 5.

Nectin-1 Knockdown and Transfection Affect Viral Entry and Cytotoxicity
(A) Nectin-1 expression (upper panels) and viral entry (lower panels) in Jurkat and THP-1 cells after knockdown of nectin-1 with shRNA. Nectin-1 expression is shown as the MFI ratio compared with cells stained with isotype-matched control mAbs. Viral entry is indicated as percentages of T-GFP-positive cells at MOI 1.0. 62933 indicates cells with nectin-1 knockdown. The data are shown as the mean ± SE of three independent measurements. Statistical analysis was conducted using a two-tailed unpaired t test. ∗p < 0.05, ∗∗p < 0.01. (B) Nectin-1 expression (left panels), viral entry (middle panels), and cytotoxic effects of T-01 (right panels) on Ramos (upper panels) and E.G7-ONA (lower panels) transfected with human nectin-1. The data are shown as the mean ± SE of three independent measurements. Statistical analysis was conducted using non-repeated-measures ANOVA followed by Dunnett’s test. ∗p < 0.05, ∗∗p < 0.01.
Nectin-1 Expression and Viral Entry Are Upregulated by Epigenetic Regulation
Given that expression of nectin-1 on tumor cells is a key factor for direct cytotoxicity of T-01, we examined whether the expression of nectin-1 can be induced or enhanced by epigenetic regulation, using epigenetic compound library screening. We treated nectin-1− cells (Ramos) and nectin-1low cells (HEL) with the compounds contained in the library and examined whether expression of nectin-1 was induced or enhanced using flow cytometry (Figure S7A). A DNA methyltransferase inhibitor (decitabine), a class IIa histone deacetylase inhibitor (LMK-235), and three inhibitors (CPI-203, I-BET151, and I-BET726) of Bromodomain and Extra-Terminal motif (BET) family proteins upregulated nectin-1 on Ramos and HEL cells without reducing cell viability substantially (Figure S7A). Another DNA methyltransferase inhibitor, azacitidine, did not upregulate nectin-1 (data not shown). I-BET726 also enhanced nectin-1 expression on nectin-1low THP-1 and SYK-11L(+) cells (Figure S7B). I-BET726 significantly enhanced viral entry into HEL, THP-1, and SYK-11L(+) cells in a dose-dependent manner (Figure S7C). LMK-235 significantly enhanced viral entry into Ramos, HEL, and THP-1 cells in a dose-dependent manner (Figure S7C). Despite the upregulation of nectin-1 on Ramos cells by decitabine (Figures S7A and S7B), it did not significantly enhance viral entry (Figure S7C) for unknown reasons. These data indicate that nectin-1 expression can be upregulated by epigenetic regulation, leading to enhancement of entry of T-01.
Expression and Activity of the cGAS-STING Pathway in Hematological Tumor Cells
We next examined whether mechanisms of cellular antiviral responses correlated with the cytotoxicity of T-01. It has been reported that the cytoplasmic DNA sensing cyclic GMP-AMP (cGAS)-stimulator of interferon genes (STING) pathway is frequently defective in colorectal cancer,21 melanoma,22 and ovarian cancer cells,23 resulting in susceptibility to oncolytic HSV-1. Thus, we examined whether the expression of cGAS and STING correlated with the susceptibility of the hematological tumor cells to T-01. Five (HuT 102, THP-1, RPMI 8226, ATL-43T, and SYK-11L(+)) of the 9 T-01-susceptible cell lines (labeled in red in Figure S8A) expressed both cGAS and STING, whereas four (GRANTA-519, Jurkat, ED-40515(+), and MT-2) of the nine T-01-susceptible cell lines expressed either of them at low or no levels (cGAS in Jurkat and STING in GRANTA-519, ED-40515(+), and MT-2). Thus, there was no apparent correlation between susceptibility to T-01 and the presence or absence of cGAS/STING in the hematological tumor cells we examined, although the low expression in several cell lines may contribute to susceptibility to T-01.
We examined whether stimulation with T-01 induces phosphorylation of downstream molecules of STING, TBK1, and IRF3 in three T-01-susceptible cell lines (Figure S8B). TBK1 was constitutively phosphorylated in all of the cell lines. After stimulation with T-01, phosphorylation of TBK1 and IRF3 was upregulated in MT-2 (although the expression of STING was low as in Figure S8A), but not in THP-1 and ATL-43T cells (Figure S8B, left panel), possibly because of the highly efficient entry of T-01 into MT-2 (Figures S2A and S2B). When THP-1 and ATL-43T cells were stimulated with HSV-60,24 a 60-bp HSV-1-derived double-stranded DNA oligonucleotide that stimulates the STING-dependent pathway, phosphorylation of TBK1 was upregulated in both cell lines, and that of IRF3 was induced in THP-1, but not in ATL-43T cells (Figure S8B, right panel). These data suggest that pathways leading to phosphorylation of TBK1 and IRF3 are preserved in THP-1 and MT-2 cells, whereas it is suppressed in ATL-43T cells at the level of phosphorylation of IRF3.
Expression levels of mRNA of IFN-β, the main antiviral molecule induced by the cGAS-STING pathway,25 positively rather than negatively correlated with the cytotoxic effect of T-01 (Figure S9), suggesting that the signaling pathway leading to IFN-β induction is at least partially preserved in many cell lines despite the susceptibility to T-01.
Collectively, the expression and activity of the cellular antiviral machinery composed of the cGAS-STING-TBK1-IRF3 axis leading to IFN-β induction did not appear to correlate with the susceptibility to T-01 of many hematological tumor cell lines we examined.
Expression and Activity of the PKR-eIF2α Pathway in Hematological Tumor Cells
After viral infection, activation of the PKR-eIF2α pathway blocks viral protein synthesis and thereby restricts viral replication in normal cells.26 ICP34.5, the product of the γ34.5 gene of HSV-1, interacts with and redirects protein phosphatase 1α to dephosphorylate eIF2α, thus enabling continued protein synthesis despite the presence of activated PKR.27 Because PKR activation is suppressed in transformed cells with an activated Ras pathway,28 γ34.5 gene-deleted HSV-1 is expected to replicate in Ras-activated tumor cells. In addition, even if there is residual activity of PKR, early expression of the Us11 protein blocks phosphorylation of eIF2α, thereby compensating for the deletion of the γ34.5 gene.29 Thus, we examined whether expression and/or activation of the PKR-eIF2α pathway correlates with the susceptibility of hematological tumor cells. PKR protein was expressed in all of the cell lines (Figure S10A). After the addition of T-01, phosphorylation of PKR and eIF2α was not enhanced in T-01-resistant cell lines (Figure S10B, black labels), consistent with the poor entry of T-01 into these cells up to 24 h, as shown in Figure S2Cii. In contrast, phosphorylation of PKR was enhanced in T-01-susceptible cell lines except for ATL-43T cells (Figure S10B, red labels). Phosphorylation of eIF2α was enhanced in GRANTA-519, Jurkat, HuT 102, RPMI 8226, and SYK-11L(+), but not in THP-1, ED-40515(+), ATL-43T, or MT-2 cells. Collectively, the expression and activity of the PKR-eIF2α pathway did not correlate with the susceptibility to T-01 of many hematological tumor cell lines we examined. The reduced phosphorylation of PKR or eIF2α in several cell lines may contribute to susceptibility to T-01.
Antitumor Effects of T-01 In Vivo in Immunodeficient and Immunocompetent Mice
We examined whether the antitumor effect of T-01 is observed in vivo using immunodeficient and immunocompetent mice. We implanted GRANTA-519 or ED-40515(+) cells subcutaneously into severe combined immunodeficiency (SCID) Beige mice and injected T-01 intratumorally (Figure 6A). T-01 exhibited strong antitumor effects on both GRANTA-519 and ED-40515(+) cells (Figure 6B), despite the low levels of T-01 replication in these cell lines in vitro (Figure 2A). These data indicate that the direct cytotoxic effect of T-01 efficiently suppresses the growth of T-01-susceptible cell lines in vivo despite the inefficient viral replication in vitro.
Figure 6.

Antitumor Effects of T-01 In Vivo in Immunodeficient Mice
(A) Treatment schema. GRANTA-519 or ED-40515(+) cells were subcutaneously implanted into the left flank of SCID Beige mice. Mock or T-01 (0.2 × 106 or 1.0 × 106 PFUs) was injected into the established tumor on days 0 and 3. The tumor size was measured every 2 or 3 days. (B) Tumor growth curves (n = 7 and 8 for GRANTA-519 and ED-40515(+), respectively). Arrows indicate injection of mock or T-01. The data are shown as the mean ± SE. Statistical analysis was conducted using non-repeated-measures ANOVA followed by Dunnett’s test. ∗∗p < 0.01.
The susceptibility of human nectin-1-transfected mouse cell line E.G7-OVA (E.G7-OVA-nectin-1) to T-01 enabled us to examine the immune-mediated effects of T-01 in vivo using immunocompetent mouse models. We implanted E.G7-OVA-nectin-1 cells subcutaneously into the right and left flanks of C57BL/6 mice and injected T-01 intratumorally only into the right flank tumor (Figure 7A). Tumor growth in T-01-injected as well as non-injected sides was significantly suppressed (Figure 7B). In line with this, OVA-specific CD8+ T cells were intensely accumulated in both T-01-injected and non-injected tumors to the same extent (Figure 7C). In contrast, OVA-specific CD8+ T cells did not significantly accumulate in the spleen. These data suggest that tumor-specific CTLs induced in T-01-injected tumors migrate and accumulate into the remote tumor and exhibit antitumor effects on both T-01-injected and non-injected tumors.
Figure 7.

Antitumor Effects of T-01 In Vivo in Immunocompetent Mice
(A) Treatment schema. E.G7-OVA-nectin-1 was subcutaneously implanted into the right and left flanks of C57BL/6 mice. Mock or T-01 (2.0 × 106 PFUs) was injected into the established right tumor on days 0 and 3. The tumor size was measured every 2 or 3 days. (B) Tumor growth curves (n = 8). Arrows indicate injection of mock or T-01. The data are shown as the mean ± SE. Statistical analysis was conducted using a two-tailed unpaired t test. ∗p < 0.05. (C) Tetramer staining of tumor-infiltrating lymphocytes (TILs) and spleen cells. TILs from the T-01-injected right-side tumor, those from the T-01-non-injected left-side tumor, and spleen cells were isolated 10 days after the second injection of mock or T-01. The frequency (%) of H-2Kb OVA tetramer-positive cells among CD8+ T cells was measured by flow cytometry. The statistical differences of the tetramer-positive cells between mock- or T-01-treated mice were analyzed (n = 4) (upper panels). The data are shown as the mean ± SE. Statistical analysis was conducted using a two-tailed unpaired t test. ∗∗∗p < 0.001. Lower panels show representative data of TILs stained with the OVA tetramer.
Discussion
In this study, we showed that oncolytic HSV-1 has the potential to treat hematological malignancies, including T, B, and myeloid lineages. The expression of nectin-1 is a decisive factor for the oncolytic effect. Local intratumor injection of HSV-1 induced systemic antitumor immune responses. This study opens the possibility of applying oncolytic HSV-1 to hematological malignancies.
It was reported recently that UV-inactivated, non-replicating HSV-1 activates human NK cells and may be used as an immunological adjuvant to activate donor-derived mononuclear cells in vitro for infusion after allogeneic stem cell transplantation.30 However, because direct oncolysis by replicating virus is expected to be an integral part of the effect of oncolytic virus therapy, it is important to investigate whether replicating HSV-1 infects and kills hematological tumor cells. Here, we showed that T-01 replicated at low levels even in highly susceptible cell lines, such as Jurkat and GRANTA-519 cells. Attenuation of the cytotoxicity by acyclovir suggests that cytotoxic activity accompanying such low levels of viral replication contributed to the killing effects.
It has been reported that the expression of nectin-1 correlates with HSV-1 entry and oncolysis in thyroid cancer,31 squamous cell carcinoma,32 and Epstein-Barr virus (EBV)-associated lymphoproliferative disease.33 However, different studies suggested that other factors, such as cellular antiviral responses,34,35 rather than the expression of nectin-1, determine the efficacy of oncolytic HSV-1. Thus, the importance of nectin-1 expression in oncolysis remains to be determined. In this study, we showed that the expression of nectin-1 is a critical factor in viral entry and oncolysis, using cell lines, freshly isolated clinical samples, and nectin-1-transfected cells. Intriguingly, all of the nectin-1-expressing, HSV-1-susceptible clinical samples were derived from relapsed patients, suggesting that progression of malignant transformation that occurred in relapsed tumors may lead to the expression of nectin-1. Cell line HL-60 was an exception, in that T-01 did not enter the cells despite high levels of nectin-1 expression. Such a cell line may have mutations in nectin-1 at indispensable residues for the binding of gD.36 Although the expression levels of 3-OS HS also positively correlated with degrees of viral entry and cytotoxicity, its contribution appears to be less than that of nectin-1.
Epigenetic compound library screening indicated that several compounds upregulated nectin-1 expression on nectin-1− or nectin-1low cell lines and viral entry. However, it did not lead to the induction or enhancement of nectin-1-mediated oncolytic effects (data not shown), possibly because the induction of nectin-1 was weak. More potent epigenetic upregulation of nectin-1 may extend the applicability of oncolytic HSV-1 therapy.
Intratumor injection of T-01 into subcutaneous tumors of GRANTA-519 and ED-40515(+) in the SCID Beige mice was highly effective despite the low levels of T-01 replication in these cell lines in vitro. The apparent discrepancy between the inefficient viral replication in tumor cells in vitro and the strong antitumor effect in immunodeficient mice in vivo is intriguing, although the reason remains unknown.
Using an immunocompetent mouse model (E.G7-OVA-nectin-1), we showed that intratumoral injection of T-01 systemically induced antigen (OVA)-specific antitumor CD8+ T cell responses. Of note, OVA-specific T cells accumulated in both injected and non-injected tumors to the same extent, whereas they scarcely accumulated in the spleen, indicating that the antigen-specific T cells were not diluted in the systemic circulation but were efficiently concentrated at tumor sites.
Various oncolytic viruses have been applied to hematological malignancies via an intravenous route.9 However, intravenously administered viruses are extensively diluted in the systemic circulation and also rapidly induce the production of neutralizing antiviral antibodies, which hinders repeated injection. Local intratumor injection of HSV-1 induces a systemic immune-mediated effect, which is augmented by combining with immune checkpoint blockade.37 In addition, oncolytic HSV-1 is not toxic to human hematopoietic cells, as used to purge contaminating solid tumor cells from an autologous bone marrow graft.12 Thus, HSV-1 may be applicable to tumor-forming hematological malignancies, such as lymphoma for intra-tumor injection, as well as, if safety is confirmed, to marrow-located malignancies, such as leukemia and myeloma for loco-regional intra-marrow injection, which is easy to perform.
In conclusion, we showed that oncolytic HSV-1 is a viable therapy for hematological malignancies. The expression of nectin-1 rather than defects in the cellular antiviral machineries is a decisive factor to make tumor cells susceptible to HSV-1 and may predict optimal efficacy. Systemic tumor-specific CD8+ T cell responses efficiently concentrate in tumor sites, suggesting that local intratumor injection of HSV-1 is expected to provoke effective immune responses even for systemic tumors such as hematological malignancies.
Materials and Methods
Cell Lines and Antibodies
Human cell lines of hematological malignancies are listed in Table S1. Culture conditions of each cell line are described in the Supplemental Materials and Methods. E.G7-OVA (ATCC) was derived from the C57BL/6 mouse T cell lymphoma cell line EL4 by transducing OVA. Antibodies used in this study are listed in Table S3.
Viruses
T-01 and T-GFP are HSV-1 strain F-based oncolytic viruses containing modification in the same three genes as in G47Δ.13 The difference between G47Δ and T-01 is that the ICP6 gene is inactivated by insertion of the LacZ gene in the former,4 whereas the ICP6 gene is replaced by the cytomegalovirus (CMV) promoter, poly(A), and the LacZ gene in the latter.13 Virus stocks were prepared as described with modifications.38 In brief, subconfluent monolayers of Vero cells were infected and cultured in VP-SFM (Thermo Fisher, Waltham, MA, USA), and infected cells were harvested when a total cytopathic effect was observed. After a freeze-thaw/sonication regimen, cell debris was removed by low-speed centrifugation, and then virus was concentrated by high-speed centrifugation. The viral pellet was resuspended in Dulbecco’s phosphate-buffered saline (DPBS; Sigma-Aldrich, St. Louis, MO, USA) containing 10% glycerol and titrated by plaque assay on Vero cells. Mock extracts were prepared identically, except that DPBS containing 10% glycerol was used in place of virus during the infection step.
Flow Cytometry Analysis
To analyze the protein expression levels on the cell membrane, we analyzed samples on the Accuri C6 Flow Cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed using FlowJo software (BD Biosciences).
Cytotoxicity Assay in a 3D Spheroid Culture Model
Cells (1 × 105) were seeded in 1.5-mL tubes and pelleted. The cells were infected with virus or “mock” in a volume of 100 μL and incubated at 37°C for 1 h. After adding 900 μL of culture medium (total 1 mL), the infected cells were seeded at densities of 1 × 104 or 2 × 104 cells/well (200 μL/well) in PrimeSurface 96-U multi-well plates (Sumitomo Bakelite, Tokyo, Japan) and incubated at 37°C for 72 h. For cell lines, the cells were stained with trypan blue, and the numbers of viable cells were counted with Countess II FL Automated Cell Counter (Thermo Fisher). Cell lines were defined as susceptible to T-01 when viability at MOI 1.0 or less decreased significantly compared with mock treatment. For clinical samples, cells were stained with propidium iodide (BioLegend, San Diego, CA, USA) and monoclonal antibodies (mAbs) to gate tumor cells, and were analyzed by flow cytometry. After gating tumor cells, viable cells were detected as propidium iodide-negative cells. Tumor cells from clinical samples were defined as susceptible to T-01, when viability at MOI 10 decreased by 20% or more compared with mock treatment. Acyclovir was purchased from Fujifilm Wako (Osaka, Japan) and dissolved in DMSO.
Real-Time PCR for the gB Gene
DNA was isolated using a QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany). The samples were run on a ViiA7 Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). Amplification was performed using TaqMan Fast Advanced Master Mix (Thermo Fisher) as follows: 95°C for 20 s followed by 40 cycles of 95°C for 3 s and 60°C for 30 s. All of the experiments were performed in triplicate. The TaqMan probe sequence for the gB gene was 5′-CACACCTGCGAAACGGTGACGTCTT-3′. Primer sequences used were: forward: 5′-GGCGCGGTCCTCAAAGAT-3′, reverse: 5′-AGAACATCGCCCCGTACAAG-3′. DNA from T-01 with a known plaque-forming unit (PFU) was used as a control to quantify the amount of gB DNA of samples with PFUs.
HSV-1 Entry Assay
Cells were seeded in 1.5-mL tubes and pelleted. The cells infected with T-GFP or mock were seeded at densities of 1 × 105 cells/well in PrimeSurface 96-U multi-well plates and incubated at 37°C for 4 h. The cells were analyzed by flow cytometry to detect entry of T-GFP.
ATP and HMGB1 Release Assay
After cell death induction, secreted extracellular ATP was measured using a CellTiter-Glo 2.0 Assay (Promega, Madison, WI, USA) in accordance with the manufacturer’s protocol. Secreted HMGB1 was detected by western blotting after concentrating proteins in the supernatants using Amicon Ultra Centrifugal Filters (Millipore, Burlington, MA, USA).
Clinical Samples
Written informed consent was obtained from all of the patients. The study was conducted in accordance with the principles of the Helsinki Declaration and was approved by Ethics Committees of each hospital (Kagawa University Hospital, Sakaide City Hospital, and Kagawa Prefectural Central Hospital).
Animal Experiments
Mouse experiments were conducted in accordance with the guidelines approved by the Animal Care and Use Committee of Kagawa University. Cell lines together with Matrigel (BD Biosciences) were implanted subcutaneously into the flank of SCID Beige mice or C57BL/6 mice (Charles River Laboratories Japan, Yokohama, Japan), and T-01 or mock was injected intratumorally. The tumor size was measured, and OVA-specific CD8+ T cells in tumors and spleen were detected using H-2Kb OVA tetramer (MBL, Nagoya, Japan), as detailed in the Supplemental Materials and Methods.
Statistical Analyses
Statistical analysis was conducted using a two-tailed unpaired t test, non-repeated-measures ANOVA followed by Dunnett’s test or Student-Newman-Keuls (SNK) test, or Spearman’s correlation, as described in the figure legends. A p value <0.05 was considered statistically significant.
Author Contributions
Conceptualization, N.K.; Methodology, T.T.; Investigation, R.I., Y.K., M.O., and S.U.; Resources, O.I., A.M., T.T., K.K., and T.H.v.K.; Writing – Original Draft, R.I. and N.K.; Writing – Reviewing & Editing, T.K., N.S., A.T.-K., and N.K.; Supervision, T.T. and N.K.; Funding Acquisition, T.T. and N.K.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Dr. Y. Ino (Teikyo University, Japan) for support and discussion; Dr. H. Arase (Osaka University, Japan) for providing us with mouse anti-human PILRα mAb; Dr. M. Nishikori (Kyoto University, Japan) for providing us with a chemical library for epigenetics research; Dr. M. Nishikori, Dr. K. Shimoda (Miyazaki University, Japan), Dr. K. Imada (Japanese Red Cross Osaka Hospital, Japan), and Dr. M. Maeda (Institute for Frontier Life and Medical Sciences, Kyoto University, Japan) for providing us with cell lines, and R. Kojima, A. Kondo, K. Fukunaga, and M. Iwai for excellent technical support. The image of the Graphical Abstract is from TogoTV (2016 DBCLS TogoTV). This study was supported by Japan Society for the Promotion of Science (JSPS) KAKENHI (grants JP15K09501 and JP18K08359); Japan Agency for Medical Research and Development (AMED) (grant JP17ck0106144); a grant from International Joint Usage/Research Center (2018-1024); The Institute of Medical Science; The University of Tokyo; and Iwadare Scholarship Foundation, Japan (R.I.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.09.041.
Supplemental Information
References
- 1.Bommareddy P.K., Shettigar M., Kaufman H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018;18:498–513. doi: 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 2.Chen D.S., Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Bommareddy P.K., Peters C., Saha D., Rabkin S.D., Kaufman H.L. Oncolytic Herpes Simplex Viruses as a Paradigm for the Treatment of Cancer. Annu. Rev. Cancer Biol. 2018;2:155–173. [Google Scholar]
- 4.Mineta T., Rabkin S.D., Yazaki T., Hunter W.D., Martuza R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 5.Todo T., Martuza R.L., Rabkin S.D., Johnson P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bierman H.R., Crile D.M., Dod K.S., Kelly K.H., Petrakis N.L., White L.P., Shimkin M.B. Remissions in leukemia of childhood following acute infectious disease: staphylococcus and streptococcus, varicella, and feline panleukopenia. Cancer. 1953;6:591–605. doi: 10.1002/1097-0142(195305)6:3<591::aid-cncr2820060317>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 7.Bluming A.Z., Ziegler J.L. Regression of Burkitt’s lymphoma in association with measles infection. Lancet. 1971;2:105–106. doi: 10.1016/s0140-6736(71)92086-1. [DOI] [PubMed] [Google Scholar]
- 8.Taqi A.M., Abdurrahman M.B., Yakubu A.M., Fleming A.F. Regression of Hodgkin’s disease after measles. Lancet. 1981;1:1112. doi: 10.1016/s0140-6736(81)92286-8. [DOI] [PubMed] [Google Scholar]
- 9.Bais S., Bartee E., Rahman M.M., McFadden G., Cogle C.R. Oncolytic virotherapy for hematological malignancies. Adv. Virol. 2012;2012:186512. doi: 10.1155/2012/186512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tazzyman S., Muthana M., Harrison J., Lewis C., Conner J., Chantry A. Development of a Trojan horse oncolytic virus for treatment of myeloma. J. Clin. Oncol. 2015;33:e14035. [Google Scholar]
- 11.Ghose J., Russell L., Caserta E., Santhanam R., Jaime-Ramirez A.C., Viola D., Krishnan A., Hofmeister C.C., Kaur B., Pichiorri F. Exploring the Possibility of Using Herpes Simplex Virus in Oncolytic Virotherapy of Multiple Myeloma. Blood. 2016;128:4467. [Google Scholar]
- 12.Wu A., Mazumder A., Martuza R.L., Liu X., Thein M., Meehan K.R., Rabkin S.D. Biological purging of breast cancer cells using an attenuated replication-competent herpes simplex virus in human hematopoietic stem cell transplantation. Cancer Res. 2001;61:3009–3015. [PubMed] [Google Scholar]
- 13.Fukuhara H., Ino Y., Kuroda T., Martuza R.L., Todo T. Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res. 2005;65:10663–10668. doi: 10.1158/0008-5472.CAN-05-2534. [DOI] [PubMed] [Google Scholar]
- 14.Kepp O., Senovilla L., Vitale I., Vacchelli E., Adjemian S., Agostinis P., Apetoh L., Aranda F., Barnaba V., Bloy N. Consensus guidelines for the detection of immunogenic cell death. OncoImmunology. 2014;3:e955691. doi: 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Geraghty R.J., Krummenacher C., Cohen G.H., Eisenberg R.J., Spear P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 16.Montgomery R.I., Warner M.S., Lum B.J., Spear P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 17.Shukla D., Liu J., Blaiklock P., Shworak N.W., Bai X., Esko J.D., Cohen G.H., Eisenberg R.J., Rosenberg R.D., Spear P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 18.Satoh T., Arii J., Suenaga T., Wang J., Kogure A., Uehori J., Arase N., Shiratori I., Tanaka S., Kawaguchi Y. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell. 2008;132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arii J., Goto H., Suenaga T., Oyama M., Kozuka-Hata H., Imai T., Minowa A., Akashi H., Arase H., Kawaoka Y., Kawaguchi Y. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature. 2010;467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 20.Arii J., Hirohata Y., Kato A., Kawaguchi Y. Nonmuscle myosin heavy chain IIb mediates herpes simplex virus 1 entry. J. Virol. 2015;89:1879–1888. doi: 10.1128/JVI.03079-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia T., Konno H., Ahn J., Barber G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016;14:282–297. doi: 10.1016/j.celrep.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia T., Konno H., Barber G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016;76:6747–6759. doi: 10.1158/0008-5472.CAN-16-1404. [DOI] [PubMed] [Google Scholar]
- 23.de Queiroz N.M.G.P., Xia T., Konno H., Barber G.N. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol. Cancer Res. 2019;17:974–986. doi: 10.1158/1541-7786.MCR-18-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unterholzner L., Keating S.E., Baran M., Horan K.A., Jensen S.B., Sharma S., Sirois C.M., Jin T., Latz E., Xiao T.S. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gale M., Jr., Katze M.G. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol. Ther. 1998;78:29–46. doi: 10.1016/s0163-7258(97)00165-4. [DOI] [PubMed] [Google Scholar]
- 27.He B., Gross M., Roizman B. The γ(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1α to dephosphorylate the α subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farassati F., Yang A.D., Lee P.W. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat. Cell Biol. 2001;3:745–750. doi: 10.1038/35087061. [DOI] [PubMed] [Google Scholar]
- 29.Cassady K.A., Gross M., Roizman B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J. Virol. 1998;72:7005–7011. doi: 10.1128/jvi.72.9.7005-7011.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samudio I., Rezvani K., Shaim H., Hofs E., Ngom M., Bu L., Liu G., Lee J.T., Imren S., Lam V. UV-inactivated HSV-1 potently activates NK cell killing of leukemic cells. Blood. 2016;127:2575–2586. doi: 10.1182/blood-2015-04-639088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y.-Y., Yu Z., Lin S.-F., Li S., Fong Y., Wong R.J. Nectin-1 is a marker of thyroid cancer sensitivity to herpes oncolytic therapy. J. Clin. Endocrinol. Metab. 2007;92:1965–1970. doi: 10.1210/jc.2007-0040. [DOI] [PubMed] [Google Scholar]
- 32.Yu Z., Adusumilli P.S., Eisenberg D.P., Darr E., Ghossein R.A., Li S., Liu S., Singh B., Shah J.P., Fong Y., Wong R.J. Nectin-1 expression by squamous cell carcinoma is a predictor of herpes oncolytic sensitivity. Mol. Ther. 2007;15:103–113. doi: 10.1038/sj.mt.6300009. [DOI] [PubMed] [Google Scholar]
- 33.Wang P.Y., Currier M.A., Hansford L., Kaplan D., Chiocca E.A., Uchida H., Goins W.F., Cohen J.B., Glorioso J.C., van Kuppevelt T.H. Expression of HSV-1 receptors in EBV-associated lymphoproliferative disease determines susceptibility to oncolytic HSV. Gene Ther. 2013;20:761–769. doi: 10.1038/gt.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson J.D., McMorris A.M., Roth J.C., Coleman J.M., Whitley R.J., Gillespie G.Y., Carroll S.L., Markert J.M., Cassady K.A. Assessment of oncolytic HSV efficacy following increased entry-receptor expression in malignant peripheral nerve sheath tumor cell lines. Gene Ther. 2014;21:984–990. doi: 10.1038/gt.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang P.Y., Swain H.M., Kunkler A.L., Chen C.Y., Hutzen B.J., Arnold M.A., Streby K.A., Collins M.H., Dipasquale B., Stanek J.R. Neuroblastomas vary widely in their sensitivities to herpes simplex virotherapy unrelated to virus receptors and susceptibility. Gene Ther. 2016;23:135–143. doi: 10.1038/gt.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Giovine P., Settembre E.C., Bhargava A.K., Luftig M.A., Lou H., Cohen G.H., Eisenberg R.J., Krummenacher C., Carfi A. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 2011;7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ribas A., Dummer R., Puzanov I., VanderWalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Todo T., Feigenbaum F., Rabkin S.D., Lakeman F., Newsome J.T., Johnson P.A., Mitchell E., Belliveau D., Ostrove J.M., Martuza R.L. Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in aotus. Mol. Ther. 2000;2:588–595. doi: 10.1006/mthe.2000.0200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.