

Abstract
Large B cell lymphoma (LBCL) is curable with standard chemo-immunotherapy in the majority of cases. However, patients with primary refractory or relapsed disease have historically had limited treatment options. Two gene-modified chimeric antigen receptor (CAR)-T cell therapies have now been approved for these indications. The clinical decisions and management surrounding these gene-modified “living drugs” are nuanced and complex. In this article, we discuss the evolving evidence supporting the use of these CAR-T cells, including patient selection, screening procedures, special populations, bridging therapy, lymphodepletion, clinical management, relapse, and follow up.
Graphical Abstract
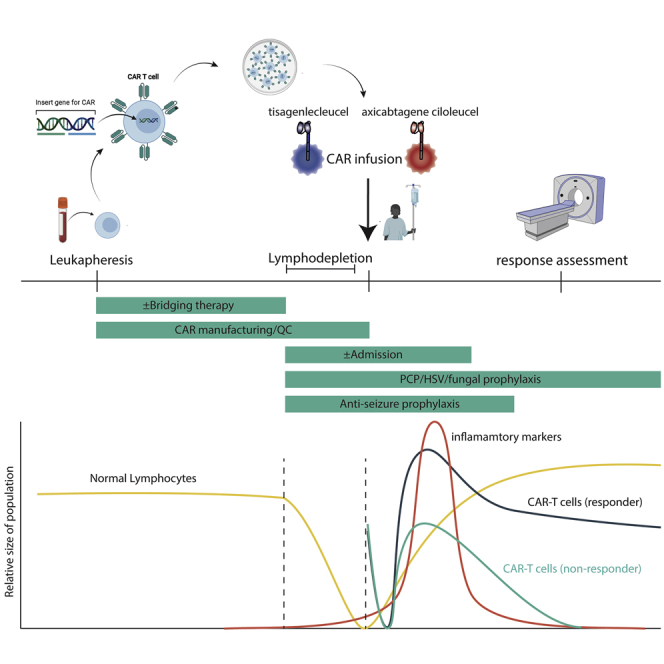
Drs. Leick, Maus, and Frigault draw from their institutional experience at Massachusetts General Hospital to provide a roadmap for the care of aggressive lymphoma patients using the two FDA-approved chimeric antigen receptor (CAR)-T cell therapies for this disease. They review the intricacies of patient screening, product selection, special populations, bridging therapy, and follow up.
Introduction
Non-Hodgkin’s lymphomas (NHLs) make up a collection of neoplasms derived from B or T cells affecting older adults, with a median age in the 60s. NHLs range from indolent, with essentially a normal life expectancy, to life-threatening, aggressive variants like diffuse large B cell lymphoma (DLBCL). DLBCL is the most common lymphoma, representing up to 40% of NHL cases globally. The standard of care until the late 1990s consisted of a combination chemotherapy regimen, including cyclophosphamide, vincristine, doxorubicin, and prednisone (known as CHOP) and was curative in about half of patients, while the addition of rituximab (R-CHOP) increased this to about 67%.1,2 The third of patients who did not achieve a cure with initial therapy were likely to succumb to their disease, representing an unmet clinical need. Until recently, the only salvage therapy was high-dose chemotherapy followed by autologous stem cell transplantation (ASCT). This option was limited to the subset of patients who relapsed with “chemo-sensitive” disease and could tolerate high-dose chemotherapy.3 SCHOLAR-1 was a large clinical study that attempted to ascertain the outcomes of refractory DLBCL (defined as unresponsiveness to first or second line therapies or relapse <12 months after ASCT) and found that just 20% of this patient population lived for 2 years.4
Adoptive cellular therapy via modulation of autologous T cells to express a chimeric antigen receptor (CAR)-T cell has resulted in a substantial improvement in the outcomes for relapsed/refractory LBCL patients, including long-term durable responses at 24 months in upward of 40% of patients.5 In a Herculean feat of logistics for this bespoke product, patients’ own T cells are collected locally, shipped to a processing center for manufacturing, transduced with the CAR transgene, expanded, and then returned to the patient for infusion. Since 2017, two FDA approvals were granted to two different gene-modified, autologous T cell products for relapsed or refractory LBCL: axicabtagene ciloleucel (axi-cel) and tisagenlecleucel (tisa-cel). Both products are specifically approved for LBCL, including DLBCL not otherwise specified (DLBCL NOS), high grade B cell lymphoma (HGBCL), and DLBCL arising from follicular lymphoma, while axi-cel also specifically references the addition of primary mediastinal B cell lymphoma (PMBCL). A third agent, lisocabtagene maraleucel, has shown efficacy in late-stage clinical trials but is not yet US Food and Drug Administration (FDA) approved and will not be discussed here. While both technologies are considered “second generation” CAR-T cell products, meaning they fuse one T cell costimulatory domain to the primary CD3ζ activation domain, they have key differences. axi-cel utilizes a CD28-costimualtory domain, which endows it with more rapid, but shorter duration, expansion kinetics and increased toxicity.6 tisa-cel has a 4-1BB costimulatory domain, leading to delayed, but longer-lived, expansion and decreased rates of toxicity.7 axi-cel also employs a gamma retroviral vector for gene modification with transgene expression driven off the endogenous long terminal repeats (LTRs), while tisa-cel employs a third-generation, self-inactivating lentiviral vector with transgene expression driven from the human EF1a promoter. The role of the different vectors in modulating the clinical differences between the two CAR-T cell products is not yet clear, and neither has resulted in oncogenic transformation or adverse integration events.8, 9, 10 Additional key manufacturing differences are in Table 1.
Table 1.
Key Differences between tisa-cel and axi-cel
| Product | tisa-cel | axi-cel |
|---|---|---|
| Costimulation | 41BB | CD28 |
| Anti-CD19 antibody clone | FMC63 (murine) | FMC63 (murine) |
| Gene transfer method | lentivirus | retrovirus |
| Collection type | frozen | manufactured from fresh (non-frozen) |
| T cell selection prior to manufacturing | yes | no |
| Infusion type | bulk CD3 (both CAR-T cells and untransduced T cells) | bulk CD3 (both CAR-T cells and untransduced T cells) |
| Approved lymphomas | DLBCL-NOS, high-grade B cell lymphoma, DLBCL arising from FL | DLBCL-NOS, high-grade B cell lymphoma, DLBCL arising from FL, PMBCL |
Registration studies for axi-cel (ZUMA-1) and tisa-cel (JULIET) reported the initial outcomes for these products. The key variables and outcomes for the clinician from the phase 1/2 trials and real world data are summarized in Table 2. ZUMA-1 was a single-arm, multicenter, phase 1/2 trial at 22 sites and included 119 patients, of which 108 were administered the CAR-T cell product (91%). The objective response rate (ORR) was 83%, with 59% achieving a complete response (CR). With a median follow up of 27.1 months, 39% of patients had ongoing responses at the last disease assessment before the data cutoff, including 37 (37%) with ongoing complete responses.11 JULIET was a single-arm multi-center phase 2 trial across centers in North America, Europe, Australia, and Japan, and included 167 patients who met eligibility criteria and underwent leukapheresis, but only 115 were infused (67%). The last reported ORR was 54%, with an initial CR rate of 40%. At a median follow up of 19.3 months, ongoing responses were seen in 36% of patients with a relapse-free probability of 64% at 12 or 18 months.12,13 Notably, in JULIET, patients could undergo enrollment, apheresis, and cryopreservation without confirming the availability of a manufacturing slot, while in ZUMA-1, patients were enrolled at time of apheresis, utilizing fresh, non-cryopreserved product for manufacturing. This led to potential survival bias (since surviving until infusion potentially indicates less aggressive disease) and may have also affected trial enrollment. However, the remarkable similarity of durable responses (39% versus 36%) that was seen in both studies is reassuring. Direct comparisons of toxicity are also difficult to make as the cytokine release syndrome (CRS) grading systems differed between the two trials. The JULIET trial utilized the Penn grading system, while ZUMA-1 utilized criteria from Lee 2014, which likely led to higher assignments of “severe” CRS in JULIET than would have been obtained with Lee 2014.14,15 Because of differences in patient selection, allowance of bridging therapy and other variables between trial comparisons are exceptionally limited.16
Table 2.
Key Phase 1/2 and Real-World Clinical Variables for axi-cel and tisa-cel
| tisa-cel | axi-cel | |
|---|---|---|
| Pivotal trial | JULIET | ZUMA1 |
| Bridging | 90% | not allowed |
| LDC | 74% flu-cy (25 mg/m2, 250 mg/m2 ×3 days, 19% benda, 7% none | 100% flu-cy (30 mg/m2, 500 mg/m2 |
| Prior ASCT | 49% | 25% |
| <3 lines of prior therapy | 49% | 31% |
| Relapsed <12 months post ASCT | 34% | 21% |
| Double/triple hit | 17% | 4% |
| Bulky disease | 8% | 16% |
| HSCT post infusion | 6% | 11% |
| Retreatment | 0% | 9% |
| Never treated | 31% | 9% |
| ORR | 54% | 83% |
| 6-month CR | 29% | 37% |
| Real world data reference | 17 | 18 |
| Number of patients (outcomes available) | 47 | 295 |
| ASTCT CRS 3+ | 4.30% | 14% |
| ASTCT ICANS 3+ | 4.30% | 39% |
| ICU stay | not reported | 30% |
| Deaths due to toxicity | 0 | 5 |
| ORR | 61%, 57%a | 70% |
| CR | 39%, 38%a | 52% |
flu, fludarabine; cy, cyclophosphamide.
CIBMTR registry data for tisa-cel was stratified by cellular viability either >80% or 60%–80%, and ORR and CR are reported respectively.
Riedell et al.19 recently presented retrospective data of outcomes from patients who underwent apheresis for commercial use of tisa-cel or axi-cel from 8 US academic centers. Notably, this analysis required that centers have both products available at the time of treatment to minimize the impact of evolving treatment strategies and patient selection and utilized the recent American Society for Transplantation and Cellular Therapy (ASTCT) 2019 consensus toxicity guidelines for grading in both products.20 In this analysis, 158 patients were treated with axi-cel and 86 with tisa-cel, of which 92% of axi-cel patients were treated inpatient versus 38% of tis-cel. Bridging therapy was utilized in 61% of axi-cel and 75% of tisa-cel patients (median time to infusion 28 versus 44 days, respectively). tisa-cel patients were older (67 versus 59 years median age) and more heavily pretreated (86% versus 73% with >3 prior therapies); however, efficacy was similar to the pivotal trials. At 90 days post-infusion, 64% of axi-cel had an objective response with 53% CR and 51% of tisa-cel had an objective response with 42% CR. The median hospital stay was 16 versus 2 days, and the ICU transfer rate was 39% versus 7%, both in favor of tisa-cel. Tocilizumab use was 61% versus 15% and corticosteroid use was 53% versus 8%, again in favor of tisa-cel.19 Additional analysis utilizing the Center for International Blood and Marrow Transplant Research (CIBMTR) registry of axi-cel included 295 patients (median age 61) and found an ORR of 70%. Incidence of greater than or equal to grade 3 CRS and immune effector cell-associated neurotoxicity syndrome (ICANS) was 14% and 39%, respectively. Five patient deaths were attributable to toxicity.18 Other post-market registry and consortium analyses have revealed similar results.21,22 In a similar CIBMTR registry analysis looking at 70 standard-of-care patients treated with tisa-cel, the ORR was comparable to the pivotal trial at 58%, and rates of grade 3 or higher CRS and ICANS were substantially lower at just 4.3% and 4.3%. No patients died from toxicity.17
Patient Selection and Screening
Given the aggressive nature of relapsed/refractory disease for most patients, early referral for CAR-T cell therapy is paramount. Ample time should be allowed, considering the referral process, apheresis, and insurance authorization. Box 1 highlights some of the key decisions that must be made when deciding on CAR-T cell therapy. The optimal timing for referral to an experienced cellular therapy center is at the time of first relapse, given the potential consideration for high-dose chemotherapy and stem cell rescue versus CAR-T cell therapy pending response to salvage therapy (Figure 1). Centers for Medicaid & Medicare Services has agreed upon coverage for CAR-T cell therapy, and multiple systems are in place to increase patient access. At our center, initial screening includes a comprehensive history and physical examination, transthoracic echocardiogram (TTE), comprehensive metabolic panel, complete blood count, glucose-6-phosphate-dehydrogenase (G6PD) levels (in the event the patient experiences tumor lysis syndrome and needs rasburicase), lymphocyte subsets for T cell quantification, and lactate dehydrogenase (LDH) levels as well as a comprehensive, nurse-driven patient education and social work evaluation to assist with housing and caregiver support (Figure 2).
Box 1. Key Decisions Regarding CAR Therapy for LCL.
Timing of collection
Choice of product
Bridging therapy
Lymphodepleting regimen
Antibiotic prophylaxis
Management of CRS/ICANS (antibiotics, tocilizumab, steroids, other agents)
Follow up screening, including IVIG
Figure 1.

Flowchart of Management Considerations
Figure 2.

Timeline of CAR-T Cell Treatment at Our Institution
Infectious Screening
Given the possibility of prolonged cytopenias, including prolonged lymphopenia and neutropenia, patients are screened for human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus per standard infectious disease guidelines. In addition, given the limited data describing long-term infectious outcomes of patients receiving CAR-T cell therapy, we routinely screen for cytomegalovirus (CMV) by polymerase chain reaction (PCR), human T-lymphotropic virus 1 (HTLV1), syphilis via rapid plasma reagin (RPR), and tuberculosis (in high risk patient populations) due to the potential need for high dose steroids and various immunomodulatory agents. Although none of these infectious comorbidities would prevent subsequent treatment, knowledge of such latent infections can assist in patient optimization prior to treatment and product selection. Thus far, patients with both active HIV and latent HBV have been successfully and safely treated with CAR-T cells.23
Other Screening
Although CD19 positivity was not shown to correlate with response to treatment in either one of the pivotal studies, insurers frequently request documentation of CD19 positivity.6,13 We have since started routine CD19 testing on patient tumors to pre-emptively demonstrate positivity. It is our hope that insurers move beyond this practice, given that it may limit patient access and delay treatment.
Choosing a Commercial Product
Logistical Considerations
A number of factors are integrated when choosing which commercial product is utilized at our institution. A critical up-front consideration is the logistics of apheresis. axi-cel collections must be scheduled with the company based on available manufacturing slots and utilizes fresh (not frozen) apheresis product. This process has reliably produced rapid and successful manufacturing.24 The timing of bridging therapy (discussed below) should be considered when timing apheresis, given concerns for optimal apheresis product. tisa-cel utilizes a cryopreserved apheresis product, which potentially allows for more flexibility around the collection timeline, given the option of cryopreservation followed by bridging therapy regardless of manufacturing availability. This also includes cryopreservation prior to salvage therapy in high-risk patients. However, due to differences in manufacturing and product release testing, tisa-cel has demonstrated longer “vein-to-vein” times, with one real-world experience median time from apheresis to CAR-T cell infusion of 28 days for axi-cel and 44 days for tisa-cel.25 These timelines continue to decrease with improvements in manufacturing capacity. Of note, tisagenlecleucel has demonstrated a higher out-of-specification rate due to differences in viability criteria between the pivotal trial and commercial approval. We routinely treat patients with tisagenlecleucel if the viability falls “out-of-spec” but there is a sufficient viable, CAR-T-positive cell dose. To date, no difference in outcomes has been demonstrated, with products greater than or less than 80% viability.17
Special Populations
Secondary CNS Lymphoma
Due to strict eligibility criteria, patients with secondary CNS lymphoma were excluded from the phase 1/2 trials that led to the US FDA approval of both products because of concern for increased risk of neurotoxicity. Recent data have suggested that ICANS may simply be a neurologic manifestation of a systemic inflammatory process, meaning that traditional CRS/ICANS risk factors, such as baseline inflammatory markers and disease burden, are more likely to predict CRS/ICANS than low volume CNS involvement.26 As such, we recently reported our institutional experience treating 8 patients with active secondary CNS lymphoma with commercial tisa-cel. tisa-cel was chosen due to the lower rates of associated neurotoxicity seen in the pivotal studies. No patient experienced greater than grade 1 ICANS and none required tocilizumab or steroids. Three patients achieved CR and another had an ongoing partial response (PR) by day +90.27 Although secondary CNS lymphoma is allowed per the FDA label (as a site of extranodal relapse), primary CNS lymphoma (PCNSL) was explicitly prohibited. As such, tisa-cel for PCNSL is currently under investigation at our institution (ClinicalTrials.gov: NCT04134117). Thus, in the cases of known or suspected CNS disease, we treat patients with tisa-cel given the dramatically lower rates of ICANS. More recently, others have presented their experience utilizing axi-cel for patients with active or a history of CNS lymphoma. Bennani et al.24 described 15 patients (5 of which had active CNS disease, while 10 had a history of treated CNS disease) that, when treated with axi-cel, demonstrated higher rates of severe ICANS in patients with active CNS disease (3 out of the 5). We therefore favor use of tisa-cel in patients with active secondary CNS disease.
Elderly or Frail
A recent retrospective analysis of tisa-cel-treated patients in a pivotal phase 1/2 trial looked at the incidence and severity of toxicities in patients older than age 65 compared to patients younger than age 65, finding no differences in toxicity or efficacy (though the difference in median age was small, median age 55 versus 69 years).28 In both the pivotal studies (ZUMA-1 versus JULIET) and the real experience registry data, severity and incidence of CRS and neurotoxicity were lower with tisa-cel than axi-cel.6,13,17,18,29 Additionally, recent data have suggested that older patients (>75 years) may have worse progression-free survival (PFS) and increased toxicities with axi-cel, although these data are evolving.30 In light of these data, we preferentially use tisa-cel in older, more frail patients as it is better tolerated in our experience. Biologic age, in and of itself, is not a contraindication. Indeed, we have successfully treated patients into their eighth decade of life with tisa-cel and prefer this product for patients for whom tolerance of toxicity is a concern. Importantly, patients who may not typically be considered eligible for an autologous stem cell transplant can be, and have been, successfully treated with CAR-T cells.
PMBCL
Given the exclusion of PBMCL patients from the pivotal study of tisa-cel and a paucity of subsequent data, we preferentially use axi-cel unless there is evidence of CNS involvement. We await further data for potential extension of tisa-cel into this subtype.
HIV
Our institution23 and others31 have successfully treated patients with HIV infection and concurrent high-grade B cell lymphoma who were excluded from pivotal trials. In our 2 patient case report, axi-cel was chosen given tisagenlecleucel manufacturing restrictions in HIV+ patients. Our first patient had a low, but detectable, viral load during apheresis. His course was complicated by CRS, requiring tocilizumab and dexamethasone, and a short period of grade 2 ICANS. He had an initial partial response at day +30, followed by improvement to CR, which persisted at the last follow up of 1 year. Our second patient had an undetectable viral load at the time of apheresis. His hospital course was uncomplicated, and he had a CR by day +30, which he has maintained. Neither patient had evidence of HIV reactivation during treatment. We feel confident that this patient subgroup can, and should, be offered these therapies given their curative potential.
Post-transplant
Although both JULIET and ZUMA-1 excluded patients with prior allogeneic stem cell transplant, the pivotal ELIANA study allowed for prior allogeneic stem cell transplant in the absence of ongoing graft-versus-host disease (GVHD). Given limited data, we therefore favor tisagenlecleucel post allogeneic stem cell transplant for LCL.32
Circulating Disease/Richter’s Transformation
Although Richter’s transformation was not allowed in the pivotal studies, it is not excluded per the product label. As such, when considering treating patients with Richter’s transformation, the presence of circulating disease will impact product selection. While tisa-cel utilizes a T cell selection step in manufacturing, axi-cel (KTE-C19) does not, leading to the potential for transduction of circulating tumor and a recently described mechanism of resistance termed “CAR-B.” In this scenario, transduced tumor is able to mask the CD19 epitope by expressing the CAR single-chain variable fragment (scFv) and develop resistance to CAR-T cell killing.33 In order to avoid this issue in mantle cell lymphoma (MCL), ZUMA-2 utilized a T cell selection process prior to CAR-T manufacturing (KTE-X19). The CAR-T cell product utilizing this strategy, brexucabtagene autoleucel, was recently approved for MCL. We therefore favor tisa-cel in patients with Richter’s transformation.
Contraindications
Patients with significant baseline pulmonary or cardiovascular compromise, such as New York Heart Association (NYHA) class III-IV, significant valvular or peripheral vascular disease, and severe chronic obstructive pulmonary disease are considered high risk for CAR-T cell therapy, given the possibility of CRS and ICANS and potential need for fluid and/or vasopressor administration. In most instances, attempts to optimize these patients in a multidisciplinary setting are pursued with the hopes of being able to offer these curative therapies. That being said, the decision to proceed is made on a patient-by-patient basis, with alternative therapeutic options considered when appropriate. For borderline patients with moderate to severe cardiovascular or pulmonary compromise, the relevant specialist is involved as early as possible during screening for patient optimization. We have retrospectively investigated the cardiac effects of CAR-T cell therapy. In a cohort of mostly axi-cel-treated patients, elevated troponin (29 of 53, 54%) and a decrease of left ventricular ejection fraction (8 of 29, 28%) were common. Cardiac events occurred in 17 of 137 patients (12%), including 6 cardiovascular deaths, 6 decompensated heart failures, and 5 arrythmias. All of the cardiac events occurred with grade 2 or greater CRS, and 95% of events occurred after elevated troponin. A shorter time from CRS onset to tocilizumab administration was associated with a lower rate of cardiovascular events.34 In a separate cohort of 145 patients with B-acute lymphoblastic leukemia (B-ALL) (25%), chronic lymphocytic leukemia (CLL) (46%), and DLBCL (30%) treated with commercial or investigational CD19 CAR-T cells, 21% experienced major adverse cardiac events (MACEs). Baseline creatinine (hazard ratio 15.5) and grade 3 or 4 CRS (hazard ratio 8 and 30 respectively) were independently associated with MACEs on multivariate analysis.35 Since we are unable to safely administer standard lymphodepleting chemotherapy (LDC) with fludarabine, due to fatal fludarabine associated neurotoxicity in the setting of low creatinine clearance and evidence that MACE and other toxicities significantly track with worsened glomerular filtration rate (GFR), we use a CrCl of less than 30 mL/min as a contraindication to CAR-T cell therapy.35,36 Finally, we use baseline eastern cooperative group (ECOG) performance status greater than 2 (not attributable to disease) as a cutoff for CAR-T cell therapy eligibility.
Apheresis and Collection
Multiple relapsed/refractory aggressive lymphoma patients may have difficulty achieving an adequate concentration of blood lymphocytes, which is necessary for producing a viable CAR-T cell product. We therefore assess for absolute CD3+ T cell counts by lymphocyte subsets at the time of CAR-T cell screening. Ideally, patients would have an absolute lymphocyte count (ALC) of >500 cells/uL; however, we typically allow for an ALC of >100 cells/uL prior to apheresis.
Bridging Therapy
One of the major considerations in moving forward with CAR-T cell therapy is the strategy to ensure that the patient maintains an adequate functional status, remains optimized for treatment, and survives long enough to receive the cellular infusion and have a subsequent response. Therefore, bridging therapy before and/or after apheresis is often required. While ZUMA-1 did not allow for any bridging therapy, 90% of patients in JULIET received bridging therapy. Some have retrospectively examined the role of bridging therapy in CAR-T cell therapy outcomes, with some evidence suggesting that those who receive bridging therapy prior to axi-cel treatment may have worse outcomes; however, it is unclear whether this is a result of more aggressive disease requiring bridging therapy or the bridging therapy itself.37 In a real-world experience, bridging therapy was given in 61% of axi-cel-receiving patients and 72% of tisa-cel patients.25 Further prospective studies are warranted. For our center, we often decide to pursue bridging therapy based upon factors such as disease sites, patient symptoms, bulky disease/elevated LDH, rapidly progressive disease, and manufacturing timeline. General considerations for what agent to use include fitness of the patient, prior responsiveness to earlier line therapies, timing of collection, and histologic features of the patients’ disease. Of note, when considering giving third line therapy prior to CAR-T cell referral, it is essential to discuss options with the referral center, as use of certain agents, such as bendamustine, may negatively impact cell manufacturing and/or delay the collection timeline due to insufficient peripheral lymphocytes. We therefore recommend close collaboration with the referring physicians to devise the optimal treatment strategy.
Newer agents have recently been approved, such Polatuzumab (a novel CD79b targeted antibody-drug conjugate [ADC]) in combination with bendamustine and Rituxan, and Tafasitamab (an anti-CD19 ADC) in combination with lenalidomide. Although promising, given the risk of prolonged lymphopenia resulting in a poor T cell function, we typically avoid bendamustine prior to apheresis but do consider such therapy following successful apheresis. Additionally, we avoid CD19-directed therapies prior to CAR infusion, as there is a theoretical risk of resistance via CD19 antigen loss.38 Both ibrutinib and revlimid have demonstrated efficacy in activated B cell type (ABC) DLBCL and are potential options as oral therapy. Ibrutinib is also one of the few agents active in DLBCL that also has reliable CNS penetration,39,40 and additional data suggest that ibrutinib may actually improve T cell fitness, resulting in more potent CAR-T cells.41, 42, 43 Furthermore, pre-clinical44,45 evidence has suggested that lenalidomide may also improve CAR-T cell function. Notably, rapid progression of disease has been noted after cessation of ibrutinib, and caution should be taken with discontinuation.46 When necessary, short courses of high-dose steroids (dexamethasone 40 mg for 4 days) can be used for short-term palliation, assuming they are held within 72 h of apheresis and infusion.
Radiation therapy (RT) represents another attractive option for bridging therapy that potentially mitigates further pharmacologic therapy in a group of typically heavily pretreated patients. Provocative hypothesis-generating data suggest that RT may be superior to cytotoxic chemotherapy for bridging purposes.47 Furthermore, CR rates appeared to be higher among RT-bridged patients than those that received no bridging therapy, and treatment-related mortality was lower, with seven patients in the study dying from bridging therapy-related toxicity (versus none with RT). Based on these data and our experience, it is our practice to consider the use of RT, when feasible, as bridging therapy. Some patients treated with limited RT fields have relapsed at sites excluded from RT, despite an initial response to CAR-T cell therapy. Thus, it is our practice to consider irradiating surrounding areas, when feasible, weighing risk versus benefit.
LDC
All patients on ZUMA-1 received fludarabine and cyclophosphamide (flu/cy) LDC, while, on JULIET, 74% received this combination, 19% received bendamustine, and 7% received none. In a post hoc analysis of the JULIET trial, patients who received flu/cy LDC had better outcomes than those who received bendamustine or no LDC.48 For this reason, we use almost exclusively flu/cy (with close attention to renal function) rather than bendamustine or no LDC.
Infectious Disease
We delay lymphodepletion for patients with serious uncontrolled infections; however, we will consider proceeding if the patient is clinically stable on an appropriate antimicrobial regimen. All patients are screened for coronavirus disease 2019 (COVID-19) and any with respiratory symptoms receive an expanded respiratory viral panel and chest imaging. Immunoglobulin (Ig) levels are tested, and patients with an IgG level <400 mg/dL receive intravenous immunoglobulin (IVIG) following infusion . On day −5, we start acyclovir or valacyclovir, continuing for at least 6 months. We don’t typically monitor for CMV reactivation, though we will if the patient received an allogeneic transplant within the prior year or requires substantial immunosuppression for GVHD or CAR-T cell toxicity. Some centers, including ours, use fluoroquinolone prophylaxis for severe neutropenia with an absolute neutrophil count (ANC) < 500 cells/mm3.49,50 For patients with anticipated neutropenia greater than 10 days or ongoing use of corticosteroids, we provide antifungal prophylaxis with fluconazole until cessation of corticosteroids or recovery of neutropenia. For patients with severe and ongoing neutropenia beyond a month or more, we consider a mold-active azole. We provide Pneumocystis jirovecii prophylaxis for all patients, starting with lymphodepletion, and continued for 6 months or T cell recovery.
Patients with a history of prior (anti-HBs/HBc) or chronic HBV (HBsAg positive or have +HBV DNA) receive entecavir daily with LDC and for 6 months after. Small case series have demonstrated the safety of this approach, though more data are needed to confirm.51 Patients with anti-HBs are at a further reduced risk of reactivation, and vaccination can be considered for those who are anti-HBs negative (regardless of anti-HBc status), if time allows.52 Notably, a case of fulminant HBV reactivation has been reported after cessation of prophylaxis, just 1 month after CAR-T cell infusion of CARs targeting B cell lineage antigens.53 Patients with HCV are referred to a subspecialist to discuss monitoring any timing of curative therapy.
Fatal infections after CAR-T cell therapy are rare, and almost all cases of bacteremia occur in the first 2 weeks after infusion. Respiratory viral infections occur, and cases of HSV/VZV have been observed in patients not taking prophylaxis. In certain series, invasive fungal infections have been seen in up to 8% of patients, including mold.54 Patients who experience CRS or who have a high number of prior regimens are at greater risk of infection likely due to prolonged cytopenias, though tocilizumab use does not appear to be associated with increased infectious risk.55 Given the inability to conclusively differentiate CRS from infection, we treat neutropenic fever aggressively, regardless of attribution to CRS. New or progressive symptoms should raise concern for a new infection, especially given that CRS is an independent risk factor for infections after CAR therapy.49 Patients with altered mental status felt to be related to neurotoxicity should undergo CSF sampling only if the pretest probability of a bacterial or viral infection is high and it is deemed clinically safe, as many of these patients are coagulopathic and thrombocytopenic at time of toxicity.
Notably, older plasma cells, which make the majority of antibodies, have relatively low CD19 expression and may escape CD19-CAR-mediated destruction and continue to produce pathogen-specific antibodies despite B cell aplasia.56 We consider giving vaccinations >6 months after CAR-T cell infusion for killed/inactivated and >1 year for live and non-live adjuvant vaccines. This recommendation is based on vaccine responses following autologous and allogeneic stem cell transplant, but there are limited data to support the efficacy of this approach. Additional studies are warranted.
Toxicity Grading and Management
CRS is a systemic inflammatory syndrome, similar to sepsis, with a constellation of clinical findings, including hyperpyrexia, hypoxia, and hypotension, which can lead to multi-organ disfunction. While typically following CRS, ICANS can occur independently and may manifest as hallucinations, encephalopathy, seizures, aphasia, headaches, and most concerningly, rapidly progressive cerebral edema, which, in rare cases, can lead to fatal cerebral herniation. Comprehensive review of the management of CAR-T cell toxicities is beyond the scope of this article and has been described in detail elsewhere.57 We have also previously discussed, in detail, management of ICANS at our institution.58 We utilize the ASTCT consensus grading system for CRS and ICANS.59 In brief, we include neuro-oncologic specialists at the earliest signs of ICANS. Levetiracetam is started day 0, before cellular infusion, and is continued for 30 days after. Brain imaging (CT/MRI) and/or electroencephalogram (EEG) is pursued with ICANS ≥2, development of a focal neurologic deficit, onset of seizure activity, or with other clinical concerns. Management of CRS has continued to evolve with earlier and earlier use of tocilizumab when comparing earlier versus more recent studies. That being said, some studies have suggested that prophylactic tocilizumab may result in improved severe CRS but with worsening ICANS.60 We therefore do not favor tocilizumab for isolated ICANS, CRS prophylaxis, or prolonged grade 1 CRS unless that patient is frail and/or has the presence of medical comorbidities. For grade 2 or higher ICANS, we favor dexamethasone at a maximum dose of 10 mg every 6 h and a rapid steroid taper, as prolonged steroid use has been associated with increased infectious complications. Fungal prophylaxis should be considered in patients receiving high-dose steroids. Although prolonged steroid use has also been associated with poorer outcomes, it is unclear if this is a result of higher risk disease features and subsequent toxicity or steroid use itself.61
Follow Up
After infusion, we follow patients weekly for the first month, followed by monthly thereafter. We obtain routine blood counts and chemistries, Ig levels, and an absolute CD4 count. PET-CT (with MRI for CNS disease) is obtained at days 30, 90, 180, and 360. Pneumocystis jirovecii prophylaxis is continued until CD4 count is greater than 200 cells/μL. HSV/VZV prophylaxis is continued for 1 year. Antiseizure prophylaxis is continued for 30 days. Survivorship is an important and active area of evolving research.62 Given the history of severe toxicities associated with CAR-T cell therapy, the risk of outpatient versus inpatient infusion is center specific. As we obtained experience with specific CAR-T cell products, we began outpatient infusions primarily utilizing tisagenlecleucel due to its delayed onset of toxicities and lower overall severity. A key consideration with outpatient CAR-T cell therapy is the infrastructure available to rapidly evaluate, admit, and treat outpatient CAR-T cell patients in the event of toxicity. Such an arrangement requires that the patients remain in close proximity to the treating center, with a caregiver who can stay with the patient at all times, and an institution with the resources to triage and bring the patients into the hospital, if needed, at any time.
Restaging
Disease response is assessed by PET-CT imaging at day +30 (and with brain MRI for patients with CNS disease), day +90, 6 months, and 1 year. Half of partial responses may convert to complete responses and demonstrate ongoing durable remissions.11,13 In general, if there is residual PET avidity for persistent disease, we attempt a biopsy (if at all possible), as the results may be informative for the next course of treatment. On biopsy, we confirm histology (ensuring no alternative malignancy, transformation to an alternate histology, progression of an underlying indolent lymphoma, or residual inflammatory infiltrate without evidence of disease) and assess PD-L1 and CD19 staining. CD19 antigen loss is common in ALL, and seemingly less so in DLBCL after CD19 CAR-T cell therapy.63,64
In the cases of clear tumor antigen loss, subsequent CD19-directed therapies are unlikely to be beneficial. Additionally, there are limited data to support reinfusion with the same, or different, CAR-T cell product. Our rescue CAR-T cell strategies instead, focus on orthogonal, non-CD19 based therapies. In the presence of ongoing CD19 expression, we consider various immunomodulatory agents, such as checkpoint blockade, ibrutinib, and revlimid; however, there are limited data suggesting their optimal practice. Additionally, consideration for clinical trial evaluation should be considered. If the patient can be successfully salvaged, we strongly consider subsequent allogeneic stem cell transplant. One exception to this rule is in the case of an isolated site of relapse after an apparent systemic response. In this case, it is possible that a single site of disease has evolved resistance and may be amenable to targeted RT. Clinical parameters, such as ongoing B cell aplasia, indicating continued CAR-T cell functionality, may be corroborative. We therefore favor localized RT for focal relapse.
Conclusions
CD19 CAR-T cell therapy for DLBCL has provided an effective treatment options for a subset of patients who previously had few. There are a number of considerations for these new therapies, and they are best utilized in the hands of an experienced center with a multidisciplinary team of physicians, nurses, and support staff.
Acknowledgments
Biorender was used in the Graphical Abstract.
Declaration of Interest
M.J.F. and M.V.M. have received sponsored research support from Kite Pharma and Novartis and are investigators and consultants on multiple industry-sponsored trials of CAR-T cells. M.V.M. holds equity in Century Therapeutics and TCR2.
References
- 1.Union for International Camcer Control . 2014. Diffuse Large B-Cell Lymphoma. 2014 Review of Cancer Medicines on the WHO List of Essential medicines.https://www.who.int/selection_medicines/committees/expert/20/applications/DiffuseLargeBCellLymphoma.pdf?ua=1 [Google Scholar]
- 2.Coiffier B., Sarkozy C. Diffuse large B-cell lymphoma: R-CHOP failure-what to do? Hematology (Am. Soc. Hematol. Educ. Program) 2016;2016:366–378. doi: 10.1182/asheducation-2016.1.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Den Neste E., Schmitz N., Mounier N., Gill D., Linch D., Trneny M., Bouadballah R., Radford J., Bargetzi M., Ribrag V. Outcomes of diffuse large B-cell lymphoma patients relapsing after autologous stem cell transplantation: an analysis of patients included in the CORAL study. Bone Marrow Transplant. 2017;52:216–221. doi: 10.1038/bmt.2016.213. [DOI] [PubMed] [Google Scholar]
- 4.Crump M., Neelapu S.S., Farooq U., Van Den Neste E., Kuruvilla J., Westin J., Link B.K., Hay A., Cerhan J.R., Zhu L. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800–1808. doi: 10.1182/blood-2017-03-769620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Locke F.L., Ghobadi A., Jacobson C.A., Miklos D.B., Lekakis L.J., Oluwole O.O. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2018 doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak Ö., Brogdon J.L., Pruteanu-Malinici I., Bhoj V., Landsburg D. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scholler J., Brady T.L., Binder-Scholl G., Hwang W.T., Plesa G., Hege K.M., Vogel A.N., Kalos M., Riley J.L., Deeks S.G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nobles C.L., Sherrill-Mix S., Everett J.K., Reddy S., Fraietta J.A., Porter D.L., Frey N., Gill S.I., Grupp S.A., Maude S.L. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J. Clin. Invest. 2020;130:673–685. doi: 10.1172/JCI130144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang G.P., Levine B.L., Binder G.K., Berry C.C., Malani N., McGarrity G., Tebas P., June C.H., Bushman F.D. Analysis of lentiviral vector integration in HIV+ study subjects receiving autologous infusions of gene modified CD4+ T cells. Mol Ther. 2009;17:844–850. doi: 10.1038/mt.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Locke F.L., Ghobadi A., Jacobson C.A., Miklos D.B., Lekakis L.J., Oluwole O.O., Lin Y., Braunschweig I., Hill B.T., Timmerman J.M. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20:31–42. doi: 10.1016/S1470-2045(18)30864-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schuster S.J., Bishop M.R., Tam C.S., Borchmann P., Jaeger U., Waller E.K. Long-Term Follow-up of Tisagenlecleucel in Adult Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma: Updated Analysis of Juliet Study. Biol. Blood Marrow Transplant. 2019;25:S20–S21. [Google Scholar]
- 13.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., JULIET Investigators Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 14.Schuster S.J., Maziarz R.T., Ericson S.G., Rusch E.S., Signorovitch J., Romanov V.V. Consensus Grading of Cytokine Release Syndrome (CRS) in Adult Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma (r/r DLBCL) Treated with Tisagenlecleucel on the JULIET Study. Blood. 2018;132:4190. 4190. [Google Scholar]
- 15.Schuster S.J., Maziarz R.T., Rusch E.S., Li J., Signorovitch J.E., Romanov V.V., Locke F.L., Maloney D.G. Grading and management of cytokine release syndrome in patients treated with tisagenlecleucel in the JULIET trial. Blood Adv. 2020;4:1432–1439. doi: 10.1182/bloodadvances.2019001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J., Li J., Ma Q., Yang H., Signorovitch J., Wu E. A Review of Two Regulatory Approved Anti-CD19 CAR T-Cell Therapies in Diffuse Large B-Cell Lymphoma: Why Are Indirect Treatment Comparisons Not Feasible? Adv. Ther. 2020;37:3040–3058. doi: 10.1007/s12325-020-01397-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaglowski S., Hu Z.-H., Zhang Y., Kamdar M., Ghosh M., Lulla P. Tisagenlecleucel Chimeric Antigen Receptor (CAR) T-Cell Therapy for Adults with Diffuse Large B-Cell Lymphoma (DLBCL): Real World Experience from the Center for International Blood & Marrow Transplant Research (CIBMTR) Cellular Therapy (CT) Registry. Blood. 2019;134:766. 766. [Google Scholar]
- 18.Pasquini M.C., Locke F.L., Herrera A.F., Siddiqi T., Ghobadi A., Komanduri K.V. Post-Marketing Use Outcomes of an Anti-CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy, Axicabtagene Ciloleucel (Axi-Cel), for the Treatment of Large B Cell Lymphoma (LBCL) in the United States (US) Blood. 2019;134:764. 764. [Google Scholar]
- 19.Riedell P.A., Walling C., Nastoupil L.J., Pennisi M., Maziarz R.T., McGuirk J.P. A Multicenter Retrospective Analysis of Outcomes and Toxicities with Commercial Axicabtagene Ciloleucel and Tisagenlecleucel for Relapsed/Refractory Aggressive B-Cell Lymphomas. Biol. Blood Marrow Transplant. 2020;26:S41–S42. [Google Scholar]
- 20.Lee D.W., Santomasso B.D., Locke F.L., Ghobadi A., Turtle C.J., Brudno J.N., Maus M.V., Park J.H., Mead E., Pavletic S. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PubMed] [Google Scholar]
- 21.Nastoupil L.J., Jain M.D., Feng L., Spiegel J.Y., Ghobadi A., Lin Y., Dahiya S., Lunning M., Lekakis L., Reagan P. Standard-of-Care Axicabtagene Ciloleucel for Relapsed or Refractory Large B-Cell Lymphoma: Results From the US Lymphoma CAR T Consortium. J Clin Oncol. 2020;38:3119–3128. doi: 10.1200/JCO.19.02104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobson C.A., Hunter B.D., Redd R., Rodig S.J., Chen P.H., Wright K., Lipschitz M., Ritz J., Kamihara Y., Armand P. Axicabtagene Ciloleucel in the Non-Trial Setting: Outcomes and Correlates of Response, Resistance, and Toxicity. J. Clin. Oncol. 2020;38:3095–3106. doi: 10.1200/JCO.19.02103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abramson J.S., Irwin K.E., Frigault M.J., Dietrich J., McGree B., Jordan J.T., Yee A.J., Chen Y.B., Raje N.S., Barnes J.A., Davis B. Successful anti-CD19 CAR T-cell therapy in HIV-infected patients with refractory high-grade B-cell lymphoma. Cancer. 2019;125:3692. doi: 10.1002/cncr.32411. 3698. [DOI] [PubMed] [Google Scholar]
- 24.Bennani N.N., Maurer M.J., Nastoupil L.J., Jain M.D., Chavez J.C., Cashen A.F. Experience with Axicabtagene Ciloleucel (Axi-cel) in Patients with Secondary CNS Involvement: Results from the US Lymphoma CAR T Consortium. Blood. 2019;134:763. 763. [Google Scholar]
- 25.Riedell P.A., Walling C., Nastoupil L.J., Pennisi M., Maziarz R.T., McGuirk J.P. A Multicenter Retrospective Analysis of Clinical Outcomes, Toxicities, and Patterns of Use in Institutions Utilizing Commercial Axicabtagene Ciloleucel and Tisagenlecleucel for Relapsed/Refractory Aggressive B-Cell Lymphomas. Blood. 2019;134:1599. 1599. [Google Scholar]
- 26.Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 27.Frigault M.J., Dietrich J., Martinez-Lage M., Leick M., Choi B.D., DeFilipp Z., Chen Y.B., Abramson J., Crombie J., Armand P. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood. 2019;134:860–866. doi: 10.1182/blood.2019001694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neelapu S.S., Jacobson C.A., Oluwole O.O., Munoz J., Deol A., Miklos D.B., Bartlett N.L., Braunschweig I., Jiang Y., Kim J.J. Outcomes of older patients in ZUMA-1, a pivotal study of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood. 2020;135:2106–2109. doi: 10.1182/blood.2019004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jacobson C.A., Hunter B., Armand P., Kamihara Y., Ritz J., Rodig S.J. Axicabtagene Ciloleucel in the Real World: Outcomes and Predictors of Response, Resistance and Toxicity. Blood. 2018;132:92. 92. [Google Scholar]
- 30.Fitzgerald L., Kittai A., Nastoupil L.J., Waller A., Jacobson C.A., Saucier A. Real-world outcomes of elderly patients with relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL) treated with chimeric antigen receptor T-cell (CAR-T) therapy. J. Clin. Oncol. 2020;38:8039. 8039. [Google Scholar]
- 31.Abbasi A., Peeke S., Shah N., Mustafa J., Khatun F., Lombardo A., Abreu M., Elkind R., Fehn K., de Castro A. Axicabtagene ciloleucel CD19 CAR-T cell therapy results in high rates of systemic and neurologic remissions in ten patients with refractory large B cell lymphoma including two with HIV and viral hepatitis. J. Hematol. Oncol. 2020;13:1. doi: 10.1186/s13045-019-0838-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruella M., Xu J., Barrett D.M., Fraietta J.A., Reich T.J., Ambrose D.E., Klichinsky M., Shestova O., Patel P.R., Kulikovskaya I. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018;24:1499–1503. doi: 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvi R.M., Frigault M.J., Fradley M.G., Jain M.D., Mahmood S.S., Awadalla M., Lee D.H., Zlotoff D.A., Zhang L., Drobni Z.D. Cardiovascular Events Among Adults Treated With Chimeric Antigen Receptor T-Cells (CAR-T) J. Am. Coll. Cardiol. 2019;74:3099–3108. doi: 10.1016/j.jacc.2019.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lefebvre B., Kang Y., Smith A.M., Frey N.V., Carver J.R., Scherrer-Crosbie M. Cardiovascular Effects of CAR T Cell Therapy: A Retrospective Study. JACC CardioOncol. 2020;2:193–203. doi: 10.1016/j.jaccao.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beitinjaneh A., McKinney A.M., Cao Q., Weisdorf D.J. Toxic leukoencephalopathy following fludarabine-associated hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2011;17:300–308. doi: 10.1016/j.bbmt.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 37.Pinnix C.C., Gunther J.R., Dabaja B.S., Strati P., Fang P., Hawkins M.C., Adkins S., Westin J., Ahmed S., Fayad L. Bridging therapy prior to axicabtagene ciloleucel for relapsed/refractory large B-cell lymphoma. Blood Adv. 2020;4:2871–2883. doi: 10.1182/bloodadvances.2020001837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salles G., Duell J., González Barca E., Tournilhac O., Jurczak W., Liberati A.M., Nagy Z., Obr A., Gaidano G., André M. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): a multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020;21:978–988. doi: 10.1016/S1470-2045(20)30225-4. [DOI] [PubMed] [Google Scholar]
- 39.Lionakis M.S., Dunleavy K., Roschewski M., Widemann B.C., Butman J.A., Schmitz R., Yang Y., Cole D.E., Melani C., Higham C.S. Inhibition of B Cell Receptor Signaling by Ibrutinib in Primary CNS Lymphoma. Cancer Cell. 2017;31:833–843.e5. doi: 10.1016/j.ccell.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang M., Fowler N., Wagner-Bartak N., Feng L., Romaguera J., Neelapu S.S., Hagemeister F., Fanale M., Oki Y., Pro B. Oral lenalidomide with rituximab in relapsed or refractory diffuse large cell, follicular and transformed lymphoma: a phase II clinical trial. Leukemia. 2013;27:1902–1909. doi: 10.1038/leu.2013.95. [DOI] [PubMed] [Google Scholar]
- 41.Gauthier J., Hirayama A.V., Purushe J., Hay K.A., Lymp J., Li D.H., Yeung C.C.S., Sheih A., Pender B.S., Hawkins R.M. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood. 2020;135:1650–1660. doi: 10.1182/blood.2019002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraietta J.A., Beckwith K.A., Patel P.R., Ruella M., Zheng Z., Barrett D.M., Lacey S.F., Melenhorst J.J., McGettigan S.E., Cook D.R. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–1127. doi: 10.1182/blood-2015-11-679134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gill S.I., Vides V., Frey N.V., Metzger S., O’Brien M., Hexner E. Prospective Clinical Trial of Anti-CD19 CAR T Cells in Combination with Ibrutinib for the Treatment of Chronic Lymphocytic Leukemia Shows a High Response Rate. Blood. 2018;132:298. doi: 10.1182/bloodadvances.2022007317. 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otáhal P., Průková D., Král V., Fabry M., Vočková P., Latečková L., Trněný M., Klener P. Lenalidomide enhances antitumor functions of chimeric antigen receptor modified T cells. OncoImmunology. 2015;5:e1115940. doi: 10.1080/2162402X.2015.1115940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pavel O., Prukova D., Král V., Jaksa R., Lateckova L., Vockova P. Immunomodulatory Agent Lenalidomide Enhances Antitumor Functions of Chimeric Receptor-Modified T Cells in Vitro and in Vivo. Blood. 2014;124:805. 805. [Google Scholar]
- 46.Hampel P.J., Chaffee K.G., Ding W., Call T., Kenderian S., Muchtar E. Rapid progression of disease following ibrutinib discontinuation in patients with chronic lymphocytic leukemia. J. Clin. Oncol. 2018;36:7525. 7525. [Google Scholar]
- 47.Pinnix C.C., Gunther J.R., Dabaja B.S., Adkins S., Hawkins M., Westin J.R. Radiation Therapy Can be an Effective Bridging Strategy Prior to Axicabtagene Ciloleucel Therapy for Relapsed/Refractory Large B-Cell Lymphoma. Blood. 2019;134:1609. 1609. [Google Scholar]
- 48.Andreadis C., Tam C.S., Borchmann P., Jaeger U., McGuirk J.P., Holte H. Correlation of Bridging and Lymphodepleting Chemotherapy with Clinical Outcomes in Patients with Relapsed/Refractory Diffuse Large B-Cell Lymphoma Treated with Tisagenlecleucel. Blood. 2019;134:2883. 2883. [Google Scholar]
- 49.Hill J.A., Li D., Hay K.A., Green M.L., Cherian S., Chen X., Riddell S.R., Maloney D.G., Boeckh M., Turtle C.J. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 2018;131:121–130. doi: 10.1182/blood-2017-07-793760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park J.H., Romero F.A., Taur Y., Sadelain M., Brentjens R.J., Hohl T.M., Seo S.K. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin. Infect. Dis. 2018;67:533–540. doi: 10.1093/cid/ciy152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strati P., Nastoupil L.J., Fayad L.E., Samaniego F., Adkins S., Neelapu S.S. Safety of CAR T-cell therapy in patients with B-cell lymphoma and chronic hepatitis B or C virus infection. Blood. 2019;133:2800–2802. doi: 10.1182/blood.2019000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hwang J.P., Torres H.A. Hepatitis B virus and hepatitis C virus infection in immunocompromised patients. Curr. Opin. Infect. Dis. 2018;31:535–541. doi: 10.1097/QCO.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 53.Wei J., Zhu X., Mao X., Huang L., Meng F., Zhou J. Severe early hepatitis B reactivation in a patient receiving anti-CD19 and anti-CD22 CAR T cells for the treatment of diffuse large B-cell lymphoma. J. Immunother. Cancer. 2019;7:315. doi: 10.1186/s40425-019-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haidar G., Dorritie K., Farah R., Bogdanovich T., Nguyen M.H., Samanta P. Invasive Mold Infections After Chimeric Antigen Receptor-Modified T-cell Therapy: A Case Series, Review of the Literature, and Implications for Prophylaxis. Clin. Infect. Dis. 2020;71:672–676. doi: 10.1093/cid/ciz1127. [DOI] [PubMed] [Google Scholar]
- 55.Frigault M.J., Nikiforow S., Mansour M.K., Hu Z.-H., Horowitz M.M., Riches M.L., Hematti P., Turtle C.J., Zhang M.J., Perales M.A., Pasquini M.C. Tocilizumab not associated with increased infection risk after CAR T-cell therapy: implications for COVID-19? Blood. 2020;136:137–139. doi: 10.1182/blood.2020006216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhoj V.G., Arhontoulis D., Wertheim G., Capobianchi J., Callahan C.A., Ellebrecht C.T., Obstfeld A.E., Lacey S.F., Melenhorst J.J., Nazimuddin F. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. 2016;128:360–370. doi: 10.1182/blood-2016-01-694356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brudno J.N., Kochenderfer J.N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karschnia P., Jordan J.T., Forst D.A., Arrillaga-Romany I.C., Batchelor T.T., Baehring J.M., Clement N.F., Gonzalez Castro L.N., Herlopian A., Maus M.V. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. 2019;133:2212–2221. doi: 10.1182/blood-2018-12-893396. [DOI] [PubMed] [Google Scholar]
- 59.Lee D.W., Santomasso B.D., Locke F.L., Ghobadi A., Turtle C.J., Brudno J.N., Maus M.V., Park J.H., Mead E., Pavletic S. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019;25:625–638. doi: 10.1016/j.bbmt.2018.12.758. [DOI] [PubMed] [Google Scholar]
- 60.Locke F., Neelapu S., Bartlett N., Lekakis L., Jacobson C., Braunschweig I. Preliminary Results of Prophylactic Tocilizumab after Axicabtageneciloleucel (axi-cel; KTE-C19) Treatment for Patients with Refractory,Aggressive Non-Hodgkin Lymphoma (NHL) Blood. 2017;130:1547. 1547. [Google Scholar]
- 61.Strati P., furqan F., Westin J., Fayad L., Ahmed S., Lee H.J., Iyer S.P., Nair R., Nastoupil L.J., Parmar S. Prognostic impact of dose, duration, and timing of corticosteroid therapy in patients with large B-cell lymphoma treated with standard of care axicabtagene ciloleucel (Axi-cel) J. SClin. Oncol. 2020;38(suppl8011):8011. 8011. [Google Scholar]
- 62.Buitrago J., Adkins S., Hawkins M., Iyamu K., Oort T. Adult Survivorship: Considerations Following CAR T-Cell Therapy. Clin. J. Oncol. Nurs. 2019;23:42–48. doi: 10.1188/19.CJON.S1.42-48. [DOI] [PubMed] [Google Scholar]
- 63.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Majzner R.G., Mackall C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]