

Abstract
Gene-mediated cytotoxic immunotherapy (GMCI) is an immuno-oncology approach involving local delivery of a replication-deficient adenovirus expressing herpes simplex thymidine kinase (AdV-tk) followed by anti-herpetic prodrug activation that promotes immunogenic tumor cell death, antigen-presenting cell activation, and T cell stimulation. This phase I dose-escalation pilot trial assessed bronchoscopic delivery of AdV-tk in patients with suspected lung cancer who were candidates for surgery. A single intra-tumoral AdV-tk injection in three dose cohorts (maximum 1012 viral particles) was performed during diagnostic staging, followed by a 14-day course of the prodrug valacyclovir, and subsequent surgery 1 week later. Twelve patients participated after appropriate informed consent. Vector-related adverse events were minimal. Immune biomarkers were evaluated in tumor and blood before and after GMCI. Significantly increased infiltration of CD8+ T cells was found in resected tumors. Expression of activation, inhibitory, and proliferation markers, such as human leukocyte antigen (HLA)-DR, CD38, Ki67, PD-1, CD39, and CTLA-4, were significantly increased in both the tumor and peripheral CD8+ T cells. Thus, intratumoral AdV-tk injection into non-small-cell lung cancer (NSCLC) proved safe and feasible, and it effectively induced CD8+ T cell activation. These data provide a foundation for additional clinical trials of GMCI for lung cancer patients with potential benefit if combined with other immune therapies.
Keywords: lung cancer, immunotherapy, gene-mediated cytotoxic immunotherapy, intratumoral immunotherapy, checkpoint inhibitor, PD-1, PD-L1, gene therapy, adenovirus, neoadjuvant clinical trial
Graphical Abstract
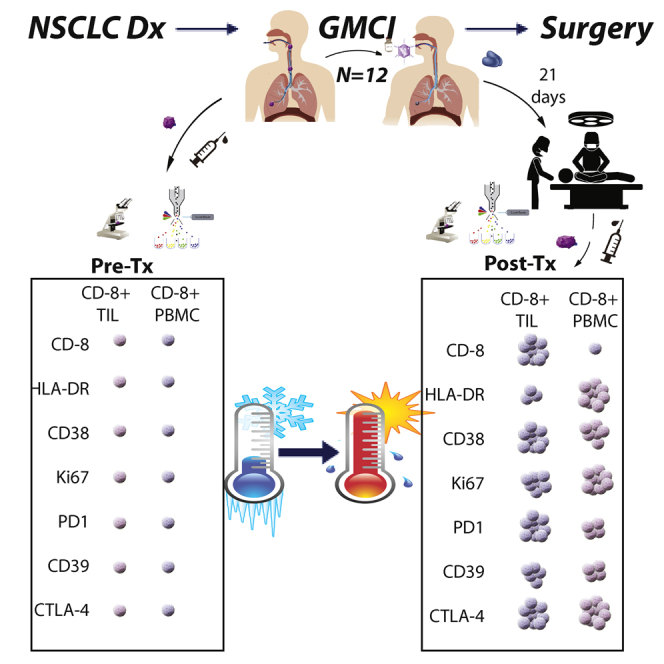
Predina and colleagues describe a phase I dose-escalation trial using bronchoscopic delivery of a replication-deficient adenovirus expressing herpes simplex thymidine kinase (AdV-tk) in lung cancer patients who underwent surgery. AdV-tk injection proved safe, feasible, and effectively induced CD8+ T cell activation in blood and in resected tumor tissues.
Introduction
Non-small-cell lung cancer (NSCLC) is the leading cause of cancer-related mortality in the United States and throughout most of the world.1 Although surgery offers the best chance for cure, a substantial proportion of resected patients, especially those with more advanced stages with nodal invasion (i.e., stages II and IIIA), recur both locally and distantly.2 The addition of adjuvant chemotherapy and radiation to surgery has only marginally improved outcomes, and thus novel approaches are needed.3,4
During the last decade, immune checkpoint inhibitors (ICIs), such as anti-CTLA-4, anti-PD-1, or PD-L1 antibodies, have emerged as a novel therapeutic option in the armamentarium against NSCLC. ICIs are thought to “remove the brakes” on existing anti-tumor T cells. Although very promising, anti-PD-1 checkpoint blockade still results in only a 30%–40% response rate in patients with PD-L1 expression. When combined with chemotherapy, the response rates are somewhat increased, but still under 50%.5,6 Increases in progression-free and overall survival have also been observed; however, 5-year survival rates remain below 20%–30%.
The absence of clinical activity for ICIs in more than 50% of patients may be due to the inability of many patients to mount an endogenous anti-tumor immune response. In this scenario, there simply is no response that can be “released.” To overcome this hurdle and expand ICI potency, it would be necessary to stimulate or generate active T cells with anti-tumor activity. These T cells either can be created exogenously and reinfused via adoptive T cell transfer or formed endogenously by vaccinating the patient with tumor or tumor-derived antigens. Unfortunately, to date, vaccination at peripheral sites has not proven successful as a therapeutic approach in solid tumors, including for NSCLC.7 Strong local tumor immunosuppression and vaccines that target only a small number of antigens are key factors that likely limit this approach.
A potential strategy to overcome some of these challenges is “in situ vaccination.” In this approach, an agent is administered directly into a tumor with the goal of killing tumor cells to release multiple tumor neo-antigens, to generate and/or recruit immune-effector cells, and to shift the tumor microenvironment from immunosuppressive to pro-inflammatory and immunostimulatory.8,9 Gene-mediated cytotoxic immunotherapy (GMCI) is an in situ vaccination approach that uses aglatimagene besadenovec (AdV-tk), an “armed” non-replicating adenoviral (Ad) vector, to induce immunogenic cancer cell death (e.g., apoptosis, autophagy, necroptosis) and an anti-tumor immune response through a multi-step process.10 Intratumoral injection of this type 5 adenovirus stimulates a robust innate immune response due to the presence of a large number of pathogen-associated molecular patterns (PAMPs), which include portions of the viral capsid and viral nucleic acids.11 The transgene protein (herpes simplex virus thymidine kinase [TK]), when combined with administration of the antiviral prodrugs ganciclovir or valacyclovir, kills tumor cells in an immunogenic fashion.12 The TK protein also acts as a super-antigen, stimulating an even more potent immune response.10 Viral and TK-mediated induction of immunogenic cell death promotes anti-tumor responses via recruitment and activation of antigen-presenting cells (APCs) and subsequent T cell activation, resulting in an acute and long-lasting memory immune response.13, 14, 15 Our work and that of others showed that direct intratumoral (i.t.) GMCI delivery can disrupt physical barriers within the tumor and modify the tumor microenvironment via induction of local anti-tumor inflammatory cytokine production.16,17 This intra-tumoral approach results in a broad, polyclonal anti-tumor immune response and improves CD8 T cell trafficking and function.
GMCI has shown safety and anti-tumor activity in multiple trials for solid tumor types, including glioblastoma,18,19 pancreatic cancer,20 prostate cancer,21 and mesothelioma/malignant pleural effusions.22 In a recent malignant pleural effusion study from our group, three of four NSCLC patients whose disease progressed through two to four prior lines of therapy, lived more than 2 years.22 In a phase II glioblastoma study, survival outcomes were significantly improved, particularly in patients where most of the immune-inhibitory primary tumor was removed.18 Interestingly, multiple preclinical studies have suggested that the impact of GMCI may be maximized when used in a neoadjuvant fashion with surgery or other debulking therapies.13,15,17 Similar advantages were seen with neoadjuvant use of an adenoviral vector expressing interferon (IFN)-β.23 In these circumstances, the debulking both removes physical barriers within the tumor microenvironment and reduces local and systemic tumor-associated immunosuppressive cytokine and cellular barriers.14,17
Based on these observations, the current study explored the use of GMCI in the context of surgically resectable NSCLC. A phase I dose-escalation trial of i.t. neoadjuvant GMCI followed by definitive resection 3 weeks after vector delivery was conducted. Clinical objectives were to evaluate the safety, feasibility, and dosing of this combination. The scientific objective was to obtain biological data to better understand the impact of GMCI on the tumor microenvironment with a specific focus on i.t. CD8 T cell activation and function while also assessing its effect on systemic immune responses. GMCI effects were evaluated by comparing post-resection specimens to (1) an internal control consisting of each patient’s own pre-treatment needle biopsy and blood samples, and (2) an external cohort of matched patients who had undergone standard surgical resection without GMCI.24 The results showed safety, feasibility, and evidence of significant immune activation. These data provide a foundation for additional clinical trials to optimize a multi-modal approach of neoadjuvant immunotherapy followed by surgery for resectable NSCLC patients.
Results
Subject Characteristics
Between May 2017 and October 2019, 22 subjects with presumed resectable NSCLC were enrolled in this phase I, dose-escalation trial of i.t. GMCI. As shown in the CONSORT diagram (Figure S1), seven patients were excluded and did not receive AdV-tk for one of the following reasons: (1) declined participation after enrollment but before receiving AdV-tk (n = 2), (2) no definitive cancer diagnosis was made at the time of staging procedure (n = 4), and (3) positive mediastinal lymph node biopsy at staging precluded future resection (n = 1). Three patients received AdV-tk but did not complete the trial, one due to refusal to take valacyclovir and two because the final pathology was other than NSCLC.
Twelve subjects completed neoadjuvant GMCI (AdV-tk + valacyclovir) and surgery with characteristics shown in Table 1. There were three patients each in cohorts 1 and 2, and six in cohort 3. The median age was 65 (range, 55–80), and the mean tumor size (largest diameter) from baseline imaging prior to injection was 5.9 cm (range, 2.6–14.8 cm). Eastern Cooperative Oncology Group (ECOG) performance status was 0 in 50% and 1 in 50%. The pre-treatment stage was based on imaging prior to lymph node interrogation during the bronchoscopic staging procedure. The final tumor-node-metastasis (TNM) and surgical stage was based on the pathology assessment from the resection surgery (Table 1). Seven tumors were squamous cell carcinomas, four were adenocarcinomas, and one was classified as a sarcomatoid carcinoma.
Table 1.
Patient Characteristics
| Patient ID | Age | Sex | ECOG | Tumor Sizea (cm) | Tumor Location | Pre-treatment TNM | Pre-treatment Stage | Histology | Delivery Method | Final TNM | Surgical Staging |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose Level 1: 2.5 × 1011vp | |||||||||||
| 1LU02 | 74 | M | 0 | 9.4 | RLL | T4N0 | IIIA | SCC | VATS | T4N2 | IIIB |
| 1LU04 | 55 | M | 1 | 4.0 | RLL | T2aN0 | IB | SCC | EBUS | T2bN0 | IIA |
| 1LU05 | 57 | M | 1 | 5.4 | LUL | T3N0 | IIB | SCC | EBUS | T3N1 | IIIA |
| Dose Level 2: 5 × 1011vp | |||||||||||
| 2LU01 | 65 | M | 1 | 9.8 | RUL | T4N0 | IIIA | SCC | EBUS | T4N0 | IIIA |
| 2LU02 | 72 | M | 1 | 3.2 | RUL | T2aN0 | IB | adeno | EBUS | T2aN0 | IB |
| 2LU04 | 77 | M | 1 | 2.6 | LLL | T1cN0 | IA | SCC | EBUS | T2aN0 | IB |
| Dose Level 3: 1 × 1012vp | |||||||||||
| 3LU01 | 65 | M | 0 | 4.3 | RLL | T2bN0 | IIA | SCC | EBUS | T3N2 | IIIB |
| 3LU02 | 70 | M | 1 | 14.8 | LLL | T4N0 | IIIA | sarco | EBUS | T4N0 | IIIA |
| 3LU06 | 80 | F | 0 | 1.8/4 | RUL/RLL | T2aN0 | IB | adeno | EBUS | T3N2 | IIIB |
| 3LU08 | 63 | F | 0 | 6.5 | RLL | T3N0 | IIB | SCC | EBUS | T3N0 | IIB |
| 3LU12 | 64 | M | 0 | 3.3 | RUL | T2aN0 | IB | adeno | EBUS | T1cN0 | IA |
| 3LU13 | 62 | F | 0 | 3.6 | RUL | T2aN0 | IB | adeno | EBUS | T3N0 | IIB |
SCC, squamous cell carcinoma; VATS, video-assisted thoracoscopic surgery; EBUS, endobronchial ultrasound; adeno, adenocarcinoma; sarco, sarcomatoid carcinoma.
Largest diameter from baseline imaging.
Feasibility
Vector was successfully injected into all patients and added fewer than 5 min to the staging procedures. One patient received AdV-tk via thoracoscopic staging while the remainder underwent vector delivery via peripheral radial ultraminature endobronchial ultrasound (EBUS) guide sheath-directed bronchoscopy (Table 1). Resections were approached via thoracotomy and included lobectomy (n = 8), bilobectomy (n = 1), pneumonectomy (n = 2), and segmentectomy (n = 1). Although inflammation and adhesions were consistently noted at the site of vector delivery, dissection of pulmonary vessels and airways was not made more challenging. Additionally, surgeons did not appreciate any off-target visible intrathoracic irregularities.
Safety
The study drug was well tolerated with no dose-limiting toxicities (DLTs) observed in any of the 12 subjects. Treatment-related adverse events (Table 2) included grade 1 fever (n = 1), flu-like symptoms (n = 1), and nausea/vomiting/diarrhea (thought to be related to the valacyclovir) (n = 1). The only grade >2 laboratory abnormalities were transient grade 3 lymphopenia (n = 2). There was no evidence of hypoxia or a pulmonary parenchymal inflammatory response following AdV-tk delivery. Importantly, as mentioned above, GMCI did not delay any of the surgical procedures. Unrelated adverse events, laboratory abnormalities, and all serious adverse events are included in Tables S2–S4, respectively,
Table 2.
Related Adverse Events and Acute Laboratory Abnormalities Greater Than Grade 1
| Adverse Event | CTC Grade |
||
|---|---|---|---|
| 1 | 2 | 3 | |
| Gastrointestinal Disorders | |||
| Diarrhea | 1 | ||
| Nausea | 1 | ||
| Vomiting | 1 | ||
| General Disorders and Administration Site Conditions | |||
| Fever | 1 | ||
| Flu-like symptoms | 1 | ||
| Laboratory Abnormalities Greater Than Grade 1 | |||
| Lymphopenia | 2 | ||
CTC, common toxicity criteria.
Clinical Responses
Five patients receiving the highest AdV-tk dose underwent imaging in the 3-week period between AdV-tk injection and surgery. Reduction in tumor size was observed in one of these five patients (Figure 1). It is noteworthy that this responding patient was quite unusual in that the tumor was classified as a sarcomatoid carcinoma. The biocorrelative analysis of this tumor was also unusual in that it showed an extremely high percentage of lymphocytes (see details below). In the other four cases, the tumor size was unchanged.
Figure 1.

CT Scans of Tumor before and after AdV-tk Administration
Thoracic CT scans from PT 3LU02P show a reduction in size of the pleural-based tumor (white arrows) when the scan from pre-AdV-tk administration (A) is compared to the scan immediately prior to surgery 3 weeks later (B).
At the time of surgery, the estimated average percentage of necrosis in the resection specimens did not differ (p = 0.5) between the treated tumors (29.4%; SEM of 8.6%) and a group of 24 control patients matched by histology and tumor size (34.6%; SEM of 7.2%)
Patients received standard of care after surgery, including adjuvant chemotherapy if indicated by final pathologic disease stage. The primary endpoints of this phase I study were safety and feasibility, and thus the sample size and study duration were not powered to evaluate efficacy. However, at the time of this manuscript submission, we contacted all patients to obtain follow-up (see Table S5 for details). As of June 2020, the median time of follow-up was 17.7 months with a range of 7.5–32 months. All patients were alive and there were three recurrences. Patient 1LU02P on dose level 1, with a large 9.4-cm tumor and a pathologic stage IIIB (T4N2), developed recurrence 26 months after surgery. Patient 3LU01P on dose level 3, with a 4.3-cm tumor and pathologic stage IIIB (T3N2), recurred 6 months after surgery. The third, patient 2LU02P on dose level 2, with stage IB NSCLC, received no adjuvant treatment and had recurrence 24 months after surgery.
Biocorrelates
Effect of Neoadjuvant GMCI on CD8 Tumor-Infiltrating Lymphocytes (TILs)
To study the effects of GMCI on TILs, portions of the surgical resections done 3 weeks after vector injection (called the “post-Rx” samples) were digested and analyzed using flow cytometry. These samples were first compared to the baseline tumor needle biopsies taken at the time of vector injection (called the “pre-Rx” sample). This analysis was performed on 7 of the 12 patients (left panels of Figures 2, 3, and 4). Four were not included in the analyses due to lack of adequate baseline biopsy samples, and one was excluded because it was a sarcomatoid carcinoma with an unusually high baseline T cell infiltration (75% of live cells; more than 2- to 3-fold higher than any other patient). To supplement the analysis, TIL data from this trial were also compared to similar data from 44 early-stage lung cancers recently reported by our group using the same flow cytometry panels and protocols (called “control” samples);24 these data are shown in the right panels of Figures 2, 3, and 4.
Figure 2.

Effect of Neoadjuvant GMCI on Types of TILs
Left panels: samples from baseline tumor needle biopsies taken at the time of vector injection (called the “pre-Rx” sample) are compared to the surgical specimens (called the “post-Rx” sample). The percentage of specific types of T cells of live cells in tumor biopsy samples are plotted. The following markers are plotted: CD3+ cells, CD8+ cells, CD4+ cells, and CD4+ regulatory T cells (CD4+/FOXP3+ cells). Paired t tests were applied to define the statistical significance (p value) of the changes. Right panels: the percentages of specific types of T cells of live cells in the tumor biopsy sample from the current study (Ad.TK) were compared with values from a recent independent study24 where TIL phenotype and function were analyzed in 44 early-stage lung cancers using the same flow cytometry panels and protocols (called “control” samples). Student’s t tests were applied to define the statistical significance (p value) of the differences.
Figure 3.

Effect of Neoadjuvant GMCI on Activation Markers and Cytokine Production in CD8+ TILs
Using the same samples as described in Figure 2, the expression of three proliferation/activation markers (Ki67, CD38, and HLA-DR) on the CD8+ TILs in the pre-Rx samples were compared to post-Rx tumor samples. Paired t tests were applied to define the statistical significance (p value) of the changes. The upper right panel shows the comparison between the Ki67 expression in this trial compared to our lung cancer control samples. A Student’s t test was applied to define the statistical significance (p value) of the differences.
Figure 4.

Effect of Neoadjuvant GMCI on Inhibitory/Activation Markers in CD8+ TILs
Left panels: using the same samples as described in Figure 2, the expression of five inhibitory/activation markers (PD-1, CD39, CTLA-4, TIM3, and TIGIT) on CD8+ T cells in the pre-Rx samples were compared to post-Rx tumor samples. Paired t tests were applied to define the statistical significance (p value) of the changes. Right panels: the percentages of specific markers on CD8+ T cells from the current study (Ad.TK) were compared with values from a recent independent study,24 where TIL phenotype and function were analyzed in 44 early-stage lung cancers using the same flow cytometry panels and protocols (control samples). Student’s t tests were applied to define the statistical significance (p value) of the differences.
A 3.5-fold average increase in the percentage of CD3+ cells (of live cells in the tumor biopsy sample) was found in the post-Rx sample compared to the pre-Rx samples (p = 0.01) (Figure 2). The most significant and consistently increased T cell subset was the CD8+ cells, with a 5.2-fold increase (p = 0.001). The percentage of CD8+ cells (of live cells in the tumor biopsy sample) was also increased by 1.3-fold in the post-AdV-tk samples compared to the control lung cancer samples (p = 0.05). CD4+ cells were increased by 2.7-fold in the post-Rx samples compared to the pre-Rx samples (p = 0.04), although these were not significantly increased compared to the control samples. The average percentage of CD4+ regulatory T (Treg) cells (of live cells in the tumor biopsy sample) was increased 2.6-fold (Figure 2); compared to control samples, the Treg cell percentage was decreased by 58% (p < 0.001). A representative example of the flow tracings (from patient 3LU13P) is shown in Figure S2.
In tumors, characterization of the CD8+ cells revealed significant increases in proliferation and activation markers compared to baseline (Figure 3). For example, the proliferation marker Ki67 showed a 4.8-fold average increase (p = 0.02) compared to baseline and a 1.8-fold increase compared to the control group (p = 0.02). Similarly, the activation marker CD38 showed a 2.5-fold increase compared to baseline (p = 0.002); this was not assessed in the control group. The average increase in human leukocyte antigen (HLA)-DR was 1.7-fold, which was not significantly different from baseline (p = 0.40), although two patients did show marked increases. A representative example of the flow tracings (from patient 3LU13P) is shown in Figure S3.
Expression of so-called “inhibitory” receptors on the CD8+ TILs, also a sign of activation, were likewise significantly increased (Figure 4). Of those analyzed, PD-1 showed an average 1.9-fold increase (p = 0.002), CD39 an average 2.9-fold increase (p = 0.04), and CTLA-4 an average 4.8-fold increase (p < 0.001) over baseline. No significant differences were seen in TIM3 or TIGIT expression levels, although one patient had a large drop in TIM3 expression. Similarly, compared to control tumors, there was a significant 1.3-fold increase in PD-1 (p = 0.014) and a 1.7-fold increase in CD39 (p = 0.018), with no significant differences in TIM3 or TIGIT. A representative example of the flow tracings (from patient 3LU13P) is shown in Figure S4.
To assess T cell functionality, we tested the ability of the CD8+ TILs from the surgical resection to produce intracellular IFN-γ or tumor necrosis factor (TNF)-α ex vivo after stimulation with plate-bound anti-CD3 antibody and compared these data with our previously reported results from normal lung tissue and similar early-stage lung cancer patients24 (Figure S5). In that previous study, we found that >20% of T cells from normal lung tissues responded to anti-CD3 antibody to produce IFN-γ and thus defined a response of <20% of T cells responding as hypofunctional. Using this definition from the prior study, only about one-third of early-stage lung cancer patients had TILs that were relatively hypofunctional. Although the mean of the percentage of cells making intracellular IFN-γ after stimulation was slightly higher in the post-AdV-tk samples compared to our historical controls, the difference was not significant, nor was the difference in the percentage of cells making intracellular TNF-α (Figure S5).
Effect of Neoadjuvant GMCI on PD-L1 Expression in Tumors
When the pre-treatment biopsy provided a sufficient sample, the extent of tumor cells expressing membranous staining for PD-L1 was assessed by a pathologist (C.D.) and compared to the surgical specimen. There was no statistically significant difference in the eight evaluable patients (p = 0.39), although one patient had a large increase (Figure S6).
Effects of Neoadjuvant GMCI on T Cells in the Blood
To assess the systemic effect of i.t. injection of AdV-tk followed by valacyclovir, baseline peripheral blood mononuclear cells (PBMCs) were obtained from patients prior to vector injection and compared to PBMCs obtained at the time of surgery, ∼3 weeks after vector delivery. As shown in Figure 5A, there were no significant differences in the percent of CD3 or CD8 T cells in circulation; however, there was a significant 20% decrease in the percent of circulating CD4 T cells (p = 0.04) and CD4+/FOXP3+ Treg cells (p = 0.02). As shown in Figure 5B, highly significant increases in three CD8 T cell activation and proliferation markers were observed, including a 3.4-fold average increase in CD38 (p = 0.006), a 4.2-fold average increase in HLA-DR (p = 0.002), and a 5.8-fold average increase in Ki67 (p = 0.017). These markers have been previously shown to indicate T cell activation after vaccination.25 Flow cytometry tracings for one representative patient are shown in Figure 6. Similarly, there were significant increases in expression of the “inhibitory” receptors we examined with an average expression increase of 2.3-fold in PD-1 (p = 0.043), 2.1-fold in CD39 (p = 0.043), and 2.9-fold in CTLA-4 (p = 0.004). There was no detectable change in TIGIT expression (Figure 5C). A representative example of the flow tracings (from patient 3LU13P) is shown in Figure S7.
Figure 5.

Effects of Neoadjuvant GMCI on T Cells in the Blood
Expression of T cell markers was assessed on baseline peripheral blood mononuclear cells (PBMCs) obtained from each patient prior to vector injection and compared to PBMCs obtained at the time of surgery (19–22 days after vector delivery). Paired t tests were applied to define the statistical significance (p value) of the change. (A) The percentages of specific types of T cells within the live PBMC population are plotted: upper left, CD3+ T cells; upper right, CD8+ T cells; lower left, CD4+ T cells; lower right, CD4+ regulatory T cells (CD4+/FOXP3+ cells). (B) The percentage of CD8+ T cells expressing specific activation/proliferation markers are plotted: upper left, CD38; upper right, HLA-DR; lower left, Ki67 (proliferation marker); lower right, 41BB (CD137). (C) The percentage of CD8+ T cells expressing specific inhibitory/activation markers are plotted: upper left, PD-1; upper right, CD39; lower left, CTLA-4; lower right, TIGIT.
Figure 6.

Flow Cytometry Tracings Showing Activation Markers on CD8+ T Cells in PBMCs from Patient 3LU13P
Examples of flow tracings from the CD8+ T cells of pre-treatment (day 0, upper tracings) and post-treatment (day 21, lower tracings) in the PBMCs from one patient (3LU13P) are shown. Expression of HLA-DR, Ki67, and CD38 are shown as marked.
In order to examine the specificity of these CD8+ T cell activation changes in this trial after i.t. AdV-tk injection, we compared our data to patient samples stored from two previous clinical trials in which we injected the same AdV-tk vector (three samples), or a similar replication-deficient type 5 adenovirus vector encoding IFN-α (two samples) into the pleural spaces of cancer patients. PBMCs from the five patients before the delivery of the vectors and then again 14 days later were analyzed using the same flow cytometry protocol used for this trial. In the PBMCs from the other adenovirus trials, smaller, but still significant increases in the pre- versus post-treatment expression of CD38 (average 1.7-fold increase; p = 0.02) and Ki67 (average 2.9-fold increase; p = 0.03) were seen without a significant increase in HLA-DR expression (Figure S8). However, in samples from those trials, no significant systemic increases were seen in expression of CD39, CTLA-4, TIGIT, or PD-1 (Figure S9).
Effect of Neoadjuvant GMCI on Anti-tumor Antibodies
Pre-AdV-tk injection serum and serum collected ∼6 weeks after surgery were available from eight subjects and used to perform immunoblots on gels containing seven different human lung cancer cell lines. New or increased bands were identified in three of the eight subjects (Figure S10).
Discussion
The rationale for a neoadjuvant in situ vaccination approach is that GMCI can release endogenous tumor antigens, alter the tumor microenvironment, stimulate endogenous TILs, and expand the tumor-specific T cell repertoire. Based on our previous preclinical and clinical data (discussed above), we postulated that administering AdV-tk in a neoadjuvant setting followed by standard-of-care complete oncologic resection would generate memory immune responses that might reduce lung cancer recurrence. To move forward with this strategy, our first step was to establish the safety and feasibility of neoadjuvant AdV-tk delivery in lung cancer and to determine the immediate immunologic effects of GMCI.
In terms of safety, our data show that direct i.t. AdV-tk administration followed by oral valacyclovir administration was very well tolerated and did not compromise the ability of the surgeon to carry out a complete oncologic resection. Off-target inflammatory responses were not obvious within the chest or lung parenchyma and did not impede the ability of the surgeon to identify and to isolate bronchovascular structures and to resect tumors safely. The safety of intra-tumoral injection of adenoviral vectors into lung cancers shown in this trial is consistent with a number of previous studies where only minimal side effects were noted. These include injection of adenoviral vectors encoding β-galactosidase and interleukin (IL)-2,26 p53,27,28 and L523S, an immunogenic lung cancer antigen.29
With regard to feasibility, the study showed that bronchoscopic AdV-tk delivery was possible at the time of an initial diagnostic and staging procedure. By coupling vector delivery with the standard-of-care diagnostic procedure, we further reduced potential risk and discomfort to the patient. In our patient population, there were no difficulties with the oral prodrug, but if needed, the intravenous (i.v.) prodrug could be used as an alternate. Although there were technical challenges that restricted vector delivery to some lesions, in the future, more advanced bronchoscopic technologies, such as robotics and cone beam computed tomography (CT) scan, should expand the lesions accessible to bronchoscopic vector delivery. In addition, the use of transthoracic needle biopsy may be an alternative vector delivery strategy. A minimally invasive video-assisted thoracoscopic injection was performed as a proof of principle, which could constitute yet another alternative in selected patients.
No maximum tolerated dose (MTD) was identified since there were no DLTs in the tested dose levels, similar to other GCMI trials. Previous clinical studies with GMCI have shown responses within the dose ranges tested in this study and shown a relatively broad therapeutic window. We did not detect significant immune response differences based on the dose used. The vector dose for future clinical studies being planned will be within the dose range tested in the present study.
The other major goal of this trial was to evaluate the immediate immune effects of GMCI. Since GMCI induces a broad, polyclonal response to undetermined tumor neoantigens, its effect cannot be easily quantified by assessing the numbers of antigen-reactive cells in blood or tumor using tetramer staining or enzyme-linked immunospot (ELISPOT) assays, as is traditionally done for antigen-specific vaccines. It is much more challenging to assess the immunological effects where the targets and responses are generated in situ and are patient specific. To identify T cell responses in this study, we used an approach initially described by Miller et al.25 that analyzed blood CD8 T cell responses after yellow fever and vaccinia vaccines in normal volunteers. In that study, using flow cytometry, they were able to demonstrate that the cellular response to the vaccine could be identified best by upregulation of the CD8 T cell activation markers CD38 and HLA-DR, along with evidence of T cell proliferation marked by increased expression of the intracellular marker Ki67. They found that the response peaked about 2 weeks after vaccination.
Although our study used i.t. injection and a systemic signal was not necessarily expected, blood samples were evaluated in a similar time window as described in the Miller study (at the time of vector delivery and ∼3 weeks afterward at the time of surgery). Surprisingly, AdV-tk directly injected into NSCLC tumors not only caused significant immunostimulation in the tumor microenvironment, but it also generated a significant systemic signal as observed in peripheral blood T cells. Although GMCI did not induce quantitative changes of CD8+ T cells in PBMCs, CD8+ T cells were functionally more activated following GMCI with significant upregulation of CD38, HLA-DR, and Ki67. An increased expression of the surface markers PD-1, CD39, and CTLA-4, which can also be associated with recent T cell stimulation, was additionally noted. There was also a small decrease in the percentage of CD4+ cells and CD4+ Treg cells. In summary, these data indicate that i.t. GMCI induced strong peripheral CD8+ T cell activation. However, we only detected anti-tumor antibody responses to allogeneic cell lines in three of our subjects. TCR sequencing was not performed because interpretation would likely be difficult due to an expected increased in T cell clones directed against adenoviral antigens that could potentially overwhelm any neoantigen clones.
A major advantage of a neoadjuvant “window of opportunity” trial, such as this, is the availability of a large tissue specimen at a controlled time point after therapy administration, in this case, ∼3 weeks after AdV-tk injection. However, in neoadjuvant studies, a major consideration is selection of appropriate comparator samples. Given the known heterogeneity of NSCLC, the ideal comparator would be a large surgical biopsy taken at the time of AdV-tk delivery. This was not possible, but we were able to obtain pre-treatment core needle biopsies suitable for immunohistochemical and flow cytometric analyses in 7 of the 12 patients. To supplement our analysis, we used an independent surgical biopsy control group (that was very similar in histology, stage, sex, and age to those operated upon in the current trial) from a large study recently conducted by our group to analyze the CD8 T cell phenotypes and function in early-stage lung cancers.24 These comparisons with our surgical samples were quite congruent with our core needle biopsy comparisons and showed a number of findings supporting the conclusion of i.t. GMCI-induced T cell activation. First, significant increases in the percentage of CD3+, CD4+, and CD8+ TILs in treated tumors were noted. Second, significant increases in the expression of the activation markers Ki67 and CD38 on the CD8+ TILs were seen, although, in contrast to blood, the increase in TIL HLA-DR was not significant. Third, significant increases in the so-called “activation/inhibitory receptors” that included PD-1, CD39, and CTLA-4 were also noted, further suggesting that the T cells had been activated. The upregulation of CD39 on the TILs is especially interesting, as a number of recent reports have suggested that CD39 is a marker of tumor-reactive TILs.30,31 Another value of the control group comparisons was exemplified by the analysis of CD4 Treg cells. In the study samples, there appeared to be a small increase in the percentage of CD4 Treg cells in the surgical specimens compared to the pre-treatment core needle biopsy samples. However, when compared to the historical control group, the percentage of CD4 Treg cells was significantly lower, suggesting that GMCI may actually have reduced the increase of CD4 Treg cells. In terms of T cell functionality, we noted no differences in the amounts of cytokines secreted by the CD8+ TILs isolated from the post-AdV-tk surgical samples from this trial compared to our previous study. However, in both cases, less than half of the T cells showed hypofunction in response to CD3 antibody stimulation (as compared to T cells obtained from non-cancerous lungs). It is possible, however, that the higher level of inhibitory receptors might have made these T cells more sensitive to inhibition in situ.
The large tumors injected in this trial had a relatively high amounts of necrosis noted on qualitative examination of the surgical specimen. We think this necrosis was not likely due to the AdV-tk injection, as we saw similar degrees of necrosis in a size-matched control group of patients. However, induction of large areas of vector-induced necrosis is not critical for efficacy, as the GMCI approach primarily depends on bystander effects caused by inducing immune responses. This does not require large-scale tumor destruction, but only enough cell killing to induce anti-tumor immune response. In our early studies of AdV-tk after intrapleural instillation in mesothelioma patients, we saw only limited amounts of gene transfer 3 days after vector instillation, yet we induced strong immune and clinical responses with a number of the patients living >3 years and two living longer than 6.5 years.32
We also assessed the pre- and post-tumor biopsies for expression of PD-L1 on the tumor cells. Unlike other AdV-tk human trials showing PD-L1 upregulation (see below), the samples in this study did not show any significant increased PD-L1 expression. This may have been impacted by the relatively large percentage of necrosis seen in these tumors.
This feasibility trial was not powered to address clinical efficacy; however, there were some interesting observations. First, none of the 12 subjects had succumbed to their disease with 7–32 months of follow-up. Furthermore, only three subjects have had recurrences. Given the advanced stage of the included subjects (see Table 1), we think that this is provocative, but we also acknowledge the need to continue follow-up and evaluate this strategy in larger studies. In the highest dosing cohort, subjects underwent baseline and post-vector CT scans to assess for effects. One patient had an impressive reduction in tumor size without any additional intervention in-between the scans (Figure 1). However, this response may be quite unusual, as this subject’s histology showed a very rare subtype (sarcomatoid carcinoma) and the subject had a very high baseline infiltration of CD8 T cells that may have made them more responsive to the GMCI.
Our data extend the results from other clinical trials using i.t. GMCI that demonstrate immune activation in injected tumors. Ayala et al.21 gave intra-prostatic AdV-tk injections followed by surgery 2–4 weeks later. They noted a significant ∼3-fold increase in CD8 T cells within treated tumors. They also observed an increase in the percentage of CD8 T cells expressing HLA-DR 2 weeks after GMCI started. Rojas-Martínez et al.33 performed a similar prostate cancer trial and anecdotally noted increased numbers of activated CD8 T cells in post-treatment biopsies. Aguilar et al.20 reported a trial of i.t. AdV-tk injection in pancreatic cancer that showed a significant increase in CD8+ T cell infiltrates, as well as tumor PD-L1 expression.
Although there is a consistent trend of GMCI-mediated immune stimulation, several questions remain. One of the presumed advantages of this approach is the ability to generate antigens in situ for an auto-vaccination to the tumor. In pre-clinical models, this has been demonstrated by same tumor challenge or passive transfer of T cells from a treated animal to a naive animal.13,14,34 However, in this clinical study, it is not known how much of the activation that we observed was due to a response to the i.t. injection of a highly inflammatory replication-deficient adenovirus11 or due to the immune stimulation caused by tumor cell death and super-antigen activation induced by the TK transgene.20 Although the question could be partially answered by using a control adenoviral vector without transgene, this was not considered justified in a clinical setting. As an imperfect alternative, the results were compared to five samples from two previous clinical trials in which we instilled similar doses of either AdV-tk or a similar replication-deficient adenoviral vector encoding IFN-α2A into the pleural spaces of patients with malignant mesothelioma or lung cancer and obtained blood about 2 weeks later.22,35 In those trials, there were also significant increases of CD38 and Ki67 in circulating CD8+ T cells, although the changes observed were smaller than those seen in the current trial. These comparisons are difficult to interpret because of the number of different variables, including tumor types, small numbers of samples, and route of administration. Nonetheless, it appears that i.t. GMCI for NSCLC induced more potent peripheral CD8+ T cell activation than the previous intra-pleural injections.
The increase in expression of “inhibitory” receptors on TILs and PBMCs, such as PD-1, CD39, and CTLA-4, is also a sign of activation, but it could limit the effectiveness of the anti-tumor immune response. An increase in checkpoint ligand expression after GMCI has been seen in other clinical trials. In the pancreatic cancer GMCI clinical trial, PD-L1 levels were increased in five of seven patients analyzed.20 There is preclinical data to support the hypothesis that a combination of adenoviral gene therapy (including oncolytic adenoviral vectors and GMCI) with checkpoint inhibitors may be synergistic.36 Woller et al.37 demonstrated that treatment of lung cancers with an oncolytic adenovirus significantly abrogated systemic resistance to PD-1 immunotherapy, leading to improved elimination of disseminated lung tumors. Kuryk et al.38 found that combination of an immunogenic oncolytic adenovirus with the anti-PD-1 antibody pembrolizumab exhibited a synergistic antitumor effect in the humanized A2058 melanoma huNOG mouse model. With regard to AdV-tk, Shin et al.39 showed that an adenovirus expressing both TK and a soluble PD-1 enhanced antitumor immunity by strengthening the CD8 T cell response. Most relevant to our study, preclinical experiments in a glioblastoma model showed synergy from the combination of GMCI and an anti-PD-1 antibody.15 Although in this clinical trial, we did not observe consistent PD-L1 increases on the NSCLC cells, it is known that PD-1-driven inhibition can also occur during interactions of CD8 T cells with dendritic cells in tumor-draining lymph nodes,40 supporting evaluation of a combined approach with a checkpoint inhibitor regimen.
In summary, this trial of GMCI in resectable NSCLC shows that i.t. AdV-tk delivery is safe, feasible, and results in systemic and intra-tumoral CD8+ T cell activation. Given these findings, additional studies in NSCLC and combination studies with checkpoint blockade should be considered to evaluate the ability to increase response rates and reduce recurrence rates through generation of GMCI-mediated memory immunologic responses.
Materials and Methods
GMCI
AdV-tk (aglatimagene besadenovec) is a non-replicating human adenovirus serotype 5 vector containing the HSV-tk gene driven by a Rous sarcoma virus long terminal repeat.41 Following transduction, the HSV-tk protein produced from AdV-tk phosphorylates anti-herpetic prodrugs (e.g., ganciclovir and valacyclovir), thus converting to a toxic agent that produces immunogenic cell death. AdV-tk was produced in accordance with current good manufacturing practices and provided by Advantagene (d.b.a. Candel Therapeutics, Needham, MA, USA). Following vector delivery, subjects were treated with 14 days of oral valacyclovir (2 g three times per day).
Study Design
This open-label, phase I dose-escalation trial was approved by the University of Pennsylvania Institutional Review Board (ClinicalTrials.gov: NCT03131037). Informed consent was obtained before enrollment. Eligible patients had presumed resectable NSCLC with lung tumors measuring 2 cm or greater. All of the patients underwent radiographic staging with 1-mm fine-cut CT and positron-emission tomography (PET) scanning. Any patient with mediastinal lymph nodes larger than 10 mm in maximal measurement and evidence of elevated standardized uptake value (SUV) (>2.5) were not eligible for the trial and underwent additional staging to determine whether they were surgical candidates. It is our institutional practice that if a patient who is deemed a surgical candidate and has no PET avid nodes and all nodes by CT scan are less than 5 mm in the short axis, then these patients do not undergo pre-resection curvilinear EBUS nodal interrogation of nodes less than 5 mm, as it is technically challenging and the probability of cancer recovery in these patients is likely less than 5%. Patients with nodes between 5 and 10 mm in size were staged with curvilinear EBUS nodal interrogation of nodes at nodal stations 3, 4, and 7. One patient had a level 5 node sampled via video-assisted thoracoscopic surgery (VATS). No mediastinoscopies were performed. These nodal needle biopsies were evaluated in real time by a cytopathologist in the operating room, and any patients with positive cytology were excluded from surgery and from the trial. By definition (as shown in Table 1), all patients were deemed to be N0 by this approach.
Other eligibility criteria stipulated a minimum age of 18 years and ECOG performance status of 0 or 1. Laboratory inclusion criteria included bilirubin ≤1.5 × upper limit of normal (ULN), serum glutamic-oxaloacetic transaminase (SGOT) (aspartate aminotransferase [AST]) ≤3 × ULN, granulocyte count (absolute neutrophil count [ANC]) ≥1,000/mm3, peripheral lymphocyte count ≥500/mm3, hemoglobin ≥9 g/dL, platelets ≥100,000/mm3, serum creatinine <2 mg/dL, and calculated creatinine clearance >30 mL/min.
On day 0, subjects underwent standard-of-care mediastinal lymph node staging via EBUS or a minimally invasive surgical approach. Any suspicious lymph nodes were examined on-site by a cytopathologist. When the absence of nodal involvement was confirmed and the primary lesion biopsy demonstrated findings consistent with NSCLC, primary lesion biopsies were performed for collection of baseline tissue for biocorrelative analyses prior to AdV-tk injection. The assigned AdV-tk dose was then administered in 2 mL using a 22G needle in up to four locations within the tumor. Vector delivery was via peripheral radial ultraminiature EBUS guide sheath-directed bronchoscopy. The peripheral ultraminiature probes are 1.4 and 1.7 mm in diameter and can be threaded through a guide sheath and advanced as far as the pleural surface. Once a lesion was identified by peripheral ultraminiature radial EBUS, the guide sheath was left in place and instruments for biopsy were passed through the guide sheath for tissue acquisition. The guide sheath remained in place and the needle for vector delivery was passed through the guide sheath into the tumor and the vector was delivered. Three dose cohorts were evaluated: cohort 1, 2.5 × 1011 vector particles (vp); cohort 2, 5 × 1011 vp; cohort 3, 1 × 1012 vp. On day 1, subjects began oral valacyclovir at 2 g three times daily for 14 days, as has been established in previous trials. Patients underwent standard-of-care surgery on day 18–24, followed by standard-of-care adjuvant therapy based on final surgical pathology staging.
Safety Monitoring
Toxicity was assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.42 DLTs were defined as any of the following GMCI-related adverse events: grade 3 or greater allergic reaction, grade 4 hematologic toxicity persisting for >7 days (except lymphopenia), grade 3 hypoxia lasting for 48 h, any grade 4 non-hematologic toxicity, or any grade 3 non-hematologic toxicity (except alanine aminotransferase [ALT], AST, alkaline phosphatase, bilirubin, or creatinine) lasting for >7 days. The MTD was defined as the highest dose level in which no more than one patient developed a DLT up to 2 weeks after surgery.
Biocorrelative and Immunologic Data
Immune responses were assessed by evaluation of serially collected pre- and post-vector tumor, serum, and PBMC samples. Baseline tumor samples were obtained by core biopsy or fine needle aspiration, and after-treatment samples were obtained from the operative specimen. Baseline biopsies and a portion of operative samples were formalin-fixed and paraffin-embedded and then compared using a combination of hematoxylin and eosin staining and immunostaining (CD8, PD-L1) as previously described by our group.35 The needle biopsy and a portion of the operative specimen were immediately digested as previously described43 and compared to baseline tumor aspirates by flow cytometry (using antibodies against CD3, CD4, CD8, FOXP3, Ki67, HLA-DR, CD38, CD39, 41BB, PD-1, CTLA-4, TIGIT, and TIM-3). Tumor digests were also treated with monensin/brefeldin, submaximally stimulated with plate-bound anti-CD3 antibodies (0.5 mg/mL) for 18 h, and then subjected to intracellular cytokine staining for TNF-α and IFN-γ as previously described in detail by our group.24,44 Antibodies used are listed in Table S1. The data from our previous analysis of early-stage lung cancer surgery patients24 was replotted and analyzed as a control group to the AdV-tk-treated patients.
PBMCs were collected at day 0 (D0) and at the day of surgery and compared by flow cytometry in a batched fashion as previously detailed.35
To detect induced humoral responses against tumor antigens, we performed immunoblotting against purified extracts from commercially purchased allogeneic lung cancer cell lines similar to the approach we have used previously for mesothelioma.35 Serum for immunoblotting was obtained from D0 and ∼D43 (before the administration of adjuvant chemotherapy). Extracts from cells were prepared and immunoblotted with patient serum diluted at 1:1,500. Multiple exposures were obtained and comparisons were made only on the exposures in which the major bands detected on pre-treatment blots were of equal intensity in post-treatment blots. Two independent, blinded observers visually scanned each blot to detect humoral responses and came to a consensus score.
For estimates of tumor necrosis, each subject’s surgical pathology specimens were reviewed and an estimate of tumor necrosis was made by a thoracic pathologist (C.D.). We analyzed the entire tumor when gross tumor size was ≤3.0 cm. For tumors ≥3.0 cm, one section per cm of tumor was submitted for evaluation using sections showing the greatest dimension for evaluation. Tumor necrosis present was then evaluated based on the size of the tumor in its largest dimension on each section and an estimate of area of tumor necrosis relative to the tumor present. These values were then averaged to estimate total percent tumor necrosis. A case-control comparison was made in which we selected two historical “matching” cases based on similar histology and tumor size for each subject and estimated the degree of necrosis.
Statistical Analysis
A standard 3+3 design was used to determine the MTD with an implicit 50% chance of further dose escalation after achievement of a toxicity rate of 30%. Correlative data are presented as mean (standard error) unless noted. Differences between two independent groups with continuous data were compared by the Student’s t test. Differences between samples from the same patient taken at different time points were compared by paired two-sided t tests. Comparisons were made using Stata: Release 14 (Stata, College Station, TX, USA). A p value of 0.05 was considered statistically significant.
Acknowledgments
J.D.P. was supported by the NIH / NCI (F32CA210409), the American Philosophical Society, and by the Association for Academic Surgery Foundation Resident Research Award. S.O. was funded by the NIH / NCI (T32CA009140). J.S. and E.E. were funded by DOD-LC140199 and NIH / NCI (R01CA187392). S.S. was supported by the NIH (R01 CA193556). S.M.A. was supported by the NIH / NCI (P01CA087971, R01CA172921). S.O., M.M., and S.M.A. were partially supported by a clinical trial agreement with Advantagene.
Author Contributions
Conceptualization, J.D.P., L.K.A., E.A.-C., S.S., and S.M.A.; Methodology, J.D.P., L.K.A., E.A.-C., S.S., A.R.H., and S.M.A.; Investigation, J.D.P., S.O., E.K.M., A.R.H., M.M., P.W., J.S., C.C., L.F.S., M.G.B., and C.D.; Resources, L.K.A., B.W.G., A.G.M., and E.A.-C.; Writing – Original Draft, J.D.P., S.M.A., L.K.A., and E.A.-C.; Writing – Review and Editing, A.R.H., C.D., C.L., L.K.A., E.A.-C., and S.S.; Visualization, J.D.P., L.K.A., B.W.G., A.G.M., and S.M.A.; Supervision, E.E., E.K.M., A.R.H., C.L., L.K.A., E.A.-C., S.S., and S.M.A.; Project Administration, J.D.P., C.C., L.F.S., M.G.B., L.K.A., B.W.G., A.G.M., E.A.-C., S.S., and S.M.A.; Funding Acquisition, S.S. and S.M.A.
Declaration of Interests
S.O., M.M., and S.M.A. were partially supported by a clinical trial agreement with Advantagene. L.K.A., B.W.G., A.G.M., and E.A.-C. are employees of Advantagene, who sponsored the trial. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.11.001.
Supplemental Information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2017. CA Cancer J. Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Aliperti L.A., Predina J.D., Vachani A., Singhal S. Local and systemic recurrence is the Achilles heel of cancer surgery. Ann. Surg. Oncol. 2011;18:603–607. doi: 10.1245/s10434-010-1442-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pignon J.P., Tribodet H., Scagliotti G.V., Douillard J.-Y., Shepherd F.A., Stephens R.J., Dunant A., Torri V., Rosell R., Seymour L., LACE Collaborative Group Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J. Clin. Oncol. 2008;26:3552–3559. doi: 10.1200/JCO.2007.13.9030. [DOI] [PubMed] [Google Scholar]
- 4.Wakelee H.A., Dahlberg S.E., Keller S.M., Tester W.J., Gandara D.R., Graziano S.L., Adjei A.A., Leighl N.B., Aisner S.C., Rothman J.M., ECOG-ACRIN Adjuvant chemotherapy with or without bevacizumab in patients with resected non-small-cell lung cancer (E1505): an open-label, multicentre, randomised, phase 3 trial. Lancet Oncol. 2017;18:1610–1623. doi: 10.1016/S1470-2045(17)30691-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reck M., Rodríguez-Abreu D., Robinson A.G., Hui R., Csőszi T., Fülöp A., Gottfried M., Peled N., Tafreshi A., Cuffe S. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J. Clin. Oncol. 2019;37:537–546. doi: 10.1200/JCO.18.00149. [DOI] [PubMed] [Google Scholar]
- 6.Gandhi L., Rodríguez-Abreu D., Gadgeel S., Esteban E., Felip E., De Angelis F., Domine M., Clingan P., Hochmair M.J., Powell S.F., KEYNOTE-189 Investigators Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N. Engl. J. Med. 2018;378:2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 7.van der Burg S.H., Arens R., Ossendorp F., van Hall T., Melief C.J.M. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat. Rev. Cancer. 2016;16:219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 8.Hammerich L., Bhardwaj N., Kohrt H.E., Brody J.D. In situ vaccination for the treatment of cancer. Immunotherapy. 2016;8:315–330. doi: 10.2217/imt.15.120. [DOI] [PubMed] [Google Scholar]
- 9.Sheen M.R., Fiering S. In situ vaccination: harvesting low hanging fruit on the cancer immunotherapy tree. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019;11:e1524. doi: 10.1002/wnan.1524. [DOI] [PubMed] [Google Scholar]
- 10.Aguilar L.K., Guzik B.W., Aguilar-Cordova E. Cytotoxic immunotherapy strategies for cancer: mechanisms and clinical development. J. Cell. Biochem. 2011;112:1969–1977. doi: 10.1002/jcb.23126. [DOI] [PubMed] [Google Scholar]
- 11.Shaw A.R., Suzuki M. Immunology of adenoviral vectors in cancer therapy. Mol. Ther. Methods Clin. Dev. 2019;15:418–429. doi: 10.1016/j.omtm.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eastham J.A., Chen S.H., Sehgal I., Yang G., Timme T.L., Hall S.J., Woo S.L., Thompson T.C. Prostate cancer gene therapy: herpes simplex virus thymidine kinase gene transduction followed by ganciclovir in mouse and human prostate cancer models. Hum. Gene Ther. 1996;7:515–523. doi: 10.1089/hum.1996.7.4-515. [DOI] [PubMed] [Google Scholar]
- 13.Predina J.D., Judy B., Aliperti L.A., Fridlender Z.G., Blouin A., Kapoor V., Laguna B., Nakagawa H., Rustgi A.K., Aguilar L. Neoadjuvant in situ gene-mediated cytotoxic immunotherapy improves postoperative outcomes in novel syngeneic esophageal carcinoma models. Cancer Gene Ther. 2011;18:871–883. doi: 10.1038/cgt.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Predina J.D., Kapoor V., Judy B.F., Cheng G., Fridlender Z.G., Albelda S.M., Singhal S. Cytoreduction surgery reduces systemic myeloid suppressor cell populations and restores intratumoral immunotherapy effectiveness. J. Hematol. Oncol. 2012;5:34–45. doi: 10.1186/1756-8722-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Speranza M.C., Passaro C., Ricklefs F., Kasai K., Klein S.R., Nakashima H., Kaufmann J.K., Ahmed A.K., Nowicki M.O., Obi P. Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro-oncol. 2018;20:225–235. doi: 10.1093/neuonc/nox139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Predina J.D., Judy B., Fridlender Z.G., Aliperti L.A., Madajewski B., Kapoor V., Cheng G., Quatromoni J., Okusanya O., Singhal S. A positive-margin resection model recreates the postsurgical tumor microenvironment and is a reliable model for adjuvant therapy evaluation. Cancer Biol. Ther. 2012;13:745–755. doi: 10.4161/cbt.20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Predina J., Eruslanov E., Judy B., Kapoor V., Cheng G., Wang L.C., Sun J., Moon E.K., Fridlender Z.G., Albelda S., Singhal S. Changes in the local tumor microenvironment in recurrent cancers may explain the failure of vaccines after surgery. Proc. Natl. Acad. Sci. USA. 2013;110:E415–E424. doi: 10.1073/pnas.1211850110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wheeler L.A., Manzanera A.G., Bell S.D., Cavaliere R., McGregor J.M., Grecula J.C., Newton H.B., Lo S.S., Badie B., Portnow J. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro-oncol. 2016;18:1137–1145. doi: 10.1093/neuonc/now002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiocca E.A., Aguilar L.K., Bell S.D., Kaur B., Hardcastle J., Cavaliere R., McGregor J., Lo S., Ray-Chaudhuri A., Chakravarti A. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 2011;29:3611–3619. doi: 10.1200/JCO.2011.35.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aguilar L.K., Shirley L.A., Chung V.M., Marsh C.L., Walker J., Coyle W., Marx H., Bekaii-Saab T., Lesinski G.B., Swanson B. Gene-mediated cytotoxic immunotherapy as adjuvant to surgery or chemoradiation for pancreatic adenocarcinoma. Cancer Immunol. Immunother. 2015;64:727–736. doi: 10.1007/s00262-015-1679-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayala G., Satoh T., Li R., Shalev M., Gdor Y., Aguilar-Cordova E., Frolov A., Wheeler T.M., Miles B.J., Rauen K. Biological response determinants in HSV-tk + ganciclovir gene therapy for prostate cancer. Mol. Ther. 2006;13:716–728. doi: 10.1016/j.ymthe.2005.11.022. [DOI] [PubMed] [Google Scholar]
- 22.Aggarwal C., Haas A.R., Metzger S., Aguilar L.K., Aguilar-Cordova E., Manzanera A.G., Gómez-Hernández G., Katz S.I., Alley E.W., Evans T.L. Phase I study of intrapleural gene-mediated cytotoxic immunotherapy in patients with malignant pleural effusion. Mol. Ther. 2018;26:1198–1205. doi: 10.1016/j.ymthe.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kruklitis R.J., Singhal S., Delong P., Kapoor V., Sterman D.H., Kaiser L.R., Albelda S.M. Immuno-gene therapy with interferon-β before surgical debulking delays recurrence and improves survival in a murine model of malignant mesothelioma. J. Thorac. Cardiovasc. Surg. 2004;127:123–130. doi: 10.1016/j.jtcvs.2003.08.034. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien S.M., Klampatsa A., Thompson J.C., Martinez M.C., Hwang W.T., Rao A.S., Standalick J.E., Kim S., Cantu E., Litzky L.A. Function of human tumor-infiltrating lymphocytes in early stage non-small cell lung cancer. Cancer Immunol. Res. 2019;7:896–909. doi: 10.1158/2326-6066.CIR-18-0713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller J.D., van der Most R.G., Akondy R.S., Glidewell J.T., Albott S., Masopust D., Murali-Krishna K., Mahar P.L., Edupuganti S., Lalor S. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 26.Griscelli F., Opolon P., Saulnier P., Mami-Chouaib F., Gautier E., Echchakir H., Angevin E., Le Chevalier T., Bataille V., Squiban P. Recombinant adenovirus shedding after intratumoral gene transfer in lung cancer patients. Gene Ther. 2003;10:386–395. doi: 10.1038/sj.gt.3301928. [DOI] [PubMed] [Google Scholar]
- 27.Nemunaitis J., Swisher S.G., Timmons T., Connors D., Mack M., Doerksen L., Weill D., Wait J., Lawrence D.D., Kemp B.L. Adenovirus-mediated p53 gene transfer in sequence with cisplatin to tumors of patients with non-small-cell lung cancer. J. Clin. Oncol. 2000;18:609–622. doi: 10.1200/JCO.2000.18.3.609. [DOI] [PubMed] [Google Scholar]
- 28.Swisher S.G., Roth J.A., Komaki R., Gu J., Lee J.J., Hicks M., Ro J.Y., Hong W.K., Merritt J.A., Ahrar K. Induction of p53-regulated genes and tumor regression in lung cancer patients after intratumoral delivery of adenoviral p53 (INGN 201) and radiation therapy. Clin. Cancer Res. 2003;9:93–101. [PubMed] [Google Scholar]
- 29.Nemunaitis J., Meyers T., Senzer N., Cunningham C., West H., Vallieres E., Anthony S., Vukelja S., Berman B., Tully H. Phase I trial of sequential administration of recombinant DNA and adenovirus expressing L523S protein in early stage non-small-cell lung cancer. Mol. Ther. 2006;13:1185–1191. doi: 10.1016/j.ymthe.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 30.Duhen T., Duhen R., Montler R., Moses J., Moudgil T., de Miranda N.F., Goodall C.P., Blair T.C., Fox B.A., McDermott J.E. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat. Commun. 2018;9:2724. doi: 10.1038/s41467-018-05072-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.-L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 32.Sterman D.H., Recio A., Vachani A., Sun J., Cheung L., DeLong P., Amin K.M., Litzky L.A., Wilson J.M., Kaiser L.R., Albelda S.M. Long-term follow-up of patients with malignant pleural mesothelioma receiving high-dose adenovirus herpes simplex thymidine kinase/ganciclovir suicide gene therapy. Clin. Cancer Res. 2005;11:7444–7453. doi: 10.1158/1078-0432.CCR-05-0405. [DOI] [PubMed] [Google Scholar]
- 33.Rojas-Martínez A., Manzanera A.G., Sukin S.W., Esteban-María J., González-Guerrero J.F., Gomez-Guerra L., Garza-Guajardo R., Flores-Gutiérrez J.P., Elizondo Riojas G., Delgado-Enciso I. Intraprostatic distribution and long-term follow-up after AdV-tk immunotherapy as neoadjuvant to surgery in patients with prostate cancer. Cancer Gene Ther. 2013;20:642–649. doi: 10.1038/cgt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall S.J., Mutchnik S.E., Chen S.H., Woo S.L., Thompson T.C. Adenovirus-mediated herpes simplex virus thymidine kinase gene and ganciclovir therapy leads to systemic activity against spontaneous and induced metastasis in an orthotopic mouse model of prostate cancer. Int. J. Cancer. 1997;70:183–187. doi: 10.1002/(sici)1097-0215(19970117)70:2<183::aid-ijc8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 35.Sterman D.H., Alley E., Stevenson J.P., Friedberg J., Metzger S., Recio A., Moon E.K., Haas A.R., Vachani A., Katz S.I. Pilot and feasibility trial evaluating immuno-gene therapy of malignant mesothelioma using intrapleural delivery of adenovirus-IFNα combined with chemotherapy. Clin. Cancer Res. 2016;22:3791–3800. doi: 10.1158/1078-0432.CCR-15-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen C.-Y., Hutzen B., Wedekind M.F., Cripe T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother. 2018;7:65–77. doi: 10.2147/OV.S145532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woller N., Gürlevik E., Fleischmann-Mundt B., Schumacher A., Knocke S., Kloos A.M., Saborowski M., Geffers R., Manns M.P., Wirth T.C. Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed t-cell responses. Mol. Ther. 2015;23:1630–1640. doi: 10.1038/mt.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuryk L., Møller A.S.W., Jaderberg M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology. 2019;8:e1532763. doi: 10.1080/2162402X.2018.1532763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin S.-P., Seo H.-H., Shin J.-H., Park H.-B., Lim D.-P., Eom H.S., Bae Y.S., Kim I.H., Choi K., Lee S.J. Adenovirus expressing both thymidine kinase and soluble PD1 enhances antitumor immunity by strengthening CD8 T-cell response. Mol. Ther. 2013;21:688–695. doi: 10.1038/mt.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fransen M.F., Schoonderwoerd M., Knopf P., Camps G.M., Hawinkels L., Keneilling M., van Hall T., Ossendorp F. Tumor draining lymph nodes are pivotal in PD-1/PD-L1 checkpoint therapy. JCI Insight. 2018;3:e124507. doi: 10.1172/jci.insight.124507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chévez-Barrios P., Chintagumpala M., Mieler W., Paysse E., Boniuk M., Kozinetz C., Hurwitz M.Y., Hurwitz R.L. Response of retinoblastoma with vitreous tumor seeding to adenovirus-mediated delivery of thymidine kinase followed by ganciclovir. J. Clin. Oncol. 2005;23:7927–7935. doi: 10.1200/JCO.2004.00.1883. [DOI] [PubMed] [Google Scholar]
- 42.National Institutes of Health . 2010. Common terminology criteria for adverse events (CTCAE) v4.0.https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm [Google Scholar]
- 43.Quatromoni J.G., Singhal S., Bhojnagarwala P., Hancock W.W., Albelda S.M., Eruslanov E. An optimized disaggregation method for human lung tumors that preserves the phenotype and function of the immune cells. J. Leukoc. Biol. 2015;97:201–209. doi: 10.1189/jlb.5TA0814-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Klampatsa A., O’Brien S.M., Thompson J.C., Rao A.S., Stadanlick J.E., Martinez M.C., Liousia M., Cantu E., Cengel K., Moon E.K. Phenotypic and functional analysis of malignant mesothelioma tumor-infiltrating lymphocytes. Oncoimmunology. 2019;8:e1638211. doi: 10.1080/2162402X.2019.1638211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.