

Abstract
Evaluation of immune responses to adeno-associated virus (AAV)-mediated gene therapies prior to and following dose administration plays a key role in determining therapeutic safety and efficacy. This report describes up to 3 years of immunogenicity data following administration of valoctocogene roxaparvovec (BMN 270), an AAV5-mediated gene therapy encoding human B domain-deleted FVIII (hFVIII-SQ) in a phase 1/2 clinical study of adult males with severe hemophilia A. Patients with pre-existing humoral immunity to AAV5 or with a history of FVIII inhibitors were excluded from the trial. Blood plasma and peripheral blood mononuclear cell (PBMC) samples were collected at regular intervals following dose administration for assessment of humoral and cellular immune responses to both the AAV5 vector and transgene-expressed hFVIII-SQ. The predominant immune response elicited by BMN 270 administration was largely limited to the development of antibodies against the AAV5 capsid that were cross-reactive with other common AAV serotypes. No FVIII inhibitor responses were observed within 3 years following dose administration. In a context of prophylactic or on-demand corticosteroid immunosuppression given after vector infusion, AAV5 and hFVIII-SQ peptide-specific cellular immune responses were intermittently detected by an interferon (IFN)-γ and tumor necrosis factor (TNF)-α FluoroSpot assay, but they were not clearly associated with detrimental safety events or changes in efficacy measures.
Keywords: adeno-associated virus vector, AAV, gene therapy, hemophilia A, clinical immunogenicity, humoral immunity, cellular immunity, AAV antibody, cross-reactivity, clinical safety
Graphical Abstract
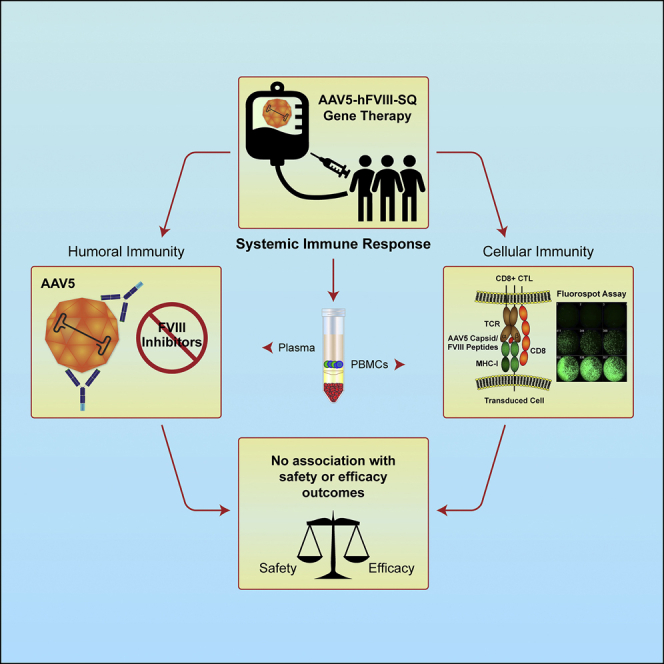
This report describes up to 3 years of immunogenicity data following administration of valoctocogene roxaparvovec, an AAV5-mediated gene therapy encoding hFVIII-SQ, in a phase 1/2 clinical study of adult males with severe hemophilia A. Immunogenicity elicited by BMN 270 administration was predominantly a humoral antibody response against the AAV5 capsid. No FVIII inhibitor responses were observed within 3 years following dose administration. No immunogenicity measures were consistently associated with detrimental safety or efficacy parameters.
Introduction
The development of adeno-associated viruses (AAVs) as vectors for delivery of gene therapies has seen significant clinical success recently for the treatment of congenital monogenic disorders, including hemophilia A (HA), hemophilia B, Duchenne muscular dystrophy, spinal muscular atrophy, and Leber congenital amaurosis, among others.1,2 HA is an X-linked recessive bleeding disorder that affects approximately 1 in 5,000 males3 and is caused by mutations in the gene that codes for coagulation factor VIII (FVIII), an essential cofactor in the coagulation pathway. Valoctocogene roxaparvovec (BMN 270) is an AAV type 5 (AAV5)-mediated gene therapy developed for the treatment of HA, and it encodes a codon-optimized, B-domain-deleted human FVIII protein (hFVIII-SQ) under control of a hybrid liver-specific promoter.4, 5, 6
Currently, treatment of HA consists of FVIII replacement therapy or novel therapies, either prophylactically or on-demand. Despite availability of these therapies, patients with HA continue to experience breakthrough bleeds and progressive, debilitating joint disease.7,8 Gene therapy offers the advantage of continuous and measurable steady-state levels of FVIII that negate the peaks and troughs observed with currently available therapies.
Comprehensive assessment of human immune responses to gene therapy candidates includes characterization of humoral and cellular immunogenicity specific for both the viral vector and the expressed transgene (protein) product before and after dose administration. BMN 270 is administered as a single intravenous infusion and, based on preclinical studies, distributes to several peripheral tissues with long-term detection of vector genomes found primarily in the liver.9,10 Tissue distribution and transduction of parenchymal cells by AAV vectors can be negatively impacted by pre-existing neutralizing antibodies, and even low titers have been reported to limit the efficacy of vector-mediated gene therapies in humans.11, 12, 13 As a consequence, all patients enrolled in the current study were required to screen negative for pre-existing immunity to AAV5 using two orthogonal measures, that is, an AAV5-specific total binding antibody (TAb) assay and a cell-based assay for evaluating the AAV5 neutralizing capacity of a patient’s plasma.14
The most significant complication arising in response to factor replacement therapy is the potential development of a neutralizing antibody response to the FVIII, commonly referred to as inhibitors. Inhibitors occur in approximately one third of HA patients following exposure to FVIII replacement products, usually within the first 75 exposure days (ED).15 As a criterion for enrollment into the current study, all patients were required to have more than 150 exposure days to FVIII replacement products without previous inhibitor development.
An additional consideration regarding post-administration immune response to AAV is the potential for cellular cytotoxicity directed against transduced hepatocytes expressing cell-associated capsid peptide epitopes.16,17 Prior studies of liver-directed gene therapy in hemophilia B (using AAV vectors encoding for human factor IX [FIX]) noted an increase in the alanine aminotransferase (ALT) that was associated with a decline in FIX expression levels, and that was temporally associated with the detection of interferon (IFN)-γ secretion by AAV peptide-stimulated peripheral blood mononuclear cells (PBMCs) in an enzyme-linked immunospot (ELISpot) assay.11, 12, 13,18 These data led to the hypothesis that, in some instances, AAV-specific cytotoxic T cells were targeting capsid peptides that were presented by transduced hepatocytes, leading to their elimination, release of ALT, and loss of FIX expression. Treatment with corticosteroids seemed to provide resolution of ALT and stabilization of FIX expression in a subset of patients, and it informed their prophylactic or on-demand use in response to ALT elevations in the current study.
BMN 270-201 is an ongoing, fully enrolled, first-in-human phase 1/2 open-label, dose escalation study in patients with severe HA. As previously reported, FVIII activity measured by chromogenic substrate assay remained below 1–3 IU/dL in both cohort 1 (6E12 vector genomes [vg]/kg) and cohort 2 (2E13 vg/kg). Cohort 3 and cohort 4 patients receiving a 6E13 and 4E13 vg/kg dose, respectively, showed gradually increasing FVIII activity measures rising above the 5 IU/dL cutoff for mild HA by week 16 in all participants. Longer term data reported that the 13 patients in cohorts 3 and 4 treated with 6E13 and 4E13 vg/kg, respectively, continue to have substantial reductions in the incidence of bleeding events with resolution of bleeding in target joints and complete cessation of FVIII prophylaxis. Mean FVIII activity levels were 33 IU/dL at the end of year 3 for cohort 3 and 15 IU/dL at the end of year 2 for cohort 4.19,20
This report examines both cellular and humoral immune responses to the AAV5 capsid and the hFVIII-SQ transgene product in participants of the phase 1/2 clinical study and assesses impact on safety and efficacy. These data cover a time period from enrollment of the first patient in September of 2015 through a data cutoff of April 1, 2019, with all dosed subjects having been followed for at least 104 weeks to a maximum of 183 weeks. Additionally, the report describes the development of cross-reactive antibodies against capsids of diverse AAV serotypes.
Results
Study Design
Patient demographics are described in Table 1, and further patient characteristics were reported previously.19,20 A total of 15 adult male participants with severe HA were enrolled into one of four dose cohorts. As of this report, all dosed subjects having been followed for at least 104 weeks to a maximum of 183 weeks. Interim results demonstrated that a single infusion of BMN 270 had a favorable safety profile and led to long-term elevation in FVIII activity in patients with severe HA, accompanied by resolution of target joints and substantial reductions in annualized bleeding rates and exogenous FVIII utilization.
Table 1.
Demographics and Dose Cohorts (All Enrolled Subjects)
| Demographic Variable | 6E12 vg/kg (n = 1) | 2E13 vg/kg (n = 1) | 4E13 vg/kg (n = 6) | 6E13 vg/kg (n = 7) | Total (n = 15) |
|---|---|---|---|---|---|
| Age at Enrollment (years) | |||||
| N | 1 | 1 | 6 | 7 | 15 |
| Mean (SD) | 25.0 (N/A) | 43.0 (N/A) | 31.3 (9.6) | 30.4 (5.8) | 31.3 (7.8) |
| Min, max | 25, 25 | 43, 43 | 22, 45 | 23, 42 | 22, 45 |
| Age at Enrollment, n (%) | |||||
| 18 to <40 years | 1 (100%) | 0 | 5 (83.3%) | 6 (85.7%) | 12 (80.0%) |
| 40 to 60 years | 0 | 1 (100%) | 1 (16.7%) | 1 (14.3%) | 3 (20.0%) |
| ≥60 years | 0 | 0 | 0 | 0 | 0 |
| Sex, n (%) | |||||
| Male | 1 (100%) | 1 (100%) | 6 (100%) | 7 (100%) | 15 (100%) |
| Female | 0 | 0 | 0 | 0 | 0 |
| Race, n (%) | |||||
| Asian | 1 (100%) | 0 | 0 | 1 (14.3%) | 2 (13.3%) |
| Black or African American | 0 | 0 | 1 (16.7%) | 0 | 1 (6.7%) |
| White | 0 | 1 (100%) | 5 (83.3%) | 6 (85.7%) | 12 (80.0%) |
The most common adverse event following dose administration was a mild and transient elevation in ALT occurring in 11 participants. With the exception of the first two subjects dosed at the lowest vector doses, most participants received a course of oral corticosteroids. All patients in the 6E13 vg/kg cohort received corticosteroids prophylactically from week 3, and four of six patients in the 4E13 vg/kg cohort received corticosteroids reactively in response to an increase in ALT of 1.5-fold the baseline value or the upper limit of normal. Detailed data on the timing, dose, and duration of corticosteroid use are provided in Figures S1 and S2.
Assessment of Humoral Immune Response to FVIII
FVIII inhibitors were measured using a Bethesda assay with Nijmegen modification at multiple time points, including screening, baseline (within 7 days prior to dose administration), then weekly through 36 weeks, and then every 2–6 weeks at later time points post-infusion out to 3 years. Inhibitor titers were reported as Bethesda units (BU), and all subjects in all dose cohorts remained FVIII inhibitor negative at all time points, using a cut-point of ≥0.6 BU for positivity.
Separately, TAbs specific for the FVIII protein (FVIII TAbs) were measured at similar time points as FVIII inhibitors. All patients tested negative at all time points, except for a single patient (patient 7) in the 6E13 vg/kg cohort screened and confirmed positive at a single time point (week 15) with a FVIII TAb titer below the minimum required dilution (MRD) (data not shown). This patient was negative at all other time points and there was no corresponding change in FVIII activity.
Assessment of Humoral Immune Response to AAV5
All patients enrolled in study 270-201 screened negative for pre-existing AAV5 immunity using two separate methods. The first method was a cell-based AAV5 transduction inhibition (TI) assay measuring the neutralizing capacity of patient plasma toward AAV5, and the second was a bridging electrochemiluminescence (ECL) assay (ECLA) measuring AAV5 total binding antibody (TAb). Samples for the AAV5 TI assay were collected during patient screening and on day 1 just prior to BMN 270 dose administration. All enrolled subjects tested negative for AAV5 TI at both time points. The TAb screening values ranged from 0.65 to 1.05 normalized electrochemiluminescence unit (ELCU) and were below the assay cut-point of 1.15. The transduction values at the minimal required dilution (1:2) ranged from 123% to 187% transduction and were above the assay cut-point of 44.9%, indicating no detectable transduction inhibitors.
AAV5 TAb was tested at baseline and then at regular intervals after treatment (Figure 1). As expected, all subjects seroconverted to AAV5 TAb positive by week 8, the first time point assessed post-dosing (Figure 1). The overall mean (SD) titer for all subjects at week 8 was 546,043 (477,612) (Table 2). Anti-AAV5 TAb titers peaked at week 40 with an overall mean (SD) of 8,791,437 (10,567,127). Following the peak, anti-AAV5 TAb titers remained stable with no meaningful reduction in titer through week 104, with a mean (SD) of 4,641,256 (4,738,982). There was no apparent dose relationship of BMN 270 and titers of post-administration anti-AAV5 TAb. Week 8 (the first time point assessed for all subjects) anti-AAV5 TAb titer mean (SD) was 103,041 (not applicable [N/A]) for cohort 1 (6.0E12 vg/kg, n = 1), 729,167 (N/A) for cohort 2 (2.0E13 vg/kg, n = 1), 353,122 (297,427) for cohort 4 (4.0E13 vg/kg, n = 6), and 782,276 (589,106) for cohort 3 (6.0E13 vg/kg, n = 7).
Figure 1.

All Subjects Develop a Sustained Anti-AAV5 Antibody Response following Dose Administration
Longitudinal assessment of AAV5 capsid-specific total binding antibody. Plasma was collected from clinical patients at regular intervals after valoctocogene roxaparvovec administration and assayed for AAV5-specific TAbs. For similar titers across dose cohorts, titers peaked at week 40 with a mean of 8.8E6, 4.6E6 at year 2, and 7.6E6 at year 3. Box plots show the interquartile range with whiskers indicating minimum and maximum values. Line indicates median, diamond indicates mean
Table 2.
AAV5 TAb Titers by Dosing Cohort
| TAb | Cohort 1, 6E12 vg/kg (n = 1) | Cohort 2, 6E13 vg/kg (n = 1) | Cohort 4, 4E13 vg/kg (n = 6) | Cohort 3, 6E13 vg/kg (n = 7) | Overall (n = 15) |
|---|---|---|---|---|---|
| Baseline | |||||
| n | 1 | 1 | 6 | 7 | 15 |
| Mean (SD) | N/A (N/A) | N/A (N/A) | N/A (N/A) | N/A (N/A) | N/A (N/A) |
| Week 8 | |||||
| n | 1 | 1 | 6 | 6 | 14 |
| Mean (SD) | 103,041 (N/A) | 729,167 (N/A) | 353,122 (297,427) | 782,276 (589,106) | 546,043 (477,612) |
| Week 16 | |||||
| n | 1 | 1 | 6 | 7 | 15 |
| Mean (SD) | 293,269 (N/A) | 7,732,372 (N/A) | 1,245,682 (389,988) | 3,747,741 (6,063,604) | 2,782,261 (4,413,894) |
| Week 20 | |||||
| n | 1 | 1 | 6 | 7 | 15 |
| Mean (SD) | 1,152,052 (N/A) | 6,718,750 (N/A) | 2,352,946 (2,324,139) | 7,529,366 (13,598,985) | 4,979,603 (9,415,725) |
| Week 24 | |||||
| n | 0 | 0 | 6 | 0 | 6 |
| Mean (SD) | N/A (N/A) | N/A (N/A) | 1,361,133 (641,098) | N/A (N/A) | 1,361,133 (641,098) |
| Week 28 | |||||
| n | 1 | 1 | 6 | 7 | 15 |
| Mean (SD) | 6,562,500 (N/A) | 1,501,820 (N/A) | 2,125,603 (1,749,443) | 3,935,335 (2,866,686) | 3,224,352 (2,528,444) |
| Week 32 | |||||
| n | 0 | 0 | 5 | 1 | 6 |
| Mean (SD) | 3,891,589 (2,747,456) | 6,541,314 (N/A) | 4,333,210 (2,684,956) | ||
| Week 36 | |||||
| n | 1 | 1 | 6 | 6 | 12 |
| Mean (SD) | 1,527,778 (N/A) | 1,551,438 (N/A) | 11,804,027 (14,432,979) | 6,166,444 (3,110,935) | 7,921,574 (9,924,045) |
| Week 40 | |||||
| n | 0 | 0 | 6 | 6 | 12 |
| Mean (SD) | 6,509,428 (8,951,513) | 11,073,446 (12,370,669) | 8,791,437 (10,567,127) | ||
| Week 44 | |||||
| n | 1 | 1 | 6 | 7 | 15 |
| Mean (SD) | 952,744 (N/A) | 4,817,708 (N/A) | 7,146,169 (6,869,530) | 5,953,550 (7,729,996) | 6,021,488 (6,704,115) |
| Week 48 | |||||
| n | 0 | 0 | 6 | 7 | 13 |
| Mean (SD) | 4,602,122 (2,228,659) | 4,555,184 (3,349,900) | 4,576,847 (2,771,472) | ||
| Week 52 | |||||
| n | 1 | 1 | 6 | 7 | 15 |
| Mean (SD) | 1,000,000 (N/A) | 4,062,500 (N/A) | 4,671,039 (2,151,715) | 3,346,982 (3,603,551) | 3,767,841 (2,865,374) |
| Week 65 | |||||
| n | 1 | 1 | 3 | 7 | 12 |
| Mean (SD) | 741,071 (N/A) | 3,717,672 (N/A) | 5,805,667 (1,361,377) | 1,888,120 (1,837,096) | 2,924,382 (2,369,785) |
| Week 78 | |||||
| n | 1 | 1 | 3 | 7 | 12 |
| Mean (SD) | 937,500 (N/A) | 1,391,267 (N/A) | 5,342,412 (5,628,484) | 2,195,763 (2,452,378) | 2,810,529 (3,395,773) |
| Week 91 | |||||
| n | 1 | 1 | 0 | 7 | 9 |
| Mean (SD) | 543,981 (N/A) | 1,284,722 (N/A) | N/A (N/A) | 5,132,010 (2,935,971) | 4,194,752 (3,155,652) |
| Week 104 | |||||
| n | 1 | 1 | 3 | 7 | 12 |
| Mean (SD) | 275,000 (N/A) | 16,885,081 (N/A) | 4,476,970 (4,337,276) | 3,586,297 (2,329,754) | 4,641,256 (4,738,982) |
| Week 117 | |||||
| n | 1 | 1 | 3 | 3 | 8 |
| Mean (SD) | 1,474,781 (N/A) | 6,496,711 (N/A) | 7,071,818 (8,917,458) | 2,916,667 (633,621) | 4,742,118 (5,343,601) |
| Week 130 | |||||
| n | 0 | 1 | 0 | 4 | 5 |
| Mean (SD) | N/A (N/A) | 6,937,500 (N/A) | N/A (N/A) | 10,960,161 (16,318,238) | 10,155,628 (14,246,053) |
| Week 156 | |||||
| n | 1 | 0 | 0 | 7 | 8 |
| Mean (SD) | 2,952,450 (N/A) | N/A (N/A) | N/A (N/A) | 8,506,411 (11,716,651) | 7,812,166 (11,023,806) |
The AAV5 TAb response was not associated with efficacy (FVIII activity) or safety (ALT) measures of BMN 270. There was no association between anti-AAV5 TAb titer at week 8, nor the maximal anti-AAV5 TAb titer and peak ALT measures (Figure 2A). Similarly, there was no association between anti-AAV5 TAb titer at week 8, nor maximal anti-AAV5 TAb titer and median FVIII activity measured at weeks 23–36 (Figure 2B).
Figure 2.

Correlation Analysis of AAV5 TAb Titer with Plasma ALT and FVIII Activity
(A and B) The maximum observed AAV5 TAb titers for each patient (upper plots) and the AAV5 TAb titer at week 8 (lower plots) show no association with the maximum observed plasma ALT value matched by patient (A), or with the median FVIII activity measures from weeks 23 to 26 after dose administration (B). Data in (B) are from cohort 3 and cohort 4 patients only. Mean FVIII activity from weeks 23–26 was not calculable for patients in cohorts 1 and 2.
Cross-Reactive Antibodies against Divergent AAV Capsids
Exploratory plasma samples from patients within the BMN 270-201 dose escalation study were assessed for antibodies against AAV2, AAV5, AAV6, AAV8, and rh10 serotype at multiple time points from screening through a maximum of 134 weeks after administration (Table 3). Despite being negative for AAV5-reactive antibodies, two patients were positive for pre-existing antibodies specific for AAV6, with one of these also being positive for antibodies to AAV2. Following administration of BMN 270, AAV5 antibody titers rose 4–5 logs by week 8 and peaked at week 40 with a mean titer of 8.8 × 106 and remained durable during 1.5–2.5 years of follow-up (Figure 3). Cross-reactive antibodies to divergent capsids developed by week 8 in all patients. These titers were generally several orders of magnitude lower than corresponding anti-AAV5 TAb titers, and after an early peak from weeks 8 to 16, they reached a plateau that remained relatively stable, or in some cases declined slightly, for the duration of the follow-up. By 1.5–2.5 years following administration, cross-reactive antibody titers were generally in the 10E2–10E4 range, which is similar to AAV5 TAb titer levels we detect in the context of natural immunity in subjects who screen positive for pre-existing antibody to AAV5.14 One notable exception occurred with a single cohort 4 patient (patient 15) with pre-existing reactivity to AAV2 and AAV6. This patient’s AAV2 and AAV6 titers remained in the 10E5 range at week 82 (titers of 322,524 and 225,573, respectively).
Table 3.
Mean AAV5 and Cross-Reactive Antibody Titers
| Serotype (LoD) | 6E12 (n = 1) |
2E13 (n = 1) |
4E13 (n = 4)a |
6E13 (n = 7) |
||||
|---|---|---|---|---|---|---|---|---|
| Week 8 | End | Week 8 | End | Week 8 | End | Week 8 | End | |
| AAV5 (33 ng/mL) | 10,504 | 87,235 | 357,751 | 8,596,124 | 748,831 | 2,896,108 | 421,222 | 3,413,365 |
| AAV2 (60 ng/mL) | 30 | <MRD | 240 | 382 | 153,226a | 53,930a | 7,011 | 360 |
| AAV6 (15 ng/mL) | 455 | 48 | 951 | 440 | 174,235a | 38,119a | 9,314 | 1,148 |
| AAV8 (70 ng/mL) | 177 | 40 | 428 | 176 | 26,802 | 1,433 | 6,275 | 440 |
| AAVrh10 (42 ng/mL) | 55 | <MRD | 377 | 287 | 17,600 | 1,408 | 6,283 | 394 |
LoD, limit of detection; End, last time point taken for cross-reactive antibody assessment ranges from week 76 to week 134; MRD, minimal required dilution.
One 4E13 patient with pre-existing antibodies to AAV2 and AAV6 developed comparatively higher antibody titers following dose administration and contributed to the elevated mean AAV2 and AAV6 titers at week 8 and the final time point.
Figure 3.

Cross-Reactive Antibody Response against Divergent Capsids
(A and B) Plots of AAV2, AAV5, AAV6, AAV8, and AAVrh10 specific TAb titers over time are displayed for individual subjects in cohort 1 (A) cohort 2 (B). (C and D) Grouped individual plots are shown for n = 7 subjects in cohort 3 (C) and n = 6 subjects in cohort 4 (D). The final available sample time point after infusion ranged from week 76 to week 134.
Cellular Immune Response to AAV5 and FVIII Are Detected in PBMCs
Cellular immune responses specific for AAV5 capsid protein and FVIII protein were assessed using a fluorogenic IFN-γ immunospot (FluoroSpot) assay for IFN-γ and tumor necrosis factor (TNF)-α secretion using cryopreserved PBMCs. Cryopreserved PBMC samples were collected at baseline and at several time points after administration that varied by cohort but were generally every 2–4 weeks during the first 6 months, 4–8 weeks through the rest of year 1, and every 3 months thereafter. FluoroSpot data were available for all patients through a minimum of 52 weeks to a maximum of 134 weeks after BMN 270 administration, and data are summarized as heatmaps indicating the magnitude of positive responses reported as spot-forming units (SFU)/106 PBMCs (Figure 4). All 15 patients tested negative for the AAV5-specific IFN-γ responses at baseline, and 4 of 15 patients tested positive (≥50 SFU/106 PBMCs) for AAV5-specific IFN-γ responses at a single time point post-administration and negative at all subsequent time points tested (Figure 4A). For FVIII-specific IFN-γ responses, 5 of 15 patients tested positive overall, with 3 of 5 testing positive at only a single time point, 1 at baseline only, and reverting to negative at the next available time point (Figure 4B). Two of five patients were positive at two or three time points, with one reverting back to negative by the last available time point. For the other, additional samples were not available.
Figure 4.

Heatmap of Cellular Immune Responses
Heatmaps show instances of IFN-γ- and TNF-α-positive responses for individual patients at baseline and each post-dose administration time point (weeks 4–52 and by quarter [Q1–Q4] as indicated for years 2 [Y2] and 3 [Y3]). Coloration signifies magnitude of response by SFU/106 PBMCs and the highest SFU value, displayed for positive responses, was used when positive responses were detected to more than one peptide pool. Results too numerous to count were interpolated to a value of 5,000 SFU/106 PBMCs. (A) Results of the IFN-γ FluoroSpot assay following stimulation with AAV5-derived peptide pools and (B) FVIII-derived peptide pools. (C) Results of the TNF-α FluoroSpot assay following stimulation with AAV5-derived peptide pools and (D) FVIII- derived peptide pools. Cells in white (blank) indicate time points where there were too few cells to test. Tested samples with a result <50 SFU/106 PBMCs are left blank. SFU counts are shown for positive responses ≥50.
Cellular immune responses were also measured through secretion of TNF-α, although data from assay characterization studies complicated the analysis as described in more detail below. Positive TNF-α responses observed in PBMCs stimulated with AAV5 capsid peptides were sparse and detected at only single time points, ranging from week 8 to year 3 (Y3Q1) in 8 of 15 patients (Figure 4C). An additional 2 of 15 patients were TNF-α positive at two or three separate time points.
Following FVIII peptide stimulation, TNF-α secretion was more frequent, with all patients having a positive response at multiple time points, including 5 of 15 patients at baseline prior to dose administration (Figure 4D). These responses did not demonstrate any clear pattern or relationship to BMN 270 administration, and interpretation of the data following FVIII peptide stimulation was confounded by a high rate of positivity in healthy donors not exposed to gene therapy (Figure S3). Responses in healthy donors demonstrated a similar pattern and magnitude of response as baseline and post-dose administration measures. This pattern of TNF-α secretion is consistent with previous findings on T cell responses to FVIII in healthy donors and HA patients,21 and it brings into question whether TNF-α responses to FVIII observed here were associated with administration of BMN 270. Overall, no clear relationship between vector infusion and T cell responses to AAV5 or FVIII was observed. However, cellular immune response measures after dose administration may have been impacted by the use of corticosteroids during the first several weeks after gene transfer in most of the subjects enrolled. The temporal relationships of AAV5 and FVIII-specific cellular immune responses with ALT, FVIII activity measures, and corticosteroid use are shown in Figure S1 (IFN-γ) and Figure S2 (TNF-α).
Positive Cellular Immune Responses Were Not Associated with ALT Rises or FVIII Expression
FVIII activity measures as well as measures of plasma ALT following BMN 270 administration were reported separately.19,20 Although there was some variability in efficacy measures by dose cohort, differences in FVIII activity and ALT are not clearly associated with positive cellular immune responses. To further assess the impact of cellular immune responses with safety and efficacy, we performed a pairwise comparison of IFN-γ and TNF-α positive and negative responses with the most proximal corresponding ALT values and FVIII activity measurements by time point. This comparison was used to determine whether positive responses were associated with an increase in ALT or change in FVIII activity measures. There was no difference in mean ALT or mean FVIII activity at time points corresponding to positive IFN-γ responses versus negative responses (Figure 5A). Similarly, TNF-α-positive responses were not associated with mean ALT or mean FVIII activity measures (Figure 5B). Overall in this analysis, no association was apparent between cellular immune responses and changes in ALT or FVIII activity measures.
Figure 5.

Distribution of FVIII Activity and ALT Levels by FluoroSpot
An IFN-γ and TNF-α FluoroSpot assay was used to measure cell-mediated immunity (CMI) specific for AAV5 or FVIII peptides. (A) Pairwise comparison of instances of IFN-γ-positive and IFN-γ-negative time points with corresponding measures of ALT or FVIII activity. Boxplots show mean of pairwise matched ALT (upper plots) or FVIII activity measures (lower plots) following stimulation with AAV5--derived peptides (left plots) or FVIII derived peptides (right plots). (B) Similar pairwise comparison of instances of TNF-α-positive and TNF-α-negative time points with corresponding mean measures of ALT or FVIII activity. Box plots show the interquartile range with whiskers indicating minimum and maximum values. Line indicates median, diamond indicates mean.
Discussion
BMN 270 is an AAV5-based gene therapy that encodes a truncated but fully functional form of hFVIII (FVIII SQ) under the control of a hybrid human liver-specific promoter restricting expression to liver hepatocytes.9 BMN 270 is administered as a single intravenous dose and is designed to achieve stable, durable expression of active FVIII in the plasma, synthesized from vector-transduced liver tissue. Administration of BMN 270 in patients with HA was safe and effective and demonstrated a clinically relevant benefit as measured by a substantial reduction in annualized bleeding rate and complete cessation of prophylactic factor VIII use in all participants who had received 4E13–6E13 vg/kg of gene therapy.19,20 The most common adverse event was a mild, transient elevation in ALT, which was mitigated through the use of corticosteroids. There was no evidence of FVIII inhibitors through 3 years of follow-up.
In preclinical studies, following intravenous administration, BMN 270 distributes to several peripheral tissues with long-term detection of vector genomes primarily in the liver.9,10 At more than 4.9 kb, the single-strand vector genome is oversized for incorporation into a single AAV particle such that separately packaged plus and minus strands are required within the same transduced cell to achieve stable, full-length transgenes with evidence of inverted terminal repeat (ITR) fused, episome-like structures.19 Formation of stable transgene structures represents the culmination of the transduction process that begins with receptor-mediated uptake into the endosome, and proceeds through endosomal escape, trafficking to the nuclear membrane, capsid uncoating, and delivery of the DNA to the nucleus.22 Each of these transduction steps can be negatively impacted by pre-existing immunity to AAV.22
Pre-existing antibodies and other inhibitors against the viral vector capsid may interfere with receptor binding and/or vector uptake and lead to reduced transduction. Several previous studies have determined that even comparatively modest titers of neutralizing antibodies specific for AAV capsid are capable of neutralizing transduction and limiting the efficacy of vector-mediated gene therapies in humans11, 12, 13 and non-human primate species.23 To avoid any potential safety or efficacy-related consequences from AAV5-specific immunogenicity, a screening strategy for detection of pre-existing AAV5 antibodies was used to select patients for enrollment. This screening strategy assessed subjects for pre-existing immunity to AAV5 using both a cell-based in vitro neutralization (TI) assay and an ECLA-based method for semi-quantitation of total AAV5-binding antibody titers. Patients testing positive in either assay were excluded from enrollment in BMN 270-201.
While cell-based NAb assays offer similar or even higher sensitivity over direct measures of specific binding antibody, such as ligand-binding assay (LBA) or ELISA formats, emerging evidence suggests that the detection of ultra-low levels of AAV5 antibodies in cell-based NAb assays may limit their predictive value for in vivo gene therapy administration. Two recent reports describing use of a highly sensitive luciferase reporter-based NAb assay evaluated the pre-treatment plasma of hemophilia B patients receiving an AAV5-FIX gene therapy. These studies showed no relationship between the presence of pre-treatment anti-AAV5 NAb and the therapeutic efficacy.24,25 In the Majowicz et al.24 study, depletion of immunoglobulin G (IgG) did reduce these NAb titers, but AAV5-specific IgG could only be directly detected using an ELISA method in pre-treatment serum samples from patients with higher NAb titers. In the report by Von Drygalski et al.,25 all three participants had NAbs to AAV5 at screening (titers 48, 44, and 25) detected by using this sensitive luciferase-based assay. However, based on ELISA data, no participants had anti-AAV5 IgG antibodies at screening or baseline. These reports note that patients from both studies with pre-existing NAb titers had sufficient and durable FIX expression following gene therapy administration. Similarly, non-clinical data from cynomolgus monkeys treated with an AAV5-FVIII gene therapy construct demonstrated diminished expression of FVIII only in animals with detectable AAV5-specific TAb.23 Animals that tested positive in a cell-based NAb (TI) assay, but without detectable TAb, all had normal levels of FVIII expression. Similar to the clinical data described above, there was a trend toward animals with higher TI titers having detectable TAb. Overall, these data suggest that the improved sensitivity of the latest generation of cell-based NAb assays may detect physiologically irrelevant ultra-low levels of antibodies, possibly in conjunction with other non-antibody-based inhibitory factors or plasma matrix effects, which decrease transduction efficiency in vitro without exhibiting any in vivo relevance for clinical patients. Therefore, these data support the use of ELISA or ECL-based detection of AAV-specific antibodies as the most appropriate screening method to identify subjects with pre-existing immunity who may receive less benefit from AAV gene therapy.
Immunogenicity against the AAV5 capsid was an expected consequence of BMN 270 dose administration, as has been observed with other AAV-based gene therapies in previous clinical trials.11, 12, 13,26 High-titer anti-AAV5 TAb responses were detected in all patients by week 8 after dose administration, demonstrating the robust humoral antigenicity of AAV capsids in humans. The generation and maturation of this antibody response could, in theory, exacerbate any ongoing capsid-specific cytotoxicity through antibody-dependent cellular cytotoxicity, complement fixation, or other mechanisms. To evaluate these potential safety and efficacy concerns, a correlation analysis was performed of antibody titers at week 8 and the maximal AAV5 TAb titer for each patient, each of which could be considered a measure of the robustness of the response across individuals, with either the maximal ALT value or median FVIII activity measures. The post-dose AAV5 TAb response was not associated with either of these safety or efficacy variables, and it may be that as the antibody response matures, insufficient quantities of capsid material remain associated with transduced cells to affect a response. The antibody response elicited against AAV5 showed detectable cross-reactivity to other AAV serotypes, including AAV2, AAV6, AAV8, and AAVrh10. Antibodies that cross-react with other capsid serotypes may be of concern for patients who require administration of other gene therapies that may use those serotypes, or closely related serotypes. Over time, the cross-reactive portion of the antibody response appeared to reach a plateau in titer in most patients that was generally several orders of magnitude lower than the AAV5-specific titer that peaked around 40 weeks following dose administration and then remained relatively stable. Although different assay methodologies are used, cross-reactive titers observed at later time points in this study are similar to cross-reactive neutralizing antibody titers recently reported in a 15-year follow-up study of systemic AAV2-FIX administration in hemophilia B subjects.27 Cross-reactive neutralizing antibody titers have been reported in several clinical and non-clinical studies and likely preclude repeat dose administration with AAV capsids for which cross-reactivity is detected.28, 29, 30, 31 It remains to be determined whether there is an AAV5-specific or cross-reactive titer level below which gene therapy dose administration or repeat administration would be successful.
Out of an abundance of caution, patients with a previous history of FVIII inhibitors were excluded from enrollment, as the safety of continuous, endogenous FVIII production in the presence of FVIII neutralizing antibody has not been established. More than 99% of previously unexposed patients (PUPs) who go on to develop FVIII inhibitors do so by 75–150 exposure days,15 and the risk of FVIII inhibitor development beyond 150 exposure days is estimated to be in the range of 2–5 per 1,000 person years.32,33 For this reason, only patients who received more than 150 exposure days of FVIII replacement therapy without development of a FVIII inhibitor response were enrolled. Following dose administration, patients were tested frequently for the development of FVIII inhibitors, as these could potentially lead to diminished efficacy of BMN 270 or the requirement for on-demand FVIII replacement therapy, bypassing agents or more novel, recently approved bispecific antibody therapies that circumvent the need for FVIII altogether.34 All subjects in all dose cohorts tested negative for FVIII inhibitors at all time points assessed. Separately, in an assay used to detect FVIII-specific TAbs, a single subject tested positive at a single time point but remained negative at all other time points. This patient did not have a worse safety or efficacy profile, with FVIII levels consistent with other responders in the trial. Low levels of FVIII-specific immune responses are not unexpected. Previous studies have detected low titers of FVIII-binding antibodies in up to 19% of healthy donors and in a substantial proportion of HA patients; however, these antibodies do not mature into FVIII inhibitors.35 Often times, these responses are of low affinity and have no demonstrable clinical consequences.36
The most common adverse event following administration of BMN 270 was a transient increase in ALT, which was managed with either on-demand or prophylactic administration of corticosteroids. A widely held hypothesis based on previously published gene therapy clinical trial data with FIX constructs suggests that a capsid-specific cellular immune response may be stimulated to target catabolized capsid antigens associated with transduced hepatocytes. Consequently, transduced hepatocytes could become targets for cellular cytotoxicity, resulting in a release of ALT and loss of transgene expression.11,12,22 Corticosteroid treatment in some of these previous studies promoted resolution of ALT increases and was associated with a loss of detectable cellular immunity and stabilization of FIX expression. In some other studies, loss of FIX expression was observed despite the administration of corticosteroids (reviewed in Verdera et al.37). Additionally, other studies have demonstrated that TNF-α appears to be the primary cytokine secreted by CD8+ T cells in response to the AAV capsid, and these cells could readily be detected in AAV2 seropositive individuals.38
In the current study, sporadic, low-level AAV5-specific cellular immune responses were detected in a minority of subjects, often at single time points, and without clear corresponding impact to ALT or FVIII activity. These responses appeared to be self-limiting and of little or no clinical significance. The administration of corticosteroids, which is commonly used to blunt lymphocyte reactions, is a confounding factor in the interpretation of the results presented herein, and may explain why, in this study, cellular immune responses detected through stimulated IFN-γ secretion were detected less often than anticipated based on the previously mentioned studies. The low incidence of detected cellular immune responses may also be in part attributable to the large number of instances in which there were too few cells to perform the assay or the cryopreserved PBMCs were of poor viability upon thawing and were not tested, another possible consequence of the administration of corticosteroids.39 Furthermore, the ability of different AAV capsids to elicit robust cellular immune responses may be variable and particularly low for AAV5. Two recent reports from clinical trials using AAV5 for the treatment of acute intermittent porphyria40 and hemophilia B41 similarly described very limited or no AAV5-specific cellular immunity detectable by an IFN-γ ELISpot assay. Similar to AAV5-specific IFN-γ responses, FVIII-specific T cell reactivity was detected only sporadically. One interesting observation in this study is the production of TNF-α detected in several trial participants in response to FVIII peptides at baseline and after vector administration, and regardless of corticosteroid administration. Both T cell activation and antibody responses to FVIII in healthy donors and HA subjects have been previously documented.21,35 This could explain the current observations and highlight the complexities of the immune system homeostasis with regard to endogenous proteins.
Importantly, experience with liver-directed gene therapy suggests that other inflammatory factors that were not measured may play a role in the observed ALT elevations, such as innate immune factors or liver-specific cellular responses (reviewed in Verdera et al.37). Ongoing inflammatory events and cellular cytotoxicity occurring in the liver or other parenchymal tissues may not always be accurately reflected by PBMC-derived T cells present in the periphery. In addition, innate inflammation factors that were not measured in these studies may be at play in the liver. At present, there is not a full mechanistic understanding of the causes of liver inflammation following AAV-mediated gene therapies, and this remains an active area of investigation. Furthermore, by requiring a minimum number of exposure days to FVIII replacement therapy without evidence of FVIII inhibitor development, this study effectively enrolled only patients with verified immunological tolerance toward FVIII. Future studies in larger patient populations and in patients with pre-existing FVIII inhibitors, or who have previously had FVIII inhibitors, will help to define the frequency of immune response against the transgene product in these higher risk subjects.
In summary, BMN 270 was well tolerated in most subjects with the main safety findings related to asymptomatic ALT elevation, which in some instances were seen to improve or resolve with concurrent corticosteroid treatment. The predominant immune response detected following AAV5-hFVIII-SQ administration was largely limited to the development of antibodies against the AAV5 vector capsid. No FVIII inhibitor responses were observed within 3 years of follow-up after AAV5-hFVIII-SQ administration. Although sporadic positive AAV5 and FVIII-specific cellular immune responses were detected in a limited number of BMN 270 patients, no temporal correlations with safety and efficacy measurements were detected.
Materials and Methods
Study Design
This study was designed by the sponsor, BioMarin Pharmaceutical. The study was conducted in accordance with good clinical practice guidelines and the principles of the Declaration of Helsinki. The study protocol was approved by relevant ethics boards. Written informed consent was provided by each participant. BioMarin Pharmaceutical oversaw the collection and analysis of data.
Measurement of Anti-AAV5 TAbs in Human Plasma by ECLA
AAV5 TAbs were measured in human plasma using a validated bridging ECLA on the Meso Scale Discovery (MSD) platform. Unlabeled BMN 270 drug product was used as a capture reagent and passively coated onto the wells of an MSD plate. After incubation at room temperature with shaking and subsequent washing, the wells were blocked with assay diluent (AD). After blocking followed by washing, quality control (QC) samples and test samples were added to the plate at the MRD of 1:20 and incubated with shaking at room temperature. Anti-AAV5 antibodies in the plasma sample will specifically bind to BMN 270 coated onto the bottom of the plate. After another wash step, anti-AAV5 antibodies were detected by adding ruthenylated BMN 270 drug product diluted in AD followed by incubation at room temperature. After a final wash, MSD read buffer T, containing the substrate tripropylamine (TPA), was added to initiate a chemical reaction with ruthenium. The plate was then electrically stimulated, resulting in an ECL signal that was detected by the MSD instrument. A confirmatory assay step was conducted by adding excess unlabeled drug to the test samples in order to compete with capture and detection reagents and to deplete the specific signal generated by anti-AAV5 antibodies. The assay reports semiquantitative titer values as a relative measure of anti-AAV5 antibody levels in positive samples. Screening, confirmation and titer cut-points, sensitivity, and selectivity were assessed in normal plasma.
Measurement of Anti-AAV2, AAV5, AAV6, AAV8, and rh10 TAbs in Human Plasma by ECLA
Analogous anti-AAV TAb assays capable of detecting antibodies against AAV2, AAV5, AAV6, AAV8, and rh10 serotypes were validated (ARUP, USA). Antibodies specific for each capsid were measured using the same bridging ECLA on the MSD platform as described for AAV5. Limits of detection (LoD) for each AAV serotype were determined utilizing affinity-purified polyclonal antibody derived against each capsid.
Measurement of AAV5 TI in Human Plasma by Cell-Based Assay
AAV5 transduction inhibitors in plasma were measured using a validated cell-based TI assay. HEK293T/17 cells were seeded at 40,000 cells per well in DMEM/10% fetal bovine serum (FBS) medium in white clear-bottom 96-well plates and grown overnight. On the next day, QC and test samples were mixed 1:1 with AAV5-cytomegalovirus (CMV)-luciferase reporter vector in serum-free DMEM supplemented with 1% BSA and incubated for 30 min at room temperature. Following this incubation, samples were added to the cells in duplicate wells at a multiplicity of infection (MOI) of 25,000 vg/cell. After a 1-h incubation at 37°C, a final concentration of 20 μM etoposide solution in DMEM/10% FBS was added to induce DNA repair synthesis. After 2 days, cell culture medium was removed and the luciferase substrate Steady-Glo (Promega) was added for 10 min at ambient temperature with shaking. Luminescence was measured using a 500-ms integration time on a Victor or Victor X microplate reader. Relative luminescence units of QC and test samples were normalized to that of a cut-point control (CC) sample (negative plate control), which consisted of pooled normal human plasma without detectable TI, and converted to percent transduction values relative to the plate-specific CC. The screening/titer cut-point for the percent transduction at or below which a test sample is considered positive was assessed in normal plasma. The assay reports semiquantitative TI titer values as a relative measure of AAV5 transduction inhibitor levels in positive samples.
Measurement of Anti-FVIII TAbs in Human Plasma by ECLA
TAbs against full-length FVIII were measured in plasma by a validated bridging ECLA on the MSD platform. QC and test plasma samples were heated at 55°C–60°C for 30 ± 5 min to remove interference from endogenous FVIII and then diluted to an MRD of 1:40 into AD. Biotinylated and ruthenylated recombinant full-length human FVIII (each prepared from Kogenate) were used as capture and detection reagents, respectively, and added to QC and test samples to bind to anti-FVIII antibodies. The samples were then added to a blocked streptavidin (SA)-coated MSD plate. The biotin enables the complex to bind to the SA-coated MSD plate, and the bridging action of the anti-FVIII antibodies allows the ruthenylated FVIII to come into close proximity to the electrode base of the plate. After a wash, MSD read buffer T, containing the substrate TPA, was added to initiate a chemical reaction with ruthenium. The plate was then electrically stimulated, resulting in an ECL signal that was detected by the MSD instrument. A confirmatory assay step was conducted by adding excess unlabeled FVIII (Kogenate) to the test samples in order to compete with capture and detection reagents and to deplete the specific signal generated by anti-FVIII antibodies. The assay reports semiquantitative titer values as a relative measure of anti-FVIII antibody levels in positive samples. The screening, confirmation, and titer cut-points were assessed in normal plasma.
Measurement of FVIII Inhibitors in Human Plasma by Nijmegen-Bethesda Assay
FVIII inhibitors were measured in a validated Bethesda assay with Nijmegen modification. The Bethesda assay is a modification of the activated partial thromboplastin time (APTT) assay in which a test sample is pre-incubated with FVIII in normal plasma and then calibrated against a normalized standard curve in a one-stage clot FVIII activity assay. The Nijmegen modification of the Bethesda assay uses buffered plasma, as the test system and was developed to reduce the incidence of false low-positive FVIII inhibitor titers. First, QC and test samples were heated at 58°C ± 2°C for 90 min in order to inactivate any endogenous FVIII activity in the test sample prior to conducting the Nijmegen-Bethesda assay. Following heat inactivation, the samples were centrifuged to remove any debris caused by the heating process. Dilutions of heat-inactivated patient plasma were prepared in buffered congenital FVIII-deficient plasma. The dilutions were then mixed with an equal volume of test base (i.e., imidazole-buffered normal pooled plasma containing FVIII at approximately 1.0 IU/mL activity). A C-tube was prepared by mixing equal volumes of buffered heat-inactivated congenital FVIII-deficient plasma and the test base. The mixtures were incubated at 37°C for 2 h and then cooled in an ice bath (2°C–8°C) for 10 min to stop further FVIII inhibition from occurring. A FVIII one-stage clot activity assay was then performed on both the mixed test samples and C-tube at the base dilution. The residual FVIII activity was determined by calculating the ratio of the FVIII activity in the test-sample mixture to the FVIII activity of the C-tube mixture. The percentage of residual FVIII activity in a sample dilution must fall between 25% and 75% to be reported. When the residual activity result was <25%, further sample dilutions were tested until the residual activities were between 25% and 75%. When the residual activity was >75% for the neat test sample, the sample was considered negative for the presence of FVIII inhibitors. If two sample dilutions resulted in residual activities between 25% and 75%, the dilution closest to 50% was used. If two sample dilutions had residual activities equidistant from 50%, the inhibitor result for the less dilute sample was reported. One BU is defined as the amount of inhibitor in a plasma test sample that will neutralize 50% of 1 U of FVIII in normal plasma after a 2-h incubation at 37°C. The normal range for a negative FVIII inhibitor result was <0.6 BU; any test sample result with ≥0.6 BU was reported as positive.
Detection of Cellular Immune Responses against AAV5 and FVIII-SQ in Human PBMCs by FluoroSpot Assay
Cellular immune responses against the AAV5 capsid and the expressed transgene product FVIII-SQ were detected in human PBMCs using a characterized FluoroSpot assay. PBMCs were collected under serum-free conditions and cryopreserved in serum-free freezing medium. PBMCs were tested using the human IFN-γ FluoroSpot kit (Cellular Technology) at a cell density of 200,000 cells/well and stimulated with 1 μg/mL AAV5 or FVIII-SQ peptide mixtures, phytohemagglutinin (PHA) (positive control), or incubated in medium only (negative control). Each AAV5 or FVIII-SQ peptide mixture consisted of approximately 70 overlapping 15-mer peptides that were synthesized based on the sequence of AAV5 capsid protein VP1 or FVIII-SQ. When, for example, antigen-specific T cells encounter their cognate peptides in the context of major histocompatibility complex (MHC) molecules on antigen-presenting cells, the T cells will secrete the cytokine IFN-γ. As the cytokine is secreted, it is immobilized by the anti-IFN-γ capture antibody coated onto the polyvinylidene fluoride (PVDF) membrane at the bottom of an ELISpot plate. After the cells are removed, the ELISpot plate was washed and fluorescently labeled (fluorescein isothiocyanate [FITC]) anti-IFN-γ detection antibody was added. The PVDF membrane was washed with water and left to dry, protected from light and moisture. Any developed spots were enumerated using an automated spot counter (ImmunoSpot CTL S6 FluoroSpot line; ImmunoCapture software, Cellular Technology). Primary results were expressed as SFU per well, and final results were reported as SFU per 1 million PBMCs by correcting the primary result based on the number of cells plated per well. Results from AAV5 or FVIII-SQ peptide-stimulated samples that fell below a pre-specified detection threshold of 50 SFU per 1 million PBMCs were reported as negative. Results from AAV5 or FVIII-SQ peptide-stimulated PBMC samples that fell at or above the detection threshold were reported as positive, unless the negative control of the sample also generated spot numbers that were equal to or greater than the detection threshold. In this case, a pre-specified confirmatory cut-point of ≥3 was applied to the response ratio (defined as the spot number of a PBMC sample after peptide stimulation divided by the spot number of the same PBMC sample under negative control conditions) to confirm a positive PBMC response to stimulation with AAV5 or FVIII-SQ peptide mixtures. Results too numerous to count were interpolated to a value of 5,000 SFU/106 PBMCs. Heatmaps of FluoroSpot responses show the highest SFU count by peptide pool stimulation (two AAV5 and four FVIII pools) when more than one pool tested positive, and were generated using GraphPad Prism version 8.4.2.
Acknowledgments
The authors would like to thank the study participants and their families, the study investigators, and study-site personnel. Funding for this work was provided by BioMarin Pharmaceutical Inc. The authors acknowledge the technical assistance of Krystal Sandza and Theresa Seitel (BioMarin Pharmaceutical Inc.), Stefan Tiefenbacher and Mary Robinson (Colorado Coagulation, A LabCorp Company), the editorial assistance of Gillian Clague, Kendra Bolt, and Lyn Garret (BioMarin Pharmaceutical Inc.), the graphic design assistance of Terry Manspeaker (Show Your Science, LLC), and Glenn F. Pierce, consultant, for helpful review of the draft manuscript. Development and implementation of cellular immunogenicity assays was supported by the European Union’s Research Council Consolidator Grant under Grant agreement no. 617432 (MoMAAV to F.M.).
Author Contributions
B.R.L., M.B.H., K.L., S.L.Z., C.V., G.M.H., and B.S. developed the immunogenicity risk assessment and devised the assay strategy. B.R.L. drafted the manuscript. F.M., K.K., R.H., and P.V. developed and characterized the FluoroSpot assay for measuring cellular immune responses, provided expertise and feedback, and contributed to sections of the manuscript. N.M. and W.Y.W. monitored the clinical study and provided expertise on patient safety and efficacy. M.L. provided statistical analysis and data analysis expertise.
Declaration of Interests
B.R.L., N.M., G.H., W.Y.W., K.L., M.L., M.B.H., S.L.Z., C.V., and B.S. are employees of BioMarin Pharmaceutical Inc. F.M. and K.K. are currently employees of Spark Therapeutics. The remaining authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.008.
Supplemental Information
References
- 1.Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–360. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- 2.High K.A., Roncarolo M.G. Gene therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 3.Iorio A., Stonebraker J.S., Chambost H., Makris M., Coffin D., Herr C., Germini F., Data and Demographics Committee of the World Federation of Hemophilia Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann. Intern. Med. 2019;171:540–546. doi: 10.7326/M19-1208. [DOI] [PubMed] [Google Scholar]
- 4.McIntosh J., Lenting P.J., Rosales C., Lee D., Rabbanian S., Raj D., Patel N., Tuddenham E.G., Christophe O.D., McVey J.H. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood. 2013;121:3335–3344. doi: 10.1182/blood-2012-10-462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiorini J.A., Kim F., Yang L., Kotin R.M. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999;73:1309–1319. doi: 10.1128/jvi.73.2.1309-1319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotin R.M., Snyder R.O. Manufacturing clinical grade recombinant adeno-associated virus using invertebrate cell lines. Hum. Gene Ther. 2017;28:350–360. doi: 10.1089/hum.2017.042. [DOI] [PubMed] [Google Scholar]
- 7.Fischer K., Collins P., Björkman S., Blanchette V., Oh M., Fritsch S., Schroth P., Spotts G., Ewenstein B. Trends in bleeding patterns during prophylaxis for severe haemophilia: observations from a series of prospective clinical trials. Haemophilia. 2011;17:433–438. doi: 10.1111/j.1365-2516.2010.02450.x. [DOI] [PubMed] [Google Scholar]
- 8.Berntorp E., Spotts G., Patrone L., Ewenstein B.M. Advancing personalized care in hemophilia A: ten years’ experience with an advanced category antihemophilic factor prepared using a plasma/albumin-free method. Biologics. 2014;8:115–127. doi: 10.2147/BTT.S53456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bunting S., Zhang L., Xie L., Bullens S., Mahimkar R., Fong S., Sandza K., Harmon D., Yates B., Handyside B. Gene therapy with BMN 270 results in therapeutic levels of FVIII in mice and primates and normalization of bleeding in hemophilic mice. Mol. Ther. 2018;26:496–509. doi: 10.1016/j.ymthe.2017.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L., Handyside B., Murphy R., Sihn C.-R., Xie L., Vitelli C., Harmon D., Sisó S., Liu S., Bullens S. Prednisolone does not regulate factor VIII expression in mice receiving AAV5-hFVIII-SQ: valoctocogene roxaparvovec. Mol. Ther. Methods Clin. Dev. 2019;17:13–20. doi: 10.1016/j.omtm.2019.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B. Successful transduction of liver in hemophilia by AAV-factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 12.Nathwani A.C., Tuddenham E.G.D., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falese L., Sandza K., Yates B., Triffault S., Gangar S., Long B., Tsuruda L., Carter B., Vettermann C., Zoog S.J., Fong S. Strategy to detect pre-existing immunity to AAV gene therapy. Gene Ther. 2017;24:768–778. doi: 10.1038/gt.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Berg H.M., Fischer K., Carcao M., Chambost H., Kenet G., Kurnik K., Königs C., Male C., Santagostino E., Ljung R., PedNet Study Group Timing of inhibitor development in more than 1000 previously untreated patients with severe hemophilia A. Blood. 2019;134:317–320. doi: 10.1182/blood.2019000658. [DOI] [PubMed] [Google Scholar]
- 16.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E.J., Ragni M.V., Manno C.S., Sommer J., Jiang H. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 17.Pien G.C., Basner-Tschakarjan E., Hui D.J., Mentlik A.N., Finn J.D., Hasbrouck N.C., Zhou S., Murphy S.L., Maus M.V., Mingozzi F. Capsid antigen presentation flags human hepatocytes for destruction after transduction by adeno-associated viral vectors. J. Clin. Invest. 2009;119:1688–1695. doi: 10.1172/JCI36891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nathwani A.C., Reiss U.M., Tuddenham E.G.D., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasi K.J., Rangarajan S., Mitchell N., Lester W., Symington E., Madan B., Laffan M., Russell C.B., Li M., Pierce G.F., Wong W.Y. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N. Engl. J. Med. 2020;382:29–40. doi: 10.1056/NEJMoa1908490. [DOI] [PubMed] [Google Scholar]
- 20.Rangarajan S., Walsh L., Lester W., Perry D., Madan B., Laffan M., Yu H., Vettermann C., Pierce G.F., Wong W.Y., Pasi K.J. AAV5-factor VIII gene transfer in severe hemophilia A. N. Engl. J. Med. 2017;377:2519–2530. doi: 10.1056/NEJMoa1708483. [DOI] [PubMed] [Google Scholar]
- 21.Hu G., Guo D., Key N.S., Conti-Fine B.M. Cytokine production by CD4+ T cells specific for coagulation factor VIII in healthy subjects and haemophilia A patients. Thromb. Haemost. 2007;97:788–794. [PubMed] [Google Scholar]
- 22.Colella P., Ronzitti G., Mingozzi F. Emerging issues in AAV-mediated in vivo gene therapy. Mol. Ther. Methods Clin. Dev. 2017;8:87–104. doi: 10.1016/j.omtm.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Long B.R., Sandza K., Holcomb J., Crockett L., Hayes G.M., Arens J., Fonck C., Tsuruda L.S., Schweighardt B., O’Neill C.A. The impact of pre-existing immunity on the non-clinical pharmacodynamics of AAV5-based gene therapy. Mol. Ther. Methods Clin. Dev. 2019;13:440–452. doi: 10.1016/j.omtm.2019.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majowicz A., Nijmeijer B., Lampen M.H., Spronck L., de Haan M., Petry H., van Deventer S.J., Meyer C., Tangelder M., Ferreira V. Therapeutic hFIX activity achieved after single AAV5-hFIX treatment in hemophilia B patients and NHPs with pre-existing anti-AAV5 NABs. Mol. Ther. Methods Clin. Dev. 2019;14:27–36. doi: 10.1016/j.omtm.2019.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Von Drygalski A., Giermasz A., Castaman G., Key N.S., Lattimore S., Leebeek F.W.G., Miesbach W., Recht M., Long A., Gut R. Etranacogene dezaparvovec (AMT-061 phase 2b): normal/near normal FIX activity and bleed cessation in hemophilia B. Blood Adv. 2019;3:3241–3247. doi: 10.1182/bloodadvances.2019000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendell J.R., Rodino-Klapac L.R., Rosales X.Q., Coley B.D., Galloway G., Lewis S., Malik V., Shilling C., Byrne B.J., Conlon T. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann. Neurol. 2010;68:629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.George L.A., Ragni M.V., Rasko J.E.J., Raffini L.J., Samelson-Jones B.J., Ozelo M., Hazbon M., Runowski A.R., Wellman J.A., Wachtel K. Long-term follow-up of the first in human intravascular delivery of AAV for gene transfer: AAV2-hFIX16 for severe hemophilia B. Mol. Ther. 2020;28:2073–2082. doi: 10.1016/j.ymthe.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calcedo R., Wilson J.M. AAV natural infection induces broad cross-neutralizing antibody responses to multiple AAV serotypes in chimpanzees. Hum. Gene Ther. Clin. Dev. 2016;27:79–82. doi: 10.1089/humc.2016.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aronson S.J., Veron P., Collaud F., Hubert A., Delahais V., Honnet G., de Knegt R.J., Junge N., Baumann U., Di Giorgio A. Prevalence and relevance of pre-existing anti-adeno-associated virus immunity in the context of gene therapy for Crigler-Najjar syndrome. Hum. Gene Ther. 2019;30:1297–1305. doi: 10.1089/hum.2019.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mingozzi F., Chen Y., Murphy S.L., Edmonson S.C., Tai A., Price S.D., Metzger M.E., Zhou S., Wright J.F., Donahue R.E. Pharmacological modulation of humoral immunity in a nonhuman primate model of AAV gene transfer for hemophilia B. Mol. Ther. 2012;20:1410–1416. doi: 10.1038/mt.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L., Bell P., Somanathan S., Wang Q., He Z., Yu H., McMenamin D., Goode T., Calcedo R., Wilson J.M. Comparative study of liver gene transfer with AAV vectors based on natural and engineered AAV capsids. Mol. Ther. 2015;23:1877–1887. doi: 10.1038/mt.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hermans C., Astermark J., De Moerloose P. Exposure to factor VIII and prediction of inhibitor development: exposure days vs. danger days, or both? J. Thromb. Haemost. 2012;10:2194–2196. doi: 10.1111/j.1538-7836.2012.04871.x. [DOI] [PubMed] [Google Scholar]
- 33.Bray G.L., Gomperts E.D., Courter S., Gruppo R., Gordon E.M., Manco-Johnson M., Shapiro A., Scheibel E., White G., 3rd, Lee M., The Recombinate Study Group A multicenter study of recombinant factor VIII (recombinate): safety, efficacy, and inhibitor risk in previously untreated patients with hemophilia A. Blood. 1994;83:2428–2435. [PubMed] [Google Scholar]
- 34.Oldenburg J., Mahlangu J.N., Kim B., Schmitt C., Callaghan M.U., Young G., Santagostino E., Kruse-Jarres R., Negrier C., Kessler C. Emicizumab prophylaxis in hemophilia A with inhibitors. N. Engl. J. Med. 2017;377:809–818. doi: 10.1056/NEJMoa1703068. [DOI] [PubMed] [Google Scholar]
- 35.Whelan S.F., Hofbauer C.J., Horling F.M., Allacher P., Wolfsegger M.J., Oldenburg J., Male C., Windyga J., Tiede A., Schwarz H.P. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. 2013;121:1039–1048. doi: 10.1182/blood-2012-07-444877. [DOI] [PubMed] [Google Scholar]
- 36.Hofbauer C.J., Whelan S.F., Hirschler M., Allacher P., Horling F.M., Lawo J.P., Oldenburg J., Tiede A., Male C., Windyga J. Affinity of FVIII-specific antibodies reveals major differences between neutralizing and nonneutralizing antibodies in humans. Blood. 2015;125:1180–1188. doi: 10.1182/blood-2014-09-598268. [DOI] [PubMed] [Google Scholar]
- 37.Verdera H.C., Kuranda K., Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol. Ther. 2020;28:723–746. doi: 10.1016/j.ymthe.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuranda K., Jean-Alphonse P., Leborgne C., Hardet R., Collaud F., Marmier S., Costa Verdera H., Ronzitti G., Veron P., Mingozzi F. Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J. Clin. Invest. 2018;128:5267–5279. doi: 10.1172/JCI122372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashwell J.D., Lu F.W., Vacchio M.S. Glucocorticoids in T cell development and function. Annu. Rev. Immunol. 2000;18:309–345. doi: 10.1146/annurev.immunol.18.1.309. [DOI] [PubMed] [Google Scholar]
- 40.D’Avola D., López-Franco E., Sangro B., Pañeda A., Grossios N., Gil-Farina I., Benito A., Twisk J., Paz M., Ruiz J. Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria. J. Hepatol. 2016;65:776–783. doi: 10.1016/j.jhep.2016.05.012. [DOI] [PubMed] [Google Scholar]
- 41.Miesbach W., Meijer K., Coppens M., Kampmann P., Klamroth R., Schutgens R., Tangelder M., Castaman G., Schwäble J., Bonig H. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131:1022–1031. doi: 10.1182/blood-2017-09-804419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.