

Abstract
Chimeric antigen receptor T cell (CAR-T) therapy has revolutionized the clinical treatment of hematological malignancies due to the prominent anti-tumor effects. B cell maturation antigen (BCMA) CAR-T cells have demonstrated promising effects in patients with relapsed/refractory multiple myeloma. However, the dynamics of CAR-T cell proliferation and cytotoxicity in clinical patients remains unexplored. Here, we longitudinally profiled the transcriptomes of 55,488 T cells including CAR-T products, CAR-T cells, and endogenous T cells at the peak and remission phases in a plasma cell leukemia (PCL) patient treated with BCMA CAR-T cells by single-cell transcriptomic analysis. Our results showed distinct CAR-T and endogenous T cell subsets indicating stage-specific expression in proliferation, cytotoxicity, and intercellular signaling pathways. Furthermore, we found that CAR-T cells at peak phase gradually convert to a highly cytotoxic state from a highly proliferative state along a development trajectory. Moreover, re-analysis of a single cell study from CD8+ CD19 CAR-T confirmed our findings. These commonalities suggest conserved mechanisms for CAR-T treatment across hematological malignancies. Taken together, our current study provides insight into CAR-T cell dynamics during CAR-T therapy and proves that both BCMA CAR-T and CD19 CAR-T have similar transcriptional characteristics, especially at the CAR-T peak phase.
Keywords: chimeric antigen receptor T cell, plasma cell leukemia, single-cell RNA sequencing
Graphical Abstract
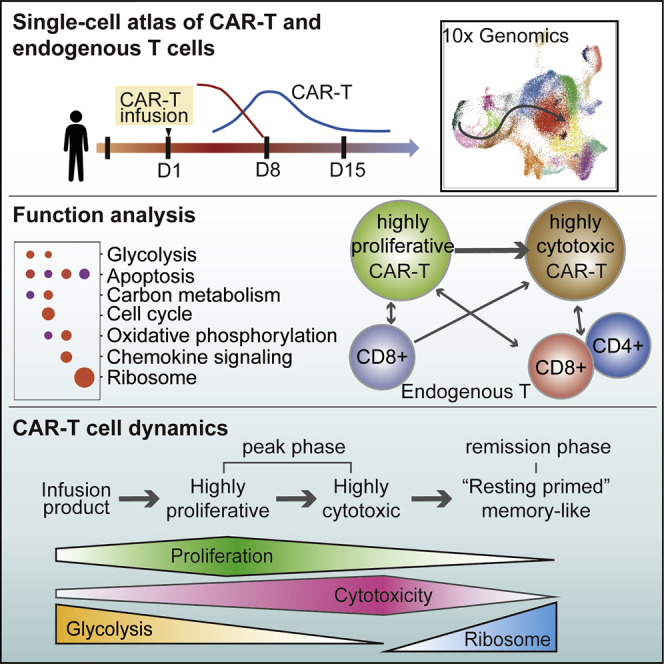
Li and colleagues have provided a developmental trajectory showing the dynamics of BCMA-CART cells since CART product infusion to disease remission. They also gave a new insight into the interaction between CAR-T cells and the endogenous immune system.
Introduction
Chimeric antigen receptor T (CAR-T) cells targeting CD19 or CD22 have emerged as a reliable treatment strategy for relapsed/refractory acute lymphoblastic leukemia (R/R ALL) and lymphoma over the last few years that have been published previously by our group and others.1, 2, 3, 4 Recently, B cell maturation antigen (BCMA) CAR-T cells have produced similarly impressive effects in patients with R/R multiple myeloma (MM).5 Clinical studies found that expansion and persistence of BCMA CAR-T cells was positively associated with favorable prognosis,6 but the underlying mechanisms remain unclear. In most MM patients, peripheral blood contains low numbers of malignant cells because most malignant cells are sequestered in the bone marrow.7 This makes the examination of the dynamic activity of BCMA CAR-T cells difficult because they act at the bone marrow and thus cannot be recovered from peripheral blood draws. Plasma cell leukemia (PCL), the most aggressive of the plasma cell dyscrasias, is a form of MM characterized by the presence of more than 20% and/or more than 2× 109/L of circulating plasma cells in the peripheral blood.8 Thus, PCL provides an accessible model for studying the dynamic activity of BCMA CAR-T cells in patient peripheral blood. Examination of these cells in their therapeutic environment is crucial for understanding the mechanisms of anti-tumor effects and interaction with plasma cells.
Single-cell RNA sequencing (scRNA-seq) technologies can systematically characterize all transplanted and endogenous cells in a patient. Recent scRNA-seq applications in CAR-T cells include analysis of cytokine profiles in CD19 CAR-T cells and a microfluidics-based analysis of CD19 CAR-T upon antigen stimulation.9,10 Both studies were performed ex vivo and concluded that CAR-T cells are polyfunctional and produce a highly diverse landscape of cytokines. A recent study of in vivo isolated CD19 CAR-T cells from ALL, chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL) patients at different phases after infusion showed clonal diversity of CAR-T cells and distinct patterns of proliferation for individual clones.11 Further, clusters of cells with high proliferation and cytotoxicity signatures at the infusion stage could successfully expand in patients.11 Whether these clusters of cells persist at the peak and remission phases and how they dynamically changed is unknown. Additionally, what transcriptional signatures dictate each cluster’s function and what is the crosstalk between CAR-T and non-CAR-T cells remain a mystery. The differences and commonalities between the driving mechanisms of BCMA versus CD19 CAR-T therapy at the single-cell level have also not been investigated.
Here, we detail the anti-tumor activity of BCMA CAR-T cells in a patient with refractory primary PCL (pPCL). Using scRNA-seq analysis, we depicted a trajectory of CAR-T cells during their journey from ex vivo infusion products to the in vivo peak phase and finally to the remission phase. Further, we defined highly proliferative and cytotoxic subsets within the peak phase and identified unique metabolic features at each phase with different subsets. Our findings appear to be consistent in both BCMA and CD19 CAR-T cells, implying conserved CAR-T cell dynamics across different targeting antigens in distinct hematological cancers.
Results
BCMA CAR-T Cell Therapy Achieved CR in a Patient with Refractory pPCL
A 66-year-old Chinese male patient was first admitted to our hospital in February 2018. His blood count showed leukocytosis of 169.5 × 109/L with 91% circulating plasma cells, hemoglobin 112 g/L, and a platelet count 201 × 109/L. Bone marrow biopsy revealed 81% infiltration of lambda clonal plasma cells confirmed by flow cytometry. The concentration of lambda free light chain (FLC) was 2,348 mg/L with a pathological FLC ratio of 2134. Fluorescence in situ hybridization (FISH) analysis revealed rearrangements of chromosome 1q21, loss of D13S319 and RB1. The diagnosis of pPCL was established based on these results. The patient received DCEP (dexamethasone, cyclophosphamide, etoposide, and cisplatin) combined with bortezomib chemotherapy and PRD (bortezomib, lenalidomide, and dexamethasone) combined with ibrutinib. No treatment response was observed, which led to the diagnosis of refractory pPCL. The patient was enrolled in a clinical trial of BCMA CAR-T treatment (ChiCTR1800017404). After FC (fludarabine 30mg/m2× 3d (D-4 to-2)+ cyclophosphamide 500mg/m2×3d(D-3 to -2)) lymphocyte-depleting chemotherapy, the patient received an infusion of BCMA CAR-T cells at a dose of 4.3 × 106 per kilogram. The patient developed fever (Figure 1A) with marked elevation of serum interleukin-6 (IL-6), IL-10, interferon-γ (IFN-γ), C reactive protein (CRP), and ferritin 2 days after the infusion (Figure S1A). CAR-T cells had engrafted and expanded in peripheral blood (Figures 1B, 1C, and S1B). Grade 3 cytokine release syndrome (CRS) was diagnosed, and high tumor lysis was indicated by an increase in lactate dehydrogenase (LDH) and uric acid content (Figure S1C). At a follow-up 15 days after CAR-T cell infusion, the patient showed complete remission (CR).
Figure 1.

The Proportion of CAR-T and Plasma Cells after CAR-T Infusion
(A) Graphs showing the daily highest temperature of the patient after CAR-T cell infusion. (B) Proportion of CAR-T cells and plasma cells in peripheral blood. (C) Flow cytometry plots showing the immune phenotype of CD4+ CAR-T and CD8+ CAR-T in PBMCs at three phases: CAR-T products before infusion, CAR-T cells on day 8 after infusion, and CAR-T cells on day 15 after infusion.
Distinct CAR-T and Endogenous T Cell Subsets Were Identified via scRNA-Seq
To better understand the dynamic features of CAR-T cells, we performed scRNA-seq on CAR-T cells (CD3+CAR+) and endogenous T cells (CD3+CAR−) isolated from fresh peripheral blood at three phases: CAR-T products, CAR-T at the peak phase on day 8, and at remission phase on day 15 (Figure 2A). After removing low-quality cells, a total of 28,516 CAR-T cell and 26,972 endogenous T cell transcriptomes were profiled. Twenty-four clusters were identified and visualized by uniform manifold approximation and projection (UMAP; Figure 2B). The expression of marker genes indicated clusters of regulatory T cells (CD4+CD25+ cluster 4 and CD4+CD25+FOXP3+ cluster 15), memory T cells (CD62L+CD44+CCR7+ clusters 3, 5, 9, 10, 21, and 23), CD4+ exhausted T cells (TIM3+PD1+LAG3+TIGIT+ cluster 19), CD8+ exhausted T cells (TIM3+PD1+LAG3+TIGIT+ clusters 8 and 14), CD8+CD160+ T cells (cluster 11), natural killer-like T cells (CD69+ZNF683+ cluster 20), CD69+IFIT1+IFIT3+ cells (cluster 22), CD8+ JUN+CD69+ T cells (cluster 0), and CD8+GZMB+ cytotoxic T cells (cluster 1, 2, 6, 7, 12, 13, 16, 18; Figures 2B, 2C, S2A, and S2B). The clustering result showed that CAR-T and endogenous T cells were distributed across several distinct clusters (Figures 2B, S2C, and S2D), and the proportion of each subset changed dynamically after infusion. CAR-T product was composed of 64.1% CD4+ T cells and 35.9% CD8+ T cells (Figure 2D), but at the peak phase, CD8+GZMB+ T cells were dominant in both CAR-T (98.1%) and endogenous T (89.1%) samples (Figure 2D). At the remission, when levels of leukemia cells were undetectable, CD8+ CAR-T cells transitioned to a more homogeneous cluster 0 (CD8+JUN+CD69+) memory-like cell state. CD4+ CAR-T cells were almost completely diminished at remission. Flow cytometry analyses of the three phases confirmed a higher fraction of CD8+ CAR-T after infusion (Figure 1C). Together, these results demonstrate a transition of CAR-T cells from mixed subsets before infusion to predominantly cytotoxic CD8+ T cells at the peak phase and finally to JUN+ memory-like T cells at remission.
Figure 2.

A Single-Cell Atlas of CAR-T and Endogenous T Cells during CAR-T Therapy
(A) Overall study design of single-cell RNA-seq. Single-cell RNA-seq is applied to CAR-T (CD3+CAR+) cells and endogenous T (CD3+CAR−) cells derived from PBMCs. (B) The UMAP visualization of 55,488 cells from CAR-T and endogenous T cell samples at three phases. 24 clusters are indicated by different colors. Sample origins are distinguished by colors as shown at top-left corner. (C) Expression patterns of CD4, CD8A, CD25, and GZMB are exhibited on UMAP plot. Color intensity represents expression level. (D) Proportions of CD4+ T cells and CD8+ T cells within each sequenced sample. Only cells expressing CD4 or CD8 are taken into account.
CAR-T Cells at the Peak Phase Transitioned from Proliferative to Cytotoxic Intermediate States along a Development Trajectory
Interestingly, at the peak phase, CAR-T cells showed dispersed clustering, although all of them were activated cytotoxic CD8+GZMB+ T cells. In addition, we identified the top 10 specific genes of each cluster and found that during the peak phase, some adjacent clusters (clusters 16, 7, 13) only differ in their expression levels (Figure S3A). Therefore, a more elaborate subpopulation analysis was necessary to define what cellular states this cluster dispersion represented. Previous studies with UMAP have shown robust developmental trajectory production,12 so we constructed a CAR-T developmental trajectory by combining cluster labels and UMAP results (Figure 3A).
Figure 3.

Highly Proliferative and Highly Cytotoxic States at the Peak Phase
(A) Proliferation score of total CAR-T cells at three phases. Bottom, boxplots of the proliferation score of CAR-T cells in each cluster. Pie chart for each cluster depicting the percentage of samples. The gray- and yellow-colored background indicates the clusters mainly from the peak phase. Two-sided unpaired Wilcoxon test is used to compare adjacent clusters; ∗p < 1e−10. (B) Bar graph showing the cytotoxicity signature genes that are most correlated with GZMB and PRF1 across all CAR-T cells. The x axis represents the Pearson correlation coefficient. (C) Cytotoxic score of all CAR-T cells at three phases. Bottom, boxplots of cytotoxicity score of CAR-T clusters, in the same format as (A). (D) Density distribution of the count of cytokine types expressed in each cell at different phases. (E) Heatmap of average expression of 16 cytokines for each of the CAR-T clusters: effector (IFNG, GZMB, PRF1, CCL3, TNF, LTA), chemoattractive (CCL5, CCL4), stimulatory (CXCL8, IL-5), regulatory (TGFB1, TNFRSF9, IL-22, IL-13, IL-4, CD40LG). Pie chart for each cluster depicting the percentage of samples.
First, to characterize the proliferative status through the CAR-T cell trajectory, we computed a proliferation score by calculating the mean expression level of cell cycle genes in each cell (Figure 3A).13 Interestingly, the proliferation score of CD8+GZMB+ cytotoxic CAR-T cells gradually decreased along the development trajectory (Wilcoxon rank-sum test: p < 1e−10; Figure 3A). CD8+ CAR-T cells obtained a higher proliferative score than CD4+ CAR-T cells (Figure S3B), which was consistent with the CD8+ T cell predomination over CD4+ T cells at the peak phase (Figure 2D). At the same point, the proliferation score of endogenous T cells was lower than that of CAR-T cells (Figure S3B), suggesting that CAR-T cells were proliferating in response to stimulation by tumor antigens.
Next, we examined the cytotoxicity status of the CAR-T cell clusters. Elevated expression of granzyme B (GZMB) and perforin (PRF1) was observed in cytotoxic endogenous T cells and CAR-T cells.14,15 To quantify the cytotoxicity of CAR-T cells, we defined a cytotoxicity score for each cell based on the mean expression of a series of GZMB and PRF1 co-expressed genes defined in our dataset.13 Following this analysis, 20 total genes correlated with GZMB and PRF1 were considered as cytotoxicity signature genes, including FGFBP2, NKG7, and CCL3, among others (Figure 3B). In contrast to the proliferation score, the cytotoxicity score of all CAR-T clusters at the peak phase gradually increased along the developmental trajectory (Wilcoxon rank-sum test, p < 1e−10; Figure 3C). Together, these results provided evidence that CAR-T cells at the peak phase shifted from a highly proliferative state to a highly cytotoxic state.
To gain deeper insight into the immunomodulatory effects of BCMA CAR-T cells, we measured 16 cytokines that had been defined as associated with different CAR-T cell states in a previous study10 in our single-cell data. Indeed, BCMA CAR-T cells and endogenous T cells exhibited a polyfunctional characteristic, which means that they produced ≥ 5 cytokines, including PRF1, CCL4, IFNG, GZMB, CCL3, and CCL5 at the peak phase (Figures 3D and 3E).10 While examining the cytokines across our pseudotemporal mapping, we found that those CAR-T clusters with higher proliferation score early in the developmental trajectory (clusters 4, 16, 7, 13) expressed relatively low levels of effector cytokines and higher levels of regulatory cytokines.10 CAR-T cells from the remission phase did not express effector cytokines (Figure 3E). Together, by combining the proliferation/cytotoxicity scores and the cytokine analysis, we discovered two intermediate states of CAR-T cells at the peak phase: the highly proliferative (clusters 16, 7, 13) and highly cytotoxic (clusters 6, 18, 12, 1) states (Figure 3E), suggesting CAR-T cells in vivo first proliferate, followed by assuming higher cytotoxic activity during the peak phase.
CAR-T Cells in the Remission Phase Had Elevated Signatures of Ribosomal Protein Genes
To obtain a broad understanding of enriched gene signatures in CAR-T cell products, highly proliferative CAR-T cells, highly cytotoxic CAR-T cells, and memory-like CAR-T cells, we performed KEGG (Kyoto Encyclopedia of Genes and Genomes) enrichment analysis (Figures 4A and 4B). The CAR-T product cluster 4 was a subgroup of CD4+ CAR-T cells that specifically express TNFRSF4, CD25 (IL2RA), and LTB (Figures 4A and S4). Previous work has proven that TNFRSF4 expression in activated T cells promotes cell division and survival.16 Pathway analyses of the top 120 specific genes in this cluster revealed a significant association with glycolysis/gluconeogenesis, which is consistent with previous findings demonstrating that T cell activation induced aerobic glycolysis to support rapid proliferation.17 In the highly proliferative state, there was a significant enrichment of genes associated with the cell cycle and DNA replication, such as TUBA1B, TUBB, RRM2, and TOP2A (Figures 4A and S4). Similar to the CAR-T product, the highly expressed genes in this state were involved in glycolysis and anabolic metabolism, such as biosynthesis of amino acid and carbon metabolism pathways. Furthermore, CAR-T cells in the highly cytotoxic state had increased expression of genes related to the immune response and chemokine signaling pathways like CCL3, CCL4, CCL5, GNLY, PRF1, and GZMB (Figures 4A and S4).
Figure 4.

Enriched Functional Analysis of CAR-T State-Specific Genes
(A) Heatmap depicting expression levels of the 20 cytotoxicity signature genes expressed in each CAR-T state in individual cells (CD4+ CAR-T products, highly proliferative, highly cytotoxic, and memory-like). (B) Dot plot depicting significant KEGG enrichment results for each CAR-T state. KEGG enrichment for the top 120 specific marker genes expressed in each of the four CAR-T states. Bubble size represents the proportion of corresponding pathway-specific genes in 120 specific genes. The color intensity of the bubble represents the adjusted p value.
Memory-like CAR-T cells were noted by high expression of IL7R, FOS, JUN, JUNB, CD69, and various ribosomal protein (RP) genes, such as RPL23, RPL6, and RPS27 (Figures 4A and S4). FOS and JUN form the AP-1 transcription factor complex, which is pivotal in the control of cell proliferation, differentiation, and T cell activation.18 Furthermore, overexpression of JUN reduces CAR-T cell exhaustion.19 Although the major function of RP is translation, recent studies have indicated that many RPs signal in immune-related pathways controlling stress sensing, anti-apoptotic, and other functions.20, 21, 22 We speculate that CAR-T cells at the remission phase were in a “resting primed” memory-like state with minimal energy and biosynthetic material consumption and are primed for further activation.
CAR-T State Dynamics Is Conserved in CD19 CAR-T Cells for CLL and NHL Treatment
To validate our findings, we re-analyzed a recent CD19-targeting CAR-T scRNA-seq dataset from two CLL and two NHL patients at four time points: CAR-T products, early, late, and very late CAR-T treatment.11 A total of twelve distinct clusters were detected in the combined datasets. Based on the sampling time and UMAP, the CD8+ CAR-T cell developmental trajectories of four patients were constructed (Figure 5A). The CAR-T product was mainly composed of clusters 2, 4, 7, and cluster 9; clusters 0, 5, and 11 were enriched in early CAR-T (7–14 days after infusion); clusters 1, 6, and 8 were enriched in late CAR-T (26–30 days after infusion); and cluster 3 and cluster 10 represented very late CAR-T cells (83–112 days after infusion). For the comparison with our own dataset, we examined the expression levels of the same 16 CAR-T-related cytokines10 in each cluster (Figure 5B). Consistent with our results, IL-5, CD40LG, IL-4, IL-13, TNF, and IL-22 were highly expressed in the CD8+ CAR-T product, and a significantly higher expression of effector cytokines, such as GZMB, PRF1, CCL3, and CCL4, was observed in the early and late time points, which corresponds to our peak phase, whereas for the very late time point, expression of most of these cytokines was significantly lowered.
Figure 5.

Validation of CAR-T Cell States from Other Blood Cancer Types
(A) The UMAP visualization of 62,167 cells from CD8+ CAR-T samples at four time points: CAR-T products, early, late, and very late. Different colors indicate annotation by time points (left) and cell clusters (right). (B) Heatmap of average expression of 16 cytokines/chemokines for each of the CD8+ CAR-T clusters as in Figure 3E. Pie chart for each cluster depicting the percentage of samples. (C) Dot plot depicting significant KEGG enrichment results for each CD8+ CAR-T clusters, same as Figure 4B. (D) Venn diagram showing overlapping RP genes between the BCMA CAR-T cells of our dataset and CD19 CAR-T cells of a previously published dataset.
KEGG enrichment further confirmed our BCMA analysis with the CD19 CD8+ CAR-T product having strong signatures of glycolysis, biosynthesis of amino acids, and carbon metabolism (Figure 5C). Additionally, our highly proliferative BCMA CAR-T cells matched the cell cycle signatures seen in clusters 9, 7, and 5 from the CD19 CAR-T product and early time points. Some clusters showed a blend of signatures from two discrete cell states, such as cluster 8, which has both cytotoxicity and ribosome signatures, which alludes to a transition from the cytotoxic state to the “resting primed” memory-like state. The late and very late CD19 CAR-T cells were also significantly enriched in RP genes, identified in the “resting primed” memory-like BCMA CAR-T cells such as RPL23, RPL6, and RPL34 (Figure 5D). Notably, RPL23 and RPL34 are thought to confer apoptotic resistance in metastatic tumor cells,23,24 and RPL6 is involved in ribosomal stress and the p53 signaling pathway.25 In addition, the same gene sets of proliferation and cytotoxicity were used to score each CD19 CAR-T cell. The results showed that the CD19 CAR-T cells with relatively high proliferation scores were enriched in CAR-T products and early CAR-T samples, while those with extremely high cytotoxicity were enriched in late CAR-T samples (Figure S5A). Together, these data showed consistent dynamics for BCMA CAR-T in the PCL patients and CD19 CAR-T in CLL/NHL patients.
Crosstalk between CAR-T Subsets and Endogenous T Cells Regulated Cytotoxicity and Proliferation
To understand the complex T cell interactions, we predicted ligand-receptor (L-R) pairs among all T cell clusters. We discovered that in addition to the strong interaction between highly proliferative and highly cytotoxic CAR-T cells, three endogenous T cell clusters (clusters 8, 11, 19) also have strong predicted interactions with these CAR-T subsets (Figure S6).26 It was speculated that there may be a regulatory relationship between transplanted CAR-T cells and endogenous T cells. To further characterize these potential interactions, we found a total of 111 L-R pairs between highly proliferative CAR-T, highly cytotoxic CAR-T, CD8+ endogenous T cells (cluster 8 and cluster 11), and CD4+ endogenous T cells (cluster 19). We divided these L-R pairs into two groups, with one containing interactions between highly proliferative and highly cytotoxic CAR-T cells (Figure 6A, left), and the other containing interactions between endogenous T cells and CAR-T cells (Figure 6A, right). Further, gene ontology (GO) enrichment analysis was performed on these two groups of L-R pairs. The results showed that the first group of pairs was mainly involved in the immune response, T cell activation, and cell-cell adhesion, while the second group of genes was enriched in tumor necrosis factor (TNF)-related pathways and the nuclear factor κB (NF-κB) signaling pathway (Figure 6A, bottom).
Figure 6.

Cell-Cell Interactions between CAR-T Subsets and Endogenous T Cells
(A) Heatmap showing the L-R pairs between CAR-T (highly proliferative and highly cytotoxic state) and endogenous T cells (clusters 8, 11, and 19). Rows represent the L-R pairs. Columns represent cell subset-cell subset pairs. The color gradient from purple to yellow indicates low to high mean values of the L-R pairs calculated by the CellPhoneDB tool. Only significant L-R pairs (p < 0.05) are colored. The colored names of L-R pairs correspond to the genes presented in (B). Bottom, L-R genes involved in GO terms. (B) Boxplot showing the selected ligand/receptor expression of in different clusters. (C) The schematic diagram indicates the relationship between CAR-T subsets and endogenous T cells at the peak phase.
Several L-R pairs were associated with T cell activation, such as CCR5-CCL5, CCL3-CCR5, CD70-CD27, and ICAM1-SPN/ITGAL. CCL5/CCR5 showed highly significant interaction between highly cytotoxic CAR-T cells and endogenous T cells. We speculate that CAR-T cells may be able to recruit endogenous T cells to establish a new immune system. Moreover, previous studies have shown that CD70/CD27 co-stimulatory factors could promote the proliferation and activation of T cells expressing CD27.27 CD70, a ligand for CD27, was strongly expressed in the highly proliferative CAR-T cells, but higher CD27 expression was observed in the cytotoxic CAR-T cells (Figure 6B). ICAM1, which is the ligand of both ITGAL and SPN, was relatively increased in the highly proliferative CAR-T cells. Interestingly, a previous report showed that the ICAM1/ITGAL interaction contributes to T cell cytotoxicity.28 Thus, these findings indicated that the highly proliferative subset of CAR-T cells might promote the activation and cytotoxicity of all CAR-T cells by signaling via the CD70-CD27 and ICAM1-SPN/ITGAL interactions. Interestingly, a portion of endogenous CD4 and CD8 T cells (cluster 19 and cluster 8) contained the above signatures, indicating that they might have also been activated by highly proliferative CAR-T cells. Several other L-R interaction pairs were observed. TNFSF12-TNFRSF25 between endogenous T clusters 19 and 8 and CAR-T along with CD160-TNFRSF14 between endogenous T cluster 11 and CAR-T were noted in particular because these interactions could induce clonal T cell expansion (Figures 6A, right, and 6B).29
Suppressive L-R pairs revealed by our analysis were also carefully examined, as they could play a key role in balancing efficacy and safety. IL-10 is an anti-inflammatory cytokine that can induce immune tolerance by binding to IL-10 receptor (IL-10R) on the surface of T cells.30,31 Previous research on CAR-T combination therapy has attempted to restore CAR-T cell activity by blocking the IL-10 signaling.32 In our dataset, L-R analysis showed that there was an IL-10-IL-0R pair formed between clusters 19 and 8 of the endogenous T cells and highly proliferative CAR-T cells (Figures 6A, right, and 6B). In combination, these analyses imply that the highly cytotoxic CAR-T cells might be activated by highly proliferative CAR-T cells via the CD70/CD27 co-stimulatory interaction, and endogenous T cells might regulate the activation of CAR-T cells by secreting IL-10.
Discussion
pPCL is a rare and aggressive plasma cell disorder with high tumor burden and poor outcomes. The use of conventional therapies exhibited low response rates and a median overall survival (OS) ranging from 2-7 months.33 Despite the development of new chemotherapy drugs and regimens, a considerable number of patients still progress to refractory or relapsed disease. Due to the rare and unique nature of the condition, most pPCL patients are excluded from clinical trials. To our knowledge, this was the first study to describe the potent anti-tumor activity of BCMA CAR-T therapy without increased risks of treatment-related toxicities in a heavily treated refractory pPCL patient.
To further understand the transcriptional program of CAR-T and endogenous T cells in this patient, we used the scRNA-seq technology to examine these cells throughout the course of CAR-T treatment. Our results from BCMA CAR-T cells in pPCL were largely consistent with the transcriptomes of CD19 CAR-T cells from NHL/CLL patients. Overall, our results showed a model of the dynamic changes of CAR-T cells in the proliferation, cytotoxicity, glycolysis, and RP gene expression characteristics (Figure 7). First, the infusion product cells were highly metabolically active, with high glycolysis and biosynthetic gene expression. Second, CAR-T cells at the peak phase exhibited two continuous intermediate states: the highly proliferative and the highly cytotoxic state. At the late remission or very late contraction stage in the CD19 CAR-T single-cell study, CAR-T cells were not proliferative but maintained their cytotoxic capacity. Notably, they assumed a signature of the chemokine-signaling pathway. Finally, the remission phase and the very late stage CAR-T cells had declined proliferation and cytotoxicity signatures but assumed a strong induction of RP genes in both CD19 and BCMA CAR-T cells, which we name as a “resting primed” memory-like state. Even though we demonstrated several conserved dynamic transitions and mechanisms of action between CD19 and BCMA CAR-T results, key differences were noted. Expression of JUN, FOS, and CD69 was observed in the CD19 CAR-T product, while these genes mainly expressed at the remission phase for BCMA CAR-T cells (Figure S5B).
Figure 7.

Schematic Model of the BCMA CAR-T Dynamic Changes
Schematic model of the dynamic changes in the proliferation, cytotoxicity, glycolytic properties, and RP transcription characteristics of CAR-T cells.
T cell activation and proliferation is tightly linked to the underlying metabolic program.34 In the CAR-T products, high glycolysis level might result from the cell’s high glucose media in culture before infusion. After infusion at the peak phase, high glycolysis is accompanied with high proliferation, and this signature disappears at remission phase, with a remarkable concurrent elevation in RP gene expression in the “resting primed” memory-like cells of both CD19 and BCMA CAR-T. We speculate that even though these cells have low proliferative and cytotoxic activity, they may have high translational activity to be prepared for stress responses or strong resistance to apoptosis, thus priming them for further activation.
In addition, we searched for hallmarks of T cell dysfunction by characterizing the CAR-T expression of PDCD1 (PD1), TIGIT, HAVCR2 (TIM3), CTLA4, and LAG3.35 The lack of expression of these signature genes indicated that the CAR-T cells did not exhibit dysfunctional characteristics during remission phase or the very late stages in both studies (Figure S5). Memory signatures were also investigated through SELL (CD62L) and IL-7R expression. Overexpression of SELL (CD62L) or IL-7R was seen at remission in our data, but this was not observed not in the CD19 data for the late or very late time points (Figure S5B).
A previous in vitro study pointed out that CD4+ CD19 CAR-T cells could kill target leukemia cells via cytotoxic mechanisms;9 another study found a large amount of CD4+ T cells in the mouse models after CAR-T infusion.36 This supports the notion that CD4+ CAR-T cells play a more central role than previously expected. However, our study showed that CD4+ BCMA CAR-T cells significantly reduced in number post-infusion and comparatively lacked strong cytotoxic activity.
There are some limitations of the current study. First, we only collected scRNA-seq data from one pPCL patient. To combat this bias and to examine the effects of different CAR-T targets, we extended our analysis to a recently published scRNA-seq dataset of CD19 CAR-T cells from CLL/NHL patients. Second, only T cells were analyzed, yet previous studies have illustrated that other types of immune cells influence CAR-T cell dynamics by secreting IL-1 and IL-6 and causing CRS.36 The crosstalk between the entire immune microenvironment and CAR-T cells remains largely unknown and requires further investigation.
Materials and Methods
Clinical Sample Collection
The protocol number of clinical trial on BCMA CAR-T treatment is ChiCTR1800017404. Informed consent from the patient has been received. The research and publication of data has been approved by the Data and Safety Monitoring Board. One refractory pPCL patient was enrolled in this study. The patient received an infusion of BCMA CAR-T cells with a dose of 4.3 × 106 CAR-T cells per kg. BCMA CAR-T cells were generated as previously reported.37 Peripheral blood mononuclear cells (PBMCs) were isolated using the FicollDpaque PLUS (TBD Science) solution according to the standard protocol. In brief, fresh peripheral blood was collected in an EDTA anticoagulant tube and subsequently layered onto FicollDpaque PLUS. After centrifugation, lymphocytes remaining above the FicollDpaque PLUS interface were transferred to a new tube and washed twice with 1× PBS. PBMCs were re-suspended with sorting buffer (PBS supplemented with 1% fetal bovine serum [FBS]) (Gibco).
Flow Cytometry
Detection of CAR-T and leukemia cells was conducted with a nine-parameter DxFLEX flow cytometer (Beckman Coulter). To determine CAR protein expression, cells were first incubated with anti-mouse immunoglobulin G (IgG) biotin (Jackson). After washing, cells were stained by fluorescein isothiocyanate (FITC) streptavidin (BioLegend). Other monoclonal antibodies and their isotype control antibodies were purchased from BioLegend, including APC-CD45 (clone 2D1, mouse IgG1, κ), PE-Cy7-CD3 (clone UCHT1, mouse IgG1, κ), PerCP-Cy5.5-CD4 (clone OKT4, mouse IgG2b, κ), PE-CD8 (clone HIT8a, mouse IgG1, κ), FITC-CD138 (clone MI15, mouse IgG1, κ), PE-BCMA (clone 19F2, mouse IgG2a, κ). FlowJo software was used to evaluate all the surface markers.
scRNA-Seq
scRNA-seq samples were collected at three phases: CAR-T products before infusion, CAR-T on day 8 after infusion, and CAR-T on day 15 after infusion. After obtaining the PBMCs for each phase, CD3+CAR+ (CAR-T) and CD3+CAR− (endogenous T) cells were collected by fluorescence-activated cell sorting (FACS; BD FACS Aria II) with anti-mouse IgG Biotin (Jackson), FITC streptavidin (BioLegend), and anti-human CD3 APC (clone UCHT1, BioLegend). Single-cell suspensions of all samples were resuspended in DPBS (Dulbecco’s phosphate-buffered saline) -0.04% BSA at 1e6 cells/mL. Then scRNA-seq libraries were generated from the 10× chromium single cell 3′ reagent kits v2 (PN-120237) according to the manufacturer’s protocol (10× Genomics). Libraries were sequenced on the Illumina Hiseq X10 platform according to the manufacturer’s instructions (Illumina). Read 1 and read 2 (paired end) were 150 bp, and the index primer was 8 bp. All samples were sequenced to an average depth of 236 million paired-end reads per sample, with 150 bp on read 1 and read 2.
Raw Data Processing and Analysis
Each scRNA-seq sample data was separately demultiplexed, aligned to the human genome (version GRCh38), and calculated unique molecular identifier (UMI) were estimated using the Cellranger toolkit (version 3.0.0; 10× Genomics), with default parameters. Cells with fewer than 200 detected genes, greater than 20,000 detected transcripts, or total mitochondrial gene expression exceeding 5% were excluded from the analysis. Genes expressed in less than 3 cells were also removed. Then, five UMI matrices from three phases were considered as three batches, and data integration was performed using the Seurat (version 3.0.0)38 R package. The data was then normalized using the NormalizeData function in Seurat (LogNormalize method with a scale factor of 10,000). Anchors were identified using FindIntegrationAnchors with 1–60 dimensions and 5,000 anchor features. An expression matrix of 55,488 cells with average 1,469 genes per cell were obtained for further analysis. Downstream analysis after integration included data feature scaling (ScaleData), principal-component analysis (PCA; RunPCA), and SNN (shared nearest neighbor) graph building (FindNeighbors). The first 60 dimensions were used for UMAP reduction by the RunUMAP function. Cells were clustered using the FindClusters function in Seurat with resolution = 1. A total of 24 clusters were defined by common markers, with marker genes identified using the FindMarkers function. Cluster 17 is a subgroup with high expression of mitochondrial gene, which was excluded from subsequent analysis.
Proliferation and Cytotoxic Score
To clarify the characteristics of single cells in proliferation and cytotoxic states, we selected two lists of signature genes to represent the corresponding states and defined the score of the corresponding signature for each single cell by averaging the expression level of the genes in the set. For the gene set of the proliferation state, we used a previously defined set of 94 cell cycle genes.39 For the gene set of the cytotoxic state, we calculated the Pearson correlation coefficients of every gene with GZMB and PRF1. The genes with a correlation coefficient with GZMB or PRF1 greater than 0.4, a total of 20 genes, were used as cytotoxicity signature genes (Figure 3B).
Identification of Differentially Expressed Genes and Pathway Enrichment Analysis
Differential expressed genes between two subgroups or multiple subgroups were calculated using the FindMarkers function from the Seurat package. The significance of the difference was determined using a Wilcoxon rank-sum test with Bonferroni correction. The specific genes were selected based on the statistical threshold (absolute log fold-change > 0.25, Wilcoxon rank-sum test p < 0.01, and adjusted p value [Bonferroni method] < 0.05). Functional enrichment analysis for each gene set was conducted by the R/Bioconductor package clusterProfiler (version 3.14.3).40 The threshold to determine the significance of the KEGG enrichment was set to an adjusted p value <0.05.
Reanalysis of CD8+ CAR-T Single-Cell Dataset
Filtered and processed CD8+ CAR-T scRNA-seq data from a previously published study was obtained from the Gene Expression Omnibus (GEO) with accession number GEO: GSE125881. The count matrix was processed using the Seurat R package (v 3.6.1). In brief, the quality control, normalization, scaling, and PCA used the same parameters as the original study. The FindNeighbors function was then run using the top 15 principal components (PCs). Using the FindClusters function with default parameters except for “resolution” = 0.4, we divided the 621,67 cells into 12 clusters. Dimensionality reduction using UMAP was performed based on the top 15 PCs. Differentially expressed markers were identified for every cluster by the FindAllMarkers function with only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25. Significant markers with an adjusted p value <0.01 in every cluster were used for KEGG enrichment analysis using the compareCluster function from the clusterProfiler package.
Cellular Interaction Analysis
The CellphoneDB tool (version 2.0) was used to predict cellular interaction with default parameters across total T cell populations.26 Significant L-R pairs were filtered with p value <0.05. These L-R pairs only between highly proliferative _highly cytotoxic, cluster-19_highly proliferative, cluster-19_highly cytotoxic, cluster-8_ highly proliferative, cluster-8_highly cytotoxic, cluster-11_highly proliferative, and cluster-11_highly cytotoxic cells were retained. KEGG pathway analysis was performed on the list of L-R genes using clusterProfiler.
Data Availability
scRNA-seq sequencing reads have been submitted to GEO under GEO: GSE151310.
Acknowledgments
The authors would like to thank Pengxu Qian and Luyi Tian for helpful discussions on data analysis. This work was supported by the Natural Science Foundation of China (grant nos. 81730008, 81770201, 81970137, 81870153) and the Key Project of Science and Technology Department of Zhejiang Province (grant no. 2019C03016,2018C03016-2). J.Z. is supported by the National Key Research and Development Program of China (2018YFA0107100, 2018YFA0107103, and 2018YFC1005002); the National Natural Science Foundation Projects of China (31871453 and 91857116); the Zhejiang Natural Science Foundation Projects of China (LR19C120001); and the Zhejiang Innovation Team grant (2019R01004).
Author Contributions
Y.H., J.Z., and H.H. conceived of and supervised the study. Xue Li and X.G. performed all the bioinformatics analysis and generated the figures. Xia Li and Y. Zhu performed the single-cell experiments and library construction. Y.H., G.W., Y. Zhang., H.X., J.H., W.W., and A.H.C provided samples and clinical contextual information. J.C. and Xia Li performed the flow cytometry and analysis. M.E.R. and H.Y. advised on bioinformatics analysis. L.D., M.S., Q.L., X.X., and X.T. participated in the preprocessing of samples. T.M.W. and H.L. provided suggestions on data analysis and writing the paper. Xue Li, J.Z., Y.H., and X.G. wrote the manuscript with support from all authors.
Declaration of Interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.11.028.
Contributor Information
Jin Zhang, Email: zhgene@zju.edu.cn.
He Huang, Email: huanghe@zju.edu.cn.
Yongxian Hu, Email: 1313016@zju.edu.cn.
Supplemental Information
References
- 1.Curran K.J., Margossian S.P., Kernan N.A., Silverman L.B., Williams D.A., Shukla N., Kobos R., Forlenza C.J., Steinherz P., Prockop S. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134:2361–2368. doi: 10.1182/blood.2019001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June C.H., Sadelain M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Y., Wu Z., Luo Y., Shi J., Yu J., Pu C., Liang Z., Wei G., Cui Q., Sun J. Potent Anti-leukemia Activities of Chimeric Antigen Receptor-Modified T Cells against CD19 in Chinese Patients with Relapsed/Refractory Acute Lymphocytic Leukemia. Clin. Cancer Res. 2017;23:3297–3306. doi: 10.1158/1078-0432.CCR-16-1799. [DOI] [PubMed] [Google Scholar]
- 4.Pan J., Niu Q., Deng B., Liu S., Wu T., Gao Z., Liu Z., Zhang Y., Qu X., Zhang Y. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33:2854–2866. doi: 10.1038/s41375-019-0488-7. [DOI] [PubMed] [Google Scholar]
- 5.D’Agostino M., Raje N. Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia. 2020;34:21–34. doi: 10.1038/s41375-019-0669-4. [DOI] [PubMed] [Google Scholar]
- 6.Brudno J.N., Maric I., Hartman S.D., Rose J.J., Wang M., Lam N., Stetler-Stevenson M., Salem D., Yuan C., Pavletic S. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018;36:2267–2280. doi: 10.1200/JCO.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Udd K.A., Spektor T.M., Berenson J.R. Monitoring multiple myeloma. Clin. Adv. Hematol. Oncol. 2017;15:951–961. [PubMed] [Google Scholar]
- 8.Gundesen M.T., Lund T., Moeller H.E.H., Abildgaard N. Plasma Cell Leukemia: Definition, Presentation, and Treatment. Curr. Oncol. Rep. 2019;21:8. doi: 10.1007/s11912-019-0754-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xhangolli I., Dura B., Lee G., Kim D., Xiao Y., Fan R. Single-cell Analysis of CAR-T Cell Activation Reveals A Mixed TH1/TH2 Response Independent of Differentiation. Genomics Proteomics Bioinformatics. 2019;17:129–139. doi: 10.1016/j.gpb.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xue Q., Bettini E., Paczkowski P., Ng C., Kaiser A., McConnell T., Kodrasi O., Quigley M.F., Heath J., Fan R. Single-cell multiplexed cytokine profiling of CD19 CAR-T cells reveals a diverse landscape of polyfunctional antigen-specific response. J. Immunother. Cancer. 2017;5:85. doi: 10.1186/s40425-017-0293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheih A., Voillet V., Hanafi L.A., DeBerg H.A., Yajima M., Hawkins R., Gersuk V., Riddell S.R., Maloney D.G., Wohlfahrt M.E. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020;11:219. doi: 10.1038/s41467-019-13880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 13.Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C.J., van den Braber M., Rozeman E.A., Haanen J., Haanen J.B.A.G. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell. 2019;176:775–789e18. doi: 10.1016/j.cell.2018.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boroughs A.C., Larson R.C., Choi B.D., Bouffard A.A., Riley L.S., Schiferle E., Kulkarni A.S., Cetrulo C.L., Ting D., Blazar B.R. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight. 2019;5:e126194. doi: 10.1172/jci.insight.126194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu J., Deng Y., Benson D.M., He S., Hughes T., Zhang J., Peng Y., Mao H., Yi L., Ghoshal K. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croft M., So T., Duan W., Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009;229:173–191. doi: 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menk A.V., Scharping N.E., Moreci R.S., Zeng X., Guy C., Salvatore S., Bae H., Xie J., Young H.A., Wendell S.G., Delgoffe G.M. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018;22:1509–1521. doi: 10.1016/j.celrep.2018.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Dam H., Castellazzi M. Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis. Oncogene. 2001;20:2453–2464. doi: 10.1038/sj.onc.1204239. [DOI] [PubMed] [Google Scholar]
- 19.Lynn R.C., Weber E.W., Sotillo E., Gennert D., Xu P., Good Z., Anbunathan H., Lattin J., Jones R., Tieu V. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576:293–300. doi: 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou X., Liao W.J., Liao J.M., Liao P., Lu H. Ribosomal proteins: functions beyond the ribosome. J. Mol. Cell Biol. 2015;7:92–104. doi: 10.1093/jmcb/mjv014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guimaraes J.C., Zavolan M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome Biol. 2016;17:236. doi: 10.1186/s13059-016-1104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murre C. Ribosomal proteins and the control of alphabeta T lineage development. Immunity. 2007;26:751–752. doi: 10.1016/j.immuni.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Qi Y., Li X., Chang C., Xu F., He Q., Zhao Y., Wu L. Ribosomal protein L23 negatively regulates cellular apoptosis via the RPL23/Miz-1/c-Myc circuit in higher-risk myelodysplastic syndrome. Sci. Rep. 2017;7:2323. doi: 10.1038/s41598-017-02403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai J., Wei W. Influence of the RPL34 gene on the growth and metastasis of oral squamous cell carcinoma cells. Arch. Oral Biol. 2017;83:40–46. doi: 10.1016/j.archoralbio.2017.06.035. [DOI] [PubMed] [Google Scholar]
- 25.Bai D., Zhang J., Xiao W., Zheng X. Regulation of the HDM2-p53 pathway by ribosomal protein L6 in response to ribosomal stress. Nucleic Acids Res. 2014;42:1799–1811. doi: 10.1093/nar/gkt971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15:1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 27.van de Ven K., Borst J. Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: rationale and potential. Immunotherapy. 2015;7:655–667. doi: 10.2217/imt.15.32. [DOI] [PubMed] [Google Scholar]
- 28.Cox M.A., Barnum S.R., Bullard D.C., Zajac A.J. ICAM-1-dependent tuning of memory CD8 T-cell responses following acute infection. Proc. Natl. Acad. Sci. USA. 2013;110:1416–1421. doi: 10.1073/pnas.1213480110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soroosh P., Doherty T.A., So T., Mehta A.K., Khorram N., Norris P.S., Scheu S., Pfeffer K., Ware C., Croft M. Herpesvirus entry mediator (TNFRSF14) regulates the persistence of T helper memory cell populations. J. Exp. Med. 2011;208:797–809. doi: 10.1084/jem.20101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Couper K.N., Blount D.G., Riley E.M. IL-10: the master regulator of immunity to infection. J. Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 31.Shouval D.S., Biswas A., Goettel J.A., McCann K., Conaway E., Redhu N.S., Mascanfroni I.D., Al Adham Z., Lavoie S., Ibourk M. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin C., Yu D., Essand M. Prospects to improve chimeric antigen receptor T-cell therapy for solid tumors. Immunotherapy. 2016;8:1355–1361. doi: 10.2217/imt-2016-0125. [DOI] [PubMed] [Google Scholar]
- 33.Katodritou E., Terpos E., Delimpasi S., Kotsopoulou M., Michalis E., Vadikolia C., Kyrtsonis M.C., Symeonidis A., Giannakoulas N., Vadikolia C. Real-world data on prognosis and outcome of primary plasma cell leukemia in the era of novel agents: a multicenter national study by the Greek Myeloma Study Group. Blood Cancer J. 2018;8:31. doi: 10.1038/s41408-018-0059-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Almeida L., Lochner M., Berod L., Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin. Immunol. 2016;28:514–524. doi: 10.1016/j.smim.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 35.Anderson A.C., Joller N., Kuchroo V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norelli M., Camisa B., Barbiera G., Falcone L., Purevdorj A., Genua M., Sanvito F., Ponzoni M., Doglioni C., Cristofori P. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018;24:739–748. doi: 10.1038/s41591-018-0036-4. [DOI] [PubMed] [Google Scholar]
- 37.Hu Y., Zhang Y., Wei G., Chang H., Huang H. Potent anti-tumor activity of BCMA cAR-T therapy against heavily treated Multiple Myeloma and dynamics of immune cell subsets using single-cell mass cytometry. Blood. 2019;134(Suppl 1):1859. [Google Scholar]
- 38.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
scRNA-seq sequencing reads have been submitted to GEO under GEO: GSE151310.