

Abstract
Single-stranded oligonucleotides have been explored as a therapeutic modality for more than 20 years. Only during the last 5 years have single-stranded oligonucleotides become a modality of choice in the fields of precision medicine and targeted therapeutics. Recently, there have been a number of development efforts involving this modality that have led to treatments for genetic diseases that were once untreatable. This review highlights key applications of single-stranded oligonucleotides that function in a sequence-dependent manner when applied to modulate precursor (pre-)mRNA splicing, gene expression, and immune pathways. These applications have been used to address diseases that range from neurological to muscular to metabolic, as well as to develop vaccines. The wide range of applications denotes the versatility of single-stranded oligonucleotides as a robust therapeutic platform. The focus of this review is centered on approved single-stranded oligonucleotide therapies and the evolution of oligonucleotide therapeutics into novel applications currently in clinical development.
Keywords: antisense oligonucleotides, splicing, RNase H, gapmer, steric blocking, CpG oligonucleotides
Graphical Abstract
Single-stranded oligonucleotide therapeutics represent a platform of targeted and selective medicines that address severe genetic diseases that were once untreatable. Continued advances in chemistry, targeted delivery, and new applications have paved the way for a future in which ASO therapeutics become a common modality to treat genetic diseases and beyond.
Main Text
Single-stranded oligonucleotides are synthetic, short, modified RNA or DNA molecules that function in either a sequence-dependent (antisense oligonucleotides [ASOs] and immune stimulatory oligonucleotides [ISOs]) or tertiary structure-dependent manner (e.g., aptamers1). In this review, we focus our attention on oligonucleotide therapeutics whose activities are dictated by their sequences; ASOs bind to their target by Watson and Crick base-pairing,2 while ISOs are recognized and bound by proteins that detect specific motifs.3
Single-stranded oligonucleotide therapeutics have evolved substantially since the use of DNA ASOs were first described in the late 1970s.4,5 The year 1998 saw the first ever approved ASO therapy, formivirsen, which consisted of a 21-mer DNA ASO with a phosphorothioate (PS) backbone (Figure 1).6 Formivirsen, much like the first ASO applications, was an antiviral molecule, designed to target cytomegalovirus. It was not until 2013 that the second ASO drug, mipomersen, obtained US Food and Drug Administration (FDA) approval to treat homozygous familial hypercholesterolemia (HoFH) via RNase H-mediated degradation of apolipoprotein B (APOB).7 Mipomersen represented a new generation of ASOs using new chemical modifications and design that led to more stable (increased nuclease resistance), safer (decreased pro-inflammatory response), and potent (increased RNA binding affinity) compounds with enhanced pharmacokinetic (PK) and pharmacodynamic (PD) properties (Figure 1). During the 15 years between the two approvals, the field of single-stranded oligonucleotide therapeutics matured, leading to an acceleration in the development of this therapeutic modality and an increase in the number of approved drugs. In the last 4 years, six new ASO therapies and one ISO therapy have obtained FDA or European Medicines Agency (EMA) approval. These therapies address a range of genetic diseases across neurological, metabolic/cardiovascular, and muscular conditions as well as potentiate the efficacy of a hepatitis B vaccine. The approved therapies utilize three major types of single-stranded oligonucleotide modalities, that is, gapmers, steric blocking, and CpGs, to modulate gene expression, precursor (pre-)mRNA splicing, and immune pathways. Target organs expanded beyond the eye and liver to the central nervous system (CNS) and muscle. Single-stranded oligonucleotides represent a broad-spectrum therapeutic platform that continues to evolve with innovations in applications, chemistry, and tissue targeting.
Figure 1.

Single-Stranded Oligonucleotide Chemistries
Chemical structures of ribose and backbone modifications used in single-stranded oligonucleotides compared to DNA and RNA structures are shown. Modifications highlighted in green are used in approved oligonucleotide drugs. DNA, deoxyribonucleic acid; RNA ribonucleic acid; Me, methyl; MOE, methoxyethyl; LNA, locked nucleic acid; cEt, constrained ethyl; BNA, bridged nucleic acid; PMO, phosphorodiamidate morpholino oligomer; PO, phosphodiester; PS phosphorothioate.
Approved Single-Stranded Oligonucleotide Therapeutics
Gapmer ASOs
Mechanism of Action
Gapmers are synthetic, single-stranded, and typically 16–20 nt long with a central stretch of 8–10 DNA nt flanked on each side by a stretch or wing of 4–5 nt containing modifications in the sugar ring. These sugar modifications in the wings increase nuclease resistance protecting the ends of the ASO, avoid or reduce an immune response, and increase binding affinity. Commonly, gapmers contain PS bonds throughout to increase stability and plasma protein binding, which improves the PK properties of the gapmer ASO (Figure 1).8 With their typical 4-8-4/5-10-5 (wing-DNA-wing) structure, gapmers bind in a sequence-specific manner by Watson and Crick base-pairing to their target RNA and form a stretch of double-stranded RNA-DNA hybrid. These RNA-DNA hybrids mimic endogenous RNA-DNA hybrids that occur naturally during DNA replication, which are recognized and disrupted via RNase H-mediated cleavage of the RNA strand. The cleavage then leads to the degradation of the RNA. Leveraging this endogenous mechanism, gapmers cause RNase H-mediated cleavage of the target pre-mRNA or mRNA at the location bound by the stretch of DNA. The target transcript is subsequently degraded, resulting in reductions of the target transcript and corresponding protein (Figure 2). As single-stranded ASOs, gapmers are taken up by cells in the CNS, eye, liver, kidney, adrenal glands, and lungs via a natural endocytic process, and they are subsequently released from endosomes into the cytoplasm, reaching the nucleus by a yet to be fully understood mechanism.3 Gapmers can be used to reduce the level of any protein or specific protein isoforms, as well as toxic proteins that result from a gain-of-function or dominant negative mutation.9
Figure 2.

Mechanism of Action of Gapmer ASOs
(A) Expression of an example target gene from transcription through translation of a wild-type or toxic protein. (B) Gapmer ASO with the typical structure of modified chemistry in the wings to protect the ends from nucleases and an internal stretch of DNA that leads to the formation of the RNA-DNA hybrid when bound to the target transcript. In this example, the gapmer ASO binds to an exon in the pre-mRNA and mRNA and recruits RNase H that recognizes the RNA/DNA hybrid and cleaves the RNA. The cleavage triggers RNA degradation, leading to reduction of wild-type or toxic protein levels. Gapmer ASOs can be designed to target other transcript regions, e.g., introns. Even though RNase H is more abundant in the nucleus, RNase H-mediated cleavage of RNA-DNA hybrids can also occur in the cytoplasm.
Mipomersen
Mipomersen (Kynamro; Ionis Pharmaceuticals/Genzyme) was approved by the FDA in January 2013 to treat HoFH, a rare and serious hereditary condition with high risk for premature coronary heart disease associated with atherosclerosis.10,11 HoFH is caused by homozygous or compound heterozygous mutations in one of three genes (APOB, LDLR, or PCSK9), leading to severely elevated low-density lipoprotein cholesterol (LDL-C) levels.10 Mipomersen is a second-generation gapmer ASO with a 5-10-5 structure, 2′-O-methoxyethyl (2′MOE) modifications in the wings (2′MOE-DNA-2′MOE), and PS backbone (Figure 1). Mipomersen was designed to bind to the coding region of the APOB-100 mRNA (an isoform encoded by the APOB gene), and elicit RNase H-mediated degradation of the mRNA and reduce ApoB protein levels (see Figure 2B for the mechanism of action).12
PK studies in preclinical species as well as humans showed that mipomersen has a 30-day half-life in plasma and target tissue (liver).7 In patients with HoFH, mipomersen is administered systemically via weekly subcutaneous injections at a dose of 200 mg and is indicated as an adjunct therapy to lipid-lowering medications and diet to reduce LDL-C, ApoB, total cholesterol, and non-high-density lipoprotein cholesterol. In humans, PD studies showed a mipomersen-mediated reduction of ApoB protein of 46%, which resulted in a 47% decrease of LDL-C.7
During clinical trials, safety concerns of hepatotoxicity as determined by increased levels of transaminases were observed leading to a black label warning for mipomersen.13 Because of the risk of hepatotoxicity, mipomersen is available only through a restricted program under a risk evaluation and mitigation strategy. Other adverse effects include injection site reaction, fever, flu-like symptoms, and fatigue.14 Mipomersen was not approved by the EMA based on safety concerns. In 2016, Ionis and Kastle Therapeutics acquired the rights to mipomersen, and in May 2018 the marketing of the drug was discontinued.
Inotersen
Inotersen (Tegsedi; Ionis Pharmaceuticals/Akcea) was approved by the FDA and EMA in 2018 for the treatment of patients with hereditary amyloid transthyretin (ATTR) amyloidosis, a rare systemic disorder that is characterized by progressive peripheral polyneuropathy, cardiomyopathy, nephropathy, and gastrointestinal dysfunction. The disorder is caused by dominant-negative mutations in the TTR gene that lead to destabilization of the tetrameric TTR protein complex, causing aggregation of monomers into extracellular amyloid deposits.15 The design and mechanism of action of inotersen are analogous to mipomersen, and it was designed to bind and elicit RNase H-mediated degradation of mutant and wild-type TTR transcripts, resulting in decreased mutant and wild-type TTR proteins (see Figure 2B for the mechanism of action).16
Analyses of preclinical species and healthy human volunteers dosed with inotersen demonstrated a more than 80% reduction of plasma TTR protein levels, which correlated with a reduction in the TTR transcript in the liver (target organ) of preclinical species.16 In patients with ATTR amyloidosis, inotersen is administered via weekly subcutaneous injection of a recommended dose of 284 mg to treat the polyneuropathy phenotype. Clinical data indicated that inotersen improved the course of the neurological disease as well as the quality of life of ATTR amyloidosis patients. During clinical trials, safety concerns included thrombocytopenia (fatal in some patients) and glomerulonephritis, which occurred independently in 3% of patients.17 Inotersen is the first approved example of a gapmer ASO that directly targets the root cause of a disease.
Volanesorsen
Volanesorsen (Waylivra; Ionis Pharmaceuticals/Akcea) received a conditional market authorization in Europe issued by the EMA in 2019 for the treatment of familial chylomicronemia syndrome (FCS), which is characterized by 10- to 100-fold increased levels of triglycerides compared to the healthy population, which leads to hypertriglyceridemia and recurrent episodes of pancreatitis. The elevated triglycerides result from reduced or lack of lipoprotein lipase activity caused predominantly by recessive mutations in the LPL gene.18 To address the lack of LPL activity, volanesorsen was designed to reduce the levels of APOC3 mRNA and apolipoprotein C (ApoC)-III protein to decrease plasma triglycerides via an LPL-independent mechanism (see Figure 2B for the mechanism of action).
Volanesorsen is another second-generation gapmer ASO that targets a primarily liver-expressed mRNA. Preclinical studies in rodents and non-human primates showed a significant reduction of ApoC-III and triglyceride plasma levels. Similar results were obtained from the administration of volanesorsen to healthy human subjects, demonstrating a good translation between preclinical species and human clinical studies.19 In patients suffering from FCS, volanesorsen treatment consists of a once weekly subcutaneous dose of 300 mg in combination with dietary restrictions. Clinical data indicated that 77% of patients treated with volanesorsen have significantly reduced levels of triglyceride in plasma, reaching values lower than 750 mg/dL, well below the 880 mg/dL required to reduce the risk of pancreatitis. In these patients, dietary restriction alone was not sufficient to reduce cholesterol levels. Similar to the other two systemically administered, approved second-generation gapmer ASOs, thrombocytopenia and site of injection reactions were common adverse events.20
Steric-Blocking ASOs
Mechanism of Action
Steric-blocking ASOs are synthetic, fully modified, single-stranded oligonucleotides that typically range from 15 to 30 nt in length and contain chemical modifications in the sugar as well as the backbone (Figure 1). Many of the chemical modifications are similar to gapmer ASOs and are also intended to increase nuclease resistance, reduce the immune response, and increase binding affinity. Unlike gapmers, steric-blocking ASOs do not elicit RNase H-mediated degradation of the target RNA, but rather bind to the target RNA via sequence-specific Watson and Crick base-pairing and either hinder the binding of trans-acting factors (e.g., small nuclear RNA [snRNA], microRNA [miRNA], long non-coding RNA [lncRNA], or RNA-binding proteins) to their cognate sequence or prevent the formation of RNA secondary structures.2,9 Their first application dates to mid-1980s in which an ASO was designed to target a 3′ splice site in the herpes simplex virus 1 pre-mRNA preventing splicing and functioning as an antiviral agent.21 Since then, steric blocking ASOs have been used to correct mutation-driven splicing defects and modulate mRNA stability and protein translation.22 Steric-blocking ASOs are thought to be taken up by cells in the CNS, eye, liver, kidney, adrenal glands, lungs, and, to a much lesser extent, to the muscle (see below for a specific example) via the same mechanism as gapmer ASOs.3
Nusinersen
Nusinersen (Spinraza; Ionis Pharmaceuticals/Biogen) was approved by the FDA in December 2016 and the EMA in 2017 for the treatment of spinal muscular atrophy (SMA), a rare neuromuscular disorder that is the most common genetic cause of infant mortality. SMA is caused by recessive mutations in the survival motor neuron 1 (SMN1) gene, and the disease severity is mitigated by a copy gene, SMN2, which produces low levels of normal SMN protein.23 SMN2 exon 7 carries a single nucleotide change with respect to SMN1 that severely decreases its inclusion in the final mRNA, leading to an unstable SMN protein (Figure 3A).24 Nusinersen is an 18-mer, fully modified 2′MOE-PS (Figure 1) that was designed to bind to position +10 of intron 7 of the SMN1/2 pre-mRNA and block access of the splicing repressor hnRNPA1 to its cognate intronic splicing silencer (Figure 3B).25 This results in increased levels of SMN2 exon 7 inclusion, leading to correctly spliced mRNA and increased levels of SMN protein, which compensates for the loss of SMN1 (Figure 3B).26,27
Figure 3.

Mechanism of Action of Nusinersen for the Treatment of Spinal Muscular Atrophy
(A) Region of SMN2 pre-mRNA containing exons 6, 7, and 8. SMN2 exon 7 carries a silent single nucleotide change with respect to SMN1 that causes exon 7 skipping, which leads to an unstable SMN protein. Only 10% of SMN2 pre-mRNA is properly spliced, resulting in an insufficient level of functional SMN protein to compensate for the loss of the SMN1 gene. (B) Nusinersen binding to intron 7 of the SMN2 pre-mRNA and promoting exon 7 inclusion, which leads to increased levels of SMN protein and improved motor neuron function.
Preclinical studies in mice indicated that nusinersen has a long-lasting effect on SMN2 splicing in the brain with continued maximal effect through the 6-month observational period following a single intracerobroventricular injection. In addition, intrathecal (i.t.) injection in non-human primates showed a widespread distribution throughout the spinal cord where motor neurons (target cell) reside.28 PK studies in patients injected i.t. with up to a 9-mg dose of nusinersen showed consistent results with preclinical species and indicated a prolonged half-life of 4–6 months in cerebrospinal fluid (CSF).29 PK/PD analysis in autopsy tissue demonstrated broad distribution of the drug in the spinal cord and brain regions, as well as increases in SMN protein in the spinal cord.30
Clinical trials have successfully demonstrated efficacy in infants and older children treated with nusinersen shown by an increase in motor function and survival.30 Nusinersen is approved to treat infants through adult SMA patients and is administered to patients directly into the CSF via i.t. injection by lumbar puncture at a dose of 12 mg in 5 mL every 4 months after an initial loading dose of 12 mg administered monthly for the first 4 months.31 Adverse reactions include lower respiratory infection, upper respiratory infection, and constipation. The label has warnings for thrombocytopenia and coagulation abnormalities as well as renal toxicity. While these warnings relate to the class of ASOs, they have not been observed for nusinersen.30 In addition to the approved patient population, nusinersen has been further tested in pre-symptomatic infants with mutations in SMN1 and two to three copies of SMN2 in the NURTURE trial, and the results underscore the proven efficacy of the treatment and the importance of neonatal diagnosis.32
Eteplirsen
Eteplirsen (Exondys 51; Sarepta Therapeutics) received an accelerated approval by the FDA in 2016 for the treatment of Duchenne muscular dystrophy (DMD), which is caused by complete loss-of-function mutations in the X-linked DMD gene and predominantly affects males. DMD is a fatal disease due to loss of muscle function caused by the lack of dystrophin protein (Figure 4A). Loss of function mutations in the DMD gene that result in partially functional albeit truncated dystrophin protein (e.g., in-frame deletions) lead to a milder disease presentation known as Becker muscular dystrophy.33 This genotype-phenotype correlation provided the rationale to skip specific DMD exons to avoid deleterious nonsense and frameshift mutations that cause DMD to restore the frame at the transcript level and produce a Becker-like truncated, partially functional dystrophin.34,35 Following this rationale, eteplirsen was designed as a 30-mer phosphorodiamidate morpholino oligomer (PMO) (Figure 1) that binds to exon 51 of the DMD gene, causing the skipping of exon 51 to avoid deleterious mutations. The skipping of exon 51 restores the mRNA frame and leads to the production of an internally truncated, partially functional dystrophin protein (Figure 4B).36 Unlike nusinersen that can treat the vast majority of SMA patients (patients with no SMN2 are very rare), eteplirsen can only address patients who are amenable to exon 51 skipping, which account for 13% of the DMD patient population.37,38
Figure 4.

Mechanism of Action of Eteplirsen for the Treatment of Duchenne Muscular Dystrophy (DMD)
(A) Effect of a deletion of exons 49–50 region in the DMD gene that causes a frameshift and leads to the introduction of a premature termination codon in exon 51. The mutant mRNA is degraded in the cytoplasm by nonsense-mediated mRNA decay (NMD), and no dystrophin protein is produced, causing DMD. (B) Eteplirsen binding to exon 51, which prevents its inclusion and restores the frame. The resulting mRNA lacking exons 49–51 is translated to generate an internally truncated dystrophin protein that retains partial function.
Proof-of-concept studies using DMD patient muscle cells and a humanized mouse model validated the exon skipping approach preclinically.36 PK studies in the mdx mouse model indicated that eteplirsen has a 6-h plasma half-life and is distributed to heart, skeletal, and diaphragm muscles (target tissue), as well as to kidney, where exposure was highest.39 In patients, muscle biopsies and western blot and immunofluorescence analyses detected a small but significant increase of dystrophin.40,41 While muscle is not a tissue that is normally targetable by unconjugated ASOs, the membranes of the muscle of DMD patients are leaky, allowing for eteplirsen to be taken up by myofibers.42
Clinical assessment of patients treated with eteplirsen concluded that there was some preservation of the distance walked in 6 min compared to patients in natural history studies. Despite the small increase in dystrophin and the small number of patients in the clinical trials, the FDA granted conditional approval for eteplirsen for the treatment of males with mutations amenable to exon 51 skipping. A weekly 30 mg/kg dose is administered systemically by intravenous (i.v.) infusion (35–60 min). Adverse reactions include balance disorder and vomiting. The label has no warnings; however, eteplirsen was not approved by the EMA in 2019, citing lack of efficacy. Follow-on studies are still underway.
Golodirsen and Viltolarsen
Golodirsen (Vyondys 53; Sarepta Therapeutics) and viltolarsen (Viltepso; NS Pharma) are treatments for DMD that were granted accelerated approval by the FDA in 2019 and 2020, respectively. The rationale for the development of golodirsen and viltolarsen and their mechanism of action is analogous to eteplirsen. Golodirsen is a 25-mer PMO that was designed to bind to exon 53 of the DMD gene and cause the skipping of exon 53 to avoid deleterious loss-of-function frameshifting mutations. Likewise, viltolarsen is a 21-mer PMO that also binds to exon 53 to avoid mutations. In both cases, the skipping of exon 53 restores the frame and leads to the production of an internally truncated, partially functional dystrophin protein (see Figure 4 for an analogous example). Both golodirsen and viltolarsen address 8% of DMD patients.
Golodirsen and viltolarsen are approved for males with mutations amenable to exon 53 skipping and are administered by i.v. infusion (35–60 min) of a weekly 30 mg/kg dose and 80 mg/kg, respectively. The following adverse reactions have been described for golodirsen: hypersensitivity reactions, including rash, pyrexia, pruritus, urticaria, dermatitis, and skin exfoliation, with some requiring treatment. The most common adverse reactions observed in patients treated with viltolarsen include upper respiratory tract infection, injection site reaction, cough, and pyrexia. In both cases, renal toxicity was observed in animals who received golodirsen or viltolarsen in preclinical studies, but not in the clinical studies.43,44
Milasen: N-of-1 ASO Therapy
The advancements and availability of next-generation sequencings have enabled the identification of pathogenic “private” mutations in a wide range of genetic diseases, especially mutations located in flanking and deep intronic sequences. The vast majority of disease-causing intronic mutations lead to aberrant pre-mRNA splicing. Depending on how disruptive these mutations are to the splicing process, their effect could range from 100% faulty mRNA to varied levels of residual properly spliced mRNA. The latter indicates that splicing is not completely “broken” by the mutation and it could be fixed. Given the targeted nature and success of nusinersen in treating SMA by correcting a splicing defect, combined with local delivery, safety profile, and manufacturing ease,45 steric blocking ASOs have become the preferred modality for personalized treatment.
The first N-of-1 drug was developed to treat a single patient with neuronal ceroid lipofuscinosis 7 (CLN7), a form of Batten’s disease. This condition is a rare and fatal neurodegenerative disease that is characterized by progressive symptoms resulting in blindness, ataxia, seizures, and developmental delay and results from recessive mutations in the MFSD8 gene. At the onset at 3 years of age, symptoms in a patient were mild, but by her sixth birthday she could barely speak. After a Batten’s disease diagnosis was made, genetic screening revealed a known pathogenic mutation in one allele of the MFSD8 gene. Using whole-genome sequencing, a deep intronic insertion of a SINE-VNTR-Alu (SVA) retrotransposon was uncovered. RT-PCR analysis of the patient’s blood cells revealed that the insertion led to the activation of a cryptic 3′ splice site in intron 6, resulting in mis-splicing and consequently the introduction of a premature termination codon. Seven ASOs were designed to target the cryptic 3′ splice site and predicted exonic splicing enhancers, of which three led to an increase in proper MFSD8 splicing. As the chemical modifications used in nusinersen proved safe for local delivery in the CNS, the 2′MOE-PS ASO (Figure 1) targeting MFSD8, which was the most efficacious hit, was selected for further evaluation. The ASO was named “milasen” after the patient’s first name, Mila. Milasen (Boston Children’s Hospital/Mila’s Miracle Foundation) is a 22-mer ASO that was shown to alleviate lysosomal dysfunction phenotypes in the patient’s derived fibroblast cells.46
Preclinical toxicology studies in rats were conducted to show that i.t. injection of a single ascending dose of milasen had no adverse effects at the lowest dose (0.06 mg), which guided the subsequent dose selection in clinical studies. As the patient’s condition continued to deteriorate, an investigational new drug application was filed with the FDA, and in 2018 clinical investigational treatment under an Expanded Access-Investigational New Drug application was initiated. Similar to nusinersen, milasen was administered via i.t. bolus injection by lumbar puncture. The initial dose of 3.5 mg was increased every 2 weeks until it reached 42 mg, which was then administered once every 3 months. The patient was monitored through the first year after the initial injection and no observable serious adverse events were detected. Neurological and neuropsychological assessments at 3 and 6 months after treatment initiation showed stabilization and improvement of Vineland Adaptive Behavior Scales subscores. Seizure frequency and duration decreased by 50% during the follow-up period after treatment initiation compared to pre-treatment measurements.46 Based on subsequent patient evaluations, the dose and dosing regimen was modified and the patient is currently receiving a dose of 70 mg every 2 months (T. Yu, personal communication).
Milasen is an example of the potential of steric-blocking ASOs as a modality applied to the rapid development of personalized treatment for individual patients with fatal genetic diseases caused by splicing mutations. The feasibility of development of N-of-1 treatments as demonstrated by milasen underscores the importance of molecular diagnosis, even if no therapies are available at the time of diagnosis.
ISOs
Mechanism of Action
In addition to splicing modulation and target knockdown, single-stranded oligonucleotides are also being used to stimulate the innate immune system in a variety of applications, including vaccine adjuvants and cancer. Unlike ASOs, ISOs do not bind to RNA, but rather they bind to proteins. They mimic molecular signatures in microorganisms known as pathogen-associated molecular patterns (PAMPs) that trigger the innate immune system via recognition by the Toll-like family of receptors (TLRs) expressed in cells such as antigen-presenting cells (APCs), dendritic cells (DCs), and B cells.47 Examples of PAMPs include double-stranded RNA (dsRNA), which can activate TLR3, single-stranded RNA (ssRNA), which can activate TLR7/8, and unmethylated CpG-containing single-stranded DNA (ssDNA) or dsDNA, which can trigger a protective immune response by activating TLR9.48, 49, 50, 51 Synthetic single-stranded oligonucleotides containing unmethylated CpG motifs can mimic bacterial unmethylated DNA and cause a similar response. There are at least three structurally distinct classes of CpG oligonucleotides, that is, CpG-A, CpG-B, and CpG-C.52,53 The three classes differ in the specific response they trigger, which is influenced by their sequence, chemistry, and higher order structure.54,55 CpG-B ASOs are ssDNA sequences with a PS backbone and contain multiple CpG motifs. They primarily promote the production of T helper (Th)1-type cytokines and type I interferons, the maturation of plasmacytoid DCs (pDCs), and the proliferation and activation of B cells, which boost the immune response to co-administered vaccines (Figure 5).51, 52, 53
Figure 5.

CpG-Containing Oligonucleotides Mediated Immune Cell Stimulation
Internalized CpG-containing ISOs are mainly recognized by TLR9, which activates a complex signaling cascade resulting in the nuclear translocation of transcription factors including AP1, IRF7, and nuclear factor κB (NF-κB). Transcriptional activation of pro-inflammatory genes regulates the maturation of pDCs, the activation and proliferation of B cells, as well as the production of type I interferons and Th1-type cytokines. CpG-mediated immune cell activation, as well as the subsequent secretion of cytokines, stimulates the immune response to the co-administered antigen (not shown), and it produces a more rapid and longer lasting antibody response when compared to alternative vaccine adjuvants. In addition to vaccine adjuvants, single-stranded CpG-containing ISOs are currently also being tested in cancer patients and to modulate the immune response in inflammatory diseases (Table 1).
The first approved vaccine containing a CpG ISO as an adjuvant was Heplisav-B (Dynavax Technologies) in 2017, a vaccine to prevent hepatitis B virus (HBV) infection in adults.52,56 Heplisav-B contains 3 mg of CpG 1018, a 22-mer DNA oligonucleotide with a uniform PS backbone, which is delivered together with 20 μg of the hepatitis B surface antigen (HbsAg) via intramuscular administration in two doses 4 weeks apart.52,56 When compared to Energix-B (GSK), a three-dose hepatitis B vaccine using aluminum hydroxide (alum) as an adjuvant, CpG 1018 induced a significantly improved immune response that was induced more rapidly and lasted longer. In the first phase 2 study comparing Heplisav-B to Energix-B, 79% of Heplisav-B recipients showed a protective antibody response 4 weeks after the first dose compared to only 12% of Energix-B recipients.57 Adverse events, including fatigue and headache, were low and similar in both groups, while mild injection site tenderness was more common in the presence of CpG 1018.57 A more rapid induction of protective antibody levels was confirmed in additional studies.58, 59, 60 Heplisav-B also showed superior long-term effects 50 weeks after the last active dose,60 and it produced seroprotection in patients with chronic kidney disease who were hyporesponsive to currently licensed alum-adjuvanted hepatitis B vaccines.61,62 CpG 1018 is currently being investigated in a phase 1 clinical trial as an adjuvant in an HIV vaccine (ClinicalTrials.gov: NCT04177355) as well as vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (ClinicalTrials.gov: NCT04450004, NCT04405908, and NCT04487210).
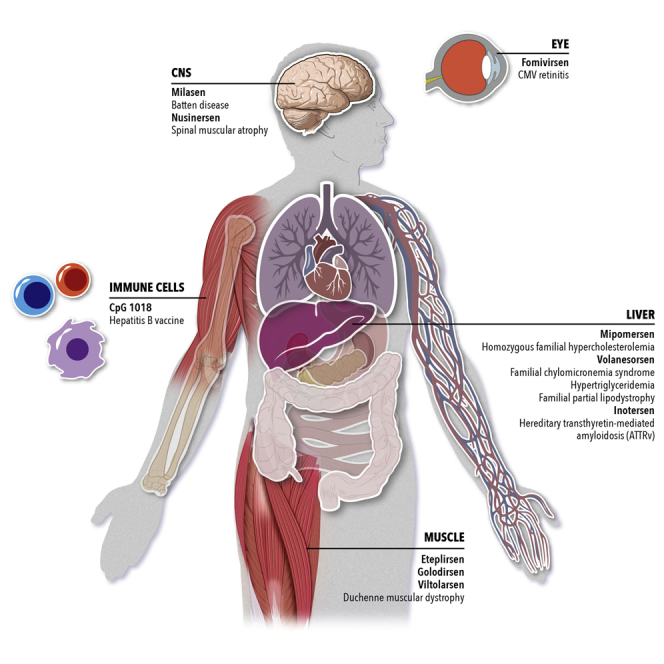
The first nine approved ASO drugs and milasen represent a broad platform with applications in five different organs (Figure 6) and a variety of mechanisms of action. These ASO therapies are administered via local (CNS and eye) or systemic delivery to treat or prevent a wide range of genetic diseases or viral infections. Locally administered ASOs in the eye and CNS have an advantage over systemically injected ASOs, given their lower doses that result in a lower concentration of plasma circulating drug and accumulation of drug in the kidneys. Generally, the higher the ASO dose is, the more likely it is to cause adverse effects. In the second part, this review focuses on some of the mitigation strategies that are underway to reduce ASO doses and improve their therapeutic index by increasing the potency and uptake efficiency of ASOs.
Figure 6.

Single-Stranded Oligonucleotide Therapies
Summary of approved drugs to date and their target tissue. Milasen is a clinical investigational treatment under an Expanded Access-Investigational New Drug application.
Innovations in the Clinic
Beginning with the first DNA oligonucleotides, and continuing through the currently approved single-stranded oligonucleotides (unconjugated DNA, 2′MOE, and PMO sequences), a great deal of understanding and innovations in the ASO field have led to improvements in the PK, distribution, and potency, as well as novel applications of this modality of nucleic acid therapeutics. However, there remains potential for further improvements in ASO potency, specificity, and safety, as well as targeted ASO delivery. Some of these innovations, as well as new applications, are currently being tested in the clinic and will be discussed in further detail.
ASO Chemistry
One of the most successful chemical modifications used in single stranded ASOs is the 2′MOE substitution in the sugar ring (Figure 1). It offers increased nuclease resistance compared to DNA, and it forms duplexes with RNA that are ∼2°C more stable per modification compared to DNA-PS/RNA hybrids.63 Structural studies and molecular dynamics simulations of fully modified MOE-RNA duplexes show that the 2′ substitution locks the sugars in a C3′-endo conformation.64,65 Enforcing a C3′-endo sugar pucker improves affinity for RNA, as well as nuclease resistance in bicyclic 2′,4′-constrained MOE (cMOE) and 2′,4′-constrained ethyl (cEt) modifications (Figure 1), leading to more potent ASOs.66 The first 2′,4′-constrained gapmer ASOs had locked nucleic acids (LNAs) (Figure 1) in their wings. However, some LNA gapmers have been associated with hepatotoxicity in animals,67 which may be linked to increased RNase H-mediated off-target effects68 or specific sequence motifs.69 A clinical trial for an LNA gapmer targeting PCSK9 was terminated due to renal tubular toxicity observed in test subjects.70 Gapmer ASOs with cEt-modified wings have improved potency when compared to 2′MOE, without increased liver toxicity.67,71 Several ASOs containing the new generation 2.5 cEt chemistry are currently being tested in clinical trials (Table 1). One of them is AZD9150 (danvatirsen; IONIS-STAT3-2.5Rx), a 16-mer gapmer with 3-nt cEt wings targeting STAT3 to treat multiple types of cancer (Table 1). Early results from a phase 1b study in patients with diffuse large B cell lymphoma (DLBCL) showed that AZD9150 is tolerated and demonstrated efficacy at a dose of 3 mg/kg in a subset of heavily pretreated patients with DLBCL.72
Table 1.
List of Recent Clinical Trials Using Gapmer and Steric Blocking ASOs and ISOs That Have Completed Trials after January 1, 2020 or Are Currently “Active,” “Recruiting,” or “Enrolling by Invitation” as Described on ClinicalTrials.gov
 |
CpG indicates unclassified CpG oligonucleotides (A. Krieg, personal communication).
ASOs with a stereodefined backbone represent another chemistry innovation currently being tested in clinical trials. Therapeutic ASOs contain PS linkages, enhancing metabolic stability as well as protein-binding properties, facilitating ASO distribution and cellular uptake.73,74 The use of PS linkages in the ASO backbone introduces a chiral center with two configurations, Sp and Rp.75 A k-mer ASO therefore has 2k-1 different stereoisomers, each potentially with a different stability and potency profile, making it challenging to comprehensively study various Sp/Rp configurations. Early work using first-generation DNA-PS ASOs showed that the Sp and Rp forms have different biophysical and biological properties affecting melting temperature (Tm), RNase H activating ability, and nuclease resistance.76,77 Wan et al.75 tested the effect of a uniform DNA gap chirality on a limited number of second-generation MOE and generation 2.5 cEt-containing gapmers. They showed that in vitro, the effect of Rp ASOs was comparable to their stereorandom counterpart, while Sp ASOs had a lower Tm and improved endonuclease resistance. In vivo, Rp and Sp compounds were both quickly metabolized, resulting in low activity, which led to the conclusion that a mixture of Sp and Rp is required to balance RNase H activity and nuclease resistance.75 A screen of stereopure gapmer compounds with one or two Rp and Sp linkages at specific positions did not yield any improvement in potency. However, combining fixed Rp/Sp linkages with a 2′MOE at gap position 2 resulted in a dramatic improvement of the therapeutic index.78 Synthesis of stereopure ASOs, pioneered by Stec et al.,79 was traditionally limited to research use due to low coupling efficiency and difficulties in removing the chiral auxiliary. Advances in the oxazaphospholidine synthesis approach address these traditional hurdles and, while still inefficient compared to phosphoramidite coupling reactions, open the path for stereopure/stereodefined ASOs in clinical applications.80, 81, 82, 83 The first stereopure compound tested in the clinic was suvodirsen (WVE-210201), an ASO targeting DMD exon 51 in DMD patients. However, the ASO was not efficacious in patients, which resulted in the termination of the phase 2/3 trial (ClinicalTrials.gov: NCT03907072). Other stereodefined compounds are currently being tested in patients with Huntington’s disease (HD) in phase 1/2 clinical trials (WVE-120101, ClinicalTrials.gov: NCT03225833; WVE-120102, ClinicalTrials.gov: NCT03225846) (Table 1). In contrast to other HTT-targeting strategies, this technology uses allele-specific knockdown by targeting SNPs linked to the disease-causing CAG-repeat expansions.84 While such an approach would require multiple treatment candidates depending on the SNPs present in the HD patient population, it could provide a safer profile than targeting mutant and wild-type HTT non-selectively. Given the immense number of possible stereoisomers for each sequence, it remains to be determined whether the compounds in development to target HTT are the safest and most efficacious. While some progress has been made to identify a stereochemical code for specific sequences,80 this uncertainty will likely remain a limitation for the development of stereodefined compounds in the near future.
Ligand-Conjugated ASOs
Naked ASOs systemically delivered in vivo are rapidly taken up by liver and kidney.8 While ASO accumulation and activity has been detected in a variety of extrahepatic/extra kidney tissues, systemic delivery to specific target organs is relatively inefficient.8,85 The development of ASO conjugates has enabled tissue-specific targeting of ASOs and short interfering RNAs (siRNAs). Such conjugates include various receptor ligands, cell-penetrating peptides (CPPs), and antibodies, some of which are already in use in the clinic.86, 87, 88, 89 In addition to improving the delivery of ASOs, conjugation led to targeted delivery, allowing for dose reduction of ASOs.
One such ligand is triantennary N-acetylgalactosamine (GN3), a sugar moiety binding to the asialoglycoprotein receptor (ASGP-R). The ASGP-R is primarily expressed on hepatocytes, and the binding of its ligand N-acetylgalactosamine (GalNAc) promotes receptor-mediated endocytosis of the ligand. Leveraging this mechanism, conjugation of ASOs and siRNAs with GN3 promotes the receptor-mediated uptake of the conjugated therapeutic agent. Upon internalization, the conjugate is metabolized by hydrolysis of phosphodiester bonds between the nucleic acid and GN3, as well as hydrolysis of the amide linkages in GN3.90 The use of GN3 as an siRNA conjugate targeting ALAS1 (givosiran) has been approved by the FDA to treat acute hepatic porphyria.88 Several GN3-conjugated gapmers are currently being evaluated in clinical trials to target AGT, ANGPTL3, CFB, GHR, KLKB1, LPA, TMPRSS6, and TTR specifically in the liver (Table 1). In preclinical studies in mice, GN3-conjugated gapmers designed against five different targets showed a 5- to 10-fold greater potency compared to the unconjugated parental gapmer, while being well tolerated. When GN3 was conjugated to a gapmer with higher affinity cEt wings, an ∼60-fold potency increase was achieved.90 Recently published early results from a clinical trial with a GN3-gapmer targeting APOA (APO(a)-LRx) in patients with hyperlipoproteinemia(a) and cardiovascular disease showed a reduction in lipoprotein(a) levels by up to 80% using much lower doses (20, 40, or 60 mg every 4 weeks, 20 mg every 2 weeks, or 20 mg every week) than that of approved unconjugated gapmers, potentially reducing unwanted adverse events such as thrombocytopenia and renal toxicity.91
While GN3 aims to limit delivery of ASOs directly to hepatocytes, other types of conjugates are being used to broaden ASO distribution in the body. For example, there are three splice-modulating ASOs approved for DMD; however, their efficacy is limited by inefficient uptake of ASOs in skeletal muscle.38 Furthermore, DMD also presents in the heart and diaphragm, two regions where ASO uptake is also limited.92 To achieve better tissue distribution, next-generation DMD-targeting ASOs are covalently linked to CPPs. The use of small cationic peptides to enhance cellular uptake of oligonucleotides was first proposed in 1987.93 CPPs are typically 5–30 aa long and can pass through the cell membrane via energy-dependent and -independent mechanisms without interaction with specific receptors.94,95 While there are only few examples where CPPs increase biological activity of negatively charged ASOs or siRNAs (reviewed in Juliano96), CPPs show dramatic effects when linked to neutrally charged peptide nucleic acids (PNAs) and PMOs.97 Muscle-specific CPPs typically contain two tandem repeats of a series of arginine (R), 6-aminohexanoic acid (X), and/or beta-alanine (B) residues with or without a short peptide linker.98,99 A 14-aa peptide, which is highly potent in the heart, diaphragm, and quadriceps, key muscles in the treatment of DMD, is the B-peptide (RXRRBR)2XB.98 In a dog model of DMD, CPP-conjugated PMOs (PPMOs) rescued dystrophin expression in the myocardium and cardiac Purkinje fibers. PPMOs also improved electrocardiogram abnormalities without apparent toxicity, leading the way toward better therapies for DMD.86 PPMOs targeting DMD exons 45 and 51 are currently being tested in patients with DMD (Table 1).
New ASO Applications
Apart from the development of new chemistries and innovations in tissue targeting, new applications of steric-blocking ASOs have also made their way into the clinic. ASO-mediated exon skipping, for example, has traditionally been used to restore the reading frame that was disrupted by frameshifting mutations. A new approach uses an exon skipping strategy to remove retinitis pigmentosa mutations in a constitutive exon of USH2A while maintaining usherin protein function. This strategy was first proposed for diseases caused by mutations in lamin A/C such as Emery-Dreifuss muscular dystrophy.100 Lamin A/C is an intermediate filament and has an α-helical central rod domain. Proteins lacking LMNA exon 5, which encodes part of the central rod domain, localize correctly and perform better in cell-based assays than do proteins harboring dominant-negative missense mutations in exon 5, suggesting that exon skipping is a viable strategy to target missense mutations in exons that encode repetitive domains. Similarly, usherin contains multiple repetitive domains that can be shortened by deleting USH2A exon 13 (exon 12 in the mouse), while maintaining protein function (Figure 7).101 Retinitis pigmentosa patients with pathogenic homozygous or compound heterozygous nonsense and/or missense mutations in USH2A exon 13 might therefore benefit from an exon skipping approach to restore protein function, a strategy currently being tested in the clinic (QR-421a, Table 1).
Figure 7.

Mechanism of Action of QR-421a, a New Application of Steric-Blocking ASOs
The figure depicts a portion of USH2A pre-mRNA containing exons 12, 13, and 14 (middle) in which exon 13 carries missense (blue sign) or premature termination codon (PTC)-introducing (nonsense or frameshift, red sign) retinitis pigmentosa (RP) mutations. Exon 13 encodes half of laminin epidermal growth factor (EGF)-like domain 4, domains 5, 6, 7, and half of laminin EGF-like domain 8 (light blue rectangles highlighted in red). Normally, missense or PTC-introducing RP mutations lead to non-functional full-length or non-functional truncated usherin, respectively (top). QR-421a (+ASO) promotes skipping of exon 13 (depicted by lines connecting exons 12 and 14), leading to the generation of an usherin protein that lacks half of laminin EGF-like domain 4, as well as domains 5, 6, 7, and half of laminin EGF-like domain 8 (bottom), but maintains proper function. Green rectangle, signal peptide; purple rectangle, laminin G-like domain; orange rectangle, laminin N-terminal domain; light blue rectangles, laminin EGF-like domains; green rectangles, fibronectin type III repeats; yellow circles, laminin G; red rectangle, transmembrane domain; blue circle, PDZ binding motif.
While skipping of constitutive exons might also be applicable to other diseases, the exon must be in-frame and code for a redundant domain, which has to be assessed via in vitro and in vivo functional studies. Such applications will therefore likely remain a small niche of therapeutic splice-modulating ASOs.
Another innovative approach, termed TANGO, leverages naturally occurring non-productive alternative splicing events to increase gene expression.102 These events lead to the introduction of a premature termination codon (PTC) in the mRNA, inducing nonsense-mediated mRNA decay (NMD) and no protein production. ASO-mediated prevention of non-productive alternative splicing increased productive mRNA and protein levels in vitro and in vivo.102 A proposed therapeutic application focused on upregulating productive mRNA and protein in autosomal dominant haploinsufficient diseases, such that upregulation of the wild-type gene copy could compensate for the loss of the mutant copy and restore protein levels. An example of this application (STK-001, Table 1) is currently being tested in clinical trials for the treatment of Dravet syndrome (DS), an autosomal dominant haploinsufficiency (Figure 8). Preclinical studies of STK-001 in a DS mouse model demonstrated that preventing the inclusion of a non-productive exon in the Scn1a gene led to increased productive mRNA and restoration of full-length fully functional Nav1.1 protein levels. Nav1.1 protein restoration significantly increased survival and reduced seizures in DS mice.103 Unlike other splicing modulating ASOs in the clinic that address specific mutations and only a subset of the patient population, the TANGO therapeutic approach leverages the wild-type gene copy in the context of autosomal dominant haploinsufficiencies and therefore has the potential to address all patients with heterozygous loss-of-function mutations.
Figure 8.

Mechanism of Action of STK-001, a New Application of Steric-Blocking ASOs
(A) Region of SCN1A wild-type pre-mRNA containing non-productive (non-coding) exon X (yellow rectangle) and flanking coding exons (brown rectangles). SCN1A pre-mRNA is alternatively spliced such that it generates a non-productive mRNA containing the non-productive exon X, which leads to the introduction of a PTC, and a productive mRNA lacking exon X. Upon export to the cytoplasm, the non-productive mRNA is degraded by nonsense-mediated mRNA decay and the productive mRNA is translated into wild-type Nav1.1 protein. The pre-mRNA carrying DS mutations undergoes the same alternative splicing processing, but the mutant productive mRNA does not produce a functional protein (not shown in the figure), leading to haploinsufficiency of Nav1.1. (B) STK-001 (ASO) binding to the non-productive exon X of the SCN1A wild-type and mutant (not shown) pre-mRNA and promotes exon X skipping, which leads to a reduction in non-productive mRNA and increased levels of productive mRNA and wild-type Nav1.1 protein to near normal levels. STK-001 leverages the wild-type gene copy to compensate for the loss-of-function mutant alleles in DS patients.
Single-stranded oligonucleotide therapeutics represent a platform of targeted and selective medicines that have been used to address severe genetic diseases that were once untreatable. With the rise of whole-exome and genome sequencing and the multigene panel mutation screening, molecular diagnosis of genetic diseases is routinely performed leading to the identification of disease-associated genes and earlier diagnosis. Continued advances in chemistry and ligand conjugation has led to the development of new generations of ASO drugs that are more potent and therefore safer. Targeted delivery of ASOs together with new ASO applications are expanding the breadth of the ASO therapeutic platform and have paved the way for a future in which ASO therapeutics become a common modality to treat genetic diseases and beyond.
Acknowledgments
This work was supported by Stoke Therapeutics. We would like to acknowledge Jennifer Fairman from Fairman Studios for the design of the figures. We would like to thank Arthur Krieg for advice on immunostimulatory oligonucleotides. We would also like to thank the reviewers for insightful comments, and Nga Tong who provided editorial support for this review.
Declaration of Interests
I.A. and J.S. are employees and hold shares of Stoke Therapeutics.
References
- 1.Zhu G., Chen X. Aptamer-based targeted therapy. Adv. Drug Deliv. Rev. 2018;134:65–78. doi: 10.1016/j.addr.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kole R., Krainer A.R., Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koller E., Vincent T.M., Chappell A., De S., Manoharan M., Bennett C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011;39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephenson M.L., Zamecnik P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA. 1978;75:285–288. doi: 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zamecnik P.C., Stephenson M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA. 1978;75:280–284. doi: 10.1073/pnas.75.1.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roehr B. Fomivirsen approved for CMV retinitis. J. Int. Assoc. Physicians AIDS Care. 1998;4:14–16. [PubMed] [Google Scholar]
- 7.Geary R.S., Baker B.F., Crooke S.T. Clinical and preclinical pharmacokinetics and pharmacodynamics of mipomersen (Kynamro®): a second-generation antisense oligonucleotide inhibitor of apolipoprotein B. Clin. Pharmacokinet. 2015;54:133–146. doi: 10.1007/s40262-014-0224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geary R.S., Norris D., Yu R., Bennett C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015;87:46–51. doi: 10.1016/j.addr.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Bennett C.F., Swayze E.E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg A.C., Hopkins P.N., Toth P.P., Ballantyne C.M., Rader D.J., Robinson J.G., Daniels S.R., Gidding S.S., de Ferranti S.D., Ito M.K., National Lipid Association Expert Panel on Familial Hypercholesterolemia Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J. Clin. Lipidol. 2011;5(3, Suppl):S1–S8. doi: 10.1016/j.jacl.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Rader D.J., Cohen J., Hobbs H.H. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J. Clin. Invest. 2003;111:1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crooke R.M., Graham M.J., Lemonidis K.M., Whipple C.P., Koo S., Perera R.J. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J. Lipid Res. 2005;46:872–884. doi: 10.1194/jlr.M400492-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Genzyme . 2013. Kynamro: full prescribing information.https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/203568s000lbl.pdf [Google Scholar]
- 14.Genzyme . 2013. Kynamro® (mipomersen sodium) injection prescribing information.https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/203568s011lbl.pdf [Google Scholar]
- 15.Sekijima Y. Transthyretin (ATTR) amyloidosis: clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry. 2015;86:1036–1043. doi: 10.1136/jnnp-2014-308724. [DOI] [PubMed] [Google Scholar]
- 16.Ackermann E.J., Guo S., Benson M.D., Booten S., Freier S., Hughes S.G., Kim T.W., Jesse Kwoh T., Matson J., Norris D. Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid. 2016;23:148–157. doi: 10.1080/13506129.2016.1191458. [DOI] [PubMed] [Google Scholar]
- 17.Benson M.D., Waddington-Cruz M., Berk J.L., Polydefkis M., Dyck P.J., Wang A.K., Planté-Bordeneuve V., Barroso F.A., Merlini G., Obici L. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:22–31. doi: 10.1056/NEJMoa1716793. [DOI] [PubMed] [Google Scholar]
- 18.Johansen C.T., Kathiresan S., Hegele R.A. Genetic determinants of plasma triglycerides. J. Lipid Res. 2011;52:189–206. doi: 10.1194/jlr.R009720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham M.J., Lee R.G., Bell T.A., 3rd, Fu W., Mullick A.E., Alexander V.J., Singleton W., Viney N., Geary R., Su J. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res. 2013;112:1479–1490. doi: 10.1161/CIRCRESAHA.111.300367. [DOI] [PubMed] [Google Scholar]
- 20.Witztum J.L., Gaudet D., Freedman S.D., Alexander V.J., Digenio A., Williams K.R., Yang Q., Hughes S.G., Geary R.S., Arca M. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N. Engl. J. Med. 2019;381:531–542. doi: 10.1056/NEJMoa1715944. [DOI] [PubMed] [Google Scholar]
- 21.Smith C.C., Aurelian L., Reddy M.P., Miller P.S., Ts’o P.O. Antiviral effect of an oligo(nucleoside methylphosphonate) complementary to the splice junction of herpes simplex virus type 1 immediate early pre-mRNAs 4 and 5. Proc. Natl. Acad. Sci. USA. 1986;83:2787–2791. doi: 10.1073/pnas.83.9.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lundin K.E., Gissberg O., Smith C.I. Oligonucleotide therapies: the past and the present. Hum. Gene Ther. 2015;26:475–485. doi: 10.1089/hum.2015.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feldkötter M., Schwarzer V., Wirth R., Wienker T.F., Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 25.Hua Y., Vickers T.A., Okunola H.L., Bennett C.F., Krainer A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hua Y., Sahashi K., Rigo F., Hung G., Horev G., Bennett C.F., Krainer A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Passini M.A., Bu J., Richards A.M., Kinnecom C., Sardi S.P., Stanek L.M., Hua Y., Rigo F., Matson J., Hung G. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rigo F., Chun S.J., Norris D.A., Hung G., Lee S., Matson J., Fey R.A., Gaus H., Hua Y., Grundy J.S. Pharmacology of a central nervous system delivered 2′-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates. J. Pharmacol. Exp. Ther. 2014;350:46–55. doi: 10.1124/jpet.113.212407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiriboga C.A., Swoboda K.J., Darras B.T., Iannaccone S.T., Montes J., De Vivo D.C., Norris D.A., Bennett C.F., Bishop K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology. 2016;86:890–897. doi: 10.1212/WNL.0000000000002445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finkel R.S., Chiriboga C.A., Vajsar J., Day J.W., Montes J., De Vivo D.C., Yamashita M., Rigo F., Hung G., Schneider E. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:3017–3026. doi: 10.1016/S0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 31.US Food and Drug Administration . 2016. FDA approves first drug for spinal muscular dystrophy.https://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy [Google Scholar]
- 32.De Vivo D.C., Bertini E., Swoboda K.J., Hwu W.L., Crawford T.O., Finkel R.S., Kirschner J., Kuntz N.L., Parsons J.A., Ryan M.M., NURTURE Study Group Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: Interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul. Disord. 2019;29:842–856. doi: 10.1016/j.nmd.2019.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koenig M., Beggs A.H., Moyer M., Scherpf S., Heindrich K., Bettecken T., Meng G., Müller C.R., Lindlöf M., Kaariainen H. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet. 1989;45:498–506. [PMC free article] [PubMed] [Google Scholar]
- 34.Aartsma-Rus A., Bremmer-Bout M., Janson A.A., den Dunnen J.T., van Ommen G.J., van Deutekom J.C. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul. Disord. 2002;12(Suppl 1):S71–S77. doi: 10.1016/s0960-8966(02)00086-x. [DOI] [PubMed] [Google Scholar]
- 35.Wilton S.D., Lloyd F., Carville K., Fletcher S., Honeyman K., Agrawal S., Kole R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999;9:330–338. doi: 10.1016/s0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 36.Arechavala-Gomeza V., Graham I.R., Popplewell L.J., Adams A.M., Aartsma-Rus A., Kinali M., Morgan J.E., van Deutekom J.C., Wilton S.D., Dickson G., Muntoni F. Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum. Gene Ther. 2007;18:798–810. doi: 10.1089/hum.2006.061. [DOI] [PubMed] [Google Scholar]
- 37.Aartsma-Rus A., Straub V., Hemmings R., Haas M., Schlosser-Weber G., Stoyanova-Beninska V., Mercuri E., Muntoni F., Sepodes B., Vroom E., Balabanov P. Development of exon skipping therapies for Duchenne muscular dystrophy: a critical review and a perspective on the outstanding issues. Nucleic Acid Ther. 2017;27:251–259. doi: 10.1089/nat.2017.0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aartsma-Rus A., Krieg A.M. FDA approves eteplirsen for Duchenne muscular dystrophy: the next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017;27:1–3. doi: 10.1089/nat.2016.0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarepta Therapeutics . 2016. Eteplirsen briefing document NDA 206488.https://www.fda.gov/media/121650/download [Google Scholar]
- 40.Charleston J.S., Schnell F.J., Dworzak J., Donoghue C., Lewis S., Chen L., Young G.D., Milici A.J., Voss J., DeAlwis U. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology. 2018;90:e2146–e2154. doi: 10.1212/WNL.0000000000005680. [DOI] [PubMed] [Google Scholar]
- 41.Mendell J.R., Goemans N., Lowes L.P., Alfano L.N., Berry K., Shao J., Kaye E.M., Mercuri E., Eteplirsen Study Group and Telethon Foundation DMD Italian Network Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016;79:257–271. doi: 10.1002/ana.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman E.P., Bronson A., Levin A.A., Takeda S., Yokota T., Baudy A.R., Connor E.M. Restoring dystrophin expression in Duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am. J. Pathol. 2011;179:12–22. doi: 10.1016/j.ajpath.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.US Food and Drug Administration . 2019. FDA grants accelerated approval to first targeted treatment for rare Duchenne muscular dystrophy mutation.https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation [Google Scholar]
- 44.US Food and Drug Administration . 2020. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation.https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation [Google Scholar]
- 45.Levin A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019;380:57–70. doi: 10.1056/NEJMra1705346. [DOI] [PubMed] [Google Scholar]
- 46.Kim J., Hu C., Moufawad El Achkar C., Black L.E., Douville J., Larson A., Pendergast M.K., Goldkind S.F., Lee E.A., Kuniholm A. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 2019;381:1644–1652. doi: 10.1056/NEJMoa1813279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Medzhitov R., Janeway C.A., Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295–298. doi: 10.1016/s0092-8674(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 48.Hong D., Kurzrock R., Kim Y., Woessner R., Younes A., Nemunaitis J., Fowler N., Zhou T., Schmidt J., Jo M. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015;7:314ra185. doi: 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krieg A.M. CpG motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 50.Takeshita F., Leifer C.A., Gursel I., Ishii K.J., Takeshita S., Gursel M., Klinman D.M. Cutting edge: role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J. Immunol. 2001;167:3555–3558. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- 51.Kayraklioglu N., Horuluoglu B., Klinman D.M. CpG oligonucleotides as vaccine adjuvants. Methods Mol. Biol. 2021;2197:51–85. doi: 10.1007/978-1-0716-0872-2_4. [DOI] [PubMed] [Google Scholar]
- 52.Campbell J.D. Development of the CpG adjuvant 1018: a case study. Methods Mol. Biol. 2017;1494:15–27. doi: 10.1007/978-1-4939-6445-1_2. [DOI] [PubMed] [Google Scholar]
- 53.Klinman D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004;4:249–258. doi: 10.1038/nri1329. [DOI] [PubMed] [Google Scholar]
- 54.Martinson J.A., Tenorio A.R., Montoya C.J., Al-Harthi L., Gichinga C.N., Krieg A.M., Baum L.L., Landay A.L. Impact of class A, B and C CpG-oligodeoxynucleotides on in vitro activation of innate immune cells in human immunodeficiency virus-1 infected individuals. Immunology. 2007;120:526–535. doi: 10.1111/j.1365-2567.2007.02530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verthelyi D., Ishii K.J., Gursel M., Takeshita F., Klinman D.M. Human peripheral blood cells differentially recognize and respond to two distinct CPG motifs. J. Immunol. 2001;166:2372–2377. doi: 10.4049/jimmunol.166.4.2372. [DOI] [PubMed] [Google Scholar]
- 56.Kaufman M.B. Pharmaceutical approval update. P&T. 2018;43:83–84. [PMC free article] [PubMed] [Google Scholar]
- 57.Halperin S.A., Dobson S., McNeil S., Langley J.M., Smith B., McCall-Sani R., Levitt D., Nest G.V., Gennevois D., Eiden J.J. Comparison of the safety and immunogenicity of hepatitis B virus surface antigen co-administered with an immunostimulatory phosphorothioate oligonucleotide and a licensed hepatitis B vaccine in healthy young adults. Vaccine. 2006;24:20–26. doi: 10.1016/j.vaccine.2005.08.095. [DOI] [PubMed] [Google Scholar]
- 58.Halperin S.A., Ward B., Cooper C., Predy G., Diaz-Mitoma F., Dionne M., Embree J., McGeer A., Zickler P., Moltz K.H. Comparison of safety and immunogenicity of two doses of investigational hepatitis B virus surface antigen co-administered with an immunostimulatory phosphorothioate oligodeoxyribonucleotide and three doses of a licensed hepatitis B vaccine in healthy adults 18–55 years of age. Vaccine. 2012;30:2556–2563. doi: 10.1016/j.vaccine.2012.01.087. [DOI] [PubMed] [Google Scholar]
- 59.Heyward W.L., Kyle M., Blumenau J., Davis M., Reisinger K., Kabongo M.L., Bennett S., Janssen R.S., Namini H., Martin J.T. Immunogenicity and safety of an investigational hepatitis B vaccine with a Toll-like receptor 9 agonist adjuvant (HBsAg-1018) compared to a licensed hepatitis B vaccine in healthy adults 40–70 years of age. Vaccine. 2013;31:5300–5305. doi: 10.1016/j.vaccine.2013.05.068. [DOI] [PubMed] [Google Scholar]
- 60.Sablan B.P., Kim D.J., Barzaga N.G., Chow W.C., Cho M., Ahn S.H., Hwang S.G., Lee J.H., Namini H., Heyward W.L. Demonstration of safety and enhanced seroprotection against hepatitis B with investigational HBsAg-1018 ISS vaccine compared to a licensed hepatitis B vaccine. Vaccine. 2012;30:2689–2696. doi: 10.1016/j.vaccine.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 61.Janssen J.M., Heyward W.L., Martin J.T., Janssen R.S. Immunogenicity and safety of an investigational hepatitis B vaccine with a Toll-like receptor 9 agonist adjuvant (HBsAg-1018) compared with a licensed hepatitis B vaccine in patients with chronic kidney disease and type 2 diabetes mellitus. Vaccine. 2015;33:833–837. doi: 10.1016/j.vaccine.2014.12.060. [DOI] [PubMed] [Google Scholar]
- 62.Janssen R.S., Mangoo-Karim R., Pergola P.E., Girndt M., Namini H., Rahman S., Bennett S.R., Heyward W.L., Martin J.T. Immunogenicity and safety of an investigational hepatitis B vaccine with a Toll-like receptor 9 agonist adjuvant (HBsAg-1018) compared with a licensed hepatitis B vaccine in patients with chronic kidney disease. Vaccine. 2013;31:5306–5313. doi: 10.1016/j.vaccine.2013.05.067. [DOI] [PubMed] [Google Scholar]
- 63.Manoharan M. 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim. Biophys. Acta. 1999;1489:117–130. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 64.Teplova M., Minasov G., Tereshko V., Inamati G.B., Cook P.D., Manoharan M., Egli M. Crystal structure and improved antisense properties of 2′-O-(2-methoxyethyl)-RNA. Nat. Struct. Biol. 1999;6:535–539. doi: 10.1038/9304. [DOI] [PubMed] [Google Scholar]
- 65.Lind K.E., Mohan V., Manoharan M., Ferguson D.M. Structural characteristics of 2′-O-(2-methoxyethyl)-modified nucleic acids from molecular dynamics simulations. Nucleic Acids Res. 1998;26:3694–3699. doi: 10.1093/nar/26.16.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pallan P.S., Allerson C.R., Berdeja A., Seth P.P., Swayze E.E., Prakash T.P., Egli M. Structure and nuclease resistance of 2′,4′-constrained 2′-O-methoxyethyl (cMOE) and 2′-O-ethyl (cEt) modified DNAs. Chem. Commun. (Camb.) 2012;48:8195–8197. doi: 10.1039/c2cc32286b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Swayze E.E., Siwkowski A.M., Wancewicz E.V., Migawa M.T., Wyrzykiewicz T.K., Hung G., Monia B.P., Bennett C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007;35:687–700. doi: 10.1093/nar/gkl1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kasuya T., Hori S., Watanabe A., Nakajima M., Gahara Y., Rokushima M., Yanagimoto T., Kugimiya A. Ribonuclease H1-dependent hepatotoxicity caused by locked nucleic acid-modified gapmer antisense oligonucleotides. Sci. Rep. 2016;6:30377. doi: 10.1038/srep30377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burdick A.D., Sciabola S., Mantena S.R., Hollingshead B.D., Stanton R., Warneke J.A., Zeng M., Martsen E., Medvedev A., Makarov S.S. Sequence motifs associated with hepatotoxicity of locked nucleic acid-modified antisense oligonucleotides. Nucleic Acids Res. 2014;42:4882–4891. doi: 10.1093/nar/gku142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Poelgeest E.P., Hodges M.R., Moerland M., Tessier Y., Levin A.A., Persson R., Lindholm M.W., Dumong Erichsen K., Ørum H., Cohen A.F., Burggraaf J. Antisense-mediated reduction of proprotein convertase subtilisin/kexin type 9 (PCSK9): a first-in-human randomized, placebo-controlled trial. Br. J. Clin. Pharmacol. 2015;80:1350–1361. doi: 10.1111/bcp.12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seth P.P., Siwkowski A., Allerson C.R., Vasquez G., Lee S., Prakash T.P., Wancewicz E.V., Witchell D., Swayze E.E. Short antisense oligonucleotides with novel 2′-4′ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J. Med. Chem. 2009;52:10–13. doi: 10.1021/jm801294h. [DOI] [PubMed] [Google Scholar]
- 72.Reilley M.J., McCoon P., Cook C., Lyne P., Kurzrock R., Kim Y., Woessner R., Younes A., Nemunaitis J., Fowler N. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J. Immunother. Cancer. 2018;6:119. doi: 10.1186/s40425-018-0436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Geary R.S., Yu R.Z., Levin A.A. Pharmacokinetics of phosphorothioate antisense oligodeoxynucleotides. Curr. Opin. Investig. Drugs. 2001;2:562–573. [PubMed] [Google Scholar]
- 74.Vosberg H.P., Eckstein F. Effect of deoxynucleoside phosphorothioates incorporated in DNA on cleavage by restriction enzymes. J. Biol. Chem. 1982;257:6595–6599. [PubMed] [Google Scholar]
- 75.Wan W.B., Migawa M.T., Vasquez G., Murray H.M., Nichols J.G., Gaus H., Berdeja A., Lee S., Hart C.E., Lima W.F. Synthesis, biophysical properties and biological activity of second generation antisense oligonucleotides containing chiral phosphorothioate linkages. Nucleic Acids Res. 2014;42:13456–13468. doi: 10.1093/nar/gku1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Koziolkiewicz M., Krakowiak A., Kwinkowski M., Boczkowska M., Stec W.J. Stereodifferentiation—the effect of P chirality of oligo(nucleoside phosphorothioates) on the activity of bacterial RNase H. Nucleic Acids Res. 1995;23:5000–5005. doi: 10.1093/nar/23.24.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu D., Kandimalla E.R., Roskey A., Zhao Q., Chen L., Chen J., Agrawal S. Stereo-enriched phosphorothioate oligodeoxynucleotides: synthesis, biophysical and biological properties. Bioorg. Med. Chem. 2000;8:275–284. doi: 10.1016/s0968-0896(99)00275-8. [DOI] [PubMed] [Google Scholar]
- 78.Østergaard M.E., De Hoyos C.L., Wan W.B., Shen W., Low A., Berdeja A., Vasquez G., Murray S., Migawa M.T., Liang X.H. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020;48:1691–1700. doi: 10.1093/nar/gkaa031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stec W.J., Grajkowski A., Koziolkiewicz M., Uznanski B. Novel route to oligo(deoxyribonucleoside phosphorothioates). Stereocontrolled synthesis of P-chiral oligo(deoxyribonucleoside phosphorothioates) Nucleic Acids Res. 1991;19:5883–5888. doi: 10.1093/nar/19.21.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iwamoto N., Butler D.C.D., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W.Y., Meena, Standley S.M., Lu G., Apponi L.H. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017;35:845–851. doi: 10.1038/nbt.3948. [DOI] [PubMed] [Google Scholar]
- 81.Iwamoto N., Oka N., Sato T., Wada T. Stereocontrolled solid-phase synthesis of oligonucleoside H-phosphonates by an oxazaphospholidine approach. Angew. Chem. Int. Ed. Engl. 2009;48:496–499. doi: 10.1002/anie.200804408. [DOI] [PubMed] [Google Scholar]
- 82.Guo M., Yu D., Iyer R.P., Agrawal S. Solid-phase stereoselective synthesis of 2′-O-methyl-oligoribonucleoside phosphorothioates using nucleoside bicyclic oxazaphospholidines. Bioorg. Med. Chem. Lett. 1998;8:2539–2544. doi: 10.1016/s0960-894x(98)00450-8. [DOI] [PubMed] [Google Scholar]
- 83.Oka N., Yamamoto M., Sato T., Wada T. Solid-phase synthesis of stereoregular oligodeoxyribonucleoside phosphorothioates using bicyclic oxazaphospholidine derivatives as monomer units. J. Am. Chem. Soc. 2008;130:16031–16037. doi: 10.1021/ja805780u. [DOI] [PubMed] [Google Scholar]
- 84.Aslesh T., Yokota T. Development of antisense oligonucleotide gapmers for the treatment of Huntington’s disease. Methods Mol. Biol. 2020;2176:57–67. doi: 10.1007/978-1-0716-0771-8_4. [DOI] [PubMed] [Google Scholar]
- 85.Hung G., Xiao X., Peralta R., Bhattacharjee G., Murray S., Norris D., Guo S., Monia B.P. Characterization of target mRNA reduction through in situ RNA hybridization in multiple organ systems following systemic antisense treatment in animals. Nucleic Acid Ther. 2013;23:369–378. doi: 10.1089/nat.2013.0443. [DOI] [PubMed] [Google Scholar]
- 86.Echigoya Y., Nakamura A., Nagata T., Urasawa N., Lim K.R.Q., Trieu N., Panesar D., Kuraoka M., Moulton H.M., Saito T. Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA. 2017;114:4213–4218. doi: 10.1073/pnas.1613203114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Levin A.A. Targeting therapeutic oligonucleotides. N. Engl. J. Med. 2017;376:86–88. doi: 10.1056/NEJMcibr1613559. [DOI] [PubMed] [Google Scholar]
- 88.Scott L.J. Givosiran: first approval. Drugs. 2020;80:335–339. doi: 10.1007/s40265-020-01269-0. [DOI] [PubMed] [Google Scholar]
- 89.Sugo T., Terada M., Oikawa T., Miyata K., Nishimura S., Kenjo E., Ogasawara-Shimizu M., Makita Y., Imaichi S., Murata S. Development of antibody-siRNA conjugate targeted to cardiac and skeletal muscles. J. Control. Release. 2016;237:1–13. doi: 10.1016/j.jconrel.2016.06.036. [DOI] [PubMed] [Google Scholar]
- 90.Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsimikas S., Karwatowska-Prokopczuk E., Gouni-Berthold I., Tardif J.C., Baum S.J., Steinhagen-Thiessen E., Shapiro M.D., Stroes E.S., Moriarty P.M., Nordestgaard B.G., AKCEA-APO(a)-LRx Study Investigators Lipoprotein(a) reduction in persons with cardiovascular disease. N. Engl. J. Med. 2020;382:244–255. doi: 10.1056/NEJMoa1905239. [DOI] [PubMed] [Google Scholar]
- 92.Jirka S.M., Heemskerk H., Tanganyika-de Winter C.L., Muilwijk D., Pang K.H., de Visser P.C., Janson A., Karnaoukh T.G., Vermue R., ’t Hoen P.A. Peptide conjugation of 2′-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acid Ther. 2014;24:25–36. doi: 10.1089/nat.2013.0448. [DOI] [PubMed] [Google Scholar]
- 93.Lemaitre M., Bayard B., Lebleu B. Specific antiviral activity of a poly(L-lysine)-conjugated oligodeoxyribonucleotide sequence complementary to vesicular stomatitis virus N protein mRNA initiation site. Proc. Natl. Acad. Sci. USA. 1987;84:648–652. doi: 10.1073/pnas.84.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Guidotti G., Brambilla L., Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol. Sci. 2017;38:406–424. doi: 10.1016/j.tips.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 95.Raucher D., Ryu J.S. Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol. Med. 2015;21:560–570. doi: 10.1016/j.molmed.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 96.Juliano R.L. Peptide-oligonucleotide conjugates for the delivery of antisense and siRNA. Curr. Opin. Mol. Ther. 2005;7:132–136. [PubMed] [Google Scholar]
- 97.Gait M.J., Arzumanov A.A., McClorey G., Godfrey C., Betts C., Hammond S., Wood M.J.A. Cell-penetrating peptide conjugates of steric blocking oligonucleotides as therapeutics for neuromuscular diseases from a historical perspective to current prospects of treatment. Nucleic Acid Ther. 2019;29:1–12. doi: 10.1089/nat.2018.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jearawiriyapaisarn N., Moulton H.M., Buckley B., Roberts J., Sazani P., Fucharoen S., Iversen P.L., Kole R. Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 2008;16:1624–1629. doi: 10.1038/mt.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yin H., Saleh A.F., Betts C., Camelliti P., Seow Y., Ashraf S., Arzumanov A., Hammond S., Merritt T., Gait M.J., Wood M.J. Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 2011;19:1295–1303. doi: 10.1038/mt.2011.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Scharner J., Figeac N., Ellis J.A., Zammit P.S. Ameliorating pathogenesis by removing an exon containing a missense mutation: a potential exon-skipping therapy for laminopathies. Gene Ther. 2015;22:503–515. doi: 10.1038/gt.2015.8. [DOI] [PubMed] [Google Scholar]
- 101.Pendse N.D., Lamas V., Pawlyk B.S., Maeder M.L., Chen Z.Y., Pierce E.A., Liu Q. In vivo assessment of potential therapeutic approaches for USH2A-associated diseases. Adv. Exp. Med. Biol. 2019;1185:91–96. doi: 10.1007/978-3-030-27378-1_15. [DOI] [PubMed] [Google Scholar]
- 102.Lim K.H., Han Z., Jeon H.Y., Kach J., Jing E., Weyn-Vanhentenryck S., Downs M., Corrionero A., Oh R., Scharner J. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat. Commun. 2020;11:3501. doi: 10.1038/s41467-020-17093-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Han Z., Chen C., Christiansen A., Ji S., Lin Q., Anumonwo C., Liu C., Leiser S.C., Meena, Aznarez I. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020;12:eaaz6100. doi: 10.1126/scitranslmed.aaz6100. [DOI] [PubMed] [Google Scholar]