

Abstract
Until recently, there was no approved treatment for a retinal degenerative disease. Subretinal injection of a recombinant adeno-associated virus (AAV) delivering the normal copy of the human RPE65 cDNA led to reversal of blindness first in animal models and then in humans. This led to the first US Food and Drug Administration (FDA)-approved gene therapy product for a genetic disease, voretigene neparvovec-rzyl (Luxturna). Luxturna was then approved by the European Medicines Association and is now available in the US through Spark Therapeutics and worldwide through Novartis. Not only has treatment with Luxturna changed the lives of people previously destined to live a life of blindness, but it has fueled interest in developing additional gene therapy reagents targeting numerous other genetic forms of inherited retinal disease. This review describes many of the considerations for administration of Luxturna and describes how lessons from experience with Luxturna could lead to additional gene-based treatments of blindness.
Graphical Abstract
Until recently, there was no treatment for inherited retinal blindness. Late in 2017, Luxturna became the first approved gene therapy product for a genetic disease in the US and in the European Union, changing the situation. This article presents current considerations regarding the administration of this treatment in the clinic.
Main Text
Inherited retinal degenerations (IRDs) are a large group of molecularly and phenotypically heterogeneous diseases. Disease behavior and severity, which often modulate common phenotypic features are used to clinically subclassify these conditions. Leber congenital amaurosis (LCA) is a subgroup of IRDs (∼5% of all IRDs, prevalence ∼1:80,000–1:200,000) characterized by severe vision loss occurring during the first year of life.1, 2, 3, 4 Patients typically present with visual inattention, inconsistent eye tracking, attraction or aversion to bright sources of light, and eye rubbing. Nystagmus, sluggish pupillary reflexes or amaurotic pupils, and a severely reduced or non-detectable electroretinogram (ERG) are sufficient to confirm this diagnosis.5 Presentations later in life but before the age of 5–10 are grouped under the name early-onset severe retinal dystrophy (EOSRD), severe early childhood onset retinal dystrophy (SECORD), early-onset retinal degeneration (EORD), or juvenile/early-onset retinitis pigmentosa, overlapping terms intended to distinguish these presentations from LCA.6,7 There are now at least 23 genes identified that, when mutated, can lead to LCA, most inherited in an autosomal recessive (AR) manner.8
Most genes implicated in LCA/SECORD encode structural or functional proteins that are expressed either in the retinal pigment epithelium (RPE) or photoreceptors. The RPE65 gene, which is expressed in the RPE, plays a key role in the retinoid cycle as it encodes retinoid isomerohydrolase.9, 10, 11, 12, 13 This enzyme regenerates 11-cis retinal, the chromophore that plays an essential role in phototransduction in photoreceptor cells.14 Bi-allelic loss-of-function mutations in RPE65 result in either a lack of RPE65 protein or protein that is non-functional. Without this protein, photoreceptors have severely impaired responses to light and ultimately degenerate. The biochemical deficit explains the early-onset symptoms of the disease.11,15, 16, 17, 18, 19, 20 RPE65-LCA became the disease model for an IRD phenotypic pattern where a relatively preserved retina is associated with a disproportionately severe vision loss.21 There are individuals with bi-allelic RPE65 mutations whose disease may be diagnosed as retinitis pigmentosa (RP) later in life, presumably due to presence of a hypomorphic allele, but who share all of the key phenotypic features of patients with LCA/EOSRD.19,22,23 RPE65-IRDs thus serve as a model of a phenotypic continuum modulated by disease severity, a relatively common occurrence in IRDs. Individuals heterozygous for these loss-of-function RPE65 mutations can have minimal fundus changes (yellow-white dots, RPE depigmentation, localized peripheral chorioretinal atrophy), but otherwise normal vision.24
There are an estimated 1,000–2,000 individuals in the US with RPE65-IRDs. The true prevalence will become apparent as more individuals with IRDs are genotyped. RPE65 mutations occur in all ethnic groups, and they are thought to account for 5%–6% of LCA. The prevalence is in the range of 2–3 per 100,000 individuals.2,25 There are examples of greater frequency of RPE65 disease due to founder effects in geographically isolated populations, including in the Netherlands and in Israel, however.26,27 As is true with other autosomal recessive conditions, there can be an increased prevalence in the disease in consanguineous populations.
Pathogenesis
Much of what is known about the pathogenesis of RPE65 disease comes from studies of naturally occurring and genetically engineered animal models.28, 29, 30, 31 The first model identified was a Swedish Briard dog, in which disease is caused by a homozygous 5-bp deletion causing a premature stop codon in the RPE65 gene.32,33 Congenital night blindness (optimistically classified at first as stationary night blindness in one study33) was identified in these animals through identification of severely abnormal visual behavior accompanied by an abnormal ERG and abnormal pupillary light reflexes.32,33 Some animals have nystagmus. The ERGs show markedly reduced to non-detectable rod- and cone-mediated responses. ERGs, however, may be clearly detectable with the use of bright stimuli, consistent with an insensitive system.28,29,34 Histopathology early on shows changes limited to the RPE, with accumulation of cytoplasmic lipid droplets that increase in size along with age of the dog. The relatively benign morphologic findings are in contrast to the severe visual dysfunction. Over time and as RPE cell hypertrophy proceeds, photoreceptor morphology degrades and, ultimately, there are severe retinal degenerative changes.32,33 By 10 years of age, optical coherence tomography (OCT) in the affected Briard dog reveals a thinning of outer nuclear layer (ONL) in untreated retina, with lack of photoreceptors subsequently confirmed histologically.35,36 Similar phenotypes (severe ERG abnormalities followed by progressive retinal degenerative changes) were noted in both spontaneous mutant and genetically engineered mouse models with Rpe65 mutations.13,21,31,37,38
RPE65-IRD Phenotype, Clinical Diagnosis, and Differential Diagnosis
RPE65-IRD generally presents before the age of 5 as LCA or an EOSRD.39 Affected infants and young children show difficulties navigating or recognizing objects, toys, and parents’ faces in dimly lit environments, but they show better functioning in brightly illuminated places.19 Staring at brightly illuminated objects (photophilia) is common.19 A small amplitude pendular nystagmus is also recognized by parents, less frequently a gross rotatory or wondering nystagmus, which is more commonly found in other molecular subtypes of LCA.40 Unlike other molecular forms of LCA, eye rubbing or eye pocking to evoke photopsias, or Franceschetti’s oculo-digital sign, is observed only in the most severely affected patients.
Visual acuity is variably impaired but is on average at around 20/200 to 20/100.41 It is not unusual for patients with RPE65-IRDs to show much better acuities up to 20/4 to 20/32.15,41, 42, 43, 44 Color vision is also variably impaired and when measurable can show a non-specific axis of confusion, a tritan defect, or be within normal limits, reflecting variable preservation of the foveal function in these patients.45 As noted above, most patients show a small amplitude pendular nystagmus. The amplitude of their nystagmus and the position of the preferred locus of fixation relates well to the level of visual acuity and overall foveal function. Fixation in untreated eyes occurs at the fovea or within 1° of the foveal center.46 Early in life the fundus examination may appear normal or show only minor epithelial changes with white-yellow dots in the midperiphery (Figure 1, P1 and P4). More advanced disease can show waxy pallor of the nerve, often a bull’s maculopathy, attenuated vessels, and pigmentary changes (Figure 1, P2).7,42,43,47, 48, 49, 50, 51, 52 Areas of relative RPE preservation can be appreciated within the central retina and superior midperipheral fundus (Figure 1, P2, asterisk), even in advanced degeneration. Lacunar atrophic lesions (Figure 1, P3, short arrow) and depigmented whitish round lesions (Figure 1, P4) may be observed in the peripheral and midperipheral fundus. Interestingly, there is a report of a family with compound heterozygous mutations in RPE65 with a phenotype indistinguishable from fundus albipunctatus, including the association dark-adaptation defects.51 The central retina appears normal earlier in the disease, but foveal and parafoveal atrophic lesions are not uncommon even in young patients (Figures 1, long arrows, and 2A).48, 49, 50,53
Figure 1.
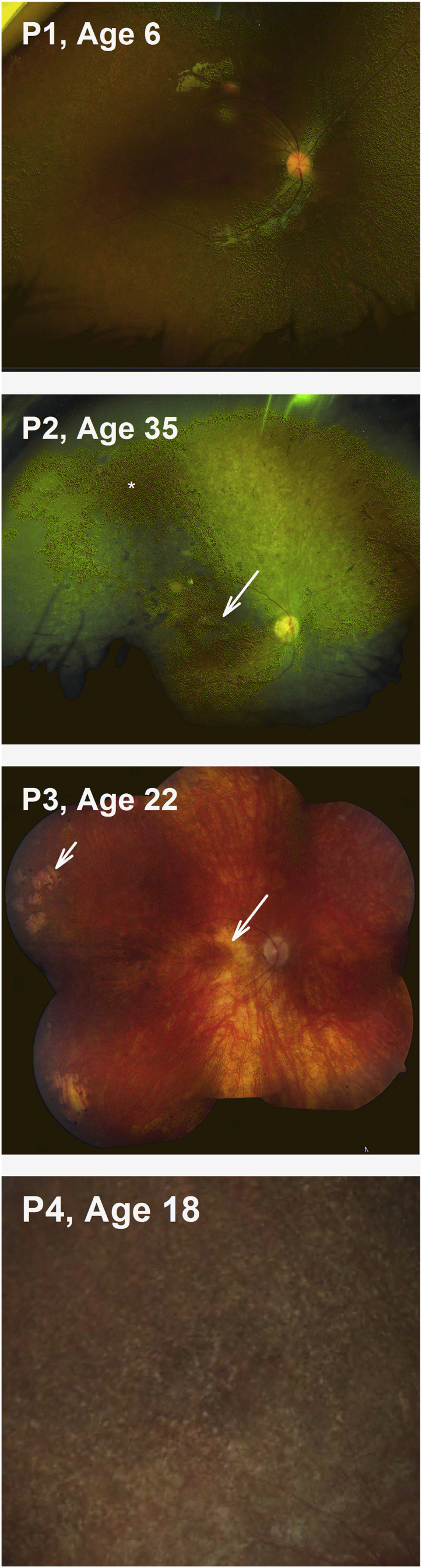
Spectrum of Funduscopic Changes in RPE65-IRD
Arrows point to areas of RPE depigmentation in a bull’s eye configuration (P2 and P3) in the parafovea and to areas of lacunar chorioretinal atrophy (P3) in the periphery. P4 shows yellow-white lesions.
Figure 2.

Structural-Functional Relationships in RPE65-IRD
(A) Color fundus images of the right eye of two of the patients. (B) Goldmann kinetic perimetry with large targets (V-4e and IV-4e) in untreated patients demonstrating limited extent of the visual fields (to the central 20°–40°) and no perception of smaller targets. (C) 7-mm-long, non-straightened, SD-OCT cross-sections along the vertical (VR21) and horizontal (VR25) meridian through the fovea in two patients. Nuclear layers are labeled (ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer). Visible outer photoreceptor/RPE sublaminae are labeled (ELM, external limiting membrane; EZ, inner segment/outer segment ellipsoid region) following conventional terminology. T, temporal retina; N, nasal retina; I, inferior retina; S, superior retina. Scale bars (bottom left), 200 μm. The images illustrate severe foveal abnormalities and the asymmetric extent of the degree photoreceptor preservation around the foveal center (T > N, S > I) at this stage in patients from this family with RPE65-LCA. Asterisk denotes points to severe foveal ONL thinning with approximation of the EZ band to the RPE (VR21) or interruption (VR25). Bar above the scan shows psychophysically determined cone (light-adapted, white stimulus). Dotted line above bar defines lower limit (mean of 2 SD) of sensitivity for control subjects. Images illustrate structural functional dissociation with severe retinal dysfunction contrasting with relatively preserved central retinal structure.
Kinetic visual fields extent measured with a V-4e target can show near full peripheral extent, although more frequently there is generalized constriction and, in more advanced cases, small central islands of vision separated from infero-temporal and/or infero-nasal peripheral remnants of vision by complete midperipheral scotomas (Figure 2B).19,44 With smaller targets the field of vision, if measurable, is often limited to the central 10° of eccentricity (Figure 2B). The relationship between visual acuity or the extent of the visual field and age has been derived mostly from cross-sectional data and limited longitudinal reports and it is not a simple one, with numerous examples of better acuities and large field expanses in the third decade of life.15,20,42,43,50,54 The course, however, is for slow progression of the visual dysfunction with worse visual acuities and fixation stability as well as smaller field extents in older patients, leading to severely impaired vision by the early third decade of life.20,44 There is severe loss of rod and cone function as measured psychophysically by automated static perimetry and full-field sensitivity testing (FST), as well as by electroretinography (Figure 2C).7,20 Most patients show severe loss of rod function often exceeding 4 log units.20 Foveal cone sensitivity is also reduced typically by at least a log unit compared to mean normal sensitivities with greater losses at increasing distance from the center (Figure 2C). The topography of the dysfunction includes peripheral sensitivity loss as the earliest abnormality followed with progression by losses in the midperiphery with relative sparing of the central and peripheral retina.20 ERGs are barely detectable or severely abnormal, consisting mainly of cone-mediated responses.17,55,56 There are, however, examples of detectable rod function by ERG in early RPE65-IRDs.7 Dark-adaptation defects have been demonstrated once sizeable rod function is restored with gene therapy as well as in a single report of a fundus albipunctatus-like condition associated with compound heterozygous mutations in RPE65.57
The total retinal and ONL thickness measured with spectral domain OCT (SD-OCT) can be within normal limits in large expanses of the retina or be limited to residual central islands.17,21,22,48,50,53,58, 59, 60, 61 The ONL thickness in the parafovea is reduced in most patients independent of age (Figures 2C and 3).21 The severity of the regional losses varies even among members of the same family and is not easily predictable by age alone. In general, the topography of the photoreceptor layer (ONL) thickness shows relative preservation within the macula and in superior and temporal pericentral and near midperipheral retina (Figures 2C and 3).21,62 RPE65-IRDs represent the first retinal degeneration where a pattern of structural-functional dissociation was described, a situation where a locally normal or relatively preserved photoreceptors are severely dysfunctional, an ideal scenario for treatments with gene augmentation (Figures 2C).21,27,63 Inner retinal thickening, which likely represents a stereotypical remodeling response to photoreceptor and RPE degeneration, is also observed in RPE65-IRDs and may lead to an underestimate of photoreceptor loss if total retinal thickness (inner + outer retina) is used alone to gauge photoreceptor and/or RPE disease topography and severity, as inner retinal thickening may mask outer retinal loss (Figure 4A).31,64 Of note, while the ONL thickness at the foveal center may be clearly detectable and often within normal limits, structural abnormalities of the foveal/parafoveal cone outer and inner segments may be recognized by SD-OCT imaging as one of the earliest disease manifestations, even in the youngest patients (Figures 2C and 3).65 Although there is variability in severity, progression of the abnormalities occurs with involvement of the foveal center, with most patients showing very limited ONL extent in the third or fourth decade of life (Figure 5). En face imaging with short-wavelength fundus autofluorescence (SW-FAF) is characteristically undetectable or barely detectable even in retinas or retinal regions with otherwise preserved photoreceptors and RPE, a nearly pathognomonic sign of a disease with a blockade in the visual cycle.22,50,56,58 The extent of relative RPE preservation may be approximated by inspection of color fundus images or more precisely by near infrared (NIR) reflectance and FAF in combination with SD-OCT (Figures 4 and 5).66
Figure 3.

Retinal Thickness Topography as a Guide for Retinal Gene Therapy
Montage built from overlapping 30o X 20o “raster” (or volume) scans vertically separated by 0.1 mm from a young patient with RPE65-IRD. Retinal thickness is mapped to a pseudocolor scale shown to the right. Overlaid are 9-mm-long SD-OCT cross-sections sampling regions with a high likelihood of demonstrating detectable photoreceptors. In young subjects the decline in thickness of a visible ONL with increasing distance from the fovea generally corresponds to the decline in overall retinal thickness.
Figure 4.

Assessing Photoreceptor and RPE Health in RPE65-IRDs
(A) SD-OCT total thickness topography (left) and near infrared fundus autofluorescence (NIR-FAF) in an RPE65-IRD patient. Overlaid dotted line defines area with detectable photoreceptors by inspection of individual SD-OCT cross-sections that may be targeted by the subretinal injection or bleb. Inset: normal NIR-FAF. f, fovea. (B) SD-OCT, 6-mm-long cross-sections through the fovea before treatment. Nuclear and outer sublaminae are labeled as in Figure 2. Inset: near infrared reflectance (NIR-REF) image with an overlaying arrow to show the position and orientation of the scans. Scale bar (bottom left), 200 μm. The ONL thickness in cross-section does not accurately match the thickness topography. The red arrow in (A) points to a thicker (warmer color) parafoveal region that does not match the even and symmetrical decline in ONL thickness with distance from the fovea into the nasal and temporal retina demonstrated in the SD-OCT cross-section in (B), suggesting, in the absence of cystoid edema, inner retinal thickening due to secondary inner retinal remodeling. A faint NIR-FAF signal near the center surrounded by background choroidal autofluorescence corresponds in lateral extent with a region of clearly detectable RPE/Bruch’s membrane (BrM), photoreceptor ONL, and EZ signals on the SD-OCT cross-section (diagonal white arrows). A very thin ONL can be traced away from the foveal center into the pericentral retina, well beyond the area of relative preservation of RPE and photoreceptors. Note the hyporeflective space between the EZ and RPE/BrM band at the foveal center that likely corresponds with sparsely distributed and shorter cone photoreceptor outer segments.
Figure 5.

Structural Details by Multimodal Retinal Imaging in Late-Stage RPE65-IRD
(A) Fundus photography: total retinal thickness topography and NIR-FAF in a patient with severe disease. White arrows points to areas of detectable RPE melanin by fundus photography (left panel) and NIR-FAF (right panel) near the foveal center and in peripapillary retina. As in Figure 4, a red arrow points to an area of increased overall retinal thickness that does not match the gradual, even decline in ONL thickness, symmetrical on either side of the fovea. (B) 16-mm-long horizontal SD-OCT cross-section through the fovea in the same patient. Red line overlaid on the NIR-REF image delineates region with detectable, albeit severely thin, ONL, as demonstrated on the SD-OCT cross-section by outlining the outer plexiform layer in yellow. The RPE/BrM is clearly detectable on the SD-OCT scan, and there are spotty signals above the apical RPE/BrM that may correspond with the abnormal photoreceptor outer segment or its remnants. (C) Fundus photography: NIR-REF and NIR-FAF in a patient with severe disease. White arrows point to areas of relatively better coloration in the parafoveal retina (left panel) possibly reflecting surviving RPE, which corresponds to a darker region on NIR-REF. There is virtually no RPE melanin autofluorescence detectable on the non-normalized NIR-FAF (right panel). The area of better coloration on the color fundus image corresponds with a darker image on the NIR-REF and NIR-FAF image suggesting detectable photoreceptors and demelanized RPE overlaying the background choroidal autofluorescence signals that are crisscrossed by large dark choroidal vessels (right panel). (D) 9-mm-long vertical SD-OCT cross-section through the fovea in the same patient. A severely thin ONL, outlined by the outer plexiform layer (in yellow), overlies a thin RPE/BrM signal that likely corresponds to a severely abnormal RPE or bare BrM devoid of overlying RPE. A severely abnormal to non-detectable RPE, even if photoreceptors are identifiable, may be considered an additional contraindication for AAV2.RPE65 augmentation treatment in RPE65-IRD.
Genetic Testing as Basic Eligibility for Treatment
Currently, the most basic criteria for eligibility for treatment with Luxturna is the demonstration of bi-allelic, disease-causing variants in RPE65. Typically, testing for RPE65 variants does not take place antenatally, unless a sibling or spouse is known to carry a disease-causing mutation. Most often, screening for retinal disease-causing variants is not done until it is apparent that a patient has significantly impaired vision and features of a retinal degenerative disease. One challenge, now that more patients are evaluated by next-generation sequencing, is that there is an increased detection of variants of uncertain significance. Some RPE65 variants may be hypomorphic or may have little effect on RPE65 isomerization activity (for example, Rpe65 C330T), in which case treatment with gene augmentation may not result in a significant benefit and where the risks and benefits of subretinal gene therapy should be carefully weighed.22,23,51,67
Dominantly inherited RPE65-associated retinal degeneration has been reported, and the efficacy of gene augmentation in such cases has not been tested.68, 69, 70, 71, 72, 73, 74, 75 Results of molecular and cellular studies suggest that heterozygous RPE65 mutation, such as the D477G mutation, can result in dual mechanisms that include RPE cellular toxicity and enzymatic deficiency.71,75 Improvement of function in RPE65-IRDs following retinoid supplementation in both recessive and dominantly inherited forms of the disease has been documented both in animal models of RPE65 deficiency as well as in patients and may be an alternative option to consider in such cases instead of, or before, attempting gene augmentation.17,76, 77, 78, 79, 80, 81, 82 Although recent documentation of improvement in retinal function following retinoid therapy in dominantly inherited RPE65 disease caused by the D477G mutation indicates that gene augmentation should be similarly effective in such cases, the complexities of the disease suggest that other approaches, such as gene editing or antisense oligonucleotides designed to target the specific mutation, may be have a role as alternative or complementary treatment.75
The challenge remains to identify those variants that are truly disease-causing. In some instances, there are spontaneous mutant or genetically engineered animal models that can be used to predict the likelihood of benefit of gene augmentation therapy for a particular genotype. In others, biochemical studies and/or in vitro mutagenesis assays may be used to confirm the pathogenicity of the variants. Yang et al.83 recently described an in vitro mutagenesis assay of RPE65 protein derived from variants identified in genetic testing of affected patients. The results demonstrated that those variants lacked RPE65 enzyme catalytic activity. The patients were treated with Luxturna and gained retinal and visual function, thereby confirming the utility of the assays.
Treatment of RPE65-IRDs
Until Luxturna became available, the only treatment options for RPE65-associated disease were supportive. These supportive treatments are still important.6 High refractive errors are not unusual in LCA and SECORD and proper refractive correction should be provided, even if severe vision loss is suspected or confirmed to reduce the potential impact of deprivation amblyopia.84 Patients are advised to increase the amount of illumination in their environments (computer screens, room lights, flashlights) to maximize use of their vision and thus their independence. A low vision consultant can provide advice about use of low vision assistive devices (software, phones, computers, lighting). A mobility expert can assist with navigational difficulties. Extra support is often needed to address learning delays or developmental delays due to poor vision. Often physical therapy, occupational therapy, and speech therapy experts can enhance the ability of the individual to function independently. Children with congenital blindness often poke their eyes to evoke phosphenes.85 Eye poking (Franceschetti’s oculo-digital sign) may cause atrophy of the periocular fat and enophthalmos, and has been implicated as an indirect cause of keratoconus (some appear to be molecularly defined) in patients with LCA.86 Eye poking can be controlled with behavioral therapy and by use of protective eyewear. A diet rich in anti-oxidants and vegetables that are rich in carotenoids and xanthophylls is recommended for all individuals with retinal dystrophies, as they may have a protective role.87, 88, 89, 90, 91 Thankfully, associated serious systemic abnormalities have not been reported in RPE65-LCA, and extra work-up and care for neurologic and renal disease commonly found in other molecular forms of LCA/SECORD are not required.
Light exposure aggravates the retinal disease in a number of animal models of IRDs, particularly those with abnormalities in the recovery of retinal function after exposure to light or adaptation abnormalities.92,93 Thus, recommendations regarding light exposure are particularly important for certain degenerations where a mechanistic link is known or suspected from studies in animal models. For example, while RPE65 mutations may confer protection against light damage, treatment has unveiled severe adaptation delays after gene therapy and in a unique form of the disease resembling fundus albipunctatus.51,57 The association between light damage and recovery abnormalities dictates that patients who have undergone gene therapy for RPE65-IRD should be particularly careful avoiding undue exposure to bright lights. Clinicians should also be mindful and limit the use of bright light sources for repeated examinations, such as SW-FAF imaging, especially since it may be tempting to use this modality to objectively confirm visual cycle restoration after gene therapy in RPE65-IRDs.59 Protocols that reduce the output of imaging devices may be an alternative.94 Caution should be exercised especially in patients with LCA and SECORD who are attracted to bright light sources. Families should be instructed to be vigilant to avoid this type of behavior, when outdoors (sun gazing), or around indoor bright light sources, such as bright LED lights. Visible blue light filtering glasses and the use of brims or hats may be recommended for use outdoors.
Gene Therapy Clinical Trials for RPE65-IRDs
Gene augmentation therapy delivers a normal copy of the native human RPE65 cDNA to the diseased RPE cells after subretinal injection of a recombinant adeno-associated virus (AAV).95, 96, 97 The transduced RPE cells then produce the RPE65 protein, the biochemical pathway leading to production of 11-cis retinal is restored, and photoreceptor function is thus improved. Proof-of-concept studies confirmed lasting restoration of retinal function in small and large animal models of RPE65 disease after subretinal delivery of AAV serotype 2 (AAV2).hRPE65 vectors.28,29,38,98, 99, 100, 101 Seven different phase 1 studies followed, which tested gene augmentation therapy for RPE65 deficiency, one of which is still currently enrolling patients (Table 1). As of September 2020, the total number of eyes injected in phase 1 studies is 101 (Table 1). These studies differed in details of the AAV vector (capsid serotype, promoter, intronic sequence, codon optimization of the transgene, presence of Kozak or WPRE sequence, presence of stuffer in the proviral plasmid, method of purification, nature of excipient), the dose and volume delivered, area of retina treated, and specific outcome measures used (Table 1). One of these early trials had cautiously excluded subjects based on presence of AAV2 neutralizing antibodies (NAbs; ≥1:1,000)102 based on prior experience in other trials targeting hemophilia with systemic AAV2 administration. Although the phase 1 studies were focused on safety, the efficacy of the treatment was obvious from the earliest reports, and it replicated in patients the gain of function expected from the pre-clinical work with dramatic increases in light sensitivity ranging from 100- to 45,000-fold (Table 1).27,57,102, 103, 104, 105,107, 108, 109, 110,112, 113, 114, 115, 116, 117, 118, 119, 120 The observations have been repeatedly confirmed since.124 None of the trials reported harmful T cell-mediated immune responses, although there were mild and transient increases in serum antibodies directed at the AAV2 capsid (but not the RPE65 protein) after subretinal injection in one trial.104 In further studies, it was found that although humans can have a high titer of anti-AAV2 antibodies in their serum, these antibodies are absent in intraocular fluid.125 Furthermore, the presence of high-titer antibodies in serum does not prevent AAV2 transduction in the subretinal space in animal models.125 These data allowed the exclusion criterion of anti-AAV2 antibodies to be eliminated in subsequent RPE65 gene therapy clinical studies.
Table 1.
Gene Augmentation Therapy Clinical Trials for Bi-allelic RPE65-IRDs
| Phase | Sponsor: Location | Trial ID: | n | Ages (Years) | Vector | Promoter | Vector Name | Efficacy Outcomes | References |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CHOP: Philadelphia, PA, USA; Naples, Italy | NCT00516477 | 12 | ≥8 | AAV2 | CBA | AAV2-hRPE65v2; voretigene neparvovec-rzyl | visual psychophysics, pupillometry, mobility | 102, 103, 104, 105, 106 |
| 1, FO | CHOP: Philadelphia, PA, USA | NCT01208389, NCT00516477 | 12a,b | ≥8 | AAV2 | CBA | AAV2-hRPE65v2; voretigene neparvovec-rzyl | visual psychophysics, pupillometry, mobility | 102, 103, 104, 105,107, 108, 109, 110, 111 |
| 1 | Applied Genetic Technologies: Gainesville, FL, USA; Philadelphia, PA, USA | NCT00481546 | 15 | ≥8 | AAV2 | CBA-shortened | AAV2-CBSB-hRPE65 | visual psychophysics, pupillometry, mobility | 57,112, 113, 114, 115 |
| 1, 2 | University College, London, UK | NCT00643747 | 12 | 5–30 | AAV2 | hRPE65 | rAAV2/2.hRPE65p.hRPE65 (tgAAG76) | visual psychophysics, ERGs, mobility | 116,117 |
| 1, 2 | Hadassah Medical Organization: Jerusalem, Israel | NCT00821340 | 3 | ≥8 | AAV2 | hRPE65 | rAAV2-hRPE65 | visual psychophysics | 27 |
| 1,2 | Applied Genetic Technologies: Portland, OR, USA; Worcester, MA, USA | NCT00749957 | 12 | ≥6 | AAV2 | CBA | rAAV2-CB-hRPE65 | visual psychophysics, ERGs | 118,119 |
| 1, 2 | Nantes University Hospital: Nantes, France | NCT01496040 | 9 | 6–50 | AAV4 | hRPE65 | rAAV2/4.hRPE65 | visual psychophysics, pupillometry, ERGs | 120 |
| 1, 2 | MeiraGTx UK II: London, UK; Ann Arbor, MI USA | NCT02781480NCT02946879 | 27 | ≥3 | AAV5 | hRPE65 (NA65p) | AAV2/5OPTIRPE65 | visual psychophysics, QOL | 117,121 |
| 3 | Spark Therapeutics: Philadelphia, PA, USA; Iowa City, IA, USA | NCT00999609 | 29b | ≥4 | AAV2 | CBA | AAV2-hRPE65v2; voretigene neparvovec-rzyl | visual psychophysics, pupillometry, MLMT | 110,111,122,123 |
Psychophysic testing includes visual acuity, full-field sensitivity testing, and visual fields (kinetic, static, and fundus-projected). CHOP, The Children’s Hospital of Philadelphia; QOL, quality of life questionnaire; NA, not available; AAV, adeno-associated virus; CBA, chicken β-actin; FO, follow-on; MLMT, multi-luminance mobility test, ERG, electroretinogram.
11 patients injected.
Bilateral sequential injections.
Continued progression of retinal degeneration in both affected dogs and patients despite evidence of efficacy after gene therapy treatment has been reported.35,36,82,114,117 Studies in the dog model strongly suggest that long-term arrest of progression after RPE65 gene therapy may only occur in retinal regions with relatively retained photoreceptors at the time of treatment.36 It is actually possible that some of the patients included in the RPE65 gene therapy clinical trials may have had locally severe photoreceptor losses exceeding the threshold needed to arrest degeneration, affecting long-term efficacy outcomes.35,115,117 Reported durability of the improvement in vision by FST and a multi-luminance mobility test (MLMT) effectively documented stability of the treatment effect for the better preserved retinal regions or less severely affected individuals, which may have not crossed the local degeneration threshold at the time of the treatment, thus reconciling the apparent contradictions in the outcomes of these trials in terms of the longevity of the treatment effect.107, 108, 109, 110,122 It will be important in going forward to further interrogate progression of retinal degeneration in patients in whom the numbers of variables have been reduced (e.g., vector, disease severity, dose, volume) in order to identify the conditions that both maximize efficacy and halt the degenerative process.
Only one group (The Children’s Hospital of Philadelphia [CHOP], ClinicalTrials.gov: NCT01208389 and NCT00516477) proceeded to studies where the same dose and volume of vector were administered to each subject. The first study was a follow-on study where the contralateral eye was treated.107 The second study was a phase 3 clinical trial (ClinicalTrials.gov: NCT00999609), the first randomized, multi-center, open-label gene therapy trial in the US, funded by Spark Therapeutics.105,107,109,110,123 The phase 3 trial evaluated effects of bilateral subretinal injection of Luxturna in a total of 29 subjects (total of 58 eyes, Table 1) who were randomized such that 9 control subjects were followed for a year without treatment and then crossed over and received the intervention.107,109,110 Twenty-one subjects received bilateral retinal injections immediately after eligibility was confirmed, although one person dropped out of the study soon after injection. Thus, a total of 29 subjects (21-1 dropout plus 9 subjects crossing over from the control group) were evaluated long-term through the trial. The inclusion criteria included age ≥4 years, presence of bi-allelic RPE65 mutations, and sufficient viable retinal cells within the posterior pole as estimated by OCT. Extensive baseline testing with retinal imaging and function studies was conducted prior to randomization to the control versus treatment group. These data later served as the comparator for year 1 post-intervention data. The primary endpoint of the phase 3 study was a change in functional vision at 1 year post-intervention reflected by an improvement in the ability of the treated subjects to orient themselves and navigate in dimmer environments compared to untreated controls. Building upon previous experience on the use of mobility as an outcome measure of functional vision, an MLMT was developed in the phase 1 and phase 1 follow-on studies, and then validated in a population of normal-sighted and visually impaired individuals.102,107,110,114,116,117,126, 127, 128 The test was developed by academic investigators after consultation with the US Food and Drug Administration (FDA) prior to formation of Spark Therapeutics. The investigators had proposed to use a change in pupillary light reflexes as a primary outcome measure, but the FDA had discouraged this measure as it does not reflect functional vision. The FDA was more receptive to a mobility test, as that would better reflect challenges in daily living in vision-impaired subjects. The test was designed to systematically measure changes in functional vision, specifically the ability of a subject to navigate an obstacle course accurately and quickly at different levels of illumination. Performance of the MLMT thus depends on a variety of aspects of vision, including visual acuity and visual field extent and sensitivity. Each of these parameters is impaired in individuals with RPE65-IRDs. The levels of illumination used in the MLMT ranged from 1 lux (the amount of light one might see outside in the countryside on a moonless summer night) to 400 lux (a typical brightly lit office).127 Scoring of MLMT performance was assessed by independent graders. A positive change score reflected the subject’s ability to carry out the MLMT test at a lower light level. Three secondary endpoints were tested: FST using white light and the change in the MLMT score for the first assigned eye; visual acuity was used as a safety measure whereby a worsening score after treatment would indicate a potential safety concern.109
The data from the study revealed robust improvement in all of the primary and secondary endpoints. MLMT performance (the primary endpoint) showed a change score of 1.6 (p = 0.0013). FST showed a >2 log units increase in light sensitivity (p = 0.0004), and MLMT performance with the first treated eye (which was the worse seeing eye at baseline) showed a change score of 1.7 (p = 0.0005). There was a non-significant trend in improvement of visual acuity (p = 0.17).109 Additional outcomes that were evaluated included visual fields and responses to a modified visual function questionnaire. There was robust improvement in visual fields after intervention as measured by either Goldmann testing (p = 0.0059) or by light-adapted automated static perimetry testing (p = 0.0005). A visual function questionnaire adapted for use in pediatric patients and in those with extremely poor vision was also used. There was a significant improvement in visual function scoring in individuals after receiving Luxturna compared to baseline (p < 0.001). Further analyses supported the reliability, validity, and responsiveness of a modified visual function questionnaire (mVFQ-25) as a measure of functional vision for use in patients with IRD due to RPE65 mutations.129 A recent durability study showed that the beneficial effects of Luxturna administration, which are nearly maximal by 30 days after administration, are durable for 4 years, with observation ongoing.110
Gene Therapy as the New Standard of Care for RPE65-IRDs
Now that Luxturna has been approved for clinical used as a drug, the procedures used during the clinical trials are being adapted to meet the demands of use as a standard of care. Numerous patients have been treated, and the numbers expand as additional treatment centers are activated around the world. Establishing the diagnosis by clinical evaluation to confirm the expected disease phenotype and by genotyping to detect the presence of bi-allelic pathogenic RPE65 mutations are the first steps toward treatment. Once an RPE65-IRD has been confirmed, the subject is further evaluated to be sure they meet the treatment indications for Luxturna administration. As per the FDA drug label, patients should be older than a year of age, have no contraindications to dilating drops and other medications that will be used during surgery and recovery, or to a pars plana vitrectomy. More importantly, they should have sufficient treatable (“viable”) photoreceptor cells as determined by SD-OCT. This basic description of the eligibility for treatment, however, may not be straightforward for the general clinician, particularly when deciding treatment for patients with the earliest or end stage of the spectrum of disease severity. As for any other medical procedure, the central question relates to risks versus benefits. That is, it is key to try to determine how early or late in the course of the disease is worth treating and to understand the potential complications and limitations of the treatment to be able to counsel patients appropriately.
Patient Eligibility for Treatment
Patient eligibility for RPE65 gene therapy clinical trials rested on detecting photoreceptors by detailed retinal imaging, including topographical mapping of the disease across large expanses of retina.21,57,65,102,104, 105, 106, 107,110,111,116,117,123 Some of the clinical trial protocols were designed not only to determine eligibility, but to define target regions for treatment with relatively preserved photoreceptors that would serve the patient’s vision best, for example, by targeting rescue of the inferior, central to midperipheral visual fields (Table 2). This ideal testing algorithm may not be possible within the time constrains of typical clinical encounters, especially in the youngest or more severely affected patients. Treatment teams should take this into consideration and adjust visits to allow sufficient time for the evaluation of potential candidates. Ancillary staff should also be familiarized with the demands of testing patients with severe retinal dysfunction, including patients with unstable and eccentric fixation. An alternative practical solution to extensive topographical mapping of the ONL is the use of long line scans at the highest speeds available on the OCT instruments and directed to the central-to-near peripheral superior and temporal retina, where remnants of photoreceptors and vision are expected to exist in patients with more severe disease (Figure 3).20,62
Table 2.
Eligibility for Gene Augmentation Therapy with Luxturna.
| Requirement/Factor | Recommendations |
|---|---|
| Biallelic disease-causing RPE65 mutations | genetic testing of proband and parents/family members to confirm segregation of the RPE65 variants |
| consider screening for involvement of additional molecular causes particularly in consanguineous families and the most severe patients | |
| >1 year of age | consider postponing inclusion to patients older than 2 years of age, unless there is evidence for severe vision loss (poor fixation, wandering nystagmus) |
| No surgical contraindications | perform comprehensive examination |
| Detectable photoreceptors | topographic mapping of the central and near midperipheral retina with OCT with segmentation of the OCT cross-sections |
| may be substituted by line profiles through main meridians | |
| best outcome should be for patients with ≤30% ONL loss | |
| topography of photoreceptor preservation dictates surgical plan and positioning of retinotomies and subretinal blebs | |
| Detectable RPE | determined by inspection of color fundus photography, near infrared reflectance, or fundus autofluorescence imaging |
| detectable RPE should co-localize with the regions of detectable photoreceptors | |
| Measurable vision | measurable classic photoreceptor function by FST and/or perimetry, complemented, if possible by ERG and/or pupillometry |
| ideally patients should be tested with perimetry and letter visual acuity quantified (Teller acuity in younger children) | |
| perimetry may be performed using large Goldmann V targets; dark-adapted perimetry will increase dynamic range | |
| poorer central vision requires consideration of mutations in other IRD genes as well as neuro-ophthalmic causes of vision loss | |
| determine whether there is structural-functional dissociation as evaluated with OCT and perimetry/FSTs | |
| Foveal health | avoid extension of the blebs to the foveal center if there is concern for early foveal/parafoveal outer segment shortening and photoreceptor loss |
ONL, outer nuclear layer; OCT, optical coherence tomography, ERG, electroretinogram; IRD, inherited retinal degeneration.
Care should be exercised when determining whether photoreceptors are still present in a given patient, particularly in end-stage disease. The clinician should be aware, for example, that total retinal thickness may overestimate the presence of photoreceptors since thickening as the result of inner retinal remodeling may mask outer retinal loss (Figure 5A, red arrow).31 Careful inspection of the SD-OCT cross-sections should be thus performed before deciding eligibility for treatment (Figures 3, 4, and 5). Likewise, using fixed cutoff values of total retinal thickness (such as central subfield thickness >100 μm) should also be avoided as proof of retained photoreceptors. Demonstration of detectable photoreceptors, however, is not the only structural prerequisite for treatment. Experimental evidence suggests degeneration may proceed after treatment if a certain threshold of photoreceptor loss has been crossed (see below).82 Loss of ∼30% of the normal complement of photoreceptors (locally or across the entire retina) is a figure that has been proposed as the threshold beyond which progression of the degeneration may occur, even in situations where the visual cycle and functional rescue have taken place.36,130 For example, it is possible that locally preserved photoreceptors may continue to degenerate after treatment when neighboring similarly treated retina has already crossed the threshold for continued degeneration. The opposite is also possible, i.e., that degeneration may be arrested locally independent of the health of other retinal regions, as demonstrated experimentally.28 Slower rates of photoreceptor degeneration, even if not total arrest, may be desirable to patients and may provide improved quality of life until the solution is found through other treatment alternatives, such as cellular replacement strategies. Longitudinal studies in a larger number of patients following this treatment are still required to confirm the different scenarios and serve as guidance to practitioners to inform and select patients for treatment.
Although there is evidence for early and severe cone loss in both patients and animal models of RPE65-IRD, photoreceptors, particularly cones, may survive in areas of severe RPE degeneration (Figure 5).56,76,99,131, 132, 133, 134 Detectable photoreceptors devoid of a supporting, relatively preserved RPE, where the basic abnormalities in RPE65-IRD reside and that is the primary cellular target of the gene transfer, would contraindicate treatment. SW-FAF is now routinely used in the clinic to delineate areas of RPE loss. This has limited utility in RPE65-IRD where FAF signals may be often undetectable, and, in addition, because of the potential for light-mediated injury on repeated examinations, especially post-treatment. The clinician may resort to the clinical examination and the inspection of retinal imaging using color photography, NIR reflectance, and/or NIR-FAF imaging, and SD-OCT to determine the extent of relative RPE preservation (Figures 4 and 5).66 Inspection of the SD-OCT cross-sections to determine whether there are detectable photoreceptors and whether RPE signals apical to Bruch’s membrane are still detectable should occur (Figure 5).
Candidates for treatment should have a comprehensive eye examination and age-appropriate measures of vision. Patients should have their color vision and extent of their visual fields documented by kinetic and/or static perimetry, as both are expected to change following treatment.44,45 Their ability to perform these tests reliably, together with the measures of photoreceptor and RPE structure mentioned above, may be factored in when considering eligibility, as a functional retina may be indicative of potential for rescue, particularly if a structural-functional dissociation is documented.21 Severe central dysfunction with nystagmus and poorer visual acuity than that of the average RPE65 patient of a given age or severity may be a valid argument for earlier intervention in children, while demonstrating measurable reliable vision mediated by classical photoreception may be a prerequisite for adult patients with severe disease.
Considerations Regarding Time Windows for Intervention
Cone photoreceptor degeneration and foveal cone photoreceptor outer segment shortening are among the earliest abnormalities in RPE65-IRD, and central atrophic lesions ensue at some point in the course of the disease (see Figure 3).50,62,65,133,134 Unambiguous improvement of foveal function was not documented after treatment in the clinical trials that measured foveal function after subfoveal injections.46,57 Some of the improvements in visual acuity were associated with changes in fixation preference toward extrafoveal locations within the regions improved after gene therapy.46 Treating the parafovea and pericentral retina early in life may allow for rescue of central visual function and prevention of deep sensory amblyopia and may be considered an argument for early intervention and for treatment of potentially fragile foveal centers. However, despite the strong evidence in support of cone photoreceptor rescue after both retinoid supplementation and gene augmentation in RPE65-IRDs, it is still unclear whether early treatment of the parafovea/fovea in patients will lead to restoration of the abnormalities and/or prevent the development of atrophic central lesions in patients in the long term.28,57,76,133,134 The surgical risks and sequelae (e.g., anesthetic risks, retinal detachments, macular holes, foveal thinning, cataracts) of the complex surgical intervention, specifically for the pediatric population, should be thus carefully considered. Specifically, foveal thinning (clinical or subclinical) and macular hole formation have been documented after the subfoveal injections used to deliver various gene therapy products, yet the factors predisposing to such an undesirable outcome have not been fully elucidated.102,112,114,120,135, 136, 137 The vulnerability of the fovea likely reflects an interplay between the foveal disease and other factors, such as vector concentration and the fluid dynamics of the subretinal injections, details that should be carefully factored in patient selection and surgical planning (see Surgical Considerations).138,139 Thus, until we know more, the treatment should not become a race to demonstrate surgical skills by treating younger patients, but instead taken as a challenge to reach a balanced decision with all risks and benefits, some still hypothetical, carefully weighed.
As noted above, there is experimental evidence in animal models and from some of the analyses of the clinical trials suggesting that there may be limits to the rescue of the degeneration that are dependent on disease severity.36 This is expected to vary from patient to patient, and locally within the retina. Thus, in theory, treatment of earlier disease (independent of age) should carry a better prognosis in terms of the ability of the treatment to not only improve vision, but to halt progression of the degenerative changes.36 A generalizing view holds that most children with RPE65-IRD may have crossed the threshold to be able to arrest local degeneration with treatment, although it is still not clear whether the extent of degeneration, in terms of the size of the retinal areas exceeding this threshold, should be factored in.36,62 Careful evaluation of photoreceptor health is thus needed before treatment for surgical planning and to best inform patients and families about treatment expectations, and after treatment, to asses both the highly reproducible vision improvements, as well as to determine long-term efficacy signals, such as the persistence of the improvements and the arrest of the photoreceptor and RPE degeneration. Attention to the various parameters affecting treatment outcome will add to our knowledge base and will help us further fine-tune this treatment. Conversely, can a patient expect improvement if treated beyond that theoretical window for treatment? Patients treated during the clinical trials that preceded the approval of Luxturna were rarely at the end stage of the disease, but rather at stages where substantial photoreceptors and RPE could be demonstrated by SD-OCT. Thus, it was unclear whether efficacy signals could be expected in such patients. Since the introduction of Luxturna to the clinic, a number of patients with end-stage disease seeking treatment to improve their functional vision have been treated after a thorough discussion and management of the expectations of the treatment in such a scenario. This has often led to an unequivocal improvement in retinal sensitivity (see below). Again, it remains to be demonstrated whether such intervention delays total photoreceptor and vision loss. The hope is that the treatment may serve as a temporizing measure until cell replacement solutions are ready for use in the clinic.
Surgical Considerations
Variable foveal thinning and macular hole formation after subfoveal injections were first observed in the RPE65 gene therapy clinical trials and have since been documented in other studies that have treated the central retina with subretinal delivery of different vectors.102,112,114,135,136 Targeting the parafoveal retina may be desirable, but avoiding the center may not be possible (see below). It is also important to manage expectations if Luxturna is to be administered, especially for older individuals with few remaining photoreceptors. Every surgery and general anesthesia come with potential risks. While the level of safety of Luxturna administration is high, there are surgical risks and the potential that a subject will not develop a meaningful response. There are a number of variables that can also potentially affect the magnitude of response, including the number and location of remaining photoreceptors, presence of amblyopia, the nature of the patient’s RPE65 mutation(s), and the presence of additional gene mutations impacting retinal function.
Baseline testing (imaging and retinal function studies, similar to those carried out in the phase 3 trial) is conducted prior to treatment with Luxturna. The surgical procedure that is used is nearly identical to that used during the phase 3 trial. Luxturna is delivered subretinally under direct visualization through the dilated pupil using standard vitreoretinal surgical techniques. In the phase 3 trial, delivery of this reagent was preceded with macular tamponade with heavier-than-water perfluorocarbon liquid that was done in an effort to buttress the fovea from hydrodynamic stresses of the injection.109 Perfluoron was then removed after the bleb had been formed. The Perfluoron step was eliminated once the drug was approved, as the Perfluoron “bubble” often rolled away from the fovea during the injection procedure, thereby not serving its intended purpose.
The patient is given oral corticosteroids (1 mg/kg prednisone/day or 40 mg/day maximum dose) for 3 days prior to injection in order to counteract any potential immunization effect of the viral vector and transgene. Only one eye is injected per procedure. If the second eye is to be injected, that procedure is carried out within the subsequent 2 weeks. An examination under anesthesia is usually performed on the first eye at the time of the second surgery. The subretinal injection, accomplished through a transvitreal and transretinal approach, is carried out while the patient is under general anesthesia, although this surgery can be done under monitored sedation in selected individuals. Subretinal injection delivers the AAV adjacent to the affected (RPE) cells and minimizes escape and/or dilution of the vector to systemic sites or the rest of the eye. The procedures are carried out with a standard three-port pars plana approach, where one port is used to control the volume of the fluid-filled eye, another is used to introduce a light source, and the third is used to deliver the active surgical devices. The pars plana is the region ∼3–4 mm from the periphery of the cornea where neural retina is thinnest (and does not contribute to vision).
Before the AAV is injected, the jelly-like material (“vitreous”) in the center of the eye is removed by simultaneously cutting it and applying gentle suction with a high-speed cutting/aspiration device (vitrector). This material is replaced with sterile saline to maintain vitreous volume. Prior to inserting the cannula, the retina is inspected for epiretinal surface membranes, which, if present, are removed manually. If there are retinal tears, these are treated with laser photocoagulation. The AAV vectors are then injected in each retina one time only using a subretinal injection cannula with a 38G to 41G blunt-tipped needle, targeting the region of the retina with evidence of residual photoreceptors (as judged by OCT).107,109,110 A small retinotomy is created during the injection, often by the injection stream itself. The cannula can be tracked under the retina and allows access to the subretinal region (which is normally occupied by tightly interdigitated photoreceptor outer segments and RPE cells). The retinotomy is carefully placed in the immediate vicinity of the central macula (≥3 mm distant from the fovea) such that subsequent injection does not apply physical stress to this structure (Figure 6). Ideally, the retinotomies should be distanced ≥2 mm from regions with clearly detectable photoreceptors expected to be rescued by the treatment to avoid scotomas resulting from the variable size of the chorioretinal scars that develop around the retinotomies. The gene therapy product is injected, creating a localized retinal detachment (“bleb”; Figure 3A, overlaid contour; Figures 6 and 7A), which should ideally include regions with detectable receptors that are of strategic importance for the patient’s vision, such as the pericentral and superior midperipheral retina that subserve inferior and central fields. The size of the bleb is in direct proportion to the volume of material injected. Typically, 300 μL of AAV solution is injected in treatment of RPE65 disease. The concentration of the compound in the bleb remains high, as it is not diluted by vitreous or other fluid, even if some of the contents reflux into the vitreous through the retinotomies.140 In fact, focus should not be on delivering an arbitrarily prescribed volume that may lead to blebs that are unnecessarily elevated, potentially increasing mechanical stresses.139 Perhaps shallower blebs that slowly expand over large expanses of rescuable retina/RPE should be the goal. Refinement of the surgical technique continues.141, 142, 143, 144 Precise control of these parameters with intraoperative OCT systems and controlled delivery of the treatment solutions will certainly help improve vector delivery and surgical outcomes, especially if the fovea is to be targeted by these treatments.140,145,146 It may be desirable to inject outside of the fovea and then exchange fluid with air following the subretinal injection (see below) in order to reduce the buoyancy of the bleb (Figure 6). That would then allow the bleb to slowly migrate secondarily into the parafovea/fovea. Frames from an intra-operative video (Videos S1A and S1B) show, as an example, migration of a bleb across the arcades and toward the fovea over a 15-min period.
Figure 6.

Surgical Details of the Subretinal Delivery of Gene Therapy
Two intraoperative snapshots demonstrating the peripheral boundaries of the subretinal bleb (arrows) in relation to the location of the retinotomy (arrowheads). Asterisk indicates foveal region.
Figure 7.

Functional Changes after Subretinal Gene Therapy for RPE65-IRD
(A) NIR-FAF, 55°-wide images of the posterior retina of the right eye of the two with RPE65-LCA treated with bilateral subretinal gene therapy (Luxturna, Sparks Therapeutics, Philadelphia, PA, USA). Red line denotes the inferior boundary of a subretinal bleb that contains the treating product, which extends from the superior retina crossing the fovea and into the inferior pericentral retina. (B) Light-adapted achromatic and dark-adapted two-color chromatic static perimetry (showing only responses to a blue 500-nm stimulus) in the patients before (dashed lines) and after (continuous line) gene therapy. Dotted lines define lower limit (mean of 2 SD) of sensitivity in control subjects. S, superior visual field; I, inferior visual field. Horizontal arrows show the improvement in sensitivity supporting a treatment effect.
Since there is typically no break in the RPE or underlying Bruch’s membrane during the injection, there is protection against systemic exposure of AAV to the underlying choroidal blood supply. This likely contributes to the relatively immune privileged characteristics of subretinal AAV delivery. After the subretinal injection, the cannula is removed, and the vitreous fluid is exchanged with air. This acts to seal the retinotomy (hole in the retina where the cannula penetrated) and to allow gravity-dependent spread of the bleb such that it will contact additional portions of the retina. The AAV infects the cells bordering the bleb and/or is endocytosed along with excipient. As the fluid is absorbed, the bleb flattens and the photoreceptors re-approximate the RPE. The patient is positioned immediately after injection so that the subretinal fluid will migrate in a gravity-assisted fashion to settle in the most dependent portion of the retina. Flattening of the retina typically occurs within a few hours after injection. The procedure is associated with minimal post-operative pain, which is usually well controlled with oral analgesics. Patients continue to take oral prednisone, which is tapered and then discontinued a few days after the second eye receives its subretinal injection. Cycloplegic, topical steroid and topical antibiotic drops are administered as is done for standard vitrectomy procedures.
Surgeons are required to attend a subretinal injection training program before they are approved to deliver Luxturna and then be deemed qualified to carry out the procedures at a “Center of Excellence.” The injection protocol calls for use of a specific instrumentation (tubing and cannula) and a trained assistant to manipulate the injection syringe while the surgeon places the injection cannula in position. Note that other subretinal AAV clinical trial protocols sometimes use modifications of this procedure, such as initiating the bleb with sterile saline and then switching to the AAV solution or using an injection cannula where pressure is applied by foot control (and thus eliminating the need of an assistant’s help in pushing the plunger).141,142
It is often difficult to precisely control the direction of the bleb. In fact, in many situations, surgeons have reported that the bleb proceeds in similar (unusual) directions in each eye of the same patient. It is possible that the unexpected behavior has to do with variable resistance to the extension of the subretinal bleb caused by intraretinal differences in RPE-photoreceptor adhesion or regional differences in the overall thickness of the retina, and thus mechanical resistance.147 As noted before, the foveal center may be vulnerable to a bleb-induced stretch. Two or more injections of smaller volumes of the gene therapy product may be administered at different sites (preferably in blind retinal regions devoid of photoreceptors) distanced from the foveal center to avoid including the fovea within the bleb but still treat the visually important pericentral and parafoveal retina, or including the foveal center within the less elevated part of the subretinal blebs. In our hands, foveal thinning in patients where blebs extended into the fovea is on average ≤10% of the baseline foveal thickness and may be subclinical, only detectable by careful segmentation of the SD-OCT cross-sections, whereas foveal thinning with vision loss is estimated to occur in about 1% of the cases. It is not known whether the exact position of the subretinal blebs shift by gravity after the injections, but an effort should be made to avoid positioning the peripheral boundary of the blebs straddling across regions that are critical for vision, such as the macula, as experimental studies in non-human primates (NHPs) suggest that structural abnormalities are expected to occur post-treatment in the vicinity of these transitional regions.139 While generating the localized detachment is usually straightforward, the retina can occasionally be challenging to lift. Some surgeons consider cutting the tip of the microcatheter subretinal cannula to create a bevel. However, the small size and relative stiffness of the cannula make entry into the subretinal space straightforward. A sharp tip is not necessary to “incise” the retina. The bleb should not be generated through delivery of air. Subretinal delivery of air can damage the RPE and photoreceptors by generating greater and less controllable mechanical forces.
One other detail relates to the guidelines for treating the second eye within a 2-week window of the first eye. The experience to date is limited to either this 2-week interval, first tested in large animal models,125 or treating the second eye at least 1 year after the first, as was done in the follow-on readministration study at CHOP.107 The 2-week time window was originally selected, as it was unlikely that the host would mount a peak immune response to the vector in this short time period. The longer interval (≥1 year) would provide time for a potential immune response to subside. The bottom line is that there are no data as of yet of the safety of readministration of subretinal gene therapy 3 weeks to <1 year following the first administration. Until such data are available, repeat administration to the contralateral eye is not recommended except for using the approved time windows.
Patients are seen in follow-up the day after injection and then between 1 and 2 weeks after the injections in order to be sure that there is no inflammation or bacterial infection, the retina has flattened, and that (if they are to take an airplane back home) the air bubble has been resorbed. Screening for potential complications such as foveal dehiscence and macular hole is performed. Those complications can result from mechanical effects of the subretinal injections (see above). Rarely, there can be permanent damage to the fovea with ONL loss despite lack of any visible trauma.112 One of the 29 individuals enrolled in the phase 3 study showed a permanent reduction in foveal function after injection, despite gaining several log units of extrafoveal sensitivity.109 Other complications that were observed soon after injection in the phase 1–3 studies mostly resolved on their own or with standard intervention. They most often included post-injection redness of the conjunctiva (“injection”), localized alteration in corneal thickness (“dellen”), or increased in intraocular pressure. These minor problems are less likely to occur with current vitrectomy systems using small-gauge trocars. Phase 1–3 patients were seen regularly after intervention (1, 3, 6, and 12 months after injection and then annually). One subject in phase 1 studies who was a high myope (extremely farsighted) showed retinal thinning in the area exposed to the bleb. Retinal thinning (“atrophic creep”) may be more frequent in high myopes148 and may be exacerbated by the stress of subretinal injection. At the later time points, cataracts were observed in a small number of subjects, a known side effect of vitrectomy in older individuals. Those patients were referred for standard cataract removal.
Now that Luxturna is approved as a drug, in addition to the early post-operative visits where the patient is evaluated for the potential complications described above as well as for short-term efficacy, patients return to clinic at the ∼4- to 6-week time point for follow-up and then can either return to the center for further follow-up or may be followed by their local ophthalmologists. A conservative model may include doubling follow-up intervals up to a year post-treatment (1–1.5, 3, 6, and 12 months), then annual follow-ups. It is important to reiterate that assessment of lasting efficacy should not rely on visual acuity measures, but instead, by at least measuring visual sensitivity and visual field extent by perimetry and FST, complemented by structural measures with OCT (see the next section). Most individuals who qualify for Luxturna treatment report perception of increased brightness within days after injection. Improved retinal function and stimulation of visual pathways in the brain has been documented by day 7 postoperatively using pupillometry and by FST as early as ∼10 days post-treatment.57,104 Subjects have reported the ability to see patterns in wood furniture/flooring, see illuminated dials, see reflections, tree branches, mirrors, light penetrating around doorways, windows, faces, recognize colors (including hair color), walk independently at night on city streets, and so forth (for the first time) within 2 weeks of injection.
Outcome Measures of Efficacy in the Clinic
During the initial clinical trials rod- and cone-specific measures of vision with two-color static perimetry documented increases in sensitivity ranging from 10- to 45,000-fold, taking their vision to near normal limits.57 The same outcome has been confirmed since the introduction of Luxturna to the clinic (Figures 7A and 7B). Although two-color dark-adapted perimetry may be implemented with standard instrumentation, such as the Humphrey field analyzer, unfortunately it has not gained traction as a measure of function in the RPE65 clinical trials or after the approval of Luxturna.149 Achromatic light-adapted perimetry such as a 30-2 protocol or other custom protocols, for example, sampling along the main meridians with a large Goldmann size V, may be an alternative.57 The tests may be performed in the dark-adapted state to extend the dynamic range and explore changes in absolute sensitivity (Figure 7B) FST measures, if available, are a valuable complement to static perimetry for its simplicity, especially in children and patients with severe vision loss or fixation instability that would not be able to perform well on perimetry (Figures 7B and 7C).20,150,151 It is important, however, to recognize that FST sensitivity estimates represent the best sensitivity within a given retina and do not offer information on the topography of the sensitivity losses or gains.152 That is, FST monitors the retinal region with the greatest sensitivity post-treatment and will not detect local improvements (or losses) of sensitivity below the maximal sensitivity (Figures 7B and 7C). Since the introduction of Luxturna in our clinic, all patients treated thus far (n = 13) have shown the expected improvement in rod- and cone-mediated function, including older, end-stage disease patients (Figures 7 and 8).
Figure 8.

Full-field Sensitivity Changes after Gene Therapy for RPE65-IRDs
FST sensitivity estimates measured with spectral stimuli (blue, 467 nm; red, 637 nm) in dark-adapted (>30 min) patients. Dotted gray line is the lower limit (mean of 2 SD) of the sensitivity to the short wavelength 467-nm stimulus in control subjects. Values are converted into positive dB values from possible negative outputs from the FST instrument. Patients are sorted left to right by age.
In children where psychophysic measures may not be possible or reliable, a standard ERG may be used not only during the initial evaluation of the patient, but also as a measure of response to the treatment.118 The hope is that some practical form of dark-adapted two-color pupillometry becomes available with clinical instruments, as they were in the research space before and during the RPE65 clinical trials.38,57,102 The technique would provide objective measures of function both in pediatric and adult patient populations. It is important to recognize that these retina-wide measures of function (ERG, FST, pupillometry) lack the ability to provide information regarding the topography of the vision loss or rescue after treatment.
Future Possibilities
The FDA approved Luxturna as a drug that could be made available to children 1 year of age and older.153 Our study team at Spark Therapeutics and CHOP had proposed approval of Luxturna for use in children 3 years of age and older, and so the recommendation for availability ages of 1 year and higher was a surprise to us. We had recommended 3 years of age and higher mainly due to issues relating to surgical access and patient cooperation. The human eye reaches ∼90% of the size of the adult eye by 2–3 years of age, thereby allowing similar surgical delivery procedures to those used in adults. There is also less risk of amblyopia, which can be caused by impaired central vision quality after injury or a procedure.
Experts had questioned the potential of offering infants as young as 1 month of age treatment with Luxturna, if the molecular defect is somehow known, for example, through prenatal screenings. Among the many concerns is the fact that important final developmental steps are still taking place in the postnatal retina months after birth, and it is still unclear how such an early intervention would impact (negatively or positively) these processes.154,155 Most families do not recognize a visual deficit this early, and a variable delay nearly always occurs between recognition of visual deficit and final clinical and molecular diagnosis, generally clearing the early critical phases of development. The complexities of delivering the vector in younger patients should also be seriously taken into account. As clinicians obtain more experience treating young children with gene therapy, and as the precise molecular diagnosis of retinal diseases becomes more widespread and less expensive, it may be possible that clinicians will become more comfortable treating, when sensible and indicated, younger infants with this and other retinal degenerations.
Refinement of the surgical techniques required to deliver the gene therapy products to the subretinal space are taking place and promise to further de-risk the procedure, allowing safe treatment of the central retina in diseases, or disease stages, at risk of macular changes post-treatment.138,139,141, 142, 143, 144,156, 157, 158, 159 Delivery of gene therapy products by intravitreal injection, while ideal as a much simpler procedure that can potentially treat wider retinal regions, is not without challenges, including limited targeting of the outer retina and RPE and the potential to trigger chronic intraocular inflammation and/or cystoid macular edema.160, 161, 162, 163, 164, 165, 166, 167, 168, 169 As noted before, 9-cis retinoids rescue the phenotype in RPE65-IRD and may be adopted as an alternative or complementary therapy if tolerability and safety concerns (see below) are overcome.17,79, 80, 81, 82,170,171 Inflammatory changes associated with retinal degenerations may also become targets for treatments.121,172, 173, 174, 175
One question is whether it will be possible to re-administer gene therapy to an eye that has already received an injection. The phase 1 follow-on and phase 3 clinical trials run by CHOP and then Spark Therapeutics showed that it is safe to readminister Luxturna to the contralateral eye despite the presence of neutralizing antibodies in the serum.105,107 While a single subretinal injection of Luxturna appears to result in durable rescue of vision (at least 5 years as shown by long-term studies after the CHOP phase 3 trial), there may be situations where same-eye readministration might be warranted. If there is a complication during the initial surgical procedure and the injection fails to target the retina, readministration at a later time point may be desirable. If there are viable cells in a region of retina unexposed to the vector through the initial injection, it may be desirable to target those in a second surgery in order to achieve additional rescue of retinal and visual function. Finally, if transgene expression dwindles over time after treatment (a theoretical outcome difficult to directly confirm in patients, unless a reversal of the correction of the classical structural-functional dissociation is documented), it may be desirable to supplement RPE65 expression in the initially treated retina. So far, same-eye readministration has not been performed in humans. However, a study in NHPs showed that ipsilateral readministration is safe.139 The primary concern relating to same-eye readministration is safety due to the potential of a cell-mediated immune response elicited during the first exposure to AAV. The cell-mediated response could be directed to the AAV2 capsid, which could expose antigen-presenting cells, and, although less likely because it is a cytoplasmic protein, to the RPE65 protein delivered through Luxturna. A harmful cell-mediated immune response can cause a loss of transduced cells, an outcome that would be devastating given that retinal photoreceptors are terminally differentiated and cannot be replenished. A second concern is the ability to obtain additional transduction events in the presence of neutralizing antibodies. Because the NHPs receiving same eye readministration of Luxturna had normal vision at baseline, it was not possible to test rescue of sight in this paradigm. However, inflammatory changes were minimal and there was immunohistochemical evidence of additional RPE65 protein in the re-injected eyes.139
Another question is whether there is another AAV serotype or transgene cassette that might confer even more robust transgene expression and rescue than the vector (AAV2-hRPE65v2) comprising Luxturna. MeiraGTx has proposed that AAV serotype 5 (AAV5) carrying a transgene cassette with a stronger RPE65 promoter than used in the initial phase 1 trial carried out at University College London117 and a codon-optimized RPE65 cDNA may lead to rescue in RPE65 deficiency.176 They have enrolled 27 subjects 3 years of age and older in their phase 1–2 study and results are pending (ClinicalTrials.gov: NCT02781480 and NCT02946879) (Table 1). Assuming they proceed to phase 3 studies, their outcome data may need to compare favorably with that delivered by Luxturna in order to be approved as a drug.
Note that there are several other interventions besides gene augmentation therapy that are under consideration for treatment of RPE65 disease. One possibility is to provide oral retinoid supplementation.17,79, 80, 81, 82,170,171 While 11-cis retinal is unstable and limits its clinical application, 9-cis retinal is stable and can substitute for 11-cis retinal in binding with rhodopsin. QLT ran phase 1 clinical trials providing oral supplementation with their test compound, QLT091001, for 1 week. Most subjects showed improvements in visual function in the first 2 months.80, 81, 82 While there were side effects (including photophobia, nausea and vomiting, and elevations in liver function tests), those were reversible. If this reagent proves efficacious and safe in phase 3 trials, there may be a synergistic role of an oral therapy with a gene therapy approach. Oral retinoid therapy may be an option for patients with severe disease with preservation of photoreceptor and/or RPE in peripheral retina or as scattered patches not easily accessible to the subretinal injections. For those in whom retinal degeneration has progressed so far that there are few remaining photoreceptors that could be rescued with gene augmentation, retinoid therapy may be used in the future as an alternative to gene augmentation or to determine potential for improvement with gene augmentation. For those patients with no potential for rescue with either gene augmentation or retinoid therapy due to total loss of classical photoreceptors, there are several other potential avenues to explore such as cell transplantation, optogenetic therapy, or device implantation.
While retinal cell transplantation studies are still in their infancy, studies have been initiated evaluating the safety and efficacy of subretinal delivery of retinal progenitor cells, embryonic stem cells, induced pluripotent stem cells (iPSCs), and mesenchymal stromal cells. Diseases under study include retinitis pigmentosa, Stargardt macular dystrophy, and macular degeneration (for example, ClinicalTrials.gov: NCT03073733, NCT03944239, NCT01344993, NCT03264976, and NCT02464956). Transplantation of normal (or gene-corrected) RPE cells would theoretically rescue vision in patients with RPE65 disease, assuming that photoreceptors remained. In end-stage disease, however, transplantation would only make sense if both RPE and photoreceptors could be delivered. Much work remains to render this approach safe and effective.
Another approach that is being evaluated for treatment of end-stage retinal degeneration is optogenetic therapy. In this approach, cells remaining in the degenerated retina are rendered light sensitive through delivery of a gene encoding a light-sensitive channel protein.177 There are two clinical trials in progress evaluating optogenetic therapy in patients with end-stage retinitis pigmentosa. Both target ganglion cells with an optogenetic gene through intravitreal injection of AAV2. One of the trials also incorporates a device that optimizes the light stimulus to stimulate the gene therapy-treated retinal cells (ClinicalTrials.gov: NCT02556736 and NCT03326336). As the optogenetic strategies mature, it may be possible to target different retinal cells (those closest to the diseased photoreceptors, such as bipolar cells) so as to take advantage of the normal computational functions of the retina.178,179
Finally, a prosthetic device was by the FDA in 2013 to treat end-stage blindness. The Argus II retinal prosthesis (SecondSight) involved pairing of a prosthesis that is implanted in the retina and around the back of the eye together with goggles with a built-in camera and processing unit that sent stimuli to the retina implant.180,181 This device reportedly allowed patients with light perception to identify objects and to enjoy improved navigation, although it did not provide high resolution vision. Production of the prosthesis was recently discontinued.
Conclusions
Until Luxturna was approved, individuals with RPE65 deficiency were destined to a life of visual impairment, with whatever poor vision they had early on slowly disappearing. The availability of a treatment has changed medical practice. Patients are now carefully screened by retinal degeneration experts for features of RPE65 disease and are also genotyped. Those with lack-of-function RPE65 mutations are referred for treatment with Luxturna or to one of the other clinical trials in progress. The medical insurance and national health systems have adapted and now cover the costs of this one-time treatment. There is even the potential that the payer receives a rebate if the treatment does not render the desired effect within a specified time period.
Prior to Luxturna, there was no path for developing treatments for such rare, debilitating diseases. Luxturna approval has had a huge impact on development of therapies for other orphan diseases and especially IRDs. The clinical trials paved the way for inclusion of pediatric subjects in gene therapy trials, an important step as most congenital diseases are genetic in origin. The trials demonstrated that statistical significance can be met in a relatively short period of time and by enrolling a small number of subjects, details that are critical to intervening with rare diseases lethal not only for a person but for the retina. Trials can be designed such that they generate data demonstrating that the disease symptoms do not spontaneously resolve or, as was seen in the phase 3 trial for Luxturna, that the disease worsens without intervention.
The growing number of gene-based clinical trials targeting retinal degenerative diseases is a reflection of the impact of the Luxturna studies. While some diseases are going to be easier to treat than others, the field is currently in an exciting phase of expanding possibilities. New approaches and reagents are gradually and carefully being de-risked, resulting in an even larger armamentarium. There are now more than 45 different retinal gene therapy clinical trials that have been initiated, with half a dozen of them in phase 3 (ClinicalTrials.gov).182, 183, 184 The vectors are diverse (including multiple different serotypes of AAV, adenovirus, lentivirus, antisense DNA, and RNA) and there are now at least three different routes of delivery being evaluated (intravitreal, subretinal, suprachoroidal).160, 161, 162, 163, 164 The disease targets include multiple forms of LCA and of retinitis pigmentosa, choroideremia, achromatopsia, XL retinoschisis, Usher syndrome (blindness and deafness), Leber’s hereditary optic neuropathy (a mitochondrial disorder), Stargardt disease, and age-related macular degeneration (AMD). The targets are gradually expanding from ultra-rare orphan diseases to those that are epidemic in nature (AMD).
In disease where the fovea requires treatment, it might be desirable to inject outside of the fovea and use the air delivered after air-fluid exchange to push the bleb, thereby expanding the area it covers to include the fovea. The initial clinical studies for the rare (and even the common) diseases can be carried out relatively rapidly by academic centers, thereby substantially reducing the costs that would typically be footed by pharmaceutical companies. Furthermore, the expertise in-house at academic centers can be tapped upon to develop appropriate outcome measures, where needed, or to explore aspects of the intervention that would typically be ignored in a commercial venture. Our Center was able, for example, to explore the effects of gene therapy treatment of the retina on the function and structure of visual pathways in the brain,108,185,186 to generate protocols evaluating pupillary light reflexes,38,102,107 to precisely map out the effects of subretinal injection on specific cone photoreceptors in the macula,187 to develop a visual function questionnaire relevant to a congenital blindness, and to develop subretinal gene therapy surgical training guidelines (https://www.med.upenn.edu/carot/resources-publications.html) while keeping focus on the predesignated outcome measures and maintaining timelines. Of course, ultimately, the costs of phase 3 trials are prohibitive for academic centers, at least for common diseases. Also, academic centers do not generally have the expertise in manufacturing and marketing held by pharmaceutical companies. Thus, the ecosystems of academia and biotechnology/pharmaceutical companies have become intertwined in a productive and mutually beneficial manner. There are still many technical and regulatory challenges to address; however, the future has never looked brighter for the development of treatments for blinding diseases.
Acknowledgments
We are grateful to the subjects who volunteered for the clinical trials. They are the true pioneers. Thanks to Erin C. O’Neil and Elizabeth Shagena for their help with data selection and processing. This work was supported by the Center for Advanced Retinal and Ocular Therapeutics (CAROT), the Brenda and Matthew Shapiro Stewardship, the Robert and Susan Heidenberg Investigative Research Fund for Ocular Gene Therapy, The Pennsylvania Lions Sight Conservation and Research Foundation, Hope For Vision, the Paul and Evanina Bell Mackall Foundation Trust, and by Research to Prevent Blindness.
Author Contributions
J.B., T.S.A., and B.P.L. searched the literature and wrote the paper; E.M.A. analyzed data and prepared figures; and A.M.M. contributed to manuscript writing and editing.
Declaration of Interests
J.B. and A.M.M. are co-authors on a patent relevant to development of gene therapy for RPE65 deficiency (US Patent #8,147,823) but waived any potential financial gain in 2002. B.P.L. serves as a consultant for Spark Therapeutics but directs his payments to the Department of Ophthalmology at the University of Gent. A.M.M. and T.S.A. carry out long-term follow-up of patients treated with Luxturna in the CHOP phase 1 to 4 (post-approval) trials under a CTA from Spark Therapeutics. E.M.A. declares no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.11.029.
References
- 1.Stone E.M., Andorf J.L., Whitmore S.S., DeLuca A.P., Giacalone J.C., Streb L.M., Braun T.A., Mullins R.F., Scheetz T.E., Sheffield V.C., Tucker B.A. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124:1314–1331. doi: 10.1016/j.ophtha.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koenekoop R.K. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv. Ophthalmol. 2004;49:379–398. doi: 10.1016/j.survophthal.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 3.Foxman S.G., Heckenlively J.R., Bateman J.B., Wirtschafter J.D. Classification of congenital and early onset retinitis pigmentosa. Arch. Ophthalmol. 1985;103:1502–1506. doi: 10.1001/archopht.1985.01050100078023. [DOI] [PubMed] [Google Scholar]
- 4.Heckenlively J., Foxmann S.G. Congenital and early-onset forms of retinitis pigmentosa. In: Heckenlively J., editor. Retinitis Pigmentosa. J.B. Lippincott; 1988. pp. 107–149. [Google Scholar]
- 5.den Hollander A.I., Roepman R., Koenekoop R.K., Cremers F.P. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Chao D.L., Burr A., Pennesi M. RPE65-related Leber congenital amaurosis / early-onset severe retinal dystrophy. In: Adam M.P., Ardinger H.H., Pagon R.A., editors. GeneReviews. University of Washington; 1993. https://www.ncbi.nim.nih.books/NBK549574/ [Google Scholar]
- 7.Weleber R.G., Michaelides M., Trzupek K.M., Stover N.B., Stone E.M. The phenotype of severe early childhood onset retinal dystrophy (SECORD) from mutation of RPE65 and differentiation from Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2011;52:292–302. doi: 10.1167/iovs.10-6106. [DOI] [PubMed] [Google Scholar]
- 8.Webb T.R., Matarin M., Gardner J.C., Kelberman D., Hassan H., Ang W., Michaelides M., Ruddle J.B., Pennell C.E., Yazar S. X-linked megalocornea caused by mutations in CHRDL1 identifies an essential role for ventroptin in anterior segment development. Am. J. Hum. Genet. 2012;90:247–259. doi: 10.1016/j.ajhg.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamel C.P., Tsilou E., Pfeffer B.A., Hooks J.J., Detrick B., Redmond T.M. Molecular cloning and expression of RPE65, a novel retinal pigment epithelium-specific microsomal protein that is post-transcriptionally regulated in vitro. J. Biol. Chem. 1993;268:15751–15757. [PubMed] [Google Scholar]
- 10.Hamel C.P., Jenkins N.A., Gilbert D.J., Copeland N.G., Redmond T.M. The gene for the retinal pigment epithelium-specific protein RPE65 is localized to human 1p31 and mouse 3. Genomics. 1994;20:509–512. doi: 10.1006/geno.1994.1212. [DOI] [PubMed] [Google Scholar]
- 11.Marlhens F., Bareil C., Griffoin J.M., Zrenner E., Amalric P., Eliaou C., Liu S.Y., Harris E., Redmond T.M., Arnaud B. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat. Genet. 1997;17:139–141. doi: 10.1038/ng1097-139. [DOI] [PubMed] [Google Scholar]
- 12.Tsilou E., Hamel C.P., Yu S., Redmond T.M. RPE65, the major retinal pigment epithelium microsomal membrane protein, associates with phospholipid liposomes. Arch. Biochem. Biophys. 1997;346:21–27. doi: 10.1006/abbi.1997.0276. [DOI] [PubMed] [Google Scholar]
- 13.Redmond T.M., Hamel C.P. Genetic analysis of RPE65: from human disease to mouse model. Methods Enzymol. 2000;316:705–724. doi: 10.1016/s0076-6879(00)16758-8. [DOI] [PubMed] [Google Scholar]
- 14.Redmond T.M., Yu S., Lee E., Bok D., Hamasaki D., Chen N., Goletz P., Ma J.X., Crouch R.K., Pfeifer K. Rpe65 is necessary for production of 11-cis-vitamin A in the retinal visual cycle. Nat. Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 15.Kumaran N., Rubin G.S., Kalitzeos A., Fujinami K., Bainbridge J.W.B., Weleber R.G., Michaelides M. A cross-sectional and longitudinal study of retinal sensitivity in RPE65-associated Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2018;59:3330–3339. doi: 10.1167/iovs.18-23873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lotery A.J., Namperumalsamy P., Jacobson S.G., Weleber R.G., Fishman G.A., Musarella M.A., Hoyt C.S., Héon E., Levin A., Jan J. Mutation analysis of 3 genes in patients with Leber congenital amaurosis. Arch. Ophthalmol. 2000;118:538–543. doi: 10.1001/archopht.118.4.538. [DOI] [PubMed] [Google Scholar]
- 17.Van Hooser J.P., Aleman T.S., He Y.G., Cideciyan A.V., Kuksa V., Pittler S.J., Stone E.M., Jacobson S.G., Palczewski K. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc. Natl. Acad. Sci. USA. 2000;97:8623–8628. doi: 10.1073/pnas.150236297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu S.M., Thompson D.A., Srikumari C.R., Lorenz B., Finckh U., Nicoletti A., Murthy K.R., Rathmann M., Kumaramanickavel G., Denton M.J., Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat. Genet. 1997;17:194–197. doi: 10.1038/ng1097-194. [DOI] [PubMed] [Google Scholar]
- 19.Lorenz B., Gyürüs P., Preising M., Bremser D., Gu S., Andrassi M., Gerth C., Gal A. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 2000;41:2735–2742. [PubMed] [Google Scholar]
- 20.Jacobson S.G., Aleman T.S., Cideciyan A.V., Roman A.J., Sumaroka A., Windsor E.A., Schwartz S.B., Heon E., Stone E.M. Defining the residual vision in Leber congenital amaurosis caused by RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 2009;50:2368–2375. doi: 10.1167/iovs.08-2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacobson S.G., Aleman T.S., Cideciyan A.V., Sumaroka A., Schwartz S.B., Windsor E.A., Traboulsi E.I., Heon E., Pittler S.J., Milam A.H. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc. Natl. Acad. Sci. USA. 2005;102:6177–6182. doi: 10.1073/pnas.0500646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz B., Poliakov E., Schambeck M., Friedburg C., Preising M.N., Redmond T.M. A comprehensive clinical and biochemical functional study of a novel RPE65 hypomorphic mutation. Invest. Ophthalmol. Vis. Sci. 2008;49:5235–5242. doi: 10.1167/iovs.07-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hull S., Holder G.E., Robson A.G., Mukherjee R., Michaelides M., Webster A.R., Moore A.T. Preserved visual function in retinal dystrophy due to hypomorphic RPE65 mutations. Br. J. Ophthalmol. 2016;100:1499–1505. doi: 10.1136/bjophthalmol-2015-308019. [DOI] [PubMed] [Google Scholar]
- 24.Galvin J.A., Fishman G.A., Stone E.M., Koenekoop R.K. Clinical phenotypes in carriers of Leber congenital amaurosis mutations. Ophthalmology. 2005;112:349–356. doi: 10.1016/j.ophtha.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 25.Stone E.M. Leber congenital amaurosis—a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 2007;144:791–811. doi: 10.1016/j.ajo.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 26.Yzer S., van den Born L.I., Schuil J., Kroes H.Y., van Genderen M.M., Boonstra F.N., van den Helm B., Brunner H.G., Koenekoop R.K., Cremers F.P. A Tyr368His RPE65 founder mutation is associated with variable expression and progression of early onset retinal dystrophy in 10 families of a genetically isolated population. J. Med. Genet. 2003;40:709–713. doi: 10.1136/jmg.40.9.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banin E., Bandah-Rozenfeld D., Obolensky A., Cideciyan A.V., Aleman T.S., Marks-Ohana D., Sela M., Boye S., Sumaroka A., Roman A.J. Molecular anthropology meets genetic medicine to treat blindness in the North African Jewish population: human gene therapy initiated in Israel. Hum. Gene Ther. 2010;21:1749–1757. doi: 10.1089/hum.2010.047. [DOI] [PubMed] [Google Scholar]
- 28.Acland G.M., Aguirre G.D., Bennett J., Aleman T.S., Cideciyan A.V., Bennicelli J., Dejneka N.S., Pearce-Kelling S.E., Maguire A.M., Palczewski K. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol. Ther. 2005;12:1072–1082. doi: 10.1016/j.ymthe.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Acland G.M., Aguirre G.D., Ray J., Zhang Q., Aleman T.S., Cideciyan A.V., Pearce-Kelling S.E., Anand V., Zeng Y., Maguire A.M. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- 30.Aguirre G.K., Komáromy A.M., Cideciyan A.V., Brainard D.H., Aleman T.S., Roman A.J., Avants B.B., Gee J.C., Korczykowski M., Hauswirth W.W. Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Med. 2007;4:e230. doi: 10.1371/journal.pmed.0040230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caruso R.C., Aleman T.S., Cideciyan A.V., Roman A.J., Sumaroka A., Mullins C.L., Boye S.L., Hauswirth W.W., Jacobson S.G. Retinal disease in Rpe65-deficient mice: comparison to human Leber congenital amaurosis due to RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 2010;51:5304–5313. doi: 10.1167/iovs.10-5559. [DOI] [PubMed] [Google Scholar]
- 32.Veske A., Nilsson S.E., Narfström K., Gal A. Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999;57:57–61. doi: 10.1006/geno.1999.5754. [DOI] [PubMed] [Google Scholar]
- 33.Aguirre G.D., Baldwin V., Pearce-Kelling S., Narfström K., Ray K., Acland G.M. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol. Vis. 1998;4:23. [PubMed] [Google Scholar]
- 34.Acland G., Aguirre G., Aleman T., Cideciyan A., Maguire A., Jacobson S., Hauswirth W.W. Continuing evaluation of gene therapy in the RPE65 mutant dog. Invest. Ophthalmol. Vis. Sci. 2002;43:4593. [Google Scholar]
- 35.Cideciyan A.V., Jacobson S.G., Beltran W.A., Sumaroka A., Swider M., Iwabe S., Roman A.J., Olivares M.B., Schwartz S.B., Komáromy A.M. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc. Natl. Acad. Sci. USA. 2013;110:E517–E525. doi: 10.1073/pnas.1218933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardiner K.L., Cideciyan A.V., Swider M., Dufour V.L., Sumaroka A., Komáromy A.M., Hauswirth W.W., Iwabe S., Jacobson S.G., Beltran W.A., Aguirre G.D. Long-term structural outcomes of late-stage RPE65 gene therapy. Mol. Ther. 2020;28:266–278. doi: 10.1016/j.ymthe.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aleman T., Cideciyan A., Bennett J., Cheung A., Glover E., Furushima M., Milam A.H., Jacobson S.G. Natural history of retinal function and structure in the Rpe65−/− murine model of Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2003;44:4920. [Google Scholar]
- 38.Aleman T.S., Jacobson S.G., Chico J.D., Scott M.L., Cheung A.Y., Windsor E.A., Furushima M., Redmond T.M., Bennett J., Palczewski K., Cideciyan A.V. Impairment of the transient pupillary light reflex in Rpe65−/− mice and humans with Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2004;45:1259–1271. doi: 10.1167/iovs.03-1230. [DOI] [PubMed] [Google Scholar]
- 39.Thompson D.A., Gyürüs P., Fleischer L.L., Bingham E.L., McHenry C.L., Apfelstedt-Sylla E., Zrenner E., Lorenz B., Richards J.E., Jacobson S.G. Genetics and phenotypes of RPE65 mutations in inherited retinal degeneration. Invest. Ophthalmol. Vis. Sci. 2000;41:4293–4299. [PubMed] [Google Scholar]
- 40.Chen N., Shin K., Millin R., Song Y., Kwon M., Tjan B.S. Cortical reorganization of peripheral vision induced by simulated central vision loss. J. Neurosci. 2019;39:3529–3536. doi: 10.1523/JNEUROSCI.2126-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walia S., Fishman G.A., Jacobson S.G., Aleman T.S., Koenekoop R.K., Traboulsi E.I., Weleber R.G., Pennesi M.E., Heon E., Drack A. Visual acuity in patients with Leber’s congenital amaurosis and early childhood-onset retinitis pigmentosa. Ophthalmology. 2010;117:1190–1198. doi: 10.1016/j.ophtha.2009.09.056. [DOI] [PubMed] [Google Scholar]
- 42.Chung D.C., Bertelsen M., Lorenz B., Pennesi M.E., Leroy B.P., Hamel C.P., Pierce E., Sallum J., Larsen M., Stieger K. The natural history of inherited retinal dystrophy due to biallelic mutations in the RPE65 gene. Am. J. Ophthalmol. 2019;199:58–70. doi: 10.1016/j.ajo.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paunescu K., Wabbels B., Preising M.N., Lorenz B. Longitudinal and cross-sectional study of patients with early-onset severe retinal dystrophy associated with RPE65 mutations. Graefes Arch. Clin. Exp. Ophthalmol. 2005;243:417–426. doi: 10.1007/s00417-004-1020-x. [DOI] [PubMed] [Google Scholar]
- 44.Roman A.J., Cideciyan A.V., Schwartz S.B., Olivares M.B., Heon E., Jacobson S.G. Intervisit variability of visual parameters in Leber congenital amaurosis caused by RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 2013;54:1378–1383. doi: 10.1167/iovs.12-11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumaran N., Ripamonti C., Kalitzeos A., Rubin G.S., Bainbridge J.W.B., Michaelides M. Severe loss of tritan color discrimination in RPE65 associated Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2018;59:85–93. doi: 10.1167/iovs.17-22905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cideciyan A.V., Aguirre G.K., Jacobson S.G., Butt O.H., Schwartz S.B., Swider M., Roman A.J., Sadigh S., Hauswirth W.W. Pseudo-fovea formation after gene therapy for RPE65-LCA. Invest. Ophthalmol. Vis. Sci. 2014;56:526–537. doi: 10.1167/iovs.14-15895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Felius J., Thompson D.A., Khan N.W., Bingham E.L., Jamison J.A., Kemp J.A., Sieving P.A. Clinical course and visual function in a family with mutations in the RPE65 gene. Arch. Ophthalmol. 2002;120:55–61. doi: 10.1001/archopht.120.1.55. [DOI] [PubMed] [Google Scholar]
- 48.Pasadhika S., Fishman G.A., Stone E.M., Lindeman M., Zelkha R., Lopez I., Koenekoop R.K., Shahidi M. Differential macular morphology in patients with RPE65-, CEP290-, GUCY2D-, and AIPL1-related Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2010;51:2608–2614. doi: 10.1167/iovs.09-3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Safari S., Zare-Abdollahi D., Bushehri A., Safari M.R., Dehghani A., Tahmasebi Z., Khorram Khorshid H.R., Ghadami M. RPE65 and retinal dystrophy: report of new and recurrent mutations. J. Gene Med. 2020;22:e3154. doi: 10.1002/jgm.3154. [DOI] [PubMed] [Google Scholar]
- 50.Pierrache L.H.M., Ghafaryasl B., Khan M.I., Yzer S., van Genderen M.M., Schuil J., Boonstra F.N., Pott J.W.R., de Faber J.T.H.N., Tjon-Fo-Sang M.J.H. Longitudinal study of RPE65-associated inherited retinal degenerations. Retina. 2020;40:1812–1828. doi: 10.1097/IAE.0000000000002681. [DOI] [PubMed] [Google Scholar]
- 51.Schatz P., Preising M., Lorenz B., Sander B., Larsen M., Rosenberg T. Fundus albipunctatus associated with compound heterozygous mutations in RPE65. Ophthalmology. 2011;118:888–894. doi: 10.1016/j.ophtha.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 52.El Matri L., Ambresin A., Schorderet D.F., Kawasaki A., Seeliger M.W., Wenzel A., Arsenijevic Y., Borruat F.X., Munier F.L. Phenotype of three consanguineous Tunisian families with early-onset retinal degeneration caused by an R91W homozygous mutation in the RPE65 gene. Graefes Arch. Clin. Exp. Ophthalmol. 2006;244:1104–1112. doi: 10.1007/s00417-005-0096-2. [DOI] [PubMed] [Google Scholar]
- 53.Simonelli F., Ziviello C., Testa F., Rossi S., Fazzi E., Bianchi P.E., Fossarello M., Signorini S., Bertone C., Galantuomo S. Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest. Ophthalmol. Vis. Sci. 2007;48:4284–4290. doi: 10.1167/iovs.07-0068. [DOI] [PubMed] [Google Scholar]
- 54.Roman A.J., Cideciyan A.V., Schwartz S.B., Olivares M.B., Heon E., Jacobson S.G. Intervisit variability of visual parameters in Leber congenital amaurosis caused by RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 2013;54:1378–1383. doi: 10.1167/iovs.12-11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poehner W.J., Fossarello M., Rapoport A.L., Aleman T.S., Cideciyan A.V., Jacobson S.G., Wright A.F., Danciger M., Farber D.B. A homozygous deletion in RPE65 in a small Sardinian family with autosomal recessive retinal dystrophy. Mol. Vis. 2000;6:192–198. [PubMed] [Google Scholar]
- 56.Jacobson S.G., Aleman T.S., Cideciyan A.V., Heon E., Golczak M., Beltran W.A., Sumaroka A., Schwartz S.B., Roman A.J., Windsor E.A. Human cone photoreceptor dependence on RPE65 isomerase. Proc. Natl. Acad. Sci. USA. 2007;104:15123–15128. doi: 10.1073/pnas.0706367104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cideciyan A.V., Aleman T.S., Boye S.L., Schwartz S.B., Kaushal S., Roman A.J., Pang J.J., Sumaroka A., Windsor E.A., Wilson J.M. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. USA. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lorenz B., Wabbels B., Wegscheider E., Hamel C.P., Drexler W., Preising M.N. Lack of fundus autofluorescence to 488 nanometers from childhood on in patients with early-onset severe retinal dystrophy associated with mutations in RPE65. Ophthalmology. 2004;111:1585–1594. doi: 10.1016/j.ophtha.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 59.Levi S.R., Oh J.K., de Carvalho J.R.L., Jr., Mahajan V.B., Tsang S.H., Sparrow J.R. Quantitative autofluorescence following gene therapy with voretigene neparvovec. JAMA Ophthalmol. 2020;138:919–921. doi: 10.1001/jamaophthalmol.2020.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Magliyah M., Saifaldein A.A., Schatz P. Late presentation of RPE65 retinopathy in three siblings. Doc. Ophthalmol. 2020;140:289–297. doi: 10.1007/s10633-019-09745-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mo G., Ding Q., Chen Z., Li Y., Yan M., Bu L., Song Y., Yin G. A novel mutation in the RPE65 gene causing Leber congenital amaurosis and its transcriptional expression in vitro. PLoS ONE. 2014;9:e112400. doi: 10.1371/journal.pone.0112400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacobson S.G., Cideciyan A.V., Aleman T.S., Sumaroka A., Windsor E.A., Schwartz S.B., Heon E., Stone E.M. Photoreceptor layer topography in children with Leber congenital amaurosis caused by RPE65 mutations. Invest. Ophthalmol. Vis. Sci. 2008;49:4573–4577. doi: 10.1167/iovs.08-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garafalo A.V., Cideciyan A.V., Héon E., Sheplock R., Pearson A., WeiYang Yu C., Sumaroka A., Aguirre G.D., Jacobson S.G. Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020;77:100827. doi: 10.1016/j.preteyeres.2019.100827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aleman T.S., Cideciyan A.V., Sumaroka A., Schwartz S.B., Roman A.J., Windsor E.A., Steinberg J.D., Branham K., Othman M., Swaroop A., Jacobson S.G. Inner retinal abnormalities in X-linked retinitis pigmentosa with RPGR mutations. Invest. Ophthalmol. Vis. Sci. 2007;48:4759–4765. doi: 10.1167/iovs.07-0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cideciyan A.V. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog. Retin. Eye Res. 2010;29:398–427. doi: 10.1016/j.preteyeres.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cideciyan A.V., Swider M., Jacobson S.G. Autofluorescence imaging with near-infrared excitation: normalization by reflectance to reduce signal from choroidal fluorophores. Invest. Ophthalmol. Vis. Sci. 2015;56:3393–3406. doi: 10.1167/iovs.15-16726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Redmond T.M., Poliakov E., Yu S., Tsai J.Y., Lu Z., Gentleman S. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc. Natl. Acad. Sci. USA. 2005;102:13658–13663. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowne S.J., Humphries M.M., Sullivan L.S., Kenna P.F., Tam L.C., Kiang A.S., Campbell M., Weinstock G.M., Koboldt D.C., Ding L. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur. J. Hum. Genet. 2011;19:1074–1081. doi: 10.1038/ejhg.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hull S., Mukherjee R., Holder G.E., Moore A.T., Webster A.R. The clinical features of retinal disease due to a dominant mutation in RPE65. Mol. Vis. 2016;22:626–635. [PMC free article] [PubMed] [Google Scholar]
- 70.Shin Y., Moiseyev G., Chakraborty D., Ma J.X. A dominant mutation in Rpe65, D477G, delays dark adaptation and disturbs the visual cycle in the mutant knock-in mice. Am. J. Pathol. 2017;187:517–527. doi: 10.1016/j.ajpath.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Choi E.H., Suh S., Sander C.L., Hernandez C.J.O., Bulman E.R., Khadka N., Dong Z., Shi W., Palczewski K., Kiser P.D. Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum. Mol. Genet. 2018;27:2225–2243. doi: 10.1093/hmg/ddy128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jauregui R., Park K.S., Tsang S.H. Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2018;39:544–549. doi: 10.1080/13816810.2018.1484929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Y., Furhang R., Ray A., Duncan T., Soucy J., Mahdi R., Chaitankar V., Gieser L., Poliakov E., Qian H. Aberrant RNA splicing is the major pathogenic effect in a knock-in mouse model of the dominantly inherited c.1430A>G human RPE65 mutation. Hum. Mutat. 2019;40:426–443. doi: 10.1002/humu.23706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jauregui R., Cho A., Oh J.K., Tanaka A.J., Sparrow J.R., Tsang S.H. Phenotypic expansion of autosomal dominant retinitis pigmentosa associated with the D477G mutation in RPE65. Cold Spring Harb. Mol. Case Stud. 2020;6 doi: 10.1101/mcs.a004952. a004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kenna P.F., Humphries M.M., Kiang A.S., Brabet P., Guillou L., Ozaki E., Campbell M., Farrar G.J., Koenekoop R., Humphries P. Advanced late-onset retinitis pigmentosa with dominant-acting D477G RPE65 mutation is responsive to oral synthetic retinoid therapy. BMJ Open Ophthalmol. 2020;5:e000462. doi: 10.1136/bmjophth-2020-000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Maeda T., Cideciyan A.V., Maeda A., Golczak M., Aleman T.S., Jacobson S.G., Palczewski K. Loss of cone photoreceptors caused by chromophore depletion is partially prevented by the artificial chromophore pro-drug, 9-cis-retinyl acetate. Hum. Mol. Genet. 2009;18:2277–2287. doi: 10.1093/hmg/ddp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maeda T., Maeda A., Casadesus G., Palczewski K., Margaron P. Evaluation of 9-cis-retinyl acetate therapy in Rpe65−/− mice. Invest. Ophthalmol. Vis. Sci. 2009;50:4368–4378. doi: 10.1167/iovs.09-3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gearhart P.M., Gearhart C., Thompson D.A., Petersen-Jones S.M. Improvement of visual performance with intravitreal administration of 9-cis-retinal in Rpe65-mutant dogs. Arch. Ophthalmol. 2010;128:1442–1448. doi: 10.1001/archophthalmol.2010.210. [DOI] [PubMed] [Google Scholar]
- 79.Maeda T., Dong Z., Jin H., Sawada O., Gao S., Utkhede D., Monk W., Palczewska G., Palczewski K. QLT091001, a 9-cis-retinal analog, is well-tolerated by retinas of mice with impaired visual cycles. Invest. Ophthalmol. Vis. Sci. 2013;54:455–466. doi: 10.1167/iovs.12-11152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koenekoop R.K., Sui R., Sallum J., van den Born L.I., Ajlan R., Khan A., den Hollander A.I., Cremers F.P., Mendola J.D., Bittner A.K. Oral 9-cis retinoid for childhood blindness due to Leber congenital amaurosis caused by RPE65 or LRAT mutations: an open-label phase 1b trial. Lancet. 2014;384:1513–1520. doi: 10.1016/S0140-6736(14)60153-7. [DOI] [PubMed] [Google Scholar]
- 81.Scholl H.P., Moore A.T., Koenekoop R.K., Wen Y., Fishman G.A., van den Born L.I., Bittner A., Bowles K., Fletcher E.C., Collison F.T., RET IRD 01 Study Group Safety and proof-of-concept study of oral QLT091001 in retinitis pigmentosa due to inherited deficiencies of retinal pigment epithelial 65 protein (RPE65) or lecithin:retinol acyltransferase (LRAT) PLoS ONE. 2015;10:e0143846. doi: 10.1371/journal.pone.0143846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jacobson S.G., Cideciyan A.V., Aguirre G.D., Roman A.J., Sumaroka A., Hauswirth W.W., Palczewski K. Improvement in vision: a new goal for treatment of hereditary retinal degenerations. Expert Opin. Orphan Drugs. 2015;3:563–575. doi: 10.1517/21678707.2015.1030393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang U., Gentleman S., Gai X., Gorin M.B., Borchert M.S., Lee T.C., Villanueva A., Koenekoop R., Maguire A.M., Bennett J. Utility of in vitro mutagenesis of RPE65 protein for verification of mutational pathogenicity before gene therapy. JAMA Ophthalmol. 2019;137:1–9. doi: 10.1001/jamaophthalmol.2019.3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hendriks M., Verhoeven V.J.M., Buitendijk G.H.S., Polling J.R., Meester-Smoor M.A., Hofman A., Kamermans M., Ingeborgh van den Born L., Klaver C.C.W., RD5000 Consortium Development of refractive errors—what can we learn from inherited retinal dystrophies? Am. J. Ophthalmol. 2017;182:81–89. doi: 10.1016/j.ajo.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 85.Tröster H., Brambring M., Beelmann A. The age dependence of stereotyped behaviours in blind infants and preschoolers. Child Care Health Dev. 1991;17:137–157. doi: 10.1111/j.1365-2214.1991.tb00684.x. [DOI] [PubMed] [Google Scholar]
- 86.Franceschetti A., Dieterle P. [Diagnostic and prognostic importance of the electroretinogram in tapetoretinal degeneration with reduction of the visual field and hemeralopia] Confin. Neurol. 1954;14:184–186. [PubMed] [Google Scholar]
- 87.Campochiaro P.A., Mir T.A. The mechanism of cone cell death in retinitis pigmentosa. Prog. Retin. Eye Res. 2018;62:24–37. doi: 10.1016/j.preteyeres.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 88.Aleman T.S., Cideciyan A.V., Windsor E.A., Schwartz S.B., Swider M., Chico J.D., Sumaroka A., Pantelyat A.Y., Duncan K.G., Gardner L.M. Macular pigment and lutein supplementation in ABCA4-associated retinal degenerations. Invest. Ophthalmol. Vis. Sci. 2007;48:1319–1329. doi: 10.1167/iovs.06-0764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aleman T.S., Duncan J.L., Bieber M.L., de Castro E., Marks D.A., Gardner L.M., Steinberg J.D., Cideciyan A.V., Maguire M.G., Jacobson S.G. Macular pigment and lutein supplementation in retinitis pigmentosa and Usher syndrome. Invest. Ophthalmol. Vis. Sci. 2001;42:1873–1881. [PubMed] [Google Scholar]
- 90.Arunkumar R., Gorusupudi A., Bernstein P.S. The macular carotenoids: a biochemical overview. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2020;1865:158617. doi: 10.1016/j.bbalip.2020.158617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bernstein P.S., Li B., Vachali P.P., Gorusupudi A., Shyam R., Henriksen B.S., Nolan J.M. Lutein, zeaxanthin, and meso-zeaxanthin: the basic and clinical science underlying carotenoid-based nutritional interventions against ocular disease. Prog. Retin. Eye Res. 2016;50:34–66. doi: 10.1016/j.preteyeres.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hunter J.J., Morgan J.I.W., Merigan W.H., Sliney D.H., Sparrow J.R., Williams D.R. The susceptibility of the retina to photochemical damage from visible light. Prog. Retin. Eye Res. 2012;31:28–42. doi: 10.1016/j.preteyeres.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cideciyan A.V., Jacobson S.G., Aleman T.S., Gu D., Pearce-Kelling S.E., Sumaroka A., Acland G.M., Aguirre G.D. In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc. Natl. Acad. Sci. USA. 2005;102:5233–5238. doi: 10.1073/pnas.0408892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cideciyan A.V., Swider M., Aleman T.S., Roman M.I., Sumaroka A., Schwartz S.B., Stone E.M., Jacobson S.G. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2007;24:1457–1467. doi: 10.1364/josaa.24.001457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bennett J., Bennicelli J., Wright V., Narfström K., Pugh E., Jacobs L.J., Komaromy G.A.A., High K., Maguire A. An optimized adeno-associated virus (AAV) for treatment of Leber congenital amaurosis (LCA-RPE65) Invest. Ophthalmol. Vis. Sci. 2007;48:1689. [Google Scholar]
- 96.Bennett J., Duan D., Engelhardt J.F., Maguire A.M. Real-time, noninvasive in vivo assessment of adeno-associated virus-mediated retinal transduction. Invest. Ophthalmol. Vis. Sci. 1997;38:2857–2863. [PubMed] [Google Scholar]
- 97.Bennett J., Maguire A.M., Cideciyan A.V., Schnell M., Glover E., Anand V., Aleman T.S., Chirmule N., Gupta A.R., Huang Y. Stable transgene expression in rod photoreceptors after recombinant adeno-associated virus-mediated gene transfer to monkey retina. Proc. Natl. Acad. Sci. USA. 1999;96:9920–9925. doi: 10.1073/pnas.96.17.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dejneka N.S., Surace E.M., Aleman T.S., Cideciyan A.V., Lyubarsky A., Savchenko A., Redmond T.M., Tang W., Wei Z., Rex T.S. In utero gene therapy rescues vision in a murine model of congenital blindness. Mol. Ther. 2004;9:182–188. doi: 10.1016/j.ymthe.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 99.Chen Y., Moiseyev G., Takahashi Y., Ma J.-X. RPE65 gene delivery restores isomerohydrolase activity and prevents early cone loss in Rpe65−/− mice. Invest. Ophthalmol. Vis. Sci. 2006;47:1177–1184. doi: 10.1167/iovs.05-0965. [DOI] [PubMed] [Google Scholar]
- 100.Ford M., Bragadóttir R., Rakoczy P.E., Narfström K. Gene transfer in the RPE65 null mutation dog: relationship between construct volume, visual behavior and electroretinographic (ERG) results. Doc. Ophthalmol. 2003;107:79–86. doi: 10.1023/a:1024431827812. [DOI] [PubMed] [Google Scholar]
- 101.Narfström K., Bragadóttir R., Redmond T.M., Rakoczy P.E., van Veen T., Bruun A. Functional and structural evaluation after AAV.RPE65 gene transfer in the canine model of Leber’s congenital amaurosis. Adv. Exp. Med. Biol. 2003;533:423–430. doi: 10.1007/978-1-4615-0067-4_54. [DOI] [PubMed] [Google Scholar]
- 102.Maguire A.M., Simonelli F., Pierce E.A., Pugh E.N., Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K.A., Testa F., Surace E.M. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bennicelli J., Wright J.F., Komaromy A., Jacobs J.B., Hauck B., Zelenaia O., Mingozzi F., Hui D., Chung D., Rex T.S. Reversal of blindness in animal models of Leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol. Ther. 2008;16:458–465. doi: 10.1038/sj.mt.6300389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maguire A.M., High K.A., Auricchio A., Wright J.F., Pierce E.A., Testa F., Mingozzi F., Bennicelli J.L., Ying G.S., Rossi S. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet. 2009;374:1597–1605. doi: 10.1016/S0140-6736(09)61836-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bennett J., Ashtari M., Wellman J., Marshall K.A., Cyckowski L.L., Chung D.C., McCague S., Pierce E.A., Chen Y., Bennicelli J.L. AAV2 gene therapy readministration in three adults with congenital blindness. Sci. Transl. Med. 2012;4:120ra15. doi: 10.1126/scitranslmed.3002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Simonelli F., Maguire A.M., Testa F., Pierce E.A., Mingozzi F., Bennicelli J.L., Rossi S., Marshall K., Banfi S., Surace E.M. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol. Ther. 2010;18:643–650. doi: 10.1038/mt.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bennett J., Wellman J., Marshall K.A., McCague S., Ashtari M., DiStefano-Pappas J., Elci O.U., Chung D.C., Sun J., Wright J.F. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388:661–672. doi: 10.1016/S0140-6736(16)30371-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ashtari M., Nikonova E.S., Marshall K.A., Young G.J., Aravand P., Pan W., Ying G.S., Willett A.E., Mahmoudian M., Maguire A.M., Bennett J. The role of the human visual cortex in assessment of the long-term durability of retinal gene therapy in follow-on RPE65 clinical trial patients. Ophthalmology. 2017;124:873–883. doi: 10.1016/j.ophtha.2017.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Russell S., Bennett J., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., McCague S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390:849–860. doi: 10.1016/S0140-6736(17)31868-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maguire A.M., Russell S., Wellman J.A., Chung D.C., Yu Z.F., Tillman A., Wittes J., Pappas J., Elci O., Marshall K.A. Efficacy, safety, and durability of voretigene neparvovec-rzyl in RPE65 mutation-associated inherited retinal dystrophy: results of phase 1 and 3 trials. Ophthalmology. 2019;126:1273–1285. doi: 10.1016/j.ophtha.2019.06.017. [DOI] [PubMed] [Google Scholar]
- 111.Russell S., Bennett J., High K., Chung D., Wellman J., Maguire A. Saudi Ophthalmologic Society; 2016. Phase 3 trial of AAV2-hRPE65v2 (SPK-RPE65) to treat RPE65 mutation-associated inherited retinal dystrophies. [Google Scholar]
- 112.Hauswirth W.W., Aleman T.S., Kaushal S., Cideciyan A.V., Schwartz S.B., Wang L., Conlon T.J., Boye S.L., Flotte T.R., Byrne B.J., Jacobson S.G. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum. Gene Ther. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cideciyan A.V., Hauswirth W.W., Aleman T.S., Kaushal S., Schwartz S.B., Boye S.L., Windsor E.A., Conlon T.J., Sumaroka A., Pang J.J. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum. Gene Ther. 2009;20:999–1004. doi: 10.1089/hum.2009.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jacobson S.G., Cideciyan A.V., Ratnakaram R., Heon E., Schwartz S.B., Roman A.J., Peden M.C., Aleman T.S., Boye S.L., Sumaroka A. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jacobson S.G., Cideciyan A.V., Roman A.J., Sumaroka A., Schwartz S.B., Heon E., Hauswirth W.W. Improvement and decline in vision with gene therapy in childhood blindness. N. Engl. J. Med. 2015;372:1920–1926. doi: 10.1056/NEJMoa1412965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bainbridge J.W., Smith A.J., Barker S.S., Robbie S., Henderson R., Balaggan K., Viswanathan A., Holder G.E., Stockman A., Tyler N. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N. Engl. J. Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- 117.Bainbridge J.W., Mehat M.S., Sundaram V., Robbie S.J., Barker S.E., Ripamonti C., Georgiadis A., Mowat F.M., Beattie S.G., Gardner P.J. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015;372:1887–1897. doi: 10.1056/NEJMoa1414221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weleber R.G., Pennesi M.E., Wilson D.J., Kaushal S., Erker L.R., Jensen L., McBride M.T., Flotte T.R., Humphries M., Calcedo R. Results at 2 years after gene therapy for RPE65-deficient Leber congenital amaurosis and severe early-childhood-onset retinal dystrophy. Ophthalmology. 2016;123:1606–1620. doi: 10.1016/j.ophtha.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 119.Pennesi M.E., Weleber R.G., Yang P., Whitebirch C., Thean B., Flotte T.R., Humphries M., Chegarnov E., Beasley K.N., Stout J.T., Chulay J.D. Results at 5 years after gene therapy for RPE65-deficient retinal dystrophy. Hum. Gene Ther. 2018;29:1428–1437. doi: 10.1089/hum.2018.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Le Meur G., Lebranchu P., Billaud F., Adjali O., Schmitt S., Bézieau S., Péréon Y., Valabregue R., Ivan C., Darmon C. Safety and long-term efficacy of AAV4 gene therapy in patients with RPE65 Leber congenital amaurosis. Mol. Ther. 2018;26:256–268. doi: 10.1016/j.ymthe.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bonilha V.L., Rayborn M.E., Li Y., Grossman G.H., Berson E.L., Hollyfield J.G. Histopathology and functional correlations in a patient with a mutation in RPE65, the gene for retinol isomerase. Invest. Ophthalmol. Vis. Sci. 2011;52:8381–8392. doi: 10.1167/iovs.11-7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Russell S., Bennett J., Wellman J., Chung D., High K., Yu Z.-F., Tillman A., Maguire A.M. Year 2 results for a phase 3 trial of voretigene neparvovec in biallelic RPE65-mediated inherited retinal disease. Invest. Ophthalmol. Vis. Sci. 2017;58:4122. [Google Scholar]
- 123.Maguire, A., Russell, S., Bennett, J., Chung, D., Wellman, J., and High, K. (2015). Phase 3 trial of AAV2-hRPE65v2 (SPK-RPE65) to treat RPE65 mutation-associated inherited retinal dystrophies. Presented at: American Academy of Ophthalmology annual meeting. Nov. 14, 2015; Las Vegas.
- 124.Miraldi Utz V., Coussa R.G., Antaki F., Traboulsi E.I. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. 2018;39:671–677. doi: 10.1080/13816810.2018.1533027. [DOI] [PubMed] [Google Scholar]
- 125.Amado D., Mingozzi F., Hui D., Bennicelli J.L., Wei Z., Chen Y., Bote E., Grant R.L., Golden J.A., Narfstrom K. Safety and efficacy of subretinal readministration of a viral vector in large animals to treat congenital blindness. Sci. Transl. Med. 2010;2:21ra16. doi: 10.1126/scitranslmed.3000659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Leat S.J., Lovie-Kitchin J.E. Measuring mobility performance: experience gained in designing a mobility course. Clin. Exp. Optom. 2006;89:215–228. doi: 10.1111/j.1444-0938.2006.00050.x. [DOI] [PubMed] [Google Scholar]
- 127.Chung D.C., McCague S., Yu Z.F., Thill S., DiStefano-Pappas J., Bennett J., Cross D., Marshall K., Wellman J., High K.A. Novel mobility test to assess functional vision in patients with inherited retinal dystrophies. Clin. Exp. Ophthalmol. 2018;46:247–259. doi: 10.1111/ceo.13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chang K.J., Dillon L.L., Deverell L., Boon M.Y., Keay L. Orientation and mobility outcome measures. Clin. Exp. Optom. 2020;103:434–448. doi: 10.1111/cxo.13004. [DOI] [PubMed] [Google Scholar]
- 129.Banhazi J., Viriato D., Spera C., Williamson N., Bradley H., Exall E., Bennett J. Psychometric evaluation of a modified version of the Visual Function Questionnaire using data from a phase III trial in biallelic RPE65 mutation-associated inherited retinal dystrophy. Invest. Ophthalmol. Vis. Sci. 2020;61:1578. [Google Scholar]
- 130.Uyhazi K., Aravand P., Bell B., Wei Z., Leo L., Serrano L., Pearson D.J., Shpylchak I., Pham J., Vasireddy V. Treatment potential for LCA5-associated Leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 2020;61:30. doi: 10.1167/iovs.61.5.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Aleman T.S., Han G., Serrano L.W., Fuerst N.M., Charlson E.S., Pearson D.J., Chung D.C., Traband A., Pan W., Ying G.S. Natural history of the central structural abnormalities in choroideremia: a prospective cross-sectional study. Ophthalmology. 2017;124:359–373. doi: 10.1016/j.ophtha.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rohrer B., Lohr H.R., Humphries P., Redmond T.M., Seeliger M.W., Crouch R.K. Cone opsin mislocalization in Rpe65−/− mice: a defect that can be corrected by 11-cis retinal. Invest. Ophthalmol. Vis. Sci. 2005;46:3876–3882. doi: 10.1167/iovs.05-0533. [DOI] [PubMed] [Google Scholar]
- 133.Mowat F.M., Breuwer A.R., Bartoe J.T., Annear M.J., Zhang Z., Smith A.J., Bainbridge J.W. RPE65 gene therapy slows cone loss in Rpe65-deficient dogs. Gene Ther. 2013;20:545–555. doi: 10.1038/gt.2012.63. [DOI] [PubMed] [Google Scholar]
- 134.Mowat F.M., Gervais K.J., Occelli L.M., Annear M.J., Querubin J., Bainbridge J.W., Smith A.J., Ali R.R., Petersen-Jones S.M. Early-onset progressive degeneration of the area centralis in RPE65-deficient dogs. Invest. Ophthalmol. Vis. Sci. 2017;58:3268–3277. doi: 10.1167/iovs.17-21930. [DOI] [PubMed] [Google Scholar]
- 135.Edwards T.L., Jolly J.K., Groppe M., Barnard A.R., Cottriall C.L., Tolmachova T., Black G.C., Webster A.R., Lotery A.J., Holder G.E. Visual acuity after retinal gene therapy for choroideremia. N. Engl. J. Med. 2016;374:1996–1998. doi: 10.1056/NEJMc1509501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Campochiaro P.A., Lauer A.K., Sohn E.H., Mir T.A., Naylor S., Anderton M.C., Kelleher M., Harrop R., Ellis S., Mitrophanous K.A. Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum. Gene Ther. 2017;28:99–111. doi: 10.1089/hum.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aleman T., Serrano L., Han G., Pearson D., McCague S., Marshall K., Chung D.C., Liu E., Wolfing Morgan J.I., Bennett J., Maguire A.M. AAV2-hCHM subretinal delivery to the macula in choroideremia: preliminary six month safety results of an ongoing phase I/II gene therapy trial. Invest. Ophthalmol. Vis. Sci. 2017;58:4485. [Google Scholar]
- 138.Ochakovski G.A., Peters T., Michalakis S., Wilhelm B., Wissinger B., Biel M., Bartz-Schmidt K.U., Fischer M.D., RD-CURE Consortium Subretinal injection for gene therapy does not cause clinically significant outer nuclear layer thinning in normal primate foveae. Invest. Ophthalmol. Vis. Sci. 2017;58:4155–4160. doi: 10.1167/iovs.17-22402. [DOI] [PubMed] [Google Scholar]
- 139.Weed L., Ammar M.J., Zhou S., Wei Z., Serrano L.W., Sun J., Lee V., Maguire A.M., Bennett J., Aleman T.S. Safety of same-eye subretinal sequential readministration of AAV2-hRPE65v2 in non-human primates. Mol. Ther. Methods Clin. Dev. 2019;15:133–148. doi: 10.1016/j.omtm.2019.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xue K., Groppe M., Salvetti A.P., MacLaren R.E. Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye (Lond.) 2017;31:1308–1316. doi: 10.1038/eye.2017.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Davis J.L. The blunt end: surgical challenges of gene therapy for inherited retinal diseases. Am. J. Ophthalmol. 2018;196 doi: 10.1016/j.ajo.2018.08.038. xxv–xxix. [DOI] [PubMed] [Google Scholar]
- 142.Davis J.L., Gregori N.Z., MacLaren R.E., Lam B.L. Surgical technique for subretinal gene therapy in humans with inherited retinal degeneration. Retina. 2019;39(Suppl 1):S2–S8. doi: 10.1097/IAE.0000000000002609. [DOI] [PubMed] [Google Scholar]
- 143.Gregori N.Z., Lam B.L., Davis J.L. Intraoperative use of microscope-integrated optical coherence tomography for subretinal gene therapy delivery. Retina. 2019;39(Suppl 1):S9–S12. doi: 10.1097/IAE.0000000000001646. [DOI] [PubMed] [Google Scholar]
- 144.Yiu G., Chung S.H., Mollhoff I.N., Nguyen U.T., Thomasy S.M., Yoo J., Taraborelli D., Noronha G. Suprachoroidal and subretinal injections of AAV using transscleral microneedles for retinal gene delivery in nonhuman primates. Mol. Ther. Methods Clin. Dev. 2020;16:179–191. doi: 10.1016/j.omtm.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ehlers J.P., Kaiser P.K., Srivastava S.K. Intraoperative optical coherence tomography using the RESCAN 700: preliminary results from the DISCOVER study. Br. J. Ophthalmol. 2014;98:1329–1332. doi: 10.1136/bjophthalmol-2014-305294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fischer M.D., Hickey D.G., Singh M.S., MacLaren R.E. Evaluation of an optimized injection system for retinal gene therapy in human patients. Hum. Gene Ther. Methods. 2016;27:150–158. doi: 10.1089/hgtb.2016.086. [DOI] [PubMed] [Google Scholar]
- 147.Hollyfield J.G., Varner H.H., Rayborn M.E., Osterfeld A.M. Retinal attachment to the pigment epithelium. Linkage through an extracellular sheath surrounding cone photoreceptors. Retina. 1989;9:59–68. [PubMed] [Google Scholar]
- 148.Matsuda S., Harino S., Iwahashi Y., Nagaya C. [Long-term enlargement of laser photocoagulation scars after treatment of choroidal neovascularization] Nippon Ganka Gakkai Zasshi. 2002;106:708–713. [PubMed] [Google Scholar]
- 149.McGuigan D.B., 3rd, Roman A.J., Cideciyan A.V., Matsui R., Gruzensky M.L., Sheplock R., Jacobson S.G. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa: filling a need to accommodate multicenter clinical trials. Invest. Ophthalmol. Vis. Sci. 2016;57:3118–3128. doi: 10.1167/iovs.16-19302. [DOI] [PubMed] [Google Scholar]
- 150.Roman A.J., Cideciyan A.V., Aleman T.S., Jacobson S.G. Full-field stimulus testing (FST) to quantify visual perception in severely blind candidates for treatment trials. Physiol. Meas. 2007;28:N51–N56. doi: 10.1088/0967-3334/28/8/N02. [DOI] [PubMed] [Google Scholar]
- 151.Klein M., Birch D.G. Psychophysical assessment of low visual function in patients with retinal degenerative diseases (RDDs) with the Diagnosys full-field stimulus threshold (D-FST) Doc. Ophthalmol. 2009;119:217–224. doi: 10.1007/s10633-009-9204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Roman A.J., Schwartz S.B., Aleman T.S., Cideciyan A.V., Chico J.D., Windsor E.A.M., Gardner L.M., Ying G.S., Smilko E.E., Maguire M.G., Jacobson S.G. Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp. Eye Res. 2005;80:259–272. doi: 10.1016/j.exer.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 153.US Food & Drug Administration . 2017. FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss.https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss#:∼:text=FDA%20News%20Release-,FDA%20approves%20novel%20gene%20therapy%20to%20treat%20patients%20with,form%20of%20inherited%20vision%20loss&text=The%20U.S.%20Food%20and%20Drug,that%20may%20result%20in%20blindness [Google Scholar]
- 154.Hendrickson A.E., Yuodelis C. The morphological development of the human fovea. Ophthalmology. 1984;91:603–612. doi: 10.1016/s0161-6420(84)34247-6. [DOI] [PubMed] [Google Scholar]
- 155.Vajzovic L., Hendrickson A.E., O’Connell R.V., Clark L.A., Tran-Viet D., Possin D., Chiu S.J., Farsiu S., Toth C.A. Maturation of the human fovea: correlation of spectral-domain optical coherence tomography findings with histology. Am. J. Ophthalmol. 2012;154:779–789.e2. doi: 10.1016/j.ajo.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ochakovski G.A., Bartz-Schmidt K.U., Fischer M.D. Retinal gene therapy: surgical vector delivery in the translation to clinical trials. Front. Neurosci. 2017;11:174. doi: 10.3389/fnins.2017.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Takahashi K., Morizane Y., Hisatomi T., Tachibana T., Kimura S., Hosokawa M.M., Shiode Y., Hirano M., Doi S., Toshima S. The influence of subretinal injection pressure on the microstructure of the monkey retina. PLoS ONE. 2018;13:e0209996. doi: 10.1371/journal.pone.0209996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nork T.M., Murphy C.J., Kim C.B., Ver Hoeve J.N., Rasmussen C.A., Miller P.E., Wabers H.D., Neider M.W., Dubielzig R.R., McCulloh R.J., Christian B.J. Functional and anatomic consequences of subretinal dosing in the cynomolgus macaque. Arch. Ophthalmol. 2012;130:65–75. doi: 10.1001/archophthalmol.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sakai T., Calderone J.B., Lewis G.P., Linberg K.A., Fisher S.K., Jacobs G.H. Cone photoreceptor recovery after experimental detachment and reattachment: an immunocytochemical, morphological, and electrophysiological study. Invest. Ophthalmol. Vis. Sci. 2003;44:416–425. doi: 10.1167/iovs.02-0633. [DOI] [PubMed] [Google Scholar]
- 160.Dalkara D., Byrne L.C., Klimczak R.R., Visel M., Yin L., Merigan W.H., Flannery J.G., Schaffer D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 161.Boye S.E., Alexander J.J., Witherspoon C.D., Boye S.L., Peterson J.J., Clark M.E., Sandefer K.J., Girkin C.A., Hauswirth W.W., Gamlin P.D. Highly efficient delivery of adeno-associated viral vectors to the primate retina. Hum. Gene Ther. 2016;27:580–597. doi: 10.1089/hum.2016.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Khabou H., Garita-Hernandez M., Chaffiol A., Reichman S., Jaillard C., Brazhnikova E., Bertin S., Forster V., Desrosiers M., Winckler C. Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight. 2018;3 doi: 10.1172/jci.insight.96029. e96029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Miller J.W., Vandenberghe L.H. Breaking and sealing barriers in retinal gene therapy. Mol. Ther. 2018;26:2081–2082. doi: 10.1016/j.ymthe.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cukras C., Wiley H.E., Jeffrey B.G., Sen H.N., Turriff A., Zeng Y., Vijayasarathy C., Marangoni D., Ziccardi L., Kjellstrom S. Retinal AAV8-RS1 gene therapy for X-linked retinoschisis: initial findings from a Phase I/IIa trial by intravitreal delivery. Mol. Ther. 2018;26:2282–2294. doi: 10.1016/j.ymthe.2018.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cideciyan A.V., Jacobson S.G., Drack A.V., Ho A.C., Charng J., Garafalo A.V., Roman A.J., Sumaroka A., Han I.C., Hochstedler M.D. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019;25:225–228. doi: 10.1038/s41591-018-0295-0. [DOI] [PubMed] [Google Scholar]
- 166.Vijayasarathy C., Zeng Y., Brooks M., Fariss R.N., Sieving P.A. Genetic rescue of X-linked retinoschisis mouse (Rs1-/y) retina induces quiescence of the retinal microglial inflammatory state following AAV8-RS1 gene transfer and identifies gene networks underlying retinal recovery. Hum. Gene Ther. 2020 doi: 10.1089/hum.2020.213. Published online October 6, 2020. 10.1089/hum.2020.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Takahashi K., Igarashi T., Miyake K., Kobayashi M., Yaguchi C., Iijima O., Yamazaki Y., Katakai Y., Miyake N., Kameya S. Improved intravitreal AAV-mediated inner retinal gene transduction after surgical internal limiting membrane peeling in cynomolgus monkeys. Mol. Ther. 2017;25:296–302. doi: 10.1016/j.ymthe.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Reichel F.F., Peters T., Wilhelm B., Biel M., Ueffing M., Wissinger B., Bartz-Schmidt K.U., Klein R., Michalakis S., Fischer M.D., RD-CURE Consortium Humoral immune response after intravitreal but not after subretinal AAV8 in primates and patients. Invest. Ophthalmol. Vis. Sci. 2018;59:1910–1915. doi: 10.1167/iovs.17-22494. [DOI] [PubMed] [Google Scholar]
- 169.Seitz I.P., Michalakis S., Wilhelm B., Reichel F.F., Ochakovski G.A., Zrenner E., Ueffing M., Biel M., Wissinger B., Bartz-Schmidt K.U., RD-CURE Consortium Superior retinal gene transfer and biodistribution profile of subretinal versus intravitreal delivery of AAV8 in nonhuman primates. Invest. Ophthalmol. Vis. Sci. 2017;58:5792–5801. doi: 10.1167/iovs.17-22473. [DOI] [PubMed] [Google Scholar]
- 170.Palczewski K., Kiser P.D. Shedding new light on the generation of the visual chromophore. Proc. Natl. Acad. Sci. USA. 2020;117:19629–19638. doi: 10.1073/pnas.2008211117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gao S., Kahremany S., Zhang J., Jastrzebska B., Querubin J., Petersen-Jones S.M., Palczewski K. Retinal-chitosan conjugates effectively deliver active chromophores to retinal photoreceptor cells in blind mice and dogs. Mol. Pharmacol. 2018;93:438–452. doi: 10.1124/mol.117.111294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hollingsworth T.J., Gross A.K. Innate and autoimmunity in the pathogenesis of inherited retinal dystrophy. Cells. 2020;9:630. doi: 10.3390/cells9030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lew D.S., Mazzoni F., Finnemann S.C. Microglia inhibition delays retinal degeneration due to MerTK phagocytosis receptor deficiency. Front. Immunol. 2020;11:1463. doi: 10.3389/fimmu.2020.01463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Iannaccone A., Radic M.Z. Increased protein citrullination as a trigger for resident immune system activation, intraretinal inflammation, and promotion of anti-retinal autoimmunity: intersecting paths in retinal degenerations of potential therapeutic relevance. Adv. Exp. Med. Biol. 2019;1185:175–179. doi: 10.1007/978-3-030-27378-1_29. [DOI] [PubMed] [Google Scholar]
- 175.Yang P., Lockard R., Titus H., Hiblar J., Weller K., Wafai D., Weleber R.G., Duvoisin R.M., Morgans C.W., Pennesi M.E. Suppression of cGMP-dependent photoreceptor cytotoxicity with mycophenolate is neuroprotective in murine models of retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 2020;61:25. doi: 10.1167/iovs.61.10.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Georgiadis A., Duran Y., Ribeiro J., Abelleira-Hervas L., Robbie S.J., Sünkel-Laing B., Fourali S., Gonzalez-Cordero A., Cristante E., Michaelides M. Development of an optimized AAV2/5 gene therapy vector for Leber congenital amaurosis owing to defects in RPE65. Gene Ther. 2016;23:857–862. doi: 10.1038/gt.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Busskamp V., Picaud S., Sahel J.A., Roska B. Optogenetic therapy for retinitis pigmentosa. Gene Ther. 2012;19:169–175. doi: 10.1038/gt.2011.155. [DOI] [PubMed] [Google Scholar]
- 178.Cronin T., Vandenberghe L.H., Hantz P., Juttner J., Reimann A., Kacsó A.E., Huckfeldt R.M., Busskamp V., Kohler H., Lagali P.S. Efficient transduction and optogenetic stimulation of retinal bipolar cells by a synthetic adeno-associated virus capsid and promoter. EMBO Mol. Med. 2014;6:1175–1190. doi: 10.15252/emmm.201404077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lagali P.S., Balya D., Awatramani G.B., Münch T.A., Kim D.S., Busskamp V., Cepko C.L., Roska B. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat. Neurosci. 2008;11:667–675. doi: 10.1038/nn.2117. [DOI] [PubMed] [Google Scholar]
- 180.da Cruz L., Dorn J.D., Humayun M.S., Dagnelie G., Handa J., Barale P.O., Sahel J.A., Stanga P.E., Hafezi F., Safran A.B., Argus II Study Group Five-year safety and performance results from the argus ii retinal prosthesis system clinical trial. Ophthalmology. 2016;123:2248–2254. doi: 10.1016/j.ophtha.2016.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ho A.C., Humayun M.S., Dorn J.D., da Cruz L., Dagnelie G., Handa J., Barale P.O., Sahel J.A., Stanga P.E., Hafezi F., Argus II Study Group Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology. 2015;122:1547–1554. doi: 10.1016/j.ophtha.2015.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Aleman, T.S., Maguire, A.M., and Bennett, J. (2020). Chapter 28: Gene therapy. In: Pediatric Retina, Third Edition, M.E. Hartnett, ed. (Wolters Kluwer), pp. 189–217.
- 183.Byrne B.J. Safety first: perspective on patient-centered development of AAV gene therapy products. Mol. Ther. 2018;26:669–671. doi: 10.1016/j.ymthe.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thompson D.A., Iannaccone A., Ali R.R., Arshavsky V.Y., Audo I., Bainbridge J.W.B., Besirli C.G., Birch D.G., Branham K.E., Cideciyan A.V., Monaciano Consortium Advancing clinical trials for inherited retinal diseases: recommendations from the second Monaciano Symposium. Transl. Vis. Sci. Technol. 2020;9:2. doi: 10.1167/tvst.9.7.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ashtari M., Cyckowski L.L., Monroe J.F., Marshall K.A., Chung D.C., Auricchio A., Simonelli F., Leroy B.P., Maguire A.M., Shindler K.S., Bennett J. The human visual cortex responds to gene therapy-mediated recovery of retinal function. J. Clin. Invest. 2011;121:2160–2168. doi: 10.1172/JCI57377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ashtari M., Zhang H., Cook P.A., Cyckowski L.L., Shindler K.S., Marshall K.A., Aravand P., Vossough A., Gee J.C., Maguire A.M. Plasticity of the human visual system after retinal gene therapy in patients with Leber’s congenital amaurosis. Sci. Transl. Med. 2015;7:296ra110. doi: 10.1126/scitranslmed.aaa8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wolfing Morgan J.I., Jiang Y.Y., Serrano L.W., Pearson D.J., Bennett J., Maguire A., Aleman T. Short-term assessment of subfoveal injection of AAV2-hCHM gene augmentation in choroideremia using adaptive optics ophthalmoscopy. Invest. Ophthalmol. Vis. Sci. 2020;61:1130. doi: 10.1001/jamaophthalmol.2022.0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.