

Abstract
Chimeric antigen receptors (CARs) are fusion proteins that contain antigen-recognition domains and T cell signaling domains. Signaling lymphocytic-activation molecule F7 (SLAMF7) is a promising target for CAR T cell therapies of the plasma cell malignancy multiple myeloma (MM) because SLAMF7 is expressed by MM but not normal nonhematopoietic cells. We designed CARs targeting SLAMF7. We transduced human T cells with anti-SLAMF7 CARs containing either CD28 or 4-1BB costimulatory domains. T cells expressing CD28-containing CARs or 4-1BB-containing CARs recognized SLAMF7 in vitro. SLAMF7-specific cytokine release was higher for T cells expressing CARs with CD28 versus 4-1BB domains. In murine solid tumor and disseminated tumor models, anti-tumor activity of T cells was superior with CD28-containing CARs versus 4-1BB-containing CARs. Because of SLAMF7 expression on some normal leukocytes, especially natural killer cells that control certain viral infections, the inclusion of a suicide gene with an anti-SLAMF7 CAR is prudent. We designed a construct with a CD28-containing anti-SLAMF7 CAR and a suicide gene. The suicide gene encoded a dimerization domain fused to a caspase-9 domain. T cells expressing the anti-SLAMF7 CAR plus suicide-gene construct specifically recognized SLAMF7 in vitro and eliminated tumors from mice. T cells expressing this construct were eliminated on demand by administering the dimerizing agent AP1903 (rimiducid).
Keywords: CAR, CAR T cells, multiple myeloma, SLAMF7, CD319, immunotherapy, T cell therapy, CD28, 4-1BB, suicide gene
Graphical Abstract
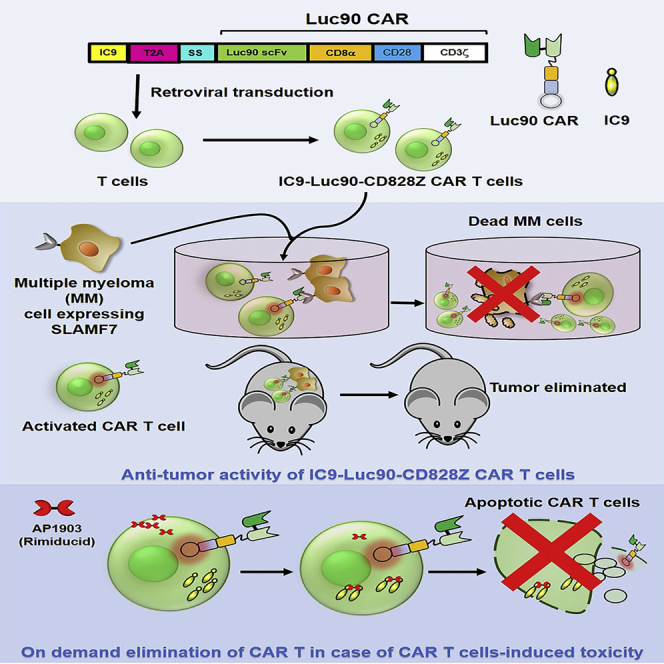
Amatya and colleagues generated safer anti-SLAMF7 CAR T cells for multiple myeloma therapy. An inducible suicide gene in the anti-SLAMF7 CAR construct allowed on-demand elimination the CAR T cells. Interestingly, the type of costimulatory domains used in the CAR construct influenced the anti-tumor activity of the CAR T cells in mice.
Introduction
Multiple myeloma (MM) is an almost-always incurable malignancy of plasma cells; new therapies are needed for MM.1,2 Chimeric antigen receptors (CARs) are artificial fusion proteins with antigen-recognition domains and T cell signaling domains.3,4 T cells expressing CARs targeting the MM-associated antigen B cell maturation antigen (BCMA) have proven clinical efficacy.5, 6, 7, 8, 9, 10 However, variable BCMA expression and loss of BCMA expression from MM after anti-BCMA CAR T cell treatment have been documented,5,8 which makes development of CAR T cell therapies targeting other antigens an important goal.
Signaling lymphocytic-activation molecule F7 (SLAMF7), also known as CD319, CD2 subset-1 (CS1), and CD2-like receptor-activating cytotoxic cells (CRACCs), is a highly glycosylated protein that belongs to the SLAMF family of cell-surface receptor proteins.11,12 SLAMF7 is a promising CAR target for MM therapy due to high and uniform expression on MM cells and lack of expression on normal nonhematopoietic cells.13, 14, 15 Although the role of SLAMF7 in MM is unclear, SLAMF7 expression is speculated to enhance growth, survival, and adhesion of MM cells to bone marrow stromal cells.16,17 Additionally, SLAMF7 is also known to be expressed on a variety of other cells.15,18,19 A CAR targeting SLAMF7 has potential to eliminate SLAMF7-expressing leukocytes, especially natural killer (NK) cells and CD8+ T cells. Thus, despite being a promising therapeutic target for MM in preclinical studies,20, 21, 22, 23 anti-SLAMF7 CAR T cells might cause cytopenias, especially NK cell deficiency that might lead to a high risk for severe viral infections, such as cytomegalovirus infections.24,25 A suicide gene could be a useful strategy to eliminate anti-SLAMF7 CAR T cells in case of severe toxicities.
The inducible caspase-9 (iCasp9 or IC9) suicide gene has been used to eradicate T cells in humans with reversal of clinical toxicity.26,27 The IC9 gene contains part of the human caspase-9 protein fused to a dimerization domain from the human FK-506-binding protein-12 (FKBP12).28 Activation of the caspase-9 domain of IC9 is dependent on dimerization of FKBP12 domains; this dimerization occurs only upon administration of the small molecule drug rimiducid (AP1903).26,27
An important consideration in CAR design is the selection of costimulatory domains. Costimulatory domains of most clinically used CARs are from either CD28 or 4-1BB.29 CD28 costimulatory domains have been reported to provide faster and stronger functional activity to CAR T cells, and 4-1BB costimulatory domains have been reported to enhance persistence of CAR T cells under some circumstances; however, the relative anti-tumor activity of CD28-containing CARs versus 4-1BB-containing CARs is unclear and might be dependent on a variety of conditions.30, 31, 32, 33, 34, 35, 36
In this paper, we performed comparisons between anti-SLAMF7 CARs with either CD28 or 4-1BB costimulatory domains. We also designed and tested constructs encoding both an anti-SLAMF7 CAR and the IC9 suicide gene.
Results
SLAMF7 Expression Is Limited to Certain Leukocytes
We assessed SLAMF7 expression in normal tissues. SLAMF7 RNA was detected by PCR in some tissues, including lymph nodes, tonsils, stomach, bone marrow, and spleen, which is likely explained by the presence of SLAMF7+ leukocytes infiltrating these organs; otherwise, SLAMF7 RNA was not detected (Figure 1A). Next, we assessed SLAMF7 protein expression by immunohistochemistry using a normal human tissue microarray (Table S1). SLAMF7 protein was not expressed in major organs except in plasma cells, lymphocytes, and macrophages. SLAMF7 was not expressed on CD34+ hematopoietic stem cells (Figure 1B). We confirmed high and uniform expression of SLAMF7 on CD38+ cells from bone marrow of MM patients (Figure 1C). We also assessed SLAMF7 expression on peripheral blood mononuclear cells (PBMCs). We found SLAMF7 expression on most CD56+ NK cells, some CD8+ T cells, and small fractions of monocytes and CD4+ T cells (Figures 1D and 1E). We assessed SLAMF7 expression on CD4+ and CD8+ T cells before and after activation with the anti-CD3 antibody OKT3 in interleukin (IL)-2-containing medium. This is the same T cell culture process used in other experiments reported in this paper and in clinical trials conducted by our group. SLAMF7 expression increased with activation of either CD4+ or CD8+ T cells. As expected, SLAMF7 expression was higher on CD8+ T cells versus CD4+ T cells (Figures 1F–1H).
Figure 1.

SLAMF7 Expression
(A) SLAMF7 RNA expression was assessed in all major normal tissues and a plasmacytoma made up of SLAMF7+ multiple myeloma (MM) cells. Quantitative PCR was performed on cDNA from each of the tissues. SLAMF7 cDNA copies were normalized to actin cDNA copies from each tissue. (B) Primary human CD34+ hematopoietic stem cells were obtained from the blood of a patient after the patient received granulocyte colony-stimulating factor to mobilize stem cells from the bone marrow. Staining with anti-CD34 and either anti-SLAMF7 or an isotype-matched control antibody was performed, followed by flow cytometry. Plots are gated on live CD34+ cells. One representative experiment of 2 experiments with different donors is shown. (C) Primary bone marrow cells from a patient with MM were stained with antibodies against CD38 alone or with antibodies against both CD38 and SLAMF7. Plots are gated on live CD3-negative cells. (D) Peripheral blood mononuclear cells (PBMCs) were stained for SLAMF7 along with antibodies against CD3, CD4, CD8, CD14, and CD56. The plot showing CD3+CD4+ cells is gated on CD3+CD4+ cells. The plot showing CD3+CD8+ cells is gated on CD3+CD8+ cells. The NK cells’ plot is gated on CD3-negative, CD56+ cells. The monocyte plot is gated on CD3-negative, CD14+ cells. For T cells and NK cells, plots were gated on lymphocyte forward-scatter and side-scatter regions. For monocytes, a more inclusive gate that included lymphocytes as well as higher forward-scatter and side-scatter events was used. All plots had dead cells excluded. Regions were set with an isotype-control antibody for SLAMF7. (E) Blood CD3+CD8+ T cells, CD3+CD4+ T cells, NK cells, and monocytes were stained and analyzed as in (D). Percentages of each cell type that expressed SLAMF7 are shown. For monocytes, n = 4 different donors. For other cell types, n = 8 different donors. Bars represent mean plus standard error of the mean (SEM). (F) Human PBMCs were cultured overnight in culture medium alone without IL-2. The PBMCs from each donor were then divided into two portions. The first portion was stained with antibodies against CD3, CD4, CD8, and SLAMF7 and analyzed by flow cytometry. Representative example plots from one of three donors are shown. The plots shown are all gated on live CD3+ lymphocytes and then on either CD4+ or CD8+ cells. The second portion of PBMCs was placed in culture with IL-2 and activated with an anti-CD3 antibody for 1 day. Activated cells were stained, and flow cytometry was performed in the same manner as before activation. (G and H) PBMCs from 3 donors were cultured and analyzed before and after activation as in (F). The mean + SEM of %SLAMF7+ cells for (G) CD4+ and (H) CD8+ T cells is shown. Statistics were by two-tailed, paired t test; p < 0.05 was considered statistically significant.
Anti-SLAMF7 CAR Expression
We constructed CARs incorporating single-chain variable fragments (scFvs) derived from either the Luc90 or Luc63 monoclonal antibodies (Figure 2A). These CARs also included hinge and transmembrane domains from CD8α, CD28 costimulatory domains and CD3ζ activation domains. The CARs were encoded by the mouse stem cell virus-based splice-gag vector (MSGV1) gamma-retroviral vector,37 and the CARs were designated Luc90-CD828Z and Luc63-CD828Z. The Luc90 scFv is derived from a murine monoclonal antibody, and the Luc63 scFv is derived from the humanized version of the monoclonal antibody Luc63, also known as elotuzumab. After transduction, Luc90-CD828Z and Luc63-CD828Z were expressed on the surface of CD4+ and CD8+ T cells (Figures 2B and 2C); both CARs had similar transduction efficiencies (Figures 2D and 2E).
Figure 2.

Comparison of Different Anti-SLAMF7 CAR Designs
(A) Luc63-CD828Z contained the Luc63 scFv, and Luc90-CD828Z contained the Luc90 scFv. Both CARs contained CD8α hinge and transmembrane domains, CD28 costimulatory domains, and CD3ζ activation domains. (B) An example of CAR expression on CD4+ T cells is shown. Whole PBMCs were stimulated on day 0 of culture with an anti-CD3 antibody and cultured in IL-2-containing medium. On day 2 of culture, transductions were carried out with MSGV1 gamma-retroviral vectors encoding either Luc63-CD828Z or Luc90-CD828Z. 5 days after transduction, flow cytometry was performed with protein L to detect CARs, and staining was also performed against CD3, CD4, and CD8. Plots are gated on live CD3+CD4+ lymphocytes. (C) CAR expression on CD8+ T cells is shown. Experiment was performed as in (B). Plots are gated on live CD3+CD8+ lymphocytes. Mean + SEM percentages of (D) CD4+ T cells or (E) CD8+ T cells expressing Luc63-CD828Z or Luc90-CD828Z is shown; n = 5 different donors for all groups. T cells were cultured, transduced, and analyzed as in (B). For both CD4+ and CD8+ T cells, there was no significant difference (N.S.) in the percentage of T cells expressing Luc63-CD828Z versus Luc90-CD828Z. (F) CD4+ T cells and (G) CD8+ T cells were assessed for SLAMF7-specific CD107a upregulation. T cells were transduced with either Luc63-CD828Z or Luc90-CD828Z or left untransduced. T cells were cultured and transduced as in (B). Assays were conducted 5 days after transduction. T cells were cultured with either SLAMF7+ SLAMF7-RPMI8226 cells or SLAMF7-negative NGFR-K562 cells for 4 h in the presence of anti-CD107a antibodies. After the incubation, T cells were stained for CD3, CD4, and CD8. Data are presented after normalization for CAR expression, and %SLAMF7-specific CD107a+ events are shown. %SLAMF7-specific CD107a+ events were calculated as %CD107a+ events with SLAMF7-RPMI8226 stimulation minus %CD107a+ events with NGFR-K562 stimulation. Bars represent mean + SEM. For both CD4+ and CD8+ T cells, there was no statistically significant difference in CD107a upregulation when Luc63-CD828Z and Luc90-CD828Z were compared; n = 5 . (D–G) Statistical testing was by paired, two-tailed t tests, and statistical significance was defined as p < 0.05. (H) T cells were cultured and transduced with Luc63-CD828Z or Luc90-CD828Z or left untransduced. 5 days after transduction, T cells were cultured overnight with target cells. SLAMF7-RPMI8226 and SLAMF7-K562 were SLAMF7+. NGFR-K562 and CCRF-CEM were SLAMF7 negative. After the overnight culture, supernatant was assayed by ELISA for IFN-γ. Bars show mean + SEM of 3 experiments with cells from 3 different donors.
Function of Luc63- and Luc90-Containing CARs
There was no difference in SLAMF7-specific degranulation when Luc90-CD828Z and Luc63-CD828Z were compared for CD4+ (Figure 2F) and CD8+ (Figure 2G). Interferon (IFN)-γ release, when T cells were cultured with SLAMF7+ target cells, was slightly higher for T cells expressing Luc90-CD828Z versus T cells expressing Luc63-CD828Z. Background IFN-γ release, when CAR T cells were cultured with SLAMF7-negative target cells and when CAR T cells were cultured alone, was slightly higher with Luc63-CD828Z versus Luc90-CD828Z (Figure 2H). Due, in part, to slightly lower nonspecific activity and slightly higher SLAMF7-specific IFN-γ release, we decided to use Luc90-CD828Z in future experiments, although functional differences between these two CARs were small.
MM patients have detectable (range, 1–80 ng/mL) serum SLAMF7.14 We sought to determine if adding soluble SLAMF7 to cultures could inhibit anti-SLAMF7 CAR T cell activity. We conducted IFN-γ ELISA by coculturing Luc90-CD828Z CAR T cells with SLAMF7+ target cells and adding varying concentrations of soluble SLAMF7. Soluble SLAMF7 did not block antigen-specific IFN-γ release by anti-SLAMF7 CAR T cells (Table S2).
Expression of CARs Containing CD28 versus 4-1BB Costimulatory Domains
We compared Luc90-CD828Z to Luc90-CD8BBZ, CARs that differ from each other only in their costimulatory domains, as shown in Figure 3A. Both Luc90-CD828Z and Luc90-CD8BBZ were expressed on T cells (Figures 3B and 3C). The percentages of transduced T cells that expressed the two CARs and the level of expression on each transduced T cell were not statistically different (Figures 3D and 3E). Interestingly, the CD4-to-CD8 ratio of Luc90-CD8BBZ-expressing T cells was higher than the CD4-to-CD8 ratio of Luc90-CD828Z-expressing T cells (Figure 3F). Analysis for memory phenotype showed that for both CD4+ and CD8+ T cells, the percentage of central memory T cells was statistically higher for Luc90-CD8BBZ-expressing T cells than Luc90-CD828Z-expressing T cells (Figure S1).
Figure 3.

Comparison of Anti-SLAMF7 CARs with Different Costimulatory Domains
(A) Luc90-CD8BBZ was identical to Luc90-CD828Z except for substitution of 4-1BB for CD28. (B and C) T cells were stimulated with anti-CD3 on day 0 and transduced on day 2 of culture. Flow cytometry with protein L staining was performed on day 7 of culture to detect CARs. (B) CAR expression on MSGV1-Luc90-CD828Z-transduced T cells. (C) CAR expression on MSGV1-Luc90-CD8BBZ-transduced T cells. (B and C) The left plot is gated on live CD3+ lymphocytes. The right plot is gated on the CD3+CAR+ population from the left plot. (D) The percentages of live CD3+ T cells expressing cell-surface Luc90-CD828Z or Luc90-CD8BBZ from cultures of transduced T cells are shown; lines connect values for each CAR from cultures of the same patient. Differences in percentages of T cells expressing Luc90-CD828Z versus Luc90-CD8BBZ were not statistically significant. (E) There was not a statistically significant difference in median fluorescence intensities (MFIs) of CD3+CAR+ cells transduced with either Luc90-CD828Z or Luc90-CD8BBZ. (F) CD4-to-CD8 ratios of T cells expressing either Luc90-CD828Z or Luc90-CD8BBZ are shown. Lines connect results from the same patient. (D–F) T cells were cultured and plots gated as in (B) and (C). (D–F) n = 7 different donors; statistics were by paired, two-tailed t test. (G and H) T cells were transduced with either Luc90-CD828Z or Luc90-CD8BBZ. T cells were labeled with CFSE and cultured with either SLAMF7-K562 cells or NGFR-K562 cells for 4 days. The changes in numbers of (G) CD4+CAR+ T cells or (H) CD8+CAR+ T cells between day 0 and day 4 of culture are shown. The change in cell numbers for Luc90-CD828Z and Luc90-CD8BBZ T cells from the same patient is connected by a line. Statistical testing was by paired, two-tailed t tests, and statistical significance was defined as p < 0.05. (I and J) At the end of the 4-day culture period, the same T cell cultures in (G) and (H) were assessed for the ratio of CFSE MFI with SLAMF7-K562 stimulation versus NGFR-K562 stimulation (SLAMF7/NGFR ratio). A decrease in CFSE MFI indicates proliferation. Lower SLAMF7/NGFR ratios indicate more SLAMF7-specific proliferation. Cultures from the same patient are connected by a line. Differences were not statistically different. (G–J) n = 5 different donors. (K–M) SLAMF7-specific cytokine production was compared between paired cultures of T cells from the same donor expressing Luc90-CD828Z or Luc90-CD8BBZ; the paired cultures are connected by lines. T cells were cultured overnight with either MM.1S cells or NGFR-K562 cells, and ELISAs were performed on supernatants. Results were normalized for CAR expression. MM.1S-specific cytokine release was calculated by subtracting cytokine levels with NGFR-K562 stimulation from cytokine levels with MM.1S stimulation. (K) IFN-γ, (L) IL-2, and (M) TNF-α. n = 6 different donors for IFN-γ; n = 5 different donors for IL-2 and TNF-α. Statistical testing was by two-tailed ratio, paired t tests; statistical significance was p < 0.05. There was a statistically significant difference for IFN-γ and IL-2. The difference for TNF-α was of borderline statistical significance.
Proliferation of Anti-SLAMF7 CARs Containing CD28 versus 4-1BB
In proliferation assays, there was not a statistically significant difference in numbers of accumulated CD4+CAR+ T cells when Luc90-CD828Z T cells and Luc90-CD8BBZ T cells were cultured for 4 days with SLAMF7+ target cells (Figure 3G). However, more CD8+ Luc90-CD828Z T cells than CD8+ Luc90-CD8BBZ T cells accumulated over a 4-day culture with SLAMF7+ target cells (Figure 3H). There was not a statistically significant difference in SLAMF7-specific proliferation for T cells expressing Luc90-CD828Z versus Luc90-CD8BBZ for either CD4+ (Figure 3I) or CD8+ (Figure 3J) T cells. We assessed activation-induced cell death (AICD) in T cells expressing either Luc90-CD828Z or Luc90-CD8BBZ. There was not a statistically significant difference in AICD for either CD4+ or CD8+ T cells; however, there was a trend toward less AICD among CD8+ T cells expressing Luc90-CD828Z versus Luc90-CD8BBZ (Figure S2).
Cytokine Release and Degranulation by T Cells Expressing CARs with CD28 versus 4-1BB
Compared with T cells expressing Luc90-CD8BBZ, T cells expressing Luc90-CD828Z released higher levels of IFN-γ (Figure 3K), IL-2 (Figure 3L), and tumor necrosis factor (TNF)-α (Figure 3M) when stimulated with MM.1S target cells in vitro. Compared with Luc90-CD828Z T cells, Luc90-CD8BBZ T cells exhibited higher levels of nonspecific IFN-γ release against SLAMF7-negative target cells in vitro (Figure S3). We did not find a statistically significant difference in SLAMF7-specific degranulation by CD107a expression when Luc90-CD828Z T cells and Luc90-CD8BBZ T cells were compared (Figure S4).
Anti-tumor Activity of CD28 CARs Was Superior to Anti-tumor Activity of 4-1BB CARs
We evaluated anti-tumor efficacy of Luc90-CD8BBZ and Luc90-CD828Z CAR T cells in NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice bearing solid tumors of the MM.1S human MM cell line. Mice were left untreated or received a single intravenous infusion of 2 × 106 T cells expressing Luc90-CD828Z, Luc90-CD8BBZ, or Hu19-CD828Z. Only T cells expressing Luc90-CD828Z eradicated all MM.1S solid tumors. The anti-tumor activity of T cells expressing Luc90-CD8BBZ was less than that of T cells expressing Luc90-CD828Z (Figures 4A and 4B). When we doubled the dose of anti-SLAMF7 CAR T cells to 4 × 106 CAR T cells per mouse, both Luc90-CD828Z and Luc90-CD8BBZ CAR T cells consistently eliminated MM.1S solid tumors (Figure S5). At the 4 × 106 CAR T cell dose, tumor elimination was superior at early time points by Luc90-CD828Z CAR T cells compared with Luc90-CD8BBZ CAR T cells.
Figure 4.

Luc90-CD828Z CAR T Cells Are Superior to Luc90-CD8BBZ in Eliminating Tumors
(A) Solid tumors were established in NSG mice by intradermal injection of 4 × 106 MM.1S cells. 7 days after tumor implantation, mice were either left untreated or infused intravenously with 2 × 106 T cells transduced with Luc90-CD828Z, Luc90-CD8BBZ, or the negative-control CAR Hu19-CD828Z. Graphs show mean tumor volume ± SEM for each time point. The graph shows combined data from three independent experiments with different donor T cells. There were five mice in each group per experiment for a total of 15 mice for each group shown on the graph. Lines end when the first mouse of the group, represented by the line, was sacrificed or when the experiment ended at day 39. (B) Kaplan-Meier survival graph of the same mice as in (A). By the log rank test, there were statistically significant differences in survival between mice receiving Hu19-CD828Z T cells and Luc90-CD828Z T cells (p < 0.0001) and mice receiving Hu19-CD828Z T cells and Luc90-CD8BBZ T cells (p < 0.0001). There was also a statistically significant difference in survival between mice receiving Luc90-CD828Z T cells and Luc90-CD8BBZ T cells (p = 0.0011). (C) Disseminated tumor burdens were established in NSG mice by injecting 8 × 106 MM.1S-luciferase cells intravenously. 13 days after tumor cell injection, mice received 4 × 106 T cells transduced with either Luc90-CD828Z or Luc90-CD8BBZ or the negative-control CAR Hu19-CD828Z. Another group of mice was left untreated. Tumor burden was evaluated by bioluminescent imaging (BLI) every 3 days. The figure is representative of one experiment out of two independent experiments with T cells from different donors. (D) Average BLI intensity was compared for mice with disseminated MM1.S-luciferase burdens receiving 4 × 106 T cells transduced with either Luc90-CD828Z or Luc90-CD8BBZ. The graph includes BLI from 10 mice from each treatment group, 12 days after CAR T cell infusion. There was a statistically significant difference in BLI between Luc90-CD828Z and Luc90-CD8BBZ (p = 0.0023; unpaired, two-tailed t test). The graph shows one point for each mouse. Bars represent mean ± SEM. Five mice in each group are from (C); an additional 5 mice from a second experiment are also included in the graph. (E) Kaplan-Meier survival plot of mice in the experiments reported in (D). The graph includes combined data from two independent experiments with different donor T cells. By a log rank test, there was a statistically significant difference in survival between mice receiving T cells expressing Hu19-CD828Z and mice receiving T cells expressing either Luc90-CD828Z or Luc90-CD8BBZ (p < 0.0001 for both comparisons). There was also a statistically significant difference in survival between mice receiving T cells that expressed Luc90-CD828Z and Luc90-CD8BBZ (p = 0.0056). There were 10 mice in each group. (F) Solid MM.1S tumors were established in NSG mice. Mice were infused with 4 × 106 CAR+ T cells. 7 days after CAR T cell infusion, tumors were excised and processed into single-cell suspensions. Quantitative PCR was performed with a PCR reaction capable of detecting both Luc90-CD828Z and Luc90-CD8BBZ to quantify the number of tumor-infiltrating T cells in the tumors of each mouse. Mice that received Luc90-CD828Z T cells had more tumor-infiltrating T cells (p = 0.0379; unpaired, two-tailed t test; n = 10 mice per group). The horizontal bars represent the medians.
Similar to solid tumors, clearance of disseminated MM.1S cells was more complete with Luc90-CD828Z T cells versus Luc90-CD8BBZ T cells (Figure 4C). On day 12 after CAR T cell infusion, when all mice in both groups were still alive, the tumor burdens of mice that received Luc90-CD828Z CAR T cells were statistically lower than the tumor burdens of mice that received Luc90-CD8BBZ CAR T cells (Figure 4D). Survival of mice with disseminated MM.1S burdens that received Luc90-CD828Z CAR T cells was significantly higher than survival of mice that received Luc90-CD8BBZ CAR T cells (Figure 4E).
We assessed CAR T cells infiltrating MM.1S tumors 7 days after infusion of CAR T cells intravenously. To avoid the issues of CAR downregulation on CAR T cells, which can be a problem with flow cytometry detection of tumor-infiltrating CAR T cells, we performed quantitative PCR (qPCR) to detect CAR+ cells. We found a statistically higher number of tumor-infiltrating CAR+ cells in mice receiving Luc90-CD828Z CAR T cells versus mice receiving Luc90-CD8BBZ CAR T cells. (Figure 4F). We recognize the limitations of only assessing tumor-infiltrating CAR+ cells at one time point, but rapid elimination of tumors in mice treated with Luc90-CD828Z T cells precluded comparison at later time points.
Anti-tumor Activity of Anti-CD19 CARs with CD28 or 4-1BB Domains
To assess whether superior anti-tumor efficacy of T cells expressing CARs with CD28 versus 4-1BB occurred with CARs targeting a different antigen, we utilized anti-CD19 CARs. We compared the CD28-containing anti-CD19 CAR Hu19-CD828Z to the 4-1BB-containing CAR Hu19-CD8BBZ. Hu19-CD8BBZ was identical to Hu19-CD828Z except for the difference in costimulatory domains. We used a model in which solid tumors of CD19+ NALM6 cells were established in NSG mice. In agreement with anti-SLAMF7 CAR results, we found that tumor treatment with Hu19-CD828Z CAR T cells was superior to treatment with Hu19-CD8BBZ CAR T cells (Figure S6). Taken together, the anti-SLAMF7 and anti-CD19 data showed that CAR T cells with CD28 domains were superior to CAR T cells with 4-1BB domains at eliminating solid tumors in the NSG mouse models used. Hu19-CD828Z T cells and Hu19-CD8BBZ T cells were specifically activated by CD19+ target cells (Table S3). As with anti-SLAMF7 CARs (Figure S3), nonspecific IFN-γ release was higher with 4-1BB-containing anti-CD19 CARs than with CD28-containing anti-CD19 CARs (Table S3). The CD28 and 4-1BB sequences in the CARs used in this work are shown in Figure S7.
T Cells Expressing an Anti-SLAMF7 CAR and a Suicide Gene Can Be Eliminated In Vitro
We designed two constructs encoding the Luc90-CD828Z CAR and the IC9 suicide gene. Diagrams of these constructs, which were designated Luc90-CD828Z-IC9 and IC9-Luc90-CD828Z are shown in Figure 5A. The Luc90-CD828Z-IC9 and the IC9-Luc90-CD828Z constructs differ only in the order of the CAR and suicide genes. The CAR and the IC9 genes were separated by a Thoseaasigna virus (T)2A ribosomal skip sequence The CARs were encoded by MSGV1. Luc90-CD828Z was expressed on T cells that were transduced with either MSGV1-Luc90-CD828Z-IC9 or MSGV1-IC9-Luc90-CD828Z (Figure 5B). Next, we evaluated the efficiency of CAR T cell elimination by adding AP1903 (rimiducid) to the cultures of T cells expressing Luc90-CD828Z-IC9 or IC9-Luc90-CD828Z (Figures 5C and 5D). Concentrations of AP1903 used in the assay were equal to concentrations of AP1903 achieved in clinical trials.26,27 We found that T cells transduced with either Luc90-CD828Z-IC9 or IC9-Luc90-CD828Z could be rapidly eliminated when AP1903 was added to the T cell cultures. Elimination of CAR+ T cells after AP1903 treatment was statistically superior with T cells expressing IC9-Luc90-CD828Z compared with T cells expressing Luc90-CD828Z-IC9 (Figure 5D). Almost all residual IC9-Luc90-CD828Z CAR+ T cells were apoptotic 6 h after AP1903 treatment by Annexin V staining (Figure 5E).
Figure 5.

Constructs Containing an Anti-SLAMF7 CAR and the IC9 Suicide Switch
(A) Schematics of the Luc90-CD828Z-IC9 and IC9-Luc90-CD828Z constructs that both encode the Luc90-CD828Z CAR and the IC9 suicide switch. IC9 was made up of a modified FKBP12 domain followed by a modified caspase-9 sequence. In each construct, CAR sequences and IC9 sequences were separated by T2A sequences. Luc90-CD828Z-IC9 and IC9-Luc90-CD828Z only differ in the order of the CAR and IC9. Both CARs were encoded by the MSGV1 gamma-retroviral vector. SS, signal sequence. (B) Representative examples of CAR expression on CD3+ T cells transduced with MSGV1-Luc90-CD828Z-IC9 or MSGV1-IC9-Luc90-CD828Z are shown. Plots are gated on live CD3+ lymphocytes. (C) 5 days after transduction, T cells transduced with either MSGV1-Luc90-CD828Z-IC9 or MSGV1-IC9-Luc90-CD828Z were exposed to the indicated concentrations of AP1903 or vehicle (DMSO) for 6 h. The plots shown are gated on CAR+, CD3+ live lymphocytes and are representative of 5 independent experiments with lymphocytes from different donors. (D) Absolute numbers of CAR+CD3+ live lymphocytes were quantified after treatment with 10 ng/mL of AP1903 or vehicle (DMSO) for 6 h. AP1903 treatment eliminated significantly more T cells expressing IC9-Luc90-CD828Z than T cells expressing Luc90-CD828Z-IC9 (p = 0.013; two-tailed, paired t test). Each bar represents mean + SEM; n = 5 different donors for all groups. (E) The plots show that most residual IC9-Luc90-CD828Z-expressing T cells from the culture treated with 10 ng/mL of AP1903 shown in (C) were Annexin V+, which indicates that the cells were apoptotic. Plots are gated on CAR+CD3+ lymphocytes. This is one of 5 experiments with similar results. (F) 6 days after transduction, T cells transduced with the indicated constructs were cultured with autologous PBMC at a 1:1 ratio for 24 h. One of the cultures containing IC9-Luc90-CD828Z CAR T cells was treated with 10 ng/mL AP1903. Plots show percent live CD3-negative, CD56+ NK cells in the upper left. (G) The graph shows percentages of NK cells from assays conducted as in (F). Results show eradication of NK cells in the presence of IC9-Luc90-CD828Z CAR T cells and prevention of NK cell eradication by eliminating IC9-Luc90-CD828Z CAR T cells with AP1903. N = 4 different donors for all groups. Bars represent mean + SEM; statistical testing was by paired, two-tailed t tests; statistical significance was p < 0.05.
Elimination of Anti-SLAMF7 CAR T Cells by AP1903 Prevents NK Cell Depletion by Anti-SLAMF7 CAR T Cells
To demonstrate elimination of NK cells by anti-SLAMF7 CAR T cells, we cocultured T cells expressing either the anti-CD19 CAR Hu19-CD828Z or IC9-Luc90-CD828Z or untransduced control T cells with autologous PBMC. In one coculture containing IC9-Luc90-CD828Z and PBMC, AP1903 was added to eliminate the CAR T cells. We then assessed NK cells by flow cytometry. NK cells were defined as CD56+CD3-negative cells (Figure 5F). NK cells were depleted from cultures containing PBMCs plus IC9-Luc90-CD828Z without AP1903 (Figure 5G). Addition of AP1903 to cultures containing IC9-Luc90-CD828Z CAR T cells prevented elimination of NK cells from cultures presumably by killing most CAR T cells (Figure 5G).
Function of T Cells Expressing IC9-Luc90-CD828Z or Luc90-CD828Z-IC9
For both CD4+ and CD8+ T cells, cell-surface CAR expression of IC9-Luc90-CD828Z and Luc90-CD828Z-IC9 was not different on day 7 of culture; however, cell-surface CAR expression of IC9-Luc90-CD828Z-transduced T cells was lower than cell-surface expression of Luc90-CD828Z-IC9-transduced T cells on day 14 of culture (Figures 6A and 6B). CAR expression on the T cell surface was lower for T cells transduced with either MSGV1-IC9-Luc90-CD828Z or MSGV1-Luc90-CD828Z-IC9 when compared with T cells transduced with MSGV1-Luc90-CD828Z that encodes the CAR without a suicide gene (Figure S8). Both CD4+ (Figure 6C) and CD8+ (Figure 6D) IC9-Luc90-CD828Z and Luc90-CD828Z-IC9 CAR T cells degranulated in a SLAMF7-specific manner; levels of degranulation were higher for CD8+ T cells than CD4+ T cells. T cells expressing either IC9-Luc90-CD828Z or Luc90-CD828Z-IC9 proliferated and accumulated during a 4-day culture with SLAMF7+ target cells (Figure 6E). There was no statistically significant difference in the degree of SLAMF7-specific proliferation between IC9-Luc90-CD828Z and Luc90-CD828Z-IC9 CAR T cells in (Figure 6F).
Figure 6.

T Cells Expressing IC9-Luc90-CD828Z Exhibit SLAMF7-Specific Reactivity In Vitro
T cells were transduced with MSGV1-IC9-Luc90-CD828Z or MSGV1-Luc90-CD828Z-IC9. On day 7 (D7) and day 14 (D14) of culture, flow cytometry was performed to detect (A) CAR+CD3+CD4+ lymphocytes or (B) CAR+CD3+CD8+ lymphocytes. Mean + SEM percentages of T cells expressing IC9-Luc90-CD828Z or Luc90-CD828Z-IC9 are shown; n = 8 for day 7; n = 6 for day 14. For CD4+ and CD8+ T cells, there was no significant difference in the percentages of T cells expressing IC9-Luc90-CD828Z versus Luc90-CD828Z-IC9 at day 7, but there was a higher level of expression for Luc90-CD828Z-IC9 versus IC9-Luc90-CD828Z at day 14. (C and D) T cells expressing IC9-Luc90-CD828Z or Luc90-CD828Z-IC9 or left untransduced were cultured with either SLAMF7+ MM.1S cells or SLAMF7-negative NGFR-K562 along with antibodies against CD107a for 4 h. Mean + SEM of %MM.1S-specific CD107a+ events are shown. %MM.1S-specific CD107a+ events were calculated as %CD107a+ events with MM.1S stimulation minus %CD107a+ events with NGFR-K562 stimulation. Data are normalized for CAR expression. For both (C) CD4+ and (D) CD8+ T cells, there was no statistical difference in CD107a upregulation between IC9-Luc90-CD828Z and Luc90-CD828Z-IC9; n = 4 different donors for all groups. (A–D) Statistical testing was by paired, two-tailed t tests; statistical significance was p < 0.05. (E) T cells were labeled with CFSE and cultured with either SLAMF7-K562 cells or NGFR-K562 cells for 4 days. Changes in numbers of CAR+ T cells during the 4-day culture are shown for T cells expressing each CAR from 3 different donors. CAR+ T cells increased from day 0 to day 4 (p = 0.017 for IC9-Luc90-CD828Z; p = 0.035 for Luc90-CD828Z-IC9 by two-tailed, paired t test). (F) Proliferation was assessed by CFSE dilution. The degree of SLAMF7-specific proliferation is presented as the ratio of MFI of CFSE-labeled T cells cultured with SLAMF7-K562 cells (SLAMF7) divided by the MFI of CFSE-labeled T cells cultured with NGFR-K562 cells (NGFR). A SLAMF7/NGFR ratio <1 indicates SLAMF7-specific proliferation. There was no statistical difference in CFSE dilution between IC9-Luc90-CD828Z and Luc90-CD828Z-IC9. (G) T cells expressing Hu19-CD828Z, Luc90-CD828Z, IC9-Luc90-CD828Z, or Luc90-CD828Z-IC9 or untransduced control T cells were cultured overnight with target cells. SLAMF7-K562 and MM.1S were SLAMF7+. NGFR-K562 and CCRF-CEM were SLAMF7-negative. Supernatant was assayed by ELISA for IFN-γ. Bars show mean + SEM. The number of experiments with cells from different donors was n = 7 for untransduced, IC9-Luc90-CD828Z, and Luc90-CD828Z; n = 4 for Luc90-CD828Z-IC9; and n = 3 for Hu19-CD828Z. (H and I) Bars show mean + SEM of 3 experiments with cells from 3 donors. After overnight culture, ELISAs were performed on culture supernatants for (H) TNF-α or (I) IL-2; data were normalized for CAR expression. (J) T cells were transduced with the indicated CARs, and a 4-h cytotoxicity assay was performed with SLAMF7+ MM.1S target cells. Mean ± SEM of duplicate wells is graphed. The experiment was conducted 2 times with cells from different donors with similar results. (K) T cells were transduced with IC9-Luc90-CD828Z, SP6-CD828Z, or left untransduced. The T cells were cocultured with primary bone marrow MM cells, SLAMF7-K562, or NGFR-K562 overnight, and an IFN-γ ELISA was performed. Bars represent average IFN-γ in duplicate wells. Two experiments that used cells from 2 different donors were conducted with similar results.
Cytokine Release and Cytotoxicity of T Cells Expressing IC9-Luc90-CD828Z
IC9-Luc90-CD828Z and Luc90-CD828Z-IC9 CAR T cells released large amounts of IFN-γ in the presence of SLAMF7+ target cells and very low levels of IFN-γ in the presence of SLAMF7-negative target cells (Figure 6G). T cells expressing IC9-Luc90-CD828Z released TNF-α (Figure 6H) and IL-2 (Figure 6I) in a SLAMF7-specific manner. T cells expressing IC9-Luc90-CD828Z specifically killed MM.1S cells (Figure 6J). In addition, IC9-Luc90-CD828Z cells did not recognize a variety of SLAMF7-negative target cells when assessed by IFN-γ ELISA (Table S4).
T cells expressing Luc90-CD828Z-IC9 and IC9-Luc90-CD828Z exhibited similar in vitro function. However, IC9-Luc90-CD828Z T cells can be eliminated slightly more effectively than Luc90-CD828Z-IC9 T cells after AP1903 treatment (Figure 5D), so IC9-Luc90-CD828Z was selected for further development
IC9-Luc90-CD828Z T Cells Specifically Recognize Primary MM Cells
T cells expressing IC9-Luc90-CD828Z released IFN-γ specifically in response to primary bone marrow cells from a patient with extensive bone marrow MM that was SLAMF7+ (Figure 6K). Similarly, IC9-Luc90-CD828Z T cells degranulated specifically in response to SLAMF7+ primary MM bone marrow cells (Figure S9).
Anti-tumor Activity of IC9-Luc90-CD828Z
T cells expressing IC9-Luc90-CD828Z have dose-dependent activity against established tumors of the MM.1S MM cell line. A single intravenous infusion of 2 × 106 or more of IC9-Luc90-CD828Z-expressing T cells/mouse eradicated MM.1S solid tumors (Figures 7A and 7B). A single intravenous dose of 2 × 106 CAR T cells/mouse of T cells expressing IC9-Luc90-CD828Z eradicated solid MM.1S tumors, whereas T cells expressing the anti-CD19 CAR Hu19-CD828Z did not have an anti-tumor effect (Figures 7C and 7D). IC9-Lu90-CD828Z T cells had activity against disseminated malignancy burdens of MM.1S cells in NSG mice. MM.1S cells reached undetectable levels within 9 days of infusion of 4 × 106 IC9-Luc90-CD828Z or Luc90-CD828Z CAR T cells/mouse (Figures 7E and 7F).
Figure 7.

Anti-SLAMF7 CAR T Cells with IC9 Eradicated Solid and Disseminated Tumor Burdens
(A) 7 days after injection of MM.1S cells into NSG mice when palpable tumors were present, the indicated doses of CAR+ T cells were infused intravenously into the mice, and tumors were measured every 3 days in a blinded manner. The graph shows mean tumor volume ± SEM for each time point. There were five mice in each group. (B) Kaplan-Meier survival plot of the same mice as in (A). (C) MM.1S solid tumors were established in NSG mice. 7 days after MM.1S cell injection, mice were either left untreated or received infusions of 2 × 106 T cells expressing Luc90-CD828Z, IC9-Luc90-CD828Z, or the negative-control CAR Hu19-CD828Z. The graph shows mean tumor volume ± SEM for each time point. There were five mice in each group. The graph is representative of one of two independent experiments that used T cells from different donors. (D) Kaplan-Meier survival plot of the same mice as in (C). p = 0.0031 for the comparisons of either IC9-Luc90-CD828Z or Luc90-CD828Z versus Hu19-CD828Z by log rank test. (E) Disseminated tumor burdens were established in NSG mice by intravenous injection of 8 × 106 MM.1S-luciferase cells. The mice were left untreated or received infusions of 4 × 106 T cells expressing Luc90-CD828Z, IC9-Luc90-CD828Z, or the negative-control CAR Hu19-828Z. Tumor burden was evaluated by BLI every 3 days. The figure shows BLI intensity of mice for each time point. The figure is representative of one experiment of two experiments with T cells from different donors. (F) Kaplan-Meier survival plots of the same mice as in (E). p = 0.0027 for the comparison of either IC9-Luc90-CD828Z or Luc90-CD828Z versus Hu19-CD828Z by log rank test.
In Vivo Assessment of Suicide Switch Function
We established MM.1S solid tumors in NSG mice and injected IC9-Luc90-CD828Z T cells intravenously with or without administration of AP1903 that activates the IC9 suicide switch. Mice were treated with AP1903 or vehicle control 3 days and 6 days after the CAR T cell infusion. Mice that received IC9-Luc90-CD828Z T cells plus AP1903 had progressive tumors; in contrast, tumors were eliminated from mice that received IC9-Luc90-CD828Z T cells plus the vehicle control (Figures 8A and 8B).
Figure 8.

AP1903 Treatment Eliminated IC9-Luc90-CD828Z T Cells
(A) MM.1S solid tumors were established in NSG mice, and then mice received 2 × 106 CAR T cells intravenously. 3 days and 6 days after CAR T cell infusions, mice were injected intraperitoneally with 5 mg/kg of AP1903 or vehicle. Tumors were measured every 3 days in a blinded manner. The mean tumor volumes ± SEM of each time point are shown. There were five mice in each group. (B) Kaplan-Meier survival plot of the same mice as shown in (A). There was a statistical difference in survival between the IC9-Luc90-CD828Z T cells plus AP1903 and IC9-Luc90-CD828Z plus vehicle; p = 0.0023 by log rank test. (C) Disseminated MM.1S cells were established in NSG mice, and the mice then received 4 × 106 T cells transduced with IC9-Luc90-CD828Z. 3 and 6 days after infusion of CAR T cells, mice were injected intraperitoneally with 5 mg/kg of AP1903 or vehicle. On day 9, mice were sacrificed. Splenocytes were isolated and stained for CAR expression on human T cells. The plots show human CD4+ (top) and CD8+ (bottom) T cells. Plots were first gated on live human CD3+ lymphocytes. The plots are from cells of one mouse and are representative results from two independent experiments performed with cells from different donors. (D) CD4+ and (E) CD8+CAR+ T cells present in spleens of mice treated with AP1903 or vehicle control as in (C) were quantified. Compared to the vehicle-control treatment group, the AP1903 treatment group had significantly lower numbers of splenic IC9-Luc90-CD828Z T cells. Each data point indicates the splenic CAR+ T cell number of an individual mouse. Horizontal bars indicate medians; n = 9 for vehicle-treated group, and n = 10 for AP1903-treated group. Statistical testing was by unpaired, two-tailed t tests. p < 0.05 was statistically significant. (F) T cells transduced with Hu19-CD828Z, Luc90-CD828Z, or IC9-Luc90-CD828Z were stained for CAR T cell expression on day 7 of culture (5 days after transduction). Plots are gated on CD3+CAR+ cells. These plots show a higher CD4-to-CD8 ratio with T cells expressing anti-SLAMF7 CARs versus T cells expressing the Hu19-CD828Z anti-CD19 CAR. Plots are representative of 9 experiments. (G) The graph shows CD4-to-CD8 ratios of T cells expressing different CARs. Experiments were performed as in (F). Each data point is the CD4-to-CD8 ratio in one culture of T cells expressing the indicated CARs. Medians are indicated on the graph by black horizontal bars; n = 9 for all groups. Statistical testing was by 1-way ANOVA, followed by Tukey test for multiple comparisons. p values for comparisons of pairs of CAR T cell cultures are shown. Statistical significance was defined as p < 0.05.
We examined AP1903-induced CAR T cell elimination in vivo. Disseminated MM.1S tumor burdens were established in NSG mice. The mice received IC9-Luc90-CD828Z T cells intravenously and were treated with two doses of AP1903 or a vehicle control. Mice were sacrificed 9 days after CAR T cell infusion. We found significantly higher numbers of CD4+ and CD8+CAR+ T cells among splenocytes of mice that received vehicle control compared with mice that received AP1903 (Figures 8C–8E).
CD4-to-CD8 Ratio of Anti-SLAMF7 CAR T Cells in Culture
The CD4:CD8 ratio of in vitro cultures of human anti-SLAMF7 CAR T cells is shown in Figures 8F and 8G. Compared to T cell cultures that were transduced with the Hu19-CD828Z anti-CD19 CAR, there was a statistically higher CD4:CD8 T cell ratio in cultures of T cells expressing anti-SLAMF7 CARs (Figure 8G). Despite possible elimination of some CD8+ T cells from cultures transduced with anti-SLAMF7 CARs, T cells in the cultures accumulated steadily (Figure S10).
Discussion
Our work focused on two important aspects of anti-SLAMF7 CAR development: first, selection of either CD28 or 4-1BB as the costimulatory molecule for the CAR; second, development of a construct encoding both an effective anti-SLAMF7 CAR and a suicide switch capable of eliminating CAR T cells on demand.
Previous articles have reported anti-SLAMF7 CARs with CD28 costimulatory domains,13,20, 21, 22 and two articles have reported 4-1BB-containing CAR constructs that targeted both SLAMF7 and BCMA.23,38 None of these prior reports compared anti-SLAMF7 CARs with CD28 versus 4-1BB costimulatory domains. We compared the CD28-containing CAR Luc90-CD828Z to Luc90-CD8BBZ, a CAR that was identical to Luc90-CD828Z except for replacement of CD28 with 4-1BB. Compared with Luc90-CD8BBZ, T cells expressing Luc90-CD828Z cleared solid tumors and disseminated tumor cells more consistently and more quickly (Figures 4A and 4C). We also compared the anti-tumor activity of T cells expressing anti-CD19 CARs with either CD28 or 4-1BB domains (Figure S6), and we found that the CD28-containing CAR had superior anti-tumor activity when compared to the 4-1BB-containing CAR against solid tumors. Our findings are in agreement with prior reports of murine experiments that showed superior anti-tumor activity of CD28-containing CARs versus 4-1BB-containing CARs;32, 33, 34 however, some prior comparisons of CD28-containing CARs and 4-1BB-containing CARs showed equivalent anti-tumor activity under some conditions and superiority of 4-1BB under other conditions.35,36 Finally, some reports have shown superior anti-tumor activity of T cells expressing 4-1BB-containing CARs versus T cells expressing CD28-containing CARs.30,31
7 days after CAR T cell infusion, when tumor regressions were starting, we found higher numbers of tumor-infiltrating T cells when the T cells expressed CD28-containing CARs versus 4-1BB-containing CARs (Figure 4F). The higher numbers of tumor-infiltrating T cells expressing the CD28-containing CAR could explain the superior clearance of tumors with Luc90-CD828Z CAR T cells versus Luc90-CD8BBZ CAR T cells. One mechanism that could be important in the greater anti-tumor activity and greater number of T cells at tumor sites could be the increased cytokine release by T cells expressing CD28-containing CARs versus 4-1BB-containing CARs (Figures 3K–3M). Cytokine production by CAR-expressing human T cells might be especially important in models, such as those used in this work, in which human T cells are infused into mice that lack human cytokines.
As a way to explain the greater numbers of tumor-infiltrating T cells when T cells expressed a CD28-containing versus a 4-1BB-containing CAR, we hypothesized that Luc90-CD828Z CAR T cells might be less prone than Luc90-CD8BBZ T cells to undergo AICD. We performed AICD assays in vitro. Although there was no statistically significant difference in AICD between Luc90-CD828Z and Luc90-CD8BBZ CAR T cells for either CD4+ or CD8+ T cells (Figure S2), there was a trend in CD8+ T cells toward more AICD in T cells expressing Luc90-CD8BBZ versus T cells expressing Luc90-CD828Z. A prior study has reported higher levels of apoptosis among T cells expressing 4-1BB-containing CARs versus CD28-containing CARs, especially when, as in the current report, a gamma-retroviral vector was used.39
We found a higher percentage of T cells with a central memory phenotype among T cells expressing a 4-1BB-containing CAR versus T cells expressing a CD28-containing CAR (Figure S1). The reason for this difference is not clear and was not assessed by our experiments. It is possible that CAR signaling caused by SLAMF7+ leukocytes in the cultures contributed to this. Other investigators have previously reported a higher percentage of central memory T cells among T cells expressing a 4-1BB-containing CAR versus a CD28-containing CAR, and these investigators also associated differences in memory phenotype with differences in cellular metabolism.40
When compared to cultures of anti-CD19 CAR T cells, the CD4:CD8 ratio was higher for cultures of anti-SLAMF7 CAR T cells (Figures 8F and 8G). This increase in CD4:CD8 ratio in cultures of T cells transduced with anti-SLAMF7 CARs suggests that SLAMF7+CD8+ T cells could have been eliminated by fratricide conducted by the anti-SLAMF7 CAR T cells against SLAMF7+CD8+ T cells. Note that SLAMF7 expression is substantially higher on CD8+ versus CD4+ T cells (Figure 1), so fratricide could preferentially eliminate CD8+ T cells.
Changes in any part of a CAR sequence might affect CAR T cell function. For example, hinge and transmembrane domains have been shown to strongly affect CAR T cell function.41,42 Anti-CD19 CARs commonly used clinically have had either CD28 or 4-1BB costimulatory domains, and 4-1BB has been thought to be an important factor promoting persistence of 4-1BB-containing CAR T cells in clinical trials;43 however, persistence of T cells expressing 4-1BB-containing CARs might not be completely attributable to 4-1BB because the CD28-containing CARs and 4-1BB-containing CARs used in clinical trials contained different hinge and transmembrane domains and differed in other parameters.43, 44, 45, 46 It is possible that increased T cell persistence attributed to 4-1BB was due, in part, to the difference in hinge and transmembrane domains.
We selected the murine Luc90 scFv rather than the humanized Luc63 scFv for three reasons. First, our results showed slightly superior recognition of SLAMF7+ target cells for Luc90-CD828Z versus Luc63-CD828Z when assessed by IFN-γ release (Figure 2H). Second, the humanized Luc63 antibody that the Luc63 scFv is derived from is the commonly used commercial monoclonal antibody elotuzumab.47 We reasoned that even though the Luc63 scFv is humanized, there is still a chance of a recipient immune response against the Luc63 scFv, particularly in patients with many prior exposures to elotuzumab. Finally, elotuzumab has modest anti-myeloma activity when used as a single agent,48 which also encouraged us to use Luc90.
Anti-SLAMF7 CAR T cells can eliminate NK cells (Figure 5F). NK cell deficiencies are associated with life-threatening viral infections, especially infections caused by members of the herpes virus family, such as cytomegalovirus.24 The risk of elimination of normal NK cells by anti-SLAMF7 CAR T cells makes inclusion of a suicide gene with an anti-SLAMF7 CAR prudent. T cells expressing IC9-Luc90-CD828Z were able to eradicate normal human NK cells from cultures, and we were able to abrogate NK cell depletion by eliminating CAR T cells with AP1903 in vitro (Figure 5F). During potential clinical use, IC9-Luc90-CD828Z T cells could be eliminated from patients experiencing severe adverse events by administration of AP1903 (rimiducid).
Previous studies reported anti-SLAMF7 CARs plus depletion systems that utilize the monoclonal antibody cetuximab.13,21 One of the main mechanisms for monoclonal antibodies to eliminate target cells is antibody-dependent cellular cytotoxicity (ADCC); NK cells are one of the main cell types participating in ADCC.49 Because anti-SLAMF7 CAR T cells and the lymphocyte-depleting chemotherapy that is normally given before clinical CAR T cell infusions will likely eliminate most NK cells in patients, use of a suicide-gene system, such as IC9, which does not depend on ADCC, is preferable.
Despite expression of SLAMF7 on some CAR-transduced CD8+ T cells, anti-SLAMF7 CAR T cells accumulated in cultures and sufficient CAR T cells for use in a clinical trial can be obtained. The preclinical results with IC9-Luc90-CD828Z have led us to initiate testing of T cells expressing this construct in a clinical trial for MM.
Materials and Methods
Patient Samples and Mice
All cells used were human cells. Use of samples from patients enrolled in National Cancer Institute (NCI) clinical trials was approved by the NCI Institutional Review Board. Informed consent was obtained from all patients. Mouse studies were carried out on protocols approved by the NCI Animal Care and Use Committee.
Cell Lines
Cell lines are described in Supplemental Materials and Methods.
PCR to Quantify SLAMF7 Expression
Detection of SLAMF7 RNA in tissues by qPCR is described in Supplemental Materials and Methods.
Tissue Staining of SLAMF7
A paraffin-fixed normal tissue microarray (number MN0661; Pantomics, Fairfield, CA) was stained with anti-SLAMF7 clone OTI3B3 (number LS-C340266; LSBio, Seattle, WA).
Design and Construction of Plasmids Encoding CARs
The sequences of Luc90-CD828Z and Luc63-CD828Z followed this pattern from N terminus to C terminus: CD8α signal sequence, anti-SLAMF7 scFv, human CD8α hinge and transmembrane domains, the cytoplasmic portion of human CD28, and the cytoplasmic portion of human CD3ζ. One CAR designated Luc90-CD828Z had a scFv derived from the murine Luc90 antibody. The Luc90 scFv had a sequence from the N terminus to C terminus of the light-chain variable domain, linker, and heavy-chain variable domain. Luc63-CD828Z had an identical sequence as Luc90-CD828Z, except it had a scFv derived from the humanized Luc63 antibody. Sequences of Luc90 and Luc63 variable regions were obtained from a patent.50 Luc90-CD8BBZ was identical to Luc90-CD828Z, except a 4-1BB sequence replaced the CD28 sequence of Luc90-CD828Z.
Combination Luc90-CD828Z plus suicide-gene constructs were designed based on prior work.26,51,52 The IC9 suicide gene used in this work includes a modified FKBP12 domain linked to a modified caspase-9 domain. The FKBP12 sequence in IC9 was based on the natural FKBP12 sequence (GenBank: AH002818). The caspase-9 sequence used in IC9 was based on the natural caspase-9 sequence (GenBank: NM_001229). The linker sequence connecting the FKBP12 and caspase-9 components was serine-glycine-glycine-glycine-serine. One CAR plus suicide-gene construct has the IC9 suicide gene 5′ to the CAR and was designated IC9-Lu90-CD828Z. The other CAR plus suicide-gene construct has IC9 3′ to the CAR and was designated Luc90-CD828Z-IC9. For both Luc90-CD828Z-IC9 and IC9-Luc90-CD828Z, the CAR and IC9 sequences were separated by a T2A sequence.53 We also utilized 3 previously reported CARs: the anti-CD19 CARs Hu19-828Z and Hu19-CD8BBZ41,42 and the SP6-CD828Z negative-control CAR.7 All CAR DNA sequences were synthesized by GeneArt/Thermo Fisher Scientific (Regensburg, Germany) and ligated into the MSGV1 gamma-retroviral vector backbone37 by standard methods.
T Cell Culture and Gamma-Retroviral Transductions
Gamma-retroviral vector supernatant was prepared as previously described.54 In brief, cultures were initiated with whole PBMC by stimulating with the anti-CD3 antibody OKT3, and transduction was performed 2 days after initiation of T cell cultures, as previously described.36 AIM V media refers to AIM V T cell culture media (Thermo Fisher Scientific) plus 5% human serum (Valley Biomedical, Winchester, VA) plus penicillin and streptomycin.
CAR Detection and T Cell Phenotyping
SLAMF7-fragment crystallizable (Fc) protein labeled with phycoerythrin (PE; Creative Biomart, Shirley, NY) or biotin-labeled protein L (GenScript, Piscataway, NJ) was used as a CAR-detection reagent in a manner similar to previous work.36,41 The percentage of CAR+ T cells was calculated as the percentage of transduced T cells stained with a CAR-detection reagent minus the percentage of untransduced T cells from the same donor stained with the CAR-detection reagent. Antibodies used are in Supplemental Methods. Dead cells were excluded with 7-amino-actinomycin D (7-AAD; BD Biosciences, San Jose, CA) in all experiments. Flow cytometry analysis was performed with FlowJo (Tree Star, Ashland, OR).
In Vitro T Cell Function Assays
CD107a degranulation assays, cytotoxicity assays, ELISA, and proliferation assays were performed as previously described.36 For normalization, ELISA (pg/mL) and CD107a (% CD107a+ events) results were divided by the fraction of CAR+ T cells for each T cell population used.
AP1903 T Cell Death Analysis
T cell cultures that were either untransduced or CAR transduced were suspended at 1 × 106 T cells/mL in AIM V media + 300 IU of IL-2. The cells were distributed to wells and treated with 1, 10, or 100 ng/mL of AP1903 (rimiducid; MedChemExpress, Monmouth Junction, NJ) or dimethyl sulfoxide (Sigma, St. Louis, MO) as a vehicle control. 6 h after these cultures were set up, the cells were counted and stained with protein L as described above. Next, the cells were incubated with Annexin V (BD Biosciences, San Diego, CA) and 7-AAD, and flow cytometry was performed immediately.
NK Cell Depletion Assay
Untransduced T cells or T cells expressing either IC9-Luc90-CD828Z or Hu19-CD828Z were cultured with autologous PBMCs in a 1:1 ratio. The cocultures were treated with either 10 ng/mL of AP1903 or vehicle control. 24 h after the cultures were set up, the cells were counted, washed, and assessed for NK cells by flow cytometry.
Solid Tumor and Disseminated Tumor Cell Mouse Experiments
Mouse tumor experiments are described in figure legends and were conducted with MM.1S cells as previously described.36 Tumor experiments with NALM6 cells were carried out as previously described.41
Real-Time qPCR for Quantifying CAR+ Cells in Tumors
Similar to previous work, we made serial 1:5 dilutions of DNA from the infused T cells of each experimental group into untransduced T cell DNA, and we made standard curves by performing qPCR on this DNA.42,55,56 We used the standard curves to determine the percentage of CAR+ cells in tumors as described in Supplemental Materials and Methods. The absolute number of CAR+ cells in each tumor was calculated by multiplying the percentage of CAR+ cells by the total number of cells in the tumor.
Solid Myeloma Tumor with AP1903 Treatment
Solid tumors of MM.1S cells were established in NSG mice. The mice were infused with 2 × 106 CAR T cells. 3 and 6 days after CAR T cell infusion, mice received intraperitoneal injections of 5 mg/kg AP1903 or vehicle. The dose of AP1903 was determined from a previous study.52
T Cell Persistence in Mice after AP1903 Treatment
Mice were injected with 8 × 106 MM.1S-luciferase cells intravenously. After 13 days, 4 × 106 CD3+CAR+ T cells/mouse were infused intravenously. Mice received an intraperitoneal injection of either 5 mg/kg of AP1903 or vehicle on days 3 and 6 after T cell infusion. Mice were sacrificed 9 days after T cell infusion. Splenocytes were counted, and flow cytometry was performed for SLAMF7-Fc-PE for CAR detection, anti-CD3, anti-CD4, and anti-CD8. To calculate the number of CAR T cells/spleen, the total number of splenocytes per mouse was multiplied by the product of the %CD3+, %CD4+, or %CD8+ and the %CAR+.
AICD Assay
T cells expressing Luc90-CD828Z or Luc90-CD8BBZ were flow sorted using a BD FACSAria. T cells were then cocultured with either MM.1S or NGFR-K562 in a 1.5:1 target-to-effector ratio for 18 h. T cells were then stained for CAR and Annexin V as described above.
Statistical Analyses
Statistical comparisons were performed as described in the figure legends with GraphPad Prism version 7.
Author Contributions
C.A. conducted experiments, planned experiments, analyzed data, and wrote the manuscript. M.A.P., N.L., C.G., D.V., S.C., and S.M.H. conducted experiments, analyzed data, and edited the manuscript. S.A.F. designed CAR constructs and edited the manuscript. J.N.K. designed CAR constructs, conducted experiments, planned experiments, analyzed data, and wrote the manuscript.
Conflicts of Interest
This work was supported by NIH, NCI, and Center for Cancer Research intramural funding. J.N.K. and S.A.F. are inventors on a patent application for the anti-SLAMF7 plus suicide switch constructs presented in this work. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Acknowledgments
We thank Bill Telford in the Experimental Transplantation and Immunotherapy Branch, NCI Flow Cytometry Facility, and Arnold Mixon and Shawn Farid in the Surgery Branch, NCI Flow Cytometry Facility. This work was supported by intramural funding of the Center for Cancer Research, National Cancer Institute, NIH.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.10.008.
Supplemental Information
References
- 1.Lonial S., Mitsiades C.S., Richardson P.G. Treatment options for relapsed and refractory multiple myeloma. Clin. Cancer Res. 2011;17:1264–1277. doi: 10.1158/1078-0432.CCR-10-1805. [DOI] [PubMed] [Google Scholar]
- 2.Branagan A., Lei M., Lou U., Raje N. Current treatment strategies for multiple myeloma. J. Oncol. Pract. 2020;16:5–14. doi: 10.1200/JOP.19.00244. [DOI] [PubMed] [Google Scholar]
- 3.Sadelain M., Brentjens R., Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eshhar Z., Waks T., Gross G., Schindler D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali S.A., Shi V., Maric I., Wang M., Stroncek D.F., Rose J.J., Brudno J.N., Stetler-Stevenson M., Feldman S.A., Hansen B.G. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688–1700. doi: 10.1182/blood-2016-04-711903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carpenter R.O., Evbuomwan M.O., Pittaluga S., Rose J.J., Raffeld M., Yang S., Gress R.E., Hakim F.T., Kochenderfer J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013;19:2048–2060. doi: 10.1158/1078-0432.CCR-12-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brudno J.N., Maric I., Hartman S.D., Rose J.J., Wang M., Lam N., Stetler-Stevenson M., Salem D., Yuan C., Pavletic S. T cells genetically modified to express an anti–B-Cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J. Clin. Oncol. 2018;36:2267–2280. doi: 10.1200/JCO.2018.77.8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen A.D., Garfall A.L., Stadtmauer E.A., Melenhorst J.J., Lacey S.F., Lancaster E., Vogl D.T., Weiss B.M., Dengel K., Nelson A. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Invest. 2019;129:2210–2221. doi: 10.1172/JCI126397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mikkilineni L., Kochenderfer J.N. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. 2017;130:2594–2602. doi: 10.1182/blood-2017-06-793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boles K.S., Mathew P.A. Molecular cloning of CS1, a novel human natural killer cell receptor belonging to the CD2 subset of the immunoglobulin superfamily. Immunogenetics. 2001;52:302–307. doi: 10.1007/s002510000274. [DOI] [PubMed] [Google Scholar]
- 12.Calpe S., Wang N., Romero X., Berger S.B., Lanyi A., Engel P., Terhorst C. The SLAM and SAP gene families control innate and adaptive immune responses. Adv. Immunol. 2008;97:177–250. doi: 10.1016/S0065-2776(08)00004-7. [DOI] [PubMed] [Google Scholar]
- 13.Wang X., Walter M., Urak R., Weng L., Huynh C., Lim L., Wong C.W., Chang W.C., Thomas S.H., Sanchez J.F. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2018;24:106–119. doi: 10.1158/1078-0432.CCR-17-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tai Y.T., Dillon M., Song W., Leiba M., Li X.F., Burger P., Lee A.I., Podar K., Hideshima T., Rice A.G. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112:1329–1337. doi: 10.1182/blood-2007-08-107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsi E.D., Steinle R., Balasa B., Szmania S., Draksharapu A., Shum B.P., Huseni M., Powers D., Nanisetti A., Zhang Y. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008;14:2775–2784. doi: 10.1158/1078-0432.CCR-07-4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai Y.T., Soydan E., Song W., Fulciniti M., Kim K., Hong F., Li X.F., Burger P., Rumizen M.J., Nahar S. CS1 promotes multiple myeloma cell adhesion, clonogenic growth, and tumorigenicity via c-maf-mediated interactions with bone marrow stromal cells. Blood. 2009;113:4309–4318. doi: 10.1182/blood-2008-10-183772. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Kikuchi J., Hori M., Iha H., Toyama-Sorimachi N., Hagiwara S., Kuroda Y., Koyama D., Izumi T., Yasui H., Suzuki A., Furukawa Y. Soluble SLAMF7 promotes the growth of myeloma cells via homophilic interaction with surface SLAMF7. Leukemia. 2020;34:180–195. doi: 10.1038/s41375-019-0525-6. [DOI] [PubMed] [Google Scholar]
- 18.Malaer J.D., Mathew P.A. CS1 (SLAMF7, CD319) is an effective immunotherapeutic target for multiple myeloma. Am. J. Cancer Res. 2017;7:1637–1641. [PMC free article] [PubMed] [Google Scholar]
- 19.Wu N., Veillette A. SLAM family receptors in normal immunity and immune pathologies. Curr. Opin. Immunol. 2016;38:45–51. doi: 10.1016/j.coi.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Chu J., He S., Deng Y., Zhang J., Peng Y., Hughes T., Yi L., Kwon C.H., Wang Q.E., Devine S.M. Genetic modification of T cells redirected toward CS1 enhances eradication of myeloma cells. Clin. Cancer Res. 2014;20:3989–4000. doi: 10.1158/1078-0432.CCR-13-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gogishvili T., Danhof S., Prommersberger S., Rydzek J., Schreder M., Brede C., Einsele H., Hudecek M. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood. 2017;130:2838–2847. doi: 10.1182/blood-2017-04-778423. [DOI] [PubMed] [Google Scholar]
- 22.Chu J., Deng Y., Benson D.M., He S., Hughes T., Zhang J., Peng Y., Mao H., Yi L., Ghoshal K. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen K.H., Wada M., Pinz K.G., Liu H., Shuai X., Chen X., Yan L.E., Petrov J.C., Salman H., Senzel L. A compound chimeric antigen receptor strategy for targeting multiple myeloma. Leukemia. 2018;32:402–412. doi: 10.1038/leu.2017.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orange J.S. Human natural killer cell deficiencies and susceptibility to infection. Microbes Infect. 2002;4:1545–1558. doi: 10.1016/s1286-4579(02)00038-2. [DOI] [PubMed] [Google Scholar]
- 25.Orange J.S. Natural killer cell deficiency. J. Allergy Clin. Immunol. 2013;132:515–525. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iuliucci J.D., Oliver S.D., Morley S., Ward C., Ward J., Dalgarno D., Clackson T., Berger H.J. Intravenous safety and pharmacokinetics of a novel dimerizer drug, AP1903, in healthy volunteers. J. Clin. Pharmacol. 2001;41:870–879. doi: 10.1177/00912700122010771. [DOI] [PubMed] [Google Scholar]
- 28.Gargett T., Brown M.P. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol. 2014;5:235. doi: 10.3389/fphar.2014.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weinkove R., George P., Dasyam N., McLellan A.D. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin. Transl. Immunology. 2019;8:e1049. doi: 10.1002/cti2.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Priceman S.J., Gerdts E.A., Tilakawardane D., Kennewick K.T., Murad J.P., Park A.K., Jeang B., Yamaguchi Y., Yang X., Urak R. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. OncoImmunology. 2017;7:e1380764. doi: 10.1080/2162402X.2017.1380764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Milone M.C., Fish J.D., Carpenito C., Carroll R.G., Binder G.K., Teachey D., Samanta M., Lakhal M., Gloss B., Danet-Desnoyers G. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carpenito C., Milone M.C., Hassan R., Simonet J.C., Lakhal M., Suhoski M.M., Varela-Rohena A., Haines K.M., Heitjan D.F., Albelda S.M. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drent E., Poels R., Ruiter R., van de Donk N.W.C.J., Zweegman S., Yuan H., de Bruijn J., Sadelain M., Lokhorst H.M., Groen R.W.J. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered t cells. Clin. Cancer Res. 2019;25:4014–4025. doi: 10.1158/1078-0432.CCR-18-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Z., Condomines M., van der Stegen S.J.C., Perna F., Kloss C.C., Gunset G., Plotkin J., Sadelain M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du H., Hirabayashi K., Ahn S., Kren N.P., Montgomery S.A., Wang X., Tiruthani K., Mirlekar B., Michaud D., Greene K. Antitumor Responses in the Absence of Toxicity in Solid Tumors by Targeting B7-H3 via Chimeric Antigen Receptor T Cells. Cancer Cell. 2019;35:221–237.e8. doi: 10.1016/j.ccell.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lam N., Trinklein N.D., Buelow B., Patterson G.H., Ojha N., Kochenderfer J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020;11:283. doi: 10.1038/s41467-019-14119-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hughes M.S., Yu Y.Y., Dudley M.E., Zheng Z., Robbins P.F., Li Y., Wunderlich J., Hawley R.G., Moayeri M., Rosenberg S.A., Morgan R.A. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum. Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zah E., Nam E., Bhuvan V., Tran U., Ji B.Y., Gosliner S.B., Wang X., Brown C.E., Chen Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020;11:2283. doi: 10.1038/s41467-020-16160-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomes-Silva D., Mukherjee M., Srinivasan M., Krenciute G., Dakhova O., Zheng Y., Cabral J.M.S., Rooney C.M., Orange J.S., Brenner M.K., Mamonkin M. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017;21:17–26. doi: 10.1016/j.celrep.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawalekar O.U., O’Connor R.S., Fraietta J.A., Guo L., McGettigan S.E., Posey A.D., Jr., Patel P.R., Guedan S., Scholler J., Keith B. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44:380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 41.Alabanza L., Pegues M., Geldres C., Shi V., Wiltzius J.J.W., Sievers S.A., Yang S., Kochenderfer J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017;25:2452–2465. doi: 10.1016/j.ymthe.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brudno J.N., Lam N., Vanasse D., Shen Y.W., Rose J.J., Rossi J., Xue A., Bot A., Scholler N., Mikkilineni L. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020;26:270–280. doi: 10.1038/s41591-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kochenderfer J.N., Dudley M.E., Feldman S.A., Wilson W.H., Spaner D.E., Maric I., Stetler-Stevenson M., Phan G.Q., Hughes M.S., Sherry R.M. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.A., Lanier B.J. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G., Bartido S., Stefanski J., Taylor C., Olszewska M. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lonial S., Dimopoulos M., Palumbo A., White D., Grosicki S., Spicka I., Walter-Croneck A., Moreau P., Mateos M.V., Magen H., ELOQUENT-2 Investigators Elotuzumab therapy for relapsed or refractory multiple myeloma. N. Engl. J. Med. 2015;373:621–631. doi: 10.1056/NEJMoa1505654. [DOI] [PubMed] [Google Scholar]
- 48.Zonder J.A., Mohrbacher A.F., Singhal S., van Rhee F., Bensinger W.I., Ding H., Fry J., Afar D.E., Singhal A.K. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood. 2012;120:552–559. doi: 10.1182/blood-2011-06-360552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka J., Miller J.S. Recent progress in and challenges in cellular therapy using NK cells for hematological malignancies. Blood Rev. 2020 doi: 10.1016/j.blre.2020.100678. Published online March 20, 2020. [DOI] [PubMed] [Google Scholar]
- 50.Williams M., Yun Tso J., Landolfi N.F., Powers D.B., DuBridge R.B., Law D., Liu G. 2006. Therapeutic Use of Anti-CS1 Antibodies. US patent application publication 2006/0024296 A1. filed November 5, 2004, and published February 2, 2006. [Google Scholar]
- 51.Clackson T., Yang W., Rozamus L.W., Hatada M., Amara J.F., Rollins C.T., Stevenson L.F., Magari S.R., Wood S.A., Courage N.L. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Straathof K.C., Pulè M.A., Yotnda P., Dotti G., Vanin E.F., Brenner M.K., Heslop H.E., Spencer D.M., Rooney C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247–4254. doi: 10.1182/blood-2004-11-4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szymczak A.L., Workman C.J., Wang Y., Vignali K.M., Dilioglou S., Vanin E.F., Vignali D.A. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat. Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 54.Kochenderfer J.N., Feldman S.A., Zhao Y., Xu H., Black M.A., Morgan R.A., Wilson W.H., Rosenberg S.A. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J. Immunother. 2009;32:689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warren E.H., Fujii N., Akatsuka Y., Chaney C.N., Mito J.K., Loeb K.R., Gooley T.A., Brown M.L., Koo K.K., Rosinski K.V. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood. 2010;115:3869–3878. doi: 10.1182/blood-2009-10-248997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.