

Abstract
Allogeneic, off-the-shelf (OTS) chimeric antigen receptor (CAR) cell therapies have the potential to reduce manufacturing costs and variability while providing broader accessibility to cancer patients and those with other diseases. However, host-versus-graft reactivity can limit the durability and efficacy of OTS cell therapies requiring new strategies to evade adaptive and innate-immune responses. Human herpes virus-8 (HHV8) maintains infection, in part, by evading host T and natural killer (NK) cell attack. The viral K3 gene encodes a membrane-tethered E3 ubiquitin ligase that discretely targets major histocompatibility complex (MHC) class I components, whereas K5 encodes a similar E3 ligase with broader specificity, including MHC-II and the MHC-like MHC class I polypeptide-related sequence A (MIC-A)- and sequence B (MIC-B)-activating ligands of NK cells. We created γ-retroviruses encoding K3 and/or K5 transgenes that efficiently transduce primary human T cells. Expression of K3 or K5 resulted in dramatic downregulation of MHC-IA (human leukocyte antigen [HLA]-A, -B, and -C) and MHC class II (HLA-DR) cell-surface expression. K3 expression was sufficient for T cells to resist exogenously loaded peptide-MHC-specific cytotoxicity, as well as recognition in one-way allogeneic mixed lymphocyte reactions. Further, in immunodeficient mice engrafted with allogeneic T cells, K3-transduced T cells selectively expanded in vivo. Ectopic K5 expression in MHC class I−, MIC-A+/B+ K562 cells also reduced targeting by primary NK cells. Coexpression of K3 in prostate stem cell antigen (PSCA)-directed, inducible MyD88/CD40 (iMC)-enhanced CAR-T cells did not impact cytotoxicity, T cell growth, or cytokine production against HPAC pancreatic tumor target cells, whereas K5-expressing cells showed a modest reduction in interleukin (IL)-2 production without effect on cytotoxicity. Together, these results support application of these E3 ligases to advance development of OTS CAR-T cell products.
Keywords: immune evasion, OTS, off-the-shelf cell therapy, CAR-T cell, iMC, HHV8, MARCH proteins, allogeneic cell therapy
Graphical Abstract
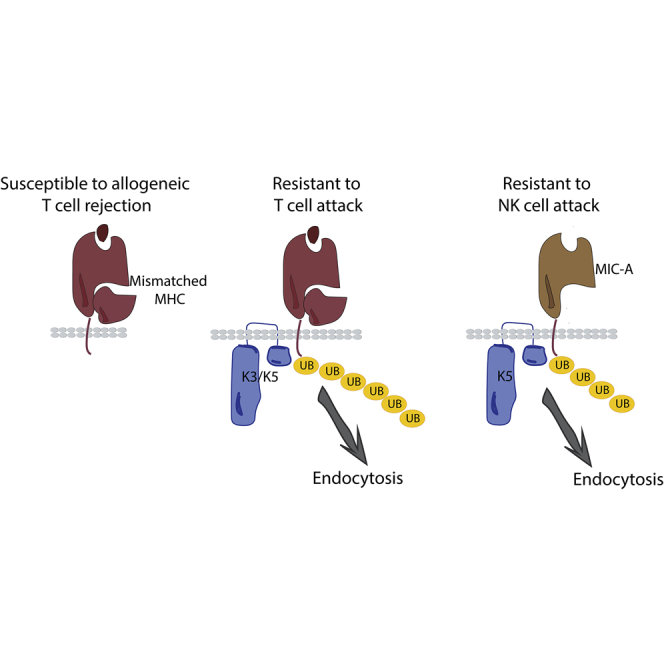
Implantation of allogeneic, off-the-shelf chimeric antigen receptor (CAR)-based anti-cancer cell therapies may reduce the cost and variation in cell quality of autologous approaches but suffers from reduced cell persistence from host immune rejection. Wang et al. report the engineering of T cells to express membrane-tethered E3 ubiquitin ligases K3 and K5 derived from HHV8. K3 and K5 target MHC and MIC-A/B complexes for lysosomal degradation, reducing allogeneic T and NK cell recognition while maintaining the anti-tumor potency of CAR-T cells.
Introduction
Adoptive transfer of engineered cell therapies, particularly chimeric antigen receptor (CAR)-T cell therapy, is transformative for the treatment of cancer.1, 2, 3, 4, 5, 6 Most CAR-T cell approaches use autologous cells, and although clinically effective, the generation of CAR-T cells on a patient-by-patient basis is expensive, highly variable, and unavailable for some patients who lack sufficient T cells or have a rapidly progressing disease. To overcome these barriers, off-the-shelf (OTS) CAR-T cell therapies derived from allogeneic or stem cell sources have been proposed, where banked CAR-T cells may be produced in large numbers, qualified, and subsequently administered to multiple cancer patients.7,8
Allogeneic immune reactions due to human leukocyte antigen (HLA) disparities, either by the graft-versus-host (GvH) or host-versus-graft (HvG), can limit OTS CAR-T cell approaches through excessive toxicity or rapid rejection of the engineered cells. Indeed, clinical outcomes of CAR-T cell therapy are strongly correlated with long-term persistence of the cells,2,4,9,10 necessitating strategies to block host responses, such as ablative preconditioning,11,12 extensive HLA matching,13 or by editing or suppressing major histocompatibility complex (MHC) expression to evade host T cell responses.7,14
Viruses that establish persistent infections use elaborate mechanisms to evade host innate and adaptive immune recognition by inhibiting the action of complement, cytokines, and antibodies, as well as through blocking peptide/MHC (pMHC) presentation.15, 16, 17 Human herpes viruse-8 (HHV8; also known as Kaposi’s sarcoma-associated herpesvirus [KSHV]) impairs MHC class I and II expression through the actions of the viral genes K3 and K5 that encode membrane-tethered E3 ubiquitin ligases that mark MHC proteins and target their secretory and endocytic trafficking away from the cell surface to the lysosome.18, 19, 20, 21, 22 The K3 and K5 protein domain architectures include an amino-terminal RING-CH domain that catalyzes ubiquitination, whereas a carboxy-terminal domain controls target selection. Subcellular localization to the endoplasmic reticulum (ER), Golgi, and plasma membrane and target identification are conferred by two transmembrane domains separated by a short luminal/extracellular linker (Figure 1A).19,23,24 K3 and K5 are the canonical members of the membrane-associated RING-CH (MARCH) family of E3 ligases, of which there are 11 members encoded in the human genome and orthologs present in poxviruses.25,26 K3 discretely targets most MHC class I components (HLA-A, -B, -C, and -E), whereas K5 only downregulates HLA-A and -B (but not -C).21,27 K5 also has broader specificity, including MHC class II and the MHC-like MHC class I polypeptide-related sequence A (MIC-A) and sequence B (MIC-B) targets of NKG2D on natural killer (NK) and cluster of differentiation (CD)8+ T cells.28, 29, 30, 31
Figure 1.

Ectopic Expression of K3 and K5 in T Cells
(A) K3 and K5 are transmembrane E3 ubiquitin ligases that target MHC proteins for polyubiquitination and endocytic trafficking to the lysosome. N, N terminus; C, C terminus of K3 or K5 protein. (B) Schematic retroviral vector designs: K3.myc-Qstalk encoding K3 fused with a myc tag, and Qstalk with the transgenes separated by P2A ribosomal skipping sequences; K3.myc encoding only K3 with a myc tag. Similar vectors were prepared for K5 expression. Qstalk only was generated as the control. (C) K3 and K5 expression in transduced T cells 9 days after transduction was probed with anti-myc antibody on an immunoblot. NT, not transduced. (D) Flow cytometry analysis to determine transduction efficiency using anti-CD34 and anti-CD3 antibodies 9 days after transduction. (E) At day 10 post-transduction, K3.myc-Qstalk- and K5.myc-Qstalk-modified T cells were subjected to magnetic sorting using anti-CD34 microbeads. Flow cytometry analysis to test the frequency of the CD34+CD3+ population post-selection is displayed.
In this study, we adapted the immune-evasion strategy of HHV8 to allogeneic CAR-T cells by expressing K3 and/or K5. Transgenic expression of either gene reduced cell-surface MHC class I expression, and K3 expression reduced allogeneic T cell receptor (TCR)-mediated T cell recognition and killing in cell culture models and in xenotransplanted mice. K5 also conferred reduced allogeneic T cell and NK cell responses. Further, K3 or K5 were coexpressed together with an inducible MyD88/CD40 (iMC)-enabled prostate stem cell antigen (PSCA)-targeted CAR platform that is regulated by the synthetic-dimerizing ligand, rimiducid (Rim).32,33 Importantly, the addition of K3 did not significantly affect CAR-T cell activation and cytotoxicity. To further reduce potential immunogenicity, we created a humanized K3 by replacing the extracellular domain with analagous residues from human MARCH4/9, which maintained the functional properties (i.e., downregulation of MHC class I) of the native K3 protein but removed potentially immunogenic epitopes. Thus, transgenic expression of HHV8-derived K3 can endow CAR-T cells with immune-evasion properties that may enable OTS cell therapies.
Results
K3 and K5 Downregulate MHC Class I Surface Expression in Human T Cells
To test whether HHV8 K3 or K5 proteins could disrupt MHC class I expression through transgenic expression, we generated γ-retroviral (γ-rv) constructs to express each protein: (1) K3 or K5 tagged with an epitope derived from human c-myc; (2) bicistronic vectors with K3-myc and K5-myc coexpressed with a transduction marker consisting of the QBEnd/10 epitope from CD34 fused with a stalk and transmembrane domain derived from human CD8α (Qstalk); and (3) a control vector expressing the Qstalk marker alone. Genes in bicistronic vectors were separated by a P2A cotranslational cleavage site derived from porcine techovirus-1 (Figure 1B).34 Transduction efficiency and transgenic expression of the E3 ligases and the Qstalk were efficient from each viral vector (Figures 1C and 1D). Moreover, Qstalk expression permitted enrichment of transduced populations to 95% by positive selection with CD34 magnetic microbeads (Figure 1E).
We initially evaluated the effects of K3 and K5 expression on human MHC-I cell-surface expression. K3- and K5-transduced T cells (as measured by CD34(Q) expression) showed markedly reduced surface levels of class 1a MHC (HLA-A, -B, and -C) (84.5% ± 6.4% with K3 measured by reduction of mean fluorescence intensity [MFI] relative to control [p = 0.0001] and 67.2% ± 8.0% [p = 0.0007] with K5; Figure 2A) compared to nontransduced (NT) T cells. Similarly, β2-microglobulin (β2-M), a marker of total MHC-I, was reduced (85.1% ± 7.6% by K3 [p < 0.0001] and 65% ± 14.8% [p = 0.001] by K5, respectively; Figure 2B). The surface level of the MHC class II molecule HLA-DR was elevated by activation of peripheral blood mononuclear cells (PBMCs) prior to transduction with γ-rv encoding the Qstalk alone, but HLA-DR expression remained low when K3 or K5 transgenes were expressed (Figure 2C). These data indicate that transgenic expression of HHV8-derived K3 and K5 proteins can reduce MHC expression on gene-modified T cells, potentially altering immune visibility.
Figure 2.

K3 and K5 Downregulate T Cell Surface Expression of MHC
K3.myc-Qstalk (K3)-, K5.myc-Qstalk (K5)-, and mock (retroviral vector encoding Qstalk marker)-transduced T cells were selected by magnetic sorting. Flow cytometry analysis to determine nontransduced (NT)-, K3-, K5-, or mock-modified T cell-surface expression of MHC class I molecules (A) by HLA-ABC clone w6/32 antibody, (B) or by human β2-microglobulin antibody. (C) MHC class II surface expression was measured with antibody against HLA-DR. Representative flow plots from two donors are shown (left). n = 4. Paired t test was used to compare indicated groups. ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; NS, not significant.
K3 and K5 Protect T Cells from Peptide-Specific TCR-Directed Cytotoxicity
We next examined whether downregulation of MHC class I by K3 and K5 was sufficient to protect transduced T cells from T cell-mediated immune recognition. Mock-transduced or K3- or K5-expressing T cells were generated from an HLA-A2+ donor. Peptides bound to MHC class I were then stripped from the T cells and replaced with HLA-A2-restricted peptides derived from cytomegalovirus (CMV) pp65 (NLV) or the tumor-associated antigen PRAME (SLL) as a specificity control. Concurrently, HLA-A2− T cells were transduced to express an HLA-A2-restricted, CMV-specific TCR (CMV-156; denoted CMV), recognizing the NLV peptide epitope (Figure 3A).
Figure 3.

K3 and K5 Protect T Cells from Peptide-Specific TCR-Directed Cytotoxicity
(A) Flow cytometry analysis to determine the surface expression of NLV peptide-specific CMV-156 TCR on HLA-A2− donor T cells (n = 3), 14 days post-transduction. (B–D) K3-, K5-, or mock (Qstalk alone)-modified HLA-A2+ donor T cells, labeled with CellTracker Green CMFDA dye and pulsed with either NLV or SLL (10 μg/mL) peptides, were cocultured with HLA-A2− donor T cells (n = 3) for 24 h at different E:T ratios. Cells were stained for viability v450, anti-HLA-A2, and subjected to flow analysis. NLV- or SLL-pulsed T cells were first gated as CMFDA+ populations. Dead cell populations were further gated as viability dye-positive cells. Two-way ANOVA was used to determine the differences among groups. For NLV-pulsed T cell killing (C), p < 0.0001; for SLL-pulsed T cell killing (D), not significant.
Peptide-loaded T cells were then fluorescently labeled and cocultured with the peptide-specific TCR transgenic T cells at decreasing effector-to-target (E:T) ratios and viability determined at 24 h (representative sample; Figure 3B). Control NLV-pulsed T cells exhibited dose-dependent sensitivity to CMV-directed T cell killing, but K3-expressing T cells were resistant even at an E:T of 20:1 (p < 0.0001; Figure 3C). Interestingly, K5-expressing T cells were only partially resistant to CMV-TCR recognition (p = 0.003, two-way ANOVA). As expected, SLL-pulsed T cells were not recognized by A2-restricted but CMV-specific effector T cells (Figure 3D). These data are consistent with the hypothesis that reduction of HLA-A2 levels by K3 expression and to a lesser extent K5 expression is sufficient to limit recognition and cytotoxic targeting by CD8 T cells.
K3 Inhibits Allogeneic Target Recognition and Activation of Human T Cells
We further examined the cloaking effect of K3 to allogeneic immune responses by determining the activation state of potentially alloreactive primary T cells exposed to transduced target T cells in a one-way mixed lymphocyte reaction (MLR). MLRs were set up by mixing irradiated K3- or Qstalk-modified T cells (CD34+-selected T cells, n = 4; Figure 4A) with CMFDA-labeled PBMCs derived from allogeneic or autologous donors (Figure 4B). The effect of K3 to permit target cells to evade allogeneic T cell activation was monitored as a failure to proliferate (and dilute CMFDA fluorescence over 5 days) (Figure 4C). Consistent with MHC downregulation, K3-modified stimulator T cells failed to activate allogeneic T cell proliferation compared with Qstalk-modified (p < 0.001) and NT groups (p < 0.001; Figure 4D). These data support the notion that masking from T cell recognition by K3 expression could effectively modulate allogeneic lymphocyte responses.
Figure 4.

K3 Suppresses Allogeneic T Cell Proliferation in a One-Way Human Mixed Lymphocyte Reaction
(A) Donor T cells (n = 4) were transduced with retroviral vector encoding Q.CD8stalk or K3-Q.CD8stalk. Transduction efficiency was determined by anti-CD3 and anti-CD34 antibodies. (B) Q.CD8stalk- or K3-Q.CD8stalk-modified T cells (stimulator) were irradiated at the dose of 25 Gy, followed by coculture with either autogeneic or allogeneic PBMCs (responder), labeled with CMFDA green dye, for 5 days. (C) The cocultures were stained for viability and anti-CD3. Divided responder T cells were gated out as a single, live CD3+ population with a dim CMFDA dye. (D) Representative flow plots showing divided CMFDA-labeled D874 (responder) after 5 days coculture with irradiated D874, D749, D754, and D912 as stimulator (left). 12 allogeneic responses and 4 autogeneic responses were plotted in NT-, Q.CD8stalk-, and K3-Q.CD8stalk-modified T cells (right). Paired t test was used to compare indicated groups. ∗∗∗p < 0.001.
K3 Expression Does Not Impair the Activation and Proliferation of CAR-T Cells
For an allogeneic CAR-T cell therapy to incorporate an immune-evasion strategy, it is essential that the methods to cloak MHC identity do not impact CAR-T cell efficacy. Upon stimulation with surface-bound αCD3 and αCD28, T cells expressing K3 or K5 were efficiently activated (Figure 5A) and induced CD25 (Figure 5B) and CD69 (Figure 5C) expression. Interestingly, among the activated CD25+CD69+ T cells, K5 demonstrated an increased CD8− population (Figure 5D).
Figure 5.

K3 and K5 Effects on T Cell Activation and CAR-T Cell Function
(A) Activated T cells were transduced with Qstalk alone (mock), K3, or K5. The cells were stained for viability dye, anti-CD3, anti-CD34, anti-CD25, anti-CD69, and anti-CD8 antibodies. Transduced T cells were gated as live CD3+CD34+ populations. T cell activations were measured by (B) CD25+ percentage and (C) CD69+ percentage. (D) Proportions of CD3+CD8− or CD3+CD8+ in CD25+CD69+ populations were determined. Paired t test was used to compare indicated groups. ∗p < 0.05. (E and F) T cells were triple transduced with retroviral vectors: one vector encoding Q.CD8stalk (mock), K3 (K3-Q.CD8stalk), or K5 (K5-Q.CD8stalk); a second CAR-encoding vector iMC-PSCA.ζ-ΔCD20; and a third vector encoding eGFPFfluc. The cells were selected with anti-CD20 magnetic beads. PSCA-expressing HPAC.RFP target cells were cocultured with gene-modified T cells at the ratio of 1 to 10 for 7 days in the presence or absence of 1 nM rimiducid (Rim). Tumor cell-killing (RFP, red) and T cell (EGFP, green) proliferation were measured by live-cell imaging in an IncuCyte imaging incubator. Two-way ANOVA was used to compare tumor killing (E) and T cell proliferation (F) among mock-, K3-, and K5-modified CAR-T cells either with Rim or without Rim. p > 0.05, not significant.
To produce CAR-T cells, K3, K5, or Qstalk was cotransduced with a CAR-encoding vector (iMC-PSCA-CAR.ζ-CD20stalk) in which costimulation of a PSCA-targeting, first-generation CAR-T cells are regulated by rimiducid-directed dimerization of iMC.32,33,35 These cells were further transduced with a marker virus to express a fusion protein of GFP and firefly luciferase (eGFPFFluc). Following enrichment of CAR-T cells by selection for the CD20stalk marker, T cells were cocultured with red fluorescent protein (RFP)-modified human pancreatic adenocarinoma (HPAC) tumor cells. Control of tumor cell expansion by K3- or K5-expressing CAR-T cells was efficient and rimiducid stimulated (Figure 5E). Further, CAR-T cell expansion was robust and also stimulated by rimiducid-mediated iMC activation, although less robust with K5 versus K3 coexpression (Figure 5F). This reduced CAR-T cell expansion did not achieve statistical significance by two-way ANOVA but correlated with a modest but significant reduction of interleukin (IL)-2 and interferon (IFN)-γ expression in K5-expressing CAR-T cell tumor cocultures (Figures S1B and S1C). We further investigated if K3 or K5 could modulate nuclear factor κB (NF-κB) activation when transduced into HEK293 cells containing an NF-κB reporter. NF-κB is a key signaling pathway to drive cytokine production that is activated by iMC.32,33,36 K3 expression did not impact reporter activity, whereas K5 showed reduced NF-κB activation (Figure S1A). Furthermore, we examined the effects of cell growth on T cells not expressing iMC or a CAR K3, K5, or the Qstalk alone. K3 expression had no effect on cell expansion in culture when measured by periodic cell counting (Figure S2A) or by real-time microscopic quantitation of activated T cells in an IncuCyte chamber, whereas K5 expression produced a modest inhibition of the growth of the activated T cells (Figure S2B).
Generation of K3-MARCH Hybrids
Whereas K3 is primarily localized internally within the ER and Golgi, plasma membrane expression has been observed,19 and the exposed K3 protein may therefore be immunogenic to the humoral compartment despite evading T cell immunity. To mitigate this possibility, we also generated chimeric proteins in which the 13 amino acid surface-exposed linker between the transmembrane domains of K3 were replaced with linker sequences derived from the related human MARCH proteins (Figure 6A). T cells expressing a K3-MARCH4 hybrid exhibited downregulation of surface MHC class I, whereas K3-MARCH1 and K3-MARCH2 hybrids failed to do so (Figures 6B and 6C). In the case of K3-MARCH1, this may have been due to poor expression or structural constraint introduced by this linker (Figure 6D). Replacement of amino acids in the MARCH4 linker with residues in the MARCH9 linker (Figure 6E) further optimized the K3-MARCH hybrid’s efficacy for MHC class I blockage (Figures 6F and 6G).
Figure 6.

Analysis of K3-MARCH Hybrid Proteins
(A) Depiction of hybrid K3 proteins in which extracellular amino acids were derived from human MARCH proteins. Schematic diagram of retroviral vector K3MARCH-Q.CD8stalk is at the right. (B) NT, wild-type K3, K3MARCH1, K3MARCH2, and K3MARCH4 gene-modified T cells were stained with anti-CD34 and anti-β2-microglobulin antibodies. β2-microglobulin with reduced surface-expression cell populations were gated. (C) Transduced T cells first gated as CD34+ populations were assessed for human β2-microglobulin cell-surface expression. Paired t test was used to compare indicated groups. ∗p < 0.05; ∗∗p < 0.01. (D) Immunoblot analysis of wild-type or various K3-MARCH chimeric protein expressions with anti-myc antibody in transduced T cells. (E) Wild-type K3, K3MARCH4, K3MARCH4-1, K3MARCH4-2, K3MARCH4-3, K3MARCH9, and K3MARCH4/9 extracellular domain sequences and expression in gene-modified T cells determined by immunoblot analysis for anti-myc. (F and G) MHC class I surface expression in NT, wild-type K3, or various versions of K3MARCH chimeric proteins as accessed by flow cytometry with (F) anti-human HLA-ABC clone w6/32 and (G) anti-human β2-microglobulin.
K3 Expression Enhances T Cell Persistence In Vivo
To determine if K3 or K3MARCH4/9 expression could allow T cells to resist allogeneic attack in vivo, we performed a modified MLR in xenotransplated mice. 8 × 106 fresh human PBMCs were transferred into immune-deficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice37 together with 2 × 106 Qstalk-only control-, K3-, or K3MARCH4/9-modified T cells derived from a separate donor. The gene-modified T cells were cotransduced with an orange-Nanolantern-Renilla luciferase (ONLRluc) marker construct, and their persistence was monitored over time by bioluminescence imaging (BLI). Both K3- and K3MARCH4/9-modified T cells demonstrated growth and enhanced persistence in the presence of an allogeneic immune system relative to cells expressing only the Qstalk (Figures 7A and S3; p < 0.05). After 3 weeks, mice were sacrificed, and human cell populations were isolated from bone marrow and spleen (Figure 7B). Populations of human cells marked with ONL human CD (hCD)45+mouse CD (mCD)45−hCD3+ONL+ T cells were readily detected from the spleen and bone marrow of mice transplanted with K3- or K3MARCH4/9-modified T cells, but cells expressing only the Qstalk were absent (Figure 7C). The selective expansion and persistence of K3-expressing T cells in the presence of an allogeneic immune system supported the hypothesis that these cells evaded alloimmune recognition and attack.
Figure 7.

K3-Modified T Cell Persistence in a Mixed Lymphocyte Reaction In Vivo
K3-, K3MARCH4/9-, or Qstalk-modified T cells (2 × 106) were labeled with ONL and mixed with freshly isolated human PBMCs (8 × 106) derived from a different donor. The cells were cotransplanted into NSG mice. (A) Gene-modified T cell persistence in vivo was monitored by IVIS imaging weekly. Two-way ANOVA was used to determine the differences among groups, p < 0.05. 18 days postengraftment, spleen and bone marrow were harvested. (B) Gene-modified T cells were identified as live single cells, hCD45+mCD45−hCD3+ONL+ populations and (C) quantified. Multiple t tests were used to determine the differences among groups. Only comparisons of hCD3+ONL+ cell counts in bone marrow of mice derived from Qstalk and K3MARCH4/9 groups were significant, p < 0.05.
K5 Expression Reduces Targeting by NK Cells
NK cells determine which cells to target through a complex integration of receptor-mediated activating and inhibitory signals. MHC class I interaction with inhibitory killer-immunoglobulin receptors (KIRs) inhibits NK cells, whereas interaction with stress-induced ligands, including MIC-A, MIC-B, activation-induced C-type lectin (AICL), and UL16 binding proteins (ULBPs) on virus-infected and transformed cells, drives NK cell targeting.38,39 Hypothetically, downregulation of MHC class I by K3 and K5 could therefore lead to NK cell targeting; however, K5 also drives endocytosis of MIC-A, MIC-B, and AICL.30,31,38 To assess the potential blockage of NK targeting by K5 expression, K562 erythroleukemia cells were transduced with γ-rv expressing K3 or K5 or a tricistronic vector expressing both K3 and K5 with the Qstalk marker (Figures 8A and S4A). K562 cells are canonical NK target cells that lack all MHC class I expression and abundantly express NK-activating ligands. Human NK cells were isolated from PBMCs of four donors, activated with IL-15, and cocultured with K562 cells at increasing E:T ratios. At ratios of 2:1 or 1:1, K5-expressing target cells resisted NK-directed killing, whereas K3-expressing cells were less resistant (Figures 8B and 8C). Expression of K5 protected marked, transduced cells from NK cell cytotoxicity relative to NT cells within the same population (Figures 8D–8F), and interestingly, expression of both K3 and K5 from a tricistronic vector similarly protected only the transduced cells (Figures S4B–S4D).
Figure 8.

K5 Expression Reduces Targeting by NK Cells
(A) Depiction of targeting of NK-activating ligands, such as MIC-A, for ubiquitination and endocytosis by K5. (B) Flow cytometry identifying K562 cells transduced to express Qstalk alone or coexpressing K3 or K5. These K562 populations were cotransduced to express an eGFP-firefly luciferase fusion reporter (see Figure S4C). (C) Transduced K562 cells were cocultured with freshly isolated human peripheral blood NK cells at decreasing E:T ratios for 24 h. Specific lysis was calculated in relation to the average level of luciferase activity of NT K562 cells at an E:T of 10:1. Multiple t tests were used to compare the difference between the mock- and K5-modified group. ∗p < 0.05; ∗∗p < 0.01. (D) Mixed populations of transduced (marked with CD34Q) and NT K562 cells were cocultured with NK cells for 3 days. Viability was determined by CMFDA exclusion. (E and F) Plot of loss of viability of the K562 cell expressing Qstalk alone or K5-Qstalk against a decreasing number of NK cells. (F) Killing of Qstalk-K562 cells within the same experimental populations as (E). Two-way ANOVA was used to determine the differences among groups, p < 0.01 (E); p = 0.68 (F).
T cells expressing K3 and thereby losing MHC class I expression become better substrates for attack by NK cells than T cells expressing the Qstalk alone (Figure S5). However, in cocultures of NK cells with T cells transduced with K5, in which caspase-3 or -7 activation was monitored in real time through release of a fluorescent DNA-binding dye, apoptosis induction was diminished relative to K3-expressing cells (Figure S5A). Importantly, coexpression of K5 with K3 also blocked the targeting of NK cells (p < 0.00001, two-way ANOVA). Similarly, Annexin V positivity in transduced T cells cocultured with NK cells was also blocked by K5 expression (p < 0.01, paired t test; Figures S2B and S2C).
Discussion
Development of OTS approaches will increase the scalability of CAR-based cell therapies toward the burden of cancer incidence. However, use of standardized donor-derived cells must overcome the host immune rejection of allogeneic implants. We present a method in which disruption of MHC expression is achieved at the protein level through the expression of factors that are easily included with CAR-expressing viral vectors. Many genes have evolved to encode products specifically to minimize viral immune recognition by T and NK cells. To mimic these biological effects, K3 and K5 from HHV8 were evaluated in this study due to their well-defined substrate specificity and mechanism of action. E3 ligases use a discrete targeting of a natural ubiquitination process that may be less likely to alter T cell physiology than other viral mechanisms for evasion of MHC-antigen presentation, such as alteration of peptide processing, alteration of G protein signaling, or ER retention.40
Other approaches to reduce immune rejection of allo-CAR-T and CAR-NK cells can be developed. For example, matching of 7/8 or 8/8 alleles for HLA-A, -B, -C, and -DRB1, as recommended for successful allogeneic stem cell transplantation, may be effective but will require elaborate qualification and banking of products if also applied to allogeneic CAR products.41 Gene editing combined with lymphoid ablation can increase CAR-T cell persistence but also subverts the anti-tumor effects of the host’s system, and conditioning regimens already used to promote engraftment also increase the incidence of opportunistic infection.42 Targeted disruption of the β2-M gene with site-directed nucleases can block all MHC class I expression but also makes cells more susceptible to NK cell targeting. Further methods to replace β2-M expression with HLA-E (or potentially HLA-G) cDNAs may block both NK and T cell immunogenicity.7,14 While ingenious, the successful editing of multiple genetic loci (β2-M, TRAC, HLA-D, etc.), while also introducing CAR constructs, presents challenges for manufacturing and qualification of CAR-T or CAR-NK cell banks for OTS use with the added safety concern of genetic mistargeting by the nuclease. An interesting, aggressive alternative to immune evasion does not evade allogeneic attack by host cells but instead redirects CAR-T cells to attack activated host NK and T cells.43
We found that K3 expression removes surface expression of MHC class I by approximately 20-fold, which was sufficient to block T cell recognition under conditions that confer very high specific peptide loading to HLA-A2 on control effector cells and in which effector cells express a pMHC-specific TCR. Of the two proteins tested, K5 was less efficient in MHC class I downregulation than K3. This finding correlates with the breadth of the described substrate specificities of these E3 ligases and perhaps their role in the HHV8 life cycle.20 K3 targets all classical MHC-I molecules (and the invariant NK T cell [iNKT] ligand CD1d) and is expressed early in the lytic cycle, whereas K5 is expressed later and is selective for HLA-A, HLA-B, but not HLA-C or HLA-E, which are ligands for NK inhibitory receptors KIRDL4 and NKG2A, respectively.20,38 We observed that K5 was less efficient than K3 for MHC class I downregulation. Based on the substrate specificity of K5, we may ascribe retained low levels of surface MHC class I in K5-expressing cells to HLA-C or HLA-E expression.
The observation of similarly reduced MHC class II levels with K3 and K5 expression in T cells was surprising based on previous observations supporting K5 but not K3 as the primary regulator of HLA-D.44,45 The observation of MHC class II blockage by K3 was supported by reduction of allogeneic recognition by both CD4+ and CD8+ T cells in MLR assays. Although K3 appears to be an ideal agent for the evasion of T cell allogeneic targeting, it is less efficient at protecting K562 cells (an ideal target) against NK cell attack, whereas K5 expression conferred a protective effect. Together, we propose that the most effective immune-evasion strategy for allogeneic cell therapies against lymphoid targeting may incorporate both K3 and K5. Whereas exposure of the foreign viral antigens themselves to T cell immunity is presumably solved by MHC downregulation, the linker between the two domains can remain exposed to the humoral compartment. This concern is mitigated by the replacement of the linker with human MARCH protein sequences that retain the K3 function. In fact, overexpression of MARCH4, MARCH8, or MARCH9 may also confer an attractive alternative immune-evasion strategy, although the potentially diverse targeting of other cellular programs may impact deleteriously CAR-T cell functionality and would require rigorous examination.
We have demonstrated that K3 and/or K5 can function in CAR-T cells. Our currently favored CAR platform integrates a 1st-generation CAR together with iMC, which comprises a fusion of the signaling domains of MyD88 with those of CD40, together with two copies of FKBP12 mutated to valine at position 36 to confer allele-specific dimerizing interaction with rimiducid.46, 47, 48 MyD88/CD40 signaling drives robust proliferation, persistence, cytokine production, and augmented cytotoxicity in T and NK cells, and dimerization of iMC with rimiducid makes this signaling tunable on demand.32,49 Addition of K3 and/or K5 to this platform makes feasible allogeneic sourcing of T or NK cells for CAR-based therapy with enhanced persistence. Despite the latent nature of iMC-expressing CAR-T cells in the absence of rimiducid, protection from persistent allogeneic CAR cells following cancer therapy can be further augmented by inclusion of a second, orthogonally regulated proapoptotic safety switch, such as rapamycin-inducible caspase-9 (iRC9).33 Together, these components represent an advance in the development of OTS CAR-based therapies, but the utility of K3/K5 (perhaps together with inducible caspase-9 switches) can be applied to increase persistence of diverse allogeneic cell therapies without compromising safety.
Materials and Methods
Cell Lines, Media, and Reagents
HEK293T, K562, and HPAC were purchased from ATCC (Manassas, VA) and maintained in media recommended by the supplier. PBMCs were isolated by a density-gradient technique (Lymphoprep; Accurate Chemical & Scientific, Westbury, NY) from buffy coat blood obtained from the Gulf Coast Blood Bank (Houston, TX). T cells generated from PBMCs were cultured in 45% RPMI 1640; 45% Click’s media (Invitrogen, Grand Island, NY), supplemented with 10% fetal bovine serum (FBS); 1% penicillin-streptomycin (pen/strep); 2 mM GlutaMAX; 5 ng/mL recombinant human IL-15; and 15 ng/mL recombinant human IL-7 (Miltenyi Biotec, San Diego, CA). Clinical-grade rimiducid was diluted in ethanol as a 100-μM stock solution for in vitro assays. Human NK cells were isolated from peripheral blood buffy coats by selection with CD56-coated magnetic beads (Miltenyi Biotec) and stimulated for 1 day with recombinant human IL-15 before culturing with gene-modified K562 cells prepared by transduction with γ-rvs.
Retroviral and Plasmid Constructs
Bicistronic SFG-based retroviral vectors were generated encoding KSHV K3 or K5 tagged with a myc epitope, together with a surface marker Q.CD8stalk comprising a signal peptide, two human CD34 QBEnd/10 minimal epitopes, the CD8α stalk, and a transmembrane domain. The SFG retroviral vector containing only K3 with a myc tag or containing K3.myc, TCRe, TCRz, and Q.CD8stalk separated by P2A and T2A was also made. These retroviral vectors were generated using appropriate restrictive enzyme digestion methods to insert synthetic DNA (Integrated DNA Technology, San Diego, CA) encoding indicated sequences. For K3-MARCH chimeric constructs, synthetic DNA (Integrated DNA Technology) encoding MARCH1, -2, or -4 extracellular sequences flanked by K3 transmembrane sequences was Gibson assembled (New England Biolab, Ipswich, MA) with pSFG-K3-Q.CD8stalk vector digested by BstZ171 and NruI. K3MARCH4-1 to -3, K3MARCH9, and K3MARCH4/9 constructs were generated using PCR-based methods using K3March4 as a template.
Generation of Gene-Modified T Cells
Retroviral supernatants were produced by transient transfection of 293T cells as previously described.32 T cells made from PBMCs were activated by anti-CD3/anti-CD28 antibodies, followed by transduction use of retronectin (Takara Bio, Mountain View, CA) and spinfection. For double transduction, T cells were transduced at day 3 and day 4 after activation. On day 14, transduced T cells were harvested for use. In certain experiments, T cells were also transduced with SFG-eGFP-firefly luciferase (eGFPFfluc) for labeling.
Immunoblotting
Details of immunoblotting were described previously.50 Antibody specific for myc (9E10) was purchased from Cell Signaling Technology (Danvers, MA). Anti-β-actin antibody was purchased from Sigma-Aldrich (St. Louis, MO).
Immunophenotyping
Gene-modified T cells were analyzed for K3 or K5 transgene expression 10–14 days post-transduction by using anti-CD3-peridinin chlorophyll protein-cyanine (PerCP-Cy)5 and anti-CD34-phycoerythrin (PE) antibodies (Abnova, Walnut, CA). To detect surface MHC class I gene expression, K3- or K5-modified T cells were also stained with anti HLA-ABC w6/32-allophycocyanin (APC)-Cy7 or anti-human β2-M-APC antibody. Gene-modified T cells were also analyzed for CD8, CD25, and CD69. All antibodies were purchased from BioLegend (San Diego, CA) except as stated otherwise. Flow cytometry was performed using a Gallios (Beckman Coulter) flow cytometer, and the data were analyzed using Kaluza (Beckman Coulter) software.
SRα-Secreted Alkaline Phosphatase (SeAP) Assay
The effect of K3 and K5 on NF-κB activity was assessed using an SRα-SeAP assay.33 Briefly, HEK293 cells were cotransfected with 1 μg of K3 and K5 or mock (control plasmid containing the same Q.CD8stalk) and the SRα-SeAP reporter plasmid. 24 h later, the cells were collected and seeded onto a 96-well plate in triplicate. A small portion of the cells was analyzed by flow cytometry for transfection efficiency. The next day, after the plate was heat inactivated at 68°C for 1 h, supernatants were transferred into a black 96-well plate (Greiner) containing 1 mM 4-methylumbelliferyl phosphate (MUP) substrate diluted in 2 M diethanolamine (pH 10.0). The plate was incubated at 37°C for 30 min, followed by fluorescence reading excited at 355 nm and emitted at 460 nm.
Cytokine Production
The effects of K3 or K5 on CAR-T cell antigen-specific cytokine production were analyzed by enzyme-linked immunosorbent assay (ELISA) using kits (eBioscience, San Diego, CA) with the manufacturer’s protocol.
Cytotoxicity and Coculture Assays
A TCR recognizing the HLA-A2-restricted NLV peptide from the pp65 protein of HCMV51,52 was cloned using the GeneRacer kit from Invitrogen (Carlsbad, CA) from oligoclonal T cells generated after 4 rounds of stimulation with peptide-pulsed autologous PBMCs. The following gene-specific primers (Sigma-Aldrich, The Woodlands, TX) were used for the rapid amplification of cDNA ends (RACE) PCR: TCRα C-region primer: 5′-TCAGCTGGACCACAGCCGCAGC-3′; TCRβ C1-region primer: 5′-TCAGAAATCCTTTCTCTTGACCATGGC-3′; TCRβ C2-region primer: 5′-CTAGCCTCTGGAATCCTTTCTCTTG-3′. The coding sequence for the α and β chains of the CMV-specific TCR was codon optimized, synthesized as gBlocks, and cloned into the SFG retrovirus (in the order TCRα-2A-TCRβ).
To access potential protective effects of K3 and K5 on TCR-mediated cytotoxicity, coculture assays were performed. Donor PBMCs were first sorted as HLA-A2+ donors and HLA-A2− donors by flow cytometry analysis. HLA-A2− donor PBMCs (effector) were activated and transduced with the γ-rv vector encoding the CMV TCR (156). HLA-A2+ donor PBMCs were transduced with the retroviral vector encoding K3myc-Q.CD8stalk and K5myc-Q.CD8stalk or a vector encoding Q.CD8stalk (mock). K3-, K5-, or mock-modified T cells were then pulsed with either the NLV peptide derived from the CMV pp65 matrix protein or the SLL (PRAME specific) peptide (10 μg/mL) at 37°C for 1 h, followed by CellTracker Green CMFDA dye (8 μM) labeling at 37°C for 30 min. The cells were washed and cocultured with HLA-A2− donor T cells modified with CMV TCR at various E:T ratios for 24 h. The cocultures were harvested and stained for viability v450, anti-HLA-A2, and subjected to flow cytometry analysis. NLV- or SLL-pulsed T cells were first gated as CMFDA+ populations. Dead cell populations were further gated out as viability dye-positive cells (peptide-pulsed T cell killing %).
Additional coculture assays were used to determine the effects of K3 and K5 on the tumor-killing efficacy of CAR-T cells. Activated T cells were triple transduced with the first retroviral vector encoding mock (retroviral vector encoding Q.CD8stalk), K3myc-Q.CD8stalk, or K5myc-Q.CD8stalk; a second retroviral vector encoding iMC.CAR(PSCA).ζ.ΔCD20; and a third retroviral vector encoding eGFPFfluc for labeling. Control T cells were only transduced with eGFPFfluc. Gene-modified T cells were selected for CD20 using Miltenyi microbeads following the manufacturer’s protocol and then cocultured with HPAC RFP cells for 7 days. Tumor cell growth (RFP, red) and T cell proliferation (eGFP, green) were monitored by real-time fluorescent microscopy (IncuCyte; Essen BioScience). For cocultures utilizing NK cells, PBMCs from different donors were incubated with magnetic αCD56 beads (Miltenyi Biotec) and selected using the manufacturer’s protocol. In T cell cocultures, NK cells were first activated by IL-15 (15 ng/mL) for 1 day and then stimulated with magnetic beads conjugated with IL-21 and 4-1BBL for 3 days prior to inclusion in cocultures. NK cells used in K562 cocultures were activated with IL-15 but not further activated with beads.
One-Way MLR
Activated donor T cells were transduced with retroviral vector encoding Q.CD8stalk or K3-Q.CD8stalk. The gene-modified T cells were irradiated at a dose of 25 Gy as to produce stimulator cell populations, followed by coculture with either autogeneic or allogeneic PBMCs (responders), labeled with CMFDA green dye previously. The MLR cocultures were performed by seeding responder cells with stimulator cells at the ratio of 1 to 5 in a round-profile, 96-well plate with a final volume of 200 μL medium for 5 days and then stained for viability and anti-CD3. Divided responder T cells were gated as a single, live CD3+ population with a dim CMFDA dye. 12 allogeneic responses and 4 autogeneic responses were recorded in NT-, Q.CD8stalk-, and K3-Q.CD8stalk-modified T cells.
In Vivo MLR
Human PBMCs were isolated from a buffy coat, obtained from the Gulf Coast Blood Bank, by a density-gradient centrifugation (Lymphoprep; Accurate Chemical & Scientific, Westbury, NY). Qstalk-, K3-, or K3MARCH4/9-modified T cells derived from a different donor were also transduced with a retroviral vector encoding ONLRluc to enable in vivo tracking of T cells. NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (Jackson Laboratory, Bar Harbor, ME) were engrafted with 2 × 106 gene-modified T cells and 8 × 106 fresh PBMCs by intravenous (i.v.) injection into the tail vein. In vivo T cell persistence was measured by BLI following intraperitoneal (i.p.) injection of 150 ng Coelenterazine-h (Perkin Elmer, Waltham, MA) and imaged using an In Vivo Imaging System (IVIS). Photon emission was analyzed by whole-body region of interest (ROI), and the signal was measured as average radiance (photons/second/square centimeter/steradian). Endpoint analysis involved fluorescence-activated cell sorting (FACS) analysis of splenocytes and bone marrow. Animal studies were performed in the Bellicum Pharmaceuticals American Association for Accreditation of Laboratory Animal Care (AAALAC)-approved vivarium and were approved by the Bellicum Pharmaceuticals Institutional Animal Care and Use Committee.
Statistics
All statistical tests (noted in figure legends) were carried out and analyzed using GraphPad Prism software (version 8.0; GraphPad). Data are presented as means ± SEM. All t tests were two tailed. All ANOVAs were two way. p Values of less than 0.05 were considered significant.
Author Contributions
X.W. designed and performed experiments described in each figure and drafted the manuscript. F.G.C. performed the animal implantations and imaging in Figure 7. K.L.S. and D.M.S. designed and supervised aspects of the project. A.E.F. and J.H.B. designed and supervised the experiments and wrote the manuscript.
Conflicts of Interest
During the conduct of this study, all authors were employees of Bellicum Pharmaceuticals and holders of stock or stock options.
Acknowledgments
The authors would like to thank Drs. MyLinh Duong, Peter Chang, Matthew Collinson-Pautz, and Eva Morschl for expert advice and Dr. Eugenio de Hostas for editing of the manuscript.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.10.019.
Contributor Information
Aaron E. Foster, Email: afoster@bellicum.com.
J. Henri Bayle, Email: jhbayle@bellicum.com.
Supplemental Information
References
- 1.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guedan S., Ruella M., June C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019;37:145–171. doi: 10.1146/annurev-immunol-042718-041407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majzner R.G., Mackall C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019;25:1341–1355. doi: 10.1038/s41591-019-0564-6. [DOI] [PubMed] [Google Scholar]
- 6.June C.H., Sadelain M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Depil S., Duchateau P., Grupp S.A., Mufti G., Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat. Rev. Drug Discov. 2020;19:185–199. doi: 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 8.Torikai H., Cooper L.J. Translational Implications for Off-the-shelf Immune Cells Expressing Chimeric Antigen Receptors. Mol. Ther. 2016;24:1178–1186. doi: 10.1038/mt.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghorashian S., Kramer A.M., Onuoha S., Wright G., Bartram J., Richardson R., Albon S.J., Casanovas-Company J., Castro F., Popova B. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019;25:1408–1414. doi: 10.1038/s41591-019-0549-5. [DOI] [PubMed] [Google Scholar]
- 11.Qasim W., Zhan H., Samarasinghe S., Adams S., Amrolia P., Stafford S., Butler K., Rivat C., Wright G., Somana K. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017;9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 12.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 13.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lanza R., Russell D.W., Nagy A. Engineering universal cells that evade immune detection. Nat. Rev. Immunol. 2019;19:723–733. doi: 10.1038/s41577-019-0200-1. [DOI] [PubMed] [Google Scholar]
- 15.Davis-Poynter N.J., Farrell H.E. Masters of deception: a review of herpesvirus immune evasion strategies. Immunol. Cell Biol. 1996;74:513–522. doi: 10.1038/icb.1996.84. [DOI] [PubMed] [Google Scholar]
- 16.Ploegh H.L. Viral strategies of immune evasion. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- 17.Vider-Shalit T., Fishbain V., Raffaeli S., Louzoun Y. Phase-dependent immune evasion of herpesviruses. J. Virol. 2007;81:9536–9545. doi: 10.1128/JVI.02636-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ganem D. KSHV infection and the pathogenesis of Kaposi’s sarcoma. Annu. Rev. Pathol. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- 19.Coscoy L., Ganem D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA. 2000;97:8051–8056. doi: 10.1073/pnas.140129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coscoy L. Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Nat. Rev. Immunol. 2007;7:391–401. doi: 10.1038/nri2076. [DOI] [PubMed] [Google Scholar]
- 21.Ishido S., Wang C., Lee B.S., Cohen G.B., Jung J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000;74:5300–5309. doi: 10.1128/jvi.74.11.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartee E., Mansouri M., Hovey Nerenberg B.T., Gouveia K., Früh K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 2004;78:1109–1120. doi: 10.1128/JVI.78.3.1109-1120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hewitt E.W., Duncan L., Mufti D., Baker J., Stevenson P.G., Lehner P.J. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J. 2002;21:2418–2429. doi: 10.1093/emboj/21.10.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin H., Li S., Shu H.B. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Front. Immunol. 2019;10:1751. doi: 10.3389/fimmu.2019.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goto E., Ishido S., Sato Y., Ohgimoto S., Ohgimoto K., Nagano-Fujii M., Hotta H. c-MIR, a human E3 ubiquitin ligase, is a functional homolog of herpesvirus proteins MIR1 and MIR2 and has similar activity. J. Biol. Chem. 2003;278:14657–14668. doi: 10.1074/jbc.M211285200. [DOI] [PubMed] [Google Scholar]
- 26.Samji T., Hong S., Means R.E. The Membrane Associated RING-CH Proteins: A Family of E3 Ligases with Diverse Roles through the Cell. Int. Sch. Res. Notices. 2014;2014:637295. doi: 10.1155/2014/637295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cadwell K., Coscoy L. The specificities of Kaposi’s sarcoma-associated herpesvirus-encoded E3 ubiquitin ligases are determined by the positions of lysine or cysteine residues within the intracytoplasmic domains of their targets. J. Virol. 2008;82:4184–4189. doi: 10.1128/JVI.02264-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartee E., McCormack A., Früh K. Quantitative membrane proteomics reveals new cellular targets of viral immune modulators. PLoS Pathog. 2006;2:e107. doi: 10.1371/journal.ppat.0020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Means R.E., Lang S.M., Jung J.U. The Kaposi’s sarcoma-associated herpesvirus K5 E3 ubiquitin ligase modulates targets by multiple molecular mechanisms. J. Virol. 2007;81:6573–6583. doi: 10.1128/JVI.02751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nice T.J., Deng W., Coscoy L., Raulet D.H. Stress-regulated targeting of the NKG2D ligand Mult1 by a membrane-associated RING-CH family E3 ligase. J. Immunol. 2010;185:5369–5376. doi: 10.4049/jimmunol.1000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas M., Boname J.M., Field S., Nejentsev S., Salio M., Cerundolo V., Wills M., Lehner P.J. Down-regulation of NKG2D and NKp80 ligands by Kaposi’s sarcoma-associated herpesvirus K5 protects against NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA. 2008;105:1656–1661. doi: 10.1073/pnas.0707883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foster A.E., Mahendravada A., Shinners N.P., Chang W.C., Crisostomo J., Lu A., Khalil M., Morschl E., Shaw J.L., Saha S. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Mol. Ther. 2017;25:2176–2188. doi: 10.1016/j.ymthe.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duong M.T., Collinson-Pautz M.R., Morschl E., Lu A., Szymanski S.P., Zhang M., Brandt M.E., Chang W.C., Sharp K.L., Toler S.M. Two-Dimensional Regulation of CAR-T Cell Therapy with Orthogonal Switches. Mol. Ther. Oncolytics. 2018;12:124–137. doi: 10.1016/j.omto.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim J.H., Lee S.R., Li L.H., Park H.J., Park J.H., Lee K.Y., Kim M.K., Shin B.A., Choi S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mata M., Gerken C., Nguyen P., Krenciute G., Spencer D.M., Gottschalk S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017;7:1306–1319. doi: 10.1158/2159-8290.CD-17-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grossmann C., Ganem D. Effects of NFkappaB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology. 2008;375:94–102. doi: 10.1016/j.virol.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shultz L.D., Lyons B.L., Burzenski L.M., Gott B., Chen X., Chaleff S., Kotb M., Gillies S.D., King M., Mangada J. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 38.Miller J.S., Lanier L.L. Natural Killer Cells in Cancer Immunotherapy. Annu. Rev. Cancer Biol. 2019;3:77–103. [Google Scholar]
- 39.Raulet D.H., Marcus A., Coscoy L. Dysregulated cellular functions and cell stress pathways provide critical cues for activating and targeting natural killer cells to transformed and infected cells. Immunol. Rev. 2017;280:93–101. doi: 10.1111/imr.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansen T.H., Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat. Rev. Immunol. 2009;9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 41.Howard C.A., Fernandez-Vina M.A., Appelbaum F.R., Confer D.L., Devine S.M., Horowitz M.M., Mendizabal A., Laport G.G., Pasquini M.C., Spellman S.R. Recommendations for donor human leukocyte antigen assessment and matching for allogeneic stem cell transplantation: consensus opinion of the Blood and Marrow Transplant Clinical Trials Network (BMT CTN) Biol. Blood Marrow Transplant. 2015;21:4–7. doi: 10.1016/j.bbmt.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hill J.A., Li D., Hay K.A., Green M.L., Cherian S., Chen X., Riddell S.R., Maloney D.G., Boeckh M., Turtle C.J. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood. 2018;131:121–130. doi: 10.1182/blood-2017-07-793760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mo F., Watanabe N., McKenna M.K., Hicks M.J., Srinivasan M., Gomes-Silva D., Atilla E., Smith T., Ataca Atilla P., Ma R. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coscoy L., Ganem D. A viral protein that selectively downregulates ICAM-1 and B7-2 and modulates T cell costimulation. J. Clin. Invest. 2001;107:1599–1606. doi: 10.1172/JCI12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lehner P.J., Hoer S., Dodd R., Duncan L.M. Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol. Rev. 2005;207:112–125. doi: 10.1111/j.0105-2896.2005.00314.x. [DOI] [PubMed] [Google Scholar]
- 46.Clackson T., Yang W., Rozamus L.W., Hatada M., Amara J.F., Rollins C.T., Stevenson L.F., Magari S.R., Wood S.A., Courage N.L. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc. Natl. Acad. Sci. USA. 1998;95:10437–10442. doi: 10.1073/pnas.95.18.10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narayanan P., Lapteva N., Seethammagari M., Levitt J.M., Slawin K.M., Spencer D.M. A composite MyD88/CD40 switch synergistically activates mouse and human dendritic cells for enhanced antitumor efficacy. J. Clin. Invest. 2011;121:1524–1534. doi: 10.1172/JCI44327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spencer D.M., Wandless T.J., Schreiber S.L., Crabtree G.R. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 49.Collinson-Pautz M.R., Chang W.C., Lu A., Khalil M., Crisostomo J.W., Lin P.Y., Mahendravada A., Shinners N.P., Brandt M.E., Zhang M. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia. 2019;33:2195–2207. doi: 10.1038/s41375-019-0417-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X., Moghimi B., Zolotukhin I., Morel L.M., Cao O., Herzog R.W. Immune tolerance induction to factor IX through B cell gene transfer: TLR9 signaling delineates between tolerogenic and immunogenic B cells. Mol. Ther. 2014;22:1139–1150. doi: 10.1038/mt.2014.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foster A.E., Marangolo M., Sartor M.M., Alexander S.I., Hu M., Bradstock K.F., Gottlieb D.J. Human CD62L- memory T cells are less responsive to alloantigen stimulation than CD62L+ naive T cells: potential for adoptive immunotherapy and allodepletion. Blood. 2004;104:2403–2409. doi: 10.1182/blood-2003-12-4431. [DOI] [PubMed] [Google Scholar]
- 52.Micklethwaite K., Hansen A., Foster A., Snape E., Antonenas V., Sartor M., Shaw P., Bradstock K., Gottlieb D. Ex vivo expansion and prophylactic infusion of CMV-pp65 peptide-specific cytotoxic T-lymphocytes following allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2007;13:707–714. doi: 10.1016/j.bbmt.2007.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.