

Abstract
Deregulation of the mRNA translational process has been observed during tumorigenesis. However, recent findings have shown that deregulation of translation also contributes specifically to cancer cell spread. During metastasis, cancer cells undergo changes in cellular state, permitting the acquisition of features necessary for cell survival, dissemination, and outgrowth. In addition, metastatic cells respond to external cues, allowing for their persistence under significant cellular and microenvironmental stresses. Recent work has revealed the importance of mRNA translation to these dynamic changes, including regulation of cell states through epithelial-to-mesenchymal transition (EMT) and tumor dormancy and as a response to external stresses such as hypoxia and immune surveillance. In this Review, we focus on examples of altered translation underlying these phenotypic changes and responses to external cues and explore how they contribute to metastatic progression. We also highlight the therapeutic opportunities presented by aberrant mRNA translation, suggesting novel ways to target metastatic tumor cells.
Introduction
mRNA translation, one of the most energetically demanding processes in a cell, is tightly regulated to match cellular demands. Like gene transcription, mRNA translation can be regulated globally or at the level of individual transcripts. As such, it is frequently deregulated during malignant transformation and metastatic progression. Tumor cells may hijack protein production to preferentially translate oncogenic transcripts and to dynamically regulate overall protein synthesis to support neoplastic growth, tumor dormancy and metastasis depending on internal and external cues. Translational dysregulation in cancer cells has been appreciated since 1896, when enlarged nucleoli were identified on histologic sections of cancer, representing aberrant ribosomal biogenesis (1). Since then, substantial evidence has highlighted the importance of translational dysregulation during tumorigenesis, including the activation of translation-promoting mTOR and MAPK signaling pathways and alterations in the eIF translation initiation factors (reviewed in detail in (2–4)). More recently, we have learned that altered translation contributes not only to tumor initiation and growth, but also to numerous steps involved in tumor cell dissemination and metastasis.
Metastasis represents the most lethal stage of cancer progression, and once cancer has metastasized, the goal of most clinical therapies is to slow further cancer growth and spread, rather than achieve cure. Hematogenous spread of cancer, the most common metastatic pathway, is a complex process: tumor cells invade into surrounding tissue, intravasate into blood vessels, survive under the physical and oxidative stress of the blood circulation, extravasate into distal tissues, and become competent to proliferate at these distant sites—all while overcoming internal apoptotic signals and avoiding immune system surveillance (5). Emerging evidence has pointed to roles for translational dysregulation at multiple stages of this process. Unlike individual genetic changes, global mRNA translation can be regulated over short timescales, and changes can have broad impact as cells adapt to diverse microenvironments. Such translational changes can modulate total protein synthesis, as well as regulate levels of specific proteins via preferential mRNA translation (3). In this Review, we focus on global and transcript-specific translational changes that modulate tumor cell states (e.g., epithelial-to-mesenchymal transition; tumor cell dormancy) and the response to microenvironmental cues (e.g., angiogenesis; immune suppression), and we will describe how these translational changes may promote progression through the metastatic cascade. We also highlight therapeutic vulnerabilities created by aberrant translation in metastatic tumors, suggesting opportunities for clinical intervention.
Translational dysregulation
mRNA translation is a highly coordinated process that involves multiple steps that can be dysregulated during cancer (Figure 1) (reviewed in (6)). A critical step in the regulation of translation occurs during translation initiation, as the 80S ribosome, comprising the 60S large and 40S small ribosomal subunits, is assembled on an mRNA transcript. Translation initiation can be broadly divided into two separate mechanisms: cap-dependent and cap-independent translation.
Figure 1:
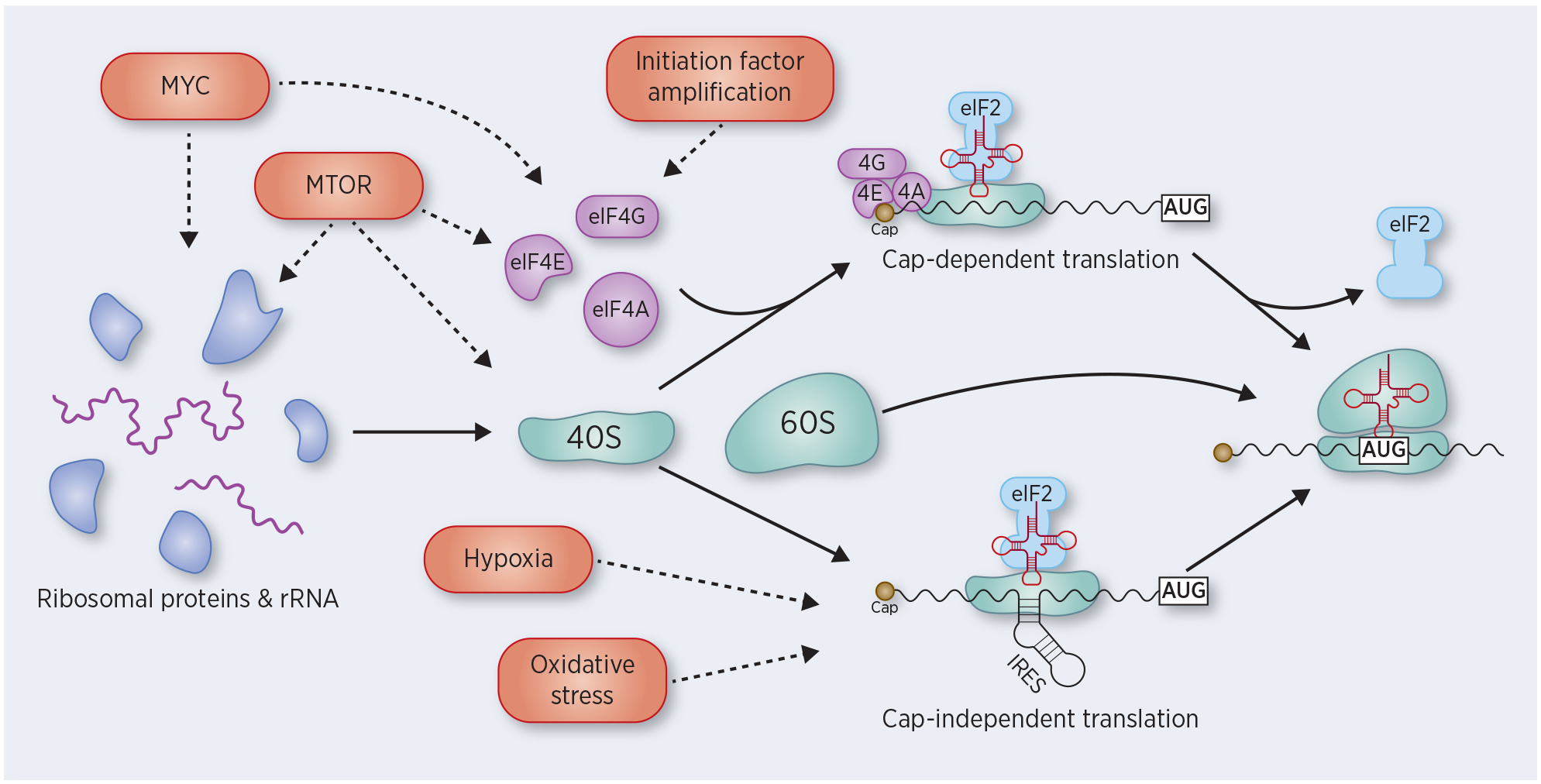
Dysregulation of translation initiation during cancer. Ribosome biogenesis includes the coordinated assembly of ribosomal proteins and rRNA to form the 40S and 60S ribosomal subunits. This process is, in part, regulated by Myc and mTOR, which are frequently aberrantly activated in cancer. Translation initiation occurs by two distinct mechanisms: cap-dependent translation and cap-independent translation. In cap-dependent translation, the initiation factors eIF4A, eIF4E, and eIF4G bind to the 5’ cap of mRNA transcripts, with subsequent recruitment of the 40S subunit. eIF4E and eIF4G are frequently amplified in cancer, and other oncogenes, including Myc and mTOR, also activate translation initiation. In cap-independent translation, an internal ribosome entry site (IRES) recruits the 40S ribosome without the initiation factors. This method of translational initiation is prevalent in situations of cellular stress, including hypoxia and oxidative stress. In both mechanisms of translational initiation, the 40S subunits scans the transcript for an AUG start site, at which point eIF2 is released, the 60S ribosome is recruited to form the full 80S ribosome, and translation elongation begins.
Cap-dependent translation is the predominant mechanism for cellular protein synthesis and is mediated by the heterotrimeric eIF4F complex (eIF4A, eIF4E, eIF4G) which binds the 7-methylguanylate cap at the 5’ end of an mRNA transcript. The eIF4F complex recruits the 40S ribosomal subunit to the 5’ end of the transcript, and the 40S ribosome then scans the transcript until it encounters an AUG start site. At the start site, eIF2 signals for recruitment of the 60S ribosomal subunit, resulting in the formation of the 80S ribosome and progression to translation elongation with formation of the nascent polypeptide chain. Assembly of the eIF4F complex is the rate-limiting step for cap-dependent translation, and members of the eIF4F complex are frequently upregulated in cancer (7), promoting nascent protein synthesis. Several oncogenic signaling pathways, including mTOR and MAPK signaling, enhance cap-dependent translation (8).
Cap-independent translation, on the other hand, is not mediated by the eIF4F complex, but instead is achieved through binding of the ribosome to a subset of mRNA transcripts containing an internal ribosome entry site (IRES). This mechanism of translation is particularly relevant under stress conditions, when global, cap-dependent protein synthesis is inhibited. Given the variety of internal and external stresses to which they are exposed, neoplastic cells often rely on IRES-mediated translation under stress conditions (9). Notably, multiple genes that promote oncogenesis are regulated by IRES-mediated translation including MYC, BCL2, FGF2, HIF1A and VEGFA (9–11).
Although translation initiation serves as the main regulatory step in translational control, dysregulation of ribosome biogenesis or of translation elongation also affects global protein synthesis. Several oncogenes—most notably Myc—promote ribosome biogenesis, leading to increased global translation (12, 13). Likewise, translation elongation can be dysregulated by aberrant Akt/mTOR signaling, leading to activation or suppression of the elongation factor eEF2 (14), or by oncogene-mediated dysregulation of tRNA levels (15).
Translational control of cell states
Epithelial-to-Mesenchymal Transition:
Epithelial-to-mesenchymal transition (EMT) is a physiological embryonic cell fate change critical to normal organogenesis. Oncogenic EMT parallels this developmental process and is associated with increased tumor cell invasion and implicated as an early step in metastasis (16). During EMT, epithelial tumor cells lose their epithelial features and acquire mesenchymal features, including reduced cell-cell adhesion, loss of cellular polarity and increased motility. In the reverse process of mesenchymal-to-epithelial transition (MET), epithelial features are reacquired. In reality, these dynamic changes in neoplastic cells present a spectrum of intermediate metastable states rather than distinct and permanent epithelial or mesenchymal phenotypes, and the ability to move between these states is known as epithelial plasticity. Mesenchymal states favor early invasion, migration and resistance of circulating tumor cells (CTCs) to anoikis in the bloodstream, while epithelial states enhance proliferation and tumor outgrowth at a distant metastatic site. While transcriptional regulation of EMT and MET has been reviewed elsewhere (16, 17), translational changes that drive epithelial plasticity are also noteworthy.
TGF-β is a classic inducer of EMT via regulation of the canonical transcriptional inducers SNAIL and TWIST (18). At the translational level, TGF-β regulates mRNA translation of DAB2 and FAM3C (also known as ILE1), transcripts necessary for induction of EMT, through a cis-regulatory element termed the TGF-β-activated translation (BAT) element (19). In the unstimulated state, the BAT is bound by a complex of hnRNP E1 and eEF1A1 which mediates translational silencing by blocking ribosomal progression. Upon treatment with TGF-β, activated Akt2 phosphorylates hnRNP E1, releasing it from the BAT element and allowing progression of the ribosome along the mRNA transcript. Knockdown of the BAT silencing protein hnRNP E1 induces EMT and metastasis in a mouse model (19). TGF-β also upregulates CELF1, an mRNA-binding protein which binds to and promotes the translation of EMT-related mRNAs, including the EMT master regulator SNAI1, likely via binding of a GU-rich 3’-UTR element and recruitment of the eukaryotic initiation factor eIF4E (20). In addition to directly promoting translation of EMT transcripts, TGF-β also activates mTOR, a master regulator of cell growth, cell size and protein synthesis (21, 22), leading to increased cell motility and invasion across many different tumor types (21–23).
mTOR activation of translation has been shown to be required for TGF-β-induced EMT by promoting cell migration and motility, leading to increased metastasis (21, 23). Furthermore, mTOR activation maintains cells in the mesenchymal state following TGF-β signaling, implicating additional regulatory mechanisms for the mTOR pathway during EMT (24). Activation of mTOR signaling alone is sufficient to induce EMT, motility and metastasis in some models (23). The effects of mTOR on EMT and metastasis have been attributed, at least in part, to mTOR-mediated activation of the translation initiation factor eIF4E and S6 kinase, subsequently increasing mRNA translation (25). In addition to activation of translation initiation, mTOR signaling also promotes the translation of specific mRNA transcripts, including those with a 5’ terminal oligopyrimidine tract (TOP)-motif, characterized by a cytosine and 7–14 pyrimidines following the 7-methylguanylate cap (26). Transcripts with 5’ TOP motifs encode translational machinery, including ribosomal proteins and translational initiation and elongation factors, as well as select additional genes such as VIM, a key mesenchymal marker associated with cell invasion (27, 28). Ribosome profiling has confirmed that mTOR signaling leads to increased translation of selected EMT transcripts, including VIM and YBX1, an RNA binding protein and key regulator of EMT; in line with these findings, the mTOR inhibitor sapanisertib (also referred to as INK128) has been shown to inhibit EMT and subsequent metastasis in mouse models of prostate cancer (Figure 2A) (4).
Figure 2:
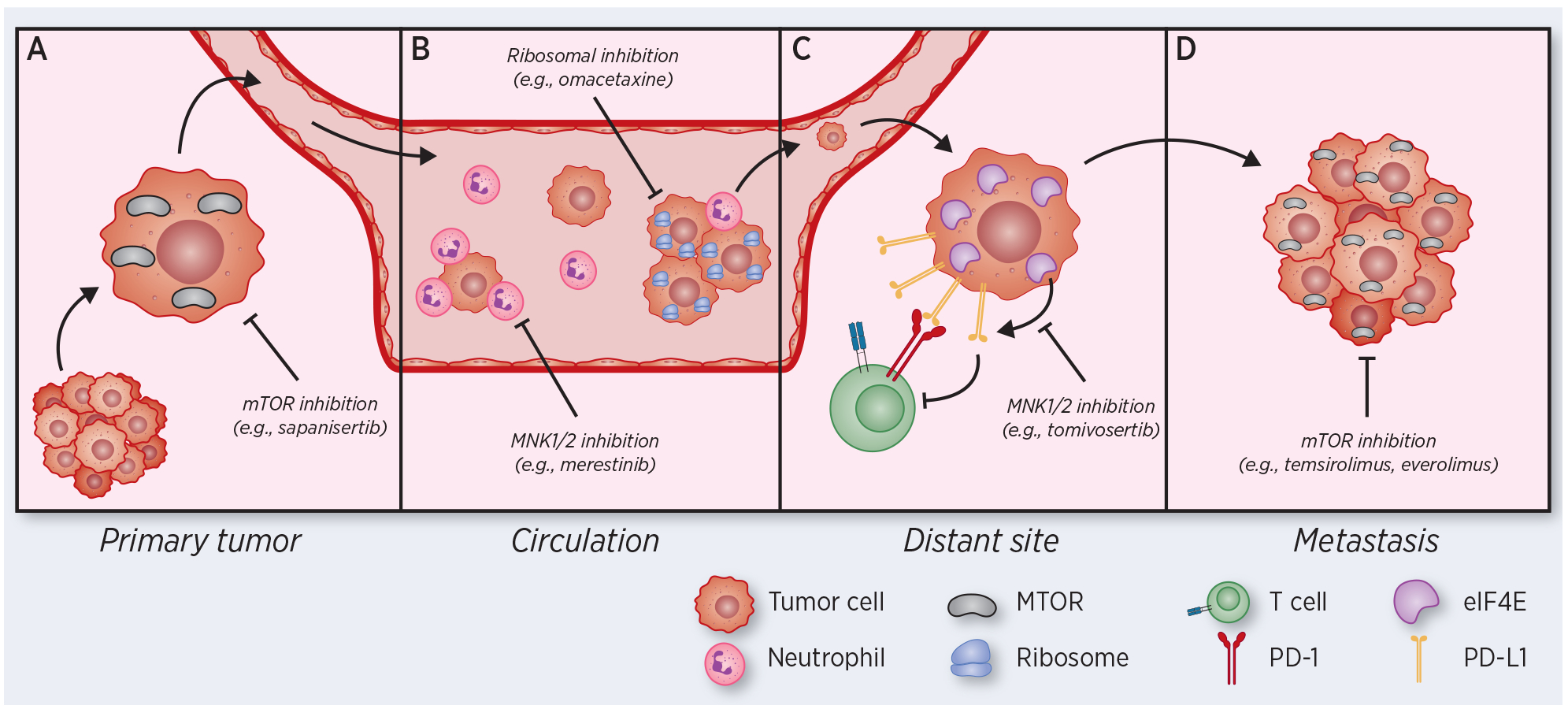
Therapeutic vulnerabilities based on translational aberrations in tumor cells progressing through the metastatic cascade. (A) Primary tumor cells activate mTOR and other translational pathways to induce epithelial plasticity, promoting escape from primary tumors and intravasation into the circulation. (B) Circulating tumor cells (CTCs) and CTC clusters with increased metastatic capacity exhibit upregulation of translational machinery. Neutrophils dependent on translation interact with CTCs to facilitate CTC extravasation. (C) Disseminated tumor cells (DTCs) translationally regulate surface expression of the immunosuppressive ligand PD-L1, promoting immune evasion. (D) Growing metastases activate mTOR and proliferation pathways, and mTOR inhibitors are approved for use in metastatic breast, renal and neuroendocrine cancers. In each panel, therapies targeting protein translation with efficacy in metastasis models or in metastatic cancer patients are highlighted.
Translational regulation during EMT extends beyond the early stages of tumorigenesis, with epithelial plasticity remaining important as CTCs travel in the bloodstream and establish distant metastases (29). Single cell RNA-sequencing of freshly isolated breast cancer patient CTCs revealed that increased expression of structural ribosomal proteins correlates with expression of epithelial markers, a higher proliferative index and poor clinical outcomes (30). Consistent with a causative role for increased ribosomal proteins in CTC-mediated metastasis, an in vivo genome-wide CRISPR activation screen identified increased ribosomal protein expression—particularly RPL15—as pro-metastatic factors in patient-derived breast CTC lines intravascularly inoculated into mice (30). In these cells, ribosomal protein expression is strongly correlated with expression of epithelial markers, and both of these confer a poor clinical outcome in women with breast cancer, suggesting that epithelial plasticity may underlie translational patterns modulating tumorigenesis and metastasis. The relationship between EMT and expression of translational machinery was confirmed by TGF-β treatment of cells, which results in global suppression of translation, with preferential reduction in the translation of ribosomal proteins. The epithelial state may thus drive a translational profile promoting CTC metastatic potential, in part by modulating ribosomal protein expression. These observations also raise the possibility that changes in expression of specific ribosomal proteins may determine global and transcript-specific translational output. Upregulation of numerous ribosomal proteins has been observed in cancer (31), and in some cases, these ribosomal proteins are associated with transformation or disease progression, including RPLP1 (32), RPL36A (33) and RPL34 (34). This heterogeneity of ribosomal protein expression in cancer raises the intriguing possibility of specialized ribosomes, with an alternative composition of ribosomal proteins, that direct translation of specific oncogenic transcripts; such specialized ribosomes have been reported in mouse embryonic stem cells (35). Further studies are needed to determine the role of specialized ribosomes in translational dysregulation in cancer.
Whereas individual tumor cells in circulation have a low probability of generating a macrometastatic lesion, multicellular clusters of CTCs in the bloodstream have greatly enhanced metastatic potential versus single CTCs (36–38). Consistent with the connection between high ribosome content and the epithelial phenotype, patient-derived CTC clusters have increased expression of ribosomal proteins and epithelial markers, as compared to single CTCs (36). Taken altogether, the heterogeneous expression of translational machinery and epithelial plasticity may be critical determinants of metastatic potential in CTCs (Figure 2B).
Tumor Dormancy and the Integrated Stress Response
After exiting the bloodstream, CTCs, now referred to as disseminated tumor cells (DTCs), may remain in a dormant state for prolonged periods of time, undetected by current screening technologies, before they initiate proliferation and give rise to metastatic lesions (39–41). A growing body of evidence suggests that this dormant state is associated with global suppression of translation, mediated by the integrated stress response (ISR). The ISR is activated upon exposure to a wide range of cellular stresses, including intrinsic factors such as accumulation of unfolded proteins (activating the unfolded protein response [UPR]) and extrinsic factors such as hypoxia, reactive oxygen species, amino acid deprivation, heme deprivation and loss of cell matrix attachments (42). ISR activation leads to phosphorylation of eIF2α, which prevents translation initiation complex formation resulting in globally reduced mRNA translation (43, 44). This global reduction in translation appears to prevent ATP depletion, accumulation of oxidative stress, and ultimately apoptosis, thereby promoting the survival of dormant DTCs (45, 46). However, despite globally reduced translation following ISR activation and eIF2α phosphorylation, certain transcripts continue to be translated through a variety of mechanisms, including IRES-mediated cap-independent translation, as described above (47, 48). Other transcripts, such as those encoding the stress response factor ATF4 contain upstream reading frames (uORFs), motifs which normally inhibit translation, but are bypassed in the setting of phosphorylated eIF2α (47). Selective translation of these transcripts leads to activation of pathways that further support stress tolerance and survival of dormant cells. For instance, ATF4 promotes autophagy, which supports the survival of DTCs under low nutrient conditions and high oxidative stress (46). This selective translation of specific transcripts critical to tumor cell survival in the setting of repressed global translation appears to be a dominant feature of tumor cell dormancy. In contrast, escape from dormancy and outgrowth of metastatic lesions likely requires reactivation of the translational machinery to match the cellular demands of rapid growth and proliferation.
Translational deregulation by tumor microenvironmental factors
In addition to promoting dynamic shifts in tumor cell states, global alterations in mRNA translation occur in response to stimuli from the tumor microenvironment. Such translational changes are evident at multiple stages of metastasis and can lead to microenvironmental remodeling in response to cues such as hypoxia and immune cell interactions.
Hypoxia and angiogenesis:
A key requirement for metastasizing tumor cells to survive is access to oxygen and nutrients via local angiogenesis and vascular remodeling. This is often mediated via secretion of the VEGF family of growth factors, which promote neovascularization (49). Hypoxia is a central driver of angiogenesis leading to transcriptional changes mediated by the HIF transcription factors, as well as a stimulus for the ISR, as discussed above (50). The hypoxic response depends on both transcriptional and translational changes in VEGF expression, with the mechanism of translational regulation dependent on an IRES in the VEGF transcript (51). In the setting of hypoxia, the ISR is activated, leading to global reductions in cap-dependent translation; however, IRES-mediated, cap-independent translation proceeds, and VEGF protein is produced despite reduced overall translation. Notably, the VEGF family member VEGF-C does not contain a HIF-binding site in its promoter, suggesting translational rather than transcriptional control in response to hypoxia (52). Indeed, IRES-mediated translation appears to be the primary mode of VEGF-C regulation in hypoxia, contributing to lymphatic metastasis (53), and similarly, IRES-mediated control of VEGF-D translation in metastasis has also been reported (54). Such translational control allows for efficient production of VEGF family members under restrictive hypoxic conditions that reduce overall protein production, enabling the initiation of angiogenic signaling; for VEGF-C and VEGF-D, this translational mechanism may also promote metastatic spread via the induction of lymphangiogenesis (55). Translation of additional angiogenic growth factors, including FGF1 and FGF2, is upregulated under hypoxic conditions through their IRES domains, highlighting the conservation of this mechanism of translational regulation across multiple angiogenic factors (56).
In addition to cap-independent translation initiation, hypoxic stress also induces translation of hypoxia-induced transcripts—including those encoding EGFR, HER2, PDGFRα, and CDH22—via assembly of alternative cap-dependent translational complexes. During hypoxia, HIF-2α binds to RBM4 at the 3’ end of hypoxia-induced transcripts (57). This complex then binds eIF4E2, eIF4G3 and eIF4A at the 5’ cap of the transcript, together forming an alternative cap-dependent complex and initiating translation. Notably, given its role in translation of hypoxia-induced transcripts, eIF4E2 may represent a translational target with a wide therapeutic window to selectively inhibit metastatic cell growth (58). Together, these hypoxia-specific mechanisms of translational control illustrate the diversity of mechanisms for regulating translation under stress conditions.
Tumor-Immune Cell Interactions:
Immune evasion is critical to tumor initiation, and recent work has highlighted translational dysregulation of immune mediators as a key mechanism by which tumor cells escape immune suppression. Ribosome profiling of liver tumors derived from a transgenic mouse model of combined expression of mutant KRAS and MYC overexpression (KRASG12D/MYC+) in a liver specific manner demonstrates selectively increased translation of the immunosuppressive ligand PD-L1 as compared to tumors expressing only KRASG12D, leading to immune evasion and increased metastatic potential (59). In these mice, increased PD-L1 translation is dependent on MYC-associated eIF2α phosphorylation and subsequent ribosomal bypass of suppressive uORFs within the PD-L1 5’-UTR. Interestingly, treatment with tomivosertib—a small molecule inhibitor of MNK1/2 which prevents phosphorylation and activation of the translation initiation factor eIF4E—selectively blocks the translational upregulation of PD-L1 in this liver cancer model, thereby sensitizing tumor cells to immune suppression and limiting metastasis (Figure 2C). Other studies have recently shown that, in the setting of global translation suppression, increased PD-L1 translation may occur as a consequence of ISR activation mediated via uORFs in the PD-L1 transcript and dependent on the alternative translation initiation factor eIF5B in the setting of eIF2α phosphorylation (60). Beyond direct translational regulation of PD-L1, the eIF4F complex selectively promotes the translation of STAT1, a PD-L1 transcriptional activator in melanoma cell lines. Treatment with the eIF4A inhibitor silvestrol leads to reduced STAT1 translation and PD-L1 transcription with subsequent immune sensitization, suggesting that targeting mRNA translation may limit tumor growth and metastasis by modulating immune surveillance pathways in multiple tumor types (61).
Translational control in circulating immune cells also appears to promote metastasis. Recent work has suggested that the metastatic potential of CTCs is modulated by circulating neutrophil translational capacity. Neutrophils are frequently found in the immune infiltrate of tumors and can be either tumor promoting or tumor suppressive depending on the signals derived from the tumor cells and the microenvironment (reviewed in (62)). During metastasis, neutrophils have been implicated in priming of the premetastatic niche (63) and retention of CTCs in target organs (64). Increased neutrophil lifespan and survival are postulated to mediate these pro-metastatic properties, and MNK1/2 activation delays neutrophil apoptosis (65). Consistent with these observations, CTC-derived lung metastases are decreased in mice with neutrophils expressing mutant, inactivated eIF4E, as compared to mice with wildtype neutrophils, presumably due to increased sensitivity of the neutrophils to apoptotic stimuli (66). Neutrophil translational activity is necessary for CTC extravasation, and inhibition of eIF4E activation using the translation inhibitor merestinib reduced neutrophil counts and metastasis formation (Figure 2B). In addition to these roles, neutrophils are the predominant white blood cells associated with CTC clusters, and the presence of neutrophils in these clusters activates a proliferative program and is correlated with reduced progression-free survival (67). Together, these findings illustrate the dynamic relationship between tumor cells and immune cells and highlight an additional role of translational regulation in shaping tumor and immune cell interactions during metastasis.
Therapeutic vulnerabilities presented by translational deregulation in cancer
The significant role of mRNA translation in the development and progression of cancer raises the intriguing possibility of targeting translation therapeutically. Given the universal role of mRNA translation, to be effective, such an approach must rely on differential oncogenic dependency providing a sufficient therapeutic window to target tumor cells progressing through critical points in the metastatic cascade, while minimizing toxicity. Of note, the translational inhibitor omacetaxine, which directly inhibits the ribosome to halt translation elongation, is FDA-approved for the treatment of chronic myelogenous leukemia (CML) (68). The effectiveness of omacetaxine in CML is thought to be related to rapid turnover of the CML-defining Bcr-Abl oncoprotein and subsequent dependency on nascent protein production (69). While this treatment is not widely used because of the efficacy of tyrosine kinase inhibitors in CML, it validates the concept that translational inhibitors are clinically-viable drugs and suggests that therapeutic vulnerabilities related to translation may be exploited. Indeed, in recent work, we have shown that ribosomal inhibition via omacetaxine, in combination with cell cycle inhibition via the CDK4/6 inhibitor palbociclib, is able to selectively inhibit the growth of translationally-activated, metastatic-competent CTCs (30).
In addition to direct targeting of the ribosome, inhibition of mTOR or other translational regulators may lead to translational inhibition with selective efficacy in malignant cells. Indeed, the first-generation, allosteric mTOR inhibitors everolimus and temsirolimus are FDA-approved for several cancer types, including metastatic breast cancer, renal cell carcinoma and neuroendocrine tumors (70) (Figure 2D). Although they are commonly used in clinical practice, these first-generation mTOR inhibitors have limited efficacy due to their partial inhibition of mTOR complex activity and to acquisition of somatic mutations increasing mTOR activity that lead to drug resistance. The development of second-generation mTOR inhibitors that bind the ATP-site (e.g., sapanisertib, vistusertib, onatasertib) may more strongly inhibit mTOR-dependent translational upregulation due to more potent inhibition of 4E-BP1 phosphorylation, as well as an additional inhibitory effect on the mTORC2 complex (4, 71, 72), and these are now in clinical trials for various malignancies. Similarly, dual PI3K/mTOR inhibitors which bind the ATP-sites of both PI3K and mTOR (e.g., voxtalixib, gedatolisib, bimiralisib, paxalisib, samotolisib, apitolisib) are also in clinical trials and may demonstrate additional translational inhibition versus mTOR-only inhibitors (73). Finally, third-generation mTOR inhibitors are under development, combining the inhibitor modalities of the first- and second-generation inhibitors by creating a bivalent mTOR inhibitor that includes a rapamycin-like domain connected to an ATP-site inhibitor domain by a long linker chain, and these may further enhance the clinical application of mTOR inhibitors (74).
The eIF4F translation initiation factors (including eIF4A, eIF4E and eIF4G) are particularly attractive targets for inhibition in cancer, since alterations in these proteins are reported in various tumors, and they interact with multiple druggable modulators. While progress on direct eIF4E inhibitors has been limited, several inhibitors have been described targeting the MNK1 and MNK2 kinases, which phosphorylate and activate eIF4E. One such MNK1/2 inhibitor, cercosporamide, attenuates pulmonary metastases in an experimental melanoma mouse model (75). Another MNK1/2 inhibitor, tomivosertib (eFT508), is particularly potent and selective and is now in clinical trials in prostate and breast cancer, as monotherapy and in combination with PD-1/PD-L1 inhibitors (NCT04261218, NCT03690141, NCT03616834) (76). Multiple inhibitors of the RNA helicase eIF4A have been reported, including hippuristanol, pateamine A and silvestrol, as well as numerous derivatives, and these compounds have demonstrated efficacy in preclinical primary and metastatic tumor models (77). Interestingly, eIF4A inhibitors have also demonstrated efficacy against MYC-induced blood cancers; MYC regulates the transcription of numerous elements of the translational apparatus including ribosomal proteins, rRNA and translation initiation and elongation factors (78), and oncogenic MYC signaling may lead to increased dependence on translational capacity (61, 79, 80). Zotatifin (eFT226), an eIF4A inhibitor with improved drug-like properties, has recently been developed and is presently in early clinical trials for advanced solid malignancies (NCT04092673) (81).
An additional avenue for affecting global translational output is modulation of eIF2α activity. The eIF2α dephosphorylation inhibitor salubrinal inhibits the protein phosphatase 1 (PP1) complex, maintaining eIF2α phosphorylation and subsequent stress response signaling (82). Conversely, inhibitors of kinases that phosphorylate eIF2α, such as the protein-like ER kinase (PERK), have also been identified and decrease metastasis in an aggressive breast cancer mouse model (83). One such PERK inhibitor, ISRIB (Integrated Stress Response InhiBitor), has been shown to inhibit breast cancer cell plasticity, especially in cells treated with the mTOR inhibitor sapanisertib, highlighting the potential of multimodal inhibition of translational plasticity (84). While no modulators of eIF2α activity are presently in clinical trials, we anticipate that, with further development, these compounds may serve as a promising approach to target metastatic spread.
Beyond inhibition of the above targets, numerous additional approaches for modulating mRNA translation are in preclinical or clinical development, including inhibitors of rRNA transcription, targeted degraders of translational regulators, and antisense oligonucleotides targeting specific transcripts (85).
Concluding remarks
Aberrant regulation of mRNA translation is a defining feature of neoplastic cells which, along with their genetic and epigenetic marks, may define many of their key functional characteristics. Cancer-associated alterations in translation are context dependent and dynamic over the course of cancer progression, often associated with reversible epithelial plasticity or cellular stress signaling. In this way, regulation of translation may allow neoplastic cells to achieve a survival advantage at different stages within the metastatic cascade or when faced with adverse environmental conditions. The hijacking by cancer cells of broad cellular regulatory mechanisms relevant to translation is consistent with their acquisition of genetic and epigenetic alterations during tumor progression, although the precise causes of these cancer-associated changes remain poorly understood. Here, we have highlighted recent insights into mechanisms of translational dysregulation during the metastatic cascade, but we note that alterations in the levels and activity of additional translational regulators, including tRNA modifications, RNA binding proteins, and miRNAs, likely play critical roles as well. Importantly, the identification of metastasis-associated translational changes suggests opportunities for therapeutic intervention, particularly at specific points within the metastatic cascade where increased dependence on altered mRNA translation may provide a therapeutic window. Further study is needed to better characterize and ultimately target such novel translational vulnerabilities, with the goal of suppressing the metastatic spread of cancer.
Acknowledgements
This work was supported by 2R01CA129933, the Breast Cancer Research Foundation, the Howard Hughes Medical Institute, and the National Foundation for Cancer Research (D.A.H.); ESSCO Breast Cancer Research (S.M.); NIH grant T32GM007753 and NIH grant 1F30CA232407-01 (R.Y.E.); and American Cancer Society grant 132140-PF-18-127-01-CSM (D.S.M). R.Y.E. has received consulting fees from Nextech Invest and nference Inc., which are not related to this work. Figures created with BioRender.com.
Footnotes
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Pianese G Beitrag zur histologie und aetiologie der carcinoma. histologische und experimentelle untersuchungen. Beitr Pathol Anat Allgem Pathol. 1896;142:1–193. [Google Scholar]
- 2.Robichaud N, Sonenberg N. Translational control and the cancer cell response to stress. Curr Opin Cell Biol. 2017;45:102–9. Epub 2017/06/06. doi: 10.1016/j.ceb.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Robichaud N, Sonenberg N, Ruggero D, Schneider RJ. Translational Control in Cancer. Cold Spring Harb Perspect Biol. 2018. Epub 2018/07/01. doi: 10.1101/cshperspect.a032896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. Epub 2012/03/01. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–92. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10(4):254–66. Epub 2010/03/25. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 7.Malka-Mahieu H, Newman M, Desaubry L, Robert C, Vagner S. Molecular Pathways: The eIF4F Translation Initiation Complex-New Opportunities for Cancer Treatment. Clin Cancer Res. 2017;23(1):21–5. Epub 2016/11/01. doi: 10.1158/1078-0432.CCR-14-2362. [DOI] [PubMed] [Google Scholar]
- 8.Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the eIF4F translation initiation complex: a critical nexus for cancer development. Cancer Res. 2015;75(2):250–63. Epub 2015/01/17. doi: 10.1158/0008-5472.CAN-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walters B, Thompson SR. Cap-Independent Translational Control of Carcinogenesis. Front Oncol. 2016;6:128 Epub 2016/06/03. doi: 10.3389/fonc.2016.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willimott S, Wagner SD. Post-transcriptional and post-translational regulation of Bcl2. Biochem Soc Trans. 2010;38(6):1571–5. Epub 2010/12/02. doi: 10.1042/BST0381571. [DOI] [PubMed] [Google Scholar]
- 11.Philippe C, Dubrac A, Quelen C, Desquesnes A, Van Den Berghe L, Segura C, et al. PERK mediates the IRES-dependent translational activation of mRNAs encoding angiogenic growth factors after ischemic stress. Sci Signal. 2016;9(426):ra44 Epub 2016/05/05. doi: 10.1126/scisignal.aaf2753. [DOI] [PubMed] [Google Scholar]
- 12.Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013;3(8). Epub 2013/08/03. doi: 10.1101/cshperspect.a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One. 2011;6(10):e26057 Epub 2011/11/01. doi: 10.1371/journal.pone.0026057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Browne GJ, Proud CG. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol Cell Biol. 2004;24(7):2986–97. Epub 2004/03/17. doi: 10.1128/mcb.24.7.2986-2997.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos M, Fidalgo A, Varanda AS, Oliveira C, Santos MAS. tRNA Deregulation and Its Consequences in Cancer. Trends Mol Med. 2019;25(10):853–65. Epub 2019/06/30. doi: 10.1016/j.molmed.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 16.Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166(1):21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 17.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Drabsch Y, ten Dijke P. TGF-beta signaling in breast cancer cell invasion and bone metastasis. J Mammary Gland Biol Neoplasia. 2011;16(2):97–108. doi: 10.1007/s10911-011-9217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussey GS, Chaudhury A, Dawson AE, Lindner DJ, Knudsen CR, Wilce MC, et al. Identification of an mRNP complex regulating tumorigenesis at the translational elongation step. Mol Cell. 2011;41(4):419–31. Epub 2011/02/19. doi: 10.1016/j.molcel.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhury A, Cheema S, Fachini JM, Kongchan N, Lu G, Simon LM, et al. CELF1 is a central node in post-transcriptional regulatory programmes underlying EMT. Nat Commun. 2016;7:13362 Epub 2016/11/22. doi: 10.1038/ncomms13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamouille S, Connolly E, Smyth JW, Akhurst RJ, Derynck R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J Cell Sci. 2012;125(Pt 5):1259–73. Epub 2012/03/09. doi: 10.1242/jcs.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol. 2007;178(3):437–51. Epub 2007/07/25. doi: 10.1083/jcb.200611146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulhati P, Bowen KA, Liu J, Stevens PD, Rychahou PG, Chen M, et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer Res. 2011;71(9):3246–56. Epub 2011/03/25. doi: 10.1158/0008-5472.CAN-10-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, et al. Chronic TGF-beta exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. 2019;12(570). Epub 2019/02/28. doi: 10.1126/scisignal.aau8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25(53):7029–40. Epub 2006/05/23. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- 26.Pichon X, Wilson LA, Stoneley M, Bastide A, King HA, Somers J, et al. RNA binding protein/RNA element interactions and the control of translation. Curr Protein Pept Sci. 2012;13(4):294–304. Epub 2012/06/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita R, Suzuki Y, Takeuchi N, Wakaguri H, Ueda T, Sugano S, et al. Comprehensive detection of human terminal oligo-pyrimidine (TOP) genes and analysis of their characteristics. Nucleic Acids Res. 2008;36(11):3707–15. Epub 2008/05/16. doi: 10.1093/nar/gkn248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meyuhas O, Kahan T. The race to decipher the top secrets of TOP mRNAs. Biochim Biophys Acta. 2015;1849(7):801–11. Epub 2014/09/23. doi: 10.1016/j.bbagrm.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 29.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339(6119):580–4. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebright RY, Lee S, Wittner BS, Niederhoffer KL, Nicholson BT, Bardia A, et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science. 2020;367(6485):1468–73. Epub 2020/02/08. doi: 10.1126/science.aay0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Las Heras-Rubio A, Perucho L, Paciucci R, Vilardell J, ME LL. Ribosomal proteins as novel players in tumorigenesis. Cancer Metastasis Rev. 2014;33(1):115–41. Epub 2014/01/01. doi: 10.1007/s10555-013-9460-6. [DOI] [PubMed] [Google Scholar]
- 32.Artero-Castro A, Kondoh H, Fernandez-Marcos PJ, Serrano M, Ramon y Cajal S, Lleonart ME. Rplp1 bypasses replicative senescence and contributes to transformation. Exp Cell Res. 2009;315(8):1372–83. Epub 2009/02/24. doi: 10.1016/j.yexcr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Kim JH, You KR, Kim IH, Cho BH, Kim CY, Kim DG. Over-expression of the ribosomal protein L36a gene is associated with cellular proliferation in hepatocellular carcinoma. Hepatology. 2004;39(1):129–38. Epub 2004/01/31. doi: 10.1002/hep.20017. [DOI] [PubMed] [Google Scholar]
- 34.Yang S, Cui J, Yang Y, Liu Z, Yan H, Tang C, et al. Over-expressed RPL34 promotes malignant proliferation of non-small cell lung cancer cells. Gene. 2016;576(1 Pt 3):421–8. Epub 2015/11/04. doi: 10.1016/j.gene.2015.10.053. [DOI] [PubMed] [Google Scholar]
- 35.Shi Z, Fujii K, Kovary KM, Genuth NR, Rost HL, Teruel MN, et al. Heterogeneous Ribosomes Preferentially Translate Distinct Subpools of mRNAs Genome-wide. Mol Cell. 2017;67(1):71–83 e7. Epub 2017/06/20. doi: 10.1016/j.molcel.2017.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158(5):1110–22. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murlidhar V, Reddy RM, Fouladdel S, Zhao L, Ishikawa MK, Grabauskiene S, et al. Poor Prognosis Indicated by Venous Circulating Tumor Cell Clusters in Early-Stage Lung Cancers. Cancer Res. 2017;77(18):5194–206. Epub 2017/07/19. doi: 10.1158/0008-5472.CAN-16-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jansson S, Bendahl PO, Larsson AM, Aaltonen KE, Ryden L. Prognostic impact of circulating tumor cell apoptosis and clusters in serial blood samples from patients with metastatic breast cancer in a prospective observational cohort. BMC Cancer. 2016;16:433 Epub 2016/07/09. doi: 10.1186/s12885-016-2406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. Epub 2008/01/03. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 40.Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14(9):611–22. Epub 2014/08/15. doi: 10.1038/nrc3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schardt JA, Meyer M, Hartmann CH, Schubert F, Schmidt-Kittler O, Fuhrmann C, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8(3):227–39. Epub 2005/09/20. doi: 10.1016/j.ccr.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–95. Epub 2016/09/16. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–4. Epub 1999/02/04. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 44.Jennings MD, Kershaw CJ, Adomavicius T, Pavitt GD. Fail-safe control of translation initiation by dissociation of eIF2alpha phosphorylated ternary complexes. Elife. 2017;6 Epub 2017/03/21. doi: 10.7554/eLife.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–90. Epub 2013/04/30. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31(17):3616–29. Epub 2011/06/29. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Young SK, Wek RC. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J Biol Chem. 2016;291(33):16927–35. Epub 2016/07/01. doi: 10.1074/jbc.R116.733899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science. 2016;352(6292):1413–6. Epub 2016/06/18. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Apte RS, Chen DS, Ferrara N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell. 2019;176(6):1248–64. Epub 2019/03/09. doi: 10.1016/j.cell.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gardner LB, Corn PG. Hypoxic regulation of mRNA expression. Cell Cycle. 2008;7(13):1916–24. Epub 2008/07/08. doi: 10.4161/cc.7.13.6203. [DOI] [PubMed] [Google Scholar]
- 51.Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol. 1998;18(6):3112–9. Epub 1998/06/20. doi: 10.1128/mcb.18.6.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chilov D, Kukk E, Taira S, Jeltsch M, Kaukonen J, Palotie A, et al. Genomic organization of human and mouse genes for vascular endothelial growth factor C. J Biol Chem. 1997;272(40):25176–83. Epub 1997/10/06. doi: 10.1074/jbc.272.40.25176. [DOI] [PubMed] [Google Scholar]
- 53.Morfoisse F, Kuchnio A, Frainay C, Gomez-Brouchet A, Delisle MB, Marzi S, et al. Hypoxia induces VEGF-C expression in metastatic tumor cells via a HIF-1alpha-independent translation-mediated mechanism. Cell Rep. 2014;6(1):155–67. Epub 2014/01/07. doi: 10.1016/j.celrep.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 54.Morfoisse F, Tatin F, Hantelys F, Adoue A, Helfer AC, Cassant-Sourdy S, et al. Nucleolin Promotes Heat Shock-Associated Translation of VEGF-D to Promote Tumor Lymphangiogenesis. Cancer Res. 2016;76(15):4394–405. Epub 2016/06/10. doi: 10.1158/0008-5472.CAN-15-3140. [DOI] [PubMed] [Google Scholar]
- 55.Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014;124(3):922–8. Epub 2014/03/05. doi: 10.1172/JCI71606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conte C, Riant E, Toutain C, Pujol F, Arnal JF, Lenfant F, et al. FGF2 translationally induced by hypoxia is involved in negative and positive feedback loops with HIF-1alpha. PLoS One. 2008;3(8):e3078 Epub 2008/08/30. doi: 10.1371/journal.pone.0003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uniacke J, Holterman CE, Lachance G, Franovic A, Jacob MD, Fabian MR, et al. An oxygen-regulated switch in the protein synthesis machinery. Nature. 2012;486(7401):126–9. Epub 2012/06/09. doi: 10.1038/nature11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Melanson G, Timpano S, Uniacke J. The eIF4E2-Directed Hypoxic Cap-Dependent Translation Machinery Reveals Novel Therapeutic Potential for Cancer Treatment. Oxid Med Cell Longev. 2017;2017:6098107 Epub 2018/01/11. doi: 10.1155/2017/6098107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25(2):301–11. Epub 2019/01/16. doi: 10.1038/s41591-018-0321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suresh S, Chen B, Zhu J, Golden RJ, Lu C, Evers BM, et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nature Cancer. 2020;1(5):533–45. doi: 10.1038/s43018-020-0056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cerezo M, Guemiri R, Druillennec S, Girault I, Malka-Mahieu H, Shen S, et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat Med. 2018;24(12):1877–86. Epub 2018/10/31. doi: 10.1038/s41591-018-0217-1. [DOI] [PubMed] [Google Scholar]
- 62.Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16(7):431–46. Epub 2016/06/11. doi: 10.1038/nrc.2016.52. [DOI] [PubMed] [Google Scholar]
- 63.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8. Epub 2015/03/31. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huh SJ, Liang S, Sharma A, Dong C, Robertson GP. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 2010;70(14):6071–82. Epub 2010/07/09. doi: 10.1158/0008-5472.CAN-09-4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pham TND, Spaulding C, Munshi HG. Controlling TIME: How MNK Kinases Function to Shape Tumor Immunity. Cancers (Basel). 2020;12(8). Epub 2020/08/01. doi: 10.3390/cancers12082096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robichaud N, Hsu BE, Istomine R, Alvarez F, Blagih J, Ma EH, et al. Translational control in the tumor microenvironment promotes lung metastasis: Phosphorylation of eIF4E in neutrophils. Proc Natl Acad Sci U S A. 2018;115(10):E2202–E9. Epub 2018/02/22. doi: 10.1073/pnas.1717439115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 2019;566(7745):553–7. Epub 2019/02/08. doi: 10.1038/s41586-019-0915-y. [DOI] [PubMed] [Google Scholar]
- 68.Wetzler M, Segal D. Omacetaxine as an anticancer therapeutic: what is old is new again. Curr Pharm Des. 2011;17(1):59–64. Epub 2011/02/08. [DOI] [PubMed] [Google Scholar]
- 69.Chen R, Gandhi V, Plunkett W. A sequential blockade strategy for the design of combination therapies to overcome oncogene addiction in chronic myelogenous leukemia. Cancer Res. 2006;66(22):10959–66. Epub 2006/11/17. doi: 10.1158/0008-5472.CAN-06-1216. [DOI] [PubMed] [Google Scholar]
- 70.Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for cancer therapy. J Hematol Oncol. 2019;12(1):71 Epub 2019/07/07. doi: 10.1186/s13045-019-0754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38 Epub 2009/02/13. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–32. Epub 2009/01/20. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X. Targeting PI3K in cancer: mechanisms and advances in clinical trials. Mol Cancer. 2019;18(1):26 Epub 2019/02/21. doi: 10.1186/s12943-019-0954-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534(7606):272–6. Epub 2016/06/10. doi: 10.1038/nature17963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Konicek BW, Stephens JR, McNulty AM, Robichaud N, Peery RB, Dumstorf CA, et al. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011;71(5):1849–57. Epub 2011/01/15. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- 76.Reich SH, Sprengeler PA, Chiang GG, Appleman JR, Chen J, Clarine J, et al. Structure-based Design of Pyridone-Aminal eFT508 Targeting Dysregulated Translation by Selective Mitogen-activated Protein Kinase Interacting Kinases 1 and 2 (MNK1/2) Inhibition. J Med Chem. 2018;61(8):3516–40. Epub 2018/03/13. doi: 10.1021/acs.jmedchem.7b01795. [DOI] [PubMed] [Google Scholar]
- 77.Heerma van Voss MR, van Diest PJ, Raman V. Targeting RNA helicases in cancer: The translation trap. Biochim Biophys Acta Rev Cancer. 2017;1868(2):510–20. Epub 2017/10/03. doi: 10.1016/j.bbcan.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruggero D The role of Myc-induced protein synthesis in cancer. Cancer Res. 2009;69(23):8839–43. Epub 2009/11/26. doi: 10.1158/0008-5472.CAN-09-1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Manier S, Huynh D, Shen YJ, Zhou J, Yusufzai T, Salem KZ, et al. Inhibiting the oncogenic translation program is an effective therapeutic strategy in multiple myeloma. Sci Transl Med. 2017;9(389). Epub 2017/05/12. doi: 10.1126/scitranslmed.aal2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen ZH, Qi JJ, Wu QN, Lu JH, Liu ZX, Wang Y, et al. Eukaryotic initiation factor 4A2 promotes experimental metastasis and oxaliplatin resistance in colorectal cancer. J Exp Clin Cancer Res. 2019;38(1):196 Epub 2019/05/16. doi: 10.1186/s13046-019-1178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ernst JT, Thompson PA, Nilewski C, Sprengeler PA, Sperry S, Packard G, et al. Design of Development Candidate eFT226, a First in Class Inhibitor of Eukaryotic Initiation Factor 4A RNA Helicase. J Med Chem. 2020;63(11):5879–955. Epub 2020/05/30. doi: 10.1021/acs.jmedchem.0c00182. [DOI] [PubMed] [Google Scholar]
- 82.da Silva DC, Valentao P, Andrade PB, Pereira DM. Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: Tools and strategies to understand its complexity. Pharmacol Res. 2020;155:104702 Epub 2020/02/19. doi: 10.1016/j.phrs.2020.104702. [DOI] [PubMed] [Google Scholar]
- 83.Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014;4(6):702–15. Epub 2014/04/08. doi: 10.1158/2159-8290.CD-13-0945. [DOI] [PubMed] [Google Scholar]
- 84.Jewer M, Lee L, Leibovitch M, Zhang G, Liu J, Findlay SD, et al. Translational control of breast cancer plasticity. Nat Commun. 2020;11(1):2498 Epub 2020/05/20. doi: 10.1038/s41467-020-16352-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laham-Karam N, Pinto GP, Poso A, Kokkonen P. Transcription and Translation Inhibitors in Cancer Treatment. Front Chem. 2020;8:276 Epub 2020/05/07. doi: 10.3389/fchem.2020.00276. [DOI] [PMC free article] [PubMed] [Google Scholar]