

Abstract
Excessive alcohol users have increased risk of developing respiratory infections in part due to oxidative stress-induced alveolar macrophage (AM) phagocytic dysfunction. Chronic ethanol exposure increases cellular oxidative stress in AM via upregulation of NADPH oxidase (Nox) 4, and treatment with the peroxisome proliferator-activated receptor gamma (PPARγ) ligand, rosiglitazone, decreased ethanol-induced Nox4. However, the mechanism by which ethanol induces Nox4 expression and PPARγ ligand reverses this defect has not been elucidated. Since microRNA (miR)-92a has been predicted to target Nox4 for destabilization, we hypothesized that ethanol exposure decreases miR-92a expression and leads to Nox4 upregulation. Previous studies have implicated mitochondrial-derived oxidative stress in AM dysfunction. We further hypothesized that ethanol increases mitochondrial-derived AM oxidative stress and dysfunction via miR-92a and that treatment with the PPARγ ligand, pioglitazone, could reverse these derangements. To test these hypotheses, a mouse AM cell line, MH-S cells, were exposed to ethanol in vitro and primary AM were isolated from a mouse model of chronic ethanol consumption to measure Nox4, mitochondrial target mRNA (qRT-PCR) and protein levels (confocal microscopy), mitochondria-derived reactive oxygen species (confocal immunofluorescence), mitochondrial fission (electron microscopy), and mitochondrial bioenergetics (extracellular flux analyzer). Ethanol exposure increased Nox4, enhanced mitochondria-derived oxidative stress, augmented mitochondrial fission, and impaired mitochondrial bioenergetics. Transfection with miR-92a mimic in vitro or pioglitazone treatment in vivo diminished Nox4 levels, resulting in improvements in these ethanol-mediated derangements. These findings provide support that pioglitazone may provide a novel therapeutic approach to mitigate ethanol-induced AM mitochondrial derangements.
Keywords: Ethanol, miR-92a, mitochondria, Nox4, alveolar macrophage, pioglitazone
Introduction
Nearly 15 million people in the United States have been diagnosed with alcohol use disorders (AUD) ("Alcohol Facts and Statistics," 2018), and excessive alcohol use increases morbidity and mortality (Moss, 2005). Compared to non-AUD individuals, chronic alcohol use increases susceptibility to respiratory infections and acute respiratory distress syndrome (Mehta & Guidot, 2012; Moss et al., 2003). Alcohol-mediated risk for developing respiratory infections in the alveolar space has been largely shown to be a consequence of alveolar macrophage (AM) dysfunction (Mehta, Yeligar, Elon, Brown, & Guidot, 2013; Yeligar, Harris, Hart, & Brown, 2012, 2014; Yeligar, Mehta, Harris, Brown, & Hart, 2016). AM are critical for innate and acquired immunity due to initiation of the immune response to fight against invading pathogens (Aderem & Underhill, 1999). Alcohol misuse suppresses AM phagocytic function and bacterial clearance (Brown, Harris, Ping, & Gauthier, 2004; Yeligar et al., 2014). Phagocytosis is a process which is energy demanding, and oxidative phosphorylation is the most efficient cellular process to generate ATP. The current study seeks to fill the gap in knowledge of whether chronic ethanol causes mitochondrial derangements in AM, which may contribute to phagocytic dysfunction and increased susceptibility to respiratory infections.
One mechanism by which ethanol-induced oxidative stress impairs AM phagocytic function is via increased expression of NADPH oxidase (Nox) 4 (Yeligar et al., 2012, 2014; Yeligar et al., 2016). Nox4 is a primary source of oxidative stress in AM, and our laboratory has shown that silencing Nox4 decreased ethanol-induced AM oxidative stress (Yeligar et al., 2012). However, it is unclear how Nox4 is regulated and its cellular localization following ethanol exposure. To elucidate the mechanism by which ethanol upregulates Nox4 levels, we examined expression of Nox4-associated microRNAs (miRs). miRs regulate gene expression at the post-transcriptional level by binding to the 3’ untranslated region (UTR) of their target mRNA, resulting in the target’s destabilization. Aberrant miR expression can dysregulate normal cellular function and contribute to the pathogenesis of various conditions, such as those characterized by inflammation and impaired phagocytosis (Morris et al., 2017; Yeligar et al., 2016). Using in silico analysis, we determined that the species conserved miR-92a seed sequence putatively binds to the 3′ UTR of Nox4. Therefore, we hypothesized that chronic ethanol exposure decreases miR-92a, thereby elevating Nox4 levels.
We have shown that loss of peroxisomal proliferator activator receptor gamma (PPARγ) increases Nox4 levels, oxidative stress, and phagocytic dysfunction. Treatment with a synthetic thiazolidinedione ligand for PPARγ, rosiglitazone, attenuated Nox4 levels and reversed ethanol-induced cellular oxidative stress and phagocytic dysfunction (Yeligar et al., 2016). Synthetic thiazolidinediones, such as rosiglitazone and pioglitazone are FDA approved drugs that have been employed to treat type 2 diabetes (Yki-Jarvinen, 2004). Although, it has been determined that long term use of rosiglitazone is linked to cardiovascular complications (Home et al., 2007; Nissen & Wolski, 2007), these adverse clinical outcomes are not known to be associated with pioglitazone (PIO) (Dormandy et al., 2005; Erdmann et al., 2007; Lincoff, Wolski, Nicholls, & Nissen, 2007; Wilcox et al., 2007). This study fills a gap in knowledge by demonstrating the mechanism by which PPARγ ligand treatment decreases Nox4 levels and confirming the benefits of PIO on mitigating ethanol-induced AM derangements.
The data outlined in this study provide novel evidence that ethanol-induced oxidative stress and mitochondrial derangements are mediated by miR-92a. We further demonstrate that ethanol exposure diminished miR-92a, resulting in increased levels of Nox4. Treatment with the pharmacological PPARγ ligand PIO was sufficient to induce expression of miR-92a, reversing ethanol-induced upregulation of Nox4, oxidative stress, and mitochondrial dysfunction in AM. As oxidative phosphorylation is the most efficient mechanism to generate ATP necessary for AM phagocytic capacity, these novel findings provide evidence into mitochondrial dysfunction as a possible mechanism by which ethanol induces AM phagocytic dysfunction. Further, these findings also provide evidence of PIO as a novel therapeutic intervention in attenuating AM mitochondrial-derived oxidative stress and improving host respiratory immune defense in individuals with a history of AUD.
Materials and Methods
Mouse Model of Chronic Ethanol Ingestion
All animal studies were carried out in accordance with the National Institutes of Health guidelines as outlined in the Guide for the Care and Use of Laboratory Animals. Further, all protocols described were reviewed and approved by the Emory University and Atlanta VA Health Care System Institutional Animal Care and Use Committees. 8-10-week-old male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were fed standard laboratory chow ad libitum. Mice were randomly divided into four groups (n=10 mice / group): control+vehicle, control+PIO, ethanol+vehicle, and ethanol+PIO. Mice in the ethanol groups received weekly increases of ethanol in the drinking water (5% w/v over 2 weeks) until reaching a final concentration of 20% w/v of ethanol, which was maintained for 10 weeks and resulted in a blood alcohol level of 0.12% (Wagner, Yeligar, Brown, & Michael Hart, 2012; Yeligar et al., 2012; Yeligar et al., 2016). During the final week of ethanol ingestion, mice were gavaged daily with PIO (10 mg/kg/day in 100 μL methylcellulose vehicle) or vehicle alone (Yeligar et al., 2016). Following euthanasia, tracheas from the mice were cannulated and a tracheotomy was performed to collect the bronchoalveolar lavage fluid. Mouse alveolar macrophages (mAM) were isolated by centrifugation at 8000 RPM for 5 minutes (Yeligar et al., 2012; Yeligar et al., 2016) and resuspended in RPMI-1640 culture medium containing 2% fetal bovine serum and 1% penicillin/streptomycin for 24 h for subsequent experimentation.
MH-S Cells Ethanol Exposure
The mouse AM cell line (MH-S) was purchased from American Type Culture Collection (Manassas, VA) and used as a model of in vitro ethanol exposure. MH-S cells were cultured in RPMI-1640 medium containing 10% fetal bovine serum, 1% penicillin/streptomycin, 11.9 mM sodium bicarbonate, gentamicin (40mg/ml) and 0.05 mM 2-mercaptoethanol. MH-S cells were cultured in the presence or absence of 0.08% ethanol (equivalent to 18 mM of ethanol) for 3 consecutive days (media changed daily) at 37°C in a humidified incubator in 5% CO2. A subset of cells were treated with or without PIO (10 μM) on the last day of ethanol exposure.
Transmission Electron Microscopy
Transmission electron microscopy studies were performed at Emory University (Atlanta, GA) at the Robert P. Apkarian Integrated Electron Microscopy Core. Control- and ethanol-treated MH-S cells were fixed with 2.5% glutaradehyde in phosphate buffered saline (PBS) for 2 hours at 4°C, followed by washing with 0.2 M cacodylate buffer in PBS. Tissue blocks were incubated in 1% osmium fixative for 1 hour at room temperature and embedded in epoxy resin and an overnight polymerization at 60-70°C as previously described (Maurice et al., 2019).
Fluorescent Microscopy
Target protein measurement was determined in MH-S cells ± ethanol and mAM isolated from control- and ethanol-fed mice. MH-S and mAM cells were then fixed with 4% paraformaldehyde and incubated with antibodies: Nox4 (Santa Cruz Biotechnology, Dallas, TX), mitochondrial transcription factor A (TFAM), mitofusin-2 (MFN2), heat shock protein family A member 9 (GRP75), and voltage-dependent anion-selective channel (VDAC), followed by incubation with fluorescent-labeled secondary antibodies. Protein values were normalized to DAPI nuclear stain.
Mitochondrial superoxide and reactive oxygen species (ROS) levels in MH-S and mAM cells were determined using MitoSOX to assess mitochondrial superoxide (Thermofisher, Waltham, WA) or MitoTrackerCMXRos to determine mitochondrial reactive oxygen species (Thermofisher), according to the manufacturer’s protocols. In short, AM were incubated in the dark for 30 minutes at room temperature with 5μM MitoSOX or MitoTrackerCMXRos. After staining with MitoTrackerCMXRos, AM were fixed with 4% paraformaldehyde. For analysis of mitochondrial number and volume, MH-S cells were incubated in the dark for 30 minutes with MitoTracker Orange (400 nM) (Thermofisher).
mAM were permeabilized with Triton X-100 and incubated with ER-Tracker, MitoTrackerCMXRos, and DAPI nuclear stain (Thermofisher) according to the manufacturer’s protocols before being fixed with 4% paraformaldehyde.
Fluorescence of all confocal images were measured using FluoView (Olympus) and were expressed as fold-change of mean relative fluorescent units (RFU) per cell ± SEM, relative to untreated control samples. RFU were measured in at least 10 cells per field, and there were 10 fields per experimental condition. Gain and gamma microscope settings were consistent for each field and experimental condition. Images were deconvoluted and analyzed using ImageJ (Yeligar et al., 2018).
microRNA and RNA isolation and quantitative RT-PCR (qRT-PCR)
microRNA was isolated from MH-S cells and mAM using the mirVana miRNA Isolation Kit (Thermofisher, Waltham, MA), and total RNA was isolated using the TRIzol reagent. Target mRNA and miRNA levels were measured and quantified by qRT-PCR with specific mouse primer sequences as outlined in Table 1. qRT-PCR was performed using the iTaq Universal SYBR Green One-Step kit (Bio-Rad, Hercules, CA) on the Applied Biosystems ABI Prism 7500 version 2.0.4 sequence detection system (Yeligar et al., 2012; Yeligar et al., 2016). Target mRNA values were normalized to housekeeping 9S mRNA levels, and miR-92a values were normalized to U5 ribosomal mRNA levels. mRNA and miRNA levels were expressed as fold-change of mean ± SEM, relative to control samples.
Table 1:
Primer sequences to measure mRNA levels using qRT-PCR.
| Gene | Forward Sequence (5’ → 3’) | Reverse Sequence (5’ → 3’) |
|---|---|---|
| NOX4 | TGTTGGGCCTAGGATTGTGTT | AGGGACCTTCTGTGATCCTCG |
| TFAM | CACCCAGATGCAAAACTTTCA | CTGTGAGCAAGTATAAAG |
| MFN2 | TCCTGGGCCCTAAGAATAGC | GAGAGGACGCTGAACCTGAT |
| GRP75 | TCCTGTGTGGCTGTTATGGA | AGGGGTAGTTCTGGCACC |
| VDAC | GGTACACTCAGACCCTAA | CACCCGCATTGACGTTCT |
| miR-92a | TATTGCACTTGTCCCGGCCTG | CAGGCCGGGACAAGTGCAATA |
| 9S | ATCCGCCAGCGCCATA | TCGATGTGCTTCTGGGAATCC |
| U5 | TCTCGTCTGATCTCGGAAGC | AGCCTACAGCACCCGGTATT |
Nox4, NADPH oxidase 4; TFAM, mitochondrial transcription factor A; MFN2, mitofusin-2; GRP75, heat shock protein family A member 9; VDAC, voltage-dependent anion-selective channel; miR-92a, microRNA-92a.
Transient Transfection of MH-S cells
miR-92a was overexpressed in MH-S cells using transient transfection of a miR-92a mimic (miR-92a-M) (Qiagen, Hilden, Germany). MH-S cells were resuspended in 100 μL of Amaxa Mouse Macrophage Nucleofector Kit solution (Lonza, Alpharetta, GA) containing 50 nM of control miR-92a scramble (miR-92a-Scr), miR-92a-M, or antisense (anti)-miR-92a (data shown in Supplemental Figure 1), followed by nucleofection according to the manufacturer’s protocol using program Y-001. Following transfection, MH-S cells were washed with media and cultured with or without 0.08% ethanol for 3 days (media changed daily).
Mitochondrial Bioenergetics
Mitochondrial bioenergetics were evaluated using a XFe96 Extracellular Flux Analyzer (Agilent Seahorse Bioscience Inc., Billerica, MA). Oxygen consumption rate (OCR) was measured in MH-S cells and mAM. Sequential injections of 2 μM of oligomycin (mitochondrial complex V inhibitor), 0.5 μM of carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP) (ATP synthase inhibitor and proton uncoupler), and 0.5 μM of rotenone (mitochondrial complex I inhibitor) plus antimycin A (mitochondrial complex III inhibitor) were given to the MH-S cells and mAM (Grunwell et al., 2018). Basal respiration, mitochondrial ATP-linked respiration, maximal respiration, and spare respiratory capacity were determined using the XF Wave 2.1 software. OCR values were normalized to MH-S or mAM protein concentration in the same sample and were expressed as mean ± SEM, relative to control samples.
Statistical Analysis
All data are presented as mean ± standard error of the mean (SEM). In studies with only two groups, Student’s t test was performed. In studies with more than two groups, statistical analyses were performed using one-way analysis of variance (ANOVA) followed by a Tukey-Kramer post hoc test to detect differences between individual experimental groups (GraphPad Prism version 8, San Diego, CA). Non-parametric statistical analysis using Kruskal-Wallis tests were performed if the data were not normally distributed. p<0.05 was considered significant.
Results
Ethanol-induced AM mitochondrial oxidative stress and fission is mediated by miR-92a modulation of Nox4 in MH-S cells
The ability to phagocytose invading pathogens is an essential immune function of AM but has high energy demands. Mitochondrial-dependent oxidative phosphorylation is the most efficient cellular process for generating the ATP needed to meet the energy demands of phagocytosis. To determine the effects of ethanol on AM mitochondria, cultured MH-S cells were either untreated (Con) or ethanol-treated (EtOH). As shown in Figure 1A, ethanol exposure altered mitochondrial morphology, where mitochondria shifted from an elongated shape in control to a spherical appearance in ethanol cells. Ethanol-induced changes in morphology correlated with increased number of mitochondria per cell while decreasing mitochondrial volume, suggesting mitochondrial fission.
Figure 1. Ethanol increases AM mitochondrial fission, mitochondrial oxidative stress, and mitochondrial Nox4 levels in vitro.

MH-S cells were exposed to either control (Con) or ethanol (EtOH; 0.08%) for three days (n=9). (A) Representative electron microscope images of mitochondria. Bar graphs represent number of mitochondria and mitochondrion volume quantified by fluorescence microscopy using MitoTracker Orange (10 fields / condition) and are expressed as mean ± SEM, relative to control. (B) mRNA levels of mitochondrial transcription factor A (TFAM), mitofusin 2 (MFN2), glucose-regulated protein 75 (GRP75) and voltage-dependent anion channels (VDAC) were measured by quantitative RT-PCR (qRT-PCR) in duplicate, normalized to 9S mRNA, and are expressed as mean ± SEM, relative to control. (C) Protein levels of TFAM, MFN2, GRP75 and VDAC were measured by fluorescence microscopy (10 fields / condition), normalized to DAPI, and are expressed as mean RFU ± SEM, relative to control. (D) Mitochondrial superoxide (MitoSOX) and (E) mitochondrial ROS (MitoTracker CMXRos) production were determined (10 fields / condition) and are expressed as mean relative fluorescence units (RFU) ± SEM, relative to control. (F) Representative confocal microscope images of Nox4 colocalization with endoplasmic reticulum (E.R.), nucleus, and the mitochondria. *p < 0.05 versus control; #p<0.05 versus ethanol.
Because ethanol exposure in MH-S cells altered mitochondrial structure suggestive of fission, we examined how ethanol affected levels of mitochondrial proteins implicated in dynamics (Yeligar et al., 2018). mRNA and protein levels of the mitochondrial proteins TFAM, MFN2, GRP75, and VDAC were measured in control or ethanol cells. Compared to control, ethanol exposure decreased the mRNA (Figure 1B) and protein (Figure 1C) levels of TFAM, a mitochondrial-specific transcription factor that regulates mitochondrial DNA copy number and respiratory chain integrity (Larsson et al., 1998); MFN2, a mitochondrial fusion protein (Park et al., 2015); as well as GRP75 and VDAC, which are important regulators of mitochondrial membrane potential (Isarangkul, Wiyakrutta, Kengkoom, Reamtong, & Ampawong, 2015; Sampson, Lovell, & Craigen, 1996). Collectively, these data show that ethanol-exposed MH-S cells exhibit increased mitochondrial fission.
Mitochondrial oxidative stress can occur due to increased mitochondrial fission (Wu, Zhou, Zhang, & Xing, 2011). Therefore, we sought to determine whether ethanol exposure affects AM mitochondrial oxidative stress. Ethanol increased mitochondrial-derived superoxide (Figure 1D) and ROS (Figure 1E) production in MH-S cells. Since Nox4 is a transmembrane protein that has been implicated in oxidative stress generation (Lassegue & Griendling, 2010), we examined whether ethanol affects Nox4 subcellular localization. Ethanol increased Nox4 colocalization with the mitochondria in mAM (Figure 1F). These results indicate that one mechanism by which ethanol increases AM mitochondrial oxidative stress in vitro is due to increased localization of Nox4 to the mitochondria.
Excessive ROS impairs AM phagocytosis (Liang, Harris, & Brown, 2014). Previous studies from our lab have established that ethanol exposure increased levels of Nox4, a primary source of ROS in AM, resulting in phagocytic dysfunction (Yeligar et al., 2012, 2014; Yeligar et al., 2016). To determine the mechanism by which ethanol induced Nox4 levels, we examined microRNAs, which are major gene regulators that destabilize their target and cause mRNA degradation or translational repression. In silico analysis of the Nox4 3’UTR identified a putative binding site for miR-92a. To determine the molecular mechanism by which Nox4 may be regulated during ethanol exposure in MH-S cells, miR-92a levels were investigated. As demonstrated in Figure 2A, miR-92a expression was diminished in ethanol-exposed MH-S cells compared to control. To study the relationship between miR-92a and Nox4, control-or ethanol-exposed MH-S cells were transfected with control miR-92a scrambled (miR-92a-Scr) or miR-92a mimic (miR-92a-M) to overexpress miR-92a in addition to the presence or absence of ethanol. miR-92a-M decreased Nox4 mRNA (Figure 2B) and protein (Figure 2C) in control MH-S cells. Ethanol-induced Nox4 mRNA (Figure 2B) and protein (Figure 2C) levels in miR-92a-Scr transfected MH-S cells were reversed with miR-92a-M, suggesting that miR-92a regulates Nox4 expression.
Figure 2. Ethanol-induced AM mitochondrial oxidative stress and fission are mediated by miR-92a modulation of Nox4 in vitro.

MH-S cells were exposed to either control (Con) or ethanol (EtOH; 0.08%) for three days (n=6). (A) miR-92a levels were measured by qRT-PCR, in duplicate, normalized to U5, and are expressed as mean ± SEM, relative to control. (B-F) MH-S transiently transfected with miR-92a scrambled (miR-92a-Scr) or miR-92a mimic (miR-92a-M) were exposed to either Con or EtOH (0.08%) for three days. (B) Nox4 mRNA was measured by qRT-PCR, in duplicate, normalized to 9S mRNA. (C) Nox4 protein levels were measured by fluorescence microscopy (10 fields / condition), normalized to DAPI, and expressed as RFU ± SEM, relative to control. Representative fluorescent and brightfield images have been provided. (D) Mitochondrial superoxide (MitoSOX) and (E) mitochondrial ROS (MitoTracker CMXRos) production were determined (10 fields / condition) and expressed as mean relative fluorescence units (RFU) ± SEM, relative to control. (F) mRNA levels of TFAM, MFN2, GRP75, and VDAC were measured by qRT-PCR, in duplicate, normalized to 9s mRNA, and expressed as mean ± SEM, relative to control. *p < 0.05 versus control; #p<0.05 versus ethanol.
Since miR-92a regulates ethanol-induced Nox4 (Figure 2B) and Nox4 is localized in AM mitochondria following ethanol exposure (Figure 1F), we examined the role of miR-92a in AM mitochondrial oxidative stress. Control-and ethanol-treated cells were transfected with miR-92a-Scr or miR-92a-M. Ethanol-exposed MH-S cells transfected with miR-92a-Scr demonstrated enhanced mitochondrial-derived superoxide (Figure 2D) and ROS (Figure 2E), which was attenuated with miR-92a-M. Since miR-92a regulates mitochondrial-derived oxidative stress, we examined the role of miR-92a on mitochondrial fission. Compared to miR-92a-Scr, miR-92a-M reversed ethanol-mediated decreases in the mRNA levels of mitochondrial dynamics mediators TFAM, MFN2, GRP75, and VDAC (Figure 2F). Together, these data show that ethanol-induced AM mitochondrial oxidative stress and fission are mediated by miR-92a in MH-S cells.
Ethanol-induced derangements in AM mitochondrial bioenergetics is mediated by miR-92a in MH-S cells
Since mitochondrial fission has been associated with metabolic dysfunction (Wai & Langer, 2016) and our data show that miR-92a plays a role in ethanol-mediated mitochondrial-derived oxidative stress and fission (Figure 2), we examined whether miR-92a-mediated alterations have functional consequences on mitochondrial bioenergetics. Ethanol exposure in MH-S cells diminished OCR bioenergetic profiling (Figure 3A), basal respiration (Figure 3B), ATP-linked respiration (Figure 3C), maximal respiration (Figure 3D), and spare respiratory capacity (Figure 3E). miR-92a-M partially restored ethanol-mediated impairments in mitochondrial bioenergetics. To provide further evidence of the importance of miR-92a in mitochondrial bioenergetics, we knocked down miR-92a in control cells and observed OCR bioenergetic profiling similar to ethanol treated MH-S cells (Supplemental Figure 1). These data demonstrate that ethanol-induced derangements in AM mitochondrial bioenergetics is mediated by miR-92a.
Figure 3. Ethanol-induced derangements in AM mitochondrial bioenergetics is mediated by miR-92a in MH-S cells.
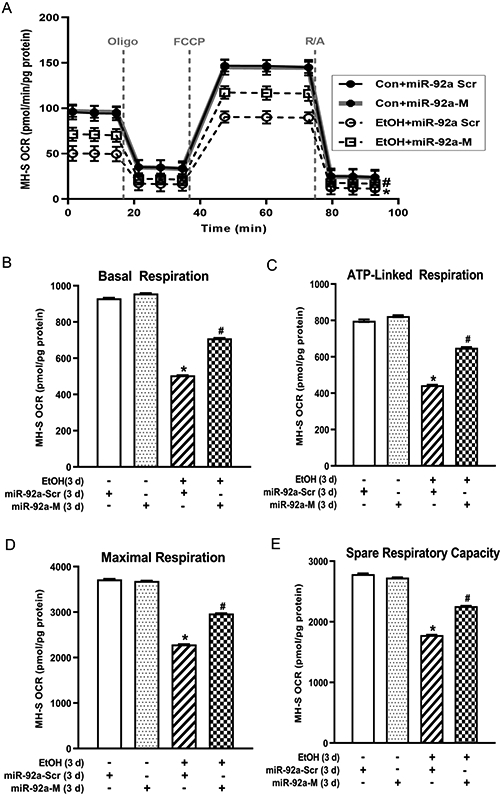
MH-S transiently transfected with miR-92a scrambled (miR-92a-Scr) or miR-92a mimic (miR-92a-M) were exposed to either control (Con) or ethanol (EtOH; 0.08%) for three days (n=9). Mitochondrial bioenergetic profiles were generated in response to oligomycin (Oligo; mitochondrial complex V inhibitor), carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP; ATP synthase inhibitor and proton uncoupler), and rotenone (mitochondrial complex I inhibitor) plus antimycin A (mitochondrial complex III inhibitor) (R/A) using an extracellular flux analyzer. (A) Oxygen consumption rate (OCR) from mitochondrial bioenergetic profiling, normalized to protein levels, are expressed as mean ± SEM, relative to control. (B) Basal respiration, (C) ATP-linked respiration, (D) maximal respiration, and (E) spare respiratory capacity are expressed as mean ± SEM, relative to control. *p <0.05 versus control; #p<0.05 versus ethanol.
Pioglitazone treatment in vivo reverses ethanol-induced AM derangements in miR-92a, mitochondrial oxidative stress, and mitochondrial fission
Previously, ethanol downregulated PPARγ expression in ethanol-exposed MH-S cells and mAM isolated from ethanol-fed mice. Loss of PPARγ upregulated Nox4, and treatment with a PPARγ ligand improved ethanol-induced oxidative stress and phagocytic function (Yeligar et al., 2016). To investigate the mechanism by which the PPARγ ligand attenuated Nox4 levels, we examined the effect of PIO on miR-92a expression, which is predicted to target Nox4. Chronic ethanol ingestion in mice diminished miR-92a expression (Figure 4A) similar to our in vitro studies in MH-S cells (Figure 2A and Supplemental Figure 2). PIO treatment partially reversed ethanol-mediated decreases in miR-92a in MH-S cells (Supplemental Figure 2). Similar to our in vitro studies, PIO treatment also partially reversed ethanol-mediated attenuation of miR-92a levels in mAM (Figure 4A). Chronic ethanol ingestion also elevated mitochondrial-derived superoxide (Figure 4B), and ROS (Figure 4C), which were attenuated with PIO treatment. Further, PIO treatment reversed chronic ethanol ingestion-mediated decreases in TFAM, MFN2, GRP75, and VDAC mRNA (Figure 4D) and protein (Figure 4E) levels. Collectively, these data illustrate that PIO treatment in vivo reverses ethanol-induced AM derangements in miR-92a and subsequent mitochondrial oxidative stress and mitochondrial fission.
Figure 4. Pioglitazone treatment in vivo reverses ethanol-induced AM derangements in miR-92a, mitochondrial oxidative stress, and mitochondrial fission.
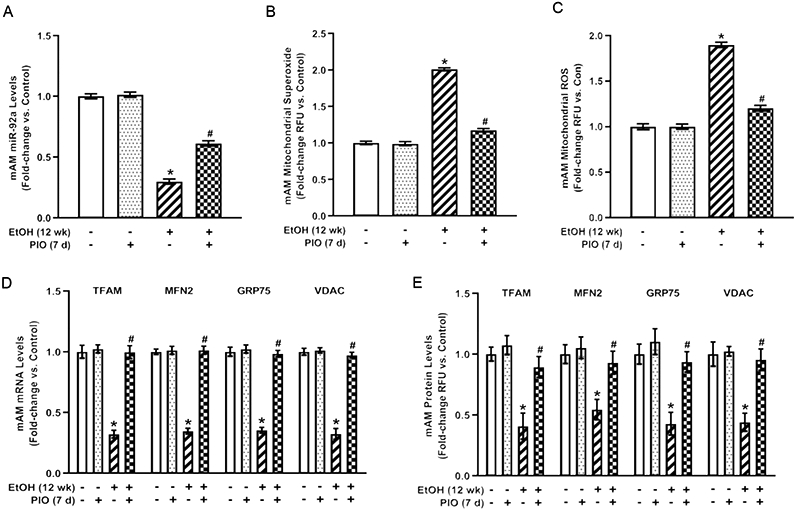
Primary mouse alveolar macrophages (mAM) were isolated from mice fed either control (Con) or ethanol (EtOH; 20% v/w in drinking water, 12 weeks) ± oral pioglitazone (PIO, last 7 days of ethanol) (n =10 /group). (A) miR-92a levels were measured by qRT-PCR, in duplicate, normalized to U5, and expressed as mean ± SEM, relative to control. (B) Mitochondrial superoxide (MitoSOX) and (C) mitochondrial ROS (MitoTracker CMXRos) production were determined (10 fields / condition) and expressed as mean relative fluorescence units (RFU) ± SEM, relative to control. (D) mRNA levels of TFAM, MFN2, GRP75, and VDAC were measured by qRT-PCR, in duplicate, normalized to 9s mRNA, and expressed as mean ± SEM, relative to control. (E) Protein levels of TFAM, MFN2, GRP75, and VDAC were measured by fluorescence microscopy (10 fields / condition), normalized to DAPI, and expressed as mean RFU ± SEM, relative to control *p < 0.05 versus control; #p<0.05 versus ethanol.
Pioglitazone treatment in vivo reverses ethanol-induced AM derangements in mitochondrial bioenergetics
As PIO partially reversed ethanol-mediated decreases in miR-92a levels (Figure 4A) and miR-92a partially improved mitochondrial bioenergetics in vitro following ethanol exposure (Figure 3), we assessed whether PIO would restore mitochondrial bioenergetics in the in vivo model. Similar to our in vitro studies, chronic ethanol ingestion decreased mAM OCR bioenergetic profile (Figure 5A), basal respiration (Figure 5B), ATP-linked respiration (Figure 5C), maximal respiration (Figure 5D), and spare respiratory capacity (Figure 5E). Treatment with oral PIO reversed ethanol-mediated impairments in mitochondrial bioenergetics. Coupled with our data that miR-92a regulates ethanol-induced Nox4 expression (Figure 2B-C), these data suggest that PIO reverses ethanol-induced mitochondrial oxidative stress, fission, and bioenergetic derangements via miR-92a modulation of Nox4.
Figure 5: Pioglitazone treatment in vivo reverses ethanol-induced AM derangements in mitochondrial bioenergetics.
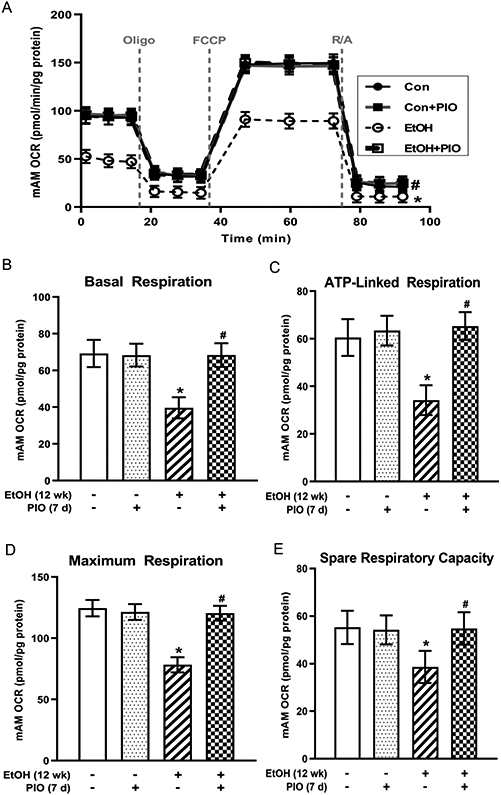
Primary mouse alveolar macrophages (mAM) were isolated from mice fed either control (Con) or ethanol (EtOH; 20% v/w in drinking water, 12 weeks) ± oral pioglitazone (PIO, last 7 days of ethanol) (n =10 /group). Mitochondrial bioenergetic profile was generated in response to oligomycin (Oligo; mitochondrial complex V inhibitor), carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP; ATP synthase inhibitor and proton uncoupler), and rotenone (mitochondrial complex I inhibitor) plus antimycin A (mitochondrial complex III inhibitor) (R/A) using an extracellular flux analyzer. (A) Oxygen consumption rate (OCR) from mitochondrial bioenergetic profiling, normalized to protein levels, are expressed as mean ± SEM, relative to control. (B) Basal respiration, (C) ATP-linked respiration, (D) maximal respiration, and (E) spare respiratory capacity are expressed as mean ± SEM, relative to control. *p <0.05 versus control; #p<0.05 versus ethanol.
Discussion
AM initiate the immune response to invading respiratory pathogens (Aderem & Underhill, 1999). Our previous studies have shown that ethanol exposure stimulates oxidative stress, in part due to Nox4 activation, and impairs AM immune functions (Brown et al., 2004; Yeligar et al., 2012, 2014; Yeligar et al., 2016). However, the mechanisms by which ethanol enhances Nox4 expression have not been fully elucidated. The current study examined whether ethanol-mediated stimulation of Nox4, mitochondrial-derived oxidative stress, and mitochondrial derangements are linked through the effects of ethanol on miR-92a, a putative Nox4 regulator. As summarized in Figure 6, our data provide evidence to show, for the first time, that ethanol decreases miR-92a levels, subsequently increasing Nox4, and mitochondrial oxidative stress and dysfunction in AM. Our novel findings additionally show that treatment with the PPARγ ligand PIO partially reversed ethanol-mediated decreases in miR-92a levels, correlative with attenuated ethanol-induced Nox4 and mitochondrial oxidative stress, leading to improved mitochondrial bioenergetic functions in AM.
Figure 6: Hypothetical schematic of ethanol-induced AM mitochondrial dysfunction.
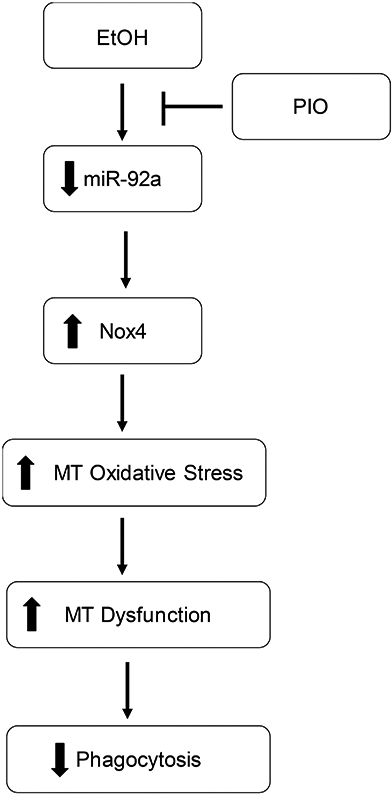
In AM, ethanol exposure diminished miR-92a, leading to increased expression of the miR-92a target Nox4. Increased Nox4 expression and localization to the mitochondria subsequently resulted in altered mitochondrial structure, enhanced mitochondrial-derived oxidative stress, and impaired mitochondrial bioenergetics. These ethanol-mediated AM derangements were mitigated with PIO treatment.
These current data provide a novel mechanistic understanding to previous studies which demonstrate that chronic ethanol exposure diminishes PPARγ and increases Nox4, resulting in AM oxidative stress and phagocytic dysfunction (Yeligar et al., 2012, 2014; Yeligar et al., 2016). Our findings used in vitro and in vivo approaches to identify a mechanism by which ethanol induces Nox4 and metabolic derangements. The major function of AM is phagocytosis of invading pathogens, which is an energy demanding process. Oxidative phosphorylation is the most efficient method of generating ATP to meet high energy demands of phagocytosis. Since mitochondrial-derived oxidative stress may induce mitochondrial dysfunction (S. Wu, Zhou, Zhang, & Xing, 2011) and impair AM phagocytic capacity (Liang et al., 2014), we investigated the effects of chronic ethanol on AM mitochondria. In a model of pulmonary hypertension, loss of PPARγ induced mitochondrial-derived ROS generation and impaired mitochondrial function in human pulmonary artery smooth muscle cells (Yeligar et al., 2018). Our novel findings herein show that ethanol enhances mitochondrial fission (Figures 1A-C and Figure 4) and mitochondrial dysfunction (Figure 3 and Figure 5) in AM. In addition to being a major source of mitochondrial oxidative stress, Nox4 has been shown to suppress mitochondrial biogenesis and bioenergetics, which were partly regulated by nuclear factor erythroid-derived 2 like 2 and TFAM expression in lung fibroblasts (Bernard et al., 2017). Taken together, these data suggest that ethanol-induced Nox4 results in mitochondrial-derived oxidative stress and mitochondrial dysfunction.
Other studies have reported that Nox4 localizes to the mitochondria under pathophysiological conditions such as diabetes and aging-associated cardiac disease (Ago et al., 2010; Block, Gorin, & Abboud, 2009; Vendrov et al., 2015), which are associated with excessive ROS levels (D. Wu & Cederbaum, 2003), altered sensitivity to insulin, and glucose tolerance (He et al., 2015; Kalyani & Egan, 2013; Zhong et al., 2012). Additionally, Nox4 subcellular localization to the mitochondria has been reported in cardiomyocytes (Kuroda et al., 2010), mesangial cells (Block et al., 2009), and neurons (Case, Li, Basu, Tian, & Zimmerman, 2013). However, for the first time, we demonstrate that chronic ethanol exposure increases Nox4 localization to the mitochondria of AM (Figure 1F).
To understand how ethanol induces Nox4 in AM, we examined microRNAs as they are cellular regulators that suppress or prevent translation by binding to seed sequences in the 3′UTR of their target mRNAs (Finnegan & Pasquinelli, 2013). miRs have been implicated as regulators of alcohol-induced disorders and in lung pathology (Natarajan, Pachunka, & Mott, 2015; Yeligar et al., 2016). In silico analysis identified miR-92a as having putative binding sites in the 3′UTR of Nox4. A previous report showed that endogenous miR-92a diminished Nox4 and hydrogen peroxide levels, while other genes involved in ROS regulation such as catalase and Nox2 were unaffected by miR-92a (Kriegel et al., 2015). Our study demonstrated that chronic ethanol exposure diminishes miR-92a expression in MH-S cells (Figure 2A) and mAM (Figure 4A). Further, our data provides evidence that Nox4 is a target of miR-92a as overexpression of miR-92a decreased ethanol-induced Nox4 and mitochondrial oxidative stress (Figures 2B-2E). We acknowledge that a limitation of our current studies is that we do not examine whether miR-92a mimic may reverse ethanol-induced alterations in mitochondrial morphology; however, this is a focus of our future studies. Since our findings show that ethanol exposure perturbed the mitochondrial bioenergetics profile, additional future studies will explore whether the cells undergo a metabolic shift from oxidative phosphorylation to a more glycolytic phenotype.
Since miRs may have multiple targets (Du & Zamore, 2007), other mitochondrial targets of miR-92a may be of interest for future studies. Other miR-92a predicted targets include those involved in the mitogen-activated protein kinase pathway (mitogen-activated protein kinase 4 and c-Jun NH2-terminal kinases 1), which could lead to increased cytochrome c release, amplified ROS generation, and mitochondrial dysfunction (Chambers & LoGrasso, 2011; Papa, Choy, & Bubici, 2019). Additionally, miR-92a is part of the miR-17/92 polycistronic cluster of miRs. To date, multiple transcription factors have been identified to target the miR-17/92 promoter, including nuclear factor kappa-light-chain-enhancer of activated B cells and c-Myc which are increased following ethanol exposure (Paice et al., 2002; Yeligar, Machida, Tsukamoto, & Kalra, 2009) and have been shown to increase inflammation and oxidative stress (Mogilyansky & Rigoutsos, 2013; Zhou, Hu, Gong, & Chen, 2010). The effect of PIO on miR-92a expression (Figure 4A) is likely influenced by PIO activating PPARγ which modulates the expression of antioxidant genes such as manganese superoxide dismutases and glutathione peroxidases (Polvani, Tarocchi, & Galli, 2012). Studies have reported that manganese superoxide dismutase (Reddy, Padmavathi, Kavitha, Saradamma, & Varadacharyulu, 2013) and glutathione peroxidases (Xiao et al., 2015) are decreased following alcohol exposure. Further investigation is required to identify whether these mechanisms may be implicated in PIO’s induction of miR-92a expression in AM.
Our lab has established that PPARγ plays an essential role in AM function. Chronic ethanol exposure decreases PPARγ levels and treatment with the PPARγ ligand rosiglitazone improved ethanol-induced Nox4 and phagocytic dysfunction (Yeligar et al., 2016). We sought to determine the mechanism by which EtOH-induced Nox4 levels. In this study, treatment with the PPARγ ligand PIO in vivo reversed ethanol-induced AM derangements by increasing expression of miR-92a which targeted Nox4 and reversed ethanol-induced mitochondrial oxidative stress, mitochondrial fission, and derangements in mitochondrial bioenergetics in AM. These data provide novel evidence that therapeutic intervention with PIO, an FDA approved drug for clinical use, may mitigate AM derangements in people with a history of AUD and potentially in individuals with other pathological conditions that are also characterized by mitochondrial dysfunction.
Supplementary Material
Highlights:
Ethanol increases mitochondrial Nox4 via decreases in miR-92a
Ethanol impairs mitochondrial bioenergetics in alveolar macrophages
Pioglitazone reverses ethanol-induced mitochondrial dysfunction
Acknowledgments
The authors would like to thank David N. Michael and Kathryn M. Crotty for their contributions in proofreading the manuscript.
Funding Sources
This study was supported by National Institute on Alcohol Abuse and Alcoholism R01 AA026086 (SMY) and National Heart, Lung, and Blood Institute T32 HL116271 (MPIs: David M. Guidot, Lou Ann S. Brown, and C. Michael Hart).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aderem A, & Underhill DM (1999). Mechanisms of phagocytosis in macrophages. Annu Rev Immunol, 17, 593–623. doi: 10.1146/annurev.immunol.17.1.593 [DOI] [PubMed] [Google Scholar]
- Ago T, Kuroda J, Pain J, Fu C, Li H, & Sadoshima J (2010). Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res, 106(7), 1253–1264. doi: 10.1161/CIRCRESAHA.109.213116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcohol Facts and Statistics. (2018). Retrieved from https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/alcohol-facts-and-statistics
- Bernard K, Logsdon NJ, Miguel V, Benavides GA, Zhang J, Carter AB, … Thannickal VJ (2017). NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J Biol Chem, 292(7), 3029–3038. doi: 10.1074/jbc.M116.752261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block K, Gorin Y, & Abboud HE (2009). Subcellular localization of Nox4 and regulation in diabetes. Proc Natl Acad Sci U S A, 106(34), 14385–14390. doi: 10.1073/pnas.0906805106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LA, Harris FL, Ping XD, & Gauthier TW (2004). Chronic ethanol ingestion and the risk of acute lung injury: a role for glutathione availability? Alcohol, 33(3), 191–197. doi: 10.1016/j.alcohol.2004.08.002 [DOI] [PubMed] [Google Scholar]
- Case AJ, Li S, Basu U, Tian J, & Zimmerman MC (2013). Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am J Physiol Heart Circ Physiol, 305(1), H19–28. doi: 10.1152/ajpheart.00974.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JW, & LoGrasso PV (2011). Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J Biol Chem, 286(18), 16052–16062. doi: 10.1074/jbc.M111.223602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, … Investigators, P. R. (2005). Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet, 366(9493), 1279–1289. doi: 10.1016/S0140-6736(05)67528-9 [DOI] [PubMed] [Google Scholar]
- Du T, & Zamore PD (2007). Beginning to understand microRNA function. Cell research, 17(8), 661–663. doi:cr200767 [pii] [DOI] [PubMed] [Google Scholar]
- Erdmann E, Dormandy JA, Charbonnel B, Massi-Benedetti M, Moules IK, Skene AM, & Investigators PR (2007). The effect of pioglitazone on recurrent myocardial infarction in 2,445 patients with type 2 diabetes and previous myocardial infarction: results from the PROactive (PROactive 05) Study. J Am Coll Cardiol, 49(17), 1772–1780. doi: 10.1016/j.jacc.2006.12.048 [DOI] [PubMed] [Google Scholar]
- Finnegan EF, & Pasquinelli AE (2013). MicroRNA biogenesis: regulating the regulators. Critical reviews in biochemistry and molecular biology, 48(1), 51–68. doi: 10.3109/10409238.2012.738643 [doi] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwell JR, Yeligar SM, Stephenson S, Ping XD, Gauthier TW, Fitzpatrick AM, & Brown LAS (2018). TGF-beta1 Suppresses the Type I IFN Response and Induces Mitochondrial Dysfunction in Alveolar Macrophages. J Immunol, 200(6), 2115–2128. doi: 10.4049/jimmunol.1701325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Li M, Zheng D, Chen Q, Liu W, & Feng L (2015). Adipose tissue hypoxia and low-grade inflammation: a possible mechanism for ethanol-related glucose intolerance? The British journal of nutrition, 113(9), 1355–1364. doi: 10.1017/S000711451500077X [doi] [DOI] [PubMed] [Google Scholar]
- Home PD, Pocock SJ, Beck-Nielsen H, Gomis R, Hanefeld M, Jones NP, … Group, R. S. (2007). Rosiglitazone evaluated for cardiovascular outcomes--an interim analysis. N Engl J Med, 357(1), 28–38. doi: 10.1056/NEJMoa073394 [DOI] [PubMed] [Google Scholar]
- Isarangkul D, Wiyakrutta S, Kengkoom K, Reamtong O, & Ampawong S (2015). Mitochondrial and cytoskeletal alterations are involved in the pathogenesis of hydronephrosis in ICR/Mlac-hydro mice. Int J Clin Exp Med, 8(6), 9192–9204. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/26309577 [PMC free article] [PubMed] [Google Scholar]
- Kalyani RR, & Egan JM (2013). Diabetes and altered glucose metabolism with aging. Endocrinol Metab Clin North Am, 42(2), 333–347. doi: 10.1016/j.ecl.2013.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegel AJ, Baker MA, Liu Y, Liu P, Cowley AW Jr., & Liang M (2015). Endogenous microRNAs in human microvascular endothelial cells regulate mRNAs encoded by hypertension-related genes. Hypertension, 66(4), 793–799. doi: 10.1161/HYPERTENSIONAHA.115.05645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, & Sadoshima J (2010). NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A, 107(35), 15565–15570. doi: 10.1073/pnas.1002178107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, … Clayton DA (1998). Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet, 18(3), 231–236. doi: 10.1038/ng0398-231 [DOI] [PubMed] [Google Scholar]
- Lassegue B, & Griendling KK (2010). NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol, 30(4), 653–661. doi: 10.1161/ATVBAHA.108.181610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Harris FL, & Brown LA (2014). Alcohol induced mitochondrial oxidative stress and alveolar macrophage dysfunction. Biomed Res Int, 2014, 371593. doi: 10.1155/2014/371593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoff AM, Wolski K, Nicholls SJ, & Nissen SE (2007). Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA, 298(10), 1180–1188. doi: 10.1001/jama.298.10.1180 [DOI] [PubMed] [Google Scholar]
- Maurice NM, Bedi B, Yuan Z, Goldberg JB, Koval M, Hart CM, & Sadikot RT (2019). Pseudomonas aeruginosa Induced Host Epithelial Cell Mitochondrial Dysfunction. Sci Rep, 9(1), 11929. doi: 10.1038/s41598-019-47457-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AJ, & Guidot DM (2012). Alcohol abuse, the alveolar macrophage and pneumonia. Am J Med Sci, 343(3), 244–247. doi: 10.1097/MAJ.0b013e31823ede77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AJ, Yeligar SM, Elon L, Brown LA, & Guidot DM (2013). Alcoholism causes alveolar macrophage zinc deficiency and immune dysfunction. Am J Respir Crit Care Med, 188(6), 716–723. doi: 10.1164/rccm.201301-0061OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilyansky E, & Rigoutsos I (2013). The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ, 20(12), 1603–1614. doi: 10.1038/cdd.2013.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris NL, Hammer AM, Cannon AR, Gagnon RC, Li X, & Choudhry MA (2017). Dysregulation of microRNA biogenesis in the small intestine after ethanol and burn injury. Biochimica et biophysica acta, 1863(10 Pt B), 2645–2653. doi:S0925-4439(17)30113-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss M (2005). Epidemiology of sepsis: race, sex, and chronic alcohol abuse. Clin Infect Dis, 41 Suppl 7, S490–497. doi: 10.1086/432003 [DOI] [PubMed] [Google Scholar]
- Moss M, Parsons PE, Steinberg KP, Hudson LD, Guidot DM, Burnham EL, … Cotsonis GA (2003). Chronic alcohol abuse is associated with an increased incidence of acute respiratory distress syndrome and severity of multiple organ dysfunction in patients with septic shock. Crit Care Med, 31(3), 869–877. doi: 10.1097/01.CCM.0000055389.64497.11 [DOI] [PubMed] [Google Scholar]
- Natarajan SK, Pachunka JM, & Mott JL (2015). Role of microRNAs in Alcohol-Induced Multi-Organ Injury. Biomolecules, 5(4), 3309–3338. doi: 10.3390/biom5043309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen SE, & Wolski K (2007). Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med, 356(24), 2457–2471. doi: 10.1056/NEJMoa072761 [DOI] [PubMed] [Google Scholar]
- Paice AG, Hesketh JE, Towner P, Hirako M, Peters TJ, & Preedy VR (2002). Alcohol increases c-myc mRNA and protein in skeletal and cardiac muscle. Metabolism, 51(10), 1285–1290. doi: 10.1053/meta.2002.34709 [DOI] [PubMed] [Google Scholar]
- Papa S, Choy PM, & Bubici C (2019). The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene, 38(13), 2223–2240. doi: 10.1038/s41388-018-0582-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Choi H, Min JS, Kim B, Lee SR, Yun JW, … Lee DS (2015). Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J Neurochem, 132(6), 687–702. doi: 10.1111/jnc.12984 [DOI] [PubMed] [Google Scholar]
- Polvani S, Tarocchi M, & Galli A (2012). PPARgamma and Oxidative Stress: Con(beta) Catenating NRF2 and FOXO. PPAR Res, 2012, 641087. doi: 10.1155/2012/641087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy VD, Padmavathi P, Kavitha G, Saradamma B, & Varadacharyulu N (2013). Alcohol-induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Molecular and cellular biochemistry, 375(1-2), 39–47. doi: 10.1007/s11010-012-1526-1 [doi] [DOI] [PubMed] [Google Scholar]
- Sampson MJ, Lovell RS, & Craigen WJ (1996). Isolation, characterization, and mapping of two mouse mitochondrial voltage-dependent anion channel isoforms. Genomics, 33(2), 283–288. doi: 10.1006/geno.1996.0193 [DOI] [PubMed] [Google Scholar]
- Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, … Madamanchi NR (2015). NOX4 NADPH Oxidase-Dependent Mitochondrial Oxidative Stress in Aging-Associated Cardiovascular Disease. Antioxid Redox Signal, 23(18), 1389–1409. doi: 10.1089/ars.2014.6221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner MC, Yeligar SM, Brown LA, & Michael Hart C (2012). PPARgamma ligands regulate NADPH oxidase, eNOS, and barrier function in the lung following chronic alcohol ingestion. Alcohol Clin Exp Res, 36(2), 197–206. doi: 10.1111/j.1530-0277.2011.01599.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox R, Bousser MG, Betteridge DJ, Schernthaner G, Pirags V, Kupfer S, … Investigators, P. R. (2007). Effects of pioglitazone in patients with type 2 diabetes with or without previous stroke: results from PROactive (PROspective pioglitAzone Clinical Trial In macroVascular Events 04). Stroke, 38(3), 865–873. doi: 10.1161/01.STR.0000257974.06317.49 [DOI] [PubMed] [Google Scholar]
- Wu D, & Cederbaum AI (2003). Alcohol, oxidative stress, and free radical damage. Alcohol Res Health, 27(4), 277–284. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/15540798 [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhou F, Zhang Z, & Xing D (2011). Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J, 278(6), 941–954. doi: 10.1111/j.1742-4658.2011.08010.x [DOI] [PubMed] [Google Scholar]
- Xiao J, Lv Y, Lin B, Tipoe GL, Youdim MB, Xing F, & Liu Y (2015). A novel antioxidant multitarget iron chelator M30 protects hepatocytes against ethanol-induced injury. Oxid Med Cell Longev, 2015, 607271. doi: 10.1155/2015/607271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeligar SM, Harris FL, Hart CM, & Brown LA (2012). Ethanol induces oxidative stress in alveolar macrophages via upregulation of NADPH oxidases. J Immunol, 188(8), 3648–3657. doi: 10.4049/jimmunol.1101278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeligar SM, Harris FL, Hart CM, & Brown LA (2014). Glutathione attenuates ethanol-induced alveolar macrophage oxidative stress and dysfunction by downregulating NADPH oxidases. Am J Physiol Lung Cell Mol Physiol, 306(5), L429–441. doi: 10.1152/ajplung.00159.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeligar SM, Kang BY, Bijli KM, Kleinhenz JM, Murphy TC, Torres G, … Hart CM (2018). PPARgamma Regulates Mitochondrial Structure and Function and Human Pulmonary Artery Smooth Muscle Cell Proliferation. Am J Respir Cell Mol Biol, 58(5), 648–657. doi: 10.1165/rcmb.2016-0293OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeligar SM, Machida K, Tsukamoto H, & Kalra VK (2009). Ethanol augments RANTES/CCL5 expression in rat liver sinusoidal endothelial cells and human endothelial cells via activation of NF-kappa B, HIF-1 alpha, and AP-1. J Immunol, 183(9), 5964–5976. doi: 10.4049/jimmunol.0901564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeligar SM, Mehta AJ, Harris FL, Brown LA, & Hart CM (2016). Peroxisome Proliferator-Activated Receptor gamma Regulates Chronic Alcohol-Induced Alveolar Macrophage Dysfunction. Am J Respir Cell Mol Biol, 55(1), 35–46. doi: 10.1165/rcmb.2015-0077OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yki-Jarvinen H (2004). Thiazolidinediones. N Engl J Med, 351(11), 1106–1118. doi: 10.1056/NEJMra041001 [DOI] [PubMed] [Google Scholar]
- Zhong W, Zhao Y, Tang Y, Wei X, Shi X, Sun W, … Zhou Z (2012). Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am J Pathol, 180(3), 998–1007. doi: 10.1016/j.ajpath.2011.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Hu G, Gong AY, & Chen XM (2010). Binding of NF-kappaB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic Acids Res, 38(10), 3222–3232. doi: 10.1093/nar/gkq056 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.