

Abstract
Purpose:
Extracellular matrix (ECM) component hyaluronan (HA) facilitates malignant phenotypes of glioblastoma (GBM), however, whether HA impacts response to GBM immunotherapies is not known. Herein, we investigated whether degradation of HA enhances oncolytic virus immunotherapy for GBM.
Methods:
Presence of HA was examined in patient and murine GBM. Hyaluronidase-expressing oncolytic adenovirus ICOVIR17, and its parental virus ICOVIR15 without transgene, were tested to determine if they increased animal survival and modulated the immune tumor microenvironment (TME) in orthotopic GBM. HA regulation of NF-kB signaling was examined in virus-infected murine macrophages. We combined ICOVIR17 with PD-1 checkpoint blockade and assessed efficacy and determined mechanistic contributions of tumor-infiltrating myeloid and T cells.
Results:
Treatment of murine orthotopic GBM with ICOVIR17 increased tumor-infiltrating CD8+ T cells and macrophages, and upregulated PD-L1 on GBM cells and macrophages, leading to prolonged animal survival, compared to control virus ICOVIR15. High-molecular weight HA inhibits adenovirus-induced NF-kB signaling in macrophages in vitro, linking HA degradation to macrophage activation. Combining ICOVIR17 with anti-PD-1 antibody further extended the survival of GBM-bearing mice, achieving long-term remission in some animals. Mechanistically, CD4+, CD8+ T cells and macrophages all contributed to the combination therapy that induced tumor-associated pro-inflammatory macrophages and tumor-specific T cell cytotoxicity locally and systemically.
Conclusions:
Our studies are the first to show that immune-modulatory ICOVIR17 has a dual role of mediating degradation of HA within GBM-ECM and subsequently modifying the immune landscape of the TME, and offers a mechanistic combination immunotherapy with PD-L1/PD-1 blockade that remodels innate and adaptive immune cells.
Introduction
Cancer is increasingly being viewed as an ecosystem, in which neoplastic cells, a variety of non-neoplastic cells, and extracellular matrix (ECM) interact with each other to sustain and promote tumor growth (1,2). Glioblastoma (GBM), the most malignant primary brain tumor in adults, represents one of the cancers in which complex crosstalk between both cellular and non-cellular components in the tumor microenvironment (TME) plays a major role in shaping the treatment-refractory nature of the tumors. Tumor-associated macrophages (TAMs) dominate the immune cells that infiltrate GBM, and contribute to the characteristic tumor-supportive, immuno-suppressive TME of GBM that is largely depleted of functional effector T cells (3–5). Lack of durable efficacy of the current multimodal standard-of-care for GBM, i.e., surgical resection followed by irradiation and chemotherapy (6), could be due in part to its inability to interfere with key interactions that occur in the GBM TME to resist therapy and sustain tumor progression.
ECM is now known to actively contribute to tumor maintenance by interacting with tumor cells as well as cellular components in the TME (7–10). Tumor ECM comprises proteins (e.g., collagen, fibronectin, and laminin) and non-proteins such as hyaluronan (HA), also known as hyaluronic acid or hyaluronate (11). The glycosaminoglycan HA is a major constituent of ECM that normally exists as a high molecular weight (HMW) form (> 1000 kDa). HA regulates proliferation and invasion of tumor cells by binding its cognate cell surface receptors, CD44 and RHAMM, and affects the activity of chemotherapy (7,12). Accumulation of HA is closely related to tumor aggressiveness and poor outcome in various malignancies such as breast (13), colorectal (14), gastric (15), prostate (16), pancreatic (17) and ovarian cancer (18). In GBM, HA is reported to be associated with tumor growth, invasion, and resistance to treatment (7). The relationship between HA and the immune system has been extensively studied in inflammatory conditions such as autoimmune diseases and chronic arthritis (19–24). HA regulates the function of macrophages via binding CD44, RHAMM and TLR2/4 (25,26). In cancer, HMW-HA has been shown to inhibit M1-polarization or induce M2-polarization in macrophages (25,27–30). However, the role of HA in GBM immune surveillance and immunotherapy has not been explored.
Oncolytic virus (OV) is an attractive modality for not only killing cancer cells but also modifying TME. The field of OV gained momentum with FDA approval of talimogene laherparepvec for advanced melanoma (31), and a variety of genetically modified viruses is under active clinical development. The fundamental mechanism-of-action of OV cancer therapy is that OV replication-mediated cell death of neoplastic cells releases tumor antigens and triggers immune and inflammatory responses, leading to the induction of anti-tumor cellular immune responses (32–37). In addition, OV can be armed with therapeutic genes of interest to increase anti-cancer potency (32). Genetically engineered oncolytic adenovirus is one of the most studied and promising OVs (38,39), and multiple clinical trials are currently ongoing for patients with GBM to investigate the safety and efficacy of oncolytic adenovirus Delta-24-RGD (DNX-2401) (NCT03714334, NCT03178032, NCT01956734, NCT02798406)(40). A phase 1 study of Delta-24-RGD in recurrent malignant glioma reported an encouraging sign of benefit, as 20% of patients survived >3 years from treatment (41). ICOVIR17 is an oncolytic adenovirus with the same modifications as Delta-24-RGD, i.e., a 24-base pair deletion in the Rb-binding domain of E1A for tumor-selective replication and an RGD-modification in the fiber for widening tropism, but with two additional modifications: insertion of E2F binding sites in the E1A promoter and the SPAM1 gene encoding PH20 hyaluronidase after the fiber, controlled by the major late promoter (42). Preclinically, we previously demonstrated that treatment of orthotopic GBM xenografts with ICOVIR17 mediated the degradation of HA in GBM ECM, resulting in increased virus spread within GBM and superior anti-tumor efficacy compared with its parental virus ICOVIR15 without the transgene (42,43). While this work revealed the function of GBM HA as a physical barrier to effective virus dispersal and tumor killing, whether HA impacts immune responses elicited by oncolytic adenovirus therapy of brain tumors remains unknown because the mice used in the studies were immunodeficient.
The major goal of the current study was to address our hypothesis that degradation of HA would enhance oncolytic adenovirus immunotherapy of GBM by overcoming the immunosuppressive functions of GBM ECM. We selected the murine GBM 005 as a suitable in vivo model since this GBM model recapitulates the critical hallmarks of human disease including GBM stem cell (GSC) properties and immuno-suppressive TME in an immunocompetent setting (44–46). Characterization of immunological changes in GBM revealed that HA degradation by ICOVIR 17 induced distinct immune activation in situ, which contributed to its therapeutic effect. This TME modification provided a mechanistic rationale to combine ICOVIR17 with immune checkpoint blockade to yield durable responses in aggressive GBM.
Materials and Methods
Viruses
Oncolytic adenoviruses ICOVIR15 and 17 were generated by Dr. Ramon Alemany’s group and described previously (42,47). Viruses were amplified in A549 cells and purified using CsCl ultracentrifuge. Virus preparation and titration was performed following (48).
Cell lines
Mouse glioma cell lines
Mouse 005 GSCs (GFP+), a gift from Dr. Inder Verma, were established from glioblastoma generated with lentiviral transduction of H-Ras and activated Akt in Cre-GFAP / p53+/− mice of a somewhat mixed (C57BL/6 and some FVB/N) background (45). They were cultured as spheres in Neurobasal medium (Invitrogen) supplemented with L-Glutamine (3 mM; Mediatech), B27 supplement (Invitrogen), N2 supplement (Invitrogen), heparin (5 μg/mL; Sigma), EGF (20 ng/mL; R and D systems), and FGF2 (20 ng/mL; Peprotec). Spheres were passaged using Accutase (Innovative Cell Technologies). Mouse CT-2A, obtained from Dr. Thomas Seyfried (49) and GL261 glioma cells, obtained from National Cancer Institute, were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum, and passaged using 0.05% Trypsin/0.53mM EDTA (Gibco).
Human glioma cell lines
Human primary GBM neurosphere lines (MGG4, MGG8) were established as described previously (50) and cultured in EF media composed of Neurobasal medium supplemented with 3 mmol/L L-Glutamine, 1% B27 supplement, 0.5% N2 supplement, 2 μg/mL heparin, 20 ng/mL recombinant human EGF, 20 ng/mL recombinant human FGF2, and 0.5% penicillin G/streptomycin sulfate/amphotericin B complex (Mediatech). Neurospheres were passaged using TrypLE (Gibco).
Cancer cell lines
Human lung carcinoma cell line A549 was obtained from American Type Culture Collection and grown in DMEM with 10% calf serum. Mouse melanoma B16.F10 cell line was provided by Dr. David Fisher at Massachusetts General Hospital (MGH). L929 cells were provided by Dr. Junying Yuan lab at Harvard Medical School, Boston, MA. 005-GFP-Fluc was generated by transduction of 005 cells with a lentivirus expressing GFP and firefly luciferase (Fluc). To generate B16.F10-mCherry-Fluc, B16.F10 cells were infected with a mCherry-Fluc lentivirus and mCherry-positive cells were sorted by a FACSAriaII flow cytometer. B16.F10-GFP was generated using a GFP-puromycin lentivirus and transduced cells were selected with puromycin. All cells were regularly confirmed to be mycoplasma-free (LookOut mycoplasma kit; Sigma).
In vivo mouse studies
Female C57BL/6 mice (aged 7–9 weeks) were obtained from Charles River Laboratories (Wilmington, MA). 005 GSCs (1.0×105 cells/mouse) were implanted stereotactically into the right striatum of the brain as described in (44,50). For survival study, mice were monitored for health status and sacrificed when neurological deficits and weight loss became significant.
Tissue processing
Brains were harvested at indicated time points and fixed in 10% formalin, embedded in paraffin and sectioned at 7 μm thickness. For frozen sections, mice were perfused with cold 4% paraformaldehyde directly into the heart, and brains were fixed in 4% paraformaldehyde overnight, replaced with 30% glucose and 7 μm thick cryo-sections prepared.
Hematoxylin and eosin staining
Sections were deparaffinized in xylene (twice, 10 minutes each), followed by gradual rehydration using 100%, 90% and 70% ethanol and PBS (5 minutes each). Slides were dipped in hematoxylin (Gill3, Millipore Sigma, 105174) for 20 sec, then 0.1% HCl for 2 sec, washed in tap water for 5 minutes, dipped into Eosin Y (1% alcoholic, Fisher Scientific, 7231) for 20 seconds, and followed by dehydration using 95% ethanol (twice, 5 minutes each) and 100% ethanol (twice, 5 minutes each). After treatment with xylene twice for 10 minutes each, they were mounted in xylene-based media (Cytoseal XYL, ThermoFisher Scientific, 8312–16E).
Immunohistochemistry (IHC)
Sections were deparaffinized in xylenes (twice, 10 minutes each), followed by gradual rehydration using 100%, 90% and 70% ethanol (5 minutes each). After 5 min PBS wash, sections were heated in 10mM Na citrate buffer (pH6.0) for 15 min with microwave for antigen retrieval, washed with PBS twice, and incubated with 3% H2O2 for 5 min to block the endogenous peroxidase or incubated with BLOXALL (Vector, SP-6000) for 10 min to block the endogenous alkaline phosphatase. After PBS washes three times, sections were incubated with blocking buffer (2.5% normal horse serum, Vector, S-2012) for 60 min at room temperature and with properly diluted primary antibodies over night at 4C. Next day, they were washed with PBS three times, and incubated with HRP or AP conjugated Ig for 30 min at room temperature. After a PBS wash, sections were incubated with DAB substrate (DAKO, K3468), followed by hematoxylin counter-staining and mounting. CD34 immunofluorescence was done on paraformaldehyde-fixed frozen sections of 005 tumors.
Histochemical staining of hyaluronan (HA staining)
Sections were deparaffinized in xylenes and dehydrated in the serial dilutions of ethanol, and the endogenous peroxidase activity was blocked by 3% H2O2. Sections were incubated with 2.5% Normal horse serum (Vector, S2012) for 60 min at room temperature for blocking, and with 5 μg/ml of a biotinylated HA-binding protein (Calbiochem, 385911, HABP-b) overnight at 4C. The specificity of HA staining was tested by pretreating an adjacent section with 20 U/ml of bovine testes hyaluronidase (Sigma, H3506) at 37 °C for 1 hour, prior to the addition of the HABP-b. The next day, sections were washed in PBS and treated with avidin–biotin–peroxidase kit (VECTASTAIN Elite ABC HRP kit, Vector, PK-6100) for 30 minutes at room temperature. After washes, sections were developed with DAB (Dako, K3468) and counterstained with hematoxylin. Quantification of HA staining was performed using Photoshop (Adobe) and Image J (NIH).
Double staining of HA and immune cells
For double IHC and HA staining, IHC for immune cell markers (CD3, CD4, CD8 or CD68) were performed first with DAB as described above, followed by three PBS washes and incubation with blocking buffer (2.5% normal horse serum) for 60 min at room temperature. The slides were then incubated with HABP-b. The subsequent HA staining procedures followed using VECTASTAIN ABC-AP kit (Vector, AK-5000) and ImmPACT red AP substrate (Vector, SK-5105).
Cell viability assay
Dissociated cells were seeded into 96-well plates, serially diluted viruses were added and cells were cultured for 3 or 7 days. To measure cell viability, 20 μl of Cell Titer Glo Luminescent cell viability reagent (Promega, G7570) was added into each well, and plates were shaken in the dark for 5 min. Luminescent values were measured by a microplate reader with the Gen5 software (BioTek). Experiments were performed in triplicate.
RT-PCR
Total RNA was isolated from cells using Trizol reagent (Invitrogen) and cDNA was synthesized by reverse transcriptase reaction with High Capacity cDNA RT kit (Applied Biosystems, 4368814). Real time PCR was conducted using SYBR select master mix (Applied Biosystems, 4472908) in a StepOnePlus Real-time PCR System (Applied Biosystems). PCR primer sequences are: PH20 (HSPAM) forward: AAACTGTTGCTCTGGGTGCT, reverse: TTTTGGCTGCTAGTGTGACG, GAPDH forward: CAATGACCCCTTCATTGACC, reverse: GACAAGCTTCCCGTTCTCAG), and mouse beta actin forward: GATCTGGCACCACACCTTCT, reverse: GGGGTGTTGAAGGTCTCAAA.
Bone marrow-derived macrophages (BMDM)
Femurs and tibias were collected from 7–8 weeks old C57BL/6 mice. Bone marrows were harvested by flashing R10 media (RPMI1640 containing 10% heat-inactivated FCS) using a 24G needle and a 5-ml syringe. Bone marrow cells were run through a 70 μm strainer, centrifuged, resuspended in BMDM media (DMEM containing 20% heat-inactivated FCS, 30% L929 cells supernatant and 1% Penicillin/Streptomycin) and seeded at 5 million cells in a 10-cm dish. Media was changed fresh on day 1, 3 and 6, and cells were split or frozen when they became 80–90% confluent (at around day7). BMDMs were used from day 6 to 14 cultures for experiments.
NF-kB signaling activation in BMDM
BMDMs were seeded on coverslips at 50,000 cells/well (24-well plate) and incubated with 2000 μg/ml of LMW-HA or HMW-HA (R and D systems, GLR-001 and 002) for 24h at 37 °C. LPS (100 pg/ml, Sigma-Aldrich, L4391) or ICOVIR15 (MOI 50) was added and incubated for 1 hour or 6 hours at 37C, respectively. Control samples were incubated without HA or stimulators. BMDMs were washed with PBS and fixed with 4% PFA, permeabilized with 0.1% Triton X and blocked with 10% normal goat serum, then incubated with anti-p65 antibody, followed by incubation with Alexa Fluor 488-conjugated secondary antibody. Nuclei were stained with DAPI. Five to eight pictures (20x magnification) were randomly captured under a fluorescence microscope and used for counting cells with and without nuclear-translocation of p65 (> 100 cells per group).
Gene expression analysis (NanoString)
C57BL/6 mice were implanted with 005 GSC (1.0×105 cells/mouse), and treated with intratumoral injection of ICOVIR17 (1.6 × 107 PFU/mouse) or PBS on days 13 and 18. Anti-mouse PD-1 antibody (BioXCell, BE0146, clone RMP1–14, 12.5 mg/kg) or isotype rat IgG control (Sigma, l4131, 12.5 mg/kg) was injected intraperitoneally (ip) on days 14, 17 and 19 (4 groups, n=3/group). On day 20, mice were euthanized and tumors were collected. Total RNA was extracted from tumor tissues using RNeasy mini kit (Qiagen, 74104) following the manufacturer’s protocol, and kept at −80 °C until analysis. Gene expression analysis was performed by the NanoString Mouse PanCancer Immune Panel (115000142) and Mouse Myeloid Innate Immunity Panel (115000181). 110 ng/mouse of total RNA was hybridized with reporter codeset and capture probeset following the protocol provided by NanoString. Hybridized samples were prepared using the automated nCounter prep station and analyzed by Set up Digital Analyzer (nCounter Max Analysis System). Data were processed using nSolver analysis software and the nCounter digital analysis module. The housekeeping genes expressions were used for the normalization of the gene expression analysis.
Immune cell depletion studies
C57BL/6 mice (N = 6/group) were implanted with 005 cells in the right hemisphere on day 0 and treated with 1.6×107 PFU of ICOVIR17 or PBS on days 9, 15 and anti-mouse PD-1 antibody (12.5 mg/kg) or isotype rat IgG (12.5 mg/kg) on days 10, 13, 16, 19. For depletion of immune cell populations, mice were administered ip injections of anti-mouse CD8a (BioXcell, BE0061, clone 2.43; 10 mg/kg), anti-mouse CD4 (BioXcell, BE0003–1, clone GK1.5; 10 mg/kg) on days 5, 8, 11, 14, 21 and 27. Clodronate liposomes (Clodrosome from Encapsula Nano Sciences, CLD8901) were given on days 8, 11, 14 and 17 at 50 mg/kg for the first injection, followed by 25 mg/kg. Mock group received isotype control rat IgG and empty liposomes (Encapsome from Encapsula Nano Sciences, CLD8910).
Multicolor flow cytometry (FCM)
Mice were implanted with 005 GSCs (1.0 × 105 cells/mouse) and treated with intratumoral injection of ICOVIR17 (day 9: 1.6 × 107 PFU/mouse, day 14: 1.0 × 107 PFU/mouse) or PBS on days 9 and 14. Anti-mouse PD-1 antibody or isotype rat IgG (as control)(12.5 mg/kg) was injected ip on days 10, 13, 16, 19. Mice were euthanized on day 21. Tumor tissues or spleens were harvested from mice and mashed through a 100 μm strainer. For splenocytes, red blood cells were lysed using Mouse RBC lysis buffer (Boston BioProducts, IBB-198). Live/dead cell discrimination was performed using Zombie UV™ Fixable viability kit (BioLegend, 423108). Cells were incubated with FcR blocking reagent (Milteneyi Biotec, 130-092-575) followed by cell surface staining with fluorochrome-conjugated anti-mouse antibodies. Staining of intracellular FOXP3 was done after fixation and permeabilization by FOXP3 Fix/Perm buffer set (BioLegend, 421403), while other intracellular staining was done using BD Cytofix/Cytoperm™ Fixation/Permeabilization kit (BD, 554714). Samples were run in an LSR II flow cytometer (BD) and data was analyzed using FlowJo software (v.10.1, Tree Star).
Antibodies are listed in Supplementary Table S1.
T cell isolation and cytotoxicity assay
Tumor infiltrated lymphocytes.
C57BL/6 mice were implanted with 005 GSC (1.0 × 105 cells/mouse), and treated with intratumoral injection of ICOVIR17 (1.6 × 107 PFU/mouse) or PBS on days 13 and 18. Anti-mouse PD-1 antibody or isotype rat IgG (both 12.5 mg/kg) were injected ip on days 14, 17, 20 and 23 (control group: n=4; combination group: n=3). On day 25, mice were euthanized and right hemisphere quadrants containing tumor were collected, and tumors in the same group were pooled. Samples were dissociated with p1000 pipetting and passed through a 100 μm strainer. After washes with PBS, cells were resuspended in cold PBS, and debris removed using Debris removal solution (Myltenyi Biotec, 130-109-398). To isolate CD4/8 positive cells, cells were resuspended in microbeads isolation buffer (PBS containing 0.5% FCS and 2 mM EDTA) and incubated with mouse CD4/CD8(TIL) MicroBeads (Miltenyi Biotec, 130-116-480) at 4°C for 15 min. After a wash with isolation buffer, magnetic isolation was performed with MiniMACS separator (Miltenyi Biotec, 130-042-102) and MS columns (Miltenyi Biotec, 130-042-201). Serial dilutions of CD4 / CD8 cells were seeded to a 96-well plate pre-seeded with 005-Fluc and B16.F10-Fluc cells at effector to target (E:T) ratios of 2:1, 10:1 and 50:1, and incubated at 37C and 5% CO2 for 24 h. Cell viability of Fluc-expressing tumor cells were measured with a microplate reader after adding 10 μl of D-luciferin (20 mM) to each well.
Splenocytes.
Mice were implanted with 005 GSCs (1.0 × 105 cells/mouse), and treated with intratumoral injection of ICOVIR17 (1.6 × 107 PFU/mouse) or PBS on days 9 and 14. Anti-mouse PD-1 antibody or isotype rat IgG (both 12.5 mg/kg) were injected ip on days 10, 13, 16, and 19. On day 21, spleens were collected, passed through a 70 μm strainer, and incubated with mouse RBC lysis buffer (Boston BioProducts, IBB-198) to remove erythrocytes. Splenocytes were cultured in RPMI with 10% FCS and IL-2 (100 U/ml) for 48 h at 37°C and 5% CO2, and seeded in 96-well plates pre-seeded with 005-Fluc or B16.F10-Fluc cells for different E: T ratios. After 24 h-co-culture, cell viability of tumor cells was measured by luciferase assay.
Intracellular FCM of splenocytes.
Mice were treated as for TILs above. Spleens were collected on day 25. Splenocytes were incubated with 100 U/ml of IL-2 for 48 hours and seeded in 6-well plates with 005 or B16.F10-GFP at the ET ratio of 16: 1. After 24 h co-culture, cells were stained with fluorophore-conjugated antibodies (surface: CD4 and CD8, intracellular: Granzyme B and interferon gamma), and subjected to FCM. Brefeldin A (GolgiPlug from BD Biosciences) was added to cultures 5 h before harvest of cells to block protein secretion.
Statistical analysis
All statistical analyses were performed using Prism 8 software (GraphPad, v8.0.2) Survival data were analyzed by Kaplan-Meier survival curves, and comparisons were performed by log-rank test followed by correction for multiple comparisons with the Bonferroni method. Cell viability data, flow cytometric data, and immunohistochemistry counts were compared using an unpaired Student’s t test (two tailed). P values of less than 0.05 were considered significant.
Study approval
All mouse procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at MGH. Excess glioma tissue at neurosurgery at MGH that would have been otherwise discarded was collected in a coded manner in accordance with Declaration of Helsinki, and approved by the Institutional Review Board at MGH.
Results
Hyaluronan is abundant in the extracellular matrix of glioblastoma with low immune cell infiltration.
We first used clinical specimens to examine the presence of HA in human brain and GBM. Immunohistochemistry (IHC) showed that HA was present in human brain tissue (Fig. 1A), as previously reported (51,52). In patient GBM specimens, HA was abundantly present with heterogeneous distribution (Fig. 1B and C). Using double staining of HA and T cell marker CD3, we observed that T lymphocytes infiltrating GBM are often present in areas where HA content is negative to low, as opposed to high HA areas typically lacking CD3+ cells (Fig. 1D), suggesting the ability of HA to modify their distribution.
Figure 1. Hyaluronan (HA) is abundant in the extracellular matrix of glioblastoma.
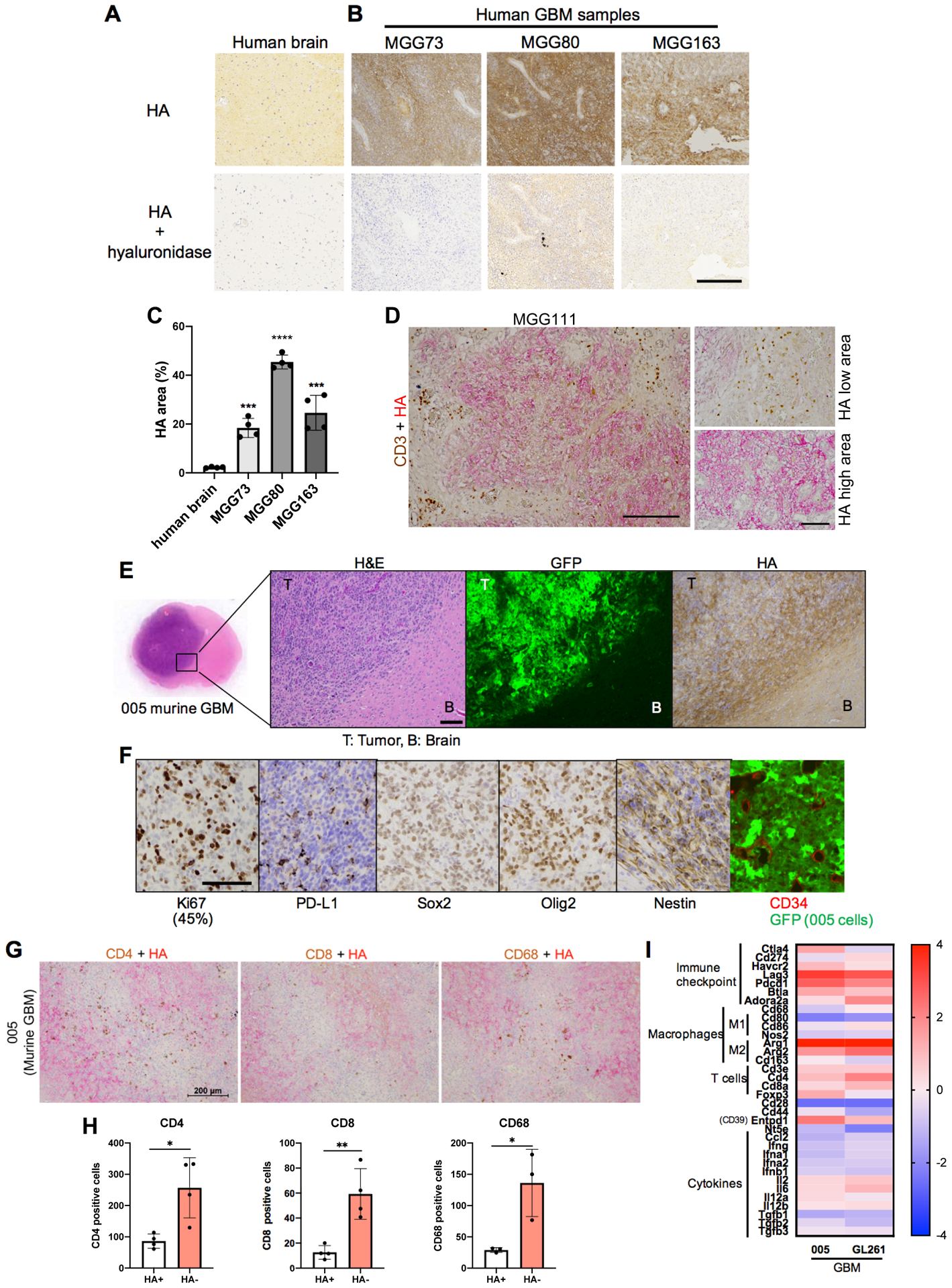
A and B, HA staining in human normal brain (A) and glioblastoma samples (B). Bovine hyaluronidase was applied before HA staining for negative control samples. Scale bar=500 μm. C, Image J quantification of HA area (%) using 4 microscopic fields per tumor shown in A and B. Data are mean ± SD. *** p<0.005, **** p<0.0001 with unpaired t-test (two-tailed) compared with human brain. D, Double staining of HA (red) and CD3 (brown) in human glioblastoma. Left, Lower magnification microscopic field containing both HA-high and HA-low areas. Scale bar=200 μm. Right, Higher magnification images showing HA-low and HA-high areas. Scale bar=100 μm. E-I, Characterization of murine 005 glioblastoma in C57BL/6 mice. E, A brain coronal section with a magnified view at the tumor border showing Hematoxylin and Eosin staining (H&E), GFP-labeled 005 cells, and HA staining. T, tumor; B, brain. Scale bar=100 μm. F, Immunohistochemistry and immunofluorescence for marker gene expression. Scale bar=100 μm. G, Double staining of immune cell markers (CD4, CD8 and CD68 in brown) and HA (in red) in murine glioblastoma 005. Nuclear counterstaining with hematoxylin. H, Quantification of immune cells based on the images shown in G. Data are mean ± SD. * p<0.05, ** p<0.01 with unpaired t-test (two-tailed). I, NanoString gene expression analysis of orthotopic murine glioblastoma models (005, GL261). Gene expression relative to that of murine melanoma (D4M3A) is presented. Scale on the right is log2 fold change.
We therefore set out to study the potential impact of HA and its degradation on OV immunotherapy for GBM. We previously showed that 005 cells have the properties of GBM stem cells and generate a GBM model in C57BL/6 mice that is suitable for the development of OV immunotherapy (44,46). 005 cells stably expressing GFP form hypercellular GBM in the brain that contains rich HA heterogeneously present within the tumor (Fig. 1E). Further phenotypic characterization of the tumor revealed a high Ki-67 proliferation index (45 %), and high levels of stem/progenitor cell markers, Sox2, olig2 and nestin, characterizing neoplastic cells (Fig. 1F). A substantive fraction of scattered PD-L1+ positive cells and CD34+ vascularity were noted within the tumor (Fig. 1F). Dual staining of HA and immune cell markers demonstrated preferential presence of CD4+, CD8+ T cells and CD68+ macrophages in areas containing no to low HA (Fig. 1G and H), as was observed in clinical GBM. We next used NanoString transcriptomic analysis to define the immunological landscape of intracerebral 005 GBM in vivo. When a murine immunocompetent melanoma model was used as a reference (53), 005 as well as the commonly used GL261 GBM model (54) were noted for higher expression of T cell immune checkpoints (e.g., Lag3 and Pdcd1), adenosine signaling (Entpd1 encoding CD39) and the M2 polarized macrophage marker Arg1, indicative of the immunosuppressive status of the TME in these GBM models (Fig. 1I). Since CD44 is one of the major cell surface receptors for HA, we examined the expression of CD44 on tumor-infiltrating immune cells. Immune cells infiltrating 005 GBM, including T cells, macrophages and microglia, as well as 005 GBM cells, were all highly positive for CD44 (Supplementary Fig. S1). Thus, HA is abundantly present in the extracellular matrix of human GBM and mouse 005 GBM, and may influence the distribution of intratumoral immune cells.
Hyaluronidase-expressing oncolytic adenovirus, ICOVIR17, has anti-GBM activity.
We tested the oncolytic properties of genetically engineered oncolytic adenoviruses ICOVIR15 and ICOVIR17 in human and murine GBM cells (42,43,47). Both ICOVIR15 and ICOVIR17 drive expression of E1A-delta24 from a promoter consisting of four palindromic E2F-binding sites and a Sp-1 binding site, conferring potency and cancer selectivity, and have a RGD modification in the fiber; ICOVIR17 additionally expresses human hyaluronidase PH20. In vitro, both viruses were comparable in killing human and murine GBM cells in a dose dependent manner, with a greater potency for human cells versus mouse cells (Supplementary Fig. S2A and B), in line with prior reports showing murine cells are less permissive to human adenovirus type 5 than human cells (55). ICOVIR17 replicated and spread much better in human cancer cells, compared with murine 005 cells, in vitro (Supplementary Fig. S2C and D). However, expression of the transgene hyualuronidase (SPAM1) was found be robust after ICOVIR17 infection of 005 cells (Supplementary Fig. S2E).
We next evaluated the anti-tumor effects of ICOVIR15 and ICOVIR17 in the orthotopic 005 GBM model in C57BL/6 mice. Because of the low susceptibility of 005 cells to the viruses, we used two injections of high titer viruses directly into the tumor (Fig. 2A). Treatment was initiated on day 11, when rapidly growing tumor was established. ICOVIR15 did not improve animal survival (median survival time (MST) 23.5 days versus 22.5 days for PBS as control, p=0.408)(Fig. 2B). Treatment with ICOVIR17 significantly increased survival to MST 28 days compared with control (p=0.037), while that did not reach statistical significance when compared with ICOVIR15 (p=0.072)(Fig. 2B). IHC analysis showed that injections of ICOVIR15 and ICOVIR17 mediated strong expression of the virus gene E1A within the tumors and that ICOVIR17, but not ICOVIR15, significantly decreased the levels of HA at the site where E1A was present, consistent with hyaluronidase-mediated degradation of HA (Fig. 2C–E). Next, we asked whether the observed anti-tumor activity of ICOVIR17 was due to its preferential spread within tumor tissue driven by HA degradation, as was seen with human GBM xenografts (43). However, we found no difference in intratumoral spread of the viruses as injections of ICOVIR15 and ICOVIR17 resulted in similarly wide-spread distribution of E1A within the tumors (Supplementary Fig. S3).
Figure 2. Intratumoral injections of ICOVIR17 degraded HA, prolonged survival, increased tumor-infiltrating immune cells and upregulated PD-L1 in mice bearing orthotopic 005 glioblastoma.
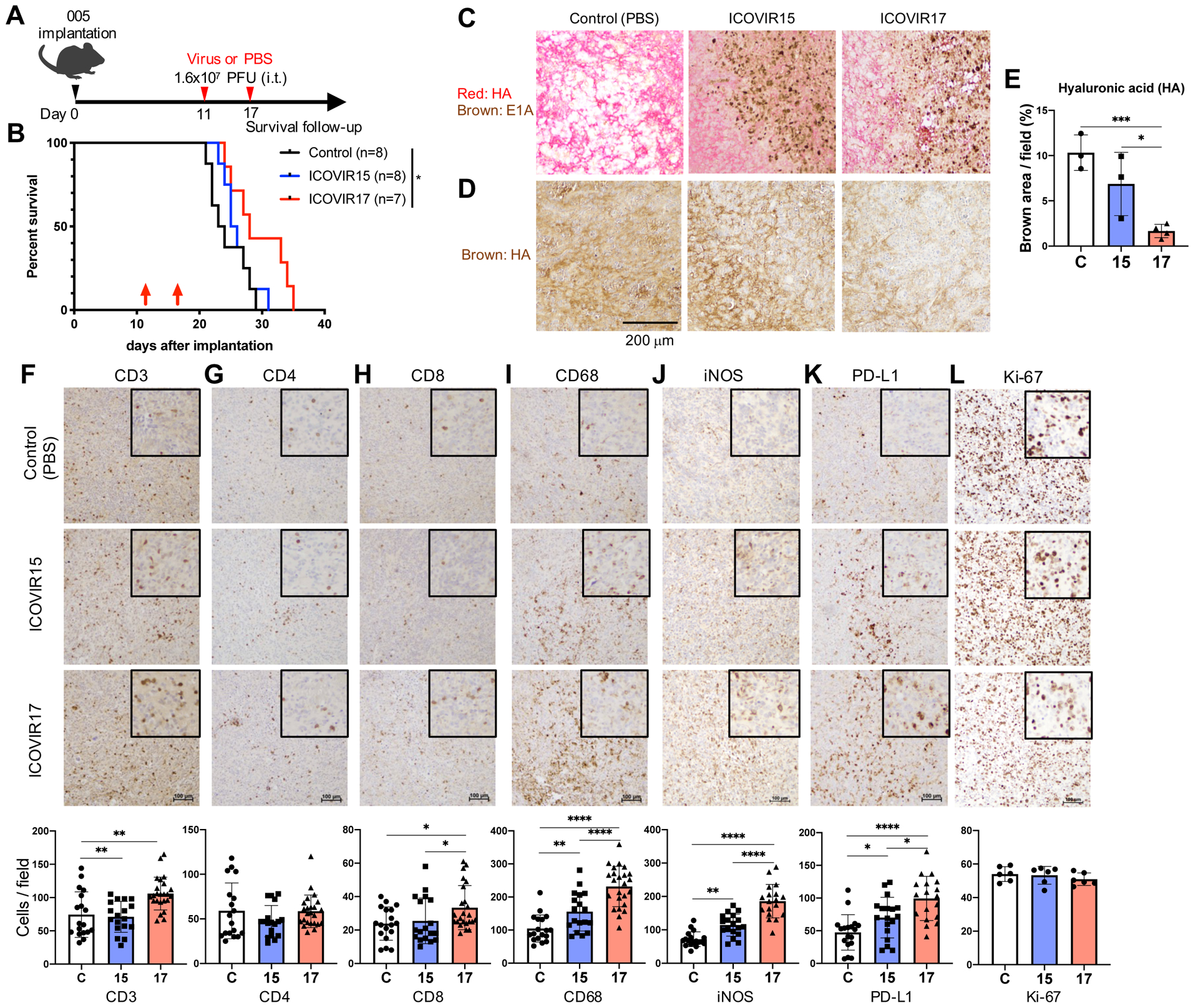
A, Treatment schema of the survival experiment. C57BL/6 mice were implanted with 005 cells into the brain (1.2 × 105 cells/mouse) on day 0. On days 11 and 17, PBS or virus (ICOVIR15 or ICOVIR17) were injected intratumorally (1.6 × 107 PFU/mouse). B, Kaplan-Meier survival analysis of 005-bearing mice treated with PBS (control), ICOVIR15, or ICOVIR17. Arrows indicate treatments. * p<0.05 (log-rank analysis) comparing control and ICOVIR17. C, Double staining of hyaluronic acid (HA, red) and E1A (adenovirus early gene, brown) at 5 days after single virus injection. Scale bar=200 μm. D, HA staining (HA, brown) Scale bar=200 μm. E, Quantification of HA area. * p<0.05, *** p<0.005 with unpaired t-test (two-tailed). F-L, Immunohistochemistry of immune markers, CD3 (F), CD4 (G), CD8 (H), CD68 (I), iNOS (J), PD-L1 (K), and Ki-67 (L), in 005 glioblastoma treated with PBS (Control), ICOVIR15, or ICOVIR17. Scale bar, 100 μm. Insets: higher magnification images to show details of staining. Quantification is shown below for each marker. Number of positive cells from randomly chosen 6 fields/tumor section/mouse were counted, and the mean ± SD of all fields across the mice (N=4/group) are presented. C: Control group, 15: ICOVIR15-treated group, 17: ICOVIR17-treated group. Data were analyzed by unpaired t test (two-tailed); * p<0.05, ** p<0.01, **** p<0.0001.
ICOVIR17 increases tumor-infiltrating immune cells and upregulates PD-L1.
We hypothesized that ICOVIR17-mediated alteration of immune TME might underlie the efficacy of its monotherapy. IHC analysis of immune markers revealed a significant increase in the numbers of tumor-infiltrating CD3 and CD8 cells, but not CD4 cells, in ICOVIR17-treated 005 GBM tumors compared to PBS control and ICOVIR15 treated tumors (Fig. 2F–H). Tumor-infiltrating macrophages marked by CD68 and iNOS-positive cells representing M1-polarized TAMs were increased by ICOVIR15, which was further increased by ICOVIR17 (Fig. 2I and J). Similarly, ICOVIR15 mediated an increase in cells immuno-positive for the immune checkpoint protein PD-L1, which was furthered by ICOVIR17 (Fig. 2K). There was no change in the index of proliferation marker Ki-67 between the groups (Fig. 2L). After treatment with ICOVIR17, CD68+ TAMs accumulated in areas lacking HA as opposed to areas that retained HA, while CD4+ and CD8+ cells were distributed similarly (Supplementary Fig. S4). Thus, ICOVIR17-mediated degradation of HA drastically altered the immune TME in this GBM model.
High-molecular weight HA inhibits virus-induced NF-kB signaling in macrophages.
Transcriptomic analysis using the Nanostring cancer immunology panel revealed that most of known LMW-HA-induced genes (56) were upregulated in ICOVIR17-treated tumors (Fig. 3A). Many of these LMW-HA-induced genes were driven by the NF-kB signaling pathway, and transcript levels of NF-kB target genes, listed at https://www.bu.edu/nf-kb/gene-resources/target-genes/ (Supplementary Table S2), were, in general, also increased in tumors after ICOVIR17 injections (Fig. 3B). Low molecular weight (LMW) HA (50–250 kDa), but not high molecular weight (HMW) HA (> 900 kDa), has been shown to act as an TLR2/4 agonist, activating NF-kB signaling pathway and triggering activation of inflammatory genes (57). These findings led us to hypothesize that virus-induced degradation of tumor HA might impact molecular signaling pathways in immune cells.
Figure 3. HMW-HA and LMW-HA differentially impact adenovirus-induced NFkB activation in bone marrow derived macrophages.
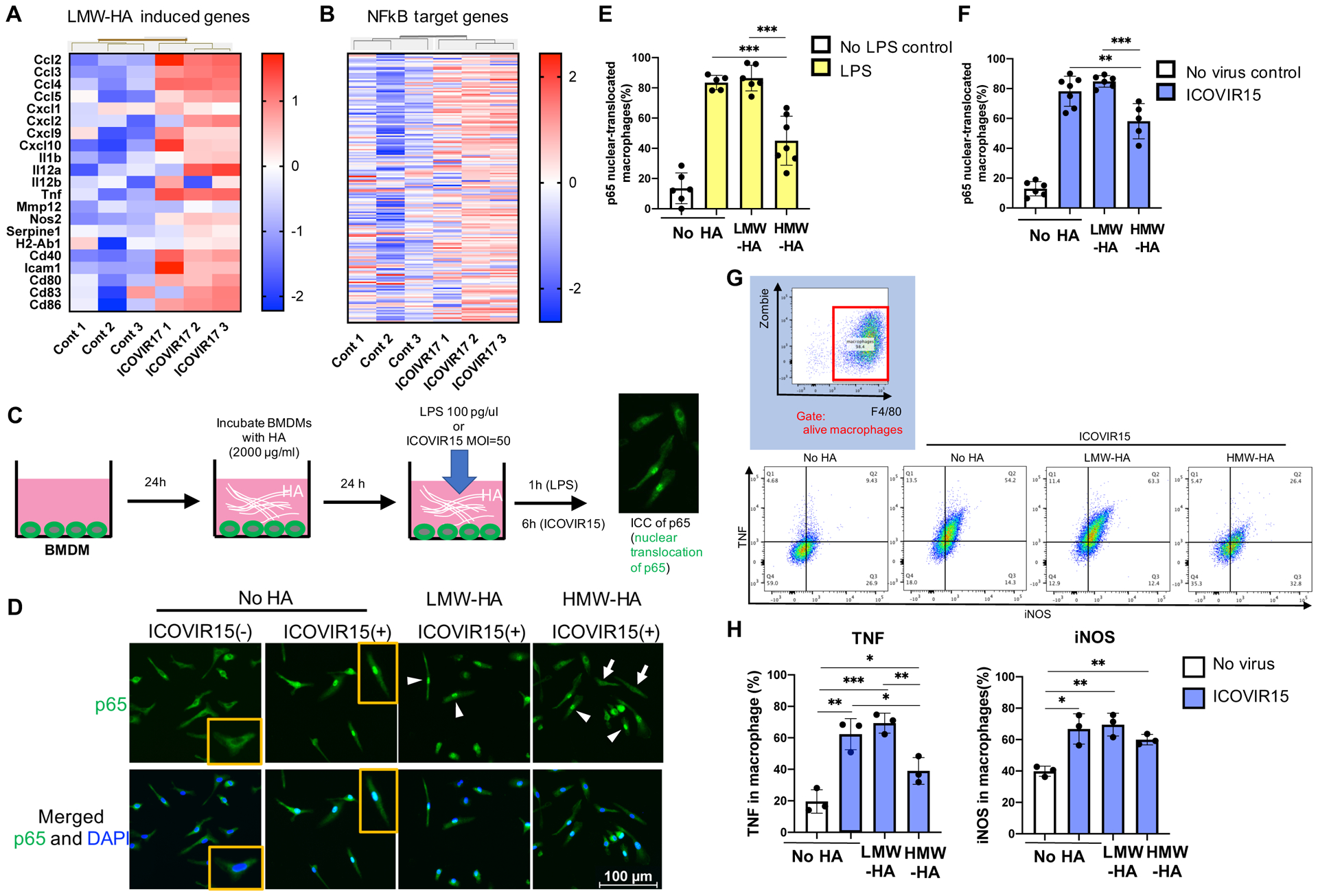
A and B, NanoString transcriptome analysis of 005 glioblastoma in the brain after treatment with PBS (Cont) or ICOVIR17. A, LMW-HA-induced genes. B, NFkB target genes. N=3 individual mice / group. C, Experimental design. BMDM, Murine bone marrow-derived macrophage. LPS, lipopolysaccharide. D-F, p65 immunofluorescence in BMDMs after stimulation with ICOVIR15 (D and F) or lipopolysaccharide (LPS, E) in the presence or absence of LMW-HA and HMW-HA. Insets in D show representative cells without and with nuclear p65 immunopositivity. Arrows and arrowheads indicate examples of cells without and with nuclear p65 translocation, respectively. E and F showing the fraction of cells having p65 in the nuclei. Student t-test (two-tailed); ** p<0.001, *** p<0.0005. G, Intracellular flow cytometry for TNF and iNOS in BMDMs. BMDMs were cultured with LMW-HA or HMW-HA for 24 hours, followed by ICOVIR15 infection. Staining was done 6 hours post infection. H, Quantification of G. TNF (left) and iNOS (right) in BMDMs after stimulation with ICOVIR15 in the presence or absence of LMW-HA and HMW-HA. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data are mean ± SD in E, F, and H.
Since ICOVIR17 treatment of 005 GBM elicited a robust accumulation of TAMs, we tested whether the size of HA affected virus-induced activation of macrophages (Fig. 3C). In vitro, both lipopolysaccharide and ICOVIR15 potently activated NF-kB signaling pathway in bone marrow-derived macrophages (BMDM) as marked by nuclear translocation of p65 (Fig. 3D–F). The presence of HMW-HA, but not LMW-HA, in BMDM culture inhibited this response (Fig. 3C–E). Intracellular flow cytometry analysis showed virus-induced upregulation of BMDM producing TNF, a canonical cytokine induced by NF-kB signaling activation (Fig. 3G and H). Again, HMW-HA, but not LMW-HA, suppressed this response significantly (Fig. 3G and H). ICOVIR15 induced iNOS in BMDM, which was not significantly inhibited by either LMW-HA or HMW-HA (Fig. 3G and H). These data reveal that adenovirus-induced activation of NF-kB signaling in macrophages is impacted by the size of extracellular HA interacting with the cells.
Combining ICOVIR17 and anti-PD1 immune checkpoint inhibitor increases survival of GBM bearing mice.
Since ICOVIR17-mediated HA degradation and OV therapy triggered cellular immune responses and increased PD-L1 levels in treated GBM, combining ICOVIR17 with blockade of the PD-L1-PD-1 immune checkpoint axis was a rational therapeutic strategy (Fig. 4A). We thus administered anti-PD-1 antibody systemically subsequent to intratumoral delivery of ICOVIR17. Combination therapy doubled the median survival time of control animals (43.5 days versus 22 days), which was significantly better than ICOVIR17 alone (32.5 days) and anti-PD-1 alone (31. 5 days), and induced a durable response and long-term cures in a fraction of the treated animals (Fig. 4B). On the other hand, combination treatment with ICOVIR15 and anti-PD-1 did not significantly increase the survival of GBM-bearing mice as compared with control or ICOVIR15 alone, despite extension of survival in a small fraction of responding animals (Supplementary Fig. S5).
Figure 4. Efficacy of combination therapy of ICOVIR17 and anti-PD-1 requires CD4+ and CD8+ T cells and macrophages.
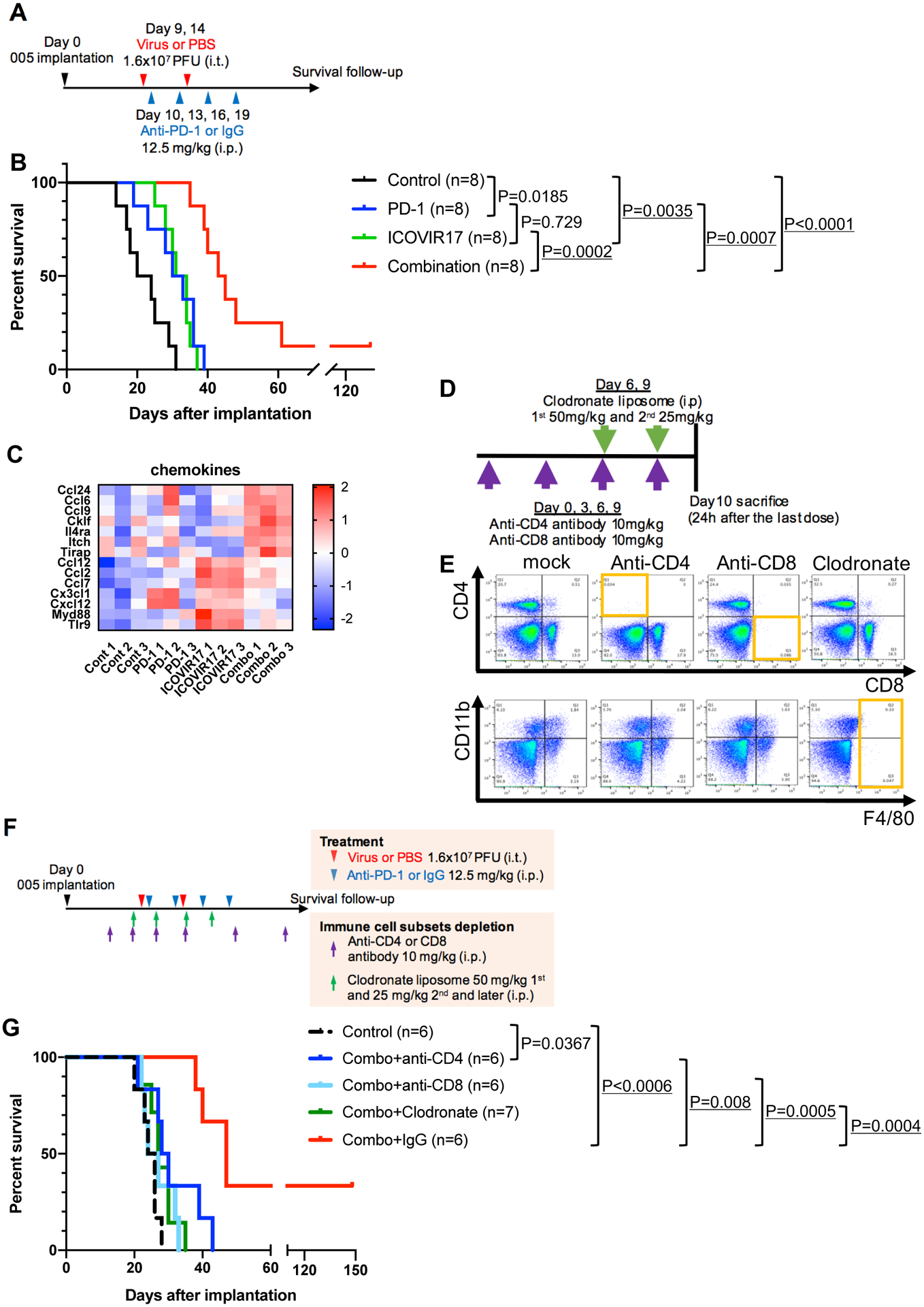
A, Treatment schema for the survival study of combination therapy of ICOVIR17 and anti-PD-1 antibody. B, Kaplan-Meier survival curves. Statistical significance was assessed between indicated groups. C, NanoString RNA analysis reveals differential expression of chemokines between ICOVIR17 alone group versus combination group (Comb). N=3 / group. D and E, In vivo depletion of immune subsets. D, Experimental schema. E, Flow cytometric analysis of splenocytes confirming successful depletion of respective target cells in the spleen. F and G, In vivo depletion of immune subsets. F, Treatment schema for the survival study of combination therapy of ICOVIR17 and anti-PD-1 antibody with and without depletion of specific immune cell subsets. G, Kaplan-Meier survival curves. Combo, combination therapy of ICOVIR17 and anti-PD-1 antibody. In B and G, P values are from log-rank test. Statistically significant differences in comparisons after the correction for multiple comparisons (Bonferroni method) are underlined.
NanoString transcriptome profiling of 1200 immune related genes in 005 GBM after different treatments revealed robust responses in innate and adaptive immunity in both the ICOVIR17 and combination groups (Supplementary Fig. S6). Overall, similar immune signatures were noted in the two groups receiving ICOVIR17 and ICOVIR17+anti-PD-1. However, there was a set of genes that were differentially expressed in the ICOVIR17 group versus the combination group (Fig. 4C, Supplementary Fig. S7). Interestingly, many chemokines were among those; for example, Ccl2, and Cxcl12 were higher in the ICOVIR17, while Ccl6 and Ccl9 were higher in the combination group, highlighting an ability of anti-PD-1 to modulate OV-induced chemokine expression (Fig. 4C).
CD4+ and CD8+ T cells and macrophages are essential for combination therapy to be effective.
We used antibody or drug-induced depletion of immune cell subsets in vivo to identify cells that mediate the effects of our combination therapy. We confirmed that administration of anti-CD4 and anti-CD8 antibodies and clodronate liposome effectively depleted CD4+, CD8+ and F4/80+ cells, respectively (Fig. 4D and E). When the combination therapy was tested in animals receiving anti-CD4, anti-CD8 or clodronate (Fig. 4F), the therapeutic efficacy was abrogated with any of these interventions, as survival was all statistically significantly different from that of combination therapy without depletion (Fig. 4G). This result indicated that CD4+ and CD8+ T cells and macrophages are all necessary for combination therapy to be functional.
Combination therapy upregulates iNOS and TNF in TAMs.
Flow cytometric analysis showed that treatments with ICOVIR17 alone and ICOVIR17+anti-PD-1 similarly increased the fraction of CD11b+, F4/80+ macrophages infiltrating 005 GBM (Fig. 5A) and decreased the fraction of 005 cells (Supplementary Fig. S8A). The proportion of PD-L1+ cells was greatly and similarly increased in the two groups receiving ICOVIR17 (Fig. 5B). 005 GBM cells and macrophages were found to be the two major cell types constituting the PD-L1+ population (Figure 5B), and the fraction of PD-L1+ cells was elevated within 005 cells as well as macrophages after treatment with ICOVIR17 and ICOVIR17+anti-PD-1 (Fig. 5B, (Supplementary Fig. S8B). Further characterization of phenotypic alterations in CD45high, CD11b+, F4/80+ TAMs revealed a significant increase in iNOS+ and TNF+ TAMs after combination therapy (Fig. 5C and D), suggesting the acquirement of an M1-like proinflammatory phenotype. However, TAMs positive for an M2-marker Arg1 and double-positive for M1 marker iNOS and M2 marker Arg1 also increased after combination therapy (Fig. 5C and D). NanoString analysis showed that both ICOVIR17 alone and combination treatment increased Nos2 and Arg1, while anti-PD-1 alone did not change either (Supplementary Fig. S8C).
Figure 5. Combination therapy increased TAMs.
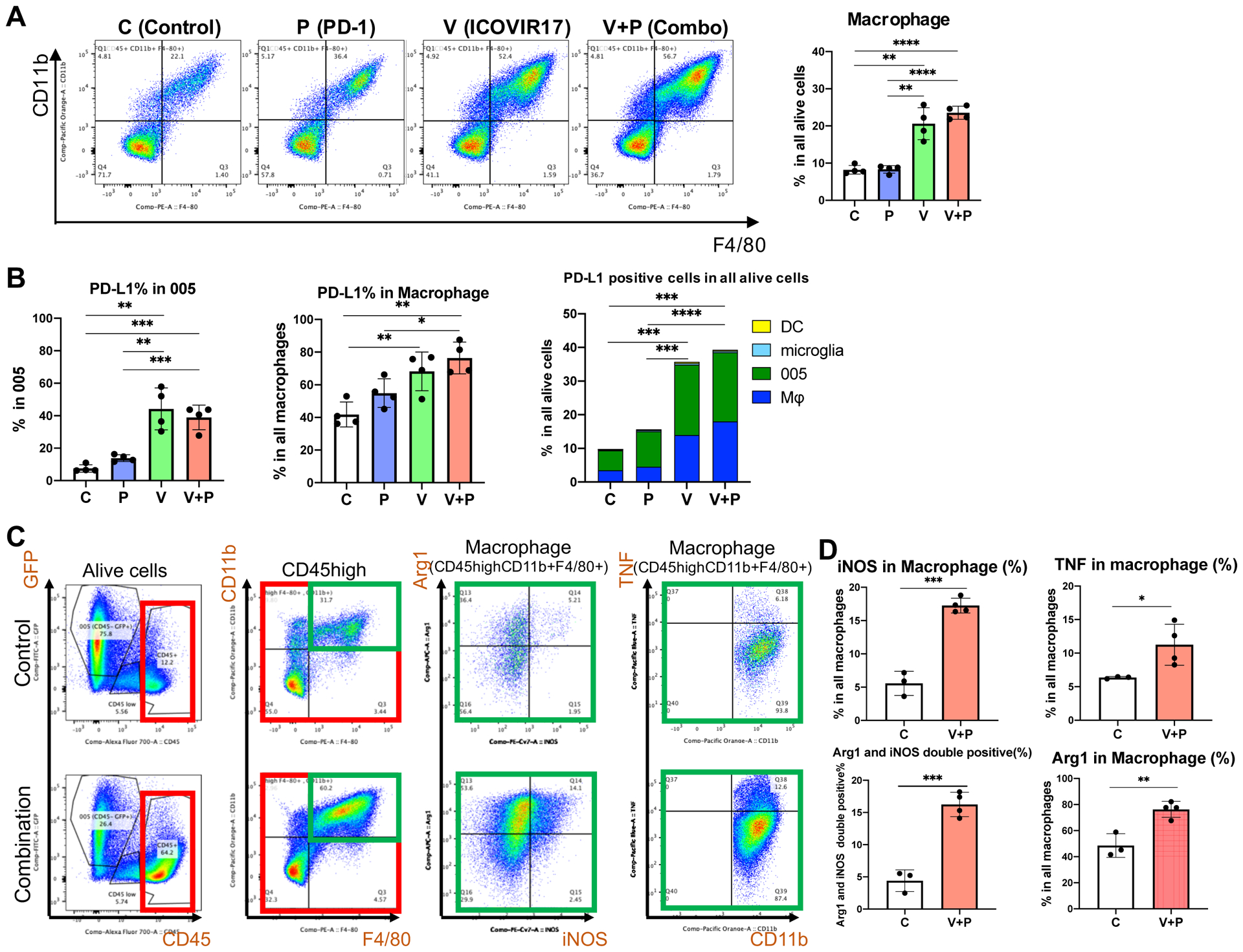
Flow cytometric analysis of 005 tumors after treatments. Representative plots and quantification of CD11b+ F4/80+ TAMs (A), PD-L1+ cells (B), and Arg1+, iNOS+, TNF+ TAMs (C). N=4 mice / group. C: control group, P: anti-PD-1 antibody monotherapy group, V: ICOVIR17 monotherapy group, V+P: ICOVIR17 and anti-PD-1 combination group. D, Quantification of C, showing % of macrophages positive for the indicated marker(s). Data are mean ± SD. *p<0.05, **p< 0.01, ***p< 0.001, ****p< 0.0001 assessed by Student’s t test (two-tailed) between indicated groups.
Combination therapy increased the CD8/Treg ratio in tumor-infiltrating T cells and induced tumor specific effector T cell responses locally and systemically.
We elucidated T cell responses that were induced by combination therapy. Flow cytometric analysis showed similar increases in tumor-infiltrating CD4+ cells in anti-PD-1, ICOVIR17 and combination groups, compared with control (Fig. 6A, Supplementary Fig. S9A). ICOVIR17 treatment increased tumor-infiltrating CD8+ cells, which was further boosted after treatment with ICOVIR17+anti-PD-1 (Fig. 6A, Supplementary Fig. S9A). Both ICOVIR17 and combination significantly decreased Foxp3+CD4+ regulatory T cell (Treg) population in the tumor (Fig. 6A, Supplementary Fig. S9A). As a result, the CD8/Treg ratio was the highest in tumors treated with combination therapy (Fig. 6A). We next examined treatment-induced changes in cell surface molecules that have been reported to mark dysfunctional or terminally exhausted T cells (58). In CD8+ T cells, the regimens involving ICOVIR17 greatly reduced the fraction positive for the immune checkpoint TIM3 (Fig. 6B). Anti-PD-1, ICOVIR17 and combination all decreased CTLA4+ CD8+ cells (Fig. 6B). Furthermore, combination therapy decreased PD-1 in CD4+ T cells (Fig. 6B).
Figure 6. Combination therapy induced tumor specific cytotoxicity in local and peripheral T cells.
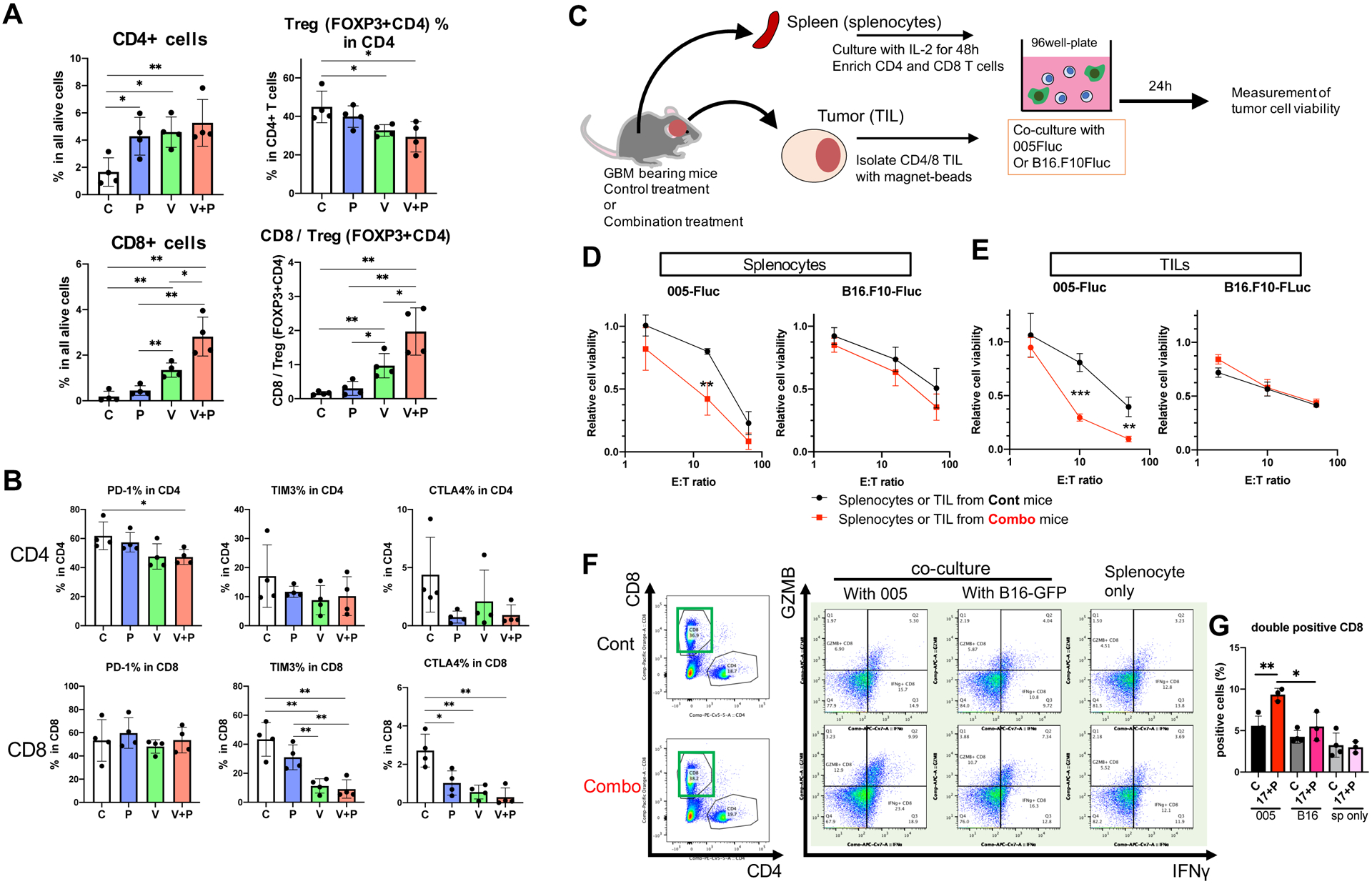
A and B, Flow cytometric analysis of tumor-infiltrating T cells (TILs) collected from orthotopic 005 glioblastoma tumors after treatment with control (C), anti-PD-1 (P), ICOVIR17 (V) or ICOVIR17+anti-PD-1 (V+P). A, analysis of total CD4+ and CD4+ FOXP3+ T regulatory cells (Treg) (upper panels) and CD8+ and CD8+/Treg ratio (lower panels). B, Analysis of T cell exhaustion/checkpoint markers (PD-1, TIM3, and CTLA4) in CD4+ cells (upper panels) and CD8+ cells (lower panels). * p< 0.05, ** p< 0.01 (two-tailed t-test) between indicated groups. C, Experimental schematic. D and E, in vitro cytotoxic assays of T cells harvested from spleens (D) and 005 glioblastomas (E) in control mice (black circles and lines) and combination therapy mice (red squares and lines). ** p< 0.01, *** p< 0.001 (two-tailed t-test). See Supplementary Figure S8B for T cell enrichment from tumors. F, Intracellular flow cytometric analysis of granzyme B (GZMB) and interferon g (IFNγ) in spleen-derived T cells after their 24h-exposure to 005 glioblastoma or B16 melanoma cells. G, Quantification of CD8+ cells double positive for GZMB and IFNγ in total CD8+ cells. C, control mice; 17+P, combination therapy mice. Sp only, splenocytes alone without tumor cells. * p< 0.05, ** p<0.01 (two-tailed t-test) between indicated groups. See Supplementary Fig. S9C for gating using FMO (fluorescence minus one controls). Data are mean ± SD.
Lastly, we sought to measure the cytotoxic function of T cells after combination therapy. Two effector cell types were prepared: 1) splenocytes that were cultured 48h in the presence of IL2, and 2) TILs: T cells isolated from 005 GBM using anti-CD4 and CD8 magnetic beads (Fig. 6C, Supplementary Fig. S9B). Twenty-four-hour co-culture of splenocytes and TILs isolated from animals that received combination therapy with 005-fluc (firefly luciferase) or B16.F10-fluc cells as targets demonstrated significantly enhanced killing of 005-fluc cells, compared with those from mice that received PBS (intratumoral) and isotype-IgG (ip) (Fig. 6D and E). Killing of irrelevant B16.F10-fluc melanoma cells were comparable between T cells derived from control and combination therapy mice (Fig. 6D and E). To support this tumor specific killing, IL2-stimulated splenocytes harvested from combination therapy group reacted to 005 cells, not B16 cells, by increasing the fraction of CD8+ cells double positive for GranzymeB and IFNγ (Fig. 6F and G, Supplementary Fig. S9C). Thus, combination therapy of ICOVIR17 and PD-1 blockade induced tumor specific effector T cell responses both locally and systemically.
Discussion
In this work, we show that GBM treatment with HA-degrading ICOVIR17 has a therapeutic effect in murine GBM that was accompanied by increases in tumor infiltrating CD8+ T cells and TAM as well as PD-L1 levels. These changes in the TME immune profile enabled ICOVIR17 to be effectively combined with anti-PD-1 antibody to induce durable response in animals bearing aggressive GBM.
The vast majority of current oncolytic adenovirus clinical trials for GBM are investigating human adenovirus serotype 5-based Delta-24-RGD (DNX-2401) (40). ICOVIR15 and ICOVIR17 drive delta24-E1A expression under the control of a promoter consisting of four palindromic E2F-binding sites and a Sp-1 binding site, making these viruses potentially more tumor selective and potent than Delta-24-RGD (42,47). Still, we found that murine GBM cells were significantly less permissive to these OVs compared to human GBM cells. The 005 GBM model we selected to study these OVs in an immunocompetent setting is highly tumorigenic in C57BL/6 mice displaying stem-like features (44,46), and recapitulated the phenotypic and immunosuppressive hallmarks of human GBM, a finding being reinforced by our time of flight cytometry (CyTOF) immune-profiling (59). Injections of HA-degrading ICOVIR17, but not its parental ICOVIR15, into 005 GBM were able to increase animal survival. Unlike what we observed previously in human GBM xenografts in immune-incompetent mice, where ICOVIR17 also had a better effect compared to ICOVIR15 (43), both viruses showed comparably, efficient distribution of infection within GBM in this immune-competent model. However, ICOVIR17-treated GBM contained elevated levels of CD8+ T cells and TAMs, and PD-L1+ 005 cells and TAMs, compared with ICOVIR15-treated tumors. Because the only difference between ICOVIR15 and ICOVIR17 is PH20 expression by ICOVIR17, the immunological changes observed in treated tumors are attributable to hyaluronidase-mediated HA degradation in the GBM TME. A similar hyaluronidase-expressing oncolytic adenovirus was shown to reduce tumor growth and induce infiltration of CD8+ T cells in a subcutaneous lung cancer model (60). In a murine model of pancreatic ductal adenocarcinoma, stromal remodeling via HA degradation increased intratumoral infiltration of effector T cells when combined with cancer vaccines (61). We showed robust accumulation of TAMs in tumor areas that were virus-positive and depleted of HA. Molecularly, our transcriptomic analysis of ICOVIR17-treated tumors in vivo revealed activation of many NF-kB target genes. Furthermore, we demonstrated in vitro that virus-induced NF-kB activation in BMDM was inhibited by HMW-HA, not by LMW-HA, a finding consistent with reports characterizing differential abilities of HMW-HA and LMW-HA to induce M2-polarized macrophages and trigger activation of the TLR2/4-NFkB signaling pathway, respectively (56,57). Thus, HA degradation in the context of oncolytic adenovirus GBM therapy activates an inflammatory M1-like phenotype in TAMs, which might have contributed to the functional activation of effector CD8+ T cells and their recruitment to the tumor site. Our current work and others’ suggest that HA depletion in the TME may improve cancer immunotherapy by removing a physical barrier for immune cell trafficking and activating their functions (61). However, further research is necessary to determine whether HA restricts immune cell penetration to and distribution within GBM.
ICOVIR17 treatment induced distinct changes in immune checkpoint levels in GBM. The most prominent was the upregulation of PD-L1 in 005 tumor cells and TAMs. In contrast, TIM3 and CTLA-4 were downregulated in CD8+ T cells. Adenovirus-induced decreases in TIM3+ cells were also reported clinically in recurrent malignant gliomas after injections of Delta-24-RGD (41). The increases in CD8+ cells and M1 TAMs, as well as selective upregulation of PD-L1 set the stage for rationally combining ICOVIR17 with blockade of the PD-L1-PD-1 axis to improve immunotherapeutic activity. Indeed, this combination therapy doubled animal survival from control, with significant prolongation over monotherapies and long-term cures in a fraction (21%) of animals. Interestingly, in vivo depletion of immune subsets revealed a pivotal role that CD4+ and CD8+ T cells and macrophages all play in mediating the activity of combination therapy. This result was analogous to what was required for an IL-12 expressing oncolytic herpes simplex virus in conjunction with dual checkpoint inhibition to exert curative activity in the same GBM model (46). Increases in iNOS+/TNF+ M1-like TAMs and in the CD8+/Treg ratio of TIL support treatment-induced activation of innate and adaptive immune cells, both likely contributing to therapeutic benefit. Finally, ex vivo functional assays confirmed systemic and local induction of tumor specific cytotoxicity in mice treated with the combination therapy. Collectively, HA-degrading oncolytic adenovirus followed by PD-1 blockade acted co-operatively to overcome the suppressive TME and boost the concerted action of innate and adaptive cellular immunity to eliminate GBM. However, durable responses were not achieved in all animals, underscoring the need to understand the mechanisms underlying heterogeneous responses and to improve efficacy. Human GBM TAMs have been reported to co-express M1 and M2 markers (62), and functional significance underlying the emergence of such TAMs after combination therapy warrants elucidation.
In summary, we identify HA abundantly present in the GBM TME as a therapeutic target in oncolytic adenovirus immunotherapy, providing an insight that could be applicable to other cancer types. ICOVIR17-mediated degradation of HA within GBM dramatically alters the immune landscape of the TME, and offers a mechanistically rational combination immunotherapy with PD-L1/PD-1 blockade. Currently, a multicenter phase II clinical trial is investigating the combinatory use of Delta-24-RGD and anti-PD-1 antibody (pembrolizumab) for recurrent GBM (Keynote-192). Our current study provides a strong basis for this combinatorial clinical translation of ICOVIR17.
Supplementary Material
Translational relevance.
Cancer immunotherapy has been a success in certain cancer types, such as melanoma and non-small cell lung cancer, but has so far proven ineffective against the malignant brain tumor glioblastoma (GBM). Glycosaminoglycan hyaluronan present in tumor extracellular matrix promotes malignant phenotypes of GBM, but how hyaluronan impacts GBM immunotherapy is unknown. Our studies are the first to show that immune-modulatory ICOVIR17 has a dual role of mediating degradation of hyaluronan within GBM and subsequently modifying the immune landscape of the TME, and propose a mechanistic combination immunotherapy with immune-checkpoint blockade that remodels both innate and adaptive immune cells. We anticipate that our findings will have a major contribution towards developing novel immune-based therapies for GBM patients and could define a new treatment paradigm for other solid tumors.
Acknowledgements
We thank Dr. Praveen Bommareddy and Dr. Maryam Rahman for sharing us with their NanoString data. We thank Dr. Robert Colvin and Ellen Acheampong at the NanoString core at MGH, and Drs. Eric Miller and Clement David of NanoString Technologies for technical assistance. This work was supported by NIH R21 NS103187 (H. Wakimoto) and NIH R01 CA204720 (to K. Shah).
Footnotes
Conflict of interest disclosore
R. Alemany reports personal fees from VCN Biosciences outside the submitted work, as well as has a patent for hyaluronidase-armed oncolytic adenoviruses issued. S.D. Rabkin reports grants from NIH during the conduct of the study, personal fees from Replimune and from Cellinta outside the submitted work, and coinventor on patents relating to oncolytic herpes simplex viruses, owned and managed by Georgetown University and Massachusetts General Hospital, which have received royalties from Amgen and ActiVec Inc. K. Shah owns equity in and is a member of the Board of Directors, AMASA Therapeutics, Inc., a company developing cell-based therapies for cancer. K. Shah’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. No disclosures were reported by the other authors.
References
- 1.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci 2012;125(Pt 23):5591–6 doi 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- 2.Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends in Cell Biology 2015;25(4):198–213 doi 10.1016/j.tcb.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol 2018;14(8):482–95 doi 10.1038/s41582-018-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia 2012;60(3):502–14 doi 10.1002/glia.21264. [DOI] [PubMed] [Google Scholar]
- 5.Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017;31(3):326–41 doi 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352(10):987–96 doi 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 7.Park J, Kwak H, Lee S. Role of hyaluronan in glioma invasion. Cell Adhesion and Migration 2008;2(3):202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kultti A, Li X, Jiang P, Thompson CB, Frost GI, Shepard HM. Therapeutic targeting of hyaluronan in the tumor stroma. Cancers (Basel) 2012;4(3):873–903 doi 10.3390/cancers4030873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silver DJ, Siebzehnrubl FA, Schildts MJ, Yachnis AT, Smith GM, Smith AA, et al. Chondroitin sulfate proteoglycans potently inhibit invasion and serve as a central organizer of the brain tumor microenvironment. J Neurosci 2013;33(39):15603–17 doi 10.1523/JNEUROSCI.3004-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chanmee T, Ontong P, Itano N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett 2016;375(1):20–30 doi 10.1016/j.canlet.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 11.Whatcott CJ, Han H, Posner RG, Hostetter G, Von Hoff DD. Targeting the tumor microenvironment in cancer: why hyaluronidase deserves a second look. Cancer Discov 2011;1(4):291–6 doi 10.1158/2159-8290.CD-11-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer 2004;4(7):528–39 doi 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- 13.Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, Johansson R, et al. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol 2000;156(2):529–36 doi 10.1016/S0002-9440(10)64757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ropponen K, Tammi M, Parkkinen J, Eskelinen M, Tammi R, Lipponen P, et al. tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Research 1998;58:342–7. [PubMed] [Google Scholar]
- 15.Setala LP, Tammi MI, Tammi RH, Eskelinen MJ, Lipponen PK, Agren UM, et al. Hyaluronan expression in gastric cancer cells is associated with local and nodal spread and reduced survival rate. Br J Cancer 1999;79(7–8):1133–8 doi 10.1038/sj.bjc.6690180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lipponen P, Aaltomaa S, Tammi R, Tammi M, Agren U, Kosma VM. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur J Cancer 2001;37(7):849–56 doi 10.1016/s0959-8049(00)00448-2. [DOI] [PubMed] [Google Scholar]
- 17.Whatcott CJ, Han H, Von Hoff DD. Orchestrating the Tumor Microenvironment to Improve Survival for Patients With Pancreatic Cancer: Normalization, Not Destruction. Cancer J 2015;21(4):299–306 doi 10.1097/PPO.0000000000000140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anttila MA, Tammi RH, Tammi MI, Syrjanen KJ, Saarikoski SV, Kosma VM. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res 2000;60(1):150–5. [PubMed] [Google Scholar]
- 19.Yang R, Yan Z, Chen F, Hansson G, Kiessling R. Hyaluronic acid and Chondroitin Sulphate a rapidly promote differentiation of immature DC with upregulation of costimulatory and antigen-presenting molecules, and enhancement of NF-κB and protein kinase activity. Scandinavian Journal of Immunology 2002;55:2–13. [DOI] [PubMed] [Google Scholar]
- 20.Gebe JA, Yadava K, Ruppert SM, Marshall P, Hill P, Falk BA, et al. Modified High-Molecular-Weight Hyaluronan Promotes Allergen-Specific Immune Tolerance. Am J Respir Cell Mol Biol 2017;56(1):109–20 doi 10.1165/rcmb.2016-0111OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qadri M, Almadani S, Jay GD, Elsaid KA. Role of CD44 in Regulating TLR2 Activation of Human Macrophages and Downstream Expression of Proinflammatory Cytokines. J Immunol 2018;200(2):758–67 doi 10.4049/jimmunol.1700713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagy N, Kuipers HF, Marshall PL, Wang E, Kaber G, Bollyky PL. Hyaluronan in immune dysregulation and autoimmune diseases. Matrix Biol 2018. doi 10.1016/j.matbio.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu J, Qu Y, Zhang YP, Deng JX, Yu QH. RHAMM induces progression of rheumatoid arthritis by enhancing the functions of fibroblast-like synoviocytes. BMC Musculoskelet Disord 2018;19(1):455 doi 10.1186/s12891-018-2370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev 2011;91(1):221–64 doi 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikitovic D, Tzardi M, Berdiaki A, Tsatsakis A, Tzanakakis GN. Cancer Microenvironment and Inflammation: Role of Hyaluronan. Frontiers in Immunology 2015;6 doi 10.3389/fimmu.2015.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front Immunol 2015;6:201 doi 10.3389/fimmu.2015.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H, Cha J, Jang M, Kim P. Hyaluronic acid-based extracellular matrix triggers spontaneous M2-like polarity of monocyte/macrophage. Biomater Sci 2019;7(6):2264–71 doi 10.1039/c9bm00155g. [DOI] [PubMed] [Google Scholar]
- 28.Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood 2007;110(2):587–95 doi 10.1182/blood-2007-01-068031. [DOI] [PubMed] [Google Scholar]
- 29.Shi Q, Zhao L, Xu C, Zhang L, Zhao H. High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation. Molecules 2019;24(9) doi 10.3390/molecules24091766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang G, Guo L, Yang C, Liu Y, He Y, Du Y, et al. A novel role of breast cancer-derived hyaluronan on inducement of M2-like tumor-associated macrophages formation. Oncoimmunology 2016;5(6):e1172154 doi 10.1080/2162402X.2016.1172154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015;33(25):2780–8 doi 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 32.Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe 2014;15(3):260–5 doi 10.1016/j.chom.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol 2018;18(8):498–513 doi 10.1038/s41577-018-0014-6. [DOI] [PubMed] [Google Scholar]
- 34.Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov 2019;18(9):689–706 doi 10.1038/s41573-019-0029-0. [DOI] [PubMed] [Google Scholar]
- 35.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nature biotechnology 2012;30(7):658–70 doi 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Quintanilla J, Seah I, Chua M, Shah K. Oncolytic viruses: overcoming translational challenges. J Clin Invest 2019;130:1407–18 doi 10.1172/JCI122287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saha D, Wakimoto H, Rabkin SD. Oncolytic herpes simplex virus interactions with the host immune system. Curr Opin Virol 2016;21:26–34 doi 10.1016/j.coviro.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foreman PM, Friedman GK, Cassady KA, Markert JM. Oncolytic Virotherapy for the Treatment of Malignant Glioma. Neurotherapeutics 2017;14(2):333–44 doi 10.1007/s13311-017-0516-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niemann J, Kuhnel F. Oncolytic viruses: adenoviruses. Virus Genes 2017;53(5):700–6 doi 10.1007/s11262-017-1488-1. [DOI] [PubMed] [Google Scholar]
- 40.Kiyokawa J, Wakimoto H. Preclinical And Clinical Development Of Oncolytic Adenovirus For The Treatment Of Malignant Glioma. Oncolytic Virother 2019;8:27–37 doi 10.2147/OV.S196403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol 2018;36(14):1419–27 doi 10.1200/JCO.2017.75.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther 2010;18(7):1275–83 doi 10.1038/mt.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martinez-Quintanilla J, He D, Wakimoto H, Alemany R, Shah K. Encapsulated stem cells loaded with hyaluronidase-expressing oncolytic virus for brain tumor therapy. Mol Ther 2015;23(1):108–18 doi 10.1038/mt.2014.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A 2013;110(29):12006–11 doi 10.1073/pnas.1307935110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med 2009;15(1):110–6 doi 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017;32(2):253–67 e5 doi 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rojas JJ, Guedan S, Searle PF, Martinez-Quintanilla J, Gil-Hoyos R, Alcayaga-Miranda F, et al. Minimal RB-responsive E1A promoter modification to attain potency, selectivity, and transgene-arming capacity in oncolytic adenoviruses. Mol Ther 2010;18(11):1960–71 doi 10.1038/mt.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davydova J, Yamamoto M. Oncolytic Adenoviruses: Design, Generation, and Experimental Procedures. Current Protocols in Human Genetics: John Wiley & Sons, Inc; 2013. p 12.4.1–.4.21. [DOI] [PubMed] [Google Scholar]
- 49.Seyfried TN, el-Abbadi M, Ecsedy JA, Bai HW, Yohe HC. Influence of host cell infiltration on the glycolipid content of mouse brain tumors. J Neurochem 1996;66(5):2026–33 doi 10.1046/j.1471-4159.1996.66052026.x. [DOI] [PubMed] [Google Scholar]
- 50.Wakimoto H, Kesari S, Farrell CJ, Curry WT Jr., Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res 2009;69(8):3472–81 doi 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bignami A, Hosley M, Dahl D. Hyaluronic acid and hyaluronic acid-binding proteins in brain extracellular matrix. Anat Embryol (Berl) 1993;188(5):419–33 doi 10.1007/bf00190136. [DOI] [PubMed] [Google Scholar]
- 52.Quirico-Santos T, Fonseca CO, Lagrota-Candido J. Brain sweet brain: importance of sugars for the cerebral microenvironment and tumor development. Arq Neuropsiquiatr 2010;68(5):799–803 doi . [DOI] [PubMed] [Google Scholar]
- 53.Bommareddy PK, Zloza A, Rabkin SD, Kaufman HL. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 2019;8(7):1591875 doi 10.1080/2162402X.2019.1591875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karachi A, Yang C, Dastmalchi F, Sayour EJ, Huang J, Azari H, et al. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro Oncol 2019;21(6):730–41 doi 10.1093/neuonc/noz015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jogler C, Hoffmann D, Theegarten D, Grunwald T, Uberla K, Wildner O. Replication properties of human adenovirus in vivo and in cultures of primary cells from different animal species. J Virol 2006;80(7):3549–58 doi 10.1128/JVI.80.7.3549-3558.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol 2006;177(2):1272–81 doi 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 57.Tavianatou AG, Caon I, Franchi M, Piperigkou Z, Galesso D, Karamanos NK. Hyaluronan: molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J 2019;286(15):2883–908 doi 10.1111/febs.14777. [DOI] [PubMed] [Google Scholar]
- 58.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019;20(3):326–36 doi 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khalsa JK, Cheng N, Keegan J, Chaudry A, Driver J, Bi WL, et al. Immune phenotyping of diverse syngeneic murine brain tumors identifies immunologically distinct types. Nat Commun 2020;11(1):3912 doi 10.1038/s41467-020-17704-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Zaher AA, Moreno R, Fajardo CA, Arias-Badia M, Farrera M, de Sostoa J, et al. Evidence of Anti-tumoral Efficacy in an Immune Competent Setting with an iRGD-Modified Hyaluronidase-Armed Oncolytic Adenovirus. Mol Ther Oncolytics 2018;8:62–70 doi 10.1016/j.omto.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blair AB, Kim VM, Muth ST, Saung MT, Lokker N, Blouw B, et al. Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer. Clin Cancer Res 2019;25(17):5351–63 doi 10.1158/1078-0432.CCR-18-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A, et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol 2017;18(1):234 doi 10.1186/s13059-017-1362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.