

Abstract
Neurosurgery for brain tumor resection or epilepsy treatment requires a craniotomy to gain access to the brain. Despite prophylactic measures, infectious complications occur at a frequency of 1–3%, with approximately half caused by Staphylococcus aureus (S. aureus) that forms a biofilm on the bone flap and is recalcitrant to antibiotics. Using scRNA-seq in a mouse model of S. aureus craniotomy infection, this study revealed the complex transcriptional heterogeneity of resident microglia and infiltrating monocytes in the brain in addition to transcriptionally diverse granulocyte subsets in the subcutaneous galea and bone flap. In the brain, trajectory analysis identified the transition of microglia from a homeostatic/anti-inflammatory to pro-inflammatory and proliferative populations, whereas granulocytes in the brain demonstrated a trajectory from a granulocyte myeloid-derived suppressor cell (G-MDSC)-like phenotype to a small population of mature neutrophils. In the galea, trajectory analysis identified the progression from two distinct G-MDSC-like populations to neutrophil clusters enriched for IFN signaling and cell cycle genes. Based on their abundance in the galea and bone flap, PMNs and MDSCs were depleted using anti-Ly6G, which resulted in increased bacterial burden. This revealed a critical role for PMNs in S. aureus containment since MDSCs were found to attenuate PMN antibacterial activity which may explain, in part, why craniotomy infection persists in the presence of PMN infiltrates. These results demonstrate the existence of a transcriptionally diverse leukocyte response that likely influences the chronicity of S. aureus craniotomy infection.
Keywords: Staphylococcus aureus, biofilm, craniotomy, microglia, monocyte, neutrophil, myeloid-derived suppressor cell, scRNA-seq
INTRODUCTION
During neurosurgical procedures that require access to the brain, such as the resection of tumors or epileptic foci, a craniotomy is performed where the piece of resected skull, or bone flap, is replaced intraoperatively. Infectious complications occur in 1–3% of craniotomies, where approximately half are caused by the gram-positive pathogen Staphylococcus aureus (S. aureus) that forms a biofilm on the bone flap surface (1, 2). Biofilms are bacterial communities encased in a matrix composed of extracellular DNA (eDNA), protein, and polysaccharide (3, 4). This complex structure creates nutrient and oxygen gradients, leading to metabolic heterogeneity within the biofilm, where a subpopulation of bacteria (referred to as persisters) are metabolically dormant (5). These persister cells are one reason why biofilm is tolerant to antibiotics (6), which is further complicated by the fact that biofilm elicits aberrant immune responses typified by an anti-inflammatory signature (7, 8). Therefore, craniotomy infections are difficult to eradicate without surgical debridement or permanent removal of the infected bone flap (9–12).
Our laboratory has developed a mouse model of S. aureus craniotomy infection that accurately reflects biofilm growth in terms of chronicity and antibiotic tolerance, both functional attributes of a biofilm (4, 13, 14). In addition, the mouse model shares features with human disease, including similarities in biofilm structure on the bone flap as revealed by scanning electron microscopy and inflammatory changes as seen by magnetic resonance imaging (MRI) (13), supporting its translational relevance. To better understand the complex immune signature associated with craniotomy biofilm infection and how this is regulated by the local tissue microenvironment, scRNA-seq was performed on CD45+ cells isolated from the brain, galea, and bone flap. This analysis revealed the preferential recruitment of monocytes and macrophages in the brain, whereas these populations were negligible in the galea and bone flap. Likewise, T and B cells and several innate lymphoid populations (i.e. NK cells, γδ T cells, NKT cells) were detected in the brain, which were largely absent from the galea and bone flap. Four distinct populations of microglia were identified in the infected brain, which were classified into homeostatic/anti-inflammatory, pro-inflammatory, and proliferative subtypes based on their transcriptional signatures and a progression between these states was inferred by trajectory analysis. The galea and bone flap were predominantly associated with granulocyte infiltrates, which clustered into several populations with distinct transcriptional profiles, whereas granulocytes were significantly less abundant in the brain. Based on the preferential recruitment of PMNs and MDSCs into the galea and bone flap, these cells were depleted using a Ly6G antibody to determine their functional importance. Ly6G depletion resulted in exaggerated S. aureus burden in not only the galea and bone flap, but also the brain. This was phenotype was attributed to the loss of PMNs, since MDSCs were found to attenuate PMN bactericidal activity, in agreement with their immune suppressive properties (15, 16). This uncovered a previously unappreciated role for PMNs in preventing S. aureus biofilm outgrowth, which is difficult to discern in the wild type setting since craniotomy infection persists in the presence of PMN infiltrates. Collectively, these findings have established the extensive transcriptional diversity of resident microglia and infiltrating leukocytes in the brain as well as the galea and bone flap, which is an important advance towards deciphering the complex interplay between these cells and S. aureus that influences biofilm persistence.
MATERIALS AND METHODS
Ethics statement.
This study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and complies with the ARRIVE guidelines. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center (#16–123-10).
Mouse model of S. aureus craniotomy-associated biofilm infection.
S. aureus craniotomy infection was established in male and female C57BL/6NCrl mice (RRID:IMSR_CRL:27) between 8–12 weeks of age as previously described, where biofilm formation on the bone flap leads to infection persistence in the subcutaneous galea and brain (13, 17). Briefly, mice were anesthetized with ketamine/xylazine and a skin incision was made to expose the skull. A high-speed pneumatic drill was used to create a bone flap (approximately 3 mm in diameter) with care taken to minimize damage to the dura. The excised bone flap was incubated with S. aureus strain USA300 LAC13c (18) for 5 min to allow for bacterial adherence (103 cfu), rinsed with PBS, and immediately reinserted into the skull, whereupon the skin incision was closed with sutures. There is no mortality associated with the craniotomy model and mice were sacrificed at the indicated time points for experimental assessments. For some experiments, mice were subjected to a sterile craniotomy using the same procedure except the bone flap was not exposed to S. aureus.
In vivo depletion studies.
To deplete PMNs and MDSCs in vivo, mice received i.p. injections of 100 μg InVivoPlus Ly6G (1A8; RRID:AB_1107721) or isotype control (rat IgG2a; RRID:AB_1107769) Abs (both from BioXCell) one day prior to S. aureus infection and every 72 h thereafter until sacrifice. Animals were euthanized at 3 or 7 days after infection to determine the impact of granulocyte depletion on S. aureus persistence and tissue-associated leukocyte infiltrates.
Tissue collection and processing for bacterial quantification.
At the indicated time points post-infection, mice were euthanized using an overdose of inhaled isoflurane and transcardially perfused with PBS. The bone flap was removed first, followed by the galea, which represents the subcutaneous tissue and associated purulent exudate. Next, the ipsilateral brain hemisphere associated with the infected bone flap was removed and placed in PBS. The bone flap was vortexed in PBS for 30 sec followed by sonication for 5 min to dislodge biofilm-associated bacteria. The galea was dissociated in PBS using the blunt end of a plunger from a 3 cc syringe and the brain was homogenized by pressing through a 70 μm cell strainer and rinsed with PBS. Once all tissues were processed, aliquots were removed to quantify bacterial burden. Titers were determined by serial dilutions on tryptic soy agar (TSA) plates supplemented with 5% sheep blood (Remel, Lenexa, KS) and are expressed as log10 colony forming units (CFUs) per mL. Blood was collected from mice via cardiac puncture in EDTA capillary collection tubes for evaluation of leukocytes by flow cytometry and scRNA-seq.
Flow Cytometry.
Following the removal of aliquots from tissues for quantifying S. aureus burden, leukocyte infiltrates in the brain, galea, and bone flap were assessed by flow cytometry. Briefly, brain homogenates were incubated in HBSS containing collagenase IV and DNase I (both from Millipore-Sigma) for 15 min at 37°C, whereupon enzymatic activity was inactivated with fetal bovine serum (FBS). Cells were added to a 25% Percoll solution (GE Healthcare; Marlborough, MA) containing 3% FBS and centrifuged at 520xg for 20 min with no brake (19). The upper myelin layer down to the pellet was discarded, and the pellet was resuspended in PBS and filtered to remove remaining particulate material. Galea samples were also centrifuged and filtered, and cells from the galea and brain as well as those recovered from the bone flap by vortexing were incubated with TruStain fcX (BioLegend) to block non-specific antibody binding and stained with either innate or adaptive immune panels. The innate immune panel included CD45-BUV805 (Cat. #748370; BD Biosciences), Ly6G-BUV395 (RRID:AB_2739417), CX3CR1-BV785 (RRID:AB_2632858), MHCII-BV605 (RRID:AB_2565894), CD11b-BV570 (RRID:AB_10896949), F4/80-BV510 (RRID:AB_2562622), CD115-PECy7 (RRID:AB_2566459), CD14-PE/Dazzle594 (RRID:AB_2721698), Ly6C-APC Cy7 (Cat. #560596; BD Biosciences), and CCR2-PE (RRID:AB_2616982). The adaptive immune panel included CD45-BUV805 (Cat. #748370; BD Biosciences), CD3ε-BUV737 (Cat. #741788, BD Biosciences), CD4-PacificBlue (RRID:AB_493374), CD8a-BV605 (RRID:AB_2562609), ɣδTCR-PE (RRID:AB_313832), NK1.1-APC-Cy7 (RRID:AB_830871), and CD19-Af700 (RRID:AB_493735). For both panels, dead cells were excluded using a Live/Dead Fixable Cell Stain Kit (Invitrogen) and analysis was performed on a BD Fortessa cytometer and analyzed with FlowJo (RRID:SCR_008520) using the gating strategy presented in Supplemental Figure 1.
T cell suppression assays.
The ability of craniotomy-associated leukocyte populations to inhibit polyclonal T cell activation was performed as previously described (20). Briefly, leukocyte populations were isolated from the brain, galea, and bone flap by FACS at days 3 and 7 post-infection using the markers for each cell population described in the Flow Cytometry section. CD4+ T cells were isolated from the spleens of naïve mice using a CD4+ T cell isolation kit (Miltenyi Biotec, Auburn, CA) and immediately labeled with eFluor 670 cell proliferation dye (eBioscience), according to the manufacturer’s instructions. Labeled CD4+ T cells were plated at 105 cells/well in a 96-well round-bottom plate in RPMI-1640 supplemented with 10% FBS and 100 ng/mL IL-2 (BioLegend). FACS-purified leukocytes were added at 1:1 ratios to CD4+ T cells that were subjected to polyclonal stimulation with CD3/CD28 Dynabeads (Life Technologies), since TCR immunodominant epitopes for S. aureus are not defined. Cells were incubated for 72 h, whereupon the extent of T cell proliferation was determined by flow cytometry using a BD LSR II (RRID:SCR_002159).
Gentamicin protection assay.
To examine the effect of MDSCs on PMN bactericidal activity, a gentamicin protection assay was performed. Mouse bone marrow-derived MDSCs were prepared as previously described (21) and PMNs were recovered from the peritoneal cavity by PBS lavage 24 h after injection of 4% sterile thioglycollate broth. Both MDSCs and PMNs were purified with a QuadroMACS Separator using anti-Ly6G MicroBeads (Miltenyi Biotec). Two co-culture paradigms were tested with direct cell contact, or separation with Transwell inserts to assess cell contact-independent effects. For direct contact assays, 1.5×105 PMNs and MDSCs were co-cultured in a 24-well plate. To assess cell contact-independent effects, PMNs were added to a 24-well plate at 3×105 cells/well and co-cultured for 2 h with 3×105 MDSCs that were separated by a 0.4 μm Transwell insert. Following the 2 h conditioning period the MDSCs in Transwells were removed and held in culture medium. At that point, PMNs or PMN-MDSC direct co-cultures were challenged with live S. aureus USA300 LAC13c at an MOI of 10:1 for 2 h. Next, plates were centrifuged and cells were washed and treated with 100 μg/mL gentamicin for 30 min to kill residual extracellular bacteria, whereupon fresh medium containing 1 μg/mL gentamicin was added. The MDSCs in Transwell inserts were then re-introduced with PMNs and intracellular bacterial burden in PMNs or PMN-MDSC co-cultures was determined at time 0 (following high dose gentamicin treatment), 2, 6, and 24 h by lysing cells in H2O. The bacterial burden of both co-culture paradigms was compared to PMNs alone.
scRNA-seq and bioinformatics.
Live CD45+ cells from the brain, galea, bone flap, and blood were collected by FACS at day 7 post-infection (6,783 to 11,874 cells collected per sample) and prepared for sequencing in the UNMC Genomics Core Facility. Briefly, sorted leukocytes were evaluated by light microscopy to determine concentration, percent viability, and to assess potential debris present in the suspension. Cells were loaded onto a 10X Genomics instrument and single cells were captured, lysed, RNA reverse transcribed, and RNA barcoded using a Chromium Single Cell 3’ Reagent Kit v3 (Cat. #PN-1000075; 10X Genomics), according to the manufacturer’s instructions. Illumina compatible cDNA libraries were created and quantified by qPCR using the KAPA Library Quant Kit (Illumina) (Cat. #KK4824; KAPA Biosystems) and were loaded at a concentration of 1.3 pM on an Illumina NextSeq550 instrument. Samples were sequenced following the parameters suggested by 10X Genomics to an average depth of approximately 50,000 reads per cell.
Single cell expression data was largely analyzed in Seurat version 3.1 (https://satijalab.org/seurat/; RRID:SCR_007322). Output from the 10XGenomics Cell Ranger pipeline was imported into Seurat through the Read10X function and the Seurat object was initialized using cells that expressed a minimum of 200 features over at least 3 individual cells. A standard quality control workflow was applied, whereby cells were selected according to the number of different features that they expressed as well as the amount of mitochondrial contamination (< 5%) that was present within the cell. Mitochondrial genes were then regressed from the dataset. All remaining variable features were used for downstream analysis. The JackStraw method was used with a num.replicate of 100 to determine the amount of statistically significant principal components. Clustering analysis was performed with the FindNeighbors and FindClusters commands using the first 20 principal components and a resolution of 0.5, respectively. Cell type identities were determined using the SingleR package (22). The ImmGen database (http://www.immgen.org/) (23) was used as the reference set for automated cell type annotation. The pruneScores function was used to remove low-quality assignments and cluster identities were renamed based on the cell type annotations provided by SingleR. Differentially expressed genes for each of the clusters were identified using the FindAllMarkers command with the following parameters: min.pct = 0.25 and logfc.threshold = 0.25, using the model-based analysis of single-cell transcriptomics (MAST) test (24).
Differentially expressed genes from each cluster that were identified by Seurat analysis were exported as comma-separated values and imported into the Ingenuity Pathway Analysis (IPA) platform (Qiagen; RRID:SCR_008653). Statistically insignificant genes (padj > 0.05) were filtered out before submission for pathway analysis. Expression data was submitted with the following adjustments to the default parameters: General Settings – direct relationships, Confidence – experimentally observed & high, Species – mouse, Tissues & Cell Lines – bone marrow cells & immune cells. Pathway analysis data was then exported as comma-separated values and plotted using the ggplot2 package in R (https://ggplot2.tidyverse.org/). The complete scRNA-seq dataset has been deposited in the GEO database (accession number GSE153435; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153435).
Trajectory analysis was performed using the dynverse family of packages in R (25). Prior to generating the dynverse object, cells of a single type were subsetted into a new Seurat object. Counts and expression data were obtained from the new object and used to create a dynverse object. Next, a new dimensionality reduction was applied to the data and a trajectory was inferred using Slingshot (RRID:SCR_017012). The newly inferred trajectory was then manually inspected to ensure that logical biological changes in gene expression were detected along the trajectory. If the trajectory appeared abnormal in terms of gene expression changes, a root was provided based on prior knowledge.
Statistics.
Significant differences between groups for flow cytometry, T cell suppression assays, gentamicin protection assays, and in vivo depletion studies were determined using an unpaired two-tailed Student’s t-test or a One-way ANOVA with Tukey’s multiple comparison test using GraphPad Prism (RRID:SCR_002798) where a p-value < 0.05 was considered statistically significant.
RESULTS
Although our earlier reports examined innate immune infiltrates during S. aureus craniotomy infection, this was with a limited panel of markers that did not assess leukocyte activation status and microglial activation or lymphocyte recruitment was also not evaluated (13, 26). Monocytes were preferentially recruited into the brain following S. aureus craniotomy infection, whereas few monocytes were associated with the galea or bone flap (Fig. 1A, C, D, and F). In contrast, PMNs and granulocytic-myeloid-derived suppressor cells (G-MDSCs) were the dominant infiltrates associated with the subcutaneous galea and bone flap, with both populations less frequent in the brain (Fig. 1B, C, D, and F). A small population of CD11bhighLy6G+Ly6C+F4/80+ cells (F4/80+) were detected primarily in the galea and bone flap whose identity remains unknown (Fig. 1B, C, D, and F). In the blood, only monocytes were increased at day 7 following S. aureus craniotomy infection, whereas PMNs and a small population of MDSCs remained unchanged (Fig. 1A, B, and E). The lack of neutrophilia suggests a more localized response to infection, in agreement with other S. aureus biofilm models (20), although alterations in peripheral blood PMNs might be seen at later intervals post-infection than those examined here.
Fig. 1. Compartmentalization of leukocyte recruitment during S. aureus craniotomy infection.
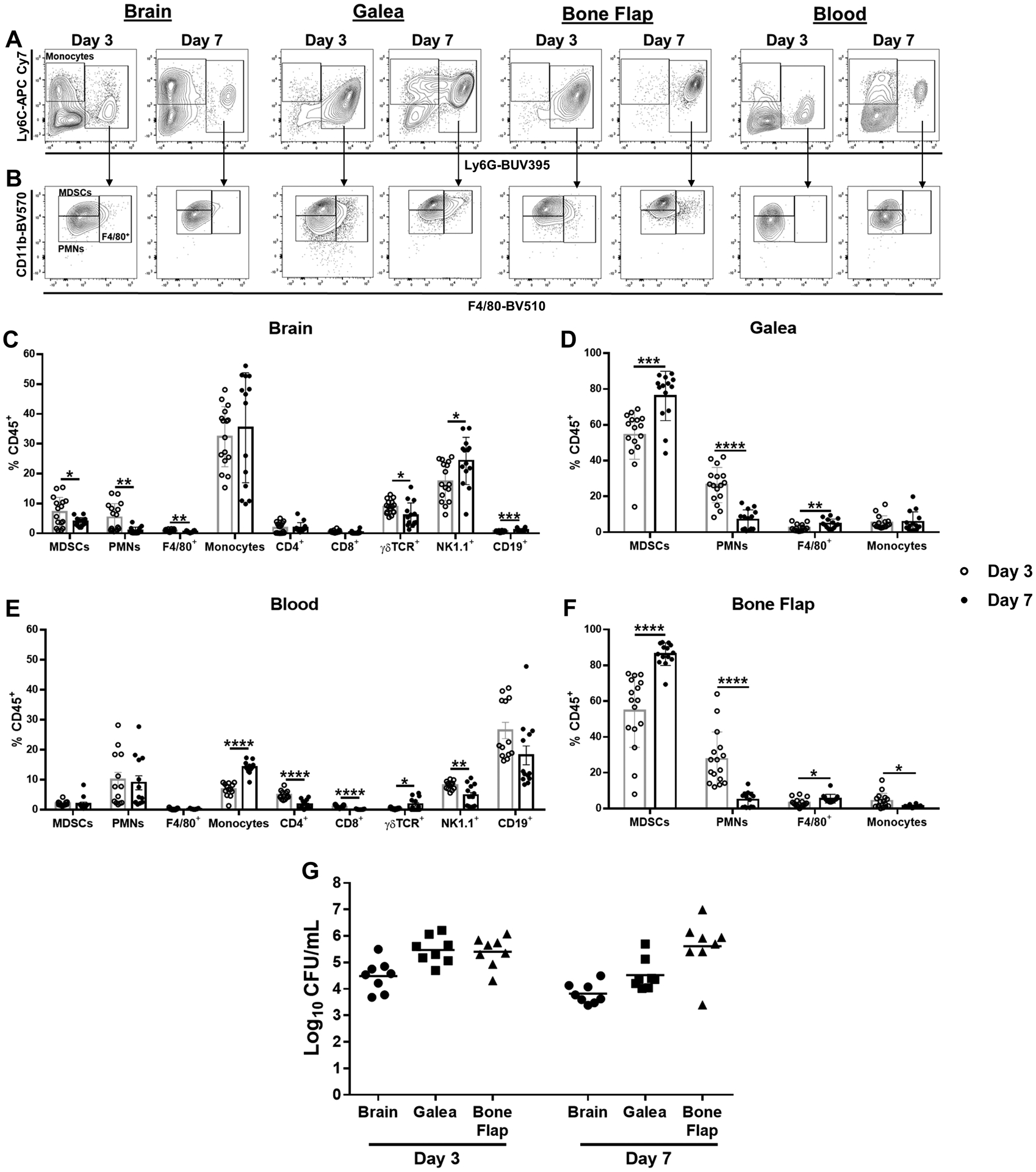
Mice were sacrificed at days 3 (n=16) or 7 (n=14) following S. aureus craniotomy infection, whereupon leukocyte infiltrates in the brain, galea, and bone flap were quantified by flow cytometry. Peripheral blood leukocytes were also examined (n=13 mice/time point). (A) Representative contour plots of Ly6G and Ly6C staining depicting Ly6G+Ly6C+ (right rectangle) and Ly6G−Ly6C+ cells (left square; monocytes) in infected tissues and blood. (B) CD11b and F4/80 expression of Ly6G+Ly6C+ cells identified in A, which differentiates MDSCs (CD11bhighLy6C+Ly6G+F4/80−) from PMNs (CD11blowLy6C+Ly6G+F4/80−) and CD11bhigh/lowLy6C+Ly6G+F4/80+ cells. Leukocyte populations were quantified in the (C) brain, (D) galea (E) blood, and (F) bone flap with (G) bacterial burden in each compartment (n=8/group). Results represent the mean ± SD from one (G) or two combined independent experiments (A-F) and significant differences between day 3 and 7 post-infection for each cell type were determined using an unpaired two-tailed Student’s t-test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001).
An examination of adaptive immune populations revealed the preferential recruitment of NK and ɣδ T cells in the brain during S. aureus craniotomy infection, whereas fewer CD3+CD4+ and CD3+CD8+ T cells or CD19+ B cells were detected (Fig. 1C). Interestingly, adaptive immune infiltrates were minimal in the galea and bone flap, which instead were dominated by granulocyte populations (Fig. 1D and F). ɣδ T cells increased in the blood between days 3 and 7 post-infection, whereas there was a contraction in CD4+, CD8+, and NK cells (Fig. 1E). Bacterial burden was higher in the galea and bone flap compared to the brain at days 3 and 7 post-infection (Fig. 1G), in agreement with our previous reports (13, 17, 26). Few leukocyte infiltrates and minimal changes in microglial activation were observed in response to a sterile craniotomy that was negative for bacterial growth.
We next examined the expression of markers indicative of leukocyte activation and identity, including CCR2, CD14, and MHC Class II. CCR2 was most highly expressed in the small population of CD11bhighLy6G+Ly6C+F4/80+ cells (F4/80+) in the galea and bone flap at day 3 as well as peripheral blood monocytes at day 7 post-infection (Fig. 2A and B). CD14 staining was the highest in CD11bhighLy6G+Ly6C+F4/80+ cells associated with the brain, galea, and bone flap at both time points and was also detected in MDSCs, PMNs, and monocytes in all three compartments, but at lower levels (Fig. 2A and C). Interestingly, the percentage of CD14+ MDSCs and PMNs on the bone flap as well as brain PMNs was reduced at day 7 post-infection compared to day 3 (Fig. 2C). MHC Class II was primarily expressed in monocytes and CD11bhighLy6G+Ly6C+F4/80+ cells (F4/80+) in the blood as well as those invading all three tissue compartments, whereas levels were minimal in PMNs and MDSCs (Fig. 2A and D). Collectively, these findings reveal extensive heterogeneity in leukocyte populations infiltrating the brain compared to the galea and bone flap during S. aureus craniotomy infection.
Fig. 2. Differential expression of leukocyte markers in tissue compartments during S. aureus craniotomy infection.
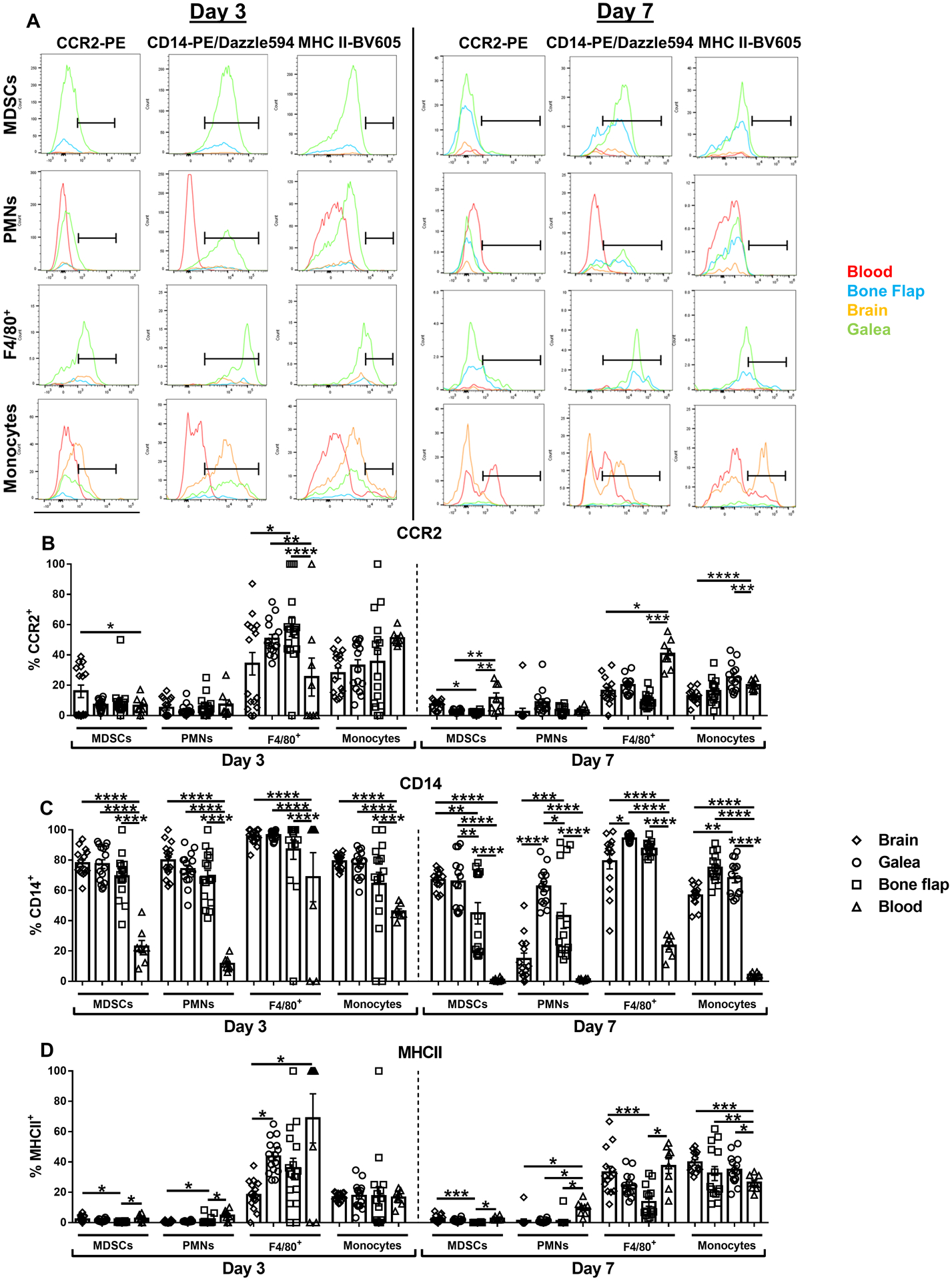
Mice were sacrificed at days 3 (n=16) or 7 (n=14) following S. aureus craniotomy infection, whereupon CCR2, CD14, and MHC Class II expression was quantified by flow cytometry. (A) Representative histograms of leukocyte populations in the brain, galea, bone flap, and blood are shown and the percentage of viable leukocytes expressing (B) CCR2, (C) CD14, and (D) MHC Class II are shown. Results represent the mean ± SEM combined from two independent experiments and analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001).
Microglial CX3CR1 expression was similar at days 3 and 7 post-infection and was significantly higher compared to peripheral blood monocytes and monocytes infiltrating the infected brain (Fig. 3A). CX3CR1 expression was homogeneous in microglia, whereas levels were heterogeneous in infiltrating brain monocytes and peripheral blood monocytes at day 3 post-infection, which became less variable in both populations by day 7 (Fig. 3C). There were no significant differences in the percentages of microglia between days 3 and 7 post-infection (Fig. 3B) and microglia expressed high levels of CD14 and MHC Class II that significantly increased from days 3 to 7 (Fig. 3D). These results are indicative of microglial activation during S. aureus craniotomy infection.
Fig. 3. Microglial characteristics during S. aureus craniotomy infection.
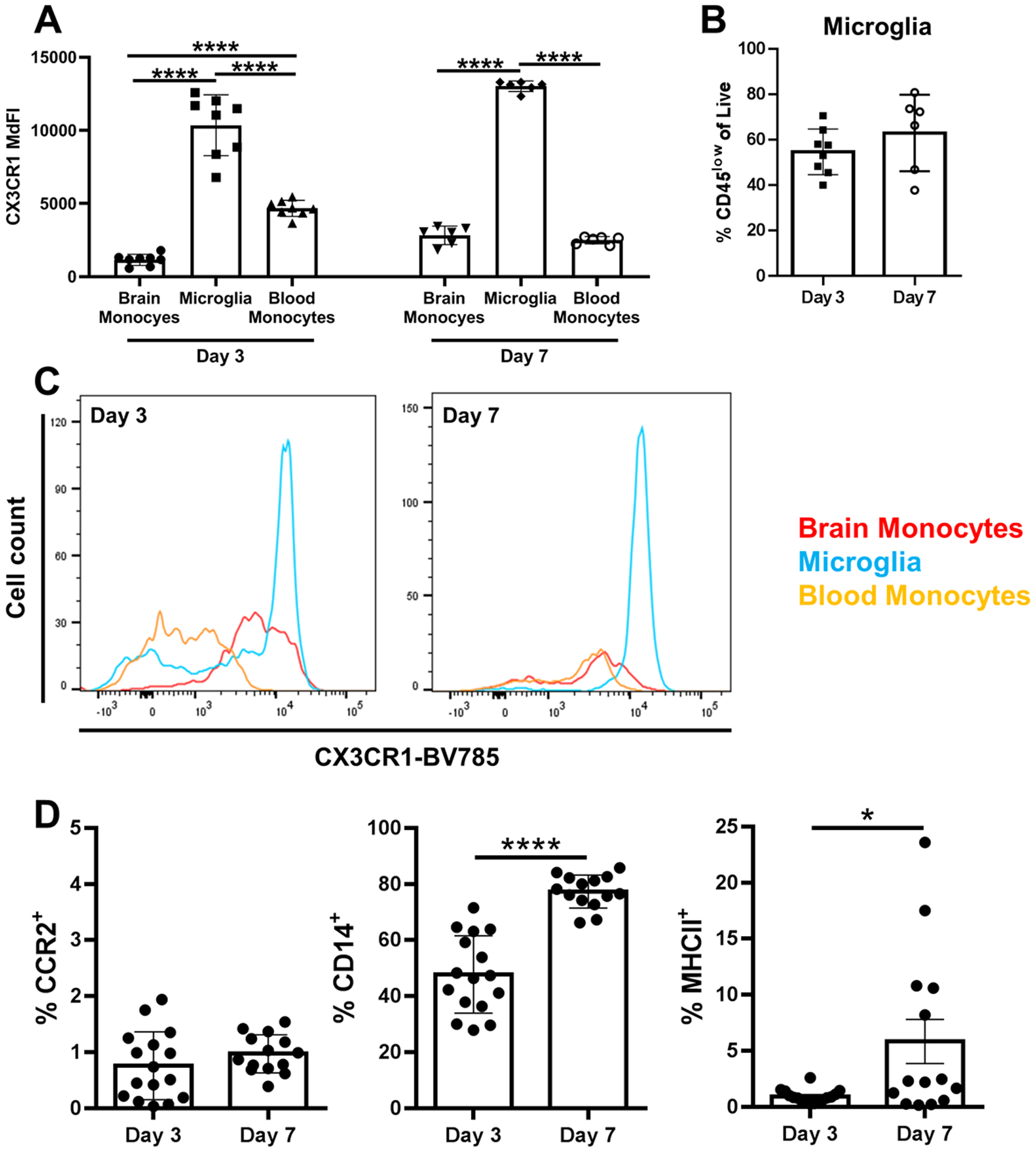
Mice were sacrificed at days 3 (n=16) or 7 (n=14) following S. aureus craniotomy infection for quantification of (A) CX3CR1 expression in microglia, infiltrating brain monocytes, and peripheral blood monocytes and (B) the percentage of microglia. (C) Representative histograms of CX3CR1 expression in microglia vs. monocytes are shown and (D) the percentage of microglia expressing CCR2, CD14, and MHC Class II. Results represent the mean ± SD combined from two independent experiments and analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05; ****, p < 0.0001).
Transcriptional heterogeneity of resident microglia and leukocyte populations infiltrating the brain during S. aureus craniotomy biofilm infection.
To explore the potential transcriptional diversity in immune subsets across the different compartments during S. aureus craniotomy infection, scRNA-seq was performed. CD45+ cells were collected from the brain, galea, bone flap, and blood at day 7 post-infection, a time corresponding to biofilm formation based on antibiotic tolerance and structure (13, 17). UMAP plots revealed the separation of CD45+ clusters in the infected brain, which was similar to the well-delineated subsets in the blood (Fig. 4C and A, respectively). In contrast, the majority of CD45+ cells in the bone flap and galea were tightly clustered, with numerous transcriptionally distinct groups identified (Fig. 4B and D, respectively). Since our study was designed by performing scRNA-seq on total CD45+ cells and not purified leukocyte populations, cluster identifications were first determined in the blood, where leukocyte frequency is well defined and would provide confidence in the subsequent accuracy of identifying transcriptional clusters in the brain, galea, and bone flap. Seventeen distinct transcriptional clusters were identified in the blood and these were classified as various leukocyte populations based on core gene signatures using SingleR (Supplemental Fig. 2). Transcriptional heterogeneity was observed for T and B cells as well as NK cells, as two distinct clusters were identified for each population (Supplemental Fig. 2). Some clusters could not be accurately identified because they lacked a dominant transcriptional signature or included a mix of multiple leukocyte types, which were left as cluster numbers (Supplemental Fig. 2). The percentages of peripheral blood leukocytes identified by scRNA-seq during S. aureus craniotomy infection roughly approximated those detected by flow cytometry (Table 1 and Fig. 1E, respectively).
Fig. 4. S. aureus craniotomy infection induces distinct transcriptional profiles in CD45+ cells.
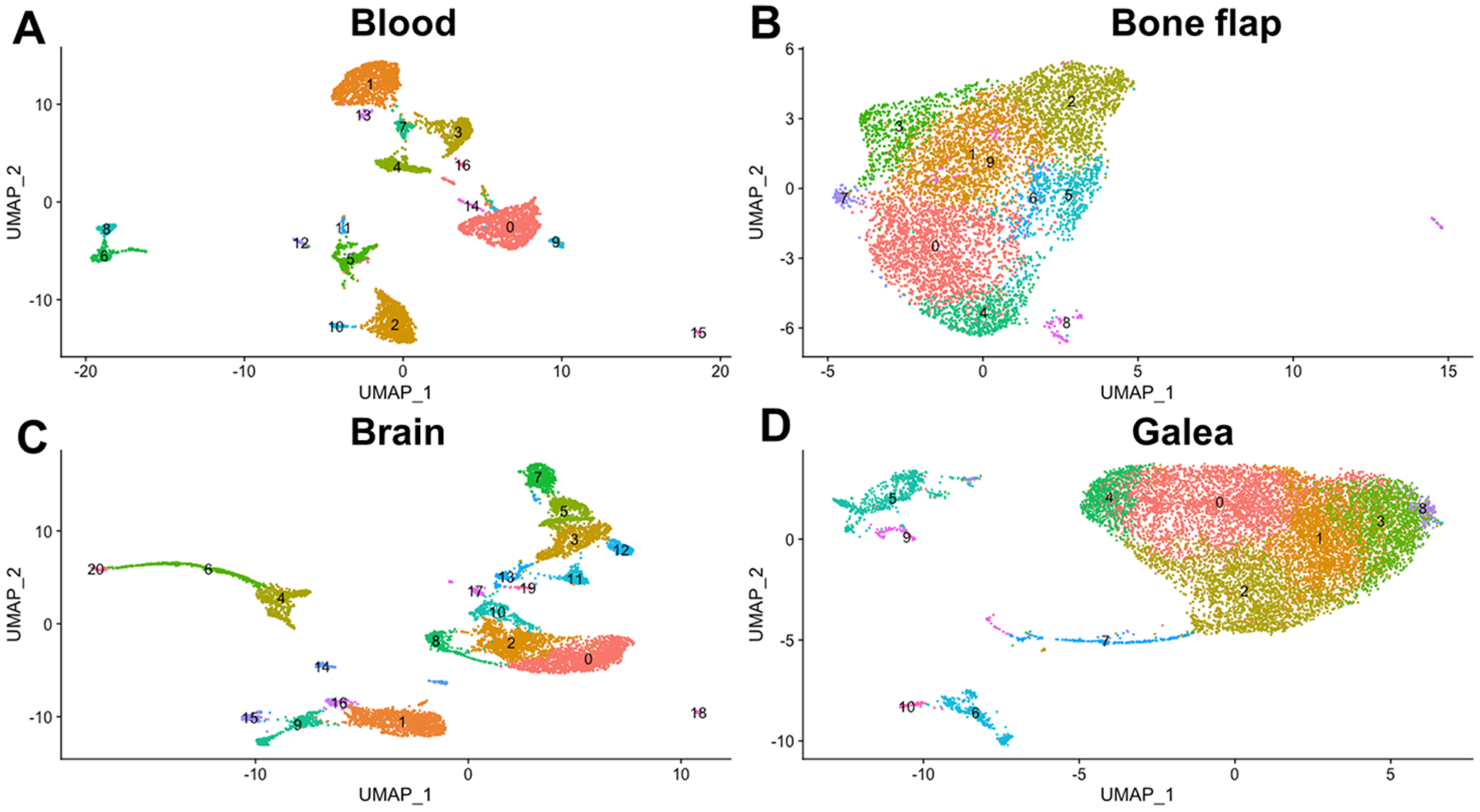
Viable CD45+ cells were isolated from the (A) blood (n= 4,914 cells), (B) bone flap (n= 6,672 cells), (C) brain (n= 9,233 cells), and (D) galea (n= 11,771 cells) of mice (n=10) at day 7 following S. aureus craniotomy infection by FACS and processed for scRNA-seq with transcriptional clusters shown in UMAP plots.
Table 1.
Leukocyte abundance during S. aureus craniotomy infection as determined by scRNA-seq
| Blood | Brain | Galea | Bone Flap | ||||
|---|---|---|---|---|---|---|---|
| Cell Type | % | % | % | % | |||
| Monocytes | 5.25 | 6.83 | 1.90 | 0.09 | |||
| Macrophages | 0.79 | 9.39 | 2.29 | 0.22 | |||
| Microglia | - | 31.20 | - | 0.03 | |||
| Dendritic Cells | 0.55 | 4.44 | 0.58 | 0.55 | |||
| Neutrophils | 22.67 | 11.98 | 90.50 | 98.08 | |||
| Eosinophils | 0.10 | 0.02 | - | 0.03 | |||
| Basophils | 0.53 | 0.23 | 0.07 | 0.01 | |||
| Mast cells | - | 0.03 | 0.01 | - | |||
| NK cells | 20.82 | 4.60 | 0.71 | 0.15 | |||
| NKT cells | 0.85 | 7.34 | 0.76 | 0.03 | |||
| Innate Lymphoid Cells | 0.53 | 3.07 | 0.58 | 0.03 | |||
| T cells | 18.64 | 8.87 | 1.17 | 0.10 | |||
| γδ T cells | 0.51 | 1.15 | 0.08 | 0.03 | |||
| Pro-B cells | 0.06 | 0.43 | 0.03 | 0.12 | |||
| B cells | 19.11 | 2.01 | 0.22 | 0.03 | |||
| Stromal cells | - | 0.02 | - | - | |||
| Stem cells | 0.26 | 0.47 | 0.08 | 0.13 | |||
| Unassigned | 9.32 | 7.91 | 1.02 | 0.90 | |||
| Total | 100.00 | 100.00 | 100.00 | 100.00 | |||
We next analyzed the CD45+ populations recovered from the brain of S. aureus infected animals. UMAP plots revealed extensive microglial heterogeneity, with four transcriptionally distinct clusters identified (Fig. 5A). There were also three granulocyte and two monocyte/macrophage clusters assigned, further evidence of diversity within a given leukocyte population during infection. Importantly, scRNA-seq confirmed the flow cytometry findings with the identification of monocytes, granulocytes, NK cells, B cells, and numerous T cell populations invading the infected brain, although the exact frequencies varied slightly (Figs. 1C and 5A and Table 1). The approach of isolating total CD45+ cells allowed us to identify leukocytes that were not captured by our flow cytometry panel, including dendritic cells and a small population of basophils/eosinophils (Fig. 5A). As was the case with blood leukocytes, the identity of two brain clusters could not be assigned, since transcripts were shared by multiple CD45+ cell types (Fig. 5A).
Fig. 5. Transcriptional heterogeneity in microglia and innate immune cells in the brain during S. aureus craniotomy infection.
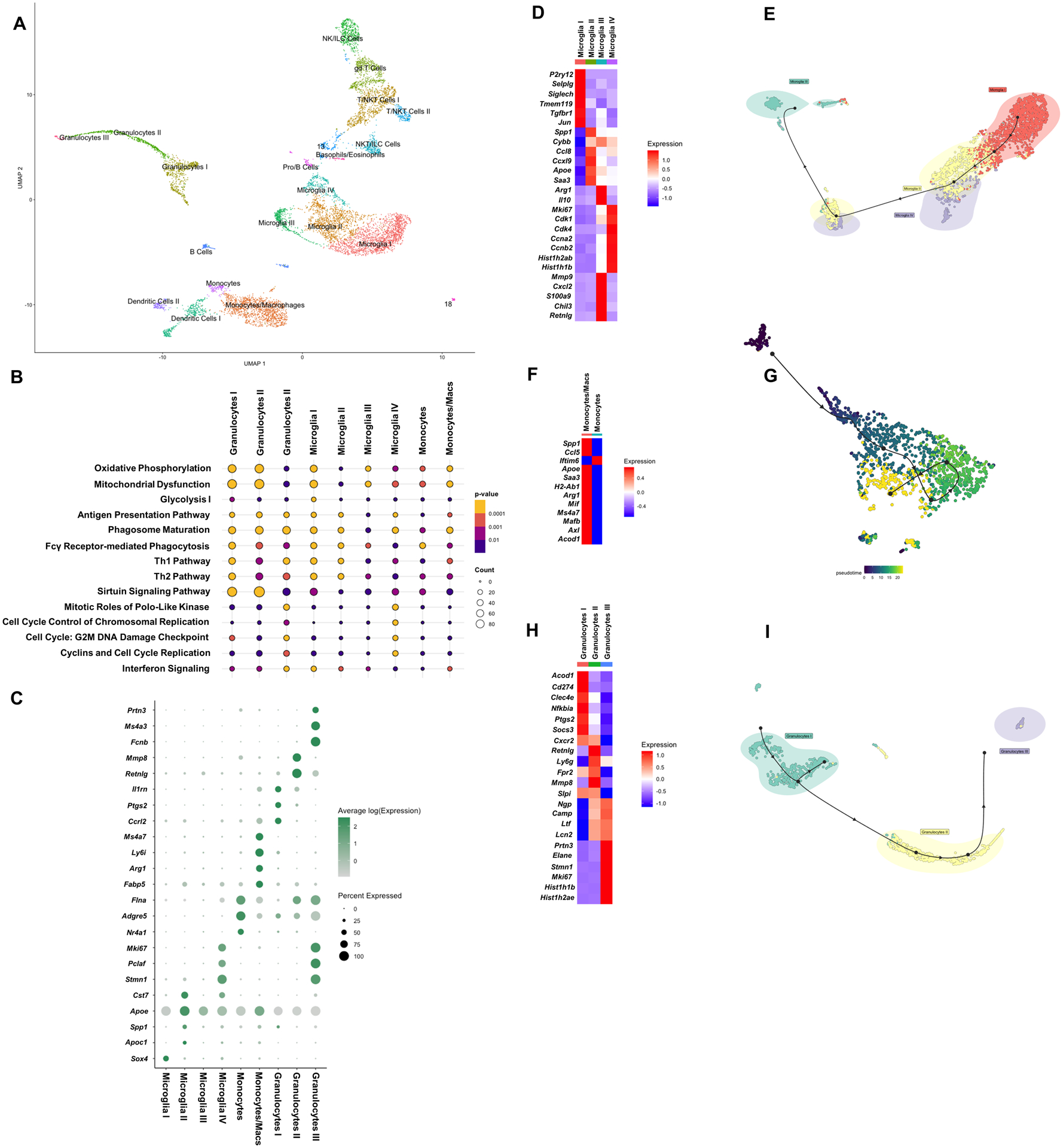
Viable CD45+ cells were isolated from the brain (n= 9,233 cells) of mice (n=10) at day 7 following S. aureus craniotomy infection by FACS and processed for scRNA-seq. (A) Transcriptional clusters were identified by SingleR and are presented as UMAP plots. (B) Ingenuity Pathway Analysis (IPA) of canonical pathways that were significantly expressed in microglia and other brain leukocyte infiltrates and (C) genes that were enriched in specific myeloid clusters in the brain (p < 0.05). Heatmaps and trajectory analysis of (D and E) microglia, (F and G) monocyte/macrophage, and (H and I) granulocyte clusters.
Four distinct clusters of microglia were identified bioinformatically, which was confirmed by their expression of well-characterized microglial markers, including Tmem119, Hexb, CX3CR1, and P2ry12 (Fig. 5A and Supplemental Fig. 3A). The assignment of microglia into clusters I-IV was based on cellular abundance of transcript similarity, where cluster I had the most (1,611 out of 9,233 or 17.5%) and cluster IV had the least (307 out of 9,233 or 3.3%) microglia. We examined the lists of significantly expressed transcripts between the microglial clusters to determine whether the inflammatory tendency of a given cell type could be identified. Microglia in cluster I displayed homeostatic and anti-inflammatory characteristics based on increased expression of p2ry12, selplg, siglech, tmem119, tgfbr1, and jun concomitant with reduced pro-inflammatory (spp1, cybb, cxcl9, apoe, and saa3) gene expression (Fig. 5D). Cluster II displayed an opposite phenotype with regard to the expression of these genes, suggesting it was more pro-inflammatory (Fig. 5D), similar to the disease-associated microglia (DAM) described in Alzheimer’s disease (27). Several cyclin-dependent kinases (cdks) and histones were enriched in cluster IV, suggesting a minor population of proliferating microglia based on their abundance (3.3% of total microglia; Fig. 5D). Cluster III did not express any clear inflammatory bias, displaying high levels of both pro- (mmp9, cxcl2, and s100a9) and anti-inflammatory (arg1, il10, chil3, and retnlg) genes (Fig. 5D). Trajectory analysis revealed the potential transition of the homeostatic/anti-inflammatory population towards the pro-inflammatory and mitotic subsets terminating on the mixed pro-/anti-inflammatory cluster (Fig. 5E).
Two monocyte/macrophage populations were identified, representing the most numerous leukocyte infiltrate in the brain (Fig. 5A and Table 1). Transcriptional patterns were virtually opposite between these subsets, with the most abundant monocyte/macrophage cluster expressing a mix of pro- (spp1, ccl5, apoe, and saa3) and anti-inflammatory (arg1, mif, ms4a7, mafb, axl, and acod1) transcripts (Fig. 5F). Trajectory analysis using pseudotime revealed a clear delineation in the transcriptome within the monocyte/macrophage population, suggesting that cells are responding to infection in an ordered manner (Fig. 5G). Finally, although less abundant (Fig. 5A and Table 1), three granulocyte populations with divergent transcriptional signatures were associated with the infected brain (Fig. 5H). Examining the expression of genes that are associated with MDSCs versus mature PMNs allowed us to identity the largest granulocyte cluster I as G-MDSC-like based on increased expression of aconitate decarboxylase 1 (acod1), cd274 (Programmed death-ligand 1; PD-L1), C-type lectin domain family 4 member E (clec4e), NF-κB inhibitor-α (nfkbia), cyclooxygenase-2 (ptgs2), and suppressor of cytokine signaling 3 (socs3) (15, 20, 28, 29) (Fig. 5H). Granulocyte cluster III was the least abundant and represented mature PMNs based on their robust expression of neutrophil granule protein (ngp), cathelicidin (camp), lactoferrin (ltf), lipocalin 2 (lcn2), proteinase 3 (prtn3), and neutrophil elastase (elane) (Fig. 5H). Interestingly, this mature PMN population (cluster III) also expressed a number of cell cycle-related genes at high levels (stathmin 1 (stmn1), marker of proliferation Ki-67 (mki67), and several histone-related genes) compared to the other granulocyte clusters (Fig. 5H). Granulocyte cluster II shared transcriptional attributes with granulocyte clusters I and III, suggesting they may represent a transitional population (Fig. 5H). This was supported by trajectory analysis, which revealed the progression from G-MDSC-like cells, to the putative transition PMN population, and terminating at mature PMNs (Fig. 5I). Ingenuity Pathway Analysis (IPA) revealed significant cell type specificity for the top pathways in microglia and infiltrating monocytes, macrophages, and granulocytes (Fig. 5B). For example, microglial cluster IV exhibited a cell division signature in agreement with the heightened expression of mitotic genes (Fig. 5D), whereas granulocyte clusters I (G-MDSC-like) and II had enriched sirtuin signaling, suggestive of epigenetic modifications (Fig. 5B).
Based on the transcriptional heterogeneity of microglia and infiltrating myeloid populations in the brain during S. aureus craniotomy infection, a goal was to identify whether unique markers could be identified to facilitate the examination of specific clusters in future studies. Bioinformatics identified a series of genes whose expression was either unique or enriched in one of the four microglial clusters or the monocyte and granulocyte subsets (Fig. 5C). In some instances, a gene was uniquely expressed in one microglial cluster (i.e. stmn1 in cluster IV) but was present in another cell type (i.e. Granulocytes III). This was still considered a useful marker since microglia and granulocytes can be discriminated based on traditional antigens. These genes may allow for the enrichment of specific microglial or infiltrating myeloid subsets to discern their role during S. aureus craniotomy infection.
Transcriptional heterogeneity of granulocytes associated with the galea and bone flap during S. aureus craniotomy biofilm infection.
CD45+ cells recovered from the galea (Fig. 6A) and bone flap (Fig. 6D) at day 7 after S. aureus craniotomy infection revealed an extensive granulocytic network that separated into distinct transcriptional clusters. Monocytes, macrophages, dendritic cells, NK cells, NKT, and T cells were detected in the galea but at very low abundance (Fig. 6A and Table 1), whereas none of these cell types were associated with the bone flap (Fig. 6D and Table 1). Each population exhibited unique transcriptional patterns, further demonstrating the extensive heterogeneity in each compartment (Fig. 6B and E). In the galea, granulocyte clusters II and IV appeared to be G-MDSC-like based on their common expression of genes enriched in MDSCs (ptgs2, clec4e, acod1, nfkbiz, and il1β), whereas cluster VI was identified as mature PMNs based on their robust expression of several granule genes including ngp, camp, ltf, prtn3, elane, and lcn2 (Fig. 6B). As was observed in the brain, the mature PMN population in the galea had high expression levels of genes associated with cell division including stmn1, mki67, and cyclin-dependent kinase-1 (cdk1) (Fig. 6B). Trajectory analysis based on transcriptional patterns suggested two divergent pathways centered at granulocytes II (G-MDSC-like), terminating on granulocytes III and mature PMN (granulocytes V) clusters (Fig. 6C).
Fig. 6. scRNA-seq reveals predominant granulocyte infiltrates in the galea and bone flap following S. aureus craniotomy infection.
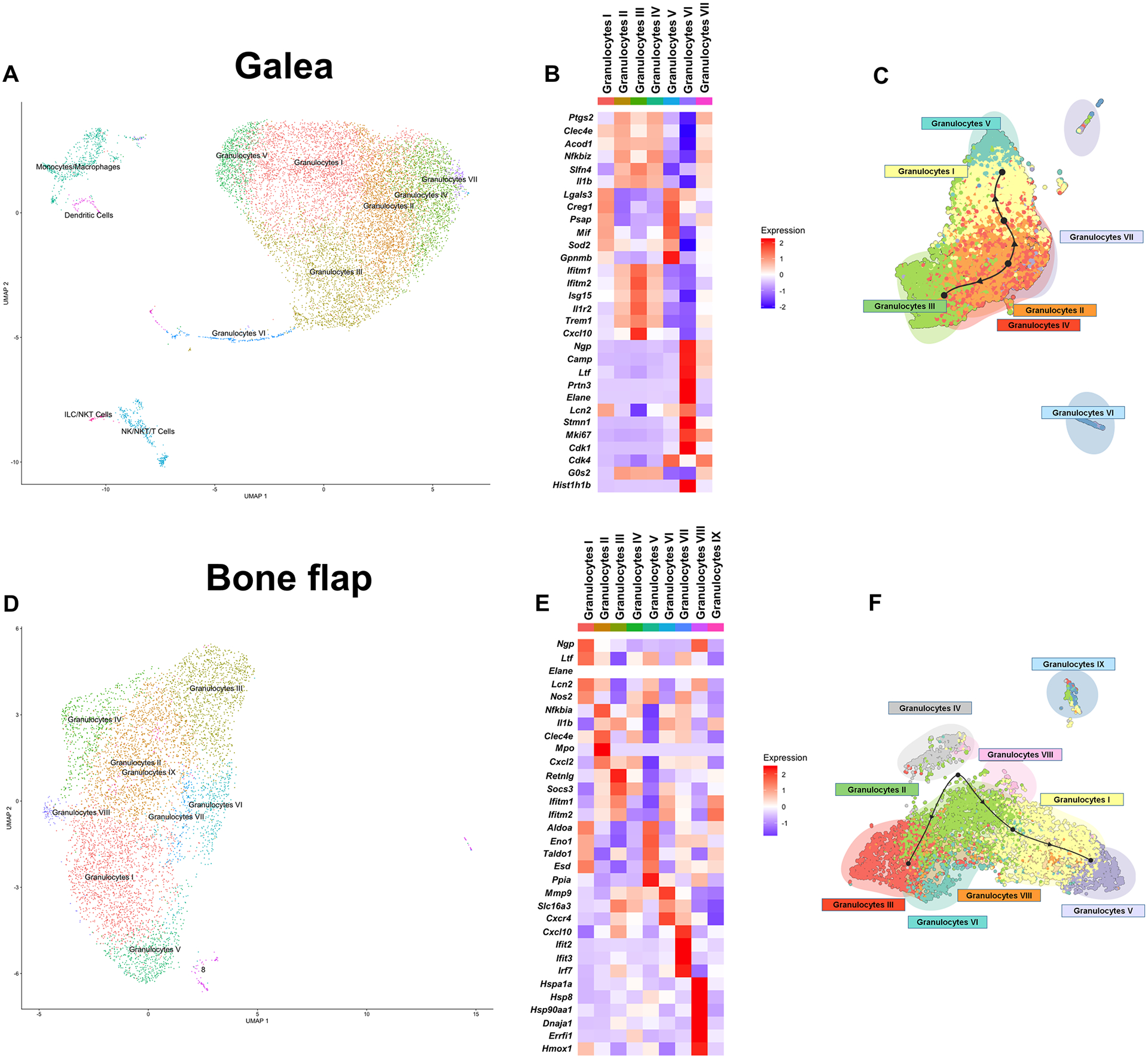
Viable CD45+ cells were isolated from the galea (n= 11,771 cells) and bone flap (n= 6,672 cells) of mice (n=10) at day 7 following S. aureus craniotomy infection by FACS and processed for scRNA-seq. Transcriptional clusters in the (A) galea and (D) bone flap were identified by SingleR and are presented as UMAP plots. Heatmaps and trajectory analysis of granulocyte clusters in the (B and C) galea and (E and F) bone flap.
The bone flap was associated with 9 distinct transcriptional clusters, 8 of which were classified as granulocytes (Fig. 6D). Granulocyte I cluster expressed higher levels of genes associated with mature PMNs (i.e. ngp, ltf, and lcn2) (Fig. 6E). This suggested that the remaining 7 clusters may represent immature granulocyte populations, which was supported by the finding that granulocyte clusters II and III expressed transcripts reflective of MDSCs although with differing patterns (Fig. 6E). Granulocytes V displayed a metabolic signature with increased expression of aldolase A (aldoa), alpha-enolase (enoI), transaldolase 1 (taldo1), S-formylglutathione hydrolase (esd), and peptidyl-prolyl cis-trans isomerase A (ppia) that was also shared with Granulocytes I, whereas granulocytes VII had significantly increased expression of IFN-regulated genes (Fig. 6E). Granulocytes VIII was a minor population but demonstrated a transcriptional signature of cellular stress depicted by elevated heat shock and hypoxia-related transcripts (Fig. 6E). The remaining granulocyte clusters did not have a transcriptional signature to ascertain their potential phenotype and unlike the galea and brain, there was no granulocyte population that was enriched for cell cycle genes. Trajectory analysis suggested a bifurcation, where some granulocyte populations progressed towards the mature PMN cluster while some remained more G-MDSC-like (Fig. 6F).
Several trends emerged from IPA analysis of the granulocyte populations in the galea and bone flap. Of particular interest was the metabolic divergence between clusters that favored more glycolytic vs. mitochondrial pathways. In general, cell types that had increased expression of oxidative phosphorylation and mitochondrial dysfunction genes had decreased glycolysis and vice versa (Fig. 7A and B). As was observed for the most abundant granulocyte clusters in the brain (I and II; Fig. 5B), sirtuin signaling was increased in some granulocyte clusters in the galea (V and VI) and bone flap (IV and V) and the sirtuin pathway was also significantly enriched in galea-associated dendritic cells, monocytes, macrophages, NK, NKT, and T cell clusters (Fig. 7A). We also compared significantly expressed pathways in each of the granulocyte clusters across the brain, galea, and bone flap to determine how the local tissue microenvironment influenced gene expression (Fig. 7C). T cell activation pathways were higher in the brain and bone flap granulocyte populations compared to granulocyte clusters in the galea, suggesting the presence of niche-specific programming. Likewise, pathways involved in phagosome maturation and antigen presentation were enriched in granulocytes in the bone flap and brain but not the galea (Fig. 7C). Collectively, these results establish the transcriptional heterogeneity within various leukocyte populations and microglia during S. aureus craniotomy infection, which are likely influenced by the local microenvironment and bacterial burden in each compartment.
Fig. 7. Pathway analysis of significantly differently expressed genes in leukocyte populations associated with the galea and bone flap during S. aureus craniotomy infection.
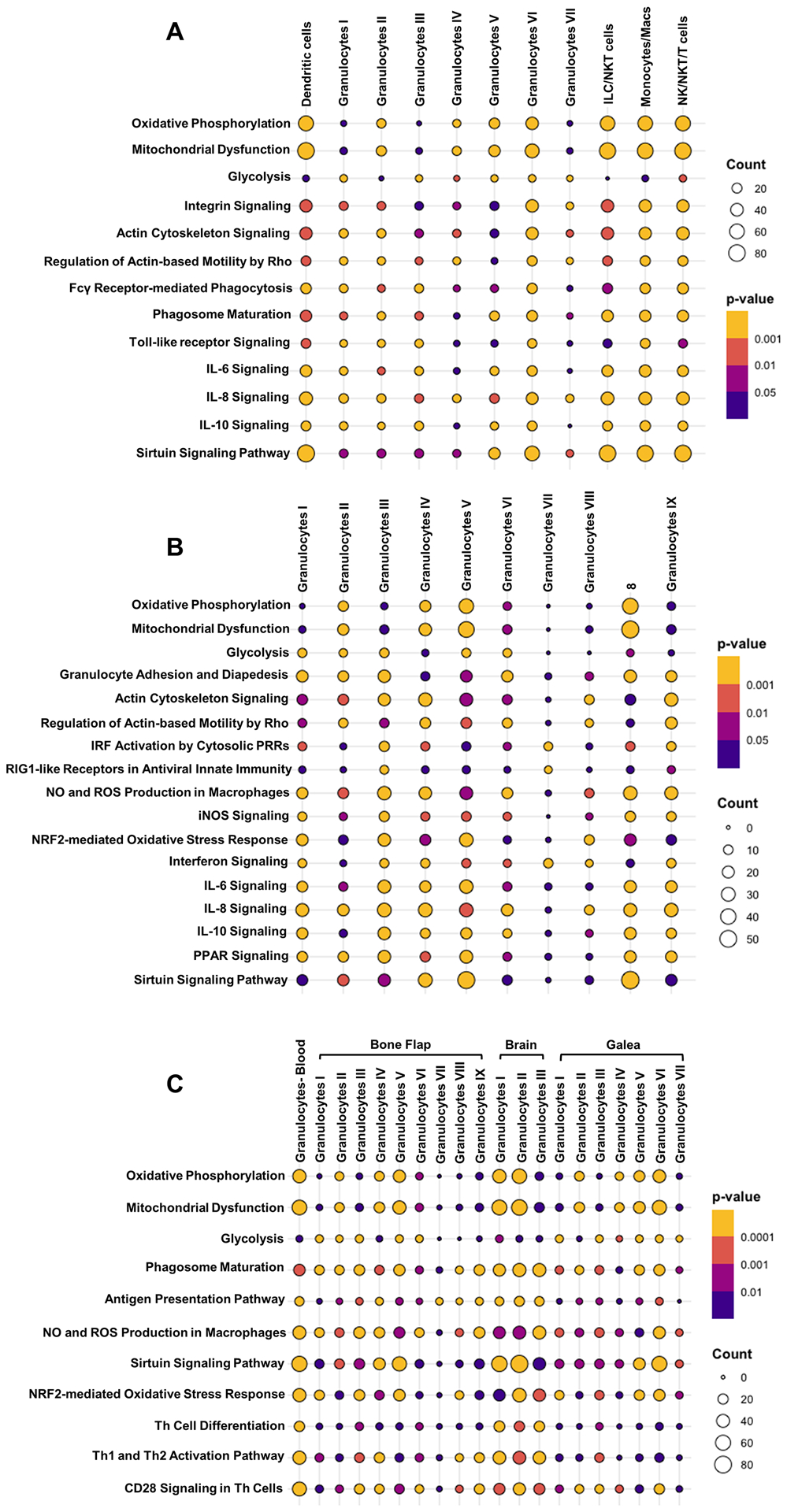
Ingenuity Pathway Analysis (IPA) of canonical pathways that were significantly expressed in the (A) galea and (B) bone flap at day 7 post-infection (p < 0.05). (C) IPA comparisons of significantly differently expressed pathways in granulocyte populations across the blood, brain, galea, and bone flap during S. aureus craniotomy infection at day 7 (p < 0.05).
Leukocytes recovered from the brain, galea, and bone flap exhibit T cell suppressive properties.
Because craniotomy infection is difficult to clear in an immune competent host without surgical intervention (10, 11, 30), we next examined whether this could be attributed, in part, to immune suppressive attributes of infiltrating leukocytes. MDSCs are known to inhibit T cell activation as a primary means of attenuating immune responses (16); therefore, we examined the ability of infiltrating MDSCs and other leukocyte subsets during craniotomy infection to inhibit CD4+ T cell activation. In the brain, MDSCs, CD11bhighLy6G+Ly6C+F4/80+ cells (F4/80+), and monocytes all inhibited T cell proliferation, which was more pronounced at day 3 vs. day 7 post-infection, whereas PMNs could not be examined because of limited cell numbers (Fig. 8). Similar findings were obtained with leukocytes recovered from the galea and bone flap; however, monocytes could not be tested in most instances due to low cell yields (Fig. 8).
Fig. 8. Leukocytes recovered from the brain, bone flap, and galea inhibit T cell proliferation.
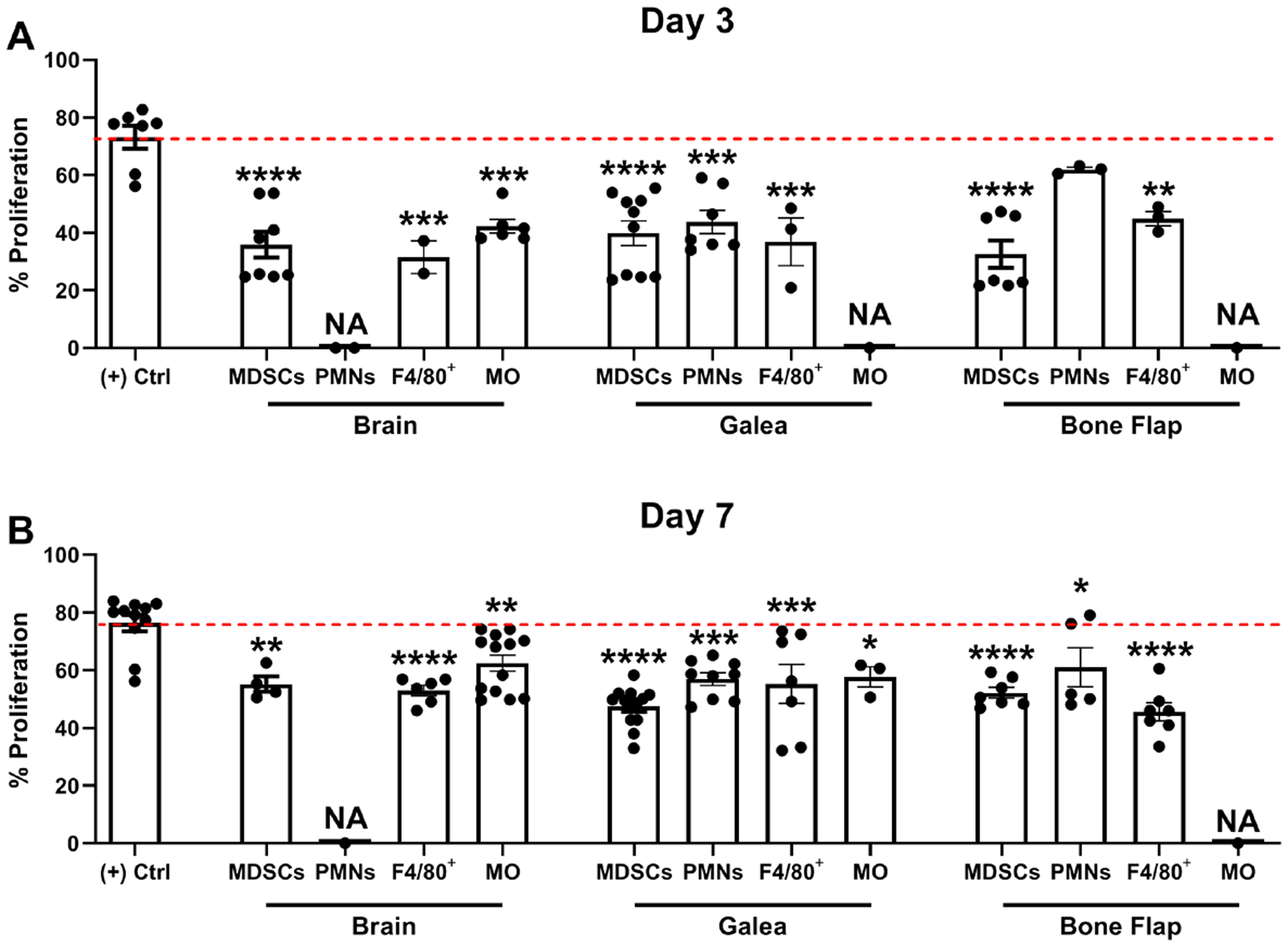
Leukocyte populations were isolated from the brain, galea, or bone flap at (A) 3 or (B) 7 days following S. aureus craniotomy infection by FACS and assessed for their ability to inhibit CD4+ T cell proliferation at a 1:1 ratio (T cell:MDSC, F4/80+, PMN, or monocyte; MO). Results are expressed as the percentage of proliferation compared to CD3/CD28-stimulated T cells (+ Control (Ctrl); red dotted line). A sufficient number of leukocytes could not be recovered from various compartments for this assay (NA, not applicable). Results represent the mean ± SD combined from three independent experiments and analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 compared to the (+) control).
Ly6G+ cells are critical for bacterial containment during S. aureus craniotomy infection.
Given the prominent granulocyte infiltrate in the galea and bone flap identified by flow cytometry and scRNA-seq, we next determined the functional importance of this population using an antibody depletion strategy. Although we previously reported that anti-Gr-1 increased S. aureus burden during craniotomy infection (13), this antibody detects both Ly6G and Ly6C (31), resulting in the depletion of not only Ly6G+ PMNs and MDSCs but also Ly6C+ monocytes. Therefore, this study utilized anti-Ly6G to target PMNs and MDSCs, leaving the monocyte population intact. Anti-Ly6G (IA8) treatment effectively depleted PMNs and MDSCs from the brain, galea, and bone flap (Fig. 9 A–C), which resulted in a significant expansion in bacterial burden in all compartments at day 7 post-infection (Fig. 9D). This finding suggests that PMNs play an important role in S. aureus biofilm containment. Although MDSCs were also depleted using this strategy, it is less likely that they are responsible for preventing bacterial outgrowth, since they are an immature and suppressive population (15, 16). To investigate this further, the effect of MDSCs on PMN bactericidal activity was assessed using gentamicin protection assays. PMNs that were co-cultured with MDSCs had higher intracellular S. aureus burden during an acute interval (2 h) compared to PMNs alone (Fig. 9E), revealing an inhibitory effect of MDSCs on PMN bactericidal activity. This is likely mediated by a soluble factor(s), the identity of which remains to be determined, since inhibition was also observed when PMNs and MDSCs were physically separated during the co-culture period using Transwell inserts. Therefore, the ability of MDSCs to attenuate PMN antimicrobial activity likely represents one mechanism to account for S. aureus persistence during craniotomy infection.
Fig. 9. Ly6G+ PMNs are critical for bacterial containment during S. aureus craniotomy infection.
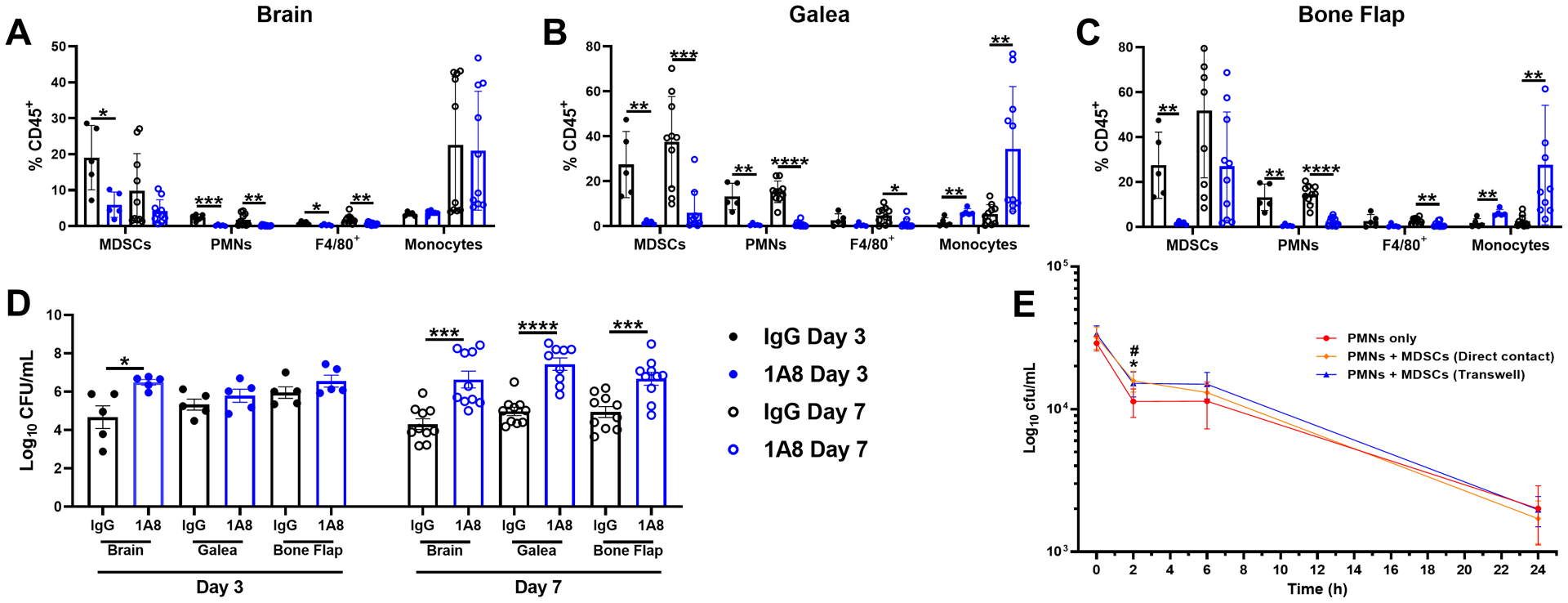
Mice received anti-Ly6G (1A8) or isotype-matched control antibody (100 μg/mouse) beginning one day prior to S. aureus craniotomy infection and every two days until animals were sacrificed. Leukocyte infiltrates in the (A) brain, (B) galea, and (C) bone flap were quantified by flow cytometry with (D) bacterial burden in each compartment determined. Results represent the mean ± SD from one (day 3; n=5 mice/group) or two combined independent experiments (day 7; n=10 mice/group) and analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001). (E) PMNs were exposed to live S. aureus USA300 LAC for 2 h at an MOI of 10:1 (bacteria:cell) in the presence or absence of MDSCs in direct contact or separated by Transwell inserts to evaluate the effects on PMN S. aureus killing by gentamicin protection assays. Results are combined from two independent experiments (n = 5–8 biological replicates) and are presented as the mean ± SD. (*, p < 0.05 for PMNs only vs. PMNs + MDSCs in direct contact; #, p < 0.05 for PMNs only vs. PMNs + MDSCs with Transwells; One-way ANOVA with Tukey’s multiple comparisons test).
DISCUSSION
This work has identified a transcriptionally diverse network of microglia and infiltrating leukocyte populations during S. aureus craniotomy infection. Transcriptional heterogeneity in the infected brain was primarily detected in microglia, monocytes, and granulocytes but interestingly, not in adaptive immune populations. Extensive microglial heterogeneity was evident, with trajectory analysis suggesting the progression of microglia from a homeostatic/anti-inflammatory subset to pro-inflammatory and mitotic clusters, terminating in a mixed pro-/anti-inflammatory population. The recruitment of innate lymphoid populations, including NK and ɣδ T cells was enriched in the brain but was negligible in the galea and bone flap, highlighting the niche-dependent differences in location. This may be explained by the fact that T cell chemokines, including CCL5, CXCL9, and CXCL10 were highly expressed in microglia, monocytes/macrophages, dendritic cells, NK cells, and ɣδ T cells in the brain, whereas these transcripts were less abundant in the granulocytic populations in the galea and bone flap. These data will serve as a valuable resource for CNS infectious disease research since comprehensive transcriptional profiling has not yet been performed in the setting of CNS S. aureus infection. Furthermore, this work has identified genes that are unique or enriched in specific transcriptional clusters of a given cell type, allowing these molecules to be leveraged in future studies to purify subsets to examine their attributes. This is an important advance since traditional markers capture the entire heterogeneous population as exemplified by the scRNA-seq data shown here.
Both flow cytometry and scRNA-seq analysis identified granulocyte populations as the predominant infiltrate in the galea and bone flap during S. aureus craniotomy infection. Although flow cytometry revealed CD14 expression in MDSCs and PMNs in both locations, levels were generally lower compared to monocytes at day 7 post-infection and the transcriptional signature of MDSCs and PMNs examined at this same time point and lack of MHC Class II supports their granulocytic origin. Depletion of these granulocyte populations with anti-Ly6G resulted in bacterial outgrowth in not only the galea and bone flap, where these populations were most abundant, but also the brain where fewer granulocytes were present. Although both PMNs and MDSCs were depleted with anti-Ly6G, PMNs were responsible for restricting S. aureus outgrowth, since as described below MDSCs were found to inhibit PMN antimicrobial activity in agreement with their immune suppressive actions (15, 16).
The fact that S. aureus craniotomy infection persists when PMNs are present suggests the action of mechanisms that limit PMN antibacterial activity. S. aureus is well recognized for its ability to thwart PMN action via the production of numerous virulence factors that target opsonophagocytosis (protein A), ROS production (catalase and superoxide dismutase), and induce PMN death (leukocidins, phenol soluble modulins, and α-toxin) (32). Another possibility could be the inhibition of PMN activity by MDSCs that were also a prominent infiltrate in the galea and bone flap. MDSCs were capable of attenuating PMN S. aureus bactericidal activity; however, this effect was only observed during an acute co-culture interval. This suggests that either MDSC-mediated inhibition is short-lived or that more dramatic differences might have been seen with an increased number of MDSCs rather than the 1:1 MDSC:PMN ratio examined here, since MDSCs outnumber PMNs in the galea and bone flap. Nevertheless, it is important to recognize the limitations of such in vitro assessments, since this reductionist approach cannot accurately model the complex microenvironment that MDSCs and PMNs encounter in the galea and bone flap during S. aureus craniotomy infection in vivo. Therefore, it is possible that more extensive crosstalk between MDSCs and PMNs exists during S. aureus craniotomy infection than demonstrated here and warrants detailed examination in future studies. Regardless, this finding provides clarity to the Ly6G depletion results and indicates that PMNs are the driving factor for preventing S. aureus outgrowth in vivo, which is not dramatically influenced by Ly6G+ G-MDSCs. Collectively, these findings may explain, in part, why PMNs are not capable of clearing S. aureus craniotomy infection due to the inhibitory action of MDSCs in concert with the arsenal of S. aureus virulence factors that target PMN activity.
Although this study has identified an important role for Ly6G+ PMNs in preventing S. aureus outgrowth during craniotomy infection, this raises the question about the functional role of monocytes/macrophages given the observed transcriptional heterogeneity in these cells as demonstrated by scRNA-seq. Although we have not directly assessed the functional role of monocytes/macrophages during craniotomy infection, we have indirect evidence suggesting that they also contribute to S. aureus containment similar to PMNs. This was supported by our prior findings that a Gr-1 antibody, which depletes both Ly6G+ PMNs and MDSCs but also Ly6C+ monocytes/macrophages, led to a significant increase in bacterial burden within 48 h post-infection and animals were extremely moribund (13). In contrast, in the current study where only PMNs and MDSCs were targeted with Ly6G antibody, the increase in S. aureus burden was not observed until day 7 post-infection and mice were not moribund. By extension, this suggests that monocytes/macrophages also play a protective role during craniotomy infection, since the consequences of infection were more severe when they were depleted concomitant with PMNs by anti-Gr-1. It will be interesting to assess the role of microglia in dictating S. aureus clearance vs. persistence during craniotomy infection, particularly given the transcriptional heterogeneity in microglia observed in this study by scRNA-seq. In practice, this will be difficult to elucidate because currently available strategies to interrogate microglial function in vivo rely on global targeting of the microglial population (i.e. CSF-1R agonists, CX3CR1-Cre systems). However, we have identified genes that are unique or enriched in particular microglial clusters and future studies will interrogate the utility of these markers for the enrichment of specific microglial subsets during craniotomy infection for an assessment of their inflammatory attributes ex vivo.
An intriguing finding was that PMNs recovered from the galea and bone flap possessed suppressive activity by inhibiting T cell proliferation at days 3 and 7 post-infection, yet PMN depletion with anti-Ly6G resulted in increased S. aureus burden in the brain, galea, and bone flap. The reason for this finding is unclear but suggests disconnect between the antimicrobial effector function of PMNs and their effects on T cell activation. We are currently examining the functional role of T cells during S. aureus craniotomy infection and the interplay between T cells and how PMNs regulate their activity. It is well appreciated that Th17-derived cytokines elicit the production of PMN chemokines (33) and ROS can inhibit T cell activation (34), which is produced by activated PMNs. This represents an interesting dichotomy because phagocyte ROS is also an important bactericidal effector. Of note, T cell infiltrates were rare or undetectable in the galea and bone flap where the majority of MDSCs were recruited, in agreement with their ability to suppress T cell activation. It was interesting that monocytes infiltrating the infected brain also inhibited T cell proliferation, which coincided with robust Arg-1 expression, which consumes extracellular arginine that is required for TCRζ expression (35–38).
In the brain, IPA identified a signature reflective of APC function and T cell crosstalk, in agreement with the preferential recruitment of lymphocytes into the brain as revealed by FACS. For example, Th1, Th2, and CD28 signaling in Th cells were enriched in microglial clusters I and II and antigen presenting cell and phagosome maturation were dominant in several microglial and granulocyte clusters. In the galea, FcR-mediated phagocytosis, phagosome maturation, and Toll-like receptor (TLR) signaling were enriched in the monocyte/macrophage/DC populations. Although these subsets were less abundant than the granulocyte clusters, these findings are in agreement with our recent study demonstrating the importance of TLR2 signaling in S. aureus containment during craniotomy infection (26). Furthermore, pathways for cytokines that are elicited by TLR signaling (i.e. IL-6, IL-8, and IL-10) were also enriched in the galea but with a broader distribution across the various clusters. The bone flap revealed extensive heterogeneity in granulocyte adhesion and diapedesis, actin cytoskeleton signaling, and regulation of actin-based motility by Rho despite the fact that all of the clusters were identified as granulocytes. IFN pathways were more significantly enriched in granulocytes III and VII, whereas oxidative and nitrogen-mediated pathways were prominent in numerous granulocyte subsets. This profile was distinct from the galea and may be influenced by the direct contact of bone flap granulocytes with S. aureus biofilm. Nevertheless, this free radicle response is not sufficient to clear the infection. Bone flap granulocyte populations also displayed a mixed inflammatory response, with a significant enrichment in both pro- (IL-6, IL-8) and anti-inflammatory (IL-10, peroxisome proliferator receptor; PPAR) pathways in the same clusters, which could be another mechanism to explain why biofilm is not eliminated.
Since granulocyte subsets were dominant in the galea and bone flap and also present in the brain, we compared pathways across these compartments to identify conserved and divergent granulocyte signatures. An opposite relationship between oxidative phosphorylation/mitochondrial dysfunction and glycolysis was observed in granulocyte clusters on the bone flap (granulocytes IV and V), the galea (granulocytes II, IV, V, and VI), and the brain (granulocytes I and II). Of note, the same relationship (i.e. increased oxidative phosphorylation/mitochondrial dysfunction and decreased glycolysis) was observed in blood granulocytes, suggesting that these CNS granulocyte populations are derived from newly recruited peripheral blood granulocytes before they become conditioned to their microenvironment to diversify transcriptionally. This was also supported by the finding that sirtuin signaling was in higher peripheral blood granulocytes and the same CNS granulocyte populations, suggestive of epigenetic regulation. Finally, T cell-related pathways were enriched in all three granulocyte subsets in the brain as well as the blood, reflective of where T cells are present. In contrast, T cell pathways were not highly abundant in the galea and only detected in minor granulocyte subsets associated with the bone flap (granulocytes IV and IX), in agreement with the paucity of lymphocytes in these compartments as validated by FACS.
Collectively, this study highlights the compartmentalization of immune responses during S. aureus craniotomy infection, which is magnified by the transcriptional diversity of microglia, monocyte, and granulocyte populations in each compartment. This information will be critical for a better understanding of pathways that dictate biofilm clearance vs. persistence, considering the distinct cellular profiles elicited in each tissue niche. This will be facilitated by the unique/enriched genes that were identified in this study for specific clusters within a given cell type. These markers can be leveraged in future studies to purify subsets to assess their inflammatory attributes and functional activity ex vivo.
Supplementary Material
KEY POINTS.
Leukocyte influx occurs in specific niches during S. aureus craniotomy infection.
S. aureus craniotomy infection elicits transcriptional diversity in myeloid cells.
PMNs are critical for S. aureus containment but not biofilm clearance.
ACKNOWLEDGEMENTS
The authors thank Rachel Fallet for managing the mouse colony.
This work was supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke R01 NS107369 (to T.K.). The UNMC Genomics Core receives partial support from the National Institute for General Medical Science (NIGMS; INBRE - P20GM103427-14 and COBRE - 1P30GM110768-01). Both the UNMC Genomics and Flow Cytometry Research Cores receive support from The Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727).
Footnotes
COMPETING INTERESTS
The authors declare that they have no competing interests.
REFERENCES
- 1.McClelland S 3rd, and Hall WA. 2007. Postoperative central nervous system infection: incidence and associated factors in 2111 neurosurgical procedures. Clin Infect Dis 45: 55–59. [DOI] [PubMed] [Google Scholar]
- 2.Chiang HY, Steelman VM, Pottinger JM, Schlueter AJ, Diekema DJ, Greenlee JD, Howard MA 3rd, and Herwaldt LA. 2011. Clinical significance of positive cranial bone flap cultures and associated risk of surgical site infection after craniotomies or craniectomies. J Neurosurg 114: 1746–1754. [DOI] [PubMed] [Google Scholar]
- 3.Flemming HC, and Wingender J. 2010. The biofilm matrix. Nat Rev Microbiol 8: 623–633. [DOI] [PubMed] [Google Scholar]
- 4.Schilcher K, and Horswill AR. 2020. Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol Mol Biol Rev 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conlon BP, Rowe SE, and Lewis K. 2015. Persister cells in biofilm associated infections. Adv Exp Med Biol 831: 1–9. [DOI] [PubMed] [Google Scholar]
- 6.Cameron DR, Shan Y, Zalis EA, Isabella V, and Lewis K. 2018. A Genetic Determinant of Persister Cell Formation in Bacterial Pathogens. J Bacteriol 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamada KJ, and Kielian T. 2019. Biofilm-Leukocyte Cross-Talk: Impact on Immune Polarization and Immunometabolism. J Innate Immun 11: 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scherr TD, Heim CE, Morrison JM, and Kielian T. 2014. Hiding in Plain Sight: Interplay between Staphylococcal Biofilms and Host Immunity. Front Immunol 5: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auguste KI, and McDermott MW. 2006. Salvage of infected craniotomy bone flaps with the wash-in, wash-out indwelling antibiotic irrigation system. Technical note and case series of 12 patients. J Neurosurg 105: 640–644. [DOI] [PubMed] [Google Scholar]
- 10.Bruce JN, and Bruce SS. 2003. Preservation of bone flaps in patients with postcraniotomy infections. J Neurosurg 98: 1203–1207. [DOI] [PubMed] [Google Scholar]
- 11.Wallace DJ, McGinity MJ, and Floyd JR 2nd. 2018. Bone flap salvage in acute surgical site infection after craniotomy for tumor resection. Neurosurg Rev 41: 1071–1077. [DOI] [PubMed] [Google Scholar]
- 12.Baumeister S, Peek A, Friedman A, Levin LS, and Marcus JR. 2008. Management of postneurosurgical bone flap loss caused by infection. Plast Reconstr Surg 122: 195e–208e. [DOI] [PubMed] [Google Scholar]
- 13.Cheatle J, Aldrich A, Thorell WE, Boska MD, and Kielian T. 2013. Compartmentalization of immune responses during Staphylococcus aureus cranial bone flap infection. Am J Pathol 183: 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Balaban NQ, Helaine S, Lewis K, Ackermann M, Aldridge B, Andersson DI, Brynildsen MP, Bumann D, Camilli A, Collins JJ, Dehio C, Fortune S, Ghigo JM, Hardt WD, Harms A, Heinemann M, Hung DT, Jenal U, Levin BR, Michiels J, Storz G, Tan MW, Tenson T, Van Melderen L, and Zinkernagel A. 2019. Definitions and guidelines for research on antibiotic persistence. Nat Rev Microbiol 17: 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medina E, and Hartl D. 2018. Myeloid-Derived Suppressor Cells in Infection: A General Overview. J Innate Immun: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talmadge JE, and Gabrilovich DI. 2013. History of myeloid-derived suppressor cells. Nat Rev Cancer 13: 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aldrich A, Kuss MA, Duan B, and Kielian T. 2019. 3D Bioprinted Scaffolds Containing Viable Macrophages and Antibiotics Promote Clearance of Staphylococcus aureus Craniotomy-Associated Biofilm Infection. ACS Appl Mater Interfaces 11: 12298–12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, Engebretsen IL, Bayles KW, Horswill AR, and Kielian T. 2011. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol 186: 6585–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LaFrance-Corey RG, and Howe CL. 2011. Isolation of brain-infiltrating leukocytes. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heim CE, West SC, Ali H, and Kielian T. 2018. Heterogeneity of Ly6G(+) Ly6C(+) Myeloid-Derived Suppressor Cell Infiltrates during Staphylococcus aureus Biofilm Infection. Infect Immun 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heim CE, Vidlak D, Scherr TD, Hartman CW, Garvin KL, and Kielian T. 2015. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J Immunol 194: 3861–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, and Bhattacharya M. 2019. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heng TS, Painter MW, and Immunological Genome Project C. 2008. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9: 1091–1094. [DOI] [PubMed] [Google Scholar]
- 24.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, Linsley PS, and Gottardo R. 2015. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 16: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saelens W, Cannoodt R, Todorov H, and Saeys Y. 2019. A comparison of single-cell trajectory inference methods. Nat Biotechnol 37: 547–554. [DOI] [PubMed] [Google Scholar]
- 26.Aldrich AL, Heim CE, Shi W, Fallet RW, Duan B, and Kielian T. 2020. TLR2 and caspase-1 signaling are critical for bacterial containment but not clearance during craniotomy-associated biofilm infection. J Neuroinflammation 17: 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, and Amit I. 2017. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169: 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- 28.Mastio J, Condamine T, Dominguez G, Kossenkov AV, Donthireddy L, Veglia F, Lin C, Wang F, Fu S, Zhou J, Viatour P, Lavilla-Alonso S, Polo AT, Tcyganov EN, Mulligan C Jr., Nam B, Bennett J, Masters G, Guarino M, Kumar A, Nefedova Y, Vonderheide RH, Languino LR, Abrams SI, and Gabrilovich DI. 2019. Identification of monocyte-like precursors of granulocytes in cancer as a mechanism for accumulation of PMN-MDSCs. J Exp Med 216: 2150–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tcyganov E, Mastio J, Chen E, and Gabrilovich DI. 2018. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol 51: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Talwar AA, Bhat DK, Heiman AJ, and Ricci JA. 2020. Outcomes of Immediate Titanium Cranioplasty Following Post-Craniotomy Infection. J Craniofac Surg. [DOI] [PubMed] [Google Scholar]
- 31.Fleming TJ, Fleming ML, and Malek TR. 1993. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6–8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol 151: 2399–2408. [PubMed] [Google Scholar]
- 32.Guerra FE, Borgogna TR, Patel DM, Sward EW, and Voyich JM. 2017. Epic Immune Battles of History: Neutrophils vs. Staphylococcus aureus. Front Cell Infect Microbiol 7: 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGeachy MJ, Cua DJ, and Gaffen SL. 2019. The IL-17 Family of Cytokines in Health and Disease. Immunity 50: 892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, and Gabrilovich DI. 2009. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 182: 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez PC, Quiceno DG, and Ochoa AC. 2007. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109: 1568–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, and Ochoa AC. 2003. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol 171: 1232–1239. [DOI] [PubMed] [Google Scholar]
- 37.Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, and Zanovello P. 2003. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol 170: 270–278. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB, and Ochoa AC. 2004. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64: 5839–5849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.