

Abstract
Cerebrovascular injuries can cause severe edema and inflammation that adversely affect human health. Here, we observed recanalization after successful endovascular thrombectomy for acute large vessel occlusion was associated with cerebral edema and poor clinical outcomes in patients who experienced hemorrhagic transformation. To understand this process, we developed a cerebrovascular injury model using transcranial ultrasound that enabled spatiotemporal evaluation of resident and peripheral myeloid cells. We discovered that injurious and reparative responses diverged based on time and cellular origin. Resident microglia initially stabilized damaged vessels in a purinergic receptor-dependent manner, which was followed by influx of myelomonocytic cells that caused severe edema. Prolonged blockade of myeloid cell recruitment with anti-adhesion molecule therapy prevented severe edema but also promoted neuronal destruction and fibrosis by interfering with vascular repair later orchestrated by pro-inflammatory monocytes and pro-angiogenic repair-associated microglia (RAM). These data demonstrate how temporally distinct myeloid cell responses can contain, exacerbate, and ultimately repair a cerebrovascular injury.
Introduction
The cerebral vasculature is sealed by a barrier system that isolates the central nervous system (CNS) from the systemic circulation1. The blood brain barrier (BBB) is comprised of endothelial cells, basement membrane, pericytes, and glia limitans that control the exchange of cells and substances between the CNS and circulation. CNS vasculature are susceptible, however, to damage resulting from mechanical forces, degenerative processes, and ischemia. Cerebrovascular diseases can have devastating outcomes, which is exemplified by ischemic disorders like strokes - the second leading cause of death and the leading cause of disability in humans worldwide2.
The importance of cerebral vasculature in maintaining CNS homeostasis is exemplified by the adverse effects associated with other barrier disruptive disorders, including traumatic brain injury (TBI), primary intracranial hemorrhage (ICH), and hemorrhagic transformation following stroke. For example, TBI can substantially disrupt the cerebrovascular network by widening of intercellular junctions between endothelial cells and promoting swelling of perivascular astrocytes as well as hemorrhage3. Vasogenic edema is another contributor to secondary damage following vascular injury and can induce cerebral herniation – a potentially fatal condition resulting from fluid-induced compression of brain tissue4. Following ischemic stroke, the initial injury is attributed to reduced regional cerebral blood flow5,6. However, secondary damage can occur upon vascular reperfusion, such as BBB breakdown, hemorrhagic transformation, and severe brain swelling associated with invasion by peripheral myelomonocytic cells5,7–10.
Vascular damage is a feature of many CNS pathologies, but most TBI and stroke models evaluate net outcomes of multiple injurious processes and attempt to extrapolate the contribution of vascular damage to these outcomes. Consequently, the isolated contribution of cerebrovascular disruption and subsequent leakage of materials from the blood into the CNS is not well understood. To better understand this process, we developed a model of isolated cerebrovascular injury using a combination of ultrasound and intravenously (i.v.) injected microbubbles that allowed us to apply injurious mechanical forces to select beds of brain vasculature. We then used this model to study the complete spatiotemporal progression of the injurious process, from the initial moments following vascular damage to the induction of angiogenic repair programming and subsequent restoration of neurological function.
Results
Intraparenchymal hemorrhage is associated with cerebral edema
Intraparenchymal hemorrhage is associated with TBI, ICH, and stroke. Evaluation of these patients by magnetic resonance imaging (MRI) revealed that intraparenchymal hemorrhage promotes T2-FLAIR hyperintensities that surround the hemorrhagic lesion – a pattern consistent with the development of edema (Fig. 1A). To gain additional insights into this pathological process, we focused on patients that develop cerebrovascular hemorrhage and edema on a more defined time scale. Specifically, we evaluated 30 patients with large vessel occlusion acute ischemic stroke who underwent endovascular thrombectomy (EVT), with largely successful recanalization (87% with TICI 2b/3) (Supplementary Table 1). In this cohort, 16 patients had no hemorrhage after embolectomy, 6 had hemorrhagic infarction (petechial blood), and 8 had parenchymal hematoma. The presence of a parenchymal hematoma was associated with an increased volume of injured tissue, as evidenced by diffusion weighted imaging (DWI) hyperintensities and elevated cerebral edema revealed by T2 FLAIR hyperintensities (Fig. 1B–D and Supplementary Table 2). Prior to EVT, the average volume of DWI hyperintensity did not differ between the groups but increased significantly post-EVT in patients who developed an parenchymal hematoma vs. those who did not (75 cm3 vs. 15.6 cm3) (Fig. 1C,D). This parameter remained elevated in parenchymal hematoma patients at 1 and 5d post-EVT. The edema volume followed a similar pattern, showing a significant increase in the parenchymal hematoma patients at both 1 and 5d post-EVT (Fig 1C, D). These data demonstrate that hemorrhagic transformation following EVT is associated with significant brain injury and edema.
Figure 1. Parenchymal hematoma is associated with generation of cerebral edema.
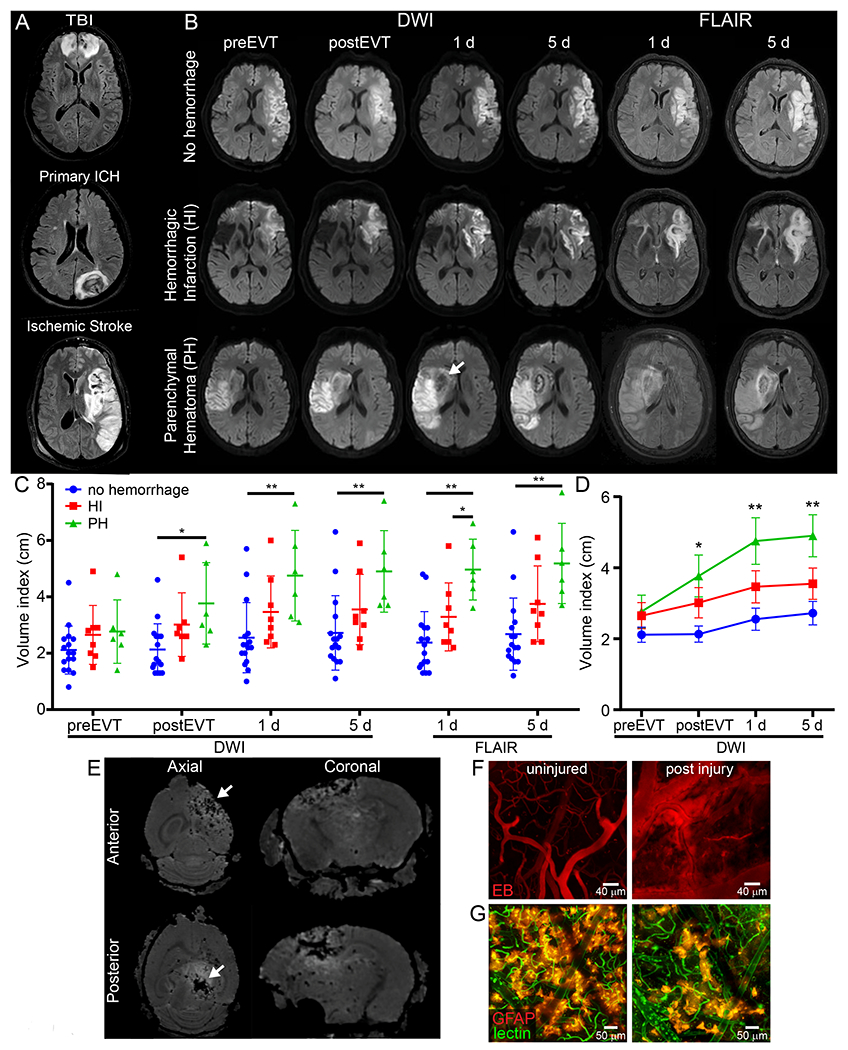
A. Representative axial T2-FLAIR MRIs of injuries associated with IPH, depicting associated injury and cerebral edema (white). B. Representative DWI and T2-FLAIR axial MRIs pre- and post-endovascular thrombectomy (EVT) (0-2 h, 1 d, 5 d) show injury and edema evolution after stroke. White arrow denotes hemorrhage. C. Dot plot shows the cube root of DWI or FLAIR hyperintensity values (mean±SD) in a cohort of n=30 patients (Supplementary Tables 1 and 2) with large vessel occlusion pre- and post-EVT (*P<0.05, **P<0.01; One-way ANOVA/Tukey test per time point). D. Scatter plot of mean±SD shows evolution of injury volume after stroke and 0-2 h, 1 d and 5 d post EVT (n=30, *P<0.05, **P<0.01; One-way ANOVA/Tukey test per time point). E. Ex-vivo T2* Multiple Gradient Echo MRI of mouse cerebrum 1 d post anterior (top row) and posterior (bottom) ultrasound injury. White arrows depict bleeding (black lesion) and surrounding white signal demonstrates edema. Images representative of 5 mice per group F. Representative maximal projections of a two-photon z-stacks viewed through the thinned skull of a naive B6 mouse 20 min after injury show vascular injury and Evans Blue (EB, red) leakage induced by sonication. Images representative of 5 mice per group (see also Video 1). G. Representative two-photon images of GFAP-creER Stopfl/wt TdTomato mice 20 min post-injury show destruction of the glia limitans superficialis and perivascularis. Astrocytes are shown in reddish orange and tomato-lectin injected intravenously in green. Images are representative of 2 independent experiments with 3 mice per group (see also Video 3).
Murine model of cerebrovascular injury
Because cerebrovascular injury and secondary edema is common in humans, we developed an animal model to study these events in which cerebrovascular damage could be controlled temporally and spatially. To this end, we applied low intensity pulse ultrasound (LIPUS) to the thinned skull windows11 of mice that received an i.v. injection of microbubbles (MB) (Extended data Fig. 1). This novel approach induced reproducible cerebrovascular damage beneath the thinned skull window (Fig. 1E,F). Following LIPUS+MB, intraparenchymal hemorrhage was immediately visible through the thinned cranial window (Extended data Fig. 2A) and was evident macroscopically (Extended data Fig. 2B). Ex vivo MRI confirmed the presence of targeted intraparenchymal hemorrhage that depended on the location of the thinned skull window. We induced damage in either the frontal lobe (anterior model) or parietal lobe and underlying midbrain (posterior model) (Fig. 1E). Intravital microscopy following sonication injury revealed destruction of capillary vessels and Evans Blue (EB) leakage within the parenchyma (Fig. 1F and Video 1), rapid cerebral swelling that distorted cortical structures (Video 2) as well as injury of glia limitans astrocytes (Fig. 1G and Video 3). Cerebrovascular damage in the frontal lobe was not fatal, whereas damage to parietal lobe / midbrain was uniformly fatal (median survival of 2 days; Extended data Fig. 2C). In both models, water content increased in the injured hemisphere (Extended data Fig. 2D), and following anterior injury, BBB leakage (measured by fluorescein extravasation) gradually decreased over 7 days (Extended data Fig. 2E). Similarly, flow cytometric analysis of Ter-119+ red blood cell (RBC) accumulation in the brain following anterior sonication demonstrated that RBCs were cleared within 6 days (Extended data Fig. 2F–H). These data demonstrate that it is possible to model cerebrovascular damage and secondary edema in rodents using transcranial ultrasound.
Microglia extend processes that contain the leaky cerebrovasculature
To study the innate immune response to cerebrovascular injury, we performed intravital microscopy. We initially focused on microglia by imaging CX3CR1gfp/wt mice. We observed immediately after injury (within 20 min) that microglia projected their processes toward injured blood vessels, forming tubular structures that completely enveloped vessels. We refer to these structures as ‘rosettes’. Over the ensuing 24h, microglia further transformed into an amoeboid morphology while maintaining their position around damaged vessels (Fig. 2A and Video 4). Microglia appeared to contain these leaking blood vessels – a theory supported by confocal images showing microglia processes walling off fibrin clots throughout injured brain tissue within an hour of injury (Fig. 2B). A functional role for microglia in this injury paradigm was established by feeding mice for a month with chow containing the CSF1R inhibitor, PLX3397. Intravital imaging studies revealed that microglia-depleted mice had 6.3-fold more EB extravasation 20min post-injury relative to untreated controls (Fig. 2C, D and Video 5). This result was replicated when a different CSF1R inhibitor, PLX5622, was used for microglia depletion (Extended data Fig. 3A, B). Microglia depletion also resulted in a loss of lesion containment, with fibrin distributing throughout the injured brain parenchyma instead of being confined to small focal clusters seen in control mice at 24h (Extended data Fig. 3C, D). In addition, enhanced extravasation of lysozyme M-GFP+ (LysMgfp/wt) myelomonocytic cells from the blood was observed immediately after injury in microglia-depleted mice (Extended data Fig. 3E, F), which was associated with elevated expression of Icam1 and Vcam1 on CNS vasculature (Extended data Fig. 3G, H) and a shift in the expression of inflammatory genes at 24h (Extended data Fig. 3I and Supplementary Table 3). Microglia depletion decreased expression of Il1rn, Il1b, Cxcl10, Ccl12, Tlr2, Ptges, Rela, Bdrkb1 and Il6st, and increased expression of Il1a, Icam1, Vcam1 and Il6 (Extended Data Fig. 3I).
Figure 2. Microglia rapidly extend processes and create a barrier surrounding injured blood vessels.
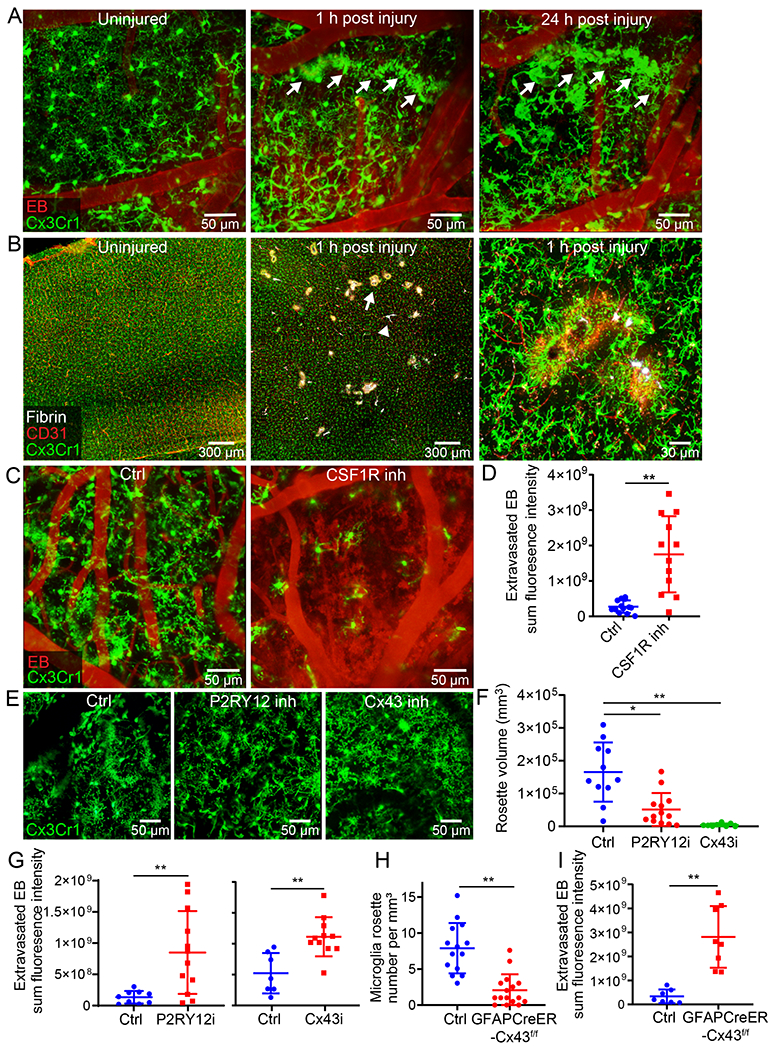
A. Intravital microscopy of CX3CR1gfp/wt mice demonstrates extension of microglia processes and rosette formation (green) 1 h after injury (white arrow denotes a microglial rosette). At 24 h, rosette-forming microglia transform into an amoeboid morphology while maintaining the pattern along the damaged vessel. EB injected i.v. is shown in red. Images are representative of 2 independent experiments with 5 mice per group (see Video 4). B. Confocal brain images of CX3CR1gfp/wt mice demonstrate petechial hemorrhages (white arrow) and clotted vessels (white arrowhead) throughout the cerebral cortex 1 h after injury. A magnified image (right) depicts microglia projections surrounding a clotted vessel. CD31 is shown in red and fluorescently tagged fibrinogen in white. Images are representative of 2 experiments with 5 mice per group. C. Intravital microscopy of CX3CR1gfp/wt (green) mice 20 min after injury shows extensive intraparenchymal EB (red) extravasation following microglia depletion (CSF1R inhibition; see Video 5). D. EB extravasation assay based on intravital microscopy time lapses depicts increased BBB leakage 20-40 min after microglia depletion. Cumulative data from 3 independent experiments (n=13 mice per group; **P=0.000062 / two-tailed Student’s t-test; 1 mouse was not processed due to poor image quality). E. Intravital microscopy of CX3CR1gfp/wt (green) mice at 20 min post-injury treated transcranially with a P2RY12 inhibitor, a Cx43 inhibitor or a vehicle control (see Video 6). F. Quantification of microglial rosettes in panel E 20 min following injury. Cumulative data from 3 independent experiments (Ctrl n=11, P2RY12i n=13, Cx43i n=9 mice; *P=0.048, **P=5.2e-6/ Kruskal-Wallis/Dunn’s test; 2 mice were not processed due to poor image quality). G. EB extravasation assay 20-40 min post-injury in mice treated transcranially with a P2RY12 inhibitor, Cx43 inhibitor or vehicle control. Left graph shows cumulative data from 2 independent experiments (Ctrl n=9, P2RY12i n=12 mice, **P=0.0048/ two-tailed Student’s t-test). Right graph shows cumulative data from 2 independent experiments (Ctrl n=7, Cx43i n=11 mice, **P=0.0015/ two-tailed Student’s t-test). (See also Extended Data Figure 4A, B and Video 7) H. Microglia rosette quantification 1 h following injury in littermate control vs. GFAPCreER-Cx43f/f mice. Cumulative data from 3 independent experiments (Ctrl n=14, GFAPCreER-Cx43f/f n=16 mice, **P=6.2e-6/ two-tailed Mann–Whitney U test; See also Extended Data Figure 4C). I. EB extravasation assay 20-40 min post-injury in littermate control vs. GFAPCreER-Cx43f/f mice. Cumulative data from 2 independent experiments (Ctrl n=7, GFAPCreER-Cx43f/f n=8 mice, **P= 0.00025/ two-tailed Student’s t-test; See also Extended Data Figure 4D and Video 8). In panels D, F, G, H, and I each symbol represents an individual mouse and graphs depict mean±SD.
To identify the mechanism underlying microglia rosette formation, we applied small molecule inhibitors transcranially to block purine signaling as described previously12. Inhibition of P2RY12 receptors, primarily found on microglia, decreased the total volume of rosette structures by 68.9%, whereas inhibition of CX43 hemichannels, required for the release of ATP by astrocytes, reduced rosette formation by 97.3% at 20min post-injury (Fig. 2E, F and Video 6). Because inhibition of P2RY12 receptors or CX43 hemichannels resulted in blockade of microglia rosette formation, we next evaluated the effect this had on BBB integrity. P2RY12 receptor or CX43 hemichannel inhibition increased EB extravasation when compared to vehicle control treated mice (Fig. 2G, Extended Data Fig. 4A, B, and Video 7). To confirm the role of Cx43 hemichannels in rosette formation, we generated GFAP-CreER x Cx43f/f mice and compared them to wild type (WT) littermate controls. Adult mice were fed tamoxifen for 4 weeks to delete Cx43 from GFAP-expressing astrocytes and then injured. At 1h post-injury, the mean number of microglia rosettes per mm2 was significantly reduced in GFAPCreER-Cx43f/f mice relative to controls (Fig. 2H and Extended Data Fig. 4C). In addition, EB leakage was increased 8.4-fold relative to the control mice (Fig. 2I, Extended Data Fig. 4D and Video 8), confirming the CX43 hemichannel inhibitor results obtained. These data demonstrate that astrocytic ATP release and subsequent P2RY12 signaling is responsible for microglia rosette formation and containment of BBB leakage following cerebrovascular injury.
Peripheral myelomonocytic cells cause cerebral edema
To determine the role of peripheral innate immune cells after cerebrovascular injury, we conducted a series of imaging and flow cytometric studies. Confocal imaging of injured brain tissue from LysMgfp/wt mice revealed clusters of parenchymal myelomonocytic cells at 24h (Fig. 3A). Intravital microscopy demonstrated that these cells rapidly entered damaged brain from the blood within 1h of injury (Fig. 3B and Video 9). To profile this infiltrate, we performed flow cytometric analyses of injured brain tissue at d1 and 6 post-injury in CX3CR1gfp/wt CCR2rfp/wt mice, which revealed an increase in inflammatory monocytes (CCR2+Ly6Chi), engrafting/patrolling monocytes (Cx3CR1hi Ly6Clow MHCII+), and neutrophils (Ly6G+ Ly6Clow) at d1. Both inflammatory monocytes and neutrophils decreased in number by d6, whereas engrafting/patrolling monocytes were similar in number to d1 (Fig. 3C and Extended Data Fig. 5A). Using intravital microscopy, we visualized rapid extravasation and migration of LysMgfp/wt myelomonocytic cells into the lesion core and perimeter that was inhibited by one bolus i.v. injection of αLFA-1 and αVLA-4 antibodies immediately after injury (Fig. 3D and Video 10). At 1h post-injury, there were on average 179 ± 131.4 LysMgfp/wt cells per field in the lesion core and 139 ± 78.2 cells in the lesion perimeter. Treatment with αLFA1/VLA4 significantly reduced these numbers to 32 ± 7.8 and 9.7 ± 6.2 cells in the lesion core and perimeter, respectively (Fig. 3E). Flow cytometric analyses performed on injured brain tissue at 24h confirmed effective inhibition of monocyte and neutrophil invasion by αLFA1/VLA4 (Fig. 3F and Extended Data Fig. 5B).
Figure 3. Vascular injury causes myelomonocytic cell invasion and subsequent cerebral edema.
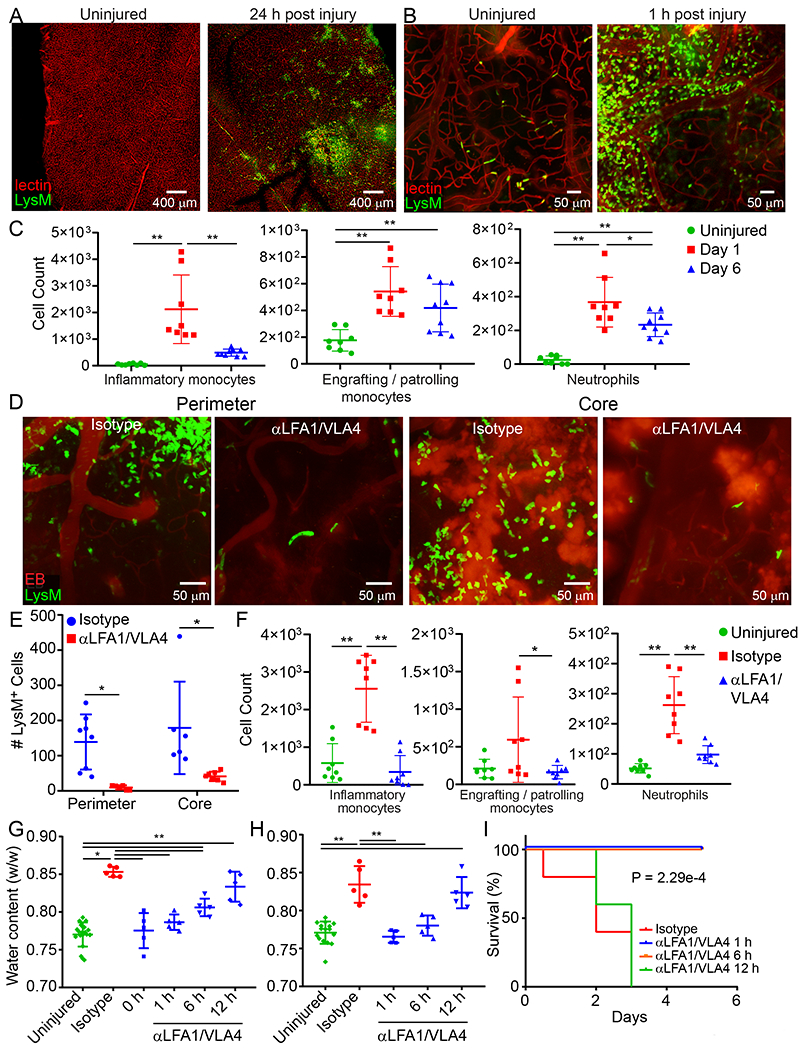
A. Confocal images of uninjured or 24 h post-injury LysMgfp/wt (green) mouse brains. Tomato-lectin is shown in red. Images are representative of 6 mice per group. B. Intravital microscopy images from LysMgfp/wt mice show rapid invasion by myelomonocytic cells (green) within 1 h of injury relative to an uninjured control. Tomato-lectin is shown in red. Images are representative of 5 mice per group (Also see Video 9). C. High parameter flow cytometric analysis of the immune landscape in the cerebral cortex of CX3CR1gfp/wt CCR2rfp/wt mice at 1 d and 6 d post-injury relative to uninjured controls was used to graph the absolute number of inflammatory monocytes (CD45+CD44+CD11b+CCR2+Ly6Chi), patrolling/engrafted monocytes (CD45+CD44+CD11b+CX3CR1+Ly6CloMHC-II+), and neutrophils (CD45+CD44+CD11b+Ly6G+Ly6Clo). Graphs show the mean±SD and are a compilation of 2 independent experiments (Ctrl n=8, d1 n=8, d6 n=9 mice, *P<0.05, **P<0.01, One-way ANOVA/Tuckey; gating strategy in Extended Figure 5A and Supplementary Figure 1B). D. Intravital microscopy images of the lesion core and perimeter at 1 h post-injury in LysMgfp/wt mice that received a bolus injection of αLFA1/VLA4 or isotype control antibodies (see also Video 10). E. Quantification of LysM+ cells that invade the lesion core and perimeter at 1 h post-injury. Graph shows the mean±SD and is representative of two independent experiments (Isotype n=8, αLFA1/VLA4 n=7 mice, *P<0.05, Perimeter P=0.025, Core P=0.019, Two-way ANOVA/Holm-Sidak test, 3 images were not processed due to poor image quality) F. High parameter flow cytometric analysis of immune landscape in the cerebral cortex of CX3CR1gfp/wtCCR2rfp/wt 1 d post-injury was used to graph the absolute number of inflammatory monocytes, patrolling monocytes, and neutrophils following a single dose of αLFA1/VLA4 or isotype control antibodies relative to uninjured control mice. Graphs show the mean±SD and represent a compilation of two independent experiments (n=8 mice per group, *P<0.05, **P<0.01, One-way ANOVA/Tuckey; gating strategy in Extended Figure 5B and Supplementary Figure 1B). G-H. Quantification of cerebral water content 1 d after anterior (G) and posterior (H) injury shows the time dependent effect of bolus αLFA1/VLA4 treatment on cerebral edema. αLFA1/VLA4 were administered at 0, 1, 6 or 12 h post-injury and compared to isotype control treated or uninjured mice (*P<0.05, **P<0.01, One-way ANOVA/Tukey test). I. Kaplan-Meier curve following posterior injury shows 100% survival of mice after αLFA1/VLA4 treatment of mice at 1 h or 6 h post-injury. The median survival is 3 d with αLFA1/VLA4 treatment 12 h after injury, and 2 d for the isotype control group (P=2.29e-4; Log-rank test). In panels G, H, and I, graphs depict the mean±SD and are representative of 2 independent experiments with n=5 mice per group. For G, H the uninjured group includes the combination of the contralateral hemispheres.
To evaluate whether myelomonocytic cell invasion contributed to cerebral edema after injury, we quantified brain water content as described previously13. Depletion of circulating myelomonocytic cells using αGr-1 prevented cerebral edema in the ipsilateral cerebral hemisphere following anterior or posterior (fatal) injury (Extended Data Fig. 6A). Importantly, myelomonocytic cell depletion also promoted survival in all mice after posterior injury, whereas the isotype control mice succumbed to the injury in 2-3d. Administration of αLFA1/VLA4 antibodies 1d prior to injury (αLFA1/VLA4 pre-treatment) yielded an identical result (Extended Data Fig. 6B). To evaluate the potential therapeutic benefit of αLFA1/VLA4, we administered a single bolus dose at 0h, 1h, 6h or 12h post-injury (αLFA1/VLA4 bolus). Relative to the isotype control group, treatment with αLFA1/VLA4 at 0h, 1h or 6h post-anterior injury significantly decreased edema, whereas treatment at 12h had no effect (Fig. 3G). Similar results were observed with αLFA1/VLA4 bolus treatment following posterior injury (Fig. 3H). In addition, treatment with αLFA1/VLA4 1 or 6h post-posterior injury promoted 100% survival, whereas treatment at 12h resulted in a similar mortality to the isotype control group (Fig. 3I). Collectively, these data indicate that fatal cerebral edema can be prevented by administering αLFA1/VLA4 up to 6h post-injury.
To identify the population(s) of myelomonocytic cells responsible for cerebral edema, we evaluated water content in CCR2−/− mice with reduced pro-inflammatory monocytes in circulation14 as well as mice pretreated with αLy6G 1d prior to injury (Extended Data Fig. 6C, D). Following both anterior and posterior injury, brain water content was reduced in CCR2−/− mice relative to WT controls but not to the same degree as observed in αLFA1/VLA4 treated mice. Moreover, survival was not significantly extended in CCR2−/− relative to WT controls (Extended Data Fig. 6C). Similar findings were obtained following depletion of neutrophils 1d prior to injury with αLy6G. Neutrophil depletion decreased cerebral edema relative to control mice but not to the same degree as αLFA1/VLA4, and this had no effect on survival (Extended Data Fig. 6D). These data suggest that the combined extravasation of monocytes and neutrophils, which are both inhibited by αLFA1/VLA4 (Fig. 3F), is responsible for fatal cerebral edema following cerebrovascular injury.
We next evaluated whether αLFA1 and αVLA4 antibodies could be used alone to achieve a therapeutic benefit. Treatment with αLFA1 or αVLA4 had a highly variable effect on brain water content relative to combined αLFA1/VLA4. This was observed in the anterior (Extended Data Fig. 7A) and posterior (Extended Data Fig. 7B) injury models. For example, in the anterior paradigm, the range of water content following αLFA1 or αVLA4 treatment was 0.71–0.86 and 0.75–0.87 w/w, respectively, compared to 0.76–0.77 w/w in the αLFA1/VLA4 group (Extended Data Fig. 7A). Consistent with this variability, administration of αLFA1 or αVLA4 alone conferred no survival benefit (Extended Data Fig. 7C).
To determine the reason for inter-sample variability following single treatment with αLFA1 or αVLA4, we quantified expression of ICAM-1 (LFA-1 ligand) and VCAM-1 (VLA-4 ligand) on cerebrovascular endothelia after injury. This was achieved in vivo by injecting fluorescently-labeled αICAM-1 and αVCAM-1 antibodies i.v. at 24h post-injury and imaging the expression pattern by intravital microscopy (Extended Data Fig. 7D). Relative to uninjured control mice, injury markedly increased cerebrovascular expression of ICAM-1 and VCAM-1 following injury (Extended Data Fig. 7D). The expression pattern of these adhesion molecules varied among vessels, with few vessel segments showing co-expression of ICAM-1 and VCAM-1. Sections of cerebrovasculature tended to express ICAM-1 or VCAM-1 but not both. In addition, there was no correlation between ICAM-1 vs. VCAM-1 expression when quantified from two-photon images (Extended Data Fig. 7F) or qPCR (Extended Data Fig. 7G). These data indicate that immune cells have at least two potential non-redundant entry mechanisms to access different segments of cerebrovasculature, which provides a potential explanation for why monotherapy with αLFA1 or αVLA4 alone is ineffective.
Myelomonocytic cells promote angiogenesis and cerebral repair
Myelomonocytic cells contribute to vascular breakdown and edema following cerebrovascular injury (Fig. 3G–I and Extended Data Fig. 6), but we postulated that they would also be involved in repair. To study the reparative process, we used intravital microscopy to image cortical vasculature at 1d and 10d post-anterior injury. Prior to imaging, we injected fluorescently-tagged tomato lectin i.v. to label vascular endothelia and EB to assess BBB integrity. At 1d post-injury, imaging revealed diffuse extravascular EB signal signifying BBB breakdown (Fig. 4A). In addition, lectin staining was sparse and not well-defined, demonstrating cerebral blood vessels were injured and often clotted shut at the injury site. In contrast, at 10d post-injury, we observed a newly formed, flowing network of cerebral blood vessels that stained with tomato lectin and did not leak EB (Fig. 4A). These vessels, however, were irregular in size and distribution. The vascular coverage at 10d was 8.1 times greater than that observed in uninjured mice, and the area between blood vessels was 0.61 times smaller (Fig. 4B).
Figure 4. Effect of myelomonocytic cells on cerebral repair and angiogenesis.
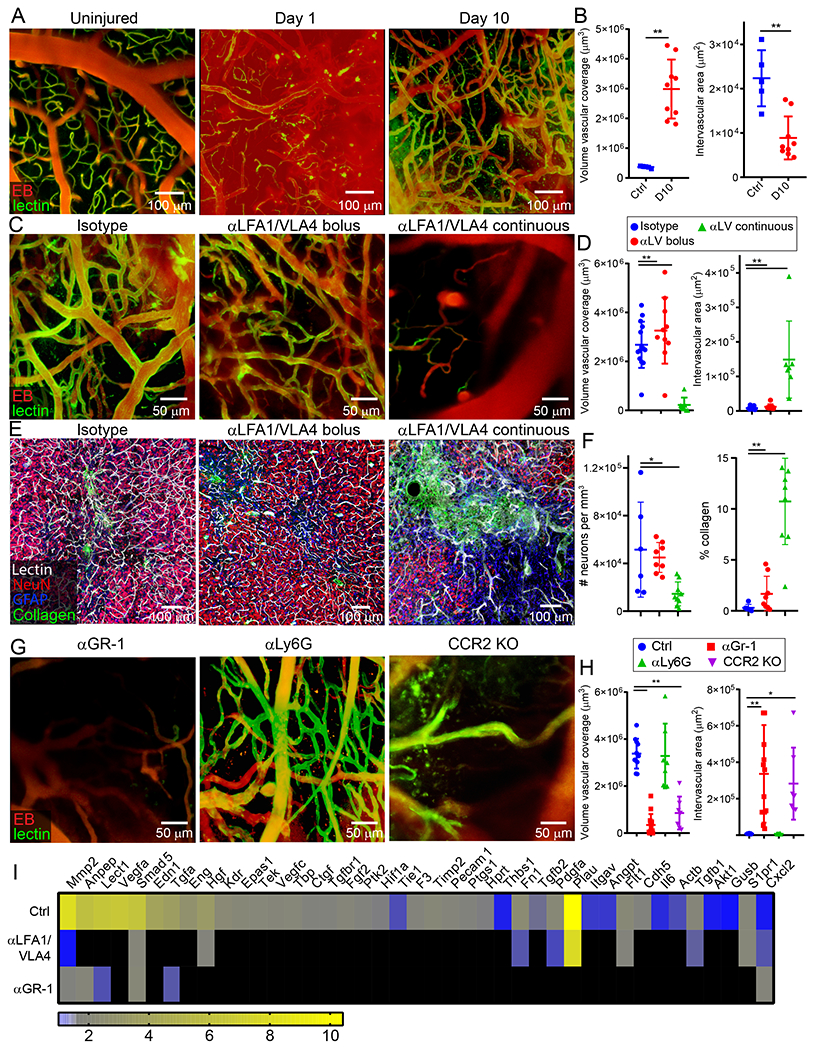
A-B. Intravital microscopy of cerebral vasculature and image-based quantification of vascular coverage and intervascular area shows development of new vessels with irregular distribution, increased diameter, and increased density at 10 d after injury relative to uninjured controls. Compilation from 2 independent experiments Ctrl n=5 and d10 n=9. C-D. A similar experiment was performed at 10 d post-injury following administration of continuous αLFA1/VLA4 (αLV), bolus αLFA1/VLA4, or isotype control antibodies. Compilation from 2 independent experiments Ctrl n=14, αLV n=10, 3 mice were not processed due to poor image quality. E-F. Confocal microscopy of brain sections from collagen-GFP mice 10 d after injury show extensive fibrosis (green) and NeuN+ neuronal (red) loss after continuous αLFA1/VLA4 administration relative to treatment with bolus αLFA1/VLA4 or isotype control antibodies. GFAP+ astrocytes are shown in blue. Compilation from 2 independent experiments Ctrl n=6, αLV n=8. G-H. Intravital microscopy of cerebral vasculature and image-based quantification of vascular coverage and intervascular area show lack of repair after continuous αGR-1 administration and in CCR2 KO mice but not after continuous αLy6G administration. Blood vessels were labeled with EB (red) and tomato-lectin (green). Compilation from 2 independent experiments Ctrl n=9, αGR-1 n=14, αLy6G, CCR2 KO n=7. In panels B, D, F, and H, graphs depict mean±SD (*P<0.05, **P<0.01, two-tailed Student’s t-test (B), One-way ANOVA/Tukey test (D,F,H)). I. Heatmap depicting qPCR analysis of genes encoding for angiogenesis-related proteins 6 d after injury shows downregulation of the angiogenesis pathway after continuous administration of αLFA1/VLA4 or αGr-1 relative to the isotype control group. Heatmap includes genes in which the difference between Ctrl and αLFA1/VLA4 had false discovery rate of Q<1% (multiple t-tests). Data are representative of 2 independent experiments with 4 mice per group per experiment. A comprehensive summary of the results and statistical analysis is provided in Extended Data Figure 9 and source data in Supplementary Table 4.
To assess the role of peripheral immune cells in this angiogenic process, we treated mice with αLFA1/VLA4 using two different regimens. Mice either received a single dose of αLFA1/VLA4 immediately post-injury (bolus) or multiple doses beginning immediately after injury and then on 1, 3, 5, 7, and 9d (continuous). Relative to the isotype control group, a single injection of αLFA1/VLA4 had no effect on the formation of new blood vessels observed at 10d. By contrast, continuous αLFA1/VLA4 administration completely blocked vascular remodeling (Fig. 4C,D). At 10d post-injury, the mean volume of vascular coverage in the continuous αLFA1/VLA4 group was 0.92 times smaller than the isotype control, and the space between blood vessels was 17.9 times greater (Fig. 4D). Confocal images confirmed the presence of highly gliotic brain tissue without vasculature after continuous αLFA1/VLA4 treatment when compared to isotype control mice in which areas of gliosis were filled with new tomato lectin+ bloods vessels (Extended Data Fig. 8A, B). Moreover, continuous αLFA1/VLA4 treatment was associated with a substantial reduction of NeuN+ neurons and an increased number of type 1 collagen+ cells in the lesion area (Fig. 4E, F). These data demonstrate that continuous but not bolus treatment with αLFA1/VLA4 impedes angiogenesis and promotes neuronal loss after cerebrovascular injury.
We next focused on identifying the specific myelomonocytic cell subset responsible for promoting angiogenesis following cerebrovascular injury. We injected B6 mice i.p. with αGr-1 (to deplete monocytes / neutrophils) or αLy6G (to deplete neutrophils) 1d prior to injury and on 1, 3, 5, 7, and 9d post-injury. We confirmed persistent depletion of monocytes and neutrophils with αGr-1 and depletion of neutrophils with αLy6G at d6 after injury (Extended Data Fig. 8C, D). Depletion of myelomonocytic cells with αGr-1 prevented cerebrovascular repair at 10d post-injury, whereas neutrophil depletion with αLy6G did not affect the reparative process (Fig. 4G, H). The importance of Gr-1+ cells was especially evident upon analysis of angiogenic gene expression in injured brain tissue by real-time qPCR (Fig. 4I, Extended Data Fig. 9 and Supplementary Table 4). Relative to uninjured mice, up-regulation of angiogenesis genes was observed in injured mice at d6 (Fig. 4I and Extended Data Fig. 9A,B). Treatment with αGr-1 completely suppressed this program (Fig. 4I and Extended Data Fig. 9C, E) to the level observed in αLFA1/VLA4 treated mice (Fig. 4I and Extended Data Fig. 9D, F), further supporting a role for myelomonocytic cells in promoting cerebrovascular repair.
Because our depletion data suggested involvement of monocytes in cerebrovascular repair, we conducted a series of experiments using monocyte-deficient CCR2−/− mice. CCR2−/− mice were unable to generate new cerebral vessels 10d after injury (Fig. 4G, H). Moreover, reconstitution of CCR2−/− mice with blood-derived CD115+ monocytes from B6 mice substantially improved cerebrovascular repair (Extended Data Fig. 8E, F). These data demonstrate the importance of CCR2+ monocytes in promoting angiogenesis following cerebrovascular injury.
Peripheral immune cells promote generation of pro-angiogenic microglia
To further understand the mechanism underlying cerebrovascular remodeling after injury, we sought to determine why interfering with the recruitment of peripheral immune cells led to failed angiogenesis. A role for CNS resident myeloid cells was uncovered in mice treated with the CSF1R inhibitors. Relative to controls, depletion of CNS myeloid cells with PLX3397 (Fig. 5A) or PLX5622 (Extended Data Fig 10A, B) blocked cerebrovascular repair and angiogenic gene expression (Fig. 5B, Extended Data Fig. 10C, D, and Supplementary Table 4) after injury. The pro-angiogenic factor vascular endothelial growth factor A (Vegfa) was among the most significantly downregulated genes at 6d post-injury in PLX3397-treated mice. Confocal microscopy of brain sections from CX3CR1gfp/wt mice at 6d post-injury revealed that VEGF-A localized to clusters of CX3CR1+ myeloid cells (Fig. 5C,D). To identify the specific cell source(s) of VEGF-A, we conducted high-parameter flow cytometry on injured brain tissue from Cx3cr1CreER/wt x Stopfl/wt TdTomato mice at 1 and 6d. These mice allowed us to distinguish brain-resident from peripheral infiltrating myeloid cells, as described previously15. Using this approach, we determined that VEGF-A expression following injury mapped to a unique subset of brain resident (Tdtomato+) CD45loCX3CR1+P2RY12+CD24+ cells we defined as repair-associated microglia (RAM) (Fig. 5E–G and Extended Data Fig. 10E). Relative to uninjured controls, these cells as well as activated microglia increased in number at 1 and 6d post-injury (Fig. 5F). In addition, blockade of peripheral immune cell infiltration by continuous treatment with αLFA1/VLA4 resulted in a near complete loss of VEGF+ microglia (Fig. 5H, I and Extended Data Fig. 10F). These data demonstrate that peripheral immune cells facilitate generation of RAM – a process impeded by continuous αLFA1/VLA4 treatment.
Figure 5. VEGF-expressing microglia are generated during angiogenesis.
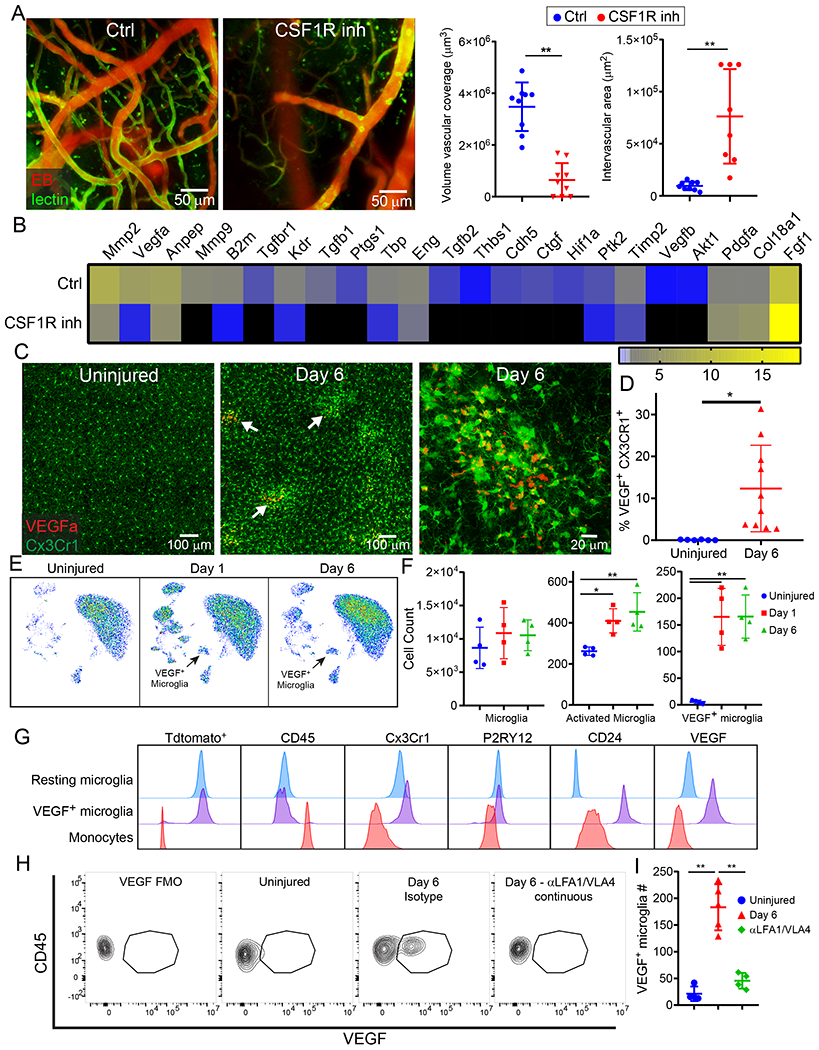
A. Intravital microscopy of cerebral vasculature 10 d after injury and image-based quantification of vascular coverage and intervascular area in PLX3397 (CSF1R inhibitor) versus vehicle control treated mice show a lack of angiogenesis after microglia depletion. Blood vessels were labeled with EB (red) and tomato-lectin (green). Graphs depict mean±SD of cumulative data from 2 independent experiments (n=9 mice per group, **P=1.4e-6 (left), P=0.0005 (right), two-tailed Student’s t-test) B. Heatmap depicting qPCR analysis of genes encoding for angiogenesis related proteins 6 d post-injury shows downregulation of the angiogenesis pathway in PLX3397-treated mice relative to the vehicle control. Heatmap includes genes in which the difference between control and PLX3397-treated mice had a false discovery rate of Q<1% (multiple t-tests). Data are representative of 2 independent experiments with 4 mice per group per experiment. A comprehensive summary of the results and statistical analysis is shown in Extended Data Figure 10A and source data in Supplementary Table 4. C. Confocal microscopy images of brain sections from Cx3Cr1gfp/wt (green) mice show VEGF-A (red) expression within myeloid cell clusters 6 d after injury (denoted with white arrows) relative to uninjured control mice. D. Graph depicts image-based quantification of VEGF-A+ Cx3Cr1+ myeloid cells. Data show mean ± SD and are representative of 2 independent experiments with Ctrl n=6 and d6 n=5 mice with two high power fields quantified per injury (*P=0.012, two-tailed Student’s t-test). E-G. High parameter flow cytometric analysis of immune landscape of Cx3cr1CreER/+ x Stopfl/+ TdTomato mice at d 1 and d 6 post-injury relative to uninjured mice. Data are representative of 2 independent experiments n=4 mice per group per experiment. E. Representative UMAP plots depict the generation of VEGF-A+ microglia after injury (black arrow). F. Scatter plots depict the absolute numbers of different microglia populations post-injury: microglia (TdTomato+CD45loCD11b+Cx3Cr1+P2RY12+), activated microglia (TdTomato+CD45intCD11b+Cx3Cr1+P2RY12lo), VEGF-A+ microglia (TdTomato+CD45intCD11b+Cx3Cr1+P2RY12loCD24+VEGF-A+) (*P<0.05, **P<0.01, One-way ANOVA/Tukey test; gating strategy in Extended Data Figure 10E and Supplementary Figure 1B). G. Fluorescence intensity histograms comparing the expression of Tdtomato, CD45, Cx3CR1, P2RY12, CD24, and VEGF-A on resting microglia, VEGF-A+ microglia, and monocytes (TdTomato−CD45hiCD11b+CD44+Ly6C+Ly6G−). Microglia were gated as described in panel F. H-I. Representative flow cytometric dot plots (H) and a corresponding graph (I) show the lack of VEGF-A+ microglia after continuous treatment with αLFA1/VLA4 relative to isotype control antibodies on d 6 post-injury. A representative flow cytometric plot is also shown for an uninjured mouse, and an FMO control is provided for VEGF-A staining at d6. Data are representative of 2 independent experiments n=4 or 5 mice per group per experiment (**P<0.01; One-way ANOVA/Tukey test; gating strategy in Extended Data Figure 10F and Supplementary Figure 1C).
Prolonged inhibition of peripheral immune cells promotes fibrosis and neurological dysfunction.
We lastly focused on the functional consequences of impeding immune-mediated cerebrovascular repair after injury. We again evaluated the effects of continuous αLFA1/VLA4 treatment because of its profound negative impact on cerebrovascular repair. Following cerebrovascular injury, mice were treated with αLFA1/VLA4 or isotype control antibodies for 10d, and gene expression was measured in injured brain tissue by RNAseq at 20d (Supplementary Table 5). Principal component analysis based on 97.3% of the detected genes revealed that injured control mice clustered near uninjured mice, whereas αLFA1/VLA4-treated mice were distinctly separate from these two groups (Fig. 6A). A Pearson correlation-based clustered heat map confirmed separate clustering of αLFA1/VLA4-treated mice from the other two groups (Fig. 6B). Use of Ingenuity Pathway Analysis to assess the concordant dysregulated genes in αLFA1/VLA4-treated mice relative to the other two groups uncovered three interconnected networks of upregulated genes suggesting brain tissue dysregulation (Fig. 6C). The most significantly enriched function that emerged from these networks was linked to fibrosis (Fig. 6D; Supplementary Table 6; p = 1.12E-11).
Figure 6. Continuous treatment with αLFA1/VLA4 promotes fibrotic repair and prevents functional recovery.
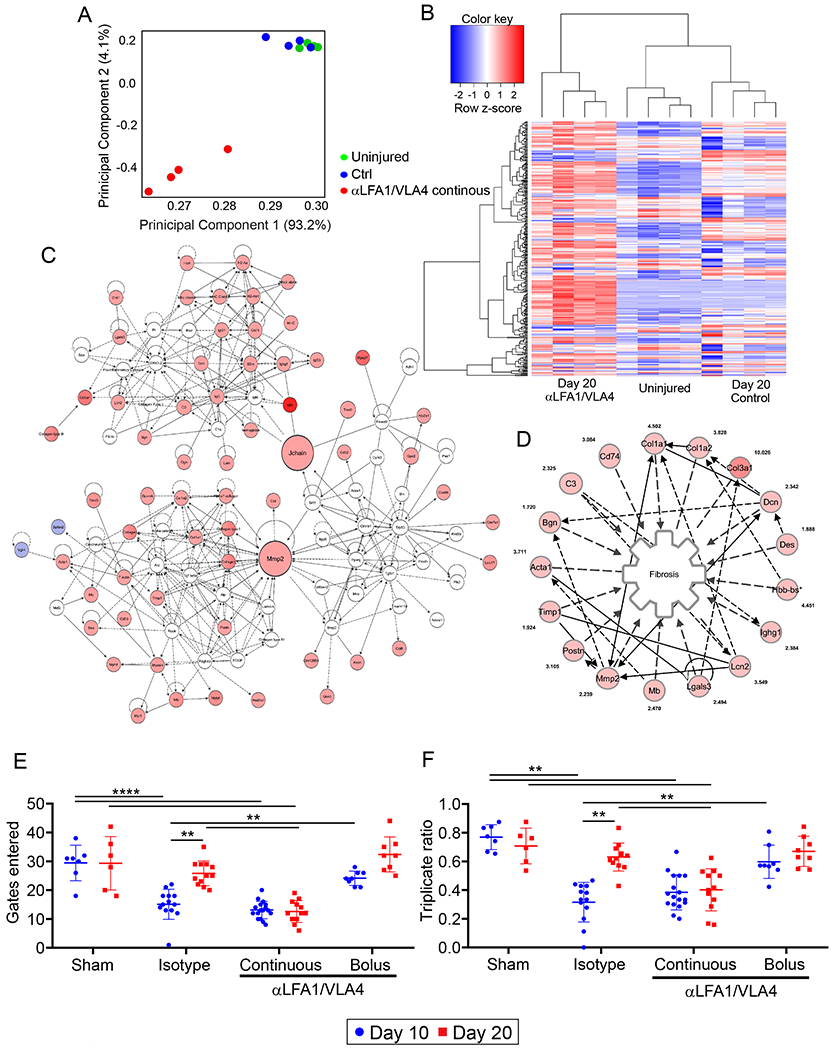
A-C. RNA sequencing results from cerebral cortex biopsies of uninjured mice versus day 20 injured mice treated continuously for 10 days with αLFA1/VLA4 or isotype control antibodies (n=4 mice per group; see also Supplementary Tables 5, 6). A. Covariance-based PCA scatterplot of first and second principal components depicts separate clustering of mice treated with αLFA1/VLA4 (red) relative to injured isotype control mice (blue) and uninjured mice (green). The latter two groups do not separate based on the first two principal components. Each dot represents a single mouse. B. Pearson correlation-based clustered heat map using the unique union of genes deemed to have a significant difference of expression between at least two classes. C. Merged enriched networks obtained using Ingenuity Pathway Analysis (IPA) tool of concordant dysregulated genes in αLFA1/VLA4 treated mice relative to injured isotype control mice and uninjured mice. Fibrosis is the enriched function of the merged enriched networks with the lowest p value (p=1.12E-11; Ingenuity Pathway Analysis tool). D. Sub-network representing genes included in the most enriched function (fibrosis). Edges representing a relationship for a node onto itself are not depicted to allow visualization of the fold-increase for each gene. E-F. Graphs of cognitive-motor function evaluation assessed by Y-maze at 10 and 20 d post-injury depict the number of gates entered (E) and the triplicate ratio (F). Injured mice were treated with isotype control antibodies, bolus αLFA1/VLA4, or continuous αLFA1/VLA4. Mice receiving a sham surgery were also included as a control for this experiment. Graphs show the mean±SD and are the compilation of 2 independent experiments (sham d10 n=7, d20 n=6; isotype d10 n=13, d20 n=12; αLV continuous d10 n=17, d20 n=13; αLV bolus n=8, **P<0.01, Two-way ANOVA/Holm-Sidak test).
The functional impact of this tissue dysregulation was evident upon evaluation of these different groups of mice using a Y-maze, which measures cognitive-motor function (Fig. 6E,F). Injured mice receiving either continuous or bolus αLFA1/VLA4 were compared to injured isotype control antibody treated mice as well as sham surgery (uninjured) mice at 10 and 20d. The number of Y-maze gates entered (Fig. 6E) and the triplicate ratio (Fig. 6F) were quantified at these two time points. When compared to sham controls, injured mice receiving isotype antibodies demonstrated a reduction in function at 10d that returned to control levels by 20d, indicating functional recovery. By contrast, mice that received continuous αLFA1/VLA4 treatment also had reduced function at 10d, but this function did not recover by 20d. Mice receiving a single bolus of αLFA1/VLA4 immediately after injury performed the best on this test, showing the lowest functional deterioration at 10d and complete recovery by 20d. Collectively, these data demonstrate that sustained interference with the peripheral immune system fosters transcriptional dysregulation, fibrosis, and an inability to functionally recover after cerebrovascular injury.
Discussion
Cerebrovascular injury and hemorrhage are serious events that can profoundly affect brain homeostasis and neurological function. By studying stroke patients that received mechanical recanalization, we observed that hemorrhagic transformation was associated with substantial cerebral edema – a potentially preventable event that accompanies neurological deterioration post-EVT16–18. The presence of cerebrovascular injury in many CNS disorders led us to develop and study a new model. Our data provide a spatiotemporal map of the damage and subsequent immune reaction that develop following isolated mechanical disruption of cerebrovasculature. We uncovered a complex network of interrelated myeloid cell responses that contributed to divergent outcomes. In the initial minutes following injury, microglia played a crucial role sealing off the damaged BBB by projecting processes and enveloping vessels. This rapid response depended on ATP release from astrocytes via Cx43 hemichannels and subsequent detection by purinergic receptors (P2RY12). This was followed by massive recruitment of myelomonocytic cells that promoted fatal cerebral edema. Recruitment depended on differential vascular expression of ICAM-1 and VCAM-1 that was inhibited by therapeutic administration of αLFA1/αVLA4. A single injection of these antibodies within 6h of injury prevented fatal cerebral edema. However, continuous antibody administration impeded cerebrovascular repair. Restoration of cerebrovasculature and preservation of neurons depended on recruitment of CCR2+ monocytes and generation of pro-angiogenic microglia at later time points. Prolonged interference with peripheral immune cell recruitment induced fibrotic wound healing associated with neuronal loss, collagen deposition, and failure to recover neurological function. These findings provide a comprehensive understanding of how microglia and peripheral myeloid cells respond to cerebrovascular injury and offer guidance for when to apply therapeutic immunomodulatory regimens to reduce cerebral edema while still permitting vascular repair.
Microglia provided the first line of protection after cerebrovascular injury by creating tube-like structures (rosettes) that encased damaged/clotted vessels, limiting the degree of leakage into the parenchyma. Microglia used their well-described capacity to detect ATP released from astrocytic Cx43 hemichannels in a P2RY12-dependent manner. Projection of microglia processes toward purines released from areas of brain damage was first described using models of focal laser injury19,20. Lou et al. also demonstrated how microglia can extend processes in a P2RY12-dependent manner to rapidly close small openings in the BBB induced by laser injury21. Our study shows how this conserved response can play out on a much larger scale after extensive cerebrovascular damage. Microglia rapidly reconstructed the BBB following vascular injury and limited the degree of damage. This mechanism may explain why microglia accumulate around cerebral microbleeds22 and prevent hemorrhage after neonatal focal arterial stroke23. We demonstrated that microglia depletion or inhibition of ATP release enhanced cerebrovascular leakage, myelomonocytic cell recruitment, parenchymal fibrin deposition, and damage. Microglia depletion also reduced expression of pro-inflammatory cytokines at 24h post-injury. These findings suggest that in the context of acute hemorrhagic vascular injury the barrier-sealing properties of microglial are protective and outweigh potential negative functions stemming from their phagocytic state or ability to release pro-inflammatory molecules24–26.
While microglia played an important early role in BBB stabilization, this was partly undone by massive recruitment of myelomonocytic cells, which induced profound cerebral edema. Massive myelomonocytic cell extravasation similarly promotes brain swelling and cerebral herniation during viral meningitis27. We observed that myelomonocytic cell extravasation following cerebrovascular injury depended almost entirely on LFA1 and VLA4, and therapeutic administration of αLFA1/αVLA4 antibodies within 6h of injury prevented fatal cerebral edema. These data indicate that there is a relatively short therapeutic window to interfere with this pathogenic event which closes once myelomonocytic cell extravasation has occurred. Several preclinical studies have inhibited diapedesis as a treatment for ischemic stroke and showed variable effects on stroke lesion volume28–31. Moreover, neutrophil inhibitory factor was used to treat patients with ischemic stroke and had no clinical benefit32. This failure could be explained by our results demonstrating that both neutrophils and inflammatory monocytes are responsible for edema following cerebrovascular injury. Depletion of neutrophils alone did not improve outcome in our model. The success of therapies designed to interfere with immune cell extravasation and subsequent brain swelling depends critically on timing and entry mechanisms. The ACTION trial found that natalizumab (αVLA4 antibody) administered up to 9h after stroke onset did not reduce infarct growth but improved secondary functional outcomes33. However, our data indicate that myelomonocytic cells can use both LFA1 and VLA4 non-redundantly to extravasate and that there is considerable variation in the vascular expression patterns of ICAM-1 and VCAM-1. Use of αLFA1 or αVLA4 alone had little therapeutic benefit in our studies. Both antibodies were required to prevent fatal cerebral edema and improve functional recovery.
Angiogenesis is critical for repair of injured brain tissue34, and previous studies have shown that an increased number of microvessels in infarcted brain tissue after stroke is associated with improved long-term survival35. While inflammatory monocytes contribute to cerebral edema in the acute phase of injury, they can also promote angiogenesis. We demonstrated that CCR2 deficiency or continuous inhibition of diapedesis prevented angiogenesis. CCR2 promotes mobilization of inflammatory monocytes from the bone marrow into the blood as well as transmigration into inflamed tissues14. A pathogenic role for inflammatory monocytes is supported by studies showing that CCR2−/− mice have reduced cerebral edema following ischemia-reperfusion injury36 and improved acute motor function following injection of blood into the brain (a model of ICH)37. However, circulating inflammatory monocytes were also shown to reduce hemorrhagic transformation, promote new vessel growth, and increase functional recovery following stroke38–40. Moreover, following ischemia-reperfusion injury, CCR2−/− mice were initially shown to have a decreased stroke volume but then did not functionally recover and had a higher mortality rate than WT mice41. Our findings clarify these contradictory results by revealing that in the acute phase following cerebrovascular injury, CCR2+ monocytes contribute to potentially fatal cerebral edema but are later required to promote angiogenesis, neuronal preservation, and functional recovery.
We found that angiogenesis following cerebrovascular injury also depended on clusters of pro-angiogenic VEGF-A-expressing microglia. Studies have begun classifying microglia based on their functional attributes and transcriptional states42. We have identified a population of CD45intCX3CR1+P2RY12+CD24+ repair-associated microglia (RAM) that clustered around damaged blood vessels and expressed VEGF-A. VEGF-A is a potent inducer of angiogenesis that facilitates endothelial cell proliferation and migration43. VEGF expression was observed in activated non-phagocytic microglia/macrophages surrounding injured areas of brain tissue from acute ischemic stroke patients as well as stroked rodents44,45. A subset of glioma associated microglia in humans was also shown to express VEGF-A46 and could promote brain tumor angiogenesis in rodents47. We observed in microglia-depleted mice that angiogenesis genes were not upregulated and that new blood vessels failed to form following cerebrovascular injury. We postulate that invading monocytes endow microglia with this pro-angiogenic function, as RAM did not develop when immune cell invasion from the blood was blocked.
While immune cells can induce CNS pathology, their role in promoting tissue repair should not be ignored. Prolonged inhibition of immune function following CNS injury can impede repair and prevent functional recovery. Continuous inhibition of peripheral immune cell invasion in our study not only prevented angiogenesis, but also changed the reparative program. In the absence of immune-mediated tissue regeneration, we observed induction of fibrotic wound healing in the injured brain associated with neuronal loss, gliosis, invading type 1 collagen+ cells, and failed functional recovery. Genomic analyses revealed that this fibrotic brain tissue was completely distinct at the transcriptional level from healthy or properly repaired tissue. In the periphery, it is known that wound repair macrophages produce many growth factors and can orchestrate reparative programs48. In addition, dysregulation of wound healing can result in an irreversible fibrotic response49. For example, Duffield et al. demonstrated in a model of liver injury that early depletion of CD11b+ myeloid cells reduced the extent of damage, whereas late depletion resulted in failure to resolve the fibrotic response50. Our studies emphasize that while the detrimental aspects of myelomonocytic cell invasion after cerebrovascular injury can be prevented with αLFA1/VLA4 antibodies, this treatment cannot be given continuously without promoting fibrosis and blocking functional recovery.
In conclusion, we offer new insights into the distinct roles of brain-resident and invading myeloid cells following cerebrovascular injury. These responses diverge based on time, cellular origin, and anatomical position. Excepting the early massive extravasation by myelomonocytic cells, the myeloid response to cerebrovascular injury is largely beneficial. The barrier sealing properties of microglia, and their eventual conversion into pro-angiogenic RAM should be promoted. Peripheral CCR2+ monocytes also play a crucial role in brain tissue remodeling and should only be blocked acutely in the initial hours post-injury. Such an acute and transient treatment might help resolve the pathogenic cerebral edema that develops following TBI, ICH, and stroke while still permitting angiogenesis and brain repair to occur. We hope that our findings improve patient outcomes by serving as a foundation for the design of future clinical trials to treat cerebrovascular injuries and will discourage approaches that indiscriminately or continuously inhibit pro-inflammatory myeloid cell responses.
Methods
Human Subjects.
The appropriate Ethics and Institutional Review Boards (NINDS/NIH IRB for Suburban Hospital, Johns Hopkins Medicine, Bethesda, MD; Medstar Washington Hospital Center, Washington Hospital Center, Washington, DC IRB) approved the study (NCT00009243). Written informed consent was obtained from all patients. Patients were included in this study if they were screened from April 2015 to July 2018 with multimodal magnetic resonance imaging (MRI) as their “baseline” scan, and if they received a clinical diagnosis of ischemic stroke involving large vessel occlusion (LVO) of the anterior circulation prior to receiving mechanical embolectomy with or without standard intravenous tPA (tissue plasminogen activator). Patients were also required to have follow-up multimodal MRI obtained at 2-hours, 24-hours and 5-days post-endovascular thrombectomy (EVT). Reperfusion following thrombectomy was scored using the thrombolysis in cerebral infarction (TICI) scale 51, and the patients included in this study had partial (TICI 2A) or successful recanalization (TICI 2B or TICI 3) following thrombectomy. Patients were consented for the NIH Natural History of Stroke Study. The appropriate Ethics and Institutional Review Boards (NINDS/NIH IRB for Suburban Hospital, Johns Hopkins Medicine, Bethesda, MD; Medstar Washington Hospital Center, Washington Hospital Center, Washington, DC IRB) approved the study (NCT00009243).
Patients were scanned with 3T MRI scanners (Siemens Skyra, Siemens AG, Munich, Germany and Philips Achieva, Philips Healthcare, Best, Netherlands) using standardized acute stroke imaging protocols. A dosage of 0.1 mmol/kg of Gd-DTPA was administered if the perfusion-weighted imaging (PWI) sequence was obtained at baseline, 2-hours, or 24-hours. Isotropic diffusion-weighted imaging (DWI) and pre-contrast fluid attenuated inversion recovery (FLAIR) sequences were required at baseline, 2-hours, 24-hours, and 5-days.
Two independent raters measured the hyperintense ischemic areas on DWI sequence at all timepoints using a validated technique as well as the hyperintense lesion areas on the pre-contrast FLAIR sequence at 24-hours and 5-days, avoiding the inclusion of hyperintense acute reperfusion injury marker (HARM) signal areas 52. The volume of these lesions was calculated and the cube root of this value (referred to as the volume index) was used for comparison of different groups. The presence of HARM was qualitatively reviewed by the independent raters, reaching consensus for any discrepancies, using the pre-contrast FLAIR images at 24-hours post-EVT. HARM was defined as enhancement in the CSF spaces and rated as: none, present but not severe, or severe if occupying 10 or more slices. The same two independent raters evaluated the gradient echo sequences (GRE) at 24-hours post-EVT compared to the baseline GRE, documenting the presence of any hemorrhagic transformation (HT) by applying the ECASS-II criteria 53 (applied to MRI) to classify patients with no hemorrhage, hemorrhagic infarction (petechial blood) and parenchymal hematoma. Discrepancies in the HARM and HT reads were resolved by consensus. The average volume for all DWI and FLAIR reads at all timepoints was used in the study analyses. A cube-root transformation producing an effective radius of the lesion (Volume Index) was used to normalize the distribution of lesion sizes for analysis and display. In addition, qualitative consensus reads for HARM and HT were used in the study analyses. Complete recanalization was defined in the interventional radiology suite as TICI scores of 2B or 3.
MRIs from representative cases of traumatic brain injury, primary intracerebral hemorrhage and hemorrhagic transformation of stroke were also used to denote the presence of edema surrounding intraparenchymal hemorrhage.
Mice.
C57BL/6J (B6) 54, B6.129P-CX3CR1tm1Litt/J (Cx3cr1gfp/gfp) 55, (Ccr2rfp/rfp; CCR2 KO) 56, B6.Cg-Tg(GFAP-cre/ERT2)505Fmv/J (GFAP-CreER) 57, B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Stopfl/fl tdTomato) 58, B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ (CX3CR1CreER/CreER) 59, and B6.129S7-Gja1tm1Dlg/J (Cx43f/wt) 60,61 mice were purchased from Jackson Laboratories. FVB.Cg-Tg(Cspg4-EGFP*)HDbe/J (NG2-mEGFP) mice were kindly provided by Dr. Dwight Bergles 62 and subsequently backcrossed onto a pure B6/J background. B6.LysMgfp/gfp mice were provided by Dr. Thomas Graf 63. Type 1 collagen-GFP mice were originally provided by Dr. David Brenner 64 and backcrossed onto a pure B6/J background. Mice were bred and maintained under specific pathogen-free conditions at the National Institute of Health. The environmental conditions in the vivarium were as follows: temperature 72 F, humidity 50% and light/dark cycle 12 h each.
CX3CR1gfp/wt mice were generated from an F1 cross of B6 and CX3CR1gfp/gfp mice. CX3CR1gfp/wt x CCR2rfp/wt double reporter mice were generated from an F1 cross of CX3CR1gfp/gfp and CCR2rfp/rfp mice. GFAPCreER x Stopfl/wt TdTomato mice were generated from an F1 cross of GFAPcre-ER/wt with Stopfl/fl tdTomato mice. LysMgfp/wt mice were generated from an F1 cross of LysMgfp/gfp with B6 mice. CX3CR1CreER/+ x Stopfl/wt TdTomato mice were generated from an F1 cross of CX3CR1CreER/CreER and Stopfl/fl tdTomato mice. GFAPCreER-Cx43f/f mice were obtained in the F2 generation after an original cross of GFAP-cre-ER and Cx43f/f mice. All mice in this study were handled in accordance with the guidelines set forth by the NIH Animal Care and Use Committee and the recommendations in the AAALAC Guide for the Care and Use of Laboratory Animals. The protocol was approved by the NINDS Animal Care and Use Committee. Male and female mice in this study were used at 10-12 weeks of age, except for CX3CR1CreER/+ Stopfl/wt TdTomato mice, which were used at 12-14 weeks of age.
Ultrasound injury.
Mouse surgeries were performed based on adapted techniques previously developed in our lab 11. Ten to 14-week old mice (weighing 24-28 g) were anesthetized using ketamine (85 mg/kg), xylazine (13 mg/kg) and acepromazine (2 mg/kg) diluted in PBS. Following an incision over the midline of the scalp, the skull was exposed, and the periosteum was removed using a microsurgical blade. A metal bracket was glued onto the skull leaving exposed a circular area of the skull, the center of which was 2 mm anterior to the lambdoid suture and 2.5 mm lateral to the sagittal suture (posterior injury) or 2 mm posterior to bregma and 2.5 mm from the sagittal suture (anterior injury). A 2 x 2 mm cranial window was carefully thinned to 20-25 μm. At this point 1 μl/g of Perflutren Protein-Type A microspheres (5 to 8 x 108 bubbles/ml with a size range of 2.0 to 4.5 μm.; Optison™, GE Healthcare, Chicago, IL) were injected intravenously via retroorbital injection. A drop of CSF was placed over the cranial window. We then applied low intensity pulse ultrasound (LIPUS) at 1MHz with peak negative pressure ~ 200KPa (2.2 W/cm2), 1 ms burst length, 10% duty cycle, and 10 sec exposure to the thinned skull using a Sonicator 740x (Mettler Electronics, Anaheim, CA) with a 5 cm2 dual frequency applicator. For intravital imaging after the injury, the skull window was further thinned to 10-15 μm.
Ex vivo MRI Imaging.
Ex vivo MRI of mouse cerebrum was performed on a vertical bore 7T Bruker BioSpin Magnetic (Billerica, MA, USA) using a volume coil (Bruker, internal diameter 2 cm). Brains were immersed in susceptibility matching fluid (Fomblin, A-Tek LLC, Longmont, CO, USA) and imaged with a T2* weighted 3-D Multiple Gradient Echo (MGE) sequence (repetition time 100 ms, echo time 5 ms (first echo)), echo spacing 5 ms, number of echos 10, flip angle=30°, field of view=30x30x30 mm and image resolution 100x100x100 μm with number of averages=6).
Macroscopic imaging.
Imaging of the skull window was completed using a 1.4 Megapixel infinity 2-1RC microscopy camera (Teledyne Lumenera, Ottawa, ON) and the Infinity capture software 6.5.2.
Tamoxifen treatment.
For induction of Cre recombinase, 6- to 8-week-old GFAP-creER Stopfl/wt TdTomato and GFAPCreER-Cx43f/f mice were treated with tamoxifen chow (80 mg per kg tamoxifen, Envigo, Huntingdon, UK) for 4 weeks. CX3CR1CreER/+ x Stopfl/wt TdTomato mice at 4 to 6 weeks of age were treated with tamoxifen chow (80 mg per kg tamoxifen, Envigo, Huntingdon, UK) for 4 weeks to activate the Cre recombinase and then switched to regular feed for 4 weeks to allow turnover of circulating CX3CR1+ cells.
In vivo cell depletions and inhibition of leukocyte extravasation.
For microglia depletion, 6- to 8-week old B6 mice were treated for 4 weeks with PLX3397 (Adooq, Irvine, CA) formulated in chow (Envigo) or treated for 4 days with PLX5562 formulated chow (Envigo) 65. For temporary myelomonocytic cell depletion, 8- to 12- week old mice were given 400 μg of rat anti-mouse Ly6G/Ly6C (Gr-1; RB6-8C5) i.p. 1 d prior to injury (αGR-1 bolus), and for persistent myelomonocytic cell depletion, mice were given 400 μg of anti-Gr-1 (RB6-8C5) i.p. 1 d prior to injury and on days 1, 3, 5, 7, and 9 post injury (αGR-1 continuous). The B6 mice in the control group of these experiments were given 400 μg of rat anti-mouse IgG2b isotype control (LTF-2) i.p. at the same time points. Similarly, for temporary neutrophil depletion, 8- to 12- week old mice were given 500 μg of rat anti-mouse Ly6G (BE0075-1) i.p. 1 d prior to injury (αLy6G bolus), and for persistent neutrophil depletion, 500 μg of rat anti-mouse Ly6G (BE0075-1) i.p. 1 d prior to injury and on days 1, 3, 5, 7, and 9 after injury (αLy6G continuous). The B6 mice in the control group of these experiments were given 500 μg of rat anti-mouse IgG2a isotype control (2A3) i.p. at the same time points. To inhibit leukocyte extravasation, 8- to 12- week old mice were given 500 μg of rat anti-mouse LFA-1α (CD11a; M17/4) i.p. and 500 μg of rat anti-mouse/human VLA-4 (CD49d; PS/2) i.p. 1 day prior to injury (αLFA1/VLA4 pre-treatment), or one dose immediately post injury (0 h, 1 h, 6 h or 12 h post-injury; αLFA1/VLA4 bolus), or one dose immediately post injury and on days 1, 3, 5, 7, and 9 after injury (αLFA1/VLA4 continuous). The B6 mice in the control group of these experiments were given 500 μg of rat anti-mouse IgG2a isotype control (2A3) i.p. and rat anti-mouse IgG2b isotype control (LTF-2) i.p. at the same time points. To evaluate the separate effects of anti-LFA-1α and anti- LA-4, we administered 500 μg of rat anti-mouse LFA-1α (CD11a; M17/4) i.p. or 500 μg of rat anti-mouse/human VLA-4 (CD49d; PS/2) i.p. 1 day prior to injury. All antibodies used for cell depletion and blocking assays were purchased from BioXcell (West Lebanon, NH).
Intravital two-photon microscopy.
Following the initial surgical approach described above, a metal bracket was glued on the skull leaving exposed a circular area of the skull centered 2 mm posterior to the coronal suture and 2.5 mm lateral to the sagittal suture (anterior injury). Mice were imaged through the thinned skull window generated for the sonication injury as described above. For uninjured mice a 2 x 2 mm cranial window was carefully thinned to 10-15 μm 11. Injured and control mice were imaged using a Leica SP8 two-photon microscope with an 8,000-Hz resonant scanner, a 25 x collar-corrected water-dipping objective (1.0 NA) or a 20 x water-dipping objective (1.0 NA), a quad HyD external detector array, a Mai Tai HP DeepSee Laser (Spectra-Physics, Santa Clara, CA) tuned to 905 nm (for GFP, Alexa Fluor 488, Dylight 488) and an Insight DS laser (Spectra-Physics) tuned to 1,050 nm (for Evans blue (EB), TdTomato) or to 1,100 nm (for PE, APC). Acquisition was completed using Leica Application Suite X (LAS X) v3.5.5.19976. Three-dimensional time-lapse movies were captured at 15 s, 30 s, 1 min and 2 min intervals using z-stacks of 20 – 72 planes (2.5 and 3-μm step size). The signal contrast was enhanced by averaging 6-8 video frames per plane in resonance scanning mode. For blood vessel visualization, 70 ul of 0.1 mg/ml Evans blue (Sigma, St. Louis, MO) and/or 70 ul of tomato lectin Dylight 488 (Vector Labs, Burlingame, CA) was injected intravenously prior to imaging. To analyze EB extravasation, 100 ul of 0.1 mg/ml EB was injected intravenously immediately after injury and prior to imaging. All intravital two-photon images obtained were subsequently imported to Imaris version 9.3 software (Bitplane, Concord, MA) for further analysis. Adobe Premiere Pro 14.0 (Adobe systems, San Jose, CA) was used to create final videos.
Transcranial inhibition of purinergic receptors and hemichannels.
Transcranial administration of inhibitors was performed as previously described by our lab 12. Following initial skull thinning to 20 – 25 μm antagonists diluted in artificial cerebral spinal fluid (aCSF; Harvard Apparatus, Cambridge, MA) were applied directly to the skull bone as a 3 mm diameter bubble for 2 h. aCSF was applied for the control group. The antagonists used were MeSAMP (P2RY12 inhibitor; 10 mM, Sigma) and carbenoxolone (CBX; connexin hemichannel inhibitor; 100 mM, Sigma). The solution was replenished as needed over the 2 h period to prevent drying. This allowed the antagonists to continuously pass through the skull bone. Subsequently, the skull was dried, and an ultrasound injury was induced. The skull was then further thinned to 10 - 15 μm, and we proceeded with intravital microscopy for 60 min. The antagonists were maintained in the submerging solution for the entire imaging experiment.
Imaging of lesion core and perimeter.
For these experiments, we used LysMgfp/wt mice. To evaluate the lesion core, we followed our standard injury protocol described above. To evaluate the perimeter of the lesion following the initial sonication through a 2 x 2 mm anterior cranial window, we created an additional thinned cranial window posteriorly and laterally to the main window. Immediately after injury, a dose of αLFA1/VLA4 was administered in one group. Three-dimensional time-lapse movies were captured at 30 sec intervals using z-stacks of 20 planes (3-μm step size) starting 30 min after the injury for approximately 60 min. We counted the number of LysM+ cells per image at 1 h post injury.
Intravital microscopy assays.
All imaging analysis was performed by a blinded investigator.
Evans Blue extravasation assay.
Mice were injected with 100 μl of 0.1 mg/ml Evans blue (Sigma) i.v. immediately following injury, and mice were subsequently imaged for 40 to 60 min. Three-dimensional time-lapse movies were captured at 1 min intervals using z-stacks of 30 planes (3-μm step size). Image series were imported into Imaris version 9.0 software, and using the volume function, the sum of intensity of EB was measured. To calculate the volume of extravasated EB, we subtracted from the volume calculated above the t=0 initial sum volume intensity of intravascular EB. We calculated the average extravasated EB from 20 min to 40 min per sample.
Rosette quantification assay.
CX3CR1gfp/wt mice were imaged for 40 – 60 min following injury. Three-dimensional time-lapse movies were captured at 2 min intervals using z-stacks of 30 planes (3-μm step size). Using the surfaces function in Imaris 9.0, we identified the microglia rosettes at time points 40 to 60 min. We extended the surfaces to the initial time points and quantified the volume of GFP+ microglial processes at time points 0 to 30 min. We report volume of microglia rosettes as an average of the time points 18, 20, and 22 min per sample.
ICAM-1 (CD54) and VCAM-1 (CD106) expression assay.
For quantification of intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), 24 h after injury, mice were intravenously injected with 10 mg of APC anti-CD106 (429 MVCAM.A, Biolegend), 10 mg PE anti-CD54 (YN1/1.7.4, Biolegend), and tomato lectin Dylight 488. One hour following injection, mice were imaged using the Leica SP8 two-photon microscope as described above. For the experiment comparing ICAM and VCAM expression in injured animals, and PLX3397-fed injured animals, the Mai Tai HP DeepSee Laser was tuned to 905 nm and the Insight DS laser was tuned to 1,050 nm. Both lasers were maintained at 10% power, and the quad HyD external detector array gain was set to 10 for both channels. For the experiment comparing ICAM and VCAM expression in uninjured animals and injured animals, the Mai Tai HP DeepSee Laser was tuned to 905 nm and the Insight DS laser was tuned to 1,050 nm. Lasers were maintained at 15% of power and the quad HyD external detector array gain was set to 20 for both channels. Images were imported to Imaris version 9.0 software, and using the volume function, we measured the sum of intensity for both anti-CD106 APC signal and anti-CD54 PE signal.
Quantification of LysM+ cells.
We imaged LysMgfp/wt mice 1 h after injury. The images were imported in Imaris 9.0, and the spots function was used to identify and quantify the number of LysM+ cells per image.
Angiogenesis assay and characterization of neo-vessels.
Mice were evaluated 10 days after initial injury. For blood vessel visualization, 70 ul of 0.1 mg/ml Evans blue and/or 70 ul of tomato lectin Dylight 488 were injected intravenously prior to imaging, and the thinned window was re-thinned to 20-25 μm. Following intravital imaging, images were imported to Imaris 9.0. The volume of vascular coverage was quantified using the surfaces function to mask Evans blue and tomato lectin Dylight 488 positive vessels and calculate their volume. The total vessel volume was then reported as a fraction of the total volume of the volume of the three-dimensional image acquired. The intervascular area was obtained by first masking the areas negative for Evans blue and tomato lectin Dylight 488 using the surfaces function. We then separated the surface of each intervascular space in the maximum intensity projection of each image and calculated the area of each intervascular space. We report the average of all intervascular spaces per image. This angiogenesis assay was used to evaluate angiogenesis following αLFA1/VLA4 bolus and continuous treatments, αGR-1 bolus and continuous treatments, as well as angiogenesis in microglia depleted mice, and CCR2rfp/rfp mice following CD115+ adoptive transfer. Angiogenesis was compared to age matched untreated B6 mice.
CD115+ adoptive transfer to CCR2 KO mice.
B6 and CCR2rfp/rfp mice underwent anterior injury. We subsequently pooled blood from B6 mice and incubated in ACK buffer at 4°C for 5 min, then washed and centrifuged at 500xg for 5 min; the process was repeated until RBCs were lysed and supernatant was clear. Cells were passed through a 40 μm cell strainer and resuspended in 100 μl of PBS with 0.5% bovine serum albumin (BSA), and 2 mM EDTA. We isolated CD115+ cells using a MACS CD115 MicroBead Kit (MACS Miltenyi Biotec, Germany). We quantified the number of cells using a hemocytometer. We administered 106 CD115+ cells per mouse immediately after injury and on days 1, 3, 5, 7 and 9 in one group of CCR2rfp/rfp mice. At day 10 following injury, angiogenesis was evaluated through intravital microscopy as described above (angiogenesis assay).
Immunohistochemistry (IHC).
Brains were harvested 1 h, 24 h, 6 d and 10 d after injury. For vascular staining, we used intravenous administration of fluorescence-conjugated tomato lectin Dylight 488 or Dylight 649 (Vector Labs, Burlingame, CA) or anti-CD31 PE (Biolegend, San Diego, CA) 5–10 min prior to euthanasia. In order to identify the fibrin distribution, we injected 100 μl of 2.5 mg/ml fibrinogen Alexa Fluor 647 (Invitrogen, Carlsbad, CA) immediately after sonication injury and 1 h prior to euthanasia. Mice were perfused with 4% paraformaldehyde. The brain was harvested and placed in 4% paraformaldehyde overnight. The brain was sectioned axially using a Compresstome Tissue Slicer (Precisionary, Greenville, NC) to produce 50 or 100 μm slices. For staining, tissues were initially blocked and permeabilized by incubating in staining buffer consisting of 0.5% Triton-X in phosphate buffered saline with Background Buster (Innovex Biosciences, Richmond, CA) as well as FcR block for 30 min in room temperature. Primary antibodies were added directly to the staining buffer and incubated at 4 °C overnight. After primary staining, meninges were washed three times in staining buffer. Secondary antibodies were added and incubated for 4 h at room temperature. After secondary staining, slices were washed again three times in staining buffer. The free-floating slices were carefully mounted on the slide and covered with one drop of FluorSave Reagent (MilliporeSigma, Burlington, MA) and a coverslip was added. The tissues were stained with the following primary antibodies: rabbit anti-Iba1 (1:500, catalog# 019-19741; Wako, Richmond, VA), chicken anti-GFAP (1:1000, catalog# ab4674; Abcam, Cambridge, UK), guinea pig polyclonal anti-NeuN (1:500, catalog# ABN09P; Millipore, Burlington, MA), and mouse VEGF120/164 antibody (1:250; Monoclonal Rat IgG2B Clone # 39917; R&D systems, Minneapolis, MN) conjugated to Alexa fluor 647 using the Alexa Fluor 647 antibody labeling kit (Invitrogen, Carlsbad, CA). Secondary antibodies (1:1000 dilution) included: donkey anti-rabbit IgG (H+L) Alexa Fluor 488 (A-21206; ThermoFisher, Waltham, MA), goat anti-guinea Pig IgG (H+L) Alexa Fluor 647 (A-21450; ThermoFisher), Donkey anti-chicken IgY (IgG) (H+L) DyLight 405 (AB_2340373; Jackson Immunoresearch, West Grove, PA), Goat anti-Rabbit IgG (H+L) Pacific Blue (P-10994, ThermoFisher), and Donkey F(ab) anti-chicken IgG (H&L) Rhod-X (Jackson Immunoresearch). For immunohistochemistry we evaluated the antibodies used in this study for the degree of non-specific binding (as measured in samples that do not express the epitope or by using isotype controls for comparison. Anti-CD31 PE was validated by the manufacturer for immunofluorescent staining and was used in previous studies 66–68. Rabbit anti-Iba1 has been used previously for immunohistochemistry 69–71. Chicken anti-GFAP was tested by the manufacturer for immunohistochemistry and was used in previous studies.72,73 Guinea pig polyclonal anti-NeuN evaluated by the manufacturer for immunohistochemistry and was used in previous studies 74. The mouse VEGF120/164 antibody conjugated to Alexa Fluor 647 was evaluated in non-injured controls as shown in Figure 5C (first panel). Additionally, the following reporter mice were used to define specific cell types: CX3CR1gfp/wt, LysMgfp/wt and collagen-GFP. Images were obtained using an Olympus FV1200 laser-scanning confocal microscope equipped with four detectors, six laser lines (405, 458, 488, 515, 559 and 635 nm) and five objectives (4 × /0.16 NA, 10 × /0.4 NA, 20 × /0.75 NA and 40 × /0.95 NA, and a chromatic aberration–corrected 60 × /1.4 NA) with the Olympus Fluoview 4.2a software. All confocal images obtained were subsequently imported to Imaris version 9.0 software for further analysis.
Image analysis of IHC images.
All image analysis was performed by a blinded investigator. To quantify the fibrin distribution, we injected CX3CR1gfp/wt mice i.v. with 100 μl of 2.5 mg/ml fibrinogen Alexa Fluor 647 immediately following sonication and 1 h prior to harvesting the brain. Using the surface function in Imaris 9.0, we measured the volume of fluorescent fibrinogen signal per image. Microglia rosettes (defined as tube-like GFP+ structures >50 μm in length) were quantified manually following Iba-1 staining. LysM+ cells were quantified in LysMgfp/wt mice using the Imaris spots function. Number of neurons 10 days following injury was evaluated following staining with guinea pig polyclonal anti-NeuN (catalog# ABN09P) also using the spots function in Imaris. The degree of fibrosis was evaluated 10 days after injury in collagen-GFP mice by using the surfaces function in Imaris 9.0 to calculate the volume of collagen-GFP signal per image.
BBB integrity assay.
In order to assess the integrity of the BBB following injury, we used mice 1 h, 1 d, 4 d, and 7 d following anterior injury. We administered 100 μl of 10% w/v Na-fluorescein (NaFl) in PBS (Sigma, St. Louis, MO) intraperitoneally and harvested a 3 x 3 x 3 mm cube of tissue from the injured area or the contralateral frontal lobe as well as 50 μl of blood, 10 min following injection. Tissues were homogenized in 7.5% trichloroacetic acid (TCA) in 5M NaOH, centrifuged at 10,000 g at 4°C, and the supernatant was then collected. We added 25 μl of 15% TCA to 25 μl of serum, centrifuged at 10,000 g at 4°C, collected 25 μl of the supernatant, and added 125 μl of 7.5% trichloroacetic acid (TCA) in 5M NaOH. Fluorescence was quantified using a microplate reader (Varioskan Flash; ThermoFisher, Waltham, MA) at an excitation at 485nm, emission at 530nm and a gain of 50. Using a standard curve, we calculated the concentrations of NaFl in the tissue and blood serum. We calculated the uptake ratio using the following formula:
This process was adjusted from our previously published BBB leakage assay 27.
Quantification of cerebral water content (edema assay).
To estimate the edema induced by injury, mice were sacrificed 24 h following an anterior injury, or when they reached the endpoint in the survival study following posterior injury. Uninjured mice or the contralateral uninjured hemisphere were used as controls. The hemisphere was isolated and weighed using an analytical balance (Mettler Toledo, Columbus, OH). Subsequently, the hemisphere was placed in overnight incubation at 80°C and the dry weight was measured.13
Survival studies.
Mice were monitored daily following injury by an investigator blinded to the groups. Mice were monitored twice a day for physical deficiencies and weight loss. Mice were sacrificed when > 25% weight loss from their original weights or sign of distress was observed. The surviving mice were sacrificed 5 days after injury.
Flow cytometric analysis.
Flow cytometry experiments were performed on day 1 and day 6 after injury. We used B6, CX3CR1gfp/wt CCR2rfp/wt double reporter and CX3CR1CreER/+ Stopfl/wt TdTomato mice. To harvest brain tissue, mice were anesthetized and perfused with cold PBS. The injured hemisphere or uninjured control hemisphere was harvested and placed in a 15 ml dounce homogenizer to create a cell suspension. We then isolated immune cells using a 70% to 30% isotonic percoll gradient and resuspended in PBS with 2% fetal bovine serum (staining buffer). For blood collection, mice were anesthetized and 20 μl of blood was collected from the retroorbital vein in a heparinized tube. Samples were incubated with ACK Lysing Buffer three times and cells were collected via centrifugation for further processing. The cells were passed through a 40 μm cell strainer. The cells were resuspended in 50 μl of staining buffer with 1:200 purified rat anti-mouse CD16/CD32 (Fc receptor block; clone 93, Biolegend) and incubated at 4°C for 10 min. We then added 50 ul of antibody cocktail mix for 15 min at 4°C and then 30 min at room temperature. Subsequently cells were washed 2 to 3 times with staining buffer and resuspended in 4% paraformaldehyde for 10 min and stored in PBS. The following anti-mouse antibodies were used: CD24 FITC (1:500, M1/69, Biolegend), Ly6C FITC (1:100, HK1.4, Biolegend), Ly6C BB790 (1:200, AL21, BD), GR-1 BV421 (1:100, RB6-8C5, Biolegend), I-A/I-E BV480 (1:450, M5/114.15.2, BD), CD11b BV570 (1:100, M1/70, Biolegend), CD115 BV605 (1:100, T38-320, BD), CD24 BV650 (1:300, M1/69, BD), Cx3Cr1 BV711 (1:100, SA011F11, Biolegend), CD11c BV785 (1:200, N418, Biolegend), P2RY12 PE (1:100, S16007D, Biolegend), Ter-119 PE/Cy5 (1:1000, Ter-119, Biolegend), CD206 PE/Cy7 (1:400, C068C2, Biolegend), CD45 BUV395 (1:200, 30-F11, BD), CD4 BUV496 (1:200, GK1.5, BD), Ly6G BUV563 (1:150, 1A8, BD), CD19 BUV661 (1:300,1D3, BD), CD44 BUV737 (1:1200, IM7, BD), CD8 BV805 (1:300, 53-6.7, BD), CD115 APC (1:100, AFS98, Biolegend), F480 APC-R700 (1:500, T45-2342, BD) and TCRβ APC/Cy7 (1:200, H57-597, Biolegend). Dead cells were excluded from the analysis using the LiveDead fixable Blue Cell Staining kit (1:1000, Invitrogen). For intracellular staining to detect VEGF, single-cell suspensions were first stained with surface antibodies and were then treated with Cytofix/Cytoperm (BD). This was followed by mouse VEGF120/164 antibody (Monoclonal Rat IgG2B Clone # 39917; R&D systems, Minneapolis, MN) directly conjugated to Alexa fluor 647 using the Alexa Fluor 647 antibody labeling kit (1:300, Invitrogen, Carlsbad, CA). Prior to analysis, 20,000 counting beads were added in the solution. For single color controls, we collected spleens from naïve mice, homogenized in a 15 ml dounce, and incubated in ACK buffer before washing and creating single cell suspensions. The CD4 antibodies used for single color controls were FITC (1:400, RM4-5, Biolegend), APC (1:1200, RM4-5, Biolegend), AF700 (1:800, RM4-5, Biolegend), APC/Cy7 (1:800, RM4-5, Biolegend), BV510 (1:400, RM4-5, Biolegend), BV563 (1:100, GK1.5, BD), BV605 (1:400, RM4-5, Biolegend), BV650 (1:400, RM4-5, Biolegend), BV 711 (1:400, RM4-5, Biolegend), BUV737 (1:400, GK1.5, BD), BV786 (1:500, RM4-5, Biolegend), PE (1:3000, RM4-5, Biolegend), PE/Dazzle 594 (1:3000, RM4-5, Biolegend), PE/Cy5 (1:3000, RM4-5, Biolegend), PE/C7 (1:3000, RM4-5, Biolegend), BUV395 (1:400, GK1.5, BD), BUV496 (1:400, GK1.5, BD), BUV563 (1:400, GK1.5, BD), BUV 805 (1:400, GK1.5, BD). Anti-mouse Ly6C BB790 (1:100, AL21, BD) and anti-mouse CD19 BUV661 (1:300,1D3, BD) were also used as single color controls. Both Biolegend and BD antibodies are quality control tested for flow cytometry. The mouse VEGF120/164 antibody (Monoclonal Rat IgG2B Clone # 39917; R&D systems) conjugated to Alexa fluor 647 using the Alexa Fluor 647 antibody labeling kit (Invitrogen) was previously validated 75. We also used fluorescence minus one (FMO) controls to determine appropriate gating for positive cells. The FMO for VEGF antibody is shown in Figure 5H. Samples were acquired using the BD FACSymphony flow cytometer (BD Biosciences, San Jose, CA) and FACSDiva 8.0.1, and data were analyzed using FlowJo software version 10.5.3 (Tree Star, Ashland, OR). For our 22-color panels in CX3CR1gfp/wt CCR2rfp/wt and CX3CR1CreER/+ Stopfl/wt TdTomato mice, we gated CD45+, live cells and subsequently used an unsupervised clustering algorithm to separate cells into subpopulations (PhenoGraph, FlowJo) and a uniform manifold approximation and projection (UMAP, FlowJo) to visualize our high parameter datasets in a two-dimensional space. Counting beads were used to calculate the absolute number of cells in each sample.
Real-time PCR analysis.
For real-time PCR analysis (qPCR) mice, a 3 x 3 x 3 mm cube of tissue was harvested and snap frozen using dry ice. Total RNA was extracted with Qiagen kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol, and RNA quantity and integrity was assessed by a spectrophotometer (Nanodrop One, Thermoscientific, Waltham, MA). cDNA was generated using an iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA). 96-well pre-made PrimePCR plates were used for qPCR experiments (Angiogenesis M96, Inflammation M96; Bio-Rad). qPCR was performed using universal SYBR Green Supermix (Bio-Rad) and cDNA template or water (non-template negative control) at an annealing temperature of 60°C with a CFX96 Real-Time PCR machine (Bio-Rad). PCR products were subjected to melt analysis to confirm purity after DNA amplification. For each gene, expression values were normalized to Gapdh housekeeping gene. The resulting relative gene expression was then expressed as a fold-change over indicated control samples. More specifically, to evaluate the inflammatory profile following microglia depletion, we harvested tissue 24 h post-injury from the frontal lobe of uninjured B6 mice, injured B6 mice, uninjured PLX3397-treated mice and injured PLX3397-treated mice. To evaluate the expression of angiogenesis related genes 6 days after injury, we harvested tissue from the frontal lobe of uninjured B6 mice, injured B6 mice, and injured B6 mice treated continuously with αLFA1/VLA4 and injured B6 mice treated continuously with αGr1.
Bulk RNA sequencing.
For RNA sequencing B6 mice were divided into 3 groups (uninjured, injured, continuous αLFA1/VLA4) with 4 mice per group. At day 20 following injury, mice were perfused with normal saline, and a 3 x 3 x 3 mm cube of injured tissue was harvested and snap frozen using dry ice. The tissues were stored at −80°C until use. Total RNA was extracted with Qiagen kit following the manufacturer’s protocol, and RNA quantity and integrity was assessed by the Bioanalyzer (Agilent, Santa Clara, CA). 500 ng of total RNA was used in conjunction with the TruSeq® Stranded Total RNA Library Prep kit (Illumina, San Diego, CA). The libraries quality was checked by the Bioanalyzer and quantitated by the Qubit (ThermoFisher Scientific). Equimolar quantities from each sample library was pooled and run on a Highoutput Next-Seq550 kit.
RNA sequencing data analysis.
Paired-end sequence files (.fastq) per sample were quality inspected using the FastQC tool 0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) then adaptor clipped (TruSeq3-PE-2.fa:2:30:10) and trimmed to remove 5’ nucleotide bias (HEADCROP:11) and low quality calls (TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:15) using the Trimmomatic tool 0.39 (http://www.usadellab.org/cms/?page=trimmomatic). Surviving intact pairs of reads per sample were then imported into the CLCbio Genomics Workbench 11 (https://www.qiagenbioinformatics.com/), down-sampled to 40M read pairs per sample, then reference mapped by sample in stranded fashion against the current instance of the mouse genome (GRCm38) using the “RNA-Seq Analysis” tool supported therein under default parameters. Expression per known annotated gene (Mus_musculus.GRCm38.83.chr.gtf) in Transcripts Per Kilobase Million (TPM) units was then exported from the Workbench and imported into R (https://cran.r-project.org/). In R, TPM expression per sample was pedestalled by 2 then Log2 transformed. Genes lacking an expression value >1 post transformation for at least one sample were discarded while expression across samples for genes not discarded were quantile normalized. To assure quality of the data post normalization and absence of sample-level outliers, exploratory inspection was performed using Tukey box plot, covariance-based PCA scatterplot and correlation-based heat map. To remove noise-biased expression values, locally weighted scatterplot smoothing (LOWESS) was applied across normalized expression for all genes by sample class (Coefficient of Variation~mean expression). LOWESS fits were then over-plotted and inspected to identify the common low-end expression value where the relationship between mean expression (i.e., “signal”) and Coefficient of Variation (i.e., “noise”) grossly deviated from linearity. Expression values were then floored to equal this value if less, while expression for genes not observed greater than this value for at least one sample were discarded as noise-biased. For genes not discarded, expression differences across sample classes were tested for using the one-factor Analysis of Variance (ANOVA) test under Benjamini–Hochberg (BH) False Discovery Rate (FDR) Multiple Comparison Correction (MCC) condition using sample class as the factor. Genes having a Type III corrected P < 0.05 by this test were then subset and the TukeyHSD post-hoc test used to generate mean differences and p-values for each possible pairwise comparison of classes. Genes having a post-hoc P < 0.05 for a specific comparison and a linear difference of means >= 1.5X for the same comparison were deemed to have expression significantly different between the compared groups. Post testing, sample-to-sample relationships were investigated via covariance-based PCA scatterplot and Pearson correlation-based clustered heat map using the unique union of genes deemed to have a significant difference of expression between at least two classes. Enriched pathways, functions, and top scoring networks for the same union set of genes were obtained using the Ingenuity Pathway Analysis (IPA) tool (https://www.qiagenbioinformatics.com/).
Cognitive-motor function evaluation.
We evaluated the cognitive-motor function of B6 mice following sham surgery as well as injured B6 mice either untreated or following αLFA1/VLA4 bolus or continuous treatment. Evaluation was completed on day 10 and day 20 after injury. A Y-maze with three white, opaque plastic arms at 120° angles from one another was used. The mice were introduced in the center of the maze and allowed to freely explore for 5 min per session. An entry was defined when all four limbs were within the arm. We recorded the total number of arm entries to assess the overall movement in the maze. We also recorded the number of times the mouse sequentially entered all 3 arms (e.g. A → B → C not A → B → A) as a measure of exploration and cognitive function. The number of triplicate A → B → C entries was divided by total number of gates entered to obtain the “triplicate ratio” (see Figure 6F). Each mouse was evaluated at one time point.
Statistical analysis.
Statistical analysis and graph design were performed using Prism 8.4 (GraphPad Software, La Jolla, CA) except for bulk RNA sequencing analysis which is described in detail above. Distribution normality was assessed using the Shapiro-Wilk normality test for n>4, for smaller sample sizes distribution was considered normal. Experiments containing two groups were analyzed using two-tailed Student’s t-test for normally distributed data or the Mann–Whitney U test for non-normally distributed data. Experiments involving more than two groups were analyzed by One-Way ANOVA followed by Tukey multiple comparison’s test for normally distributed data or Kruskal-Wallis test followed by Dunn’s multiple comparisons test for non-normally distributed tests. Data groups based on more than one nominal variable were analyzed using a Two-way ANOVA followed by the Holm-Sidak multiple comparisons method. Groups were considered statistically different at p < 0.05. All data are displayed as the mean ± standard deviation (SD). For the qPCR data analysis, we used multiple t-test using the Benjamini, Krieger and Yekutieli method, to correct for the false discovery rate, with a desired Q value of 1% or 5%. Survival studies were analyzed using Log-rank test. Source data for graphs are provided in Source Data Tables 1–12 and statistical analysis for each graph provided in Supplementary Table 7. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications.12,76 Animal littermates were randomly assigned to each group and samples were randomly selected for data acquisition and analysis, no active randomization protocol was used.
Ethics statement.
Human studies were approved by the appropriate Ethics and Institutional Review Boards (NINDS/NIH IRB for Suburban Hospital, Johns Hopkins Medicine, Bethesda, MD; Medstar Washington Hospital Center, Washington Hospital Center, Washington, DC IRB) and written informed consent was obtained from all patients. The animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the NINDS Animal Care and Use Committee (Protocol Number: 1295–17).
Extended Data
Extended Data Fig. 1. Model of ultrasound-induced injury.
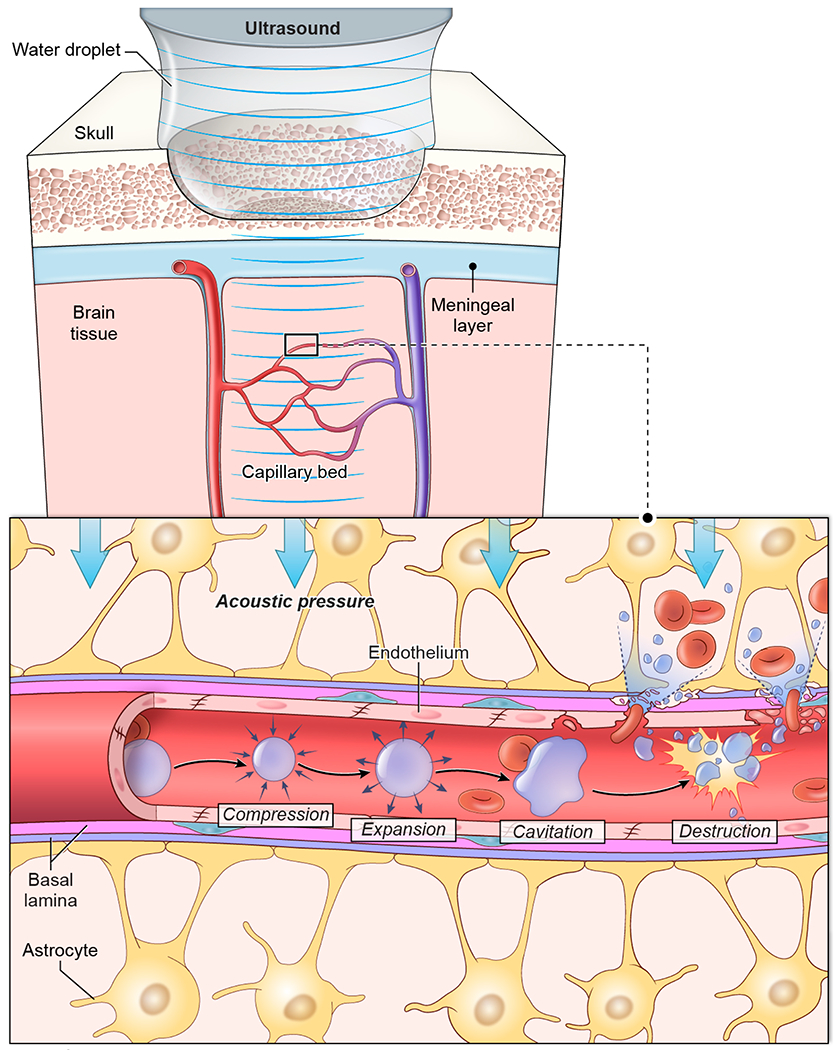
Following surgical generation of a 2mm x 2mm x 15μm thinned skull window, microbubbles were injected intravenously and a drop of aCSF was placed atop the thinned skull bone. Through this aCSF we applied low intensity pulse ultrasound (LIPUS) using a Mettler Sonicator 740x with a 5 cm2 planar dual frequency applicator operating at 1MHz, ~200KPa peak negative pressure with duty cycle 10% and 1ms burst. LIPUS induced acoustic cavitation of the microbubbles. Microbubble oscillation, inertial cavitation, and explosion caused internal injury of blood vessel walls, exposing the brain parenchyma to blood contents. This injury creates a relative column of injury in the brain tissue beneath the thinned skull window, as the ultrasound waves are not strong enough to pass through the surrounding intact bone.
Extended Data Fig. 2. Characterization of the cerebrovascular injury model.
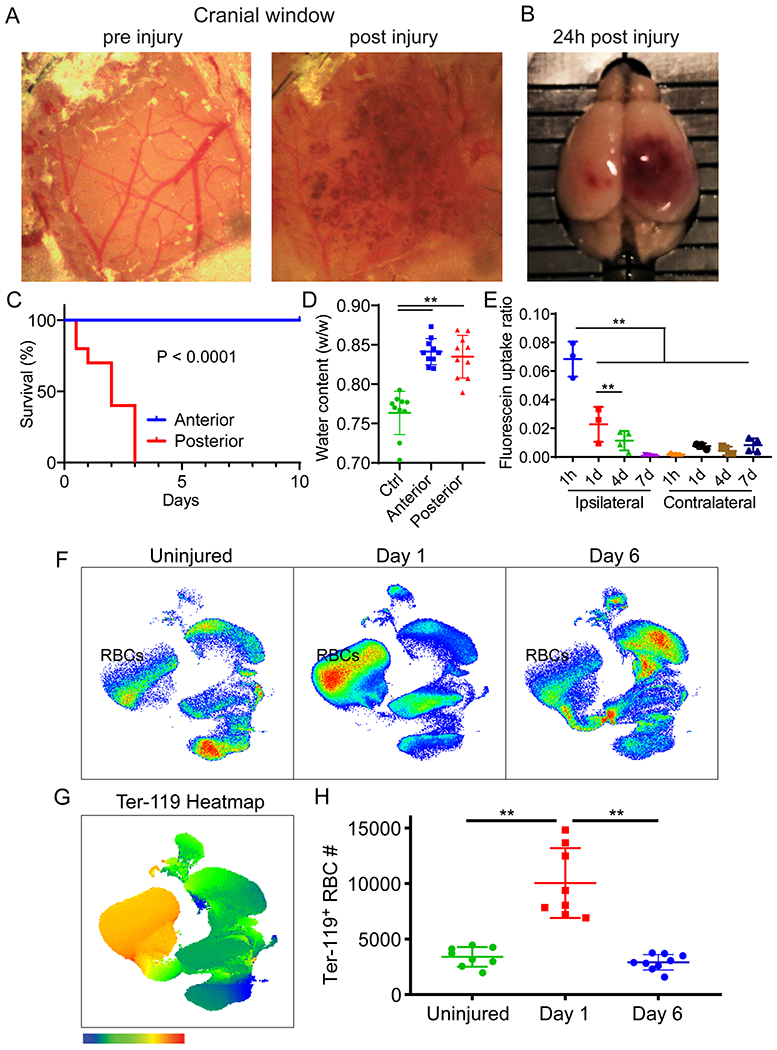
A. Magnified images of the 2mm x 2mm x 15μm thinned skull window pre- and post-injury depict petechial intraparenchymal hemorrhages at 10 min post-injury. B. Macroscopic depiction of a mouse brain 24 h following posterior sonication injury. C. Kaplan-Meier curve demonstrates a median survival of 2 days after posterior sonication injury. Anterior sonication injury does not result in fatalities. Cumulative data are shown from 2 independent experiments with 10 mice per group (P=2.96e-10, Log-rank test). D. A graph showing quantification of cerebral water content demonstrates increased edema 24 h after sonication with 7.7% and 7.1% increase in water content after anterior and posterior injury, respectively, relative to uninjured control mice (**P<0.01, anterior P=2.9e-7, posterior p=1.3e-6, One-way ANOVA/Tukey test). Cumulative data are shown from 2 independent experiments with 5 mice per group per experiment. E. A graph showing quantification of fluorescein extravasation into the ipsilateral versus contralateral brain hemisphere at the denoted time points post-injury (**P<0.01, One-way ANOVA/Tukey test). Data are representative of 2 independent experiments with 4 mice per group per experiment, 2 samples were above the detection limit and not included. F-H. High parameter flow cytometric analysis of brain biopsies from mice at d1 and d6 post-injury relative to uninjured controls. A UMAP plot of concatenated live cells from each group is shown in panel F. A heatmap of Ter-119 signal on a UMAP plot reflecting the concatenated cell populations from a single experiment is shown in panel G. Panel H shows a scatter plot depicting the absolute Ter-119+ RBCs. Cumulative data are shown from 2 independent experiments (Uninjured n=8, d1 n=8, d6 n=9, **P<0.01, One-way ANOVA/Tukey test; gating strategy in Supplementary Figure 1A). Graphs D, E, H show the mean±SD.
Extended Data Fig. 3. Microglia depletion increases BBB leakage, intraparenchymal hemorrhage, myeloid cell invasion and vascular endothelium activation.
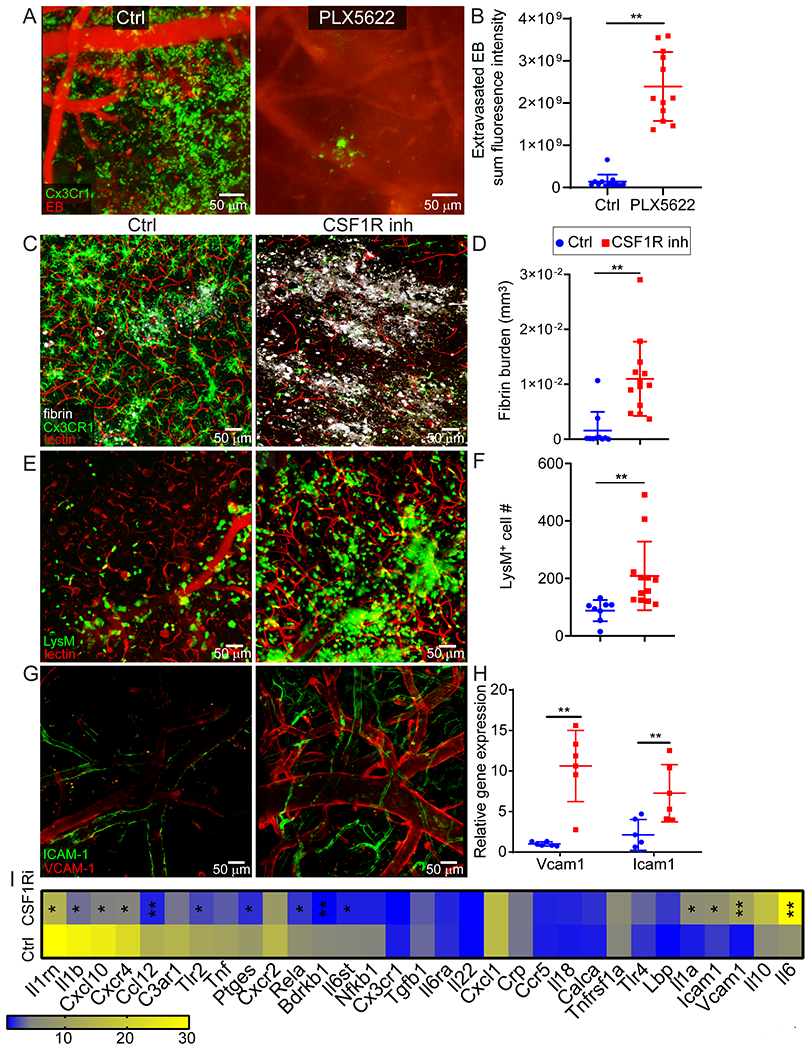
A. Intravital microscopy of CX3CR1gfp/wt (green) mice 20 min after injury shows extensive intraparenchymal EB (red) extravasation following microglia depletion using an alternate CSF1R inhibitor, PLX5622, to that used in Fig 2C, D. B. EB extravasation assay based on intravital microscopy time lapses depicts increased BBB leakage 20-40 min after microglia depletion using PLX5622. Graph depicts mean±SD of cumulative data from 2 independent experiments (n=12 mice per group, **P=3.9e-9, two-tailed Student’s t-test). C. Confocal microscopy images of cortical brain sections from of naive and microglia depleted Cx3CR1gfp/wt (green) mice 1 h following injury. Mice received an i.v. injection of fluorescent fibrinogen (white) and tomato-lectin (red). Larger and more diffuse intraparenchymal fibrinogen is observed in mice treated with PLX3397 (CSF1R inhibitor). D. Image-based quantification of fibrin burden in the brain parenchyma. Graph depicts mean±SD of cumulative data from 2 independent experiments (Ctrl n=10, CSF1R inh n=13, **P=0.00017, Mann–Whitney U test). E. Two-photon microscopy images captured in the cerebral cortex of injured LysMgfp/wt mice treated with vehicle or PLX3397 show LysM+ myelomonocytic cell (green) invasion at 20 min post-injury. Tomato-lectin is shown in red. F. Image based quantification of myelomonocytic infiltration. Graph depicts mean±SD of cumulative data from 2 independent experiments (Ctrl n=8, CSF1R inh n=12, **P=0.0002, Mann–Whitney U test). G. Intravital microscopy images in the cerebral cortex of vehicle versus PLX3397 treated B6 mice at 24 h post-injury. Prior to imaging, mice received an i.v. injection of APC-anti-CD106 (VCAM-1; red) and PE-anti-CD54 (ICAM-1; green), which revealed increased endothelial expression in PLX3397 treated mice. Representative images are from 2 independent experiments with 3 mice per group. H. qPCR analysis of ICAM and VCAM expression in vehicle vs. PLX3397 treated B6 mice at 24 h post-injury. Graph depicts mean±SD of cumulative data from 2 independent experiments (n=6 mice per group, Vcam1 P=3.58e-5, Icam1 P=0.0072, Two-way ANOVA/Holm-Sidak test). I. Heatmap shows qPCR analysis of genes encoding for acute inflammation-related proteins in vehicle vs. PLX3397-treated B6 mice. The fold increase in gene expression was calculated relative to the uninjured contralateral hemisphere for each mouse at 24 h post-injury. Data are representative of 2 independent experiments with 5 mice per group per experiment (*P<0.05, **P<0.01, multiple t tests with Holm-Sidak multiple comparisons correction, source data in Supplementary Table S3). CSF1R inh refers to PLX3397.
Extended Data Fig. 4. Representative images establishing a role for P2RY12 receptor and Cx43 hemichannels in microglial rosetting.
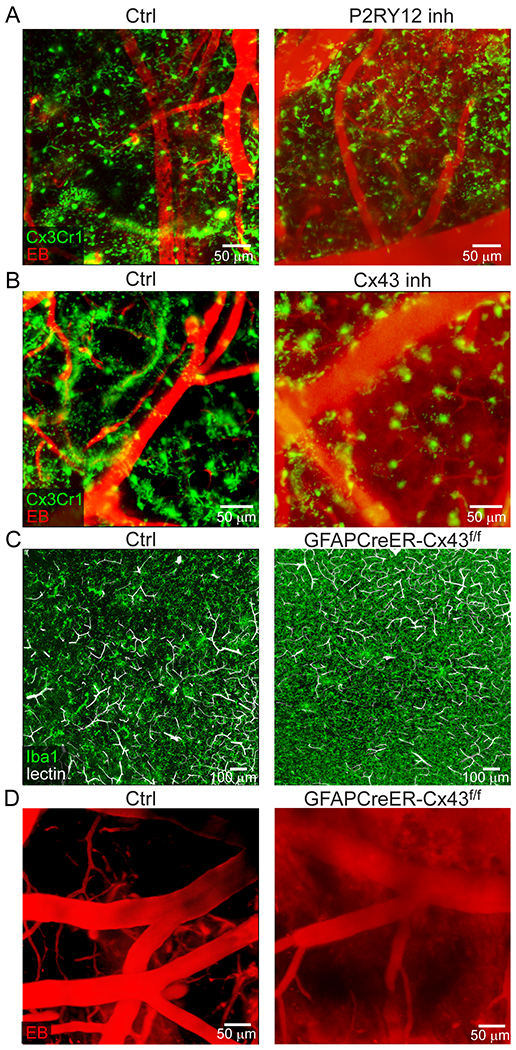
Data from these experiments are provided in Figures 2G, H and I. A. Intravital microscopy images in the cerebral cortex of Cx3CR1gfp/wt (green) mice 20 min post-injury. Mice were treated transcranially with a vehicle or a P2RY12 inhibitor (MeSAMP). Intravenous injection of EB (red) revealed increased intraparenchymal extravasation following pre-treatment with the P2RY12 inhibitor. Images are representative of experimental data graphed in Figure 2G. B. Intravital microscopy images in the cerebral cortex of Cx3CR1gfp/wt (green) mice 20 min post-injury. Mice were treated transcranially with a vehicle or a Cx43 inhibitor (carbenoxolone). Intravenous injection of EB (red) revealed extensive intraparenchymal extravasation following pre-treatment with the Cx43 inhibitor. Images are representative of experimental data graphed in Figure 2G. C. Confocal microscopy images of brain sections from littermate control (GFAPCreER-Cx43f/wt and GFAPCreER-Cx43wt/wt) vs. GFAPCreER-Cx43f/f mice 1 h after injury show decreased microglia rosetting in GFAPCreER-Cx43f/f mice. Microglia rosettes were identified with Iba1 staining (green). Tomato-lectin+ blood vessels are shown in white. Images are representative of experimental data graphed in Figure 2H. D. Intravital microscopy images from the cerebral cortex of control (GFAPCreER-Cx43f/wt, GFAPCreER-Cx43wt/wt) vs. GFAPCreER-Cx43f/f mice 20 min after injury. Intravenous injection of EB (red) revealed extensive intraparenchymal extravasation in GFAPCreER-Cx43f/f mice. Images are representative of experimental data graphed in Figure 2I.
Extended Data Fig. 5. Immune landscape in the cerebral cortex following cerebrovascular injury in Cx3cr1gfp/wtCcr2rfp/wt mice.
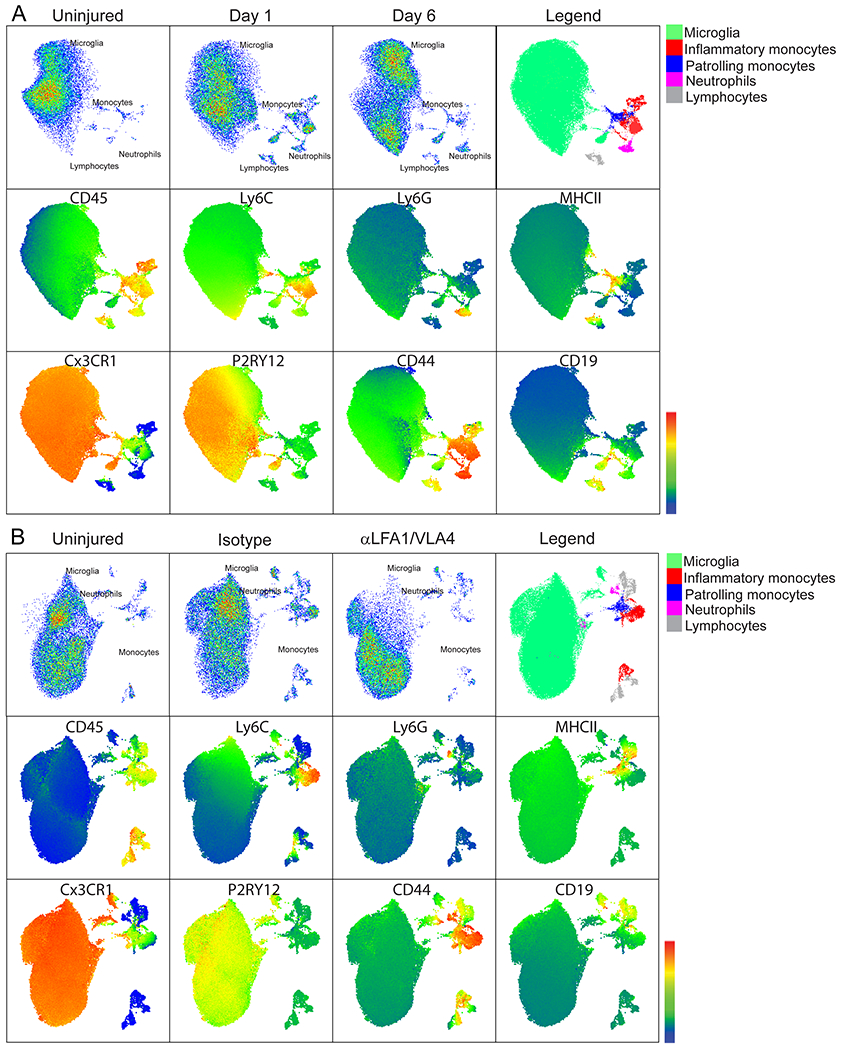
Quantification of these flow cytometric experiments is provided in Figures 3C and 3F, gating strategy in Supplementary Figure 1B. The panel used for these experiments includes: Cx3CR1-GFP, Ly6C BB790, MHCII BV480, CD11b BV570, CD115 BV605, CD24 BV650, CD11c BV785, P2RY12 PE, CCR2-RFP, Ter-119 PE/Cy5, CD206 PE/Cy7, CD45 BUV395, CD4 BUV496, Ly6G BUV563, CD19 BUV661, CD44 BUV737, CD8 BUV805, F4/80 APC-R700, TCRb APC/Cy7 and live/dead fixable blue cell staining kit. Plots were pre-gated for CD45+Ter119- live cells and subsequently analyzed using an unsupervised clustering algorithm to group data into subpopulations (PhenoGraph) and visualized using UMAP. For each experiment, the first row depicts the concatenated samples of 4 independent mice per group, and the legend shows the combined phenograph clusters corresponding to different immune cell populations. The second and third rows show six representative heatmaps of different markers used to identify the different immune cell populations. A. Immune landscape at 1 d and 6 d post-injury compared to uninjured mice. B. Immune landscape 1 d after injury in mice treated with bolus αLFA1/VLA4 or isotype control antibodies relative to uninjured mice.
Extended Data Fig. 6. Effect of myelomonocytic cell invasion on cerebral edema and survival after injury.
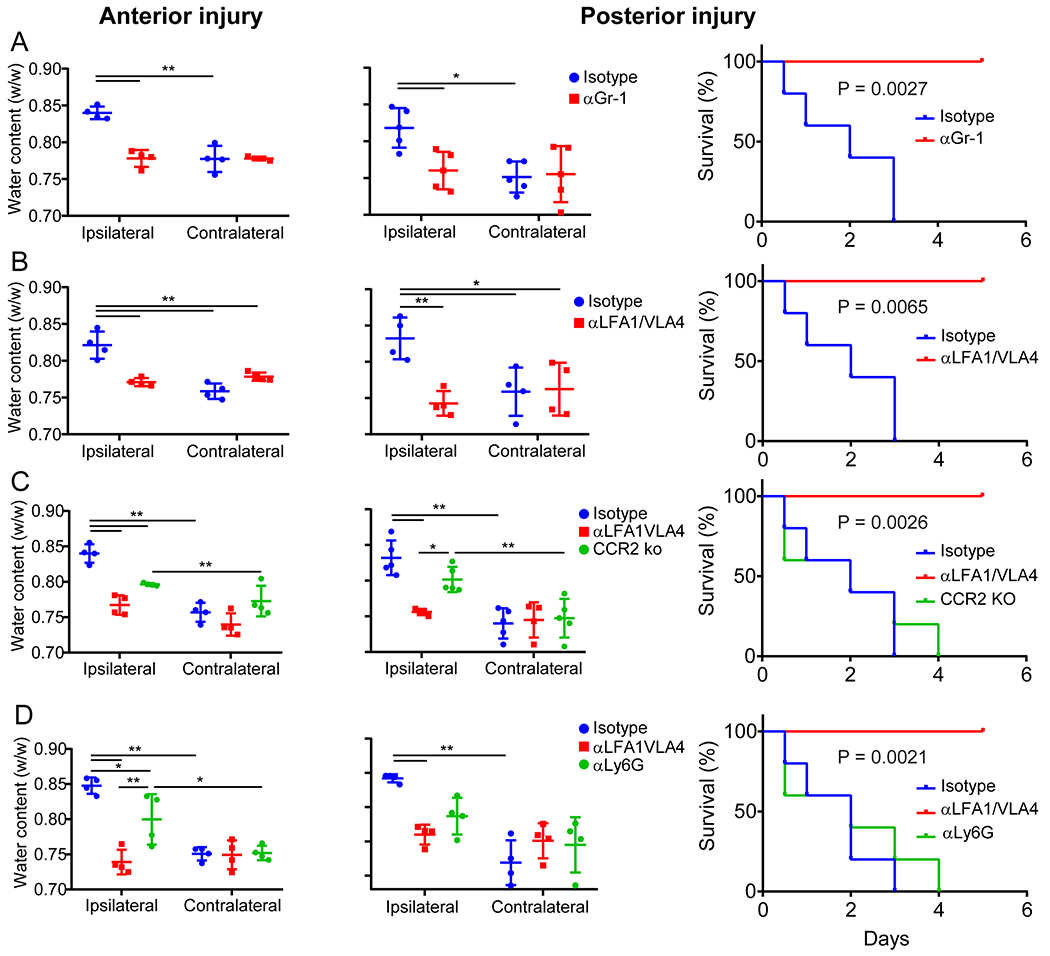
This figure depicts quantification of cerebral water content following anterior and posterior injury as well as survival after posterior injury in four different experimental paradigms. Left column shows quantification of cerebral water content in the ipsilateral hemisphere 1 d after anterior injury in the treatment vs control group compared to the contralateral hemisphere. The middle column shows quantification of cerebral water content after posterior injury when mice reached the survival end point or 5 d post-injury in the treatment vs. control group relative to the contralateral hemisphere (*P<0.05, **P<0.01, Two-way ANOVA/Holm-Sidak test). Graphs depict mean±SD. The right column shows Kaplan-Meier survival curves after posterior injury in treatment vs. control groups (Log-rank test). A. Effect of αGr-1 vs. isotype control bolus administration 24 h prior to injury. B. Effect of αLFA1/VLA4 vs. isotype control bolus administration 24 h prior to injury. C. Water content and survival after injury in CCR2 KO mice compared to B6 mice treated with isotype or αLFA1/VLA4 24 h prior to injury. D. Effect of αLy6G or αLFA1/VLA4 vs. isotype control administration 24 h prior to injury. Data are representative of 2 independent experiments with 4 or 5 mice per group.
Extended Data Fig. 7. Combined αLFA1 and αVLA4 treatment is required to prevent cerebral edema and death.
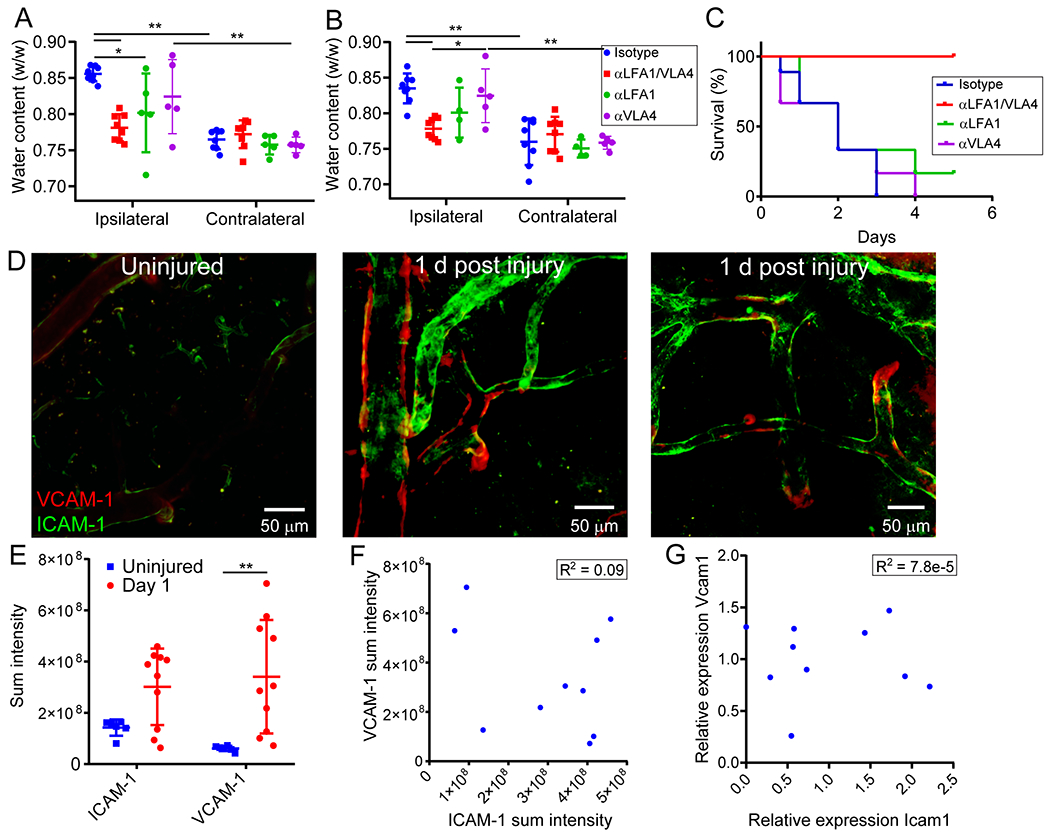
A-C. Effect of bolus treatment with αLFA1/VLA4, αLFA1, αVLA4 or isotype control on cerebral water content 24 h after anterior (A) or posterior (B, C) injury. Data compilation from 2 independent experiments. The antibodies were administered 24 h before injury. Cerebral water content was determined 1d after anterior injury (A) and 5d or when mice reached the survival endpoint after posterior injury (B). Graphs depict mean±SD (isotype n=8, αLV n=8, αLFA1 n=4 or 5, αVLA4 n=5, *P<0.05, **P<0.01, Two-way ANOVA/Holm-Sidak test). Panel C demonstrates Kaplan-Meier survival curve following posterior injury (isotype n=9, αLV n=10, αLFA1 n=6, αVLA4 n=6, Logrank test P=3.4e-5). D. Intravital microscopy images of the cerebral cortex from uninjured vs. injured B6 mice 1 h following i.v. administration of APC-anti-CD106 (VCAM-1; red) and PE-anti-CD54 (ICAM-1; green) show increased endothelial ICAM and VCAM expression 24 h after injury. Two representative images from the injured mice depict inter-sample and inter-vessel variability in VCAM-1 vs. ICAM-1 expression. Representative images are from 2 independent experiments with 3 and 5 mice per group. E. Image based quantification of vascular ICAM and VCAM expression. Graph depicts mean±SD of cumulative data from 2 independent experiments (uninjured n=6, d1 n=10, **P<0.01, Two-way ANOVA/Holm-Sidak test). F. Scatter plot of sum intensity of ICAM vs VCAM expression depicting the lack of a correlation between the two variables. Each dot represents a single mouse, and the graph is a representation of data points shown in Extended data Figure 7E, (R2 = 0.089, P = 0.4, Pearson’s product moment correlation test). G. Scatter plot of ICAM vs. VCAM gene expression determined by qPCR in the brain 24 h post-injury. No correlation was observed between the two variables. Each dot represents one mouse. Graphs depict cumulative data from 2 independent experiments with 5 mice per experiment, (R2 = 0.000078, P = 0.98, Pearson’s product moment correlation test).
Extended Data Fig. 8. Effect of myelomonocytic cells on cerebral repair and angiogenesis.
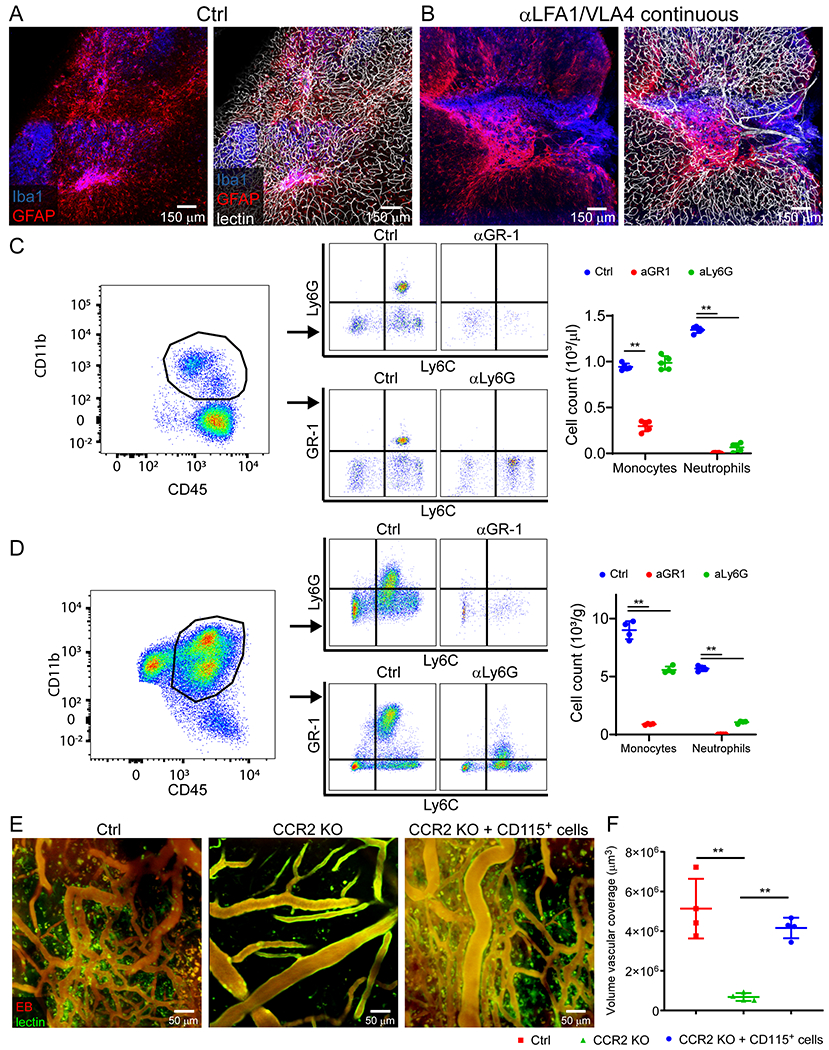
A, B. Confocal microscopy of cerebral cortex 10 days after injury shows areas of gliosis (GFAP, blue) and microglia clustering (Iba-1, red) relative to tomato-lectin+ vasculature. Mice were treated continuously with either isotype control (A) or αLFA1/VLA4 (B) antibodies. Continuous treatment with αLFA1/VLA4 results in large areas of brain tissue with blood vessels. Images are representative of 5 mice per group. C, D. Flow cytometric analysis and gating strategy of monocyte and neutrophil depletion in blood (C) and brain (D) following continuous treatment with GR-1 and Ly6G antibodies 6 d post-injury compared to isotype treated controls. The following panel was used: Ly6C FITC, GR-1 BV421, CD11b BV570, Cx3CR1 BV711, P2RY12 PE, CD45 BUV395, Ly6G BUV563, CD44 BUV737, CD115 APC and live/dead fixable blue cell staining kit. Plots were pre-gated for single, live cells and subsequently for CD45+CD11b+ cells. In brain samples gated, microglia were identified as CD45lowCx3Cr1+P2RY12+CD44− cells. Monocytes were identified as CD45+CD44+CD115+Ly6G-GR1low and neutrophils as CD45+CD44+CD115-Ly6G+GR1hi. In mice treated with Ly6G, Ly6G was not used to characterize cells flow cytometrically. Moreover, in mice treated with GR-1, GR-1 was not used to characterize the cells. Graphs show the mean±SD and are representative of 2 independent experiments with n=5 (C) and n=4 (D) mice per group (**P<0.01, Two-way ANOVA/Holm-Sidak test). E, F. Intravital microscopy of cerebral vasculature and image-based quantification of vascular coverage in naïve B6 mice, CCR2 KO mice and CCR2 KO mice with CD115+ monocyte adoptive transfer 10 d post-injury. Adoptive transfer of CD115+ monocytes from B6 mice partially reconstitutes the angiogenic process. Graph shows mean±SD and is representative of two independent experiments with (n=4 mice per group, **P<0.01, Kruskal-Wallis/Dunn’s test).
Extended Data Fig. 9. qPCR analysis of genes encoding for angiogenesis related proteins.
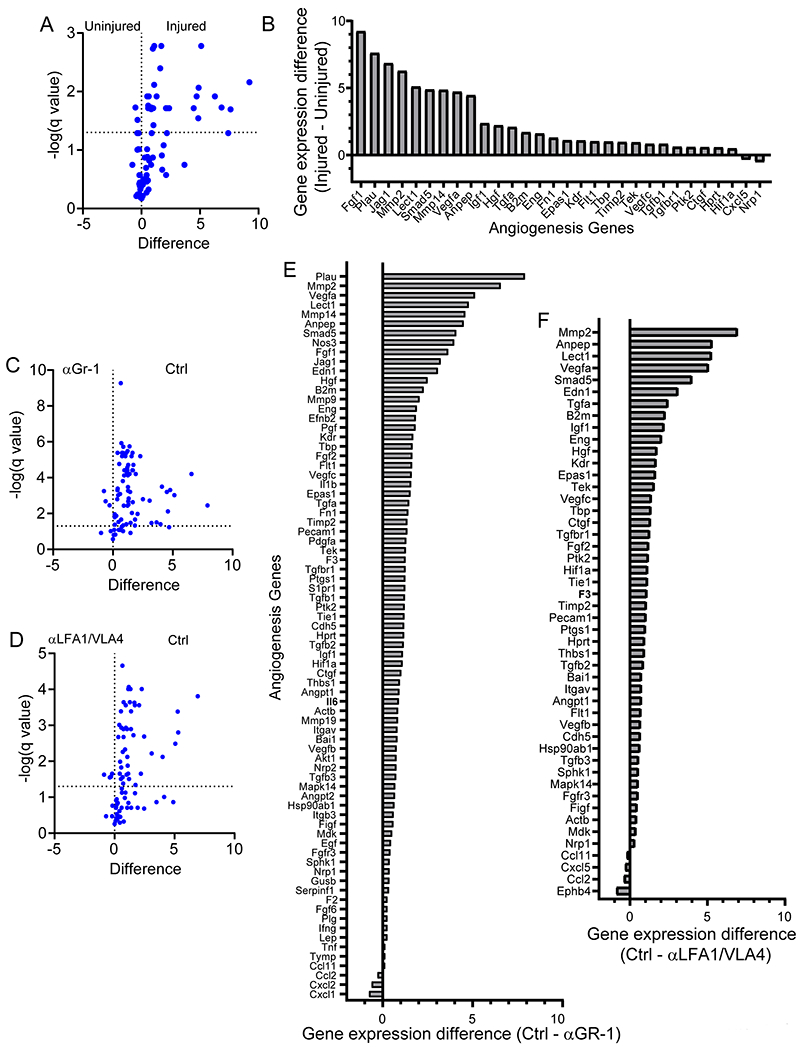
qPCR analysis of genes encoding for angiogenesis related proteins. Data from these experiments are represented in Figure 4I. A. Volcano plot of angiogenesis related gene expression between uninjured mice and mice d6 after injury. B. Bar graph of gene expression differences between injured and uninjured mice for genes with Q<5%. C-D. Volcano plot of angiogenesis related gene expression after continuous αGr-1 (C) or αLFA1/VLA4 administration (D). E-F. Bar graphs of gene expression difference for genes with Q<5%. Data are representative of 2 independent experiments with 4 mice per group. Statistical analysis was performed using multiple t-tests and the Benjamini, Krieger and Yekutieli method to correct for the false discovery rate, with a desired Q value of 5%. Data are representative of 2 independent experiments with 4 mice per group per experiment, source data in Supplementary Table 4.
Extended Data Fig. 10. VEGF-expressing microglia are involved in angiogenesis.
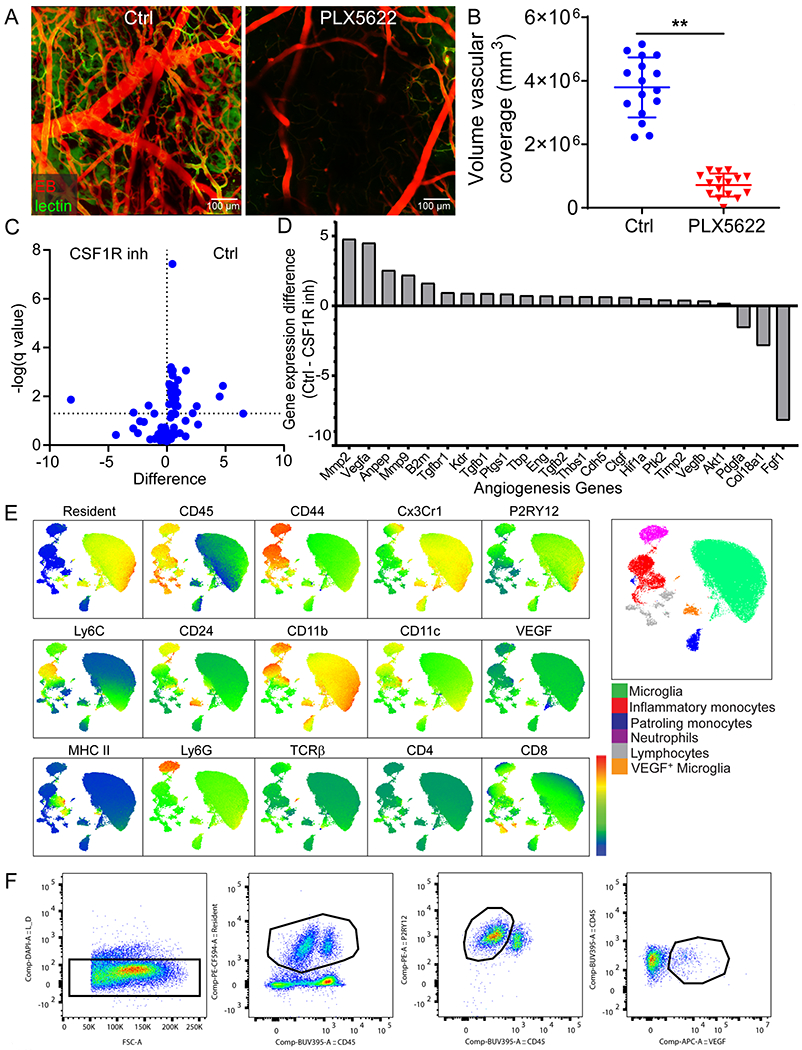
VEGF-expressing microglia are involved in angiogenesis. A, B. Intravital microscopy of cerebral vasculature 10 d after injury (A) and image-based quantification of vascular coverage (B) in PLX5622 versus vehicle control treated mice show a lack of angiogenesis after microglia depletion. Blood vessels were labeled with EB (red) and tomato-lectin (green). Graphs depict mean±SD of cumulative data from 3 independent experiments (n=16 mice per group, **P=4e-13, two-tailed Student’s t-test). C. Volcano plot of angiogenesis related gene expression in vehicle vs. PLX3397 treated (CSF1R inhibitor) mice 6 d post-injury D. Bar graph of gene expression difference for genes with Q<5%. Data are representative of 2 independent experiments with 4 mice per group. Statistical analysis was performed using multiple t-test and the Benjamini, Krieger and Yekutieli method to correct for the false discovery rate, with a desired Q value of 5%. A heatmap of the data from panels A and B is shown in Figure 5B, source data in Supplementary Table 4. E. Representative heatmaps used to identify different immune cell populations following high parameter flow cytometric analysis of the immune landscape in the cerebral cortex d1 and d6 following injury in Cx3cr1CreER/+ x Stopfl/+ TdTomato mice. The following panel was used: Ly6C BB790, MHCII BV480, CD11b BV570, CD115 BV605, CD24 BV650, Cx3Cr1 BV711, CD11c BV785, P2RY12 PE, Ter-119 PE/Cy5, CD206 PE/Cy7, CD45 BUV395, CD4 BUV496, Ly6G BUV563, CD19 BUV661, CD44 BUV737, CD8 BUV805, F4/80 APC-R700, TCRβ APC/Cy7, VEGF-A AF647, TdTomato and live/dead fixable blue cell staining kit. Data were pre-gated on CD45+Ter119- live cells, subsequently analyzed using an unsupervised clustering algorithm to group data into subpopulations (PhenoGraph) and visualized using UMAP. Quantification of these experiments is provided in Figure 5F. F. Gating strategy for flow cytometry experiment demonstrating a lack of VEGF-A+ microglia in mice treated with continuous αLFA1/VLA4 after injury. The following panel was used: CD24 FITC, CD11b BV570, P2RY12 PE, TdTomato, CD45 BUV395, VEGF-A AF647 and live/dead fixable blue cell staining kit. Quantification of these experiments is provided in Figure 5H–I and gating strategy in Supplementary Figure 1B.
Supplementary Material
Acknowledgements
This research was supported the intramural program at the National Institute of Neurological Disorders & Stroke, National Institutes of Health. We thank Alan Hoofring in the NIH Medical Arts Design Section for generating the illustration shown in Extended Data Fig. 1. We thank Abdel Elkahloun and Weiwei Wu in the National Human Genome Research Institute Microarray core for their assistance with the RNAseq experiment.
Footnotes
Data availability
The data that support the findings of this study are available from the corresponding author upon request. There are no restrictions in data availability. The following figures have associated source data in the supplementary section: Figures 1C, 1D, 2D, 2G, 2H, 2I, 3C, 3E, 3F, 3G, 3H, 3I, 4B, 4D, 4F, 4H, 4H, 5A, 5D, 5F, 5H, 6E, 6F Extended Data Figures 2D, 2E, 2H, 3B, 3D, 3F, 3H, 6A, 6B, 6C, 6D, 7A, 7B, 7C, 7E, 7F, 7G, 8C, 8D, 8F, 10B. Bulk RNA sequencing data are available in the NCBI Gene Expression Omnibus.
Accession codes
Our bulk RNA sequencing data are available in the NCBI Gene Expression Omnibus with access code GSE161424.
Competing interest statement
The authors declare no competing interests.
Reporting Summary. Additional information on research design is available in the Nature Research Reporting Summary associated with this article.
References
- 1.Mastorakos P & McGavern D The anatomy and immunology of vasculature in the central nervous system. Sci Immunol 4(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Donkor ES Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res Treat 2018, 3238165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maxwell WL, Irvine A, Adams JH, Graham DI & Gennarelli TA Response of cerebral microvasculature to brain injury. J Pathol 155, 327–335 (1988). [DOI] [PubMed] [Google Scholar]
- 4.Marcolini E, Stretz C & DeWitt KM Intracranial Hemorrhage and Intracranial Hypertension. Emerg Med Clin North Am 37, 529–544 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Simard JM, Kent TA, Chen M, Tarasov KV & Gerzanich V Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6, 258–268 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mestre H, et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 367(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kriz J Inflammation in ischemic brain injury: timing is important. Crit Rev Neurobiol 18, 145–157 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Yang Y & Rosenberg GA Matrix metalloproteinases as therapeutic targets for stroke. Brain Res 1623, 30–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadareishvili Z, et al. An MRI Hyperintense Acute Reperfusion Marker Is Related to Elevated Peripheral Monocyte Count in Acute Ischemic Stroke. J Neuroimaging 28, 57–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hermann DM, Kleinschnitz C & Gunzer M Implications of polymorphonuclear neutrophils for ischemic stroke and intracerebral hemorrhage: Predictive value, pathophysiological consequences and utility as therapeutic target. J Neuroimmunol 321, 138–143 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Manglani M & McGavern DB Intravital Imaging of Neuroimmune Interactions Through a Thinned Skull. Curr Protoc Immunol 120, 24 22 21–24 22 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth TL, et al. Transcranial amelioration of inflammation and cell death after brain injury. Nature 505, 223–228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Promeneur D, Lunde LK, Amiry-Moghaddam M & Agre P Protective role of brain water channel AQP4 in murine cerebral malaria. Proc Natl Acad Sci U S A 110, 1035–1040 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serbina NV & Pamer EG Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7, 311–317 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Rua R, et al. Infection drives meningeal engraftment by inflammatory monocytes that impairs CNS immunity. Nat Immunol 20, 407–419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang YB, et al. Early Neurological Deterioration after Recanalization Treatment in Patients with Acute Ischemic Stroke: A Retrospective Study. Chin Med J (Engl) 131, 137–143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peng G, et al. Risk factors for decompressive craniectomy after endovascular treatment in acute ischemic stroke. Neurosurg Rev (2019). [DOI] [PubMed] [Google Scholar]
- 18.Chen X, et al. A prediction model of brain edema after endovascular treatment in patients with acute ischemic stroke. J Neurol Sci 407, 116507 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Nimmerjahn A, Kirchhoff F & Helmchen F Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Davalos D, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8, 752–758 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Lou N, et al. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc Natl Acad Sci U S A 113, 1074–1079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahn SJ, Anrather J, Nishimura N & Schaffer CB Diverse Inflammatory Response After Cerebral Microbleeds Includes Coordinated Microglial Migration and Proliferation. Stroke 49, 1719–1726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez-Lopez D, et al. Microglial Cells Prevent Hemorrhage in Neonatal Focal Arterial Stroke. J Neurosci 36, 2881–2893 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fumagalli S, Perego C, Ortolano F & De Simoni MG CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia 61, 827–842 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Tang Z, et al. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation 11, 26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jolivel V, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol 129, 279–295 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Kim JV, Kang SS, Dustin ML & McGavern DB Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 457, 191–195 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Relton JK, et al. Inhibition of alpha4 integrin protects against transient focal cerebral ischemia in normotensive and hypertensive rats. Stroke 32, 199–205 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, et al. Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke 34, 1790–1795 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Langhauser F, et al. Blocking of alpha4 integrin does not protect from acute ischemic stroke in mice. Stroke 45, 1799–1806 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Llovera G, et al. Results of a preclinical randomized controlled multicenter trial (pRCT): Anti-CD49d treatment for acute brain ischemia. Sci Transl Med 7, 299ra121 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Krams M, et al. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke 34, 2543–2548 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Elkins J, et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (ACTION): a randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol 16, 217–226 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Xiong Y, Mahmood A & Chopp M Angiogenesis, neurogenesis and brain recovery of function following injury. Curr Opin Investig Drugs 11, 298–308 (2010). [PMC free article] [PubMed] [Google Scholar]
- 35.Krupinski J, Kaluza J, Kumar P, Kumar S & Wang JM Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 25, 1794–1798 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Dimitrijevic OB, Stamatovic SM, Keep RF & Andjelkovic AV Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 38, 1345–1353 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Hammond MD, et al. CCR2+ Ly6C(hi) inflammatory monocyte recruitment exacerbates acute disability following intracerebral hemorrhage. J Neurosci 34, 3901–3909 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gliem M, et al. Macrophages prevent hemorrhagic infarct transformation in murine stroke models. Ann Neurol 71, 743–752 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Gliem M, et al. Macrophage-derived osteopontin induces reactive astrocyte polarization and promotes re-establishment of the blood brain barrier after ischemic stroke. Glia 63, 2198–2207 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Wattananit S, et al. Monocyte-Derived Macrophages Contribute to Spontaneous Long-Term Functional Recovery after Stroke in Mice. J Neurosci 36, 4182–4195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang W, et al. CCR2-dependent monocytes/macrophages exacerbate acute brain injury but promote functional recovery after ischemic stroke in mice. Theranostics 8, 3530–3543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keren-Shaul H, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Apte RS, Chen DS & Ferrara N VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 176, 1248–1264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Margaritescu O, Pirici D & Margaritescu C VEGF expression in human brain tissue after acute ischemic stroke. Rom J Morphol Embryol 52, 1283–1292 (2011). [PubMed] [Google Scholar]
- 45.Xie L, Mao X, Jin K & Greenberg DA Vascular endothelial growth factor-B expression in postischemic rat brain. Vasc Cell 5, 8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sankowski R, et al. Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci 22, 2098–2110 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Brandenburg S, et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol 131, 365–378 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Vannella KM & Wynn TA Mechanisms of Organ Injury and Repair by Macrophages. Annu Rev Physiol 79, 593–617 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Wynn TA & Ramalingam TR Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18, 1028–1040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duffield JS, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 115, 56–65 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Higashida RT, et al. Trial design and reporting standards for intra-arterial cerebral thrombolysis for acute ischemic stroke. Stroke 34, e109–137 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Luby M, Bykowski JL, Schellinger PD, Merino JG & Warach S Intra- and interrater reliability of ischemic lesion volume measurements on diffusion-weighted, mean transit time and fluid-attenuated inversion recovery MRI. Stroke 37, 2951–2956 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hacke W, et al. Randomised double-blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II). Second European-Australasian Acute Stroke Study Investigators. Lancet 352, 1245–1251 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Paigen B, Morrow A, Brandon C, Mitchell D & Holmes P Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis 57, 65–73 (1985). [DOI] [PubMed] [Google Scholar]
- 55.Jung S, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20, 4106–4114 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saederup N, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5, e13693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ganat YM, et al. Early postnatal astroglial cells produce multilineage precursors and neural stem cells in vivo. J Neurosci 26, 8609–8621 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yona S, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao Y, Day KH, Damon DN & Duling BR Endothelial cell-specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc Natl Acad Sci U S A 98, 9989–9994 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calera MR, et al. Connexin43 is required for production of the aqueous humor in the murine eye. J Cell Sci 119, 4510–4519 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Hughes EG, Kang SH, Fukaya M & Bergles DE Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci 16, 668–676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faust N, Varas F, Kelly LM, Heck S & Graf T Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96, 719–726 (2000). [PubMed] [Google Scholar]
- 64.Yata Y, et al. DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells. Hepatology 37, 267–276 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Spangenberg E, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun 10, 3758 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wyder L, et al. Increased expression of H/T-cadherin in tumor-penetrating blood vessels. Cancer Res 60, 4682–4688 (2000). [PubMed] [Google Scholar]
- 67.Wiewrodt R, et al. Size-dependent intracellular immunotargeting of therapeutic cargoes into endothelial cells. Blood 99, 912–922 (2002). [DOI] [PubMed] [Google Scholar]
- 68.McQualter JL, et al. Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the sca-1 positive cell fraction. Stem Cells 27, 623–633 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Ohsawa K, Imai Y, Kanazawa H, Sasaki Y & Kohsaka S Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci 113 ( Pt 17), 3073–3084 (2000). [DOI] [PubMed] [Google Scholar]
- 70.Sasaki Y, Ohsawa K, Kanazawa H, Kohsaka S & Imai Y Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem Biophys Res Commun 286, 292–297 (2001). [DOI] [PubMed] [Google Scholar]
- 71.Kanazawa H, Ohsawa K, Sasaki Y, Kohsaka S & Imai Y Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-gamma -dependent pathway. J Biol Chem 277, 20026–20032 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Linker KE, et al. Microglial activation increases cocaine self-administration following adolescent nicotine exposure. Nat Commun 11, 306 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Skorkowska A, et al. Effect of Combined Prenatal and Adult Benzophenone-3 Dermal Exposure on Factors Regulating Neurodegenerative Processes, Blood Hormone Levels, and Hematological Parameters in Female Rats. Neurotox Res 37, 683–701 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lentz MR, et al. Diffusion tensor and volumetric magnetic resonance measures as biomarkers of brain damage in a small animal model of HIV. PLoS One 9, e105752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Krause TA, Alex AF, Engel DR, Kurts C & Eter N VEGF-production by CCR2-dependent macrophages contributes to laser-induced choroidal neovascularization. PLoS One 9, e94313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Russo MV & McGavern DB Inflammatory neuroprotection following traumatic brain injury. Science 353, 783–785 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.