

Abstract
Lung cancer is the leading cause of cancer-related deaths, demanding improvement in current treatment modalities to reduce the mortality rates. Lung cancer is divided into two major classes with non-small cell lung cancer representing ~84% of lung cancer cases. One strategy widely used to treat non-small cell lung cancer patients includes targeting the epidermal growth factor receptor (EGFR) using EGFR-inhibitors, such as erlotinib, gefitinib, and afatinib. However, most patients develop resistance to EGFR-inhibitors within a year post-treatment. Although some mechanisms that drive resistance to EGFR-inhibitors have been identified, there are many cases in which the mechanisms are unknown. Thus, in this study, we examined the role of microRNAs in driving EGFR-inhibitor resistance. As mediators of critical pro-growth pathways, microRNAs are severely dysregulated in multiple diseases, including non-small cell lung cancer where microRNA dysregulation also contributes to drug resistance. In this work, through screening of 2019 mature microRNAs, multiple microRNAs were identified that drive EGFR-inhibitor resistance in non-small cell lung cancer cell lines, including miR-432–5p.
Keywords: Erlotinib, microRNAs, Non-small cell lung cancer, miR-432, EGFR inhibitor resistance
1. Introduction
Lung cancer is expected to take the lives of 135,720 people in 2020 alone, more than that of breast, prostate, and colon cancer combined [1]. Of the two major subclasses of lung cancer, non-small lung cancer (NSCLC) constitutes approximately 84% of lung cancer cases [1]. Due to a lack of early diagnostic technologies, NSCLC patients suffer from metastatic disease, thus surgical resection of tumors is inadequate. Therefore, radiotherapy, chemotherapy, and targeted therapies are the preferred treatment strategies. Targeted therapies are designed based on the molecular drivers of the cancer, i.e. genes that cancer cells are dependent on for growth and survival. A few such drivers in NSCLC include the proto-oncogenes EGFR, MET, HER2, KRAS, and MEK that when amplified or mutated become oncogenic, driving pro-survival signaling pathways [2–7].
Epidermal Growth Factor Receptor (EGFR) is an integral membrane protein that belongs to a class of receptor tyrosine kinases known as ErbB1 or HER1. In 10–35% of NSCLC patients, EGFR is overactivated [8–10], serving as a driver of the cancer. EGFR signaling is upregulated via amplification of wildtype EGFR, through amino acid substitution (L858R), or via an in-frame deletion in exon 19, which together account for > 90% of EGFR mutations in NSCLC [11–13]. Such mutations are classified as activating mutations and patients whose tumors have these mutations are treated with targeted agents to abrogate EGFR mediated signal transduction, collectively referred to as EGFR-inhibitors (EGFR-i). EGFR-i are commonly used as a first-line therapy, or have been used as a second-line therapy after conventional chemotherapies have failed to clinically benefit the patient [14–17]. Based on the targeted mechanism of action, currently three generations of EGFR-i are used as standard-of-care therapeutics for NSCLC patients. Erlotinib belongs to the first generation of EGFR-i that clinically benefits patients with either EGFR-wildtype or EGFR-mutant tumors [11,13–16,18,19].
NSCLC tumors regress rapidly when patients are treated with erlotinib; however, within a year post-treatment, the majority of patients develop resistance. This is currently the major drawback with using erlotinib as a standard-of-care intervention. Molecularly, resistance to erlotinib occurs when the drug exposure is inadequate at targeting active EGFR or the cell no longer depends on EGFR activity for survival. Genomic profiling of tumors and cell-free DNA from patients post-erlotinib treatment reveals that erlotinib resistance in NSCLC cells is mediated via two major mechanisms [20–26]: i) acquisition of secondary mutations that change the conformation of EGFR, rendering it insensitive to erlotinib, and ii) activation of alternate mechanisms of growth and proliferation that circumvent the need to use the EGFR pathway, referred to as bypass tracks [21,27].
In approximately 60% of erlotinib treated patients, resistance results due to a secondary mutation in EGFR, T790M [11,13,18,28,29]. Whereas in 20% of patients, tumors revert to the use of common bypass tracks to evade EGFR inhibition. The common bypass tracks include activation of oncoproteins such as MET, BRAF, HER2, or PIK3CA, induction of cellular transformation including conversion of NSCLC into small cell lung cancer (SCLC), or through epithelial to mesenchymal transition (EMT) [22–26]. Many of the mechanisms involved in activation of these bypass tracks remain largely unknown. Additionally, in 15–20% of NSCLC patients the molecular mechanisms that drive erlotinib resistance remain undiscovered [27,30].
Although recent investigations have identified roles for multiple oncogenic and tumor suppressive proteins as mediators of erlotinib resistance in NSCLC [31], with additional knowledge including the involvement of epigenetic modifiers and microRNAs in nearly all cellular processes, it can be speculated that more mediators are yet to be identified. MicroRNAs (miRNAs) are small non-coding RNAs that generally function by negatively regulating protein-coding transcripts. Currently, over 2656 mature miRNAs have been identified (miRbase, version 22), many of which are misregulated in disease [32]. MiRNAs that become misexpressed, leading to the development of cancer, regarded as oncomiRs or tumor-suppressive miRNAs, have been extensively studied in cancer development, maintenance, resistance to therapeutics, and as therapeutic agents [33–36]. Indeed, the oncomiR, miR-21 is severely upregulated in erlotinib-resistant NSCLC cells, resulting in downregulation of PTEN and PDCD4, causing increased cell proliferation via activation of the AKT pro-growth pathway [37,38]. In contrast, the bona fide tumor-suppressive miRNA, miR-34 is recurrently downregulated in patients resistant to various EGFR-i, including erlotinib [39–41]. Indeed, restoring miR-34 to treat NSCLC is under active investigation, either alone [39,42,43] or in combination with erlotinib or other miRNAs that synergize with miR-34 to induce cell-cycle arrest and apoptosis [40,41,44,45] While both miR-21 and miR-34a are correlated with erlotinib resistance, and altering their intracellular concentration can sensitize resistant cells to erlotinib, whether or not they can drive the process of resistance has not yet been determined. Indeed, downregulation of miR-21 can re-sensitize NSCLC cells to one of the first-generation EGFR-i, gefitinib [37,38], and miR-147b is capable of driving resistance to a third-generation EGFR-i, osimertinib via altering a key metabolic pathway, the TCA cycle [46]. In the case of miRNAs that function as direct mediators of erlotinib resistance, miR-17–5p and miR-641 belong to bel this small class. Overexpression of miR-17–5p resensitizes NSCLC cells to erlotinib via targeting EZH1 [47], while increased expression of miR-641 mediates erlotinib resistance via downregulating NF1 [48]. Based on these individual evaluations, we hypothesized that other miRNAs can function as drivers of EGFR-i resistance via altering various cellular processes. To test our hypothesis, a miRNA library containing > 2000 human-encoded miRNAs was screened to identify miRNAs that can convert erlotinib-sensitive cells into resistant cells. Top candidates that drove resistance were validated in additional erlotinib sensitive cell lines and against human NSCLC data. The data presented support the involvement of miRNAs in EGFR-i resistance and may help identify i) tumors that are non-responsive to EGFR-i and ii) future miRNA antagonists that can be used to sensitize patients to EGFR-i.
2. Materials and methods
2.1. Cell culture
All cell lines used in the study were obtained from American Type Culture Collection (ATCC), cultured under standard conditions and were confirmed to be free of mycoplasma. Parental cell lines and cell lines generated during the study were authenticated by ATCC Cell Line Authentication and were maintained in RPMI media (Fisher Scientific, 27-016-021) supplemented with 10% FBS (Atlanta Biologicals, S12450) and 1% penicillin/streptomycin (Fisher, SV300-10). Cell lines generated during this study, EKVX-pmiR and H322M-pmiR, were continuously cultured in media containing 16 or 8 μg/mL G418 (Fisher, 10-131-027), respectively. During the screen, media was changed to phenol red free RPMI media (Life Technologies, 11835030) supplemented with 10% FBS and 1% penicillin/streptomycin.
2.2. Generation and characterization of cell lines
Erlotinib sensitive cell lines, EKVX and H322M were forward transfected with 2 μg of linearized pmiRGLO plasmid (Promega, E1330) using lipofectamine 2000 (Thermo Fisher Scientific, 11-668-019), as per manufacturer’s instructions. Forty-eight hours later, cells were selected using 100 μg/mL G418 and clones were isolated and tested for luciferase response. Briefly, ten-thousand cells for each single clone were plated into individual wells in a 96-well plate (Fisher, CLS3596) in replicates of six, and thirty-two hours post-plating, firefly and renilla activities were measured using the Dual-GLO Luciferase assay kit (Promega, E2920) following the manufacturer’s protocol. Renilla activity of EKVX-pmiR clone 2 and H322M-pmiR clone 1 (further regarded as EKVX-pmiR and H322M-pmiR, respectively) were evaluated for linearity with regard to cell number by plating increasing numbers of cells in individual wells of a 384-well plate (Corning, 3707) and assaying using the Dual-GLO Luciferase kit. Additionally, both cell lines were evaluated for siRNA-mediated targeting of LUC2, the gene encoding firefly, which was used to assess transfection efficiency such that cell growth between wells could be normalized. EKVX-pmiR cells or H322M-pmiR cells were seeded into individual wells of a 384-well plate in replicates of six and were reverse co-transfected with 0.6 nM silencing RNA targeting luciferase (siLUC2, Life Tech, Catalog # AM4629) or a negative control (sicont, Life Tech, Catalog # 4390846) and with 6 nM premiR-control as a miRNA negative control (Life Tech, Catalog # AM17111) using lipofectamine RNAiMAX (Thermo Fisher Scientific, 13-778-150), following the manufacturer’s protocol. Erlotinib dose response of the clones relative to parental cells was determined, described below in Erlotinib dose response.
2.3. Drug preparation for in vitro studies
Erlotinib (Selleck Chemicals, S7786), was dissolved in DMSO (Sigma Aldrich, D2650–100ML) to prepare 0.4 M stock solutions, which were aliquoted and stored at −80 °C. Working concentration of all the drugs was 200 μM prepared in complete medium and diluted as necessary for in vitro experiments.
2.4. Selection of controls for the overexpression screen
MiR-21 (mirVana miRNA mimic, Life Tech, Catalog # 4464066, Assay ID # MC10206) or miR-17 antagomir (Anti-miR miRNA Inhibitor, Life Tech, Catalog # AM12412, Assay ID # AM17000), which are reported mediators of erlotinib resistance were inconsistent at inducing erlotinib resistance in EKVX-pmiR cells between experiments; therefore, the following experiment was conducted to select appropriate positive controls for the screen. The human mirVana library of miRNAs (Invitrogen; based on miRBase v.21) contains 2019 miRNAs individually arrayed in 96-well plates. One of 23 plates that make up the library was randomly selected (Hs Mimic v19-A4–4) and the screening procedure described below was conducted in three biological replicates. Two miRNAs (miR-219b-3p and miR-4749–5p) that enhanced cell growth greater than four-times the standard deviation of the negative control in four replicates were selected as positive controls to be used in the screen and to monitor plate-to-plate variability.
2.5. MiRNA overexpression screen
EKVX-pmiR cells (2 × 103) were reverse co-transfected with 6 nM premiR-control (negative control), miR-219b-3p, miR-4749–5p (positive controls), or each of the individual miRNAs from the human mirVana library along with 0.6 nM siLUC2 in a 384-well plate using 0.1 μL lipofectamine RNAiMAX in a final volume of 10 μL media, in 6 replicates. Transfection with siLUC2 was used to determine the transfection efficiency which was used for normalizing cell growth between wells. Each plate included the positive and negative controls. Twenty-four hours post-transfection, media containing a final concentration of 75% growth inhibitory (GI75) concentration of erlotinib or equivalent DMSO (negative control) was added to the positive and negative control transfected wells to validate efficacy of the drug and the pro-growth effect of positive control miRNAs. Simultaneously, the same GI75 erlotinib containing media was added to the miRNA transfected wells. Seventy-two hours post-treatment, firefly and renilla activities were measured using the Dual-GLO Luciferase assay kit (E2920, Promega). Transfection efficiency for each well was calculated using the following equation:
Following which, renilla activity of transfected cells was calculated. The growth of erlotinib-treated miRNA-transfected cells is represented relative to that of negative control transfected cells. Results from the 23 individual mirVana plates were compiled, and miRNAs that enhanced cell growth greater than four-times the standard deviation of the positive controls were further evaluated.
2.6. Validation of candidates – overexpression and knockdown experiments
To evaluate the effect of miR-432–5p in mediating erlotinib resistance in sensitive NSCLC cells, 6 nM of the miR-432–5p mimic (mirVana miRNA mimic, Life Tech, Catalog# 4464066, Assay ID: MC10941) was reverse transfected using lipofectamine RNAiMAX. Erlotinib treatment and calculation of the fifty percent growth inhibitory concentration of erlotinib (GI50) values were conducted as described below.
2.7. Bioinformatic analysis
Ingenuity pathway analysis (IPA), v01–12 was performed using experimentally validated targets of the top 60 miRNAs, or for miR-432–5p. Lung cancer Illumina hiseq (miRNA seq) data was retrieved from TCGA (LUAD and LUSC) using R x64 v3.3.3. and plotted using GraphPad Prism version 8 software (GraphPad Software). MiRNA target prediction software, TargetScan Human [49] and miRmap [50] were used to identify miR-432–5p putative targets.
2.8. RNA isolation and quantitative real time PCR (qRT-PCR)
Seventy-two hours following transfection, total RNA was isolated from 1.5 ×105 cells cultured in 6-well plates or 1 ×106 cells grown in 10-cm plates (Fisher, FB092124) using the miRNeasy Kit (Qiagen, 217004) with DNase I (Qiagen, 79254) digestion. RNA integrity was evaluated on a 1.5% agarose gel (Sigma, A9539–500G), and total RNA quantified using a nanodrop. cDNA was then synthesized from 1 μg of total RNA isolated from cells using the miScript Reverse Transcriptase kit (Qiagen, 218161), as indicated by the manufacturer’s protocol. MiScript SYBR Green PCR Kit (Qiagen, 218075) was used as indicated by the manufacturer to quantify miR-432–5p (Qiagen, MS00031850), which was normalized to the housekeeping gene, RNU6B (control, Qiagen, MS00033740).
2.9. Erlotinib dose response
The protocol followed to evaluate erlotinib dose response of various NSCLC cell lines and additional cell lines generated in this study was as per the NCI-60 Cell Five-Dose Screen (NCI-60, DTP) [51]. Briefly, the sulforhodamine B (SRB, Sigma-Aldrich, S1402–5G) colorimetric assay was performed by exposing cells to varying concentrations of erlotinib or the highest equivalent volume of DMSO (negative control) containing media for 72 h [52]. Post data normalization, as described in figure legends, the fifty percent growth inhibitory concentration of erlotinib (GI50) was calculated from the respective dose curves (NCI-60, DTP) [51].
2.10. Statistical analysis
All data were analyzed using GraphPad Prism version 8 software (GraphPad Software) and are presented as mean values ± SD. Student’s t-test or one-way ANOVA statistical analyses were performed as specified in the figure legends. p-values of < 0.05 were considered significant.
3. Results
3.1. Generation of cell lines used to conduct the overexpression screen
Two cell lines, EKVX and H322M, reported to be sensitive to erlotinib by a study conducted by NCI-DTP [51] and validated in our laboratory, were used in this study. For the purpose of the screen the cells were engineered such that growth and transfection efficiency could easily be quantified. To accomplish this, EKVX and H322M cells were generated to stably express pmiRGLO (Promega), a plasmid that co-expresses both renilla and firefly luciferase. Renilla was used as a proxy for cell number, which was corrected for based on calculated transfection efficiency using an siRNA targeting firefly. Clonally derived cells were evaluated for firefly and renilla activities (Fig. 1A, B) and EKVX-pmiR clone 2 and H322M-pmiR clone 1, both with high levels of firefly and renilla were further characterized (referred to as EKVX-pmiR and H322M-pmiR, respectively). To confirm that renilla activity is a suitable proxy for cell number, pmiRGLO expressing cells were plated at increasing numbers and the resulting renilla activity was measured (Fig. 1C). Seeding densities between 1,000 and 8,000 cells were found to be within the linear range of renilla signal for both cell lines. EKVX-pmiR and H322M-pmiR cells were also evaluated for their response to erlotinib, which closely recapitulated that of the sensitive parental EKVX and H322M cells (Fig. 1D). EKVX-pmiR cells were used to perform the overexpression screen, whereas both EKVX-pmiR and H322M-pmiR cells were used to conduct validation studies for the candidate miRNAs.
Fig. 1.
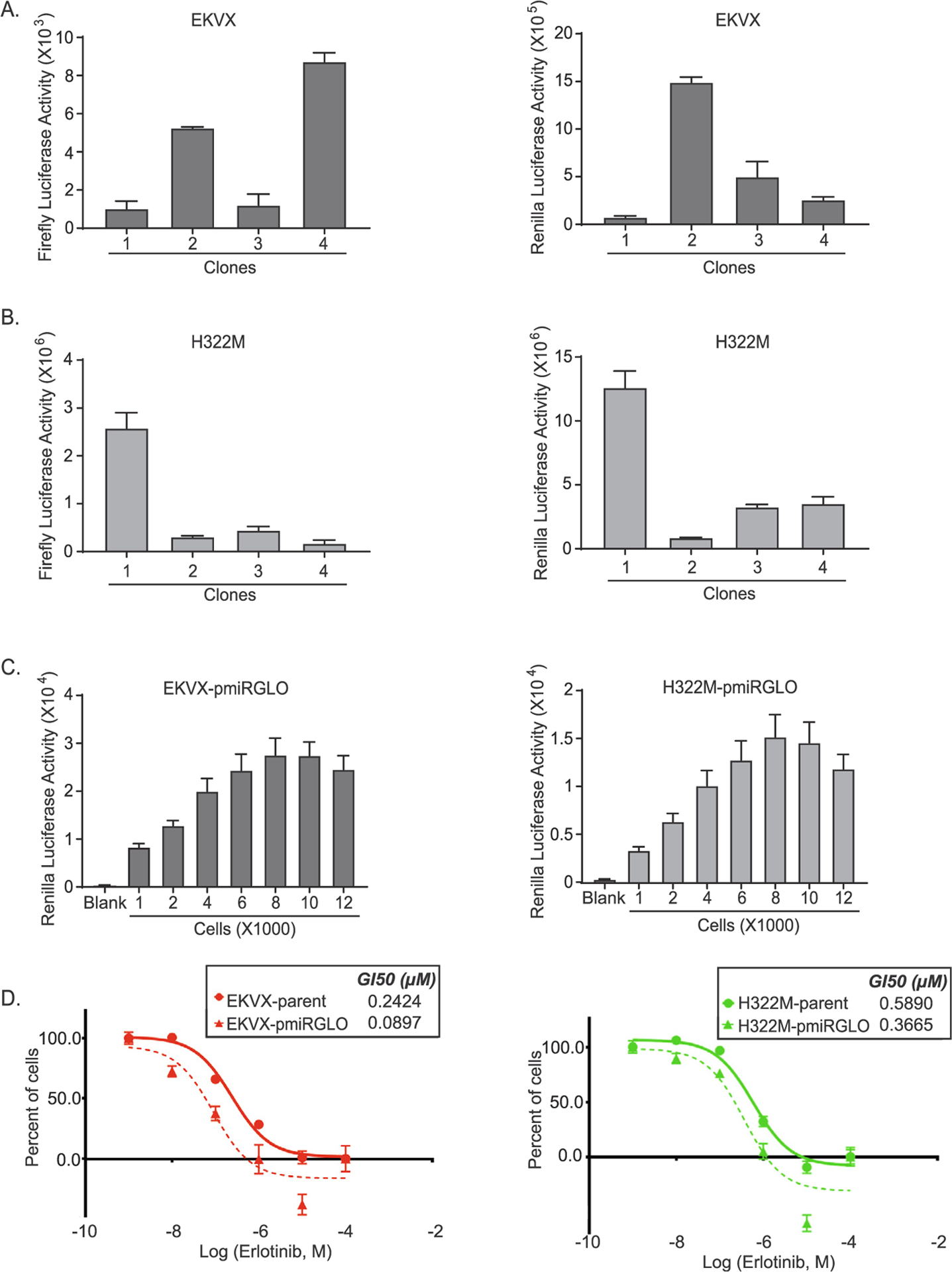
Clonal selection and characterization of EKVX and H322M cells stably expressing the pmiRGLO reporter. Clonally selected A) EKVX or B) H322M cells stably expressing pmiRGLO were evaluated for their firefly and renilla activities. C) Use of renilla activity as a proxy for cell number linearity for EKVX-pmiR clone 2 and H322M-pmiR clone 1 was determined eighteen hours post seeding. D) Erlotinib dose response via SRB assay was evaluated by exposing the parental cells or the clonally-derived cells to varying concentrations of erlotinib or the highest equivalent volume of dimethyl sulfoxide (DMSO, negative control) containing media for 72 h. For percent of cells calculation, the number of cells at the time of addition of erlotinib or DMSO (i.e. time zero or tz) was first corrected for, followed by normalization of cell number to respective corrected DMSO values. Fifty percent growth inhibitory concentration of erlotinib (GI50) was calculated from the respective dose curves (as per NCI-60 Cell Five-Dose Screen, NCI-60, DTP) [51].
Due to the nature of the screen it was critical to have a well-to-well normalizer for transfection efficiency so that miRNAs that truly altered growth in the presence of erlotinib could be identified. While renilla serves as a proxy for cell number (growth), the integrated firefly gene and transfection of an siRNA targeting the gene (siLUC2) were used to calculate the transfection efficiency. In this case, 6 nM of premiRcontrol (miRNA negative control) along with 0.6 nM (10% total transfection cocktail) of siLUC2 or a negative control siRNA (siCont) were co-transfected and firefly reporter activities were quantified. The average transfection efficiencies obtained for EKVX-pmiR cells and H233M-pmiR cells were 77.8% and 71.02%, respectively (Fig. 2A). Prior to moving forward with the screen, various ratios of premiRcontrol to siLUC2 were tested using either 0.05 or 0.1 μL of lipofectamine RNAiMAX to determine the ratio at which maximum efficiency was achieved. Based on this analysis, 6 nM premiRcontrol co-transfected with 0.6 nM siLUC2 using 0.1 μL lipofectamine RNAiMAX was determined to be the best ratio, and thus, was used to conduct the miRNA overexpression screen (Fig. 2B).
Fig. 2.
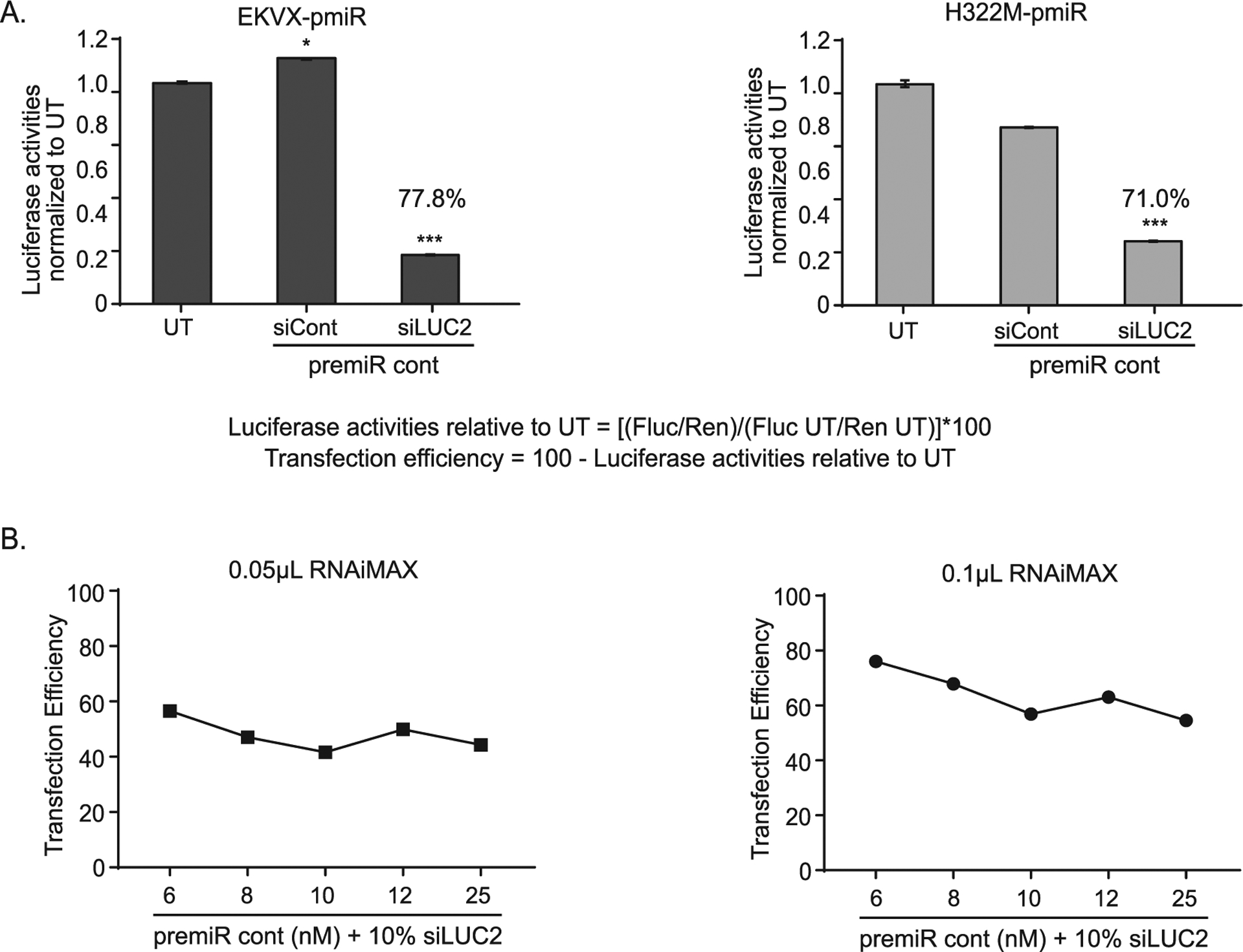
Determination of transfection efficiency of EKVX-pmiR and H322M-pmiR cells. A) EKVX-pmiR or H322M-pmiR cells were co-transfected with 6 nM premiRcontrol (premiRcont) and 0.6 nM (10%) siRNA control (sicont) or siRNA to firefly luciferase (siLUC2) using 0.1 μL of RNAiMAX. Thirty-two hours post-transfection, reporter activity was measured. UT = untransfected. Values of the siLUC2 + premiRcont bars indicate calculated transfection efficiencies as per the inset equation: Transfection efficiency of EKVX-pmiR cells relative to UT = 100 − (0.222*100) = 77.8%. Transfection efficiency of H322M-pmiR cells relative to UT = 100 − (0.290*100) = 71.0%. B) EKVX-pmiR cells were transfected with various ratios of premiRcont:siLUC2 using either 0.05 or 0.1 μL lipofectamine RNAiMAX. Raw data were evaluated using the equation in A and are represented as transfection efficiency.
3.2. Identification of positive controls
Overexpression of miR-21 is reported to cause acquired resistance to gefitinib in NSCLC and is correlated with resistance to erlotinib [37,38]. Conversely, loss of miR-17 is reported to mediate erlotinib resistance [47]. However, in this study, neither overexpressing miR-21 nor downregulating miR-17 resulted in reproducible erlotinib sensitization. Therefore, a pilot screen was conducted to identify at least two miRNAs that consistently enhanced growth of EKVX-pmiR cells in the presence of erlotinib. One plate was randomly selected from the library and the miRNAs in the plate were evaluated for their effect on promoting resistance to erlotinib. Two miRNAs, miR-219b-3p and miR-4749 generated a substantial and significant increase in cell growth in the presence of erlotinib when tested three independent times, and thus, were selected as positive control and controls to monitor plate-to-plate variability for the screen (Fig. 3A).
Fig. 3.
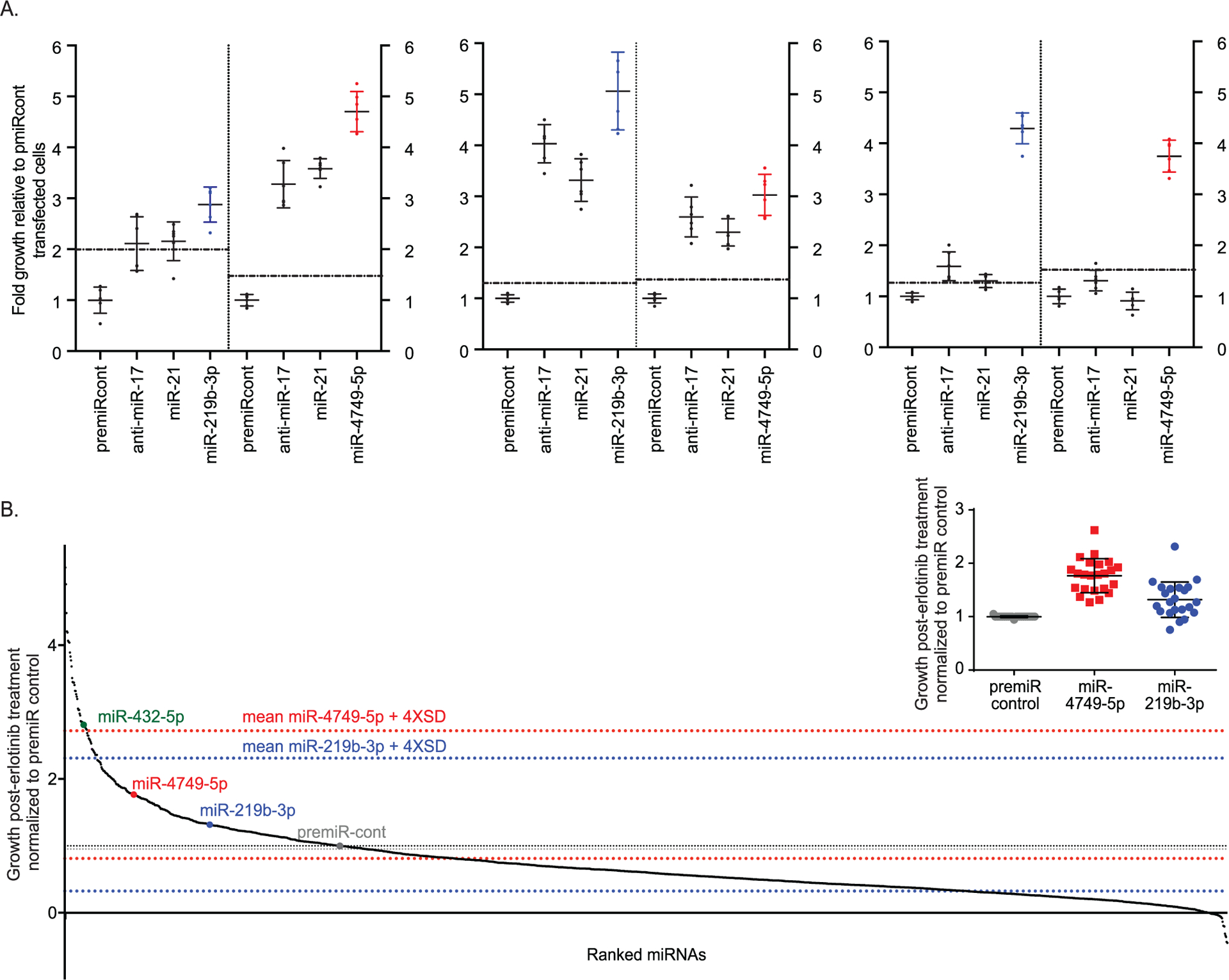
Overexpression of miRNAs alters the response of cells to erlotinib. A) Selection of positive controls for miRNA overexpression screen. EKVX-pmiR cells were transfected with 6 nM premiRcontrol, anti-miR-17, miR-21, or miRNAs from mirVana library (plate ID: Hs Mimic v19-A4–4). At the same time, all wells were transfected with 0.6 nM siLUC2 for normalization. Twenty-four hours post-transfection, cells were exposed to 75% growth inhibitory (GI75) concentration of erlotinib. Seventy-two hours post-treatment, reporter activities were measured. Following normalization for transfection, the growth of erlotinib treated, miRNA transfected cells was represented relative to that of negative control transfected cells. The means plus four-times the standard deviation of the negative control are depicted with a dotted line. Three individual replicates are shown. Right and left side of each graph include data obtained from two separate plates. Respective controls are included in each plate and the mean indicated. For clarity, only miR-219b (blue) and miR-4749–5p (red) from the library are shown. B) EKVX-pmiR cells were co-transfected with 6 nM premiRcont, miRNAs from the mirVana library, or positive controls, and 0.6 nM siLUC2. Twenty-four hours post-transfection, cells were exposed to the GI75 concentration of erlotinib. Seventy-two hours post-treatment, reporter activity was measured. Fold growth of transfected cells in the presence of erlotinib is represented relative to cells transfected with the negative control (premiRcont), in ranked order on the x-axis. Grey dotted lines represent the mean plus and minus four-times the standard deviation of the negative control, while red and blue dotted lines represent the mean plus and minus four-times the standard deviation for miR-4749 and miR-219b-3p, respectively. Inset graph depicts fold growth of cells transfected with premiRcontrol or positive controls (miR-4749 and miR-219b-3p) in the presence of erlotinib from all 23 plates used for the screen.
3.3. MiRNA overexpression screen results
To test our hypothesis that various miRNAs can function as mediators of erlotinib resistance, 2019 miRNAs were individually evaluated for their ability to stimulate cell growth in the presence of erlotinib. EKVX-pmiR cells were seeded in 384-well format and were transfected with 6 nM of each individual miRNA and 0.6 nM of siLUC2. Twenty-four hours later, the cells were exposed to the GI75 concentration of erlotinib (1.26 μM) for 72 h. MiRNAs that promoted resistance greater than four-times the standard deviation of the positive controls (Fig. 3B) are indicated in Table 1. These 60 miRNAs were reevaluated in the same cell line, EKVX-pmiR, for their ability to promote resistance, and were validated in a second erlotinib sensitive NSCLC reporter line, H322M-pmiR cells. Similar to the primary screen, reporter cells were co-transfected with the respective miRNA and 1/10th the concentration of siLUC2 followed by exposure to the GI75 concentration of erlotinib for 72 h. Renilla and firefly levels as well as fold changes with respect to the negative control were quantified. Data obtained from both cell lines included in the validation is represented in Table 1. Of the top seven miRNAs identified from each cell line, five were overlapping between the two, suggesting that these five miRNAs may be bona fide drivers of erlotinib resistance.
Table 1.
The top 60 miRNAs validated for growth response in the presence of erlotinib, in EKVX-pmiR and H322M-pmiR cells. Fold growth > 1 indicates miRNAs that can promote resistance, values < 1 indicates lack of driving resistance. MiRNAs in bold resulted in heightened resistance to erlotinib in both cell lines and constitute the top-five miRNAs selected for further validation.
| EKVX-pmiR cells | H322M-pmiR cells | ||
|---|---|---|---|
| Top 60 miRNAs | Fold growth of transfected cells | Top 60 miRNAs | Fold growth of transfected cells |
| hsa-miR-5693 | 4.806839 | hsa-miR-5693 | 6.980146 |
| hsa-miR-3618 | 4.128858 | hsa-miR-432-5p | 5.680975 |
| hsa-miR-3657 | 3.53778 | hsa-miR-3618 | 5.201033 |
| hsa-miR-432-5p | 3.447473 | hsa-miR-4787-5p | 3.098385 |
| hsa-miR-4435 | 3.369496 | hsa-miR-3198 | 2.666229 |
| hsa-miR-588 | 3.115648 | hsa-miR-4435 | 2.599998 |
| hsa-miR-4701-3p | 3.096875 | hsa-miR-588 | 2.331268 |
| hsa-miR-4787-5p | 2.550594 | hsa-miR-4693-5p | 2.305733 |
| hsa-miR-4521 | 2.406701 | hsa-miR-4797-5p | 2.231674 |
| hsa-miR-4499 | 2.176414 | hsa-miR-4775 | 2.208845 |
| hsa-miR-9-5p | 2.139334 | hsa-miR-4521 | 2.115477 |
| hsa-miR-4643 | 2.074379 | hsa-miR-4643 | 2.072277 |
| hsa-miR-4486 | 1.758585 | hsa-miR-4781-3p | 1.938728 |
| hsa-miR-548ac | 1.743734 | hsa-miR-4757-5p | 1.797722 |
| hsa-miR-4757-5p | 1.71694 | hsa-miR-4328 | 1.77053 |
| hsa-miR-3926 | 1.614334 | hsa-miR-4638-3p | 1.730049 |
| hsa-miR-4760-5p | 1.552469 | hsa-miR-4329 | 1.706815 |
| hsa-miR-4748 | 1.508298 | hsa-miR-4634 | 1.588003 |
| hsa-miR-4792 | 1.494049 | hsa-miR-4499 | 1.475899 |
| hsa-miR-3198 | 1.474083 | hsa-miR-4791 | 1.467733 |
| hsa-miR-5694 | 1.472514 | hsa-miR-4792 | 1.452575 |
| hsa-miR-4778-5p | 1.463761 | hsa-miR-3665 | 1.405999 |
| hsa-miR-4638-3p | 1.358205 | hsa-miR-4701-3p | 1.387164 |
| hsa-miR-3677-5p | 1.339528 | hsa-miR-4753-3p | 1.364724 |
| hsa-miR-4794 | 1.313135 | hsa-miR-4793-5p | 1.352131 |
| hsa-miR-4693-5p | 1.313076 | hsa-miR-4706 | 1.338108 |
| hsa-miR-4797-5p | 1.298536 | hsa-miR-4698 | 1.306059 |
| hsa-miR-4328 | 1.276663 | hsa-miR-4488 | 1.281768 |
| hsa-miR-4753-3p | 1.207611 | hsa-miR-4778-5p | 1.251342 |
| hsa-miR-4781-3p | 1.206249 | hsa-miR-4740-3p | 1.224541 |
| hsa-miR-4488 | 1.182468 | hsa-miR-4330 | 1.168158 |
| hsa-miR-4740-3p | 1.181966 | hsa-miR-4748 | 1.116061 |
| hsa-miR-548ab | 1.140096 | hsa-miR-3677-5p | 1.110286 |
| hsa-miR-4793-5p | 1.126522 | hsa-miR-4794 | 1.103088 |
| hsa-miR-4634 | 1.121035 | hsa-miR-4705 | 1.09914 |
| hsa-miR-4705 | 1.119791 | hsa-miR-4760-5p | 1.035345 |
| hsa-miR-4791 | 1.107927 | hsa-miR-4728-5p | 0.980355 |
| hsa-miR-4706 | 1.104596 | hsa-miR-548ak | 0.893768 |
| hsa-miR-4522 | 1.043842 | hsa-miR-3926 | 0.810352 |
| hsa-miR-4775 | 1.017418 | hsa-miR-5694 | 0.804238 |
| hsa-miR-548ak | 0.971604 | hsa-miR-9-5p | 0.732981 |
| hsa-miR-4468 | 0.943862 | hsa-miR-548ac | 0.313635 |
| hsa-miR-3665 | 0.934691 | hsa-miR-548ab | 0.232357 |
| hsa-miR-548aj-3p | 0.926283 | hsa-miR-4676-3p | 0.222049 |
| hsa-miR-4751 | 0.891378 | hsa-miR-4654 | 0.120642 |
| hsa-miR-4330 | 0.883232 | hsa-miR-4661-3p | 0.244 |
| hsa-miR-4654 | 0.851533 | hsa-miR-4741 | 0.065 |
| hsa-miR-4661-3p | 0.844637 | hsa-miR-4516 | −0.189 |
| hsa-miR-4329 | 0.841927 | hsa-miR-4751 | 0.040 |
| hsa-miR-4698 | 0.837794 | hsa-miR-204-3p | −0.128 |
| hsa-miR-4799-3p | 0.821699 | hsa-miR-4690-3p | −0.318 |
| hsa-miR-4728-5p | 0.798065 | hsa-miR-4486 | −0.225 |
| hsa-miR-4534 | 0.788234 | hsa-miR-548ad | −0.360 |
| hsa-miR-548ad | 0.7357 | hsa-miR-4468 | −0.590 |
| hsa-miR-4762-3p | 0.661747 | hsa-miR-4522 | 0.797 |
| hsa-miR-4516 | 0.544357 | hsa-miR-4762-3p | −0.248 |
| hsa-miR-4741 | 0.529365 | hsa-miR-3657 | −0.908 |
| hsa-miR-204-3p | 0.424363 | hsa-miR-548aj-3p | −0.128 |
| hsa-miR-4690-3p | 0.383958 | hsa-miR-4799-3p | −1.572 |
| hsa-miR-4676-3p | 0.331065 | hsa-miR-4534 | −1.743 |
In addition to miRNAs that drove resistance, there were miRNAs that clearly reduced growth of erlotinib-treated EKVX-pmiR cells. Approximately, 524 miRNAs were four standard deviations below the effect of the negative control. Indeed, transfection of multiple miRNAs that caused cell death in the presence of erlotinib (Fig. 3B, values below zero) include tumor suppressive miRNAs such as miR-642b-3p [53] (ranked 2017 out of 2019 tested miRNAs), miR-1304–3p [54] (2013/2019), miR-127 that targets BCL6 [55] (2012/2019), and others. Likewise, in a complementary study we conducted where genetic knockouts were screened for their ability to promote resistance, many of the miRNAs that antagonized resistance in this work were found to drive resistance when knocked out [31], suggesting that they may sensitize cells to erlotinib. Nonetheless, i) since the cell line used for this screen was already sensitive to erlotinib, ii) because validation studies were not conducted, and iii) because the effect of the miRNA in the absence of erlotinib was not determined (the tumor suppressive effect may be independent of synergizing with erlotinib), the additive or synergistic capacity of these miRNAs cannot be determined. Therefore, we chose to focus on the miRNAs that when overexpressed drove resistance in a sensitive cell line, a situation that likely occurs following the exposure of human tumors to erlotinib.
3.4. Bioinformatic and functional analysis of key pathways regulated by the top 60 miRNAs
Ingenuity pathway analysis (IPA), a software that enables analysis of interactions and pathways that can be mediated by genes and miRNAs, was performed for the 60 miRNAs identified from the initial screen as potentiators of erlotinib resistance. The IPA function used for this analysis was “experimentally validated mRNA targets of miRNAs.” Using this analysis, it was determined that multiple cancer related pathways are predicted to be regulated by the targets of the identified miRNAs. For the top networks regulated by genes that were experimentally validated to be targeted by these miRNAs, cell death and survival, cancer, and cell cycle were the highest scoring cell functions (Table 2 – top). The top toxic functions included cellular movement, cell death and survival, and cell growth and proliferation, with at least 21 molecules targeted in each of the top four functional group (Table 2 – middle). Of the pathways predicted to be perturbed by these miRNAs, the most significant was cancer drug resistance by drug efflux (Table 2 – bottom). At least 25 of 49 molecules involved in the cancer drug efflux pathway are predicted to be targeted by the miRNAs that were identified as mediators of erlotinib resistance. Of the 60 miRNAs identified as mediators of erlotinib resistance in EKVX cells, 38 are predicted to be involved in promoting cancer drug resistance by efflux pathway including four out of the top five validated miRNAs, indicated in bold in Table 3. Additional pathways involved in resistance to erlotinib, specifically the PTEN and PI3K/Akt pathways [23,56] are also altered by the top 60 miRNAs (Table 2 – bottom).
Table 2.
Bioinformatic analysis of key networks, functions, and pathways regulated by the top 60 miRNAs. Ingenuity Pathway Analysis (IPA) was used to analyze the pathways and cellular functions predicted to be altered by experimentally validated targets of the top 60 miRNAs. Score is defined as −log10(p-value).
| Top Networks | ||||||
|---|---|---|---|---|---|---|
| Associated Network Functions | Score | |||||
| Cell Death and Survival, Cancer, Cell Cycle | 39 | |||||
| Cell Signaling, Cardiovascular Disease, Dermatological Diseases and Conditions | 12 | |||||
| Cell Death and Survival, Cellular Development, Cellular Growth and Proliferation | 4 | |||||
| Top Tox Functions | ||||||
|---|---|---|---|---|---|---|
| Molecular and cellular functions | p-value | #Molecules | ||||
| Cellular Movement | 6.54E-05 – 9.58E-18 | 21 | ||||
| Cell Death and Survival | 6.95E-05 – 1.41E-17 | 21 | ||||
| Cellular Development | 6.23E-05 – 2.26E-16 | 22 | ||||
| Cellular Growth and Proliferation | 6.95E-05 – 2.26E-16 | 21 | ||||
| Cell Morphology | 6.95E-05 – 6.37E-15 | 17 | ||||
| Top Canonical Pathways | |||||
|---|---|---|---|---|---|
| Molecular and cellular functions | p-value | % Overlap, #Molecules | |||
| Cancer Drug Resistance by Drug Efflux | 4.59E-69 | 51.0%, 25/49 | |||
| PI3K/AKT Signaling | 8.68E-41 | 16.3%, 20/123 | |||
| Prostate Cancer Signaling | 2.54E-40 | 20.0%, 19/95 | |||
| Melanoma Signaling | 2.67E-39 | 30.9%, 17/55 | |||
| PTEN Signaling | 2.75E-38 | 16.0%. 19/199 | |||
Overlap percentage is generated from the number of genes targeted relative to the number of genes involved in the canonical pathways (indicated on the right).
Table 3.
IPA curated 38 of 60 miRNAs known to be involved in cancer drug resistance by efflux pathway. MiRNAs in bold represent four of the top five miRNAs experimentally validated to promote erlotinib resistance from Table 1.
| MiRNAs involved in cancer drug resistance by efflux pathway |
|---|
| hsa-miR-204-3p |
| hsa-miR-3198 |
| hsa-miR-3618 |
| hsa-miR-432-5p |
| hsa-miR-4328 |
| hsa-miR-4329 |
| hsa-miR-4435 |
| hsa-miR-4468 |
| hsa-miR-4488 |
| hsa-miR-4499 |
| hsa-miR-4516 |
| hsa-miR-4521 |
| hsa-miR-4522 |
| hsa-miR-4534 |
| hsa-miR-4638-3p |
| hsa-miR-4654 |
| hsa-miR-4661-3p |
| hsa-miR-4690-3p |
| hsa-miR-4693-5p |
| hsa-miR-4706 |
| hsa-miR-4728-5p |
| hsa-miR-4740-3p |
| hsa-miR-4741 |
| hsa-miR-4751 |
| hsa-miR-4753-3p |
| hsa-miR-4757-5p |
| hsa-miR-4778-5p |
| hsa-miR-4781-3p |
| hsa-miR-4787-5p |
| hsa-miR-4792 |
| hsa-miR-4793-5p |
| hsa-miR-4794 |
| hsa-miR-548ab |
| hsa-miR-548ac |
| hsa-miR-548aj-3p |
| hsa-miR-5693 |
| hsa-miR-5694 |
| hsa-miR-9-5p |
3.5. The top five candidate miRNAs promote resistance to erlotinib and other EGFR-i
The top five miRNAs that promoted erlotinib resistance in both EKVX and H322M cells (Table 1), were evaluated for their effects in the presence and absence of erlotinib. As previously observed, all of the miRNAs enhanced growth of EKVX-pmiR cells in the presence of erlotinib (Fig. 4A) and three of five enhanced growth in H322M-pmiR cells when cells were cultured in erlotinib containing media (Fig. 4B). Interestingly, miRNAs that validated in both cell lines, miR-5693, miR-3618, and miR-432–5p were identified by IPA as regulators of cancer drug resistance and efflux (Table 3). Apart from erlotinib, miRNAs can also drive resistance to additional EGFR-i including the other first-generation inhibitor gefitinib [38,46]. Therefore, the top five miRNAs were evaluated for their ability to enhance growth in the presence of other EGFR-i, namely gefitinib and the second-generation inhibitor afatinib. Three of five miRNAs, miR-5693, miR-3618, and miR-432–5p consistently and significantly enhanced cell growth in the presence of all three drugs regardless of the cell line used (Fig. 4A, B).
Fig. 4.
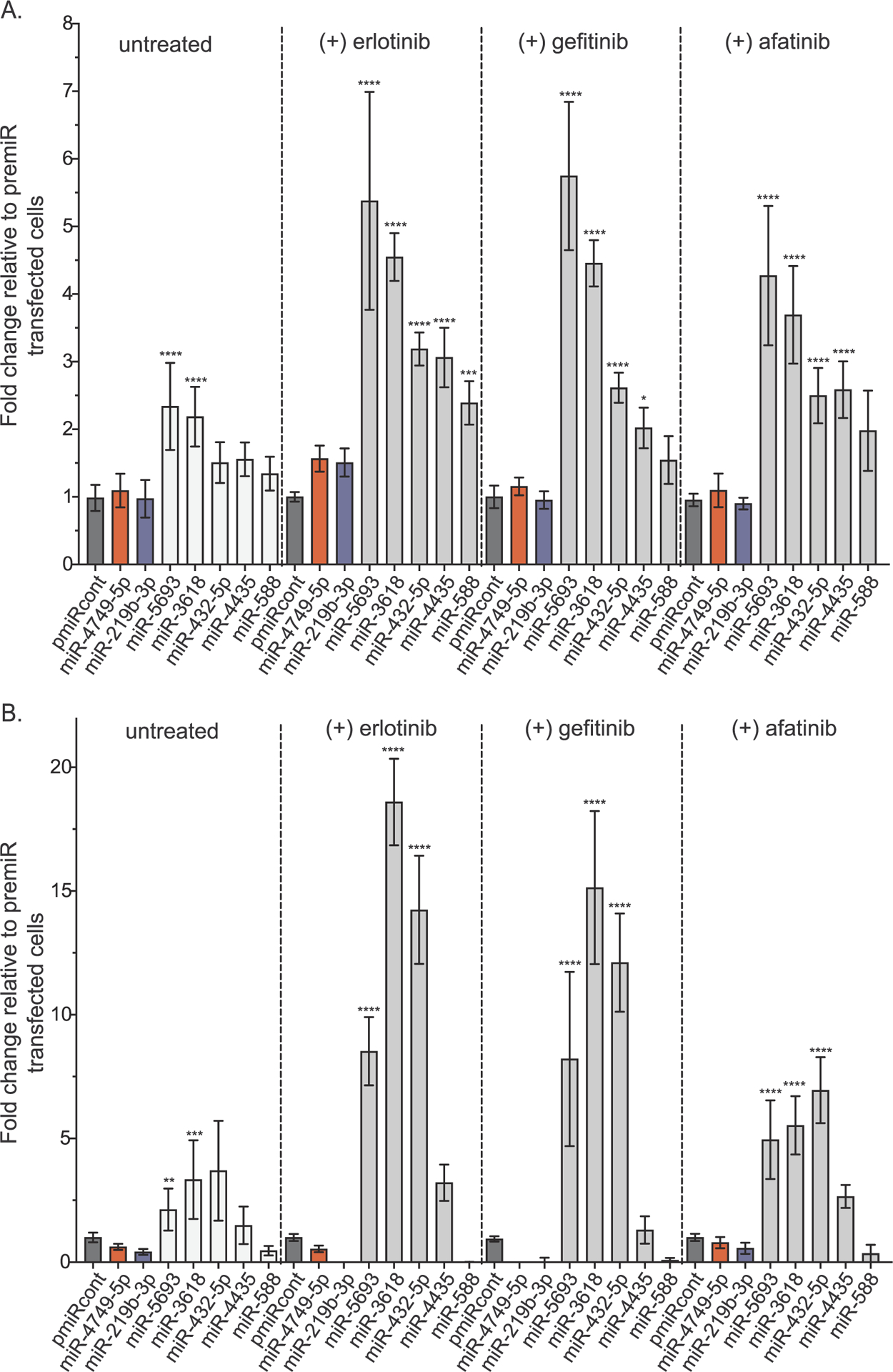
miRNAs promote resistance to erlotinib and other EGFR-is. A) EKVX-pmiR or B) H322M-pmiR cells were co-transfected with 6 nM premiRcontrol (indicated in dark grey) or 6 nM of the indicated miRNAs and 0.6 nM siLUC2. The positive controls, miR-4749 and miR-219 were also included, indicated in red and blue respectively. Twenty-four hours post-transfection, cells were exposed to either GI75 erlotinib, GI75 afatinib, or GI75 gefitinib, or equivalent amount of DMSO (negative control). Seventy-two hours post-treatment, reporter activity was measured. Fold change in growth of transfected cells in the presence or absence of each drug is represented relative to the respective negative control (premiRcont).
3.6. MiR-432 is elevated in NSCLC patient samples and mediates development of resistance in erlotinib sensitive NSCLC cells
For miRNAs with expression data available, expression was evaluated from The Cancer Genome Atlas (TCGA) from both lung adenocarcinoma and lung squamous cell carcinoma (LUAD and LUSC, respectively). MiRNAs with high annotation numbers, such as those in the 4000–5000 range, were unavailable. The heatmap in Fig. 5A shows that both miR-204–5p and miR-432–5p are highly expressed in LUAD and LUSC patient samples, while data in Fig. 5B highlights a cohort of tumors that have elevations in miR-432–5p relative to non-tumorigenic tissue. Although the initial screen identified miR-204 as a miRNA that promoted erlotinib resistance, it was not pursued for two reasons. Firstly, the miRNA that was identified in the screen was miR-204–3p, which is annotated as the passenger strand, while the mature strand, or miR-204–5p is included in the TCGA data. Secondly, the extensive validation studies we conducted did not support a role for miR-204–3p in promoting resistance. Mir-204–3p did not enhance growth in the presence of erlotinib in either of the erlotinib sensitive reporter lines. Hence, based on these findings, we further focused on the role of miR-432 as a mediator of erlotinib resistance in NSCLC.
Fig. 5.
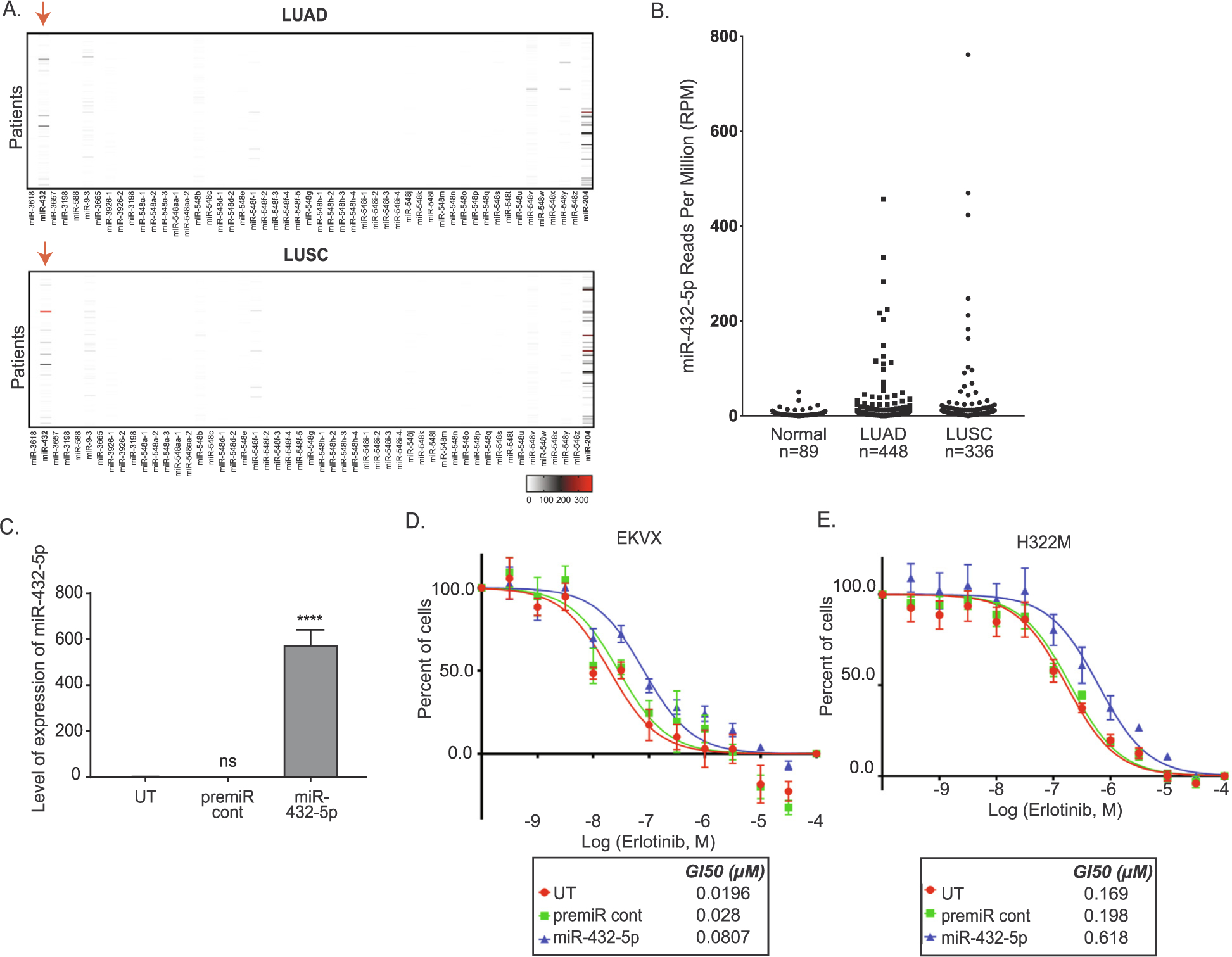
miR-432 is high in NCSLC tumors and promotes erlotinib resistance in NSCLC sensitive cell lines. A) Expression of top 60 miRNAs in patient samples. Patient miRNA sequenced data from LUAD and LUSC were retrieved from TCGA. The Reads Per Million (RPM) miRNA mapped were graphed using GraphPad prism version 8 software to visualize the reads for each miRNA in each patient. The red arrow represents miR-432–5p expression in LUAD and LUSC patients. The scale bars represent the read counts from undetected or zero (white), to moderately elevated (grey), or high (red). B) Quantification of miR-432–5p levels in EKVX-pmiR cells post-transfection of miR-432–5p. One-way ANOVA analysis was used to calculate statistical significance. C) EKVX-pmiR cells or D) H322M-pmiR cells were untransfected (UT) or were reverse transfected with 6 nM premiRcont or miR-432–5p. Cells were exposed to varying concentrations of erlotinib or the highest equivalent volume of DMSO (negative control) containing media for 72 h. Erlotinib dose response was conducted using the SRB assay. For percent of cells calculation, the absorbance obtained from the cells at the time of addition of erlotinib or DMSO (i.e. time zero or tz) was first corrected for, followed by normalization of absorbance to the respective corrected DMSO values. GI50 erlotinib was calculated from the respective dose curves (as per the NCI-60 Cell Five-Dose Screen, NCI-60, DTP) [51].
MiR-432 was transfected into EKVX-pmiR cells resulting in a strong induction of miR-432–5p (Fig. 5C), and cell proliferation in the presence of increasing concentrations of erlotinib was analyzed using a secondary assay, sulforhodamine B (SRB, Fig. 5D). This assay confirmed that miR-432 was not altering the luciferase reporter activity in the original screen through off-targeting, but that the reduction in renilla activity was a reflection of miR-432 driving erlotinib resistance. The growth promoting effect of miR-432 overexpression was also evaluated by SRB assay in H322M-pmiR cells (Fig. 5D). In both cases overexpressing miR-432 promoted a significant change in erlotinib response increasing the GI50 erlotinib concentration by ~2–3 fold.
4. Discussion
Currently, there are over 2,656 mature human miRNAs annotated in miRbase (version 22) [32]. However, functions for over half of these miRNAs remain unknown [32]. From the work reported herein, it can be inferred that several of these miRNAs have a role in altering specific phenotypes in cancer, such as resistance to therapeutics. Indeed, the results obtained from the overexpression screen are significantly supported by bioinformatic evaluation, where targets for many of the miRNAs that promote resistance belong to cancer drug resistance and efflux pathways (Table 3).
Three miRNAs identified and validated in this work, miR-5693, miR-3618, and miR-432–5p were reported to promote resistance to all EGFR-i tested, including erlotinib, gefitinib, and afatinib (Fig. 4). These same three miRNAs are predicted to function in the drug efflux pathway. Indeed, the ATP Binding Cassette (ABC) transporters that contribute to resistance via pumping drugs out of cancer cells have been shown to efflux both gefitinib and erlotinib [57,58]. If these miRNAs are sensitizing cells to erlotinib through altering the drug efflux pathwy remains to be evaluated, but is an active area of on-going reserch.
Of the three miRNAs that promoted resistance to all EGFR-i, miR-432–5p was the only miRNA that was determined to be upregulated in a cohort of NSCLC tumors (Fig. 5A, B), albeit data for miR-5693 was not available from the TCGA data. Mechanistically, the targets of miR-432–5p involved in this process have yet to be determined. However, in addition to targets in the drug efflux pathway, several metabolic pathways were also predicted to be regulated by miR-432–5p. Therefore, it is possible that miR-432–5p may mediate erlotinib resistance by triggering metabolic reprogramming in cancer cells [59]. Interestingly, a few of the canonical pathways that are significantly associated with targets of miR-432–5p are involved in the development of resistance to erlotinib [60] or other EGFR-i [46]. For example, miR-432–5p targets a component of the branched-chain amino acid (BCAA) pathway that when upregulated promotes erlotinib resistance in NSCLC [60].
Collectively this work identified, in an unbiased fashion, miRNAs that antagonize the efficacy of erlotinib – converting an otherwise sensitive cell line into a resistant state. Whether these miRNAs are involved in mediating resistance in human patients has yet to be discovered, datasets with matched tumor tissue pre- and post-treatment would be required to conduct such an analysis and are currently scarce; however, these samples are actively being procured. Future analysis of these samples to determine if these miRNAs are truly upregulated following acquired resistance to EGFR-i would be useful for generating combinatorial therapeutic agents that could ultimately be used to reduce resistance.
Acknowledgements
Funding for this work was provided by NIH R01CA205420 (ALK). ASP was funded by a Purdue Research Foundation (PRF) Research Grant award by the Department of Biological Sciences, Purdue University, a SIRG grant administered through the Purdue Center for Cancer Research, Purdue University, a Cancer Prevention Internship Program Graduate Research Assistantship funded by Purdue University, and a Bilsland Dissertation Fellowship awarded by the Department of Biological Sciences, Purdue University.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].2020. Lung Cancer Statistics | How Common is Lung Cancer. https://www.cancer.org/cancer/lung-cancer/about/key-statistics.html.
- [2].Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, et al. , Highly parallel identification of essential genes in cancer cells, Proc. Natl. Acad. Sci. U.S.A 105 (2008) 20380–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hirsch FR, Varella-Garcia M, Bunn PA Jr., Di Maria MV, Veve R, Bremmes RM, et al. , Epidermal growth factor receptor in non-small-cell lung carcinomas: correlation between gene copy number and protein expression and impact on prognosis, J. Clin. Oncol 21 (2003) 3798–3807. [DOI] [PubMed] [Google Scholar]
- [4].Onitsuka T, Uramoto H, Ono K, Takenoyama M, Hanagiri T, Oyama T, et al. , Comprehensive molecular analyses of lung adenocarcinoma with regard to the epidermal growth factor receptor, K-ras, MET, and hepatocyte growth factor status, J. Thorac. Oncol 5 (2010) 591–596. [DOI] [PubMed] [Google Scholar]
- [5].Howe LR, Leevers SJ, Gomez N, Nakielny S, Cohen P, Marshall CJ, Activation of the MAP kinase pathway by the protein kinase raf, Cell 71 (1992) 335–342. [DOI] [PubMed] [Google Scholar]
- [6].Riely GJ, Marks J, Pao W, KRAS mutations in non-small cell lung cancer, Proc. Am. Thorac. Soc 6 (2009) 201–205. [DOI] [PubMed] [Google Scholar]
- [7].Lee JW, Soung YH, Kim SY, Nam SW, Park WS, Wang YP, et al. , ERBB2 kinase domain mutation in the lung squamous cell carcinoma, Cancer Lett. 237 (2006) 89–94. [DOI] [PubMed] [Google Scholar]
- [8].Schabath MB, Cote ML, Cancer Progress and priorities: lung cancer, Cancer Epidemiol. Biomarkers Prev. 28 (2019) 1563–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].2020. Non-Small Cell Lung Carcinoma - My Cancer Genome. https://www.mycancergenome.org/content/disease/non-small-cell-lung-carcinoma/.
- [10].2014. Lung Cancer 101. https://lungevity.org/for-patients-caregivers/lung-cancer-101.
- [11].Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. , EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy, Science 304 (2004) 1497–1500. [DOI] [PubMed] [Google Scholar]
- [12].Sharma SV, Bell DW, Settleman J, Haber DA, Epidermal growth factor receptor mutations in lung cancer, Nat. Rev. Cancer 7 (2007) 169–181. [DOI] [PubMed] [Google Scholar]
- [13].Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. , Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib, N. Engl. J. Med 350 (2004) 2129–2139. [DOI] [PubMed] [Google Scholar]
- [14].Osarogiagbon RU, Cappuzzo F, Ciuleanu T, Leon L, Klughammer B, Erlotinib therapy after initial platinum doublet therapy in patients with EGFR wild type non-small cell lung cancer: results of a combined patient-level analysis of the NCIC CTG BR.21 and SATURN trials. Transl Lung, Cancer Res. 4 (2015) 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hirai F, Edagawa M, Shimamatsu S, Toyozawa R, Toyokawa G, Nosaki K, et al. , Evaluation of erlotinib for the treatment of patients with non-small cell lung cancer with epidermal growth factor receptor wild type, Oncol. Lett 14 (2017) 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Inno A, Di Noia V, Martini M, D’Argento E, Di Salvatore M, Arena V, et al. , Erlotinib for patients with EGFR wild-type metastatic NSCLC: a retrospective bio-markers analysis, Pathol. Oncol. Res 25 (2019) 513–520. [DOI] [PubMed] [Google Scholar]
- [17].Perol M, Chouaid C, Perol D, Barlesi F, Gervais R, Westeel V, et al. , Randomized, phase III study of gemcitabine or erlotinib maintenance therapy versus observation, with predefined second-line treatment, after cisplatin-gemcitabine induction chemotherapy in advanced non-small-cell lung cancer, J. Clin. Oncol 30 (2012) 3516–3524. [DOI] [PubMed] [Google Scholar]
- [18].Bonomi PD, Buckingham L, Coon J, Selecting patients for treatment with epidermal growth factor tyrosine kinase inhibitors, Clin. Cancer Res 13 (2007) s4606–s4612. [DOI] [PubMed] [Google Scholar]
- [19].Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. , Erlotinib in previously treated non-small-cell lung cancer, N. Engl. J. Med 353 (2005) 123–132. [DOI] [PubMed] [Google Scholar]
- [20].Demuth C, Madsen AT, Weber B, Wu L, Meldgaard P, Sorensen BS, The T790M resistance mutation in EGFR is only found in cfDNA from erlotinib-treated NSCLC patients that harbored an activating EGFR mutation before treatment, BMC Cancer 18 (2018) 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tetsu O, Hangauer MJ, Phuchareon J, Eisele DW, McCormick F, Drug Resistance to EGFR Inhibitors in Lung Cancer, Chemotherapy 61 (2016) 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. , The T790M mutation in EGFR kinase causes drug resistance by increasing the af-finity for ATP, Proc. Natl. Acad. Sci. U.S.A 105 (2008) 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. , RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer, Nat. Commun 6 (2015) 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. , Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors, Sci. Transl. Med 3 (2011) 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shien K, Toyooka S, Yamamoto H, Soh J, Jida M, Thu KL, et al. , Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells, Cancer Res. 73 (2013) 3051–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jakobsen KR, Demuth C, Madsen AT, Hussmann D, Vad-Nielsen J, Nielsen AL, et al. , MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells, Oncogenesis 6 (2017) e307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Camidge DR, Pao W, Sequist LV, Acquired resistance to TKIs in solid tumours: learning from lung cancer, Nat. Rev. Clin. Oncol 11 (2014) 473–481. [DOI] [PubMed] [Google Scholar]
- [28].Joshi M, Rizvi SM, Belani CP, Afatinib for the treatment of metastatic non-small cell lung cancer, Cancer Manage. Res 7 (2015) 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu SG, Liu YN, Tsai MF, Chang YL, Yu CJ, Yang PC, et al. , The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients, Oncotarget 7 (2016) 12404–12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Liao BC, Griesing S, Yang JC, Second-line treatment of EGFR T790M-negative non-small cell lung cancer patients, Ther. Adv. Med. Oncol 11 (2019) 1758835919890286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pal AS, Agredo AM, Lanman NA, Clingerman J, Gates K, Kasinski AL. Loss of SUV420H2 promotes EGFR inhibitor resistance in NSCLC through upregulation of MET via LINC01510. 2020. [DOI] [PMC free article] [PubMed]
- [32].Kozomara A, School of Biological Sciences FoB, Medicine and Health, University of Manchester, Manchester M13 9PT, UK, Birgaoanu M, School of Biological Sciences FoB, Medicine and Health, University of Manchester, Manchester M13 9PT, UK, Griffiths-Jones S, School of Biological Sciences FoB, Medicine and Health, University of Manchester, Manchester M13 9PT, UK. miRBase: from microRNA sequences to function. Nucleic Acids Research 2020;47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kasinski AL, Slack FJ, MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy, Nat. Rev. Cancer 11 (2011) 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Orellana EA, Kasinski AL, MicroRNAs in cancer: a historical perspective on the path from discovery to therapy, Cancers (Basel) 7 (2015) 1388–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pal AS, Kasinski AL, Animal models to study microRNA function, Adv. Cancer Res 135 (2017) 53–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bing Li SR, Li Xuefei, Wanga Yongsheng, Garfield David, Zhoua Songwen, Chena Xiaoxia, Sua Chunxia, Chena Mo, Kuanga Peng, Gaoa Guanghui, Hea Yayi, Fana Lihong, Feia Ke, Zhoua Caicun, Schmit-Bindertd Gerald. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. 2014;83:146–53. [DOI] [PubMed] [Google Scholar]
- [37].Shen H, Zhu F, Liu J, Xu T, Pei D, Wang R, et al. , Alteration in Mir-21/PTEN expression modulates gefitinib resistance in non-small cell lung cancer, PLoS ONE 9 (2014) e103305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou JY, Chen X, Zhao J, Bao Z, Zhang P, Liu ZF, MicroRNA-34a overcomes HGF-mediated gefitinib resistance in EGFR mutant lung cancer cells partly by targeting MET, Cancer Lett. 351 (2014) 265–271. [DOI] [PubMed] [Google Scholar]
- [39].Stahlhut C, Slack FJ, Combinatorial action of MicroRNAs let-7 and miR-34 effectively synergizes with erlotinib to suppress non-small cell lung cancer cell proliferation, Cell Cycle 14 (13) (2015) 2171–2180, 10.1080/15384101.2014.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhao J, Kelnar K, Bader AG, In-depth analysis shows synergy between erlotinib and miR-34a, PLoS ONE 9 (2014) e89105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kasinski AL, Slack FJ, miRNA-34 prevents cancer initiation and progression in a therapeutically-resistant K-ras and p53-induced mouse model of lung adenocarcinoma, Cancer Res. 72 (2012) 5576–5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Orellana EA, Tenneti S, Rangasamy L, Lyle LT, Low PS, Kasinski AL, FolamiRs: ligand-targeted, vehicle-free delivery of microRNAs for the treatment of cancer, Sci. Transl. Med 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kasinski AL, Kelnar K, Stahlhut C, Orellana E, Zhao J, Shimer E, et al. , A combinatorial microRNA therapeutics approach to suppressing non-small cell lung cancer, Oncogene 34 (2015) 3547–3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Orellana EA, Li C, Lisevick A, Kasinski AL, Identification and validation of microRNAs that synergize with miR-34a – a basis for combinatorial microRNA therapeutics, Cell Cycle 18 (2019) 1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhang WC, Wells JM, Chow KH, Huang H, Yuan M, Saxena T, et al. , miR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma, Nat. Metab 1 (2019) 460–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang W, Lin J, Wang P, Sun J, miR-17–5p down-regulation contributes to erlotinib resistance in non-small cell lung cancer cells, J. Drug Target 25 (2017) 125–131. [DOI] [PubMed] [Google Scholar]
- [47].Chen J, Cui JD, Guo XT, Cao X, Li Q, Increased expression of miR-641 contributes to erlotinib resistance in non-small-cell lung cancer cells by targeting NF1, Cancer Med. 7 (2018) 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].2015. TargetScanHuman 5.2 Custom. http://www.targetscan.org/vert_50/seed-match.html.
- [49].mirmap. 2020. miRmap web. https://mirmap.ezlab.org/app/.
- [50].NCI-60 D. 2020. NCI-60 Human Tumor Cell Lines Screen | Discovery & Development Services | Developmental Therapeutics Program (DTP). https://dtp.cancer.gov/discovery_development/nci-60/. [Google Scholar]
- [51].Orellana EA, Kasinski AL, Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation, Biol. Protoc 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang Y, Luo J, Wang X, Wang HL, Zhang XL, Gan TQ, et al. , A comprehensive analysis of the predicted targets of miR-642b-3p associated with the long non-coding RNA HOXA11-AS in NSCLC cells, Oncol. Lett 15 (2018) 6147–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li C, Pu M, Li C, Gao M, Liu M, Yu C, et al. , MicroRNA-1304 suppresses human non-small cell lung cancer cell growth in vitro by targeting heme oxygenase-1, Acta Pharmacol. Sin 38 (2017) 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].C J, W M, G M, X Y, C YS. miR-127 Regulates Cell Proliferation and Senescence by Targeting BCL6. PloS one 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. 2011. [DOI] [PMC free article] [PubMed]
- [56].M S, dV NA, B T, B MJ, vE MA, B JH, et al. Effect of the ATP-binding Cassette Drug Transporters ABCB1, ABCG2, and ABCC2 on Erlotinib Hydrochloride (Tarceva) Disposition in in Vitro and in Vivo Pharmacokinetic Studies Employing Bcrp1−/−/Mdr1a/1b−/− (Triple-Knockout) and Wild-Type Mice. Molecular cancer therapeutics 2008;7. [DOI] [PubMed] [Google Scholar]
- [57].Lemos C, Jansen G, Peters GJ, Drug transporters: recent advances concerning BCRP and tyrosine kinase inhibitors, Br. J. Cancer 98 (2008) 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Beloueche-Babari M, Box C, Arunan V, Parkes HG, Valenti M, De Haven Brandon A, et al. , Acquired resistance to EGFR tyrosine kinase inhibitors alters the metabolism of human head and neck squamous carcinoma cells and xenograft tumours, Br. J. Cancer (2015) 1206–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang Y, Zhang J, Ren S, Sun D, Huang HY, Wang H, et al. , Branched-chain amino acid metabolic reprogramming orchestrates drug resistance to EGFR tyrosine kinase inhibitors, Cell Rep. 28 (2019) 512–525.e6. [DOI] [PubMed] [Google Scholar]
- [60].de Bruin EC, Cowell C, Warne PH, Jiang M, Saunders RE, Melnick MA, et al. , Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer, Cancer Discov. 4 (2014) 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]