

Abstract
The function of a T cell depends on its subtype and activation state. Here, we show that the imaging of autofluorescence-lifetime signals from quiescent and activated T cells can be used to classify the cells. T cells isolated from human peripheral blood and activated in culture via a tetrameric antibody against the surface ligands CD2, CD3 and CD28 showed specific activation-state-dependent patterns of autofluorescence lifetime. Logistic-regression models and random-forest models classified T cells according to activation state with 97–99% accuracy, and according to activation state (quiescent or activated) and subtype (CD3+ CD8+ or CD3+ CD4+) with 97% accuracy. Autofluorescence-lifetime imaging could be used to non-destructively determine T-cell function.
T cells are an important component of the adaptive immune response and have diverse cytotoxic and immune-modulating, or “helper” activities, upon activation. The two main T cell subtypes are CD3+CD8+ T cells that engage in cell-mediated cytotoxicity and release toxic cytokines and CD3+CD4+ T cells that can be further divided into additional subtypes with differing pro- and anti- inflammatory functions due to chemokine and cytokine production[1, 2]. T cells are a promising target for immunotherapies because of these diverse functions. Immunotherapies that directly increase T cell cytotoxic activity, such as immune checkpoint blockade therapies and adoptive cell transfer therapies, are currently used clinically for cancer treatment and are in development for additional diseases including human immunodeficiency virus (HIV)[3, 4]. Immunotherapies that enhance regulatory T cell (TREG) behaviours are in development to treat transplant rejection and autoimmune diseases, including diabetes and Crohn’s disease [5, 6].
New tools that are non-destructive and label-free are needed to fully characterize T cells for assessment of immunotherapies. Currently, T cell subtype and function is determined from expression of surface proteins (e.g. CD3, CD4, CD8, CD45RA, etc.) and cytokine production (e.g. interferon-gamma (IFN-γ), transforming growth factor beta (TGF-β), interleukin (IL)-2, IL-4, IL-17, etc.) by antibody-based methods such as flow cytometry, immunohistochemistry, or immunofluorescence, or by transgenic fluorophore expression. However, these methods require exogenous contrast agents, and flow cytometry and immunohistochemistry require tissue dissociation and fixation, respectively. Autofluorescence imaging is an attractive method to probe immune cell behaviours because it is non-destructive, relies on endogenous contrast, and provides high spatial and temporal resolution. Therefore, autofluorescence imaging overcomes the single-use limitation of label-based methods, is not influenced by confounding label-related factors such as concentration and label-dependent alterations of the sample, and allows kinetic measurements on living T cells. Autofluorescence imaging can also provide functional information on limited blood volumes (e.g. neonates) that are inadequate for conventional assessments and assist with quality control on the same cells injected into patients for cellular immunotherapies.
Fluorescence imaging of the endogenous metabolic co-enzymes reduced nicotinamide adenine dinucleotide (NAD(P)H) and flavin adenine dinucleotide (FAD) provides quantitative endpoints of cellular metabolism [7–9]. (NADH and NADPH florescence are indistinguishable; therefore, NAD(P)H is used to represent the combined fluorescence signal [10].) The optical redox ratio is the relative fluorescence intensities of NAD(P)H and FAD and provides an optical measurement of the redox state of the cell [7, 11, 12]. Although there are multiple definitions of the optical redox ratio with either NAD(P)H or FAD in the numerator used in the literature, NAD(P)H/(NAD(P)H+FAD) is used here because this metric is standardized between 0 and 1 and an increase corresponds to T cell activation.
The fluorescence lifetime, the time the fluorophore is in the excited state before returning to ground state, provides information on the protein binding of NAD(P)H and FAD [8, 13]. NAD(P)H and FAD can both exist in two conformations: a quenched and unquenched form, with a short and long lifetime, respectively. NAD(P)H has a short lifetime in the free state and a long lifetime in its protein-bound state [8]. Conversely, FAD has a short lifetime when bound to an enzyme and a long lifetime when free [13]. Fluorescence lifetime imaging (FLIM) allows quantification of the short (τ1) and long (τ2) lifetime values, the fraction of free and protein-bound co-enzyme (α1 and α2, respectively, for NAD(P)H, and α2 and α1, respectively, for FAD), and the mean lifetime (the weighted average of the short and long lifetimes, τm = α1*τ1+ α2*τ2). The fluorescence intensity and lifetime of NAD(P)H and FAD are sensitive to metabolic differences between neoplasias and malignant tissues, anti-cancer drug effects in cancer cells, and differentiating stem cells [14–19]. Autofluorescence imaging has been used previously to identify macrophages in vivo and detect metabolic changes due to macrophage polarization [20–22]. Altogether, fluorescence lifetime imaging of NAD(P)H and FAD provide quantitative and functional endpoints of cellular metabolism.
Upon activation, T cells have increased metabolic demands to support cell growth, proliferation, and differentiation [23]. This metabolic state of increased aerobic glycolysis is required for T cells to maintain effector function [23–25]. Therefore, this study tests the hypothesis that fluorescence lifetime imaging of NAD(P)H and FAD provides quantitative endpoints to identify activated T cells. To test this hypothesis, we isolated T cells from the blood of healthy donors, activated the cells in an antigen-independent manner with a tetrameric antibody (anti-CD2/CD3/CD28) and imaged the NAD(P)H and FAD fluorescence intensity and lifetime of quiescent and activated T cells. This study demonstrates autofluorescence lifetime differences between quiescent and activated T cells and accurate classification of T cell activation state by machine learning models using quantitative endpoints from autofluorescence lifetime images.
Results
Autofluorescence imaging reveals differences with activation in T cells
Blood isolations for CD3+ (pan-T cell marker) and CD3+CD8+ cells were used to study all T cells, as might be utilized in adoptive cell transfer therapies, and the cytotoxic CD3+CD8+ sub-population, respectively. NAD(P)H and FAD autofluorescence imaging reveals metabolic differences in quiescent and activated T cells (Fig. 1, S1). In the autofluorescence images, the nucleus remains dark as NAD(P)H is primarily located in the cytosol and mitochondria, and FAD is primarily in the mitochondria. Immunofluorescence labelling of CD4, CD8, or CD69 surface proteins verified cell type and activation (Fig. S2). There were significant differences in cell size, optical redox ratio, NAD(P)H τm, NAD(P)H α1, and FAD α1 between quiescent and activated T cells of 6 donors (p<0.001, Fig. 1B–F). Significant changes (p<0.001) in FAD τm between quiescent and activated T cells were found only for T cells within the bulk CD3+ T cell population (Fig. 1E). The differences in autofluorescence endpoints were consistent across the 6 donors (Fig. 1, S1), at 24 and 48 hr of exposure to the activating antibodies (Fig. S3), and between experiments from two different blood draws (183 days apart) from the same donor (Fig. S4). A slight increase in FAD τ1 was found in both quiescent and activated CD3+ T cells, suggesting a slight change in the microenvironment of bound FAD between CD3+ T cells of the same donor from two blood draws; however, no other autofluorescence endpoints were significantly different between the two blood draws.
Fig. 1 |. NAD(P)H and FAD autofluorescence imaging reveals metabolic differences between quiescent and activated T cells.
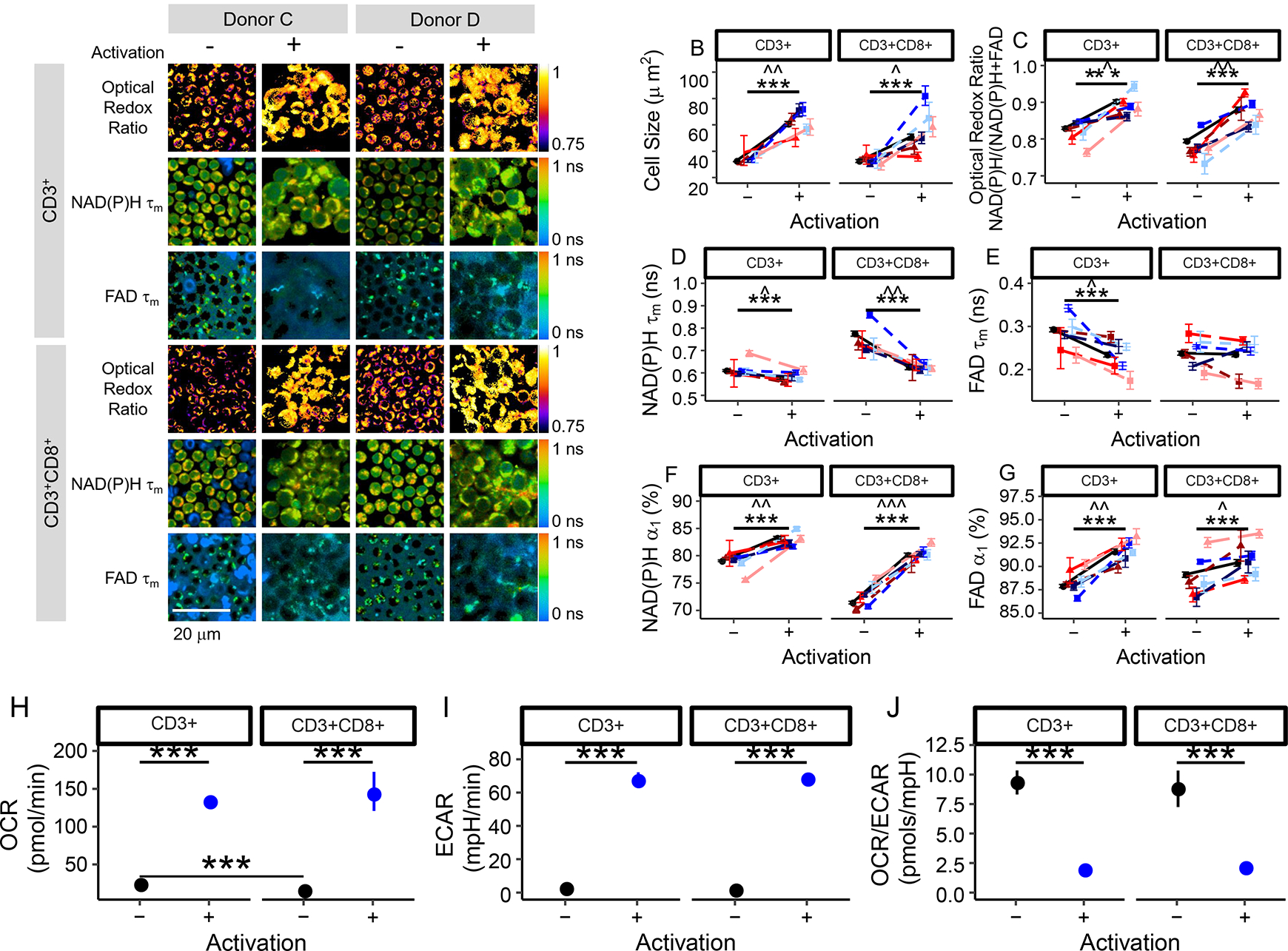
(a) Representative optical redox ratio, NAD(P)H τm, and FAD τm images (4 images selected out of 202 images acquired from 6 different donors with similar results) of quiescent (columns 1, 3) and activated (columns 2, 4) CD3+ (rows 1–3) and CD3+CD8+ (row 4–6) T cells from two different donors. Scale bar is 20 μm. Cell size (b), optical redox ratio (c), NAD(P)H τm (d), FAD τm (e), NAD(P)H α1 (f), and FAD α1 (g) of quiescent and activated CD3+ and CD3+CD8+ T cells. Black circles represent mean of all data (6 donors), triangles (donors A [dark red], B [medium red], and F [light red]) represent data from female donors, squares (donors C [dark blue], D [medium blue], and E [light blue]) represent data from male donors. Each colour shade represents data from an individual donor. Data are mean +/− 99% CI. Horizontal lines indicate statistical comparisons, Stars (*** p<0.001) indicate statistical comparisons at the cellular level (n=4,877 biologically independent CD3+ T cells from 6 donors and n=3,478 biologically independent CD3+CD8+ T cells from 6 donors) from a two-sided logistic regression, generalized linear model using an α significance level of 0.05. Carets (B: ˆˆ p=0.001, ˆ=0.016; C: ˆ p=0.011, ˆˆ p=0.002; D: ˆ p=0.015, ˆˆ p=0.002; E: ˆ p=0.022; F: ˆˆ p=0.008, ˆˆˆ p<0.001; G: ˆˆ p=0.002, ˆ p=0.021) indicate significance at the donor level, n=6 biologically independent donors. Aggregated cellular data was compared using a double-sided paired t-test. (h-j) Cellular respiration increases in activated T cells. The oxygen consumption rate (OCR; panel H) and extracellular acidification rate (ECAR, panel I) are increased in activated bulk CD3+ and isolated CD3+CD8+ T cells. The ratio of OCR to ECAR (J) is significantly decreased in activated bulk CD3+ and isolated CD3+CD8+ T cells as compared with that of quiescent T cells. *** p<1*10−5, horizontal lines indicate statistical comparisons, double-sided, Student’s t-test, n=6 wells/group CD3+CD8+ isolation, n=12 wells/group CD3+ isolation (1 donor). Mean +/− 95% CI. Error bars smaller than the symbol for the mean are not shown.
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurements confirm increased metabolic rates of the activated T cells from 1 donor (p<0.001, Fig. 1H–J), in accordance with previously published studies of activated T cells [24, 26–28]. In a metabolic inhibitor experiment of T cells from 1 donor (Fig. S5), the redox ratio of activated T cells decreased (p<0.001) with a glycolysis inhibitor (2-deoxy-d-glucose), and the redox ratio of quiescent T cells increased (p<0.001) with oxidative phosphorylation inhibitors (antimycin A and rotenone). Additionally, the glutaminolysis inhibitor BPTES (Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide) significantly decreased (p<0.001) the optical redox ratio, NAD(P)H τm, and FAD τm of both quiescent and activated T cells, suggesting a significant contribution of glutaminolysis to the metabolism of quiescent and activated T cells (Fig. S5).
Autofluorescence imaging endpoints allow classification of quiescent and activated T cells with high accuracy
Uniform Manifold Approximate and Projection (UMAP) [29], a dimension reduction technique similar to t-distributed Stochastic Neighbor Embedding (t-SNE), was used to visualize how cells cluster from autofluorescence measurements. Neighbours were defined through a cosine distance function computed across the autofluorescence endpoints (optical redox ratio, NAD(P)H τm, NAD(P)H τ1, NAD(P)H τ2, NAD(P)H α1, FAD τm, FAD τ1, FAD τ2, and FAD α1) and cell size for each cell. UMAP was chosen over other techniques, notably principle component analysis (PCA) or t-SNE, for its speed, ability to include non-metric distance functions, and performance on preserving the global structure of the data. UMAP representations of the autofluorescence imaging data of the T cells from 6 donors reveals separation of quiescent and activated T cells (Fig. 2A–B). The gain ratio of autofluorescence endpoints indicates that NAD(P)H α1, cell size, and optical redox ratio are the most important features for classification of activation state of CD3+ T cells (Fig. 2C), and NAD(P)H α1, optical redox ratio, and NAD(P)H τm are the most important features for classification of activation state of CD3+CD8+ T cells (Fig. 2C). The order of feature importance was consistent across multiple feature selection methods including information gain, χ2, and random forest (Fig. S6). Correlation analysis revealed that NAD(P)H α1, cell size, and the optical redox ratio are not significantly correlated (Fig. S7), suggesting these features provide complementary information for classification. NAD(P)H α1 and τm are significantly correlated (Fig. S7), as expected, given that τm is computed from α1. Similar feature weight and order of importance were observed from analysis without NAD(P)H τm and FAD τm (Fig. S8), indicating that the multivariate models were not significantly affected by the correlations between the mean lifetimes and the lifetime components.
Fig 2 |. Autofluorescence imaging endpoints allow classification of quiescent and activated T cells.
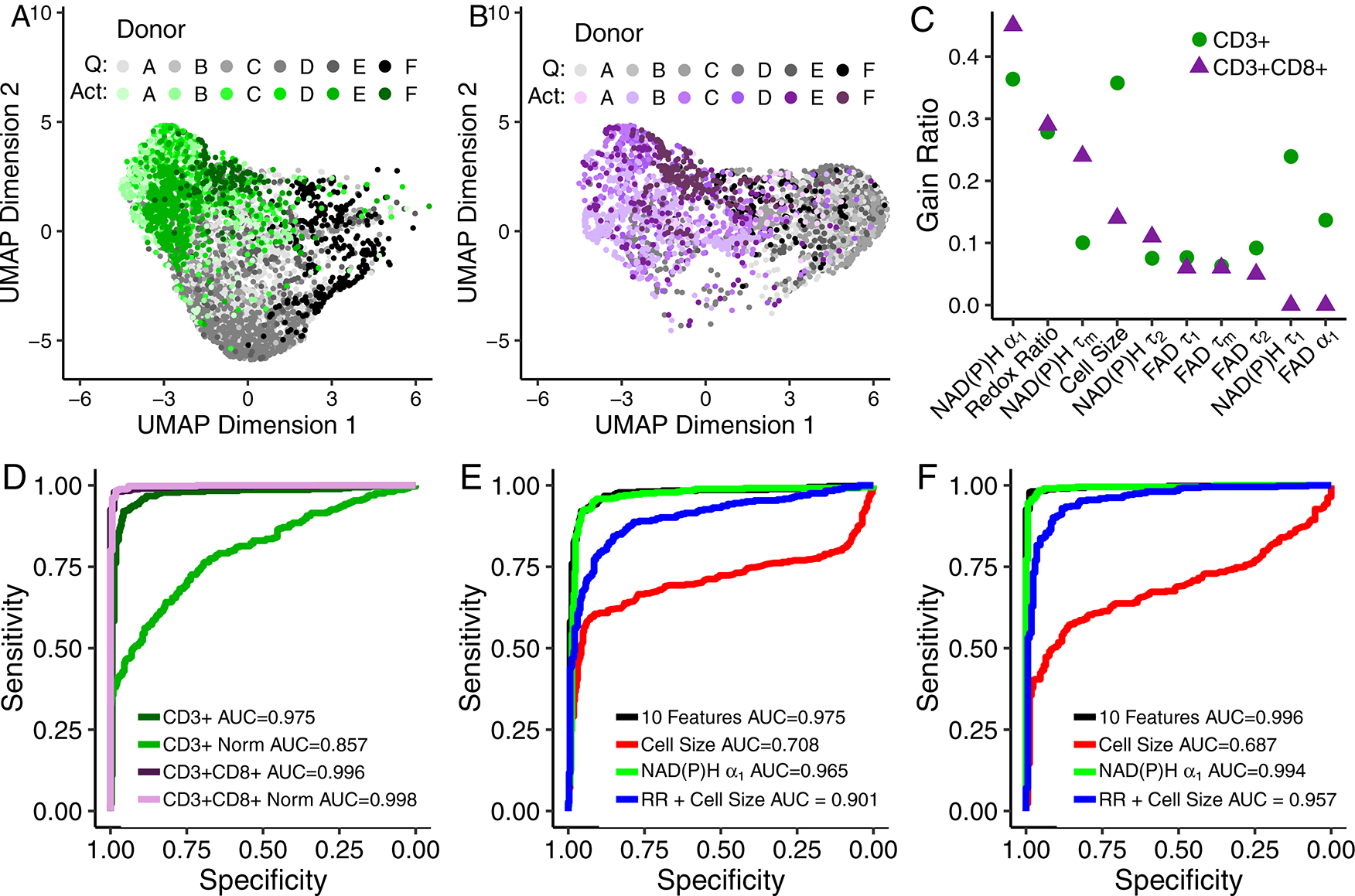
(A-B) UMAP data reduction technique allows visual representation of the separation between quiescent (“Q”) and activated (“Act”) bulk CD3+ (A) and isolated CD3+CD8+ (B) T cells. Each colour corresponds to a different donor, greys correspond to quiescent cells and green or purple to activated CD3+ or CD3+CD8+ T cells, respectively. Data are from 6 donors. Each dot represents a single cell, n=4,877 CD3+ T cells and n=3,478 CD3+CD8+ T cells. (C) Feature weights for classification of quiescent versus activated T cells by the gain ratio method. Analysis was performed at the cellular level with data from 6 donors. (D) ROC curves of the test data for logistic regression models for classification of activation state within bulk CD3+ T cells, bulk CD3+ T cells normalized within each donor (CD3+ Norm), isolated CD3+CD8+ T cells, and isolated CD3+CD8+ T cells normalized within each donor (CD3+CD8+ Norm). (E-F) ROC curves of the test data for logistic regression classification models computed using different features for the classification of (E) quiescent or activated bulk CD3+ or (F) isolated CD3+CD8+ T cells. Models for subfigures D-F were trained on cells that lacked same cell validation data from donors A, B, C, and D but were known to be quiescent or activated by culture conditions (n = 4,131 biologically independent CD3+ cells, n=2,655 biological independent CD3+CD8+ cells), and cells from donors B, E, and F with CD69 validation of activation state were used to test the models (n = 696 biologically independent CD3+ cells, n=595 biologically independent CD3+CD8+ cells). Redox Ratio = NAD(P)H/(NAD(P)H+FAD).
Classification models were developed to predict T cell activation state from NAD(P)H and FAD autofluorescence imaging endpoints (Fig. 2D–F). To protect against over-fitting, logistic regression models were trained on data from 4 donors with activation state assigned from culture conditions and tested on data with same-cell CD69 expression immunofluorescence validation from 3 donors (completely independent and non-overlapping observations). Receiver operator characteristic (ROC) curves of the test data reveal high classification accuracy for predicting activation in bulk CD3+ (AUC = 0.975) and isolated CD3+CD8+ (area under the curve (AUC) = 0.996) T cells, when the models use all autofluorescence endpoints (optical redox ratio, cell size, NAD(P)H τm, NAD(P)H τ1, NAD(P)H τ2, NAD(P)H α1, FAD τm, FAD τ1, FAD τ2, and FAD α1). When the NAD(P)H and FAD autofluorescence imaging endpoints of the T cells are normalized within a donor to the mean value of the quiescent CD3+ population, the ROC AUC decreases to 0.857 for CD3+ T cells (Fig. 2D) and increases slightly to 0.998 for isolated CD3+CD8+ T cells. While all 10 NAD(P)H and FAD autofluorescence features achieved the highest classification accuracy (AUC = 0.975) for activation of CD3+ T cells, a logistic regression model using only NAD(P)H α1 achieved a slightly lower accuracy of 0.965 (Fig. 2E). Logistic regression models that include cell size or cell size and the optical redox ratio, endpoints that can be obtained from fluorescence intensity images, were less effective at accurately predicting activation of bulk CD3+ T cells with ROC AUCs of 0.708 and 0.901, respectively (Fig. 2E). Similar results were obtained for the isolated CD3+CD8+ T cells, with the highest ROC AUC values achieved for logistic regression classification models using all 10 autofluorescence imaging endpoints and NAD(P)H α1 alone, AUC = 0.996 and 0.994, respectively (Fig. 2F). Similar classification accuracy was achieved with random forest and support vector machine models using all 10 autofluorescence imaging endpoints (Fig. S9).
Autofluorescence imaging reveals T cell heterogeneity within and across donors
T cell heterogeneity was assessed within and across the 6 donors (Fig. 3). Heatmap representation (Fig. 3A) of the z-score of autofluorescence imaging endpoint values at the donor level (each row is the mean data of a single donor, cell type, and activation) reveals that the T cells cluster by activation state (i.e. quiescent and activated cluster separately) and isolation (bulk CD3+ or isolated CD3+CD8+). Corresponding coefficient of variation heatmaps highlight the high intra-donor variability of the size of activated T cells and low intra-donor heterogeneity of the autofluorescence endpoints (Fig. S10).
Fig 3 |. Autofluorescence imaging reveals inter- and intra-donor T cell heterogeneity.
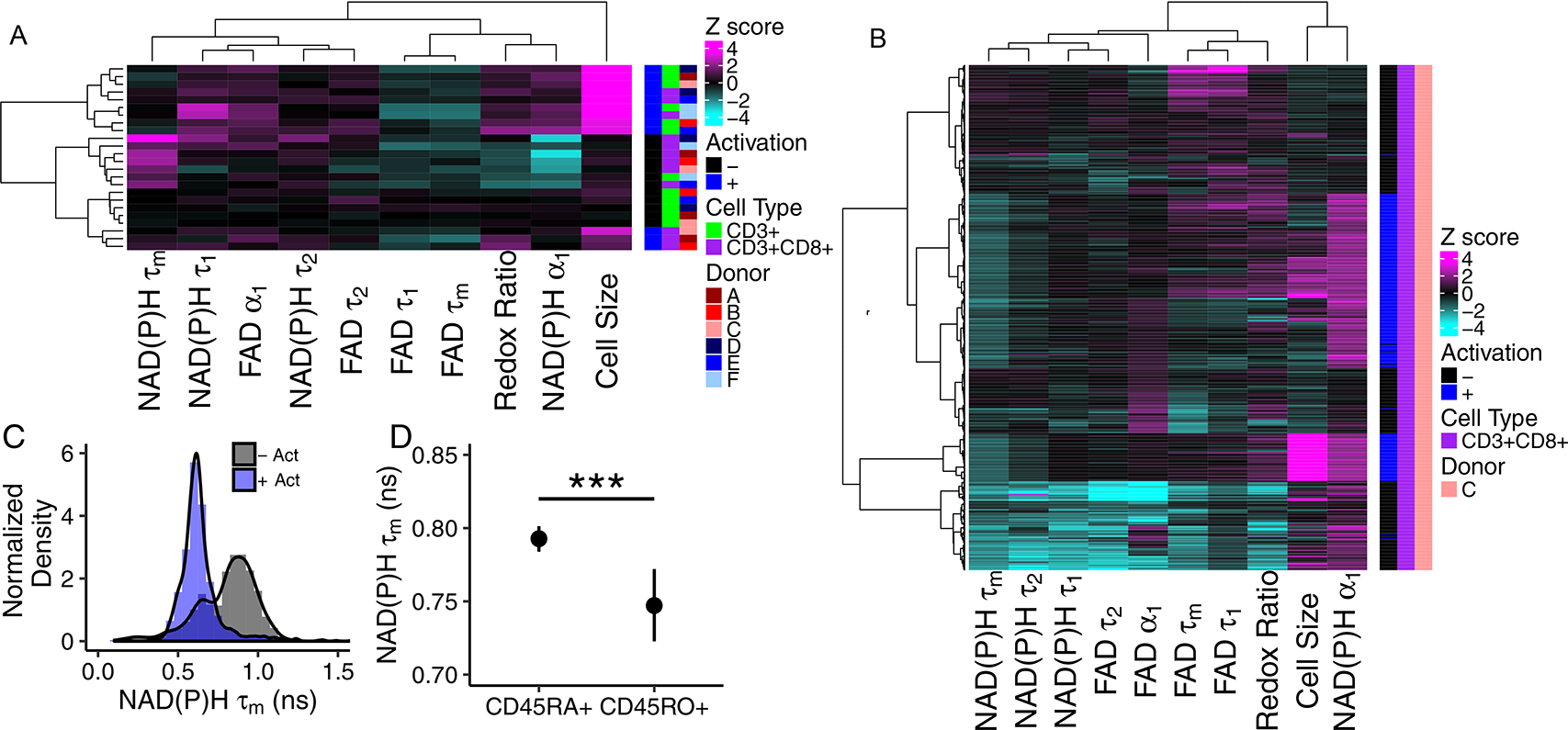
(A) Heatmap of z-scores of NAD(P)H and FAD autofluorescence imaging endpoints where each row is the mean data aggregating all cells from a single donor, subtype (CD3+ or CD3+CD8+), and activation, n=6 biologically independent donors. Data clusters by activation state and isolation (bulk CD3+ or isolated CD3+CD8+). (B) Heatmap of z-scores of NAD(P)H and FAD autofluorescence imaging endpoints of CD3+CD8+ T cells from a single donor, each row is a single cell (n=635 cells). Distinct clusters are identified within the quiescent and activated CD3+CD8+ T cells. (C) Histogram analysis of NAD(P)H τm reveals two populations in quiescent CD3+CD8+ T cells across all 6 donors (n=2126 quiescent cells, 1352 activated cells, - Act = quiescent cells, + Act = cells exposed to anti-CD3/CD2/CD28 for 48hr). (D) NAD(P)H τm is decreased in CD45RO+ CD3+CD8+ T cells compared to NAD(P)H τm of CD45RA+ CD3+CD8+ T cells (CD45RA+ n=265 cells, CD45RO+ n=33 cells from 3 donors, *** p=0.00058, two-sided logistic regression, generalized linear model. p>0.05 for data aggregated to the donor level, two-sided paired t-test.) Mean +/− 95% confidence interval. Redox Ratio = NAD(P)H/(NAD(P)H+FAD).
A representative z score heatmap where each row is a single cell from one donor reveals distinct clusters of T cells by autofluorescence imaging endpoints within the quiescent and activated CD3+CD8+ T cell populations (Fig. 3B). Multiple quiescent and activated T cell populations were observed across all six donors and arise from varied distributions of autofluorescence imaging endpoints within the T cell populations (Fig. 3C, S11–14). For example, histograms of the NAD(P)H τm values of quiescent and activated CD3+CD8+ T cells reveals a bimodal population within the quiescent CD3+CD8+ T cells, with one peak of the quiescent cells consistent with the peak of the activated cells (Fig. 3C).
We hypothesized that memory and naïve T cells within the quiescent population contributed to the observed heterogeneity within the quiescent CD3+CD8+ T cell population (Fig. 3B–C, S11–14) (i.e. the multiple clusters of quiescent CD3+CD8+ cells within the heatmaps and bimodal distribution of the NAD(P)H τm of quiescent CD3+CD8+ T cells). To test this, we co-stained quiescent CD3+CD8+ T cells with antibodies against CD45RA, a marker of naïve T cells, and CD45RO, a marker of memory T cells. NAD(P)H τm was significantly decreased in CD45RO+ cells as compared with NAD(P)H τm of CD45RA+ cells (Fig. 3D; 3 donors). Additionally, the optical redox ratio (p=0.007) and NAD(P)H α1 (p=0.0009) were increased in CD45RO+ CD3+CD8+ T cells as compared to CD45RA+ cells (Fig. S15).
Culture with CD3+CD4+ T cells affects the autofluorescence of CD3+CD8+ T cells
NAD(P)H and FAD autofluorescence imaging endpoints reveal metabolic differences between CD3+CD8+ T cells cultured as an isolated population and CD3+CD8+ T cells cultured with CD3+CD4+ T cells (bulk CD3+ isolation). A UMAP (data dimension reduction) representation of NAD(P)H and FAD autofluorescence imaging endpoints reveals that CD3+CD8+ T cells cultured from the CD3+CD8+ specific T cell isolations cluster separately from CD3+CD8+ T cells within bulk CD3+ T cell populations (Fig. 4A; 3 donors). The optical redox ratio and NAD(P)H α1 are decreased in both quiescent and activated CD3+CD8+ T cells of the isolated CD3+CD8+ population as compared to the corresponding values of quiescent and activated CD3+CD8+ T cells, respectively, within the bulk CD3+ population (Fig. 4B–C). Additional differences in NAD(P)H and FAD autofluorescence lifetime endpoints were observed between CD3+CD8+ T cells within the bulk CD3+ population and the isolated CD3+CD8+ population (Fig. S16).
Fig 4 |. T cell population composition affects T cell autofluorescence.
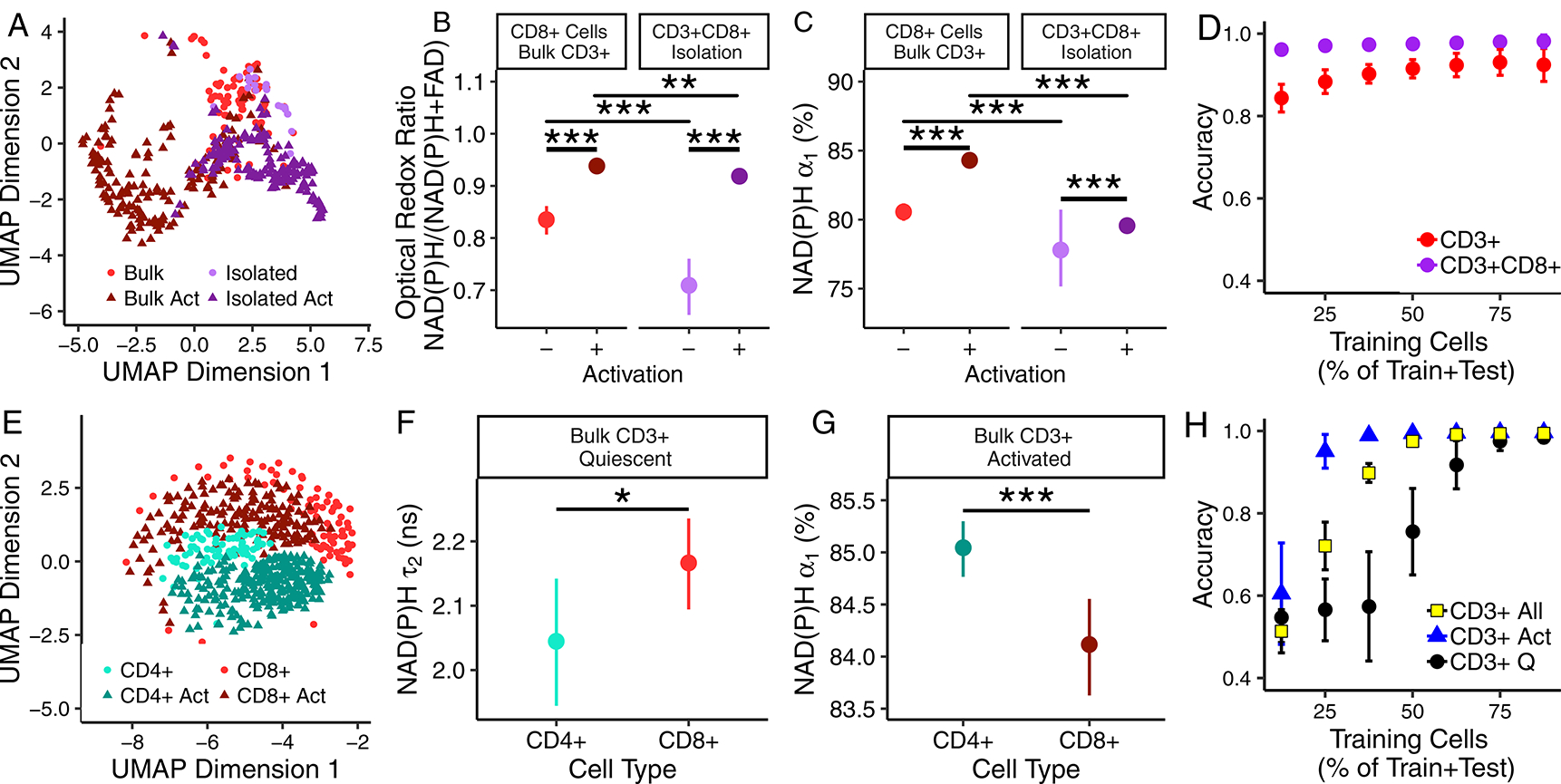
(A) UMAP of NAD(P)H and FAD autofluorescence endpoints of quiescent and activated (“Act”) CD3+CD8+ T cells identified within bulk CD3+ and specific CD3+CD8+ isolations, n=477 biologically independent cells from 3 donors. (B) Optical redox ratio and (C) NAD(P)H α1 of CD3+CD8+ T cells cultured as an isolated population (CD3+CD8+ specific isolation, n=39 quiescent cells, n=174 activated cells, from 3 donors) and with CD3+CD4+ T cells (bulk CD3+ isolation, n=83 quiescent cells, n=170 activated cells, from 3 donors). Mean +/− 95% confidence interval. Horizontal lines indicate statistical comparisons, ** p=0.002, *** p<0.001 for cell level comparisons using a two sided logistic regression, linear generalized model. Donor level p-values provided in Supplementary Tables 2–4. (D) Accuracy of random forest classification of quiescent versus activated CD3+CD8+ T cells from CD3+CD8+ specific isolation (n=213 cells, 3 donors) and bulk CD3+ isolation (n=253 cells, 3 donors). Mean +/− 95% confidence interval for 50 iterations. (E) UMAP of NAD(P)H and FAD autofluorescence imaging endpoints of quiescent and activated CD3+CD4+ and CD3+CD8+ cells identified within bulk CD3+ populations, n=583 biologically independent cells from 3 donors. (F) NAD(P)H τ2 of quiescent CD3+CD4+ and CD3+CD8+ cells (bulk CD3+ isolation, n=66 quiescent CD3+CD4+ T cells, n=83 quiescent CD3+CD8+ T cells from 3 donors, * p=0.04, two-sided logistic regression, generalized linear model; p>0.05 for two sided paired t-test at the donor level, mean +/− 95% confidence interval). (G) NAD(P)H α1 of activated CD3+CD4+ and CD3+CD8+ cells (bulk CD3+ isolation, n=264 activated CD3+CD4+ T cells, n=170 activated CD3+CD8+ T cells from 3 donors, *** p=0.0004, two-sided logistic regression, generalized linear model; p>0.05 for two sided paired t-test at the donor level, mean +/− 95% confidence interval. (H) Accuracy of random forest classification of CD3+CD4+ and CD3+CD8+ T cells from quiescent (2 group classification, “CD3+ Q”), activated (2 group classification, “CD3+ Act”), or both quiescent and activated T cells (4 group classification, “CD3+ All”) within bulk CD3+ isolations, total observations include 66 quiescent CD3+CD4+ T cells, 83 quiescent CD3+CD8+ T cells, 264 activated CD3+CD4+ T cells, and 170 activated CD3+CD8+ T cells from 3 donors. Mean +/− 95% confidence interval for 50 iterations.
Despite these differences between CD3+CD8+ T cells of CD3+CD8+ specific isolations and bulk CD3+ isolations, significant changes in NAD(P)H and FAD autofluorescence endpoints due to activation are maintained, and classification models predict activation status of CD3+CD8+ cells with high accuracy regardless of isolation (Fig. 4D). Random forest feature selection revealed that NAD(P)H α1 is the most important feature for classification of quiescent from activated CD3+ or CD3+CD8+ T cells (Fig. S17A).
Autofluorescence endpoints classify CD3+CD4+ from CD3+CD8+ T cells within bulk CD3+ populations
Heterogeneity in NAD(P)H and FAD autofluorescence endpoints between CD3+CD4+ and CD3+CD8+ T cells is observed within the bulk CD3+ isolation. CD4+ or CD8+ expression was verified in parallel T cell populations of 3 donors by immunofluorescence labelling with anti-CD4-peridinin-chlorophyll-protein complex (PerCP) or anti-CD8-PerCP (Fig. S2). A UMAP representation of the autofluorescence data allows visualization of quiescent and activated CD3+CD4+ and CD3+CD8+ T cells within the bulk CD3+ isolation (Fig. 4E). Significant differences in NAD(P)H and FAD endpoints, including NAD(P)H τ2 and NAD(P)H α1, are observed between CD3+CD4+ and CD3+CD8+ T cells (p<0.001, Fig. 4F–G, S18). Random forest models to classify T cell subtype (CD3+CD4+ or CD3+CD8+) within the bulk CD3+ T cell isolation have average test data predictions of 97.5% and 99.7% for separate predictions on subsets of quiescent or activated T cells, respectively, and 99.4% for all four groups, when trained on 75% of the T cell data set and tested on the remaining 25% (Fig. 4H). Classification accuracy scales with number of cells in the train versus test groups (Fig. 4H). Random forest feature analysis revealed that NAD(P)H τ2 is the highest weighted feature for the classification of activated CD3+CD4+ T cells from activated CD3+CD8+ T cells, and FAD τ1 is the highest weighted feature for classification of quiescent CD3+CD4+ T cells from quiescent CD3+CD8+ T cells (Fig. S17B).
Autofluorescence imaging allows classification of activated T cells in cultures of combined quiescent and activated T cells
NAD(P)H and FAD autofluorescence imaging allows label-free imaging and classification of T cell activation in T cell cultures with combined quiescent and activated cells. A representative NAD(P)H α1 image with CD69 immunofluorescence overlaid in pink, demonstrates the difference in NAD(P)H α1 between quiescent (CD69−) and activated (CD69+) T cells (Fig. 5A). UMAP visualization of the autofluorescence imaging data reveals separation of quiescent and activated CD3+ T cells within this population of combined quiescent and activated cells (Fig. 5B). When cultured in isolated populations, quiescent and activated T cells have significantly different NAD(P)H and FAD imaging endpoints, including the optical redox ratio and NAD(P)H α1, then their respective counterpart from a combined (quiescent with activated T cells) population (Fig. 5C–D, Fig. S19). Random forest feature selection for classification of activation status of T cells within a combined, quiescent and activated, T cell population reveals that NAD(P)H α1 is the most important feature for classification, followed by NAD(P)H τm (Fig. S20). Logistic regression models to predict activation status of T cells in a combined, quiescent and activated, CD3+ T cell culture achieves test data ROC AUCs of 0.95 when all 10 NAD(P)H and FAD imaging endpoints are included, 0.95 and 0.68 when only predicting from NAD(P)H α1 or cell size, respectively, and 0.67 for redox ratio and cell size (Fig. 5E).
Fig 5 |. Autofluorescence imaging allows classification of quiescent and activated T cells within combined quiescent and activated T cell populations.
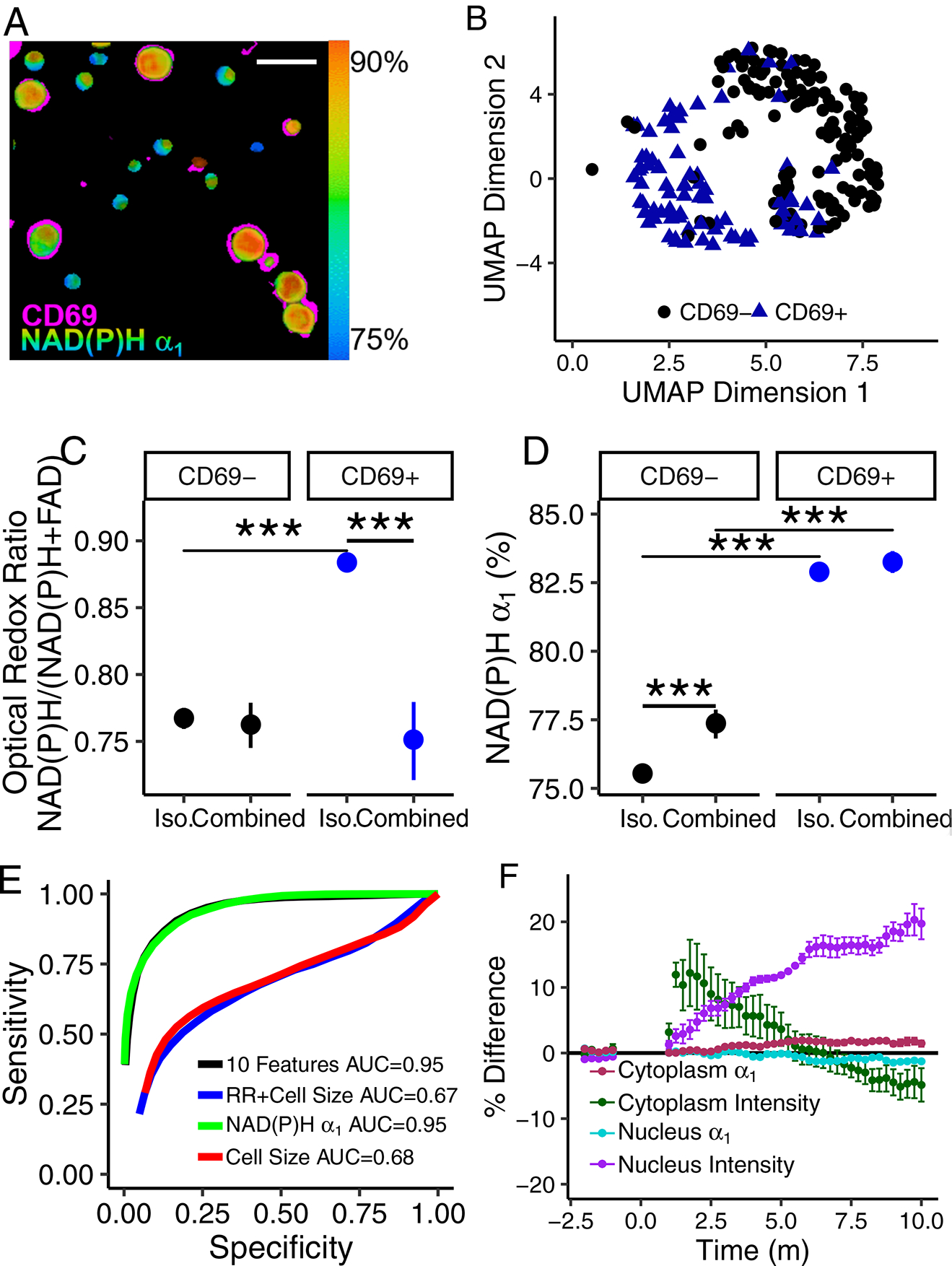
(A) Representative NAD(P)H α1 image of 4 images acquired with similar results of combined quiescent (CD69−) and activated (CD69+) T cells with CD69 immunofluorescence overlaid in pink. Scale bar is 30 μm. CD69 image is shifted to account for cell movement between frames. (B) UMAP representation of NAD(P)H and FAD imaging endpoints of CD69− and CD69+ CD3+ T cells from a combined population of quiescent and activated T cells, n=265 biologically independent cells from 1 donor. (C) Optical redox ratio and (D) NAD(P)H α1 of isolated (“Iso.”) and combined quiescent (CD69−) and activated (CD69+) CD3+ T cells. Mean +/− 95% confidence interval. Horizontal lines indicate statistical comparisons, *** p<1*10−9 for cell level analysis using a two sided logistic regression, generalized linear model, n=733 biologically independent cells, single donor. (E) ROC curves of logistic regression classification of quiescent and activated CD3+ T cells from a combined population of CD69− and CD69+ T cells from a single donor, n=250 biologically independent cells. (F) Percent difference of NAD(P)H α1 and fluorescence intensity in CD3+ T cell nuclei and cytoplasms over time. Anti-CD2/CD3/CD28 added at t=0 m. Mean +/− SE of n=94 biologically independent cells from 3 different donors.
Autofluorescence imaging resolves temporal changes in T cells with activation
Metabolic changes occur rapidly within T cells upon activation [26]; therefore, we hypothesized that time-course imaging of T cells would resolve changes in T cell autofluorescence. NAD(P)H fluorescence lifetime images were acquired from CD3+ quiescent T cells immediately after exposure to the activating tetrameric antibody (anti-CD2/CD3/CD28). The NAD(P)H intensity of the nucleus increased by 10% relative to the pre-activator values, within a few minutes of addition of the activator, and remained consistently higher than the average pre-activation NAD(P)H intensity throughout the 10 minute time-course (Fig. 5F). NAD(P)H intensity within the nucleus may indicate increased transcription [30]. The NAD(P)H intensity in the cytoplasm initially increased (t<1 m) and then decreased, relative to the pre-activation NAD(P)H intensity of the cytoplasm. NAD(P)H α1 increased significantly in the cytoplasm by 2% at t=6 minutes post addition of the activator and remained significantly increased until t=8.75 m. These autofluorescence changes observed early, within minutes of activation, indicate that autofluorescence lifetime imaging is sensitive to robust transcription and metabolic changes that occur with activation in T cells [26]. A 24 h time course of the optical redox ratio revealed an initial decrease in the optical redox ratio of activated versus control T cells at 0.5 and 1 h and then an increased optical redox ratio at 2+ h (Fig. S21). This finding is consistent with a previous study [26] that found an increase in OCR (oxygen consumption suggests increased FAD production and a reduced redox ratio [31]) within 10 minutes of stimulation of T cells by anti-CD3/CD28 beads and an increase in ECAR (consistent with an increase in redox ratio) at 20–40 minutes post-stimulation.
Discussion
T cells are an important component of the adaptive immune response with direct cytotoxic and immune-modulating behaviours. Immunotherapies that directly modify T cell behaviour show promise for treating a variety of conditions including cancer and autoimmune disease. In this study, we develop autofluorescence lifetime imaging-based methods for determination of T cell activation at the single cell level. Autofluorescence lifetime imaging is non-destructive, label-free, and has high spatial and temporal resolution that is amenable with live cell assessment, longitudinal studies, and in vivo imaging. However, autofluorescence imaging is limited by light penetration depth and thus in vivo applications are limited to surface tissues and preclinical window chamber models.
Although several endogenous molecules are autofluorescent, including NAD(P)H, FAD and other flavins, tryptophan, retinol, haemoglobin, melanin, collagen, elastin, and lipofuscin, not all of these molecules are present in T cells. In this study, NAD(P)H and FAD fluorescence are isolated by using a purified sample of T cells (which lack high concentrations of intracellular collagen, retinol, melanin, elastin, and lipofuscin) and by using specific excitation and emission wavelengths. Analysis of the lifetime data confirms isolation of NAD(P)H and FAD signals, since endogenous fluorophores have distinctive lifetimes and contributions from additional fluorophores are not observed. Additionally, the metabolic inhibitor experiment (Fig. S5) confirms isolation of NAD(P)H and FAD fluorescence as the correct trends in optical redox ratio values are observed for specific metabolic pathway inhibition: an increase in the optical redox ratio of quiescent cells with rotenone+antimycin A treatment, and the opposite effect, a reduced optical redox ratio, with 2-deoxy-d-gluocse (2DG) treatment of activated T cells.
Different redox ratio definitions with either NAD(P)H or FAD in the numerator can be found in the literature [7, 11, 12]. Here, we are using NAD(P)H/(NAD(P)H+FAD) to improve the intuitive interpretation of the results, where an increase corresponds to T cell activation. Additional commentary on the redox ratio definition is included with the supplementary materials.
Upon activation, T cell metabolism switches from tricarboxylic acid oxidation of glucose and β-oxidation of fatty acids to glycolysis and glutaminolysis [23–25, 32, 33]. T cells with high glycolytic activity in vitro show poor persistence, low recall responses and low proliferation rates that lead to poor effector activity in vivo, whereas T cells with high fatty acid oxidation show increased persistence, recall responses and proliferation leading to better effector activity within the tumour [34]. Changes in NAD(P)H and FAD autofluorescence imaging endpoints, including the increased optical redox ratio and increased free NAD(P)H (α1) observed in activated T cells relative to the corresponding endpoints of quiescent T cells, reflect a shift towards glycolysis in activated T cells (Fig. 1, S1, S18–19)[35]. An increase in glycolysis of activated T cells was verified by the Seahorse assay and metabolic inhibitor experiment (Fig. 1H–J, S5). Significant changes in the lifetimes of NAD(P)H and FAD (Fig. S1) indicate differences in the protein binding partners and microenvironment of NAD(P)H and FAD [36].
Contributions from multiple metabolic pathways can alter the optical endpoints and make interpretation of the results challenging. To aid in data interpretation, the metabolic inhibitor experiment (Fig. S5) demonstrates the effects of glycolysis, oxidative phosphorylation, fatty acid synthesis, and glutaminolysis inhibition on the optical endpoints of quiescent and activated T cells. BPTES reduced the redox ratio of both quiescent and activated T cells relative to control quiescent or activated T cells, respectively. This suggests that both quiescent and activated T cells utilize glutaminolysis. Rotenone and antimycin A selectively enhanced the optical redox ratio of quiescent cells, while 2-de-oxy-D-glucose selectively reduced the optical redox ratio of activated T cells, indicating that the optical redox ratio alterations due to activation in T cells are primarily due to alterations in glycolysis and oxidative phosphorylation.
T cells are known to be highly heterogeneous, with phenotypic heterogeneity of surface proteins and effector function observed for CD3+CD4+ and CD3+CD8+ T cells [37]. This heterogeneity can arise from the strength of the activating event, the microenvironment of the T cell, and differences in gene regulation at the time of activation [38–40]. Heterogeneity analysis, by heatmaps and histograms, revealed heterogeneous clustering of T cells within the autofluorescence imaging dataset. A difference in the mean NAD(P)H lifetime was found to be due to naïve (CD45RO+) and memory (CD45RA+) CD3+CD8+ T cells (Fig. 3C–D), which are known to have differing metabolic states: memory T cells have increased glycolytic capacity and mitochondrial mass as compared with naïve T cells [26]. An additional subpopulation was identified within the activated T cell subset and characterized by larger than average cells (Fig. 3B, S6–7). These large cells may be actively dividing cells, a condition which is also accompanied by metabolic and autofluorescence differences [41, 42].
Machine learning approaches have been used on extracted morphological features of phase-contrast images to identify cancer cells from immune cells, on brightfield images to assess cell cycle, and on phase contrast and autofluorescence images to classify macrophage exposure to lipopolysaccharide (LPS) [22, 43, 44]. Here, high ROC AUCs (0.95+) were achieved for test data sets using machine learning techniques to classify T cells as activated or quiescent using the autofluorescence imaging endpoints (optical redox ratio, cell size, NAD(P)H τm, NAD(P)H τ1, NAD(P)H τ2, NAD(P)H α1, FAD τm, FAD τ1, FAD τ2, and FAD α1) quantified for each cell. Classification of activation of T cells from CD3+CD8+ specific isolations was slightly higher than that of T cells from bulk CD3+ isolations as might be expected for a homogeneous population (CD3+CD8+) rather than a heterogeneous population (bulk CD3+ populations contain CD4+ and CD8+ subsets). Although multiple classification models were found to have similar performance, logistic regression was the best fitting model, suggesting that the predicted probability of activation is a linear combination of all 10 of the autofluorescence imaging endpoints. Interestingly, donor normalization (Fig. 2D) of the autofluorescence imaging endpoints did not improve classification accuracy, suggesting that the autofluorescence endpoints reflect changes in T cells with activation that are consistent across donors so generalized models can be used for unspecified donors or patients, which is beneficial for robust implementation of autofluorescence imaging as a universal tool to evaluate T cell activation.
The models for classification of activation in T cells reported here have higher ROC AUC values than the previously reported accuracy of 84–87% found for binary logistic regression classification of morphological and Raman spectra features of control and LPS-exposed murine RAW264 macrophages [22]. Murine macrophage activation by LPS inhibits inducible nitric oxide synthase (iNOS), stimulates the mammalian target of rapamycin (mTOR), increases expression of ubiquitous 6-phosphofructo-2-kinase (u-PFK2), and inhibits adenosine monophosphate-activated protein kinase (AMPK) leading to increased glycolysis and decreased oxidative phosphorylation [45]. Macrophages also display a spectrum of phenotypes upon activation [45] and therefore, the activated macrophage phenotype may be more heterogeneous than the antigen-independent activation of T cells by CD2/CD3/CD28. These differences in molecular changes upon activation may explain the observed differences in label-free classification accuracy between T cells and macrophages. Although high classification accuracy was achieved with the machine learning approaches, deep learning methods such as neural networks may achieve improvements in classification accuracy, as has been demonstrated for the classification of cancer cells from immune cells in phase-contrast images [43].
NAD(P)H α1 was consistently identified as the most important feature for differentiation of quiescent and activated T cells across different feature selection methods (including gain ratio, information gain, χ2, and random forest), and different subsets of CD3+, CD3+CD8+, and CD69+/CD69− T cells (Fig. 2C, S6, S19). Although the redox ratio and cell size also significantly increased with activation and were highly weighted features, NAD(P)H α1 has a lower variance within and across donors. Higher variability in intensity measurements may be due to the confounding factors of intensity levels (throughput due to laser power, detector gain, inner filter effects, etc.) that are not factors in the self-referenced (i.e., independent of absolute photon counts) fluorescence lifetime measurements. The feature weights observed here are specific for NAD(P)H and FAD autofluorescence endpoints and may change if excitation or emission wavelengths are altered to include additional or different fluorophores. The classification analysis also revealed that while models trained on all 10 autofluorescence imaging endpoints yielded the highest accuracy for classification of activation state of T cells, logistic regression using only NAD(P)H α1 yielded comparably high ROC AUCs. NAD(P)H α1 was also more accurate for predicting T cell activation than cell size alone (Fig. 2E), or fluorescence intensity measurements (cell size + redox ratio), which can be obtained by wide-field or confocal fluorescence microscopy. Slight differences (0.2–1%) in classification accuracy can result in many (104-106) cells misclassified if 107 cells need to be evaluated, as would be the case for a therapeutic dose of chimeric antigen receptor (CAR) T cells. Additional label-free methods, including third harmonic generation imaging and Raman spectroscopy of quiescent and activated splenic-derived murine T cells have revealed a significant increase in cell size and lipid content in activated T cells [46]. However, we observed a high variance in cell size within and across patients, which makes it a less important predictor than NAD(P)H lifetime values that change with activation and have lower variance (Fig. S10).
CD3+CD4+ T cells have a variety of immune-modulating behaviours. While not necessary for activation of CD3+CD8+ T cells, the presence of CD3+CD4+ T cells during activation is required for the development of memory CD3+CD8+ T cells [47]. Additionally, TREGS (CD3+CD4+FoxP3+ T cells, 5–10% of peripheral CD3+CD4+ population) suppress the activation and proliferation of other T cells [48, 49]. Differences in the NAD(P)H and FAD autofluorescence imaging endpoints (Fig. 4, S16) between CD3+CD8+ T cells cultured with and without CD3+CD4+ T cells were observed, suggesting autofluorescence imaging is sensitive to CD3+CD4+ induced changes in CD3+CD8+ T cells (Fig. 4). However, despite these differences, NAD(P)H α1 remains the highest weighted feature for classification of activation state (Fig. S17), and activation state of CD3+CD8+ T cells can be classified from autofluorescence imaging endpoints with high accuracy, regardless of T cell population (Fig. 4D).
Due to the differing physiological functions of CD3+CD4+ and CD3+CD8+ T cells [1, 50], we explored whether machine learning methods could use autofluorescence imaging data to distinguish between CD3+CD8+ and CD3+CD4+ T cells within bulk CD3+ populations. Significant differences in NAD(P)H fluorescence lifetime values between CD3+CD4+ and CD3+CD8+ T cells suggests variations in metabolic activity upon activation of CD3+CD4+ and CD3+CD8+ T cells, which is consistent with previously observed differences in T cell activation of these subtypes. Activation of CD3+CD4+ T cells occurs through Myc, estrogen-related receptor alpha (ERRα), and mTOR, while CD3+CD8+ T cells activate through Akt and mTOR [51]. High classification accuracy was achieved through random forest classification of T cell autofluorescence endpoints for all four groups: activated CD3+CD4+ T cells, quiescent CD3+CD4+ T cells, activated CD3+CD8+ T cells, and quiescent CD3+CD8+ T cells (Fig. 4H). Additionally, the differences in the metabolic pathways used by activated CD3+CD4+ and CD3+CD8+ T cells may contribute to the different autofluorescence feature weights observed for CD3+ and CD3+CD8+ T cells (Fig. 2C). Memory T cells and TREGS, populations likely within the CD3+ isolation, use more oxidative metabolism and less glycolytic metabolism as compared with activated CD3+CD8+ T cells [51]. This heterogeneity in metabolic pathway utilization by cells within the CD3+ population may contribute to the differences in optical endpoints observed between the CD3+ and CD3+CD8+ populations, increased intra-donor variability in CD3+ populations, and reduced differences in optical endpoints between quiescent and activated cells in the CD3+ isolation, as compared to CD3+CD8+ isolation.
Autofluorescence lifetime imaging has spatial and temporal resolution advantages over traditional assays to survey T cell activation and function. Autofluorescence imaging can be high resolution to allow measurements at the single cell level, allowing insights into metabolic heterogeneity within T cell populations. Additionally, the high spatial resolution and non-destructive nature of autofluorescence imaging maintains the spatial integrity of immune cells, allowing high fidelity measurements on neighbouring cells as demonstrated in the combined population of quiescent and activated T cells (Fig. 5A). Finally, autofluorescence imaging also has high temporal resolution (Fig. 5F) allowing time-course study of T cell activation.
Altogether, autofluorescence lifetime imaging of NAD(P)H and FAD of T cells, combined with machine learning for classification, is an accurate tool for non-destructive, label-free assessment of activation status of T cells. NAD(P)H and FAD autofluorescence lifetime imaging is provides high spatial, temporal, and functional information of cell metabolism, which makes it an attractive tool to evaluate T cells. Autofluorescence lifetime imaging can be used to characterize T cells in vivo in preclinical models, in clinical applications including small blood samples (e.g., paediatric samples) where antibody labelling is limited, or in cultured T cells such as those used in biomanufactured T cell therapies.
Methods
T cell isolation and culture
This study was approved by the Institutional Review Board of the University of Wisconsin-Madison (#2018–0103), and informed consent was obtained from all donors. Peripheral blood was drawn from 6 healthy donors into sterile syringes containing heparin. Two blood draws, 183 days apart, were performed on one donor to evaluate the consistency of the experimental protocol and imaging endpoints. The data from the second blood draw is only included in the figure that directly compares the results from the two blood draws, Fig. S4. Bulk CD3+ T cells or an isolated CD3+CD8+ T cell subset were extracted from whole blood using negative selection methods (RosetteSep, StemCell Technologies). Blood was transferred from the blood draw syringe to a 50 ml centrifuge tube. Following the RosetteSep Protocol, the CD3+ or CD8+ RosetteSep Cocktail (StemCell Technologies) was added to the blood (50 μl per ml of blood), mixed, and incubated at room temperature for 10 minutes. The sample volume was doubled with phosphate buffered saline (PBS) + 2% foetal bovine serum (FBS). SepMate tubes (StemCell Technologies) were prepared with a 3.5 ml bottom layer of a density gradient medium (Lymphoprep, StemCell Technologies) and a top layer of diluted blood, according to the RosetteSep instructions. The tubes were centrifuged at 1200 g for 10 minutes, brake on. The enriched cell layer within the supernatant was poured into a new 50 ml centrifuge tube, and washed with PBS+ 2% FBS with 300 g, 10 minute centrifugation, 2 times. Enriched cells were cultured in ImmunoCult-XF T cell Expansion Medium (StemCell Technologies). Approximately 24 hours post-isolation, the T cells were divided into two groups, a “quiescent” population that was grown in medium without activating antibodies, and an “activated” population that was cultured in medium supplemented with 25 μl/ml tetrameric antibody against CD2/CD3/CD28 (StemCell Technologies). Quiescent and activated T cell populations were cultured separately for 48 hours at 37°C, 5% CO2, and 99% humidity before imaging and subsequent experiments, unless otherwise noted. Prior to imaging, T cells were plated at approximately 200,000 cells/200 μl media on 35 mm poly-d-lysine coated glass bottom dishes (MatTek). To ensure that autofluorescence imaging and the classification models extend for mixed populations of quiescent and activated T cells, a subset of quiescent and activated T cells from 1 donor (48hr of culture with activating antibody) were combined and plated together in a dish 1 hour before imaging.
Autofluorescence imaging of NAD(P)H and FAD
Fluorescence images were acquired using an Ultima (Bruker Fluorescence Microscopy) two-photon microscope coupled to an inverted microscope body (TiE, Nikon) with an Insight DS+ (Spectra Physics) as the excitation source. A 100X objective (Nikon Plan Apo Lambda, numerical aperture (NA) 1.45), lending an approximate field of view of 110 μm, was used in all experiments with the laser tuned to 750 nm for NAD(P)H two-photon excitation and 890 nm for FAD two-photon excitation. NAD(P)H and FAD images were acquired sequentially through 440/80 nm and 550/100 nm bandpass filters (Chroma), respectively, by Gallium arsenide phosphide (GaAsP) photomultiplier tubes (PMTs; H7422, Hamamatsu). The laser power at the sample was 3.0–3.2 mW for NAD(P)H and 4.1–4.3 mW for FAD. Lifetime imaging was performed within Prairie View (Bruker Fluorescence Microscopy) using time-correlated single photon counting electronics (SPC-150, Becker & Hickl, Berlin, Germany). Fluorescence lifetime decays with 256 time bins were acquired across 256×256 pixel images with a pixel dwell time of 4.6 μs and an integration time of 60 s. Photon count rates were ˜1–5×105 and monitored during image acquisition to ensure that no photobleaching occurred. The short lifetime of red blood cell fluorescence at 890 nm was used as the instrument response function and had a full width at half maximum of 240 ps. A YG fluorescent bead (τ = 2.13 +/− 0.03 ns, n=6) was imaged daily as a fluorescence lifetime standard [14, 18, 52]. Four to six images per group were acquired.
Antibody validation
Antibodies against CD4 (clone OKT4, PerCP-conjugated, Biolegend Item #317431, Lot B198303), CD8 (clone SK1, PerCP-conjugated, Biolegend Item #344707, Lot B204988), CD69 (clone FN50, PerCP-conjugated, Biolegend Item #310927, Lot B180058), CD45RA (clone HI100, Alexa 647-conjugated, Biolegend Item #304153, Lot B220325), and CD45RO (clone UCHL1, PerCP-conjugated, Biolegend Item #304251, Lot B219295) were used for validation of cell type and activation. CD4, CD8, and CD69 antibody labelling was performed on T cells for 3 donors (B, E, and F) in parallel imaging dishes, each with a different immunofluorescence antibody. CD45RA and CD45RO antibody labelling was performed on populations of quiescent CD3+CD8+ T cells for 3 donors. Cells (30,000–200,000 per condition) were stained with 5 μl antibody/106 cells in 50 μl of ImmunoCult-XF T cell Expansion Medium for 30 minutes in the dark at room temperature. Cells were washed with ImmunoCult 1–2 times, resuspended in 50–200 μl of media, and added to the centre of a 35 mm poly-d-lysine coated glass bottom dish (MatTek). Cells were kept in a 37°C, 5% CO2, humidified environment until imaging. All cells were imaged within 3 hours of staining. NAD(P)H and FAD fluorescence lifetime images were acquired as described. To identify PerCP positive cells, an additional fluorescence intensity image was acquired with the laser was tuned to 1040 nm and a 690/45 nm bandpass filter before the PMT. For evaluation of Alexa647 fluorescence, the laser was tuned to 1300 nm for excitation, and a 690/45 nm bandpass filter was used to filter emitted light.
Data analysis
Fluorescence lifetime decays were analysed to extract fluorescence lifetime components (SPCImage, Becker & Hickl). A bin of 9 surrounding pixels (3×3) was used to increase the fluorescence counts in each decay. A threshold was used to exclude pixels with low fluorescence signal (i.e. background). Fluorescence lifetime decays were deconvolved from the instrument response function and fit to a 2 component exponential decay model, , where I(t) is the fluorescence intensity as a function of time, t, after the laser pulse, α1 and α2 are the fractional contributions of the short and long lifetime components, respectively (i.e., α1 + α2 = 1), τ1 and τ2 are the short and long lifetime components, respectively, and C accounts for background light. Both NAD(P)H and FAD can exist in quenched (short lifetime) and unquenched (long lifetime) configurations [8, 13]; therefore, the fluorescence decays of NAD(P)H and FAD are fit to two components.
Images were analysed at the single cell level to evaluate cellular heterogeneity [53]. NAD(P)H intensity images were segmented into cytoplasm and nucleus using edge detect and thresholding methods in CellProfiler using a customized image processing routine [54]. The CellProfiler routine applied the following steps to segment the images: first, the unprocessed NAD(P)H intensity images were rescaled between 0 and 1 by dividing the image by the value of the brightest pixel within the image. Next, a Sobel edge detect computation was applied across the image. The resulting image was thresholded to remove areas of high edge-values (i.e. cell cytoplasms), and inverted to create a mask. This mask was multiplied by the rescaled NAD(P)H intensity image leaving background and nuclear (low intensity) pixels. Primary objects (nuclei) were identified from this image using CellProfiler’s default object identification and a manually set threshold (0.04) to remove the non-cellular background. Secondary objects (cells) were then identified by outward propagation of the primary objects. Cytoplasm masks were determined by subtracting the nucleus mask from the cell mask.
Images of the optical redox ratio (fluorescence intensity of NAD(P)H divided by the summed intensity of NAD(P)H and FAD) and mean fluorescence lifetimes (τm =α1τ1 + α2τ2) of NAD(P)H and FAD were computed (MATLAB). NAD(P)H and FAD autofluorescence imaging endpoints, including the optical redox ratio, NAD(P)H τm, NAD(P)H τ1, NAD(P)H τ2, NAD(P)H α1, FAD τm, FAD τ1, FAD τ2, and FAD α1 were averaged across all pixels within a cell cytoplasm for each segmented cell, thus the fluorescence endpoints are independent of cell size. Unless otherwise stated, presented results are for endpoints averaged across all cytoplasm pixels for each cell. Cell size in μm2 was also computed from the segmented images using the number of pixels within the two-dimensional image of the cell * 0.167 μm2 (which is the pixel dimension).
Statistics
Statistical analysis and data representation were performed in R. For each donor, one dish of cells was prepared for each experimental condition. Four to six fields of view separated by at least 250 μm were acquired from each dish. Each field of view contained 25–400 cells with a mean of 143 cells. Each cell was considered an independent sample because the perturbation (“activation”) occurred at the cellular level. For statistical comparisons at the cellular level, a generalized linear, logistic regression model was used to evaluate significant differences of autofluorescence imaging endpoints between quiescent and activated T cells, CD45RA+ and CD45RO+ cells (Fig. 3), and CD3+CD4+ and CD3+CD8+ T cells. A donor interaction term was included in the model to account for donor; the general equation used was Outcome˜ Imaging Variable*Donor. P values were computed for two-tailed tests, and an alpha significance level of 0.05 was used to indicate significance. Cellular data was also aggregated to the donor level, and a two-tailed, simple paired t-test was performed to assess donor-level significance (α = 0.05) for all comparisons with three or more donors. Presented boxplots are constructed from the median (central line) and first and third quartiles (lower and upper hinges, respectively). The whiskers extend to the farthest data points that are no further than 1.5* the interquartile range. Dots represent data points beyond 1.5* the interquartile range from the hinge.
Classification
Uniform Manifold Approximate and Projection (UMAP), a dimension reduction technique [29], and z-score heatmaps were used to visualize clustering within autofluorescence imaging data sets (Python and R, respectively). Machine learning classification models and training/testing data sets are summarized in Table S1. Random forest, logistic regression, and support vector machine classification methods were trained to classify activated and quiescent T cells within either the bulk CD3+ FLIM data or the isolated CD3+CD8+ FLIM data (R). For both data sets, gain ratio, χ2, and random forest feature selection methods were employed to evaluate the contribution of the NAD(P)H and FAD autofluorescence endpoints to the accuracy of classification of quiescent versus activated T cells. These models were trained on data from donors A, B, C, and D because these cells lacked immunofluorescence CD69 validation but were known to be quiescent or activated by culture conditions (n = 4131 CD3+ cells, n=2655 CD3+CD8+ cells). Models were tested on data from T cells from donors B, E, and F with CD69 immunofluorescence validation of activation state (n = 696 CD3+ cells, n=595 CD3+CD8+ cells) and the presented ROC curves reflect this test data. Random forest models were developed to classify CD3+CD4+ from CD3+CD8+ T cells, and cells were randomly assigned to train and test data sets for a range of train/test proportions from 12.5% to 87.5%. Each model was replicated 50 times with new train and test data generated before each iteration. Logistic regression models were also estimated for the classification of T cell activation from imaging endpoints of combined quiescent and activated CD3+ T cells (both conditions together within the images). Observations were randomly divided into training and testing data sets (90%/10%, respectively), and presented ROC curves are the average of 1000 iterations of randomly data randomly selected to training and test sets. All ROC curves displayed were constructed from the test data sets using the model generated from the training data sets.
Seahorse assay
Quiescent and activated T cells were plated at 5×106 cells/ml on a Seahorse 96-well plate in unbuffered RPMI medium without serum. Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) measurements were obtained every 6.5 minutes for 5 cycles. A generalized linear model was used to determine statistical significance (α = 0.05) within OCR and ECAR measurements between control and activated T cells.
Metabolic inhibitors
Quiescent and activated (48 hr) CD3+ T cells were plated on poly-d-lysine coated 35 mm glass bottom dishes at a concentration of 200,000 cells/200 μl ImmunoCult T cell Expansion Medium as previously described (T cell Isolation and Culture). The metabolic inhibitors antimycin A (1 μM), rotenone (1 μM), 2-deoxy-d-glucose (2DG, 50 mM), Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES, 20 μM), and 5-(Tetradecyloxy)-2-furoic acid (TOFA, 50 μg/ml) were added singly, except for antimycin A and rotenone which were added together, to the dishes prior to imaging. Cells were incubated with antimycin A and rotenone for ten minutes, 2DG for ten minutes, BPTES for 1 hour, and TOFA for 1 hour. Fluorescence lifetime images of NAD(P)H and FAD were acquired for 6 random fields of view as described above. A generalized linear model was used to determine autofluorescence imaging endpoints with statistical significance (α = 0.05) between control and inhibitor-exposed cells.
10-minute activation time course
Quiescent CD3+ T cells were isolated and plated for imaging as previously described. NAD(P)H lifetime images were acquired as described but with an image size of 128×128 pixels and an integration time of 15s. Images were acquired sequentially for 2 minutes (8 frames), then 5 μl PBS was added to the cells as a mock treatment, and NAD(P)H fluorescence lifetime images were acquired for 10 minutes (40 frames). Subsequently, 5 μl of activating tetrameric antibody (anti-CD2/CD3/CD28) was added and NAD(P)H fluorescence lifetime images were acquired for 10 minutes (40 frames). NAD(P)H FLIM images were analysed in SPCImage as described. Individual cells and cell compartments (nucleus, cytoplasm) were manually segmented (author I.J.), and the autofluorescence imaging endpoints were averaged across all pixels within the segmented region (ImageJ). This procedure was repeated for 3 dishes of T cells from 3 different donors for a total of 30–34 analysed cells per donor.
24-hour activation time course
Quiescent CD3+ T cells were isolated from a healthy donor and plated for imaging as previously described with 200,000 cells/200 μl ImmunoCult T cell Expansion Medium on glass bottom petri dishes. NAD(P)H and FAD lifetime images were acquired as described with an image size of 256×256 pixels and an integration time of 60 s. Four to five images were acquired from both control (5 μl PBS added to the 200 μl of cells) and activated (5 μl anti-CD2/CD3/CD28 antibody added to the 200 μl of cells) dishes of T cells at 0.5, 1, 2, 3, 6, 12, and 24 h post-antibody exposure. Optical redox ratio images were computed as described. Cell cytoplasms were segmented as described. The optical redox ratio of the cells in the activated condition was computed as normalized to the mean redox ratio of the cells in the parallel control dish within each time point. This procedure was repeated for T cells from 3 different donors.
Supplementary Material
Acknowledgements
The authors would like to thank Arezoo Movaghar for insightful discussions of feature selection and machine learning classification methods and Rebecca Schmitz for her assistance with formatting of paper figures. This work was funded by the NIH R01 CA185747, R01 CA205101, R01 CA211082 (to M.C.S); the Biotechnology Training Program of the National Institute of General Medical Sciences of the National Institutes of Health, award #T32GM008349 (to K.S.); NIH awards R01DK098672 and R35GM131795 (to D.J.P.) and T32DK007665 (to N.M.N.); the NSF Graduate Research Fellowship Program, DGE-1747503 (to K.M); and the National Science Foundation under Grant No. EEC-1648035 (to K.S.).
Footnotes
Reporting summary. Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The main data supporting the results in this study are available within the paper and its Supplementary Information. The raw and analysed datasets generated during the study are too large to be publicly shared, yet they are available for research purposes from the corresponding authors on reasonable request.
Code availability
All code and algorithms generated during the study are available at https://github.com/walshlab/T-cell-Activation-Paper.
Competing interests
A.W. and M.S. are mentioned as co-inventors in a patent application (“Systems and methods for sorting T cells by activation state”, #62/724428, August 2018) covering devices and methods to sort T cells based on fluorescence-lifetime components.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-01X-XXXX-X.
References
- 1.Mosmann TR & Coffman RL in Advances in Immunology Volume 46 111–147 (Elsevier, 1989). [DOI] [PubMed] [Google Scholar]
- 2.Bettelli E, Korn T & Kuchroo VK Th17: the third member of the effector T cell trilogy. Current Opinion in Immunology 19, 652–657 (Dec. 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nature Reviews Cancer 12, 252–264 (Apr. 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Restifo NP, Dudley ME & Rosenberg SA Adoptive immunotherapy for cancer: harnessing the T cell response. Nature Reviews Immunology 12, 269–281 (Apr. 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canavan JB et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut 65, 584–594 (Feb. 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marek-Trzonkowska N et al. Administration of CD4+CD25highCD127- Regulatory T Cells Preserves-Cell Function in Type 1 Diabetes in Children. Diabetes Care 35, 1817–1820 (June 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chance B, Schoener B, Oshino R, Itshak F & Nakase Y Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals. Journal of Biological Chemistry 254, 4764–4771 (1979). [PubMed] [Google Scholar]
- 8.Lakowicz JR, Szmacinski H, Nowaczyk K & Johnson ML Fluorescence lifetime imaging of free and protein-bound NADH. Proceedings of the National Academy of Sciences 89, 1271–1275 (Feb. 1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Georgakoudi I & Quinn KP Optical imaging using endogenous contrast to assess metabolic state. Annual review of biomedical engineering 14, 351–367 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Huang S, Heikal AA & Webb WW Two-Photon Fluorescence Spectroscopy and Microscopy of NAD(P)H and Flavoprotein. Biophysical Journal 82, 2811–2825 (May 2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varone A et al. Endogenous Two-Photon Fluorescence Imaging Elucidates Metabolic Changes Related to Enhanced Glycolysis and Glutamine Consumption in Precancerous Epithelial Tissues. Cancer Research 74, 3067–3075 (Mar. 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostrander JH et al. Optical Redox Ratio Differentiates Breast Cancer Cell Lines Based on Estrogen Receptor Status. Cancer Research 70, 4759–4766 (May 2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakashima N, Yoshihara K, Tanaka F & Yagi K Picosecond fluorescence lifetime of the coenzyme of D-amino acid oxidase. Journal of Biological Chemistry 255, 5261–5263 (1980). [PubMed] [Google Scholar]
- 14.Skala MC et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proceedings of the National Academy of Sciences 104, 19494–19499 (Nov. 2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quinn KP et al. Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation. Scientific Reports 3 (Dec. 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walsh AJ et al. Quantitative Optical Imaging of Primary Tumor Organoid Metabolism Predicts Drug Response in Breast Cancer. Cancer Research 74, 5184–5194 (Aug. 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walsh AJ, Castellanos JA, Nagathihalli NS, Merchant NB & Skala MC Optical Imaging of Drug-Induced Metabolism Changes in Murine and Human Pancreatic Cancer Organoids Reveals Heterogeneous Drug Response. Pancreas 45, 863–869 (July 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh AJ et al. Optical Metabolic Imaging Identifies Glycolytic Levels, Subtypes, and Early Treatment Response in Breast Cancer. Cancer Research 73, 6164–6174 (Oct. 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stringari C et al. Phasor approach to fluorescence lifetime microscopy distinguishes different metabolic states of germ cells in a live tissue. Proceedings of the National Academy of Sciences 108, 13582–13587 (Aug. 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alfonso-Garcia A et al. Label-free identification of macrophage phenotype by fluorescence lifetime imaging microscopy. Journal of Biomedical Optics 21, 046005 (Apr. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szulczewski JM et al. In Vivo Visualization of Stromal Macrophages via label-free FLIM-based metabolite imaging. Scientific Reports 6 (May 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pavillon N, Hobro AJ, Akira S & Smith NI Noninvasive detection of macrophage activation with single-cell resolution through machine learning. Proceedings of the National Academy of Sciences 115, E2676–E2685 (Mar. 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frauwirth KA et al. The CD28 Signaling Pathway Regulates Glucose Metabolism. Immunity 16, 769–777 (June 2002). [DOI] [PubMed] [Google Scholar]
- 24.Chang C-H et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 153, 1239–1251 (June 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michalek RD et al. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. The Journal of Immunology 186, 3299–3303 (Feb. 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van der Windt GJW et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proceedings of the National Academy of Sciences 110, 14336{14341 (Aug. 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarasenko TN et al. Cytochrome c Oxidase Activity Is a Metabolic Checkpoint that Regulates Cell Fate Decisions During T Cell Activation and Differentiation. Cell Metabolism 25, 1254–1268.e7 (June 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gubser PM et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nature Immunology 14, 1064–1072 (Aug. 2013). [DOI] [PubMed] [Google Scholar]
- 29.McInnes L & Healy J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv: http://arxiv.org/abs/1802.03426v1 (Feb. 9, 2018). [Google Scholar]
- 30.Zhang Q, Piston DW & Goodman RH Regulation of Corepressor Function by Nuclear NADH. Science 295, 1895–1897. (2002). [DOI] [PubMed] [Google Scholar]
- 31.Hou J et al. Correlating two-photon excited fluorescence imaging of breast cancer cellular redox state with seahorse flux analysis of normalized cellular oxygen consumption. Journal of Biomedical Optics [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang R et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 35, 871–882 (Dec. 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang R & Green DR Metabolic checkpoints in activated T cells. Nature Immunology 13, 907–915 (Sept. 2012). [DOI] [PubMed] [Google Scholar]
- 34.Kishton RJ, Sukumar M & Restifo NP Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metabolism 26, 94–109 (July 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z et al. Mapping metabolic changes by noninvasive, multiparametric, high-resolution imaging using endogenous contrast. Science Advances 4 9302 (Mar. 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharick JT et al. Protein-bound NAD(P)H Lifetime is Sensitive to Multiple Fates of Glucose Carbon. Scientific Reports 8 (Apr. 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang JT, Wherry EJ & Goldrath AW Molecular regulation of effector and memory T cell differentiation. Nature Immunology 15, 1104–1115 (Dec. 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaech SM & Cui W Transcriptional control of effector and memory CD8+ T cell differentiation. Nature Reviews Immunology 12, 749–761 (Oct. 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palmer MJ, Mahajan VS, Chen J, Irvine DJ & Lauffenburger DA Signaling thresholds govern heterogeneity in IL-7-receptor-mediated responses of naive CD8+ T cells. Immunology and Cell Biology 89, 581–594 (Feb. 2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tubo NJ et al. Single Naive CD4+ T Cells from a Diverse Repertoire Produce Different Effector Cell Types during Infection. Cell 153, 785–796 (May 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krylov SN et al. Correlating cell cycle with metabolism in single cells: Combination of image and metabolic cytometry. Cytometry 37, 14–20 (Sept. 1999). [DOI] [PubMed] [Google Scholar]
- 42.Heaster TM,Walsh AJ, Zhao Y, Hiebert SW & Skala MC Autofluorescence imaging identifies tumor cell-cycle status on a single-cell level. Journal of Biophotonics 11, e201600276 (May 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CL et al. Deep Learning in Label-free Cell Classification. Scientific Reports 6 (Mar. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blasi T et al. Label-free cell cycle analysis for high-throughput imaging flow cytometry. Nature Communications 7, 10256 (Jan. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelly B & O’Neill LA Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Research 25, 771–784 (June 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gavgiotaki E et al. Detection of the T cell activation state using non-linear optical microscopy. Journal of Biophotonics, e201800277 (Oct. 2018). [DOI] [PubMed] [Google Scholar]
- 47.Janssen EM et al. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 421, 852–856 (Feb. 2003). [DOI] [PubMed] [Google Scholar]
- 48.Takahashi T et al. Immunologic Self-Tolerance Maintained by Cd25+Cd4+Regulatory T Cells Constitutively Expressing Cytotoxic T Lymphocyte-Associated Antigen 4. The Journal of Experimental Medicine 192, 303–310 (July 2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dieckmann D, Plottner H, Berchtold S, Berger T & Schuler G Ex Vivo Isolation and Characterization of Cd4+Cd25+T Cells with Regulatory Properties from Human Blood. The Journal of Experimental Medicine 193, 1303–1310 (June 2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naito Y et al. CD8+ T Cells Infiltrated within Cancer Cell Nests as a Prognostic Factor in Human Colorectal Cancer. Cancer Research 58, 3491–3494 (1998). [PubMed] [Google Scholar]
- 51.Gerriets VA & Rathmell JC Metabolic pathways in T cell fate and function. Trends in Immunology 33, 168–173 (Apr. 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bird DK et al. Metabolic Mapping of MCF10A Human Breast Cells via Multiphoton Fluorescence Lifetime Imaging of the Coenzyme NADH. Cancer Research 65, 8766–8773 (Oct. 2005). [DOI] [PubMed] [Google Scholar]
- 53.Walsh AJ & Skala MC Optical metabolic imaging quantifies heterogeneous cell populations. Biomedical Optics Express 6, 559 (Jan. 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walsh AJ & Skala MC An automated image processing routine for segmentation of cell cytoplasms in high-resolution autofluorescence images in Multiphoton Microscopy in the Biomedical Sciences XIV (SPIE, Feb. 2014).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.