

Abstract
The glycoprotein (GP)Ib-IX receptor complex plays a critical role in platelet physiology and pathology. Its interaction with von Willebrand factor (VWF) on the subendothelial matrix instigates platelet arrest at the site of vascular injury, and is vital to primary hemostasis. Its reception to other ligands and counter-receptors in the blood stream also contribute to various processes of platelet biology that are still being discovered. While its basic composition and its link to congenital bleeding disorders were well documented and firmly established more than 25 years ago, recent years have witnessed critical advances in the organization, dynamics, activation, regulation and functions of the GPIb-IX complex. This review summarizes important findings and identifies questions that remain about this unique platelet mechanoreceptor complex.
Keywords: Platelet, Thrombosis, Thrombocytopenia, Mechanoreceptor, Glycoprotein Ib
Introduction
Platelets play an invaluable role in hemostasis. After vessel injury, platelets arrest, activate, and form a platelet plug essential for sealing the site of insult and preventing excessive blood loss. However, insufficient or excessive platelet activation can both lead to pathologies. Therefore, platelet activity is tightly regulated. Platelets express a wide variety of receptors that enable their response to diverse physiological and pathological stimulants. The glycoprotein (GP)Ib-IX complex is the second most abundant platelet surface receptor [1, 2]. GPIb-IX is a major platelet mechanoreceptor and participates in several diverse functions including adhesion, activation, clearance, and thrombopoiesis. This review covers GPIb-IX’s structure and function, with an emphasis on advances made in the last decade.
Structure and organization of GPIb-IX
GPIb-IX is a highly integrated hetero-tetrameric receptor complex containing three unique subunits: GPIbα, GPIbβ, and GPIX, arranged in a 1:2:1 stoichiometry[3]. Each subunit is an independently expressed transmembrane protein with a short cytoplasmic tail, a single transmembrane domain, and a glycosylated extracellular domain[4]. Efficient expression of the GPIb-IX complex on the platelet membrane depends on co-expression of all subunits[5, 6]. GPIb-IX also associates with GPV, likely at a 1:1 stoichiometry, but the association is relatively weak and can be disrupted by nonionic detergents[7].
GPIbα is the “business end” of the complex, by far the largest subunit, and responsible for binding to almost all known ligands of the complex. At its extracellular N-terminus, GPIbα begins with a ~45-kDa domain containing 7 leucine-rich repeats (LRR), also known as the ligand-binding domain (LBD) (Fig. 1). The C-terminal portion of the LBD contains a small “thumb” region crucial for effective binding to the A1 domain of von Willebrand factor (VWF)[8]. Following the LBD is a short anionic stretch involved in thrombin binding and a flexible stalk known as the macroglycopeptide or sialomucin region, spanning 30–40 nm[4]. The sialomucin region is characterized by a variable number of tandem repeats and its excessive O-glycosylation which, by some estimates, accounts for as much as 70% of the entire sialic acid content on the platelet surface[9]. It helps raise the LBD high above the platelet membrane and facilitates its interaction with various ligands and counter-receptors in circulation. The stretch closest to the platelet membrane contains the quasi-stable mechanosensory domain (MSD), the structure of which remains to be determined[10, 11]. The MSD contains the shedding cleavage site by ADAM17, the physiological sheddase of GPIbα[12–14]. The extracellular domains of GPIbβ and GPIX are smaller than that of GPIbα; each of them contains one LRR sequence[15].
Figure 1. Organization and structure of GPIb-IX.
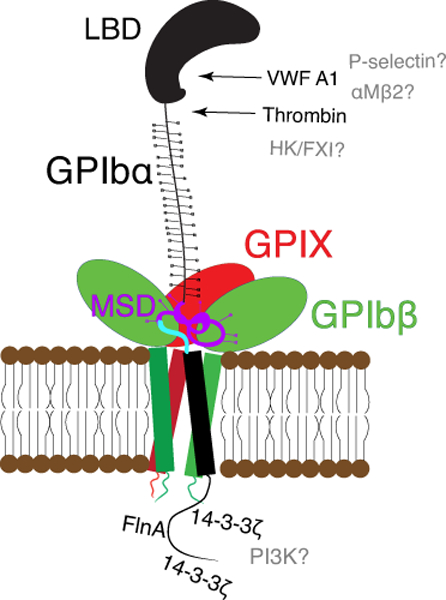
Cartoon illustration of the GPIb-IX complex including GPIbα (black), GPIbβ (green), GPIX (red). The N-terminal LBD of GPIbα is labeled, the membrane-proximal MSD is highlighted in purple, and the trigger sequence therein is highlighted in blue. The complex is held together by strong associations among the transmembrane domains as well as weak associations between GPIbβ and GPIX extracellular domains and potentially the MSD of GPIbα. Binding partners of the GPIbα LBD are listed to the right, including thrombin and VWF-A1. Intracellularly, 14-3-3ζ interacts with the intracellular tails of GPIbβ and GPIbα. FlnA binds to the tail of GPIbα. Binding partners for which a specific binding site has not been identified are listed in gray.
The transmembrane (TM) domains of GPIb-IX’s constituent subunits are highly conserved and contribute to the structural organization of the complex. The organization and assembly of GPIb-IX have been reviewed extensively elsewhere[16]. Briefly, the TM domains of GPIbα, GPIbβ, and GPIX associate to form a four-helical bundle, stabilizing the complex and facilitating formation of disulfide linkages between GPIbα and each GPIbβ subunit[3, 17]. GPIX and GPIbβ are also associated through noncovalent interactions between their extracellular domains[15]. Aside from stability granted by this interaction, disruption of the specific interfaces between GPIX and GPIbβ significantly decreases surface expression of the complex[15, 18]. The GPIbβ and GPIX extracellular domains are directly adjacent to the MSD of GPIbα (Fig. 1). Their association in the complex was recently proposed to be of significant functional consequence[19], although direct evidence for such an association is still missing. A summary of the structural features and binding partners for GPIb-IX is included in Figure 1.
GPIb-IX mutations in congenital diseases
The earliest recognition of GPIb-IX’s importance in hemostasis dates back to 1948 and the first description of the rare but severe bleeding condition, Bernard-Soulier syndrome (BSS), a congenital disorder presenting as bleeding and macrothrombocytopenia[20]. However, it was not until the 1970s that deficiency or dysfunction of GPIb-IX platelet was identified as the causative factor in BSS[21]. The etiological link between GPIb-IX deficiency and BSS was further cemented when BSS-like phenotypes were observed in knockout mice missing either GPIbα or GPIbβ[22–24]. BSS platelets are characterized by impaired ristocetin- and thrombin-induced aggregation. Genetic sequencing of BSS patients has identified mutations in genes encoding all three GPIb-IX subunits. Overall, the symptoms in BSS patients and the phenotypes of knockout mice are consistent with the requirement of expression of all subunits for efficient expression of GPIb-IX in the plasma membrane of transfected mammalian cells[5].
Pulse labeling studies indicate that GPIb-IX assembles initially in the endoplasmic reticulum (ER), with additional glycosylation modification including sialylation occurring in the Golgi compartment[25, 26]. Unassembled subunits, particularly GPIbα, are targeted for rapid degradation in the lysosome[25]. Recent studies have begun to identify the cellular machinery involved in folding and assembly of GPIb-IX, or degradation of misassembled GPIb-IX. For instance, mice with an inducible knockout of heat shock protein gp96/grp94, a molecular chaperone in the ER and a critical component of the unfolded protein response (UPR), present a phenotype indistinguishable from that of BSS[27]. Most mice missing core 1 β1,3-galactosyltransferase in their hematopoietic system and therefore extended or branched O-glycans on their platelets die perinatally from hemorrhage[28, 29], but the few survivors exhibit BSS-like phenotypes including bleeding, macrothrombocytopenia and markedly reduced expression of GPIbα on platelets[28]. The importance of O-glycans to GPIb-IX expression in platelets can be attributed to the stabilization of the MSD in GPIbα by sialic acids on these O-glycans, as removal of sialic acids by neuraminidase results in unfolding of the MSD and increased ectodomain shedding of GPIbα[30, 31].
Rapid degradation of newly synthesized GPIbα in the absence of other subunits suggests that unassembled GPIbα contains a region that can be recognized by the cellular machinery but is masked by the associated GPIbβ and GPIX. Recent studies suggest that such region is located in the MSD, since removal or replacement of the MSD, but not any other domains, resulted in significant increase of GPIbα expression in transfected mammalian cells without GPIbβ and GPIX[6, 19]. As the MSD is quasi-stable, it is conceivable that, in the absence of adjoining GPIbβ and GPIX extracellular domains, the isolated MSD could readily unfold and thus induce the unfolded protein response (UPR) in the ER[19].
Investigations of GPIb-IX mutations in BSS patient platelets and transfected cells suggest three general types of mutations that reduce the expression and/or function of GPIb-IX. Type 1 mutations disrupt the interaction between GPIb-IX subunits and include all frameshift or nonsense mutations in extracellular (including signal sequence) or TM domains of GPIb-IX. These mutations abolish the interaction between the TM domains and prevent stable assembly of the native complex[16]. Type 2 mutations include missense mutations in the LBD of GPIbα that impair ligand-binding activity. Many of these mutations interfere with the folding or stability of the LBD, which presumably induces UPR and markedly reduces GPIbα expression[32]. Type 3 mutations do not impact GPIb-IX expression and assembly, but instead abolish signaling. To-date, the sole example of this type is a homozygous nonsense mutation at residue Gln545 in the GPIbα cytoplasmic domain[33]. Platelets from the BSS patient bearing this mutation exhibit normal levels of GPIb-IX, consistent with earlier findings that the cytoplasmic domain of GPIbα does not participate in complex assembly or expression. Like other BSS patients, this patient responds poorly to ristocetin or thrombin stimulation. In this case, poor response is likely due to defects in GPIb-IX signal transduction, as the truncation at Gln545 eliminates the binding site for Filamin A (FlnA) (Fig. 1b) and possibly other signaling molecules[33].
In most cases BSS is inherited as an autosomal recessive disorder, presumably because 50% of the normal gene dose is enough to sustain platelet genesis and function. However, for several missense mutations in the LBD of GPIbα and a number of Type 1 mutations in GPIbβ, BSS is transmitted in an autosomal dominant manner[34, 35]. Many of these patients present mild macrothrombocytopenia and markedly reduced but detectable expression level of GPIb-IX. On the other hand, patients with 22q11 deletion syndrome (22q11DS) lack 0.7–3 million base pairs in their eponymous chromosome, which encompasses the GP1BB gene that encodes GPIbβ. Typically being hemizygotes for GP1BB, these patients have larger platelets, lower platelet counts, and bleed more excessively after reparative cardiac surgery[36]. However, a recent analysis found no correlation of the GP1BB copy number with either macrothrombocytopenia or bleeding[37].
Mutations in GPIbα can also lead to alterations in its binding to VWF. Under normal physiological conditions, plasma VWF is auto-inhibited and does not spontaneously bind to GPIbα and platelets. However, in platelet-type von Willebrand disease (PT-VWD), mutations in GPIbα cause spontaneous association with plasma VWF, somehow overcoming VWF autoinhibition. Four reported PT-VWD mutations are localized to the thumb region of GPIbα’s LBD[38]. Mouse models expressing these mutations largely recapitulate major symptoms observed in human patients[39, 40]. While these mutations likely cause structural or conformational alteration in the LBD[41], a fifth mutation, a 27-bp deletion at the junction of the sialomucin region and MSD of GPIbα, was also reported in one patient diagnosed with PT-VWD[42]. However, no follow-up studies have been reported, and the structural basis for this mutation’s effect on VWF binding remains to be seen. For comparison, a large deletion of the sialomucin region including the region in question does not alter ristocetin-mediated VWF binding under static conditions[43].
Platelet mechanosensation via GPIb-IX
When the endothelium suffers damage, the wound must be quickly sealed. In order to prevent significant blood loss, platelets must recognize endothelial damage, arrest, and remain in place long enough to begin forming a platelet plug under significant shear from blood flow. Although platelets activate readily at the site of injury, it is also critical that they remain inert in circulation. In mammalian platelets, GPIb-IX facilitates binding to damaged endothelia and mediates a force-dependent response. Although several receptors on platelets stabilize the adhesion of platelets to subendothelial matrix, these receptors cannot initiate thrombus formation without an initial interaction between GPIb-IX and collagen-tethered VWF, especially in high-shear regions of the vasculature[44, 45].
Although the GPIb-IX/VWF interaction has been recognized as the key event in sensing and responding to shear stress for over 25 years[1], a unified mechanism for GPIb-IX activation by ligand binding under shear has remained elusive until recently. In the early 2000s, a “clustering model” of GPIb-IX activation was proposed[46]. This model explained the observation that monoclonal antibodies (MAbs) targeting the LBD of GPIbα can activate GPIb-IX, provided they are bivalent (not monomeric Fab fragments)[47, 48]. Under this model, MAbs binding to two copies of GPIbα (one with each Fab) induces lateral dimerization or “clustering” of GPIb-IX and subsequent clearance[49]. However, the clustering model falls short of unifying all available evidence regarding GPIb-IX mechanosensation and activation. It offers no explanation for why MAbs targeting other regions of GPIb-IX don’t induce receptor activation[50], does not account for the well-established requirement of shear in VWF-mediated GPIb-IX signaling[1], and does not address numerous observations that the extracellular domains of GPIbβ participate in GPIb-IX signaling[14, 24, 51]. Thus, evidence for clustering of GPIb-IX in the membrane alone may not be sufficient to demonstrate activation of the receptor complex.
Recent studies using single-molecule optical tweezers to “pull” on the LBD of GPIbα have identified a force-sensitive domain in GPIbα’s extracellular domain that unfolds when pulling force is applied to anchored GPIb-IX from the LBD. Unfolding was localized to the juxtamembrane mechanosensory domain (MSD) between the sialomucin region and the TM domain[10]. The MSD unfolds under a continuous pulling force of ~15 pN[11, 14, 52]. Although the precise boundary of the MSD is not settled, estimates based on mutagenesis and fitting of single-molecule data estimate that the MSD spans about 60 residues. Recent studies indicate that this domain is structured and quasi-stable, and its stability is altered by O-glycosylation therein[10, 30].
The trigger model of GPIb-IX activation
In the mid-1990s, Kroll et al. described the mechanism of platelet shear-sensitivity as follows, “The initial shear-induced triggering event has so far been elusive…one hypothesis proposed to explain the mechanism of shear-induced platelet activation states that platelet GPIbα, following shear-induced binding of vWF, undergoes a conformational change that…triggers signals for cellular activation.” In their 2016 study, Deng et al. proposed the trigger model of GPIb-IX activation. Applying physiological shear stress to platelet-rich plasma, they demonstrate that VWF binding to the LBD leads to shear-dependent MSD unfolding and platelet signaling including elevation of intracellular calcium, P-selectin exposure, and surface desialylation[14]. Within the MSD, which is poorly conserved between species, a short 12-residue segment immediately preceding the TM domain exhibits remarkable sequence conservation in mammals (Fig 2). Exposure of this region, dubbed the “trigger sequence”, following MSD unfolding appears to be the crucial step in GPIb-IX activation. Deletion of the MSD leaving only the trigger sequence leads to constitutive ligand-free activation of GPIb-IX in CHO cells expressing this mutant[14]. IL4R-IbαTg mice express a chimeric GPIbα in which most of the extracellular domain has been replaced by that of the α-subunit of the interleukin-4 receptor (IL4R) leaving only the trigger sequence. These mice have a significantly lower platelet count and their platelets have a higher base level of activation[14, 53]. Utilizing a biomembrane force probe instrument to measure force and cell signaling at the single-cell and single-molecule level, the Zhu group characterized pulling force regimens for recombinant A1 domain or anti-GPIbα antibodies binding to an immobilized platelet. An extension event consistent with unfolding of the MSD for a certain time period, and sometimes in conjunction with unfolding of the LBD, was required to induce intracellular calcium flux in the platelet, thus providing additional evidence linking MSD unfolding to GPIb-IX signaling[52]. Together, these data support a trigger model of GPIb-IX signaling, wherein a pulling force exerted on GPIbα through the LBD leads to unfolding of the MSD, exposure of the trigger sequence, activation of the receptor, and subsequent platelet signaling and/or clearance (Fig. 3). Under this model, collagen-anchored VWF binds to the LBD and shear from blood flow may provide the force required to unfold the MSD, thereby activating GPIb-IX.
Figure 2. Multiple sequence alignment of the GPIbα MSD.

(A) MSD sequence of GPIbα orthologs from various species. The MSD, transmembrane domain (TMD), and trigger sequence are demarcated. Residues with identity match to the human sequence are listed in red. The triangle denotes the ADAM17 shedding cleavage site in human GPIbα. (B) Phylogenetic tree constructed from multiple sequence alignment of trigger sequences for each of the species in (a).
Figure 3. Activation and regulation of GPIb-IX signaling.
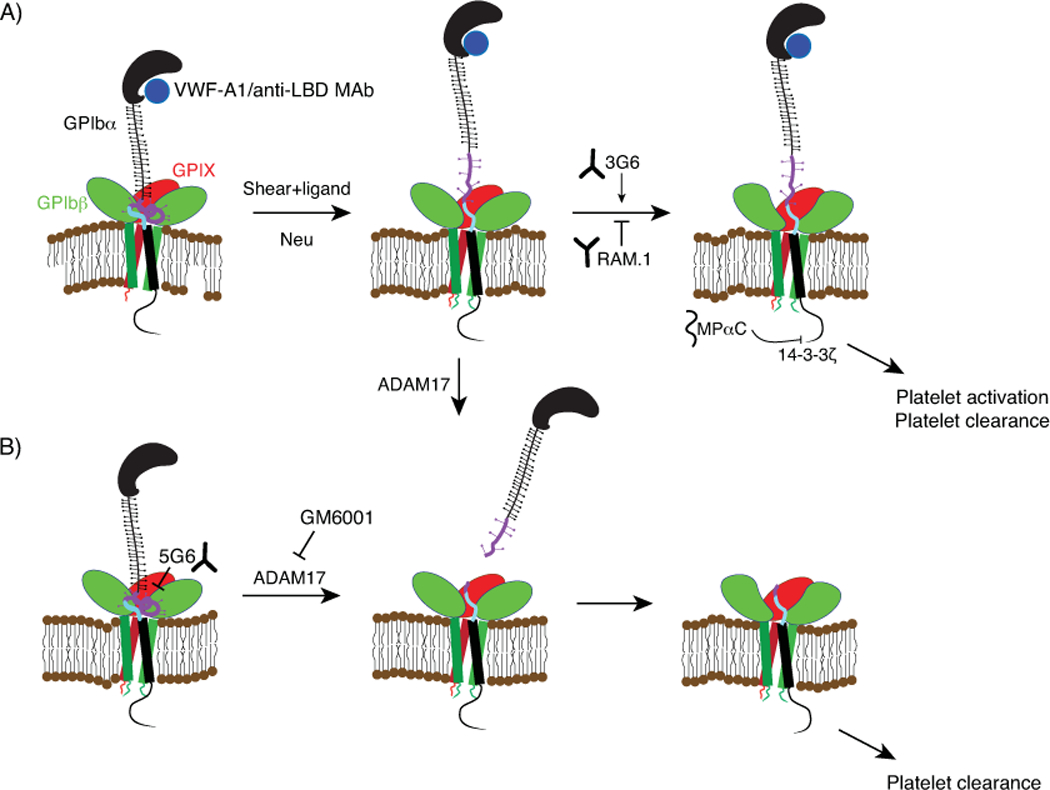
(A) The Trigger model of GPIb-IX activation. Binding of VWF or anti-LBD antibodies to the LBD of GPIbα under physiological shear induces MSD unfolding. Neuraminidase (Neu) treatment in the absence of shear also induces MSD unfolding. Unfolding of the MSD leads to exposure of the trigger sequence therein, likely inducing a conformational change in the adjoining GPIbβ/GPIX and subsequent GPIb-IX signaling into the cell. This activation is modulated by anti-GPIbβ MAbs 3G6 (which potentiates activation) and RAM.1 (which inhibits activation). MPαC competitively inhibits 14-3-3ζ binding to the intracellular tail of GPIbα, diminishes its downstream signaling, and reduces cell adhesion to VWF. The membrane-proximal MSD is highlighted in purple, and the trigger sequence therein is highlighted in blue (B) Ectodomain shedding of GPIb-IX induced by ADAM17 cleavage of a site within the MSD induces shear-independent activation. Shedding can be inhibited by the anti-MSD MAb 5G6 or metalloproteinase inhibitors such as GM6001. Note that unfolding of the MSD facilitates ADAM17-mediated shedding of GPIbα.
Triggering thrombocytopenic disorders
In addition to GPIb-IX’s role in normal hemostasis, the trigger model also explains several GPIb-IX-centric disease states. GPIb-IX is a common target for autoantibodies in patients with immune thrombocytopenia (ITP). The presence of antibodies against GPIb-IX is strongly associated with refractoriness to common first-line immunosuppressive treatments like intravenous immunoglobulin (IVIg) and corticosteroids[54]. Antibodies targeting the LBD of GPIbα can activate the receptor and cause platelet desialylation[55, 56]. In the case of IVIg-resistant ITP, the dimeric structure of activating anti-LBD MAbs permits them to crosslink platelets via GPIb-IX[56]. Under physiological shear, this generates a pulling force on GPIbα, activating GPIb-IX via MSD unfolding and inducing immune-independent clearance. Furthermore, if an antibody’s unbinding force from the LBD is insufficient to sustain the force applied by physiological shear, it will not induce GPIb-IX-mediated clearance[56]. Thus, it appears that the defining characteristic of an activating ligand to GPIb-IX is the ability to bind to the LBD and sustain at least 15 pN of force, the amount required to unfold the MSD.
Association of VWF and GPIbα underlies many thrombocytopenic and thrombotic disorders. In contexts where VWF binds spontaneously to GPIbα in bloodstream, multimeric VWF may act as a crosslinking ligand capable of forming VWF-platelet complex and activating GPIb-IX therein. Ristocetin (used as an antibiotic) can induce the VWF-GPIbα interaction in the absence of shear and was pulled from clinical use because it caused thrombocytopenia[57]. Injection of botrocetin, a snake venom that causes spontaneous VWF-GPIbα interaction, induces acute thrombocytopenia in animals[58]. Patients with type 2B VWD exhibit spontaneous VWF-GPIbα binding and, similar to the effects of anti-GPIb-IX antibodies in ITP patients, present with accelerated platelet clearance, reduced thrombopoiesis, and thrombocytopenia[59]. Transgenic mice expressing type 2B VWF or PT-VWD mutant GPIbα exhibit thrombocytopenia partly due to clearance of large VWF-platelet complexes[40, 60]. The etiology of the bleeding disorder thrombotic thrombocytopenia purpura (TTP) follows a similar pattern. In individuals with TTP, a deficiency of functional ADAMTS13 prevents cleavage of ultra-long (UL)VWF, permitting spontaneous binding to GPIbα and subsequent thrombocytopenia. Not unrelatedly, increased or altered hemodynamic shear produced by mechanic pumps in circulatory support devices such as ventricular assist devices and extracorporeal membrane oxygenation machine likely activate VWF, induce formation of VWF-platelet complexes, and result in undesired thrombocytopenic and thrombotic complications[61, 62]. Moreover, pathological binding of VWF to GPIbα also appears to be a mechanism of thrombocytopenia during infection of malaria parasite or dengue virus [63, 64]. It should be nonetheless noted that, although desialylated VWF binds GPIbα spontaneously, neuraminidase can induce platelet clearance in a VWF-independent manner by directly desialylate O-glycans in GPIbα[30].
Ligand- and shear-free mechanisms of GPIb-IX activation
Recent studies suggest that GPIbα ectodomain shedding mediated by ADAM17 and desialylation-dependent unfolding of the MSD are unique mechanisms of GPIb-IX activation which proceed in congruence with the trigger model of GPIb-IX activation, but without the requirement of shear.
In addition to the trigger sequence, the MSD of GPIbα also contains the cleavage site for the metalloproteinase ADAM17 (Fig. 2), which continuously sheds the majority of the extracellular domain of GPIbα (known as glycocalicin) from the platelet surface[12]. Although the shedding cleavage site is still accessible when the MSD is folded, consistent with constant shedding of glycocalicin from resting platelets, shear-induced MSD unfolding may further expose this sequence to ADAM17[14]. Since glycocalicin contains the LBD, ectodomain shedding of GPIbα via ADAM17 reduces the association of VWF multimer with the platelet and is thought as a means to down-regulate formation of the VWF-platelet complex. On the other hand, as the shedding cleavage site is N-terminal to the trigger sequence, upon cleavage by ADAM17, the structure of the MSD may be disrupted, exposing the trigger sequence and inducing GPIb-IX signaling (Fig. 3B). Indeed, GPIbα mutants disrupting the MSD’s structure and exposing the trigger sequence lead to ligand-free signaling and platelet clearance[14]. Shedding of GPIbα is an important event linked to platelet clearance, especially in the context of platelet storage[65]. The extent of GPIbα shedding is tightly correlated to the platelet storage lesion and inhibition of shedding has been proposed as a potential strategy for improving the survival of stored platelets. Blocking GPIbα shedding with metalloproteinase inhibitors or a GPIbα-specific MAb 5G6 improves the survival of in vitro aged platelets[66, 67] (Fig. 3B).
As previously mentioned, the MSD of GPIbα contains several O-glycosylation sites. Neuraminidase, a glycolytic enzyme released by some bacterial infections and from platelets endogenously, hydrolyzes the glycosidic linkages to sialic acids in branched glycans and exposes the penultimate galactoses. Injection with exogenous neuraminidase leads to thrombocytopenia in animal models[68], and platelet neuraminidase appears to be critically involved in clearance[31, 55, 69]. Galactoses exposed by neuraminidase can mark platelets for clearance in the liver, in which the Ashwell-Morell receptor, macrophage galactose lectin, and other receptors have been implicated[29, 31, 70, 71]. Given the high percent of total platelet sialic acid that is bound to GPIbα, it is conceivable that galactoses on desialylated GPIbα may be important for recognition by clearance receptors. However, no direct evidence for the interaction of desialylated GPIbα with a clearance receptor has been reported. It is noteworthy that while most of these clearance receptors are oligomeric and prefer to bind multiple galactoses on N-glycans with high affinity, murine GPIbα does not contain the canonical N-glycosylation sites (i.e. NxS/T). A recent study has identified that neuraminidase-mediated desialylation of O-glycans on GPIbα induces MSD unfolding and GPIb-IX signaling[30]. Here, GPIb-IX-mediated intracellular signaling includes further platelet desialylation, which could conceivably lead to desialylation of N-glycans of other glycoproteins on the platelet membrane, which can be recognized by clearance receptors.
Signaling of GPIb-IX
In addition to its adhesive functions, GPIb-IX activation and associated signaling are vital to the initiation of platelet activation during primary hemostasis. During this phase, VWF-dependent GPIb-IX activation leads to inside-out activation of the platelet integrin αIIbβ3[72, 73], formation of platelet microparticles[74, 75], TXA2 synthesis and release[76, 77], degranulation[77], desialylation via NEU1[55], and many other procoagulant phenomena. Many of these phenomena have also been observed when GPIb-IX is activated by anti-LBD MAbs[55, 56].
Several downstream signaling mediators of GPIb-IX have been identified including: Ca2+, Src family kinases, phospholipase C, PI3K; mitogen-activated protein kinase (MAPK) pathway; and LIM kinase pathway[78]. In particular, numerous studies have been published investigating the role of the regulatory protein 14-3-3ζ, which binds to the cytoplasmic tails of both GPIbα and GPIbβ[79, 80](Fig. 1b). Multiple binding sites for 14-3-3ζ have been found in the GPIbα cytoplasmic domain, and some of them may overlap or be in proximity to binding sites for PI3K and FlnA [78, 81]. Studies of transfected cells expressing GPIb-IX and αIIbβ3 suggest that 14-3-3ζ’s interaction with GPIbα is necessary for VWF-induced activation of αIIbβ3 binding to fibrinogen[72]. Consistently, addition of membrane-permeable peptides that are derived from the GPIbα intracellular tail and competitively inhibit 14-3-3ζ’s interaction with GPIbα to platelets inhibits ristocetin-induced platelet aggregation [82, 83]. It appears that 14-3-3ζ is involved in GPIb-IX-mediated platelet signaling, while another study suggests that sequestration of 14-3-3ζ by GPIbα may counteract αIIbβ3 activation[84]. It has also been reported that GPIbα-bound 14-3-3ζ regulates adhesion to VWF [81–83, 85, 86] and platelet apoptosis[87, 88]. The distinctions between GPIb-IX signaling pathways leading to platelet clearance and platelet activation remain unclear.
Additional functions of GPIb-IX
The best-established and most extensively studied functions of GPIb-IX are in the context of hemostasis, where it mediates binding to vascular damage via VWF and initiates platelet signaling. However, GPIb-IX interacts with several other ligands and counter-receptors (Fig. 1), and ongoing work continues to reveal diverse roles for GPIb-IX in thrombosis, inflammation, and platelet genesis[74].
The leukocyte integrin αMβ2 (Mac-1) interacts with GPIbα, allowing leukocytes to adhere and migrate along sites where of vascular injury where platelets are accumulating[89]. Inhibition of this binding inhibits stable interactions between leukocytes and platelets, reducing leukocyte accumulation at the site of injury[90]. This implies that the αMβ2-GPIbα interaction is vital to leukocyte adhesion and the inflammatory response to vascular injury. Alternatively, mice with mutant αMβ2 deficient in GPIbα binding or αMβ2 knockout mice have delayed thrombosis, and it has been suggested that inhibition of the αMβ2-GPIbα interaction has therapeutic potential as an anti-thrombotic[91].
GPIbα is the high-affinity platelet receptor of thrombin[92]. GPIbα binding to thrombin promotes thrombin’s cleavage and activation of PAR1, its low-affinity platelet receptor, and GPIb-IX-mediated signaling plays a role in cooperation with PAR1 signaling[93, 94]. Relatedly, recent animal studies suggest that the modest increase in acute coronary syndromes associated with the use of oral thrombin inhibitors involves the GPIbα-thrombin interaction[95].
GPIb-IX has been implicated in platelet generation and regulation of platelet size. The primary role for GPIb-IX in platelet genesis appears to be in thrombopoiesis, rather than megakaryopoiesis. Although BSS mice develop normal megakaryocytes (MKs), their proplatelet formation is impaired[96]. Shear flow is important for the development of proplatelets from MKs, especially in the context of in vitro platelet production. Interestingly, VWF binding to GPIbα under shear has been implicated in this process, and inhibition of GPIbα ectodomain shedding in MK cultures improves yields of functional platelets[97]. Thrombopoiesis is also altered in the context of type 2B VWD, wherein patients’ MKs have disordered demarcation membrane systems, smaller proplatelets, and abnormal granule distribution[98]. In a mouse model of type 2B VWD, proplatelet formation is significantly reduced, likely as a result of upregulation of the LIM kinase pathway and actin disorganization[99]. Altogether these data suggest an important role for GPIb-IX, and specifically the VWF-GPIbα interaction, in MK response to shear and regulation of proplatelet formation and fragmentation.
Recent studies have also utilized the IL4R-IbαTg mice to implicate GPIbα in the process of platelet accumulation to the liver in the context of liver production of thrombopoietin or non-alcoholic fatty liver diseases[100, 101]. In these cases, GPIbα appears to mediate association of platelets with cells in the liver, such as hepatocytes and Kupffer cells. Curiously, mice lacking P-selectin, VWF, or Mac-1, all of which are known ligands of GPIbα, do not show delays in the onset of liver diseases[101]. This suggests on these cells the existence of a new counter-receptor for GPIbα, the identity of which remains to be discovered.
Platelets have diverse roles in the progression of several cancers, interacting with tumor cells in many capacities. Studies in the 1980s and 1990s implicated GPIb-IX (specifically its interaction with VWF) in tumor cell-induced platelet aggregation (TCIPA), indicating that inhibition of the GPIbα-VWF interaction interferes with TCIPA[102, 103]. Further, VWF−/− mice or IL4R-IbαTg mice show reduced metastasis[104, 105]. A comprehensive model of cancer cell expression of VWF, binding to platelets, and the mechanism by which these interactions promote metastasis is an ongoing area of research.
Modulation of GPIb-IX
Many adhesion receptor complexes mediate bidirectional signal transduction, as exemplified by the outside-in and inside-out activation of integrins. In this review, “GPIb-IX activation” refers to outside-in activation - the induction of GPIb-IX-mediated intracellular signaling as a result of its binding to ligands. Thus far, GPIbα is the only subunit of GPIb-IX demonstrated to mediate receptor activation, containing both the MSD and the binding site for all known ligands. Unfolding of the MSD appears as a critical step in this process, converting the ligand-binding status under shear into a distinct conformational state that could be detected by and propagated through the rest of GPIb-IX. The extracellular domains of GPIX and GPIbβ are proximal to the MSD of GPIbα (Fig. 3), and even assuming full extension of the MSD, ~10 residues of the Trigger sequence could be in direct contact with them[15, 16]. However, the contributions of GPIbβ and GPIX to receptor activation and signaling remain ambiguous. Recent evidence suggests that GPIbβ and GPIX extracellular domains are malleable and could undergo conformational changes[18]. Several studies indicate that RAM.1, a MAb targeting the extracellular domain of GPIbβ, abolishes GPIb-IX signaling including intracellular Ca2+ flux and changes in morphology without an effect on ligand binding[14, 24, 51] (Fig. 3A). Data from our lab indicate that another anti-GPIbβ MAb, 3G6, can amplify or “potentiate” GPIb-IX activation by VWF and anti-LBD MAbs including degranulation, desialylation, and morphological changes (Quach et al. manuscript submitted). Although these findings clearly implicate GPIbβ in the modulation of GPIb-IX signaling, the precise role of GPIbβ in GPIb-IX signal transduction, and the nature of the contacts between GPIbβ and the MSD remain elusive. Similarly, the role of GPV in GPIb-IX function is another area where our understanding is currently evolving. Genetic ablation of GPV accelerates GPIbα/thrombin-dependent platelet activation, and GPV−/− mice exhibit faster occlusion times than wild-type[106, 107]. Thus, GPV may dampen GPIb-IX activation and signaling, but the underlying molecular mechanism remains elusive.
Is there inside-out modulation of GPIb-IX activity?
Inside-out activation of an adhesion receptor was first reported for integrin αIIbβ3 when its binding affinity (not avidity) for ligands was substantially increased as a result of mutations in its cytoplasmic domains. This inside-out activation, in which conformational changes in the membrane-proximal portion of the integrin extend allosterically to its membrane-distal ligand-binding domain, is an important physiological step during platelet activation. It was reported in early 2000’s that transfected cells expressing GPIb-IX with mutations in the cytoplasmic domains to perturb its interaction with 14-3-3 proteins exhibited altered binding to VWF in the presence of ristocetin or altered adhesion to VWF under flow conditions[82, 108, 109]. Based on these results, a toggle switch model in which switching of 14-3-3ζ binding between GPIbα and GPIbβ determines the binding affinity of GPIbα, or its accessibility, to VWF by an inside-out mechanism was proposed in 2005 [78, 82]. However, as of the time of this publication, this model has not seen any follow-up in the literature, including details describing the changes in interactions between 14-3-3ζ and its multiple binding sites in GPIbα and GPIbβ. Unlike the case of αIIbβ3 inside-out activation, it remains unclear how a change in the GPIbα cytoplasmic domain induces a conformational change in the LBD or a change in its accessibility, particularly through the long sialomucin region. Thusfar, there have been no reports of the LBD exhibiting two distinct binding affinities to VWF or a specific shielding mechanism for the LBD.
It is noteworthy that 14-3-3ζ’s association with GPIb-IX affects its binding to VWF multimers but not recombinant A1 domain of VWF [108]. In addition, RAM.1 significantly reduces adhesion of platelets or cells expressing wild-type GPIb-IX, but not cells expressing GPIb-IX with certain cytoplasmic mutations affecting 14-3-3ζ association, to VWF under flow conditions[85]. Yet RAM.1 does not affect the binding affinity of purified GPIb-IX to VWF or recombinant A1-A2-A3 fragment[110]. Thus, it appears that 14-3-3ζ modulates the binding avidity, rather than affinity, of GPIb-IX to VWF. As 14-3-3ζ mediates GPIb-IX signaling, perturbing its association with GPIb-IX may alter the morphology of the host cell, the spatial distribution of GPIb-IX therein, and subsequently cell association to VWF multimers or cell adhesion to VWF. In other words, 14-3-3ζ’s effect on VWF binding/adhesion may be due to GPIb-IX-mediated signaling rather than inside-out regulation of GPIb-IX.
Conclusion
The GPIb-IX complex on the platelet surface is critical to many aspects of platelet physiology, including platelet recruitment to vascular injuries, platelet activation, and platelet clearance. It is also implicated in thrombosis, inflammation, and many other associated pathologies. In particular, spontaneous or aberrant association of VWF with GPIbα is a key feature in many pathological contexts, including type 2B VWD, TTP, and likely other thrombotic thrombocytopenic disorders. Recent elucidation of interactions between GPIbα and the other subunits of GPIb-IX, particularly the identification and characterization of the MSD therein, has provided a new framework for future investigations of functions and regulations of GPIb-IX.
Acknowledgements
This work was supported in part by NIH grants HL082808, HL143794, and HL146299. MEQ was supported in part by HL134241.
Footnotes
Addendum
MEQ and RL wrote the manuscript.
The authors declare no conflicts of interest.
References
- 1.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996; 88: 1525–41. [PubMed] [Google Scholar]
- 2.Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012; 120: e73–82. 10.1182/blood-2012-04-416594. [DOI] [PubMed] [Google Scholar]
- 3.Luo SZ, Mo X, Afshar-Kharghan V, Srinivasan S, Lopez JA, Li R. Glycoprotein Ibalpha forms disulfide bonds with 2 glycoprotein Ibbeta subunits in the resting platelet. Blood. 2007; 109: 603–9. 10.1182/blood-2006-05-024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez JA. The platelet glycoprotein Ib-IX complex. Blood Coagul Fibrinolysis. 1994; 5: 97–119. [PubMed] [Google Scholar]
- 5.Lopez JA, Leung B, Reynolds CC, Li CQ, Fox JEB. Efficient plasma membrane expression of a functional platelet glycoprotein Ib-IX complex requires the presence of its three subunits. J Biol Chem. 1992; 267: 12851–9. [PubMed] [Google Scholar]
- 6.Mo X, Lu N, Padilla A, López JA, Li R. The transmembrane domain of glycoprotein Ibβ is critical to efficient expression of glycoprotein Ib-IX complex in the plasma membrane. J Biol Chem. 2006; 281: 23050–9. [DOI] [PubMed] [Google Scholar]
- 7.Mo X, Liu L, Lopez JA, Li R. Transmembrane domains are critical to the interaction between platelet glycoprotein V and glycoprotein Ib-IX complex. J Thromb Haemost. 2012; 10: 1875–86. 10.1111/j.1538-7836.2012.04841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ, Gros P. Structures of glycoprotein Iba and its complex with von Willebrand factor A1 domain. Science. 2002; 297: 1176–9. [DOI] [PubMed] [Google Scholar]
- 9.Grottum KA, Solum NO. Congenital thrombocytopenia with giant platelets: a defect in the platelet membrane. Br J Haematol. 1969; 16: 277–90. [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, Deng W, Zhou L, Xu Y, Yang W, Liang X, Wang Y, Kulman JD, Zhang XF, Li R. Identification of a juxtamembrane mechanosensitive domain in the platelet mechanosensor glycoprotein Ib-IX complex. Blood. 2015; 125: 562–9. 10.1182/blood-2014-07-589507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang XF, Zhang W, Quach ME, Deng W, Li R. Force-regulated refolding of the mechanosensory domain in the platelet glycoprotein Ib-IX complex. Biophys J. 2019; 116: 1960–9. 10.1016/j.bpj.2019.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergmeier W, Piffath CL, Cheng G, Dole VS, Zhang Y, von Andrian UH, Wagner DD. Tumor necrosis factor-α-converting enzyme (ADAM17) mediates GPIbα shedding from platelets in vitro and in vivo. Circ Res. 2004; 95: 677–83. [DOI] [PubMed] [Google Scholar]
- 13.Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by ADAM family metalloproteinases. J Thromb Haemost. 2007; 5: 1530–7. [DOI] [PubMed] [Google Scholar]
- 14.Deng W, Xu Y, Chen W, Paul DS, Syed AK, Dragovich MA, Liang X, Zakas P, Berndt MC, Di Paola J, Ware J, Lanza F, Doering CB, Bergmeier W, Zhang XF, Li R. Platelet clearance via shear-induced unfolding of a membrane mechanoreceptor. Nat Commun. 2016; 7: 12863. 10.1038/ncomms12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McEwan PA, Yang W, Carr KH, Mo X, Zheng X, Li R, Emsley J. Quaternary organization of GPIb-IX complex and insights into Bernard-Soulier syndrome revealed by the structures of GPIbβ and a GPIbβ/GPIX chimera. Blood. 2011; 118: 5292–301. 10.1182/blood-2011-05-356253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, Emsley J. The organizing principle of the platelet glycoprotein Ib-IX-V complex. J Thromb Haemost. 2013; 11: 605–14. 10.1111/jth.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo SZ, Li R. Specific heteromeric association of four transmembrane peptides derived from platelet glycoprotein Ib-IX complex. J Mol Biol. 2008; 382: 448–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou L, Yang W, Li R. Analysis of inter-subunit contacts reveals the structural malleability of extracellular domains in platelet glycoprotein Ib-IX complex. J Thromb Haemost. 2014; 12: 82–9. 10.1111/jth.12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao Y, Gan C, Zhang X, Liu L, Zakas PM, Doering CB, Mo X, Li R. Unaccompanied mechanosensory domain mediates low expression of glycoprotein Ibalpha: implications for Bernard-Soulier syndrome. J Thromb Haemost. 2020; 18: 510–7. 10.1111/jth.14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bernard J, Soulier JP. Sur une nouvelle variété de dystrophie thrombocytaire-hémorragipare congénitale. Sem Hop. 1948; 24: 3217–23. [PubMed] [Google Scholar]
- 21.Jenkins CS, Phillips DR, Clemetson KJ, Meyer D, Larrieu MJ, Luscher EF. Platelet membrane glycoproteins implicated in ristocetin-induced aggregation. Studies of the proteins on platelets from patients with Bernard-Soulier syndrome and von Willebrand’s disease. J Clin Invest. 1976; 57: 112–24. 10.1172/JCI108251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ware J, Russell S, Ruggeri ZM. Generation and rescue of a murine model of platelet dysfunction: the Bernard-Soulier syndrome. Proc Natl Acad Sci USA. 2000; 97: 2803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato K, Martinez C, Russell S, Nurden P, Nurden A, Fiering S, Ware J. Genetic deletion of mouse platelet glycoprotein Ibβ produces a Bernard-Soulier phenotype with increased α-granule size. Blood. 2004; 104: 2339–44. [DOI] [PubMed] [Google Scholar]
- 24.Ravanat C, Strassel C, Hechler B, Schuhler S, Chicanne G, Payrastre B, Gachet C, Lanza F. A central role of GPIb-IX in the procoagulant function of platelets that is independent of the 45-kDa GPIbα N-terminal extracellular domain. Blood. 2010; 116: 1157–64. 10.1182/blood-2010-01-266080. [DOI] [PubMed] [Google Scholar]
- 25.Dong JF, Gao S, Lopez JA. Synthesis, assembly, and intracellular transport of the platelet glycoprotein Ib-IX-V complex. J Biol Chem. 1998; 273: 31449–54. [DOI] [PubMed] [Google Scholar]
- 26.Ulsemer P, Strassel C, Baas MJ, Salamero J, Chasserot-Golaz S, Cazenave JP, De La Salle C, Lanza F. Biosynthesis and intracellular post-translational processing of normal and mutant platelet glycoprotein GPIb-IX. Biochem J. 2001; 358: 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staron M, Wu S, Hong F, Stojanovic A, Du X, Bona R, Liu B, Li Z. Heat-shock protein gp96/grp94 is an essential chaperone for the platelet glycoprotein Ib-IX-V complex. Blood. 2011; 117: 7136–44. 10.1182/blood-2011-01-330464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Jobe SM, Ding X, Choo H, Archer DR, Mi R, Ju T, Cummings RD. Platelet biogenesis and functions require correct protein O-glycosylation. Proc Natl Acad Sci USA. 2012; 109: 16143–8. 10.1073/pnas.1208253109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Fu J, Ling Y, Yago T, McDaniel JM, Song J, Bai X, Kondo Y, Qin Y, Hoover C, McGee S, Shao B, Liu Z, Sonon R, Azadi P, Marth JD, McEver RP, Ruan C, Xia L. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proc Natl Acad Sci U S A. 2017; 114: 8360–5. 10.1073/pnas.1707662114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Chen W, Zhang W, Lee-Sundlov MM, Casari C, Berndt MC, Lanza F, Bergmeier W, Hoffmeister KM, Zhang XF, Li R. Desialylation of O-glycans on glycoprotein Ibalpha drives receptor signaling and platelet clearance. Haematologica. 2020: Epub ahead of print. 10.3324/haematol.2019.240440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jansen AJ, Josefsson EC, Rumjantseva V, Liu QP, Falet H, Bergmeier W, Cifuni SM, Sackstein R, von Andrian UH, Wagner DD, Hartwig JH, Hoffmeister KM. Desialylation accelerates platelet clearance after refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice. Blood. 2012; 119: 1263–73. 10.1182/blood-2011-05-355628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenberg N, Lalezari S, Landau M, Shenkman B, Seligsohn U, Izraeli S. Trp207Gly in platelet glycoprotein Ibα is a novel mutation that disrupts the connection between the leucine-rich repeat domain and the disulfide loop structure and causes Bernard-Soulier syndrome. J Thromb Haemost. 2007; 5: 378–86. 10.1111/j.1538-7836.2007.02298.x. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto N, Akamatsu N, Sakuraba H, Matsuno K, Hosoya R, Nogami H, Kasahara K, Mitsuyama S, Arai M. Novel Bernard-Soulier syndrome variants caused by compound heterozygous mutations (case I) or a cytoplasmic tail truncation (case II) of GPIbalpha. Thromb Res. 2013; 131: e160–7. 10.1016/j.thromres.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 34.Savoia A, Balduini CL, Savino M, Noris P, Del Vecchio M, Perrotta S, Belletti S, Poggi V, Iolascon A. Autosomal dominant macrothrombocytopenia in Italy is most frequently a type of heterozygous Bernard-Soulier syndrome. Blood. 2001; 97: 1330–5. [DOI] [PubMed] [Google Scholar]
- 35.Sivapalaratnam S, Westbury SK, Stephens JC, Greene D, Downes K, Kelly AM, Lentaigne C, Astle WJ, Huizinga EG, Nurden P, Papadia S, Peerlinck K, Penkett CJ, Perry DJ, Roughley C, Simeoni I, Stirrups K, Hart DP, Tait RC, Mumford AD, BioResource N, Laffan MA, Freson K, Ouwehand WH, Kunishima S, Turro E. Rare variants in GP1BB are responsible for autosomal dominant macrothrombocytopenia. Blood. 2017; 129: 520–4. 10.1182/blood-2016-08-732248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brenner MK, Clarke S, Mahnke DK, Simpson P, Bercovitz RS, Tomita-Mitchell A, Mitchell ME, Newman DK. Effect of 22q11.2 deletion on bleeding and transfusion utilization in children with congenital heart disease undergoing cardiac surgery. Pediatr Res. 2016; 79: 318–24. 10.1038/pr.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zwifelhofer NMJ, Bercovitz RS, Weik LA, Moroi A, LaRose S, Newman PJ, Newman DK. Hemizygosity for the gene encoding glycoprotein Ibbeta is not responsible for macrothrombocytopenia and bleeding in patients with 22q11 deletion syndrome. J Thromb Haemost. 2019; 17: 295–305. 10.1111/jth.14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Othman M, Kaur H, Emsley J. Platelet-type von Willebrand disease: new insights into the molecular pathophysiology of a unique platelet defect. Semin Thromb Hemost. 2013; 39: 663–73. 10.1055/s-0033-1353442. [DOI] [PubMed] [Google Scholar]
- 39.Guerrero JA, Kyei M, Russell S, Liu J, Gartner TK, Storrie B, Ware J. Visualizing the von Willebrand factor/glycoprotein Ib-IX axis with a platelet-type von Willebrand disease mutation. Blood. 2009; 114: 5541–6. 10.1182/blood-2009-03-210823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bury L, Malara A, Momi S, Petito E, Balduini A, Gresele P. Mechanisms of thrombocytopenia in platelet-type von Willebrand disease. Haematologica. 2019; 104: 1473–81. 10.3324/haematol.2018.200378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dumas JJ, Kumar R, McDonagh T, Sullivan F, Stahl ML, Somers WS, Mosyak L. Crystal structure of the wild-type von Willebrand factor A1-glycoprotein Iba complex reveals conformation differences with a complex bearing von Willebrand disease mutations. J Biol Chem. 2004; 279: 23327–34. [DOI] [PubMed] [Google Scholar]
- 42.Othman M, Notley C, Lavender FL, White H, Byrne CD, Lillicrap D, O’Shaughnessy DF. Identification and functional characterization of a novel 27-bp deletion in the macroglycopeptide-coding region of the GPIbα gene resulting in platelet-type von Willebrand disease. Blood. 2005; 105: 4330–6. [DOI] [PubMed] [Google Scholar]
- 43.Li CQ, Dong JF, Lopez JA. The mucin-like macroglycopeptide region of glycoprotein Iba is required for cell adhesion to immobilized von Willebrand factor (VWF) under flow but not for static VWF binding. Thromb Haemost. 2002; 88: 673–7. [PubMed] [Google Scholar]
- 44.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998; 94: 657–66. [DOI] [PubMed] [Google Scholar]
- 45.Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996; 84: 289–97. [DOI] [PubMed] [Google Scholar]
- 46.Shrimpton CN, Borthakur G, Larrucea S, Cruz MA, Dong JF, Lopez JA. Localization of the adhesion receptor glycoprotein Ib-IX-V complex to lipid rafts is required for platelet adhesion and activation. J Exp Med. 2002; 196: 1057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nieswandt B, Bergmeier W, Rackebrandt K, Gessner JE, Zirngibl H. Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood. 2000; 96: 2520–7. [PubMed] [Google Scholar]
- 48.Cauwenberghs N, Meiring M, Vauterin S, van Wyk V, Lamprecht S, Roodt JP, Novak L, Harsfalvi J, Deckmyn H, Kotze HF. Antithrombotic effect of platelet glycoprotein Ib-blocking monoclonal antibody Fab fragments in nonhuman primates. Arterioscler Thromb Vasc Biol. 2000; 20: 1347–53. [DOI] [PubMed] [Google Scholar]
- 49.Yan R, Chen M, Ma N, Zhao L, Cao L, Zhang Y, Zhang J, Yu Z, Wang Z, Xia L, Ruan C, Dai K. Glycoprotein Ibalpha clustering induces macrophage-mediated platelet clearance in the liver. Thromb Haemost. 2015; 113: 107–17. 10.1160/TH14-03-0217. [DOI] [PubMed] [Google Scholar]
- 50.Liang X, Syed AK, Russell SR, Ware J, Li R. Dimerization of glycoprotein Ibα is not sufficient to induce platelet clearance. J Thromb Haemost. 2016; 14: 381–6. 10.1111/jth.13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maurer E, Tang C, Schaff M, Bourdon C, Receveur N, Ravanat C, Eckly A, Hechler B, Gachet C, Lanza F, Mangin PH. Targeting platelet GPIbβ reduces platelet adhesion, GPIb signaling and thrombin generation and prevents arterial thrombosis. Arterioscler Thromb Vasc Biol. 2013; 33: 1221–9. 10.1161/ATVBAHA.112.301013. [DOI] [PubMed] [Google Scholar]
- 52.Ju L, Chen Y, Xue L, Du X, Zhu C. Cooperative unfolding of distinctive mechanoreceptor domains transduces force into signals. Elife. 2016; 5: e15447. 10.7554/eLife.15447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanaji T, Russell S, Ware J. Amelioration of the macrothrombocytopenia associated with the murine Bernard-Soulier syndrome. Blood. 2002; 100: 2102–7. [DOI] [PubMed] [Google Scholar]
- 54.Peng J, Ma SH, Liu J, Hou Y, Liu XM, Niu T, Xu RR, Guo CS, Wang XM, Cheng YF, Ni H, Hou M. Association of autoantibody specificity and response to intravenous immunoglobulin G therapy in immune thrombocytopenia: a multicenter cohort study. J Thromb Haemost. 2014; 12: 497–504. 10.1111/jth.12524. [DOI] [PubMed] [Google Scholar]
- 55.Li J, van der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, Vadasz B, Carrim N, Grozovsky R, Ruan M, Zhu L, Zeng Q, Tao L, Zhai ZM, Peng J, Hou M, Leytin V, Freedman J, Hoffmeister KM, Ni H. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat Commun. 2015; 6: 7737 10.1038/ncomms8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quach ME, Dragovich MA, Chen W, Syed AK, Cao W, Liang X, Deng W, De Meyer SF, Zhu G, Peng J, Ni H, Bennett CM, Hou M, Ware J, Deckmyn H, Zhang XF, Li R. Fc-independent immune thrombocytopenia via mechanomolecular signaling in platelets. Blood. 2018; 131: 787–96. 10.1182/blood-2017-05-784975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gangarosa EJ, Landerman NS, Rosch PJ, Herndon EG, Jr. Hematologic complications arising during ristocetin therapy; relation between dose and toxicity. N Engl J Med. 1958; 259: 156–61. 10.1056/NEJM195807242590402. [DOI] [PubMed] [Google Scholar]
- 58.Sanders WE, Read MS, Reddick RL, Garris JB, Brinkhous KM. Thrombotic thrombocytopenia with von Willebrand factor deficiency induced by botrocetin. An animal model. Laboratory investigation; a journal of technical methods and pathology. 1988; 59: 443–52. [PubMed] [Google Scholar]
- 59.Lillicrap D von Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Blood. 2013; 122: 3735–40. 10.1182/blood-2013-06-498303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Casari C, Du V, Wu YP, Kauskot A, de Groot PG, Christophe OD, Denis CV, de Laat B, Lenting PJ. Accelerated uptake of VWF/platelet complexes in macrophages contributes to VWD type 2B-associated thrombocytopenia. Blood. 2013; 122: 2893–902. 10.1182/blood-2013-03-493312. [DOI] [PubMed] [Google Scholar]
- 61.Nascimbene A, Neelamegham S, Frazier OH, Moake JL, Dong JF. Acquired von Willebrand syndrome associated with left ventricular assist device. Blood. 2016; 127: 3133–41. 10.1182/blood-2015-10-636480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalbhenn J, Schlagenhauf A, Rosenfelder S, Schmutz A, Zieger B. Acquired von Willebrand syndrome and impaired platelet function during venovenous extracorporeal membrane oxygenation: Rapid onset and fast recovery. J Heart Lung Transplant. 2018; 37: 985–91. 10.1016/j.healun.2018.03.013. [DOI] [PubMed] [Google Scholar]
- 63.Larkin D, de Laat B, Jenkins PV, Bunn J, Craig AG, Terraube V, Preston RJ, Donkor C, Grau GE, van Mourik JA, O’Donnell JS. Severe Plasmodium falciparum malaria is associated with circulating ultra-large von Willebrand multimers and ADAMTS13 inhibition. PLoS Pathog. 2009; 5: e1000349. 10.1371/journal.ppat.1000349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Riswari SF, Tunjungputri RN, Kullaya V, Garishah FM, Utari GSR, Farhanah N, Overheul GJ, Alisjahbana B, Gasem MH, Urbanus RT, de Groot PG, Lefeber DJ, van Rij RP, van der Ven A, de Mast Q. Desialylation of platelets induced by Von Willebrand Factor is a novel mechanism of platelet clearance in dengue. PLoS Pathog. 2019; 15: e1007500. 10.1371/journal.ppat.1007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hartley PS, Savill J, Brown SB. The death of human platelets during incubation in citrated plasma involves shedding of CD42b and aggregation of dead platelets. Thromb Haemost. 2006; 95: 100–6. [PubMed] [Google Scholar]
- 66.Bergmeier W, Burger PC, Piffath CL, Hoffmeister KM, Hartwig JH, Nieswandt B, Wagner DD. Metalloproteinase inhibitors improve the recovery and hemostatic function of in vitro-aged or -injured mouse platelets. Blood. 2003; 102: 4229–35. [DOI] [PubMed] [Google Scholar]
- 67.Chen W, Liang X, Syed AK, Jessup P, Church WR, Ware J, Josephson CD, Li R. Inhibiting GPIbα shedding preserves post-transfusion recovery and hemostatic function of platelets after prolonged storage. Arterioscler Thromb Vasc Biol. 2016; Epub ahead of print. 10.1161/ATVBAHA.116.307639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi SI, Simone JV, Jorney LJ. Neuraminidase-induced thrombocytopenia in rats. Br J Haematol. 1972; 22: 93–101. [DOI] [PubMed] [Google Scholar]
- 69.Chen W, Druzak SA, Wang Y, Josephson CD, Hoffmeister KM, Ware J, Li R. Refrigeration-induced binding of von Willebrand factor facilitates fast clearance of refrigerated platelets. Arterioscler Thromb Vasc Biol. 2017; 37: 2271–9. 10.1161/ATVBAHA.117.310062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grewal PK, Aziz PV, Uchiyama S, Rubio GR, Lardone RD, Le D, Varki NM, Nizet V, Marth JD. Inducing host protection in pneumococcal sepsis by preactivation of the Ashwell-Morell receptor. Proc Natl Acad Sci USA. 2013; 110: 20218–23. 10.1073/pnas.1313905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deppermann C, Kratofil RM, Peiseler M, David BA, Zindel J, Castanheira F, van der Wal F, Carestia A, Jenne CN, Marth JD, Kubes P. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J Exp Med. 2020; 217: e20190723. 10.1084/jem.20190723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gu M, Xi X, Englund GD, Berndt MC, Du X. Analysis of the roles of 14-3-3 in the platelet glycoprotein Ib-IX-mediated activation of integrin αIIbβ3 using a reconstituted mammalian cell expression model. J Cell Biol. 1999; 147: 1085–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mazzucato M, Pradella P, Cozzi MR, De Marco L, Ruggeri ZM. Sequential cytoplasmic calcium signals in a 2-stage platelet activation process induced by the glycoprotein Ibalpha mechanoreceptor. Blood. 2002; 100: 2793–800. 10.1182/blood-2002-02-0514. [DOI] [PubMed] [Google Scholar]
- 74.Bergmeier W, Piffath CL, Goerge T, Cifuni SM, Ruggeri ZM, Ware J, Wagner DD. The role of platelet adhesion receptor GPIbα far exceeds that of its main ligand, von Willebrand factor, in arterial thrombosis. Proc Natl Acad Sci USA. 2006; 103: 16900–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reininger AJ, Heijnen HF, Schumann H, Specht HM, Schramm W, Ruggeri ZM. Mechanism of platelet adhesion to von Willebrand factor and microparticle formation under high shear stress. Blood. 2006; 107: 3537–45. 10.1182/blood-2005-02-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garcia A, Quinton TM, Dorsam RT, Kunapuli SP. Src family kinase-mediated and Erk-mediated thromboxane A2 generation are essential for VWF/GPIb-induced fibrinogen receptor activation in human platelets. Blood. 2005; 106: 3410–4. 10.1182/blood-2005-05-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu J, Pestina TI, Berndt MC, Jackson CW, Gartner TK. Botrocetin/VWF-induced signaling through GPIb-IX-V produces TxA2 in an alphaIIbbeta3- and aggregation-independent manner. Blood. 2005; 106: 2750–6. 10.1182/blood-2005-04-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen Y, Ruggeri ZM, Du X. 14-3-3 proteins in platelet biology and glycoprotein Ib-IX signaling. Blood. 2018; 131: 2436–48. 10.1182/blood-2017-09-742650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Du X, Harris SJ, Tetaz TJ, Ginsberg MH, Berndt MC. Association of a phospholipase A2 (14-3-3 protein) with the platelet glycoprotein Ib-IX complex. J Biol Chem. 1994; 269: 18287–90. [PubMed] [Google Scholar]
- 80.Calverley DC, Kavanagh TJ, Roth GJ. Human signaling protein 14-3-3z interacts with platelet glycoprotein Ib subunits Iba and Ibb. Blood. 1998; 91: 1295–303. [PubMed] [Google Scholar]
- 81.Mu FT, Andrews RK, Arthur JF, Munday AD, Cranmer SL, Jackson SP, Stomski FC, Lopez AF, Berndt MC. A functional 14-3-3ζ-independent association of PI3-kinase with glycoprotein Ibα, the major ligand-binding subunit of the platelet glycoprotein Ib-IX-V complex. Blood. 2008; 111: 4580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dai K, Bodnar R, Berndt MC, Du X. A critical role for 14-3-3ζ protein in regulating the VWF binding function of platelet glycoprotein Ib-IX and its therapeutic implications. Blood. 2005; 106: 1975–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuan Y, Zhang W, Yan R, Liao Y, Zhao L, Ruan C, Du X, Dai K. Identification of a novel 14-3-3ζ binding site within the cytoplasmic domain of platelet glycoprotein Ibα that plays a key role in regulating the von Willebrand factor binding function of glycoprotein Ib-IX. Circ Res. 2009; 105: 1177–85. 10.1161/CIRCRESAHA.109.204669. [DOI] [PubMed] [Google Scholar]
- 84.Bialkowska K, Zaffran Y, Meyer SC, Fox JE. 14-3-3 zeta mediates integrin-induced activation of Cdc42 and Rac. Platelet glycoprotein Ib-IX regulates integrin-induced signaling by sequestering 14-3-3 zeta. J Biol Chem. 2003; 278: 33342–50. 10.1074/jbc.M301217200. [DOI] [PubMed] [Google Scholar]
- 85.Perrault C, Mangin P, Santer M, Baas MJ, Moog S, Cranmer SL, Pikovski I, Williamson D, Jackson SP, Cazenave JP, Lanza F. Role of the intracellular domains of GPIb in controlling the adhesive properties of the platelet GPIb/V/IX complex. Blood. 2003; 101: 3477–84. [DOI] [PubMed] [Google Scholar]
- 86.Cranmer SL, Ulsemer P, Cooke BM, Salem HH, de la Salle C, Lanza F, Jackson SP. Glycoprotein (GP) Ib-IX-transfected cells roll on a von Willebrand factor matrix under flow. Importance of the GPib/actin-binding protein (ABP-280) interaction in maintaining adhesion under high shear. J Biol Chem. 1999; 274: 6097–106. 10.1074/jbc.274.10.6097. [DOI] [PubMed] [Google Scholar]
- 87.van der Wal DE, Gitz E, Du VX, Lo KS, Koekman CA, Versteeg S, Akkerman JW. Arachidonic acid depletion extends survival of cold-stored platelets by interfering with the [glycoprotein Ibalpha−−14-3-3zeta] association. Haematologica. 2012; 97: 1514–22. 10.3324/haematol.2011.059956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li S, Wang Z, Liao Y, Zhang W, Shi Q, Yan R, Ruan C, Dai K. The glycoprotein Ibα-von Willebrand factor interaction induces platelet apoptosis. J Thromb Haemost. 2010; 8: 341–50. 10.1111/j.1538-7836.2009.03653.x. [DOI] [PubMed] [Google Scholar]
- 89.Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, Lopez JA. Platelet glycoprotein Ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. 2000; 192: 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, Zago AC, Lopez J, Andre P, Plow E, Simon DI. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. 2005; 112: 2993–3000. [DOI] [PubMed] [Google Scholar]
- 91.Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, D AS, Bledzka K, Ustinov V, Zhu L, Qin J, Munday AD, Lopez J, Plow E, Simon DI. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat Commun. 2017; 8: 15559. 10.1038/ncomms15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Celikel R, McClintock RA, Roberts JR, Mendolicchio GL, Ware J, Varughese KI, Ruggeri ZM. Modulation of a-thrombin function by distinct interactions with platelet glycoprotein Iba. Science. 2003; 301: 218–21. [DOI] [PubMed] [Google Scholar]
- 93.De Candia E, Hall SW, Rutella S, Landolfi R, Andrews RK, De Cristofaro R. Binding of thrombin to glycoprotein Ib accelerates the hydrolysis of Par-1 on intact platelets. J Biol Chem. 2001; 276: 4692–8. 10.1074/jbc.M008160200. [DOI] [PubMed] [Google Scholar]
- 94.Estevez B, Kim K, Delaney MK, Stojanovic-Terpo A, Shen B, Ruan C, Cho J, Ruggeri ZM, Du X. Signaling-mediated cooperativity between glycoprotein Ib-IX and protease-activated receptors in thrombin-induced platelet activation. Blood. 2016; 127: 626–36. 10.1182/blood-2015-04-638387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Petzold T, Thienel M, Konrad I, Schubert I, Regenauer R, Hoppe B, Lorenz M, Eckart A, Chandraratne S, Lennerz C, Kolb C, Braun D, Jamasbi J, Brandl R, Braun S, Siess W, Schulz C, Massberg S. Oral thrombin inhibitor aggravates platelet adhesion and aggregation during arterial thrombosis. Sci Transl Med. 2016; 8: 367ra168. 10.1126/scitranslmed.aad6712. [DOI] [PubMed] [Google Scholar]
- 96.Strassel C, Eckly A, Leon C, Petitjean C, Freund M, Cazenave JP, Gachet C, Lanza F. Intrinsic impaired proplatelet formation and microtubule coil assembly of megakaryocytes in a mouse model of Bernard-Soulier syndrome. Haematologica. 2009; 94: 800–10. 10.3324/haematol.2008.001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dunois-Larde C, Capron C, Fichelson S, Bauer T, Cramer-Borde E, Baruch D. Exposure of human megakaryocytes to high shear rates accelerates platelet production. Blood. 2009; 114: 1875–83. 10.1182/blood-2009-03-209205. [DOI] [PubMed] [Google Scholar]
- 98.Nurden P, Gobbi G, Nurden A, Enouf J, Youlyouz-Marfak I, Carubbi C, La Marca S, Punzo M, Baronciani L, De Marco L, Vitale M, Federici AB. Abnormal VWF modifies megakaryocytopoiesis: studies of platelets and megakaryocyte cultures from patients with von Willebrand disease type 2B. Blood. 2010; 115: 2649–56. 10.1182/blood-2009-07-231886. [DOI] [PubMed] [Google Scholar]
- 99.Kauskot A, Poirault-Chassac S, Adam F, Muczynski V, Ayme G, Casari C, Bordet JC, Soukaseum C, Rothschild C, Proulle V, Pietrzyk-Nivau A, Berrou E, Christophe OD, Rosa JP, Lenting PJ, Bryckaert M, Denis CV, Baruch D. LIM kinase/cofilin dysregulation promotes macrothrombocytopenia in severe von Willebrand disease-type 2B. JCI Insight. 2016; 1: e88643. 10.1172/jci.insight.88643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu M, Li J, Neves MAD, Zhu G, Carrim N, Yu R, Gupta S, Marshall J, Rotstein O, Peng J, Hou M, Kunishima S, Ware J, Branch DR, Lazarus AH, Ruggeri ZM, Freedman J, Ni H. GPIbalpha is required for platelet-mediated hepatic thrombopoietin generation. Blood. 2018; 132: 622–34. 10.1182/blood-2017-12-820779. [DOI] [PubMed] [Google Scholar]
- 101.Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, Kotsiliti E, Leone V, Peiseler M, Surewaard BGJ, Rath D, Ali A, Wolf MJ, Drescher H, Healy ME, Dauch D, Kroy D, Krenkel O, Kohlhepp M, Engleitner T, Olkus A, Sijmonsma T, Volz J, Deppermann C, Stegner D, Helbling P, Nombela-Arrieta C, Rafiei A, Hinterleitner M, Rall M, Baku F, Borst O, Wilson CL, Leslie J, O’Connor T, Weston CJ, Adams DH, Sheriff L, Teijeiro A, Prinz M, Bogeska R, Anstee N, Bongers MN, Notohamiprodjo M, Geisler T, Withers DJ, Ware J, Mann DA, Augustin HG, Vegiopoulos A, Milsom MD, Rose AJ, Lalor PF, Llovet JM, Pinyol R, Tacke F, Rad R, Matter M, Djouder N, Kubes P, Knolle PA, Unger K, Zender L, Nieswandt B, Gawaz M, Weber A, Heikenwalder M. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019; 25: 641–55. 10.1038/s41591-019-0379-5. [DOI] [PubMed] [Google Scholar]
- 102.Oleksowicz L, Mrowiec Z, Schwartz E, Khorshidi M, Dutcher JP, Puszkin E. Characterization of tumor-induced platelet aggregation: the role of immunorelated GPIb and GPIIb/IIIa expression by MCF-7 breast cancer cells. Thromb Res. 1995; 79: 261–74. [DOI] [PubMed] [Google Scholar]
- 103.Bastida E, Almirall L, Ordinas A. Tumor-cell-induced platelet aggregation is a glycoprotein-dependent and lipoxygenase-associated process. Int J Cancer. 1987; 39: 760–3. 10.1002/ijc.2910390617. [DOI] [PubMed] [Google Scholar]
- 104.Jain S, Zuka M, Liu J, Russell S, Dent J, Guerrero JA, Forsyth J, Maruszak B, Gartner TK, Felding-Habermann B, Ware J. Platelet glycoprotein Ibα supports experimental lung metastasis. Proc Natl Acad Sci USA. 2007; 104: 9024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Terraube V, Marx I, Denis CV. Role of von Willebrand factor in tumor metastasis. Thromb Res. 2007; 120 Suppl 2: S64–70. 10.1016/S0049-3848(07)70132-9. [DOI] [PubMed] [Google Scholar]
- 106.Ramakrishnan V, Reeves PS, DeGuzman F, Deshpande U, Ministri-Madrid K, DuBridge RB, Phillips DR. Increased thrombin responsiveness in platelets from mice lacking glycoprotein V. Proc Natl Acad Sci USA. 1999; 96: 13336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ni H, Ramakrishnan V, Ruggeri ZM, Papalia JM, Phillips DR, Wagner DD. Increased thrombogenesis and embolus formation in mice lacking glycoprotein V. Blood. 2001; 98: 368–73. [DOI] [PubMed] [Google Scholar]
- 108.Englund GD, Bodnar RJ, Li Z, Ruggeri ZM, Du X. Regulation of von Willebrand factor binding to the platelet glycoprotein Ib-IX by a membrane skeleton-dependent inside-out signal. J Biol Chem. 2001; 276: 16952–9. [DOI] [PubMed] [Google Scholar]
- 109.Bodnar RJ, Xi X, Li Z, Berndt MC, Du X. Regulation of glycoprotein Ib-IX-von Willebrand factor interaction by cAMP-dependent protein kinase-mediated phosphorylation at Ser 166 of glycoprotein Ibβ. J Biol Chem. 2002; 277: 47080–7. [DOI] [PubMed] [Google Scholar]
- 110.Yan R, Mo X, Paredes AM, Dai K, Lanza F, Cruz MA, Li R. Reconstitution of platelet glycoprotein Ib-IX complex in phospholipid bilayer nanodiscs. Biochemistry. 2011; 50: 10598–606. 10.1021/bi201351d. [DOI] [PMC free article] [PubMed] [Google Scholar]