

Abstract
Neuroendocrine (NE) tumors include a diverse spectrum of hormone-secreting neoplasms that arise from the endocrine and nervous systems. Current chemo- and radio- therapies have marginal curative benefits. The goal of this study was to develop an innovative antibody-drug conjugate (ADC) to effectively treat NE tumors (NETs). First, we confirmed that somatostatin receptor 2 (SSTR2) is an ideal cancer cell surface target by analyzing 38 patient-derived NET tissues, 33 normal organs, and 3 NET cell lines. Then, we developed a new monoclonal antibody (mAb, IgG1 and kappa) to target two extracellular domains of SSTR2, which showed strong and specific surface binding to NETs. The ADC was constructed by conjugating the anti-SSTR2 mAb and antimitotic monomethyl auristatin E. In vitro evaluations indicated that the ADC can effectively bind, internalize, release payload, and kill NET cells. Finally, the ADC was evaluated in vivo using a NET xenograft mouse model to assess cancer-specific targeting, tolerated dosage, pharmacokinetics, and anti-tumor efficacy. The anti-SSTR2 ADC exclusively targeted and killed NET cells with minimal toxicity and high stability in vivo. This study demonstrates that the anti-SSTR2 ADC has a high therapeutic potential for NET therapy.
Introduction
Neuroendocrine (NE) tumors, such as carcinoids, pancreatic islet cell tumors, and medullary thyroid cancer (MTC), arise from cells within the neuroendocrine system that often harbor inherited or sporadic genetic mutations.(1, 2) The prevalence of NE tumor (NET) patients in the United States is in excess of 100,000, with at least 16,000 new diagnoses each year and an estimate of 200,000-plus undiagnosed cases.(3, 4) Patients living with untreatable NET liver metastases have a 5-year survival rate of 13–54%.(5) The fact that 40–95% of patients with NETs are metastatic at the time of initial diagnosis makes complete surgical resections nearly impossible.(3, 6–10) Chemotherapies utilized for NET (e.g., the mTOR inhibitor “everolimus” and the multikinase inhibitor “sunitinib”) have shown limited efficacy and can cause systemic toxicities.(11–18) Somatostatin receptor (SSTR)-targeting analogs (e.g., octreotide and lanreotide) or FDA-approved peptide receptor radionuclide therapy (Lutathera®) for gastroenteropancreatic NET treatment can extend patient’ survival but have relatively poor impact on rapidly proliferating tumors.(19, 20) Thus, it is imperative to develop new treatment strategies for this disease.
Five SSTR subtypes (SSTR1–5) belonging to the G-protein coupled receptor (GPCR) family are expressed in tumor or normal tissues.(21) NET patients overexpress SSTR2 and SSTR3 at a high density.(22–25) The membrane expression of SSTR2 in NET cells is approximately 20-fold higher than that of normal cells.(22–24) Moreover, our immunohistochemistry (IHC) analysis performed on a patient tissue microarray (TMA) demonstrated that over 70% of NET patients express SSTR2. Therefore, SSTR2 is a potential target for the development of a new therapeutic approach to treat NETs.
Targeted therapies, such as monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs) have been applied to treat cancers with minimal side effects on normal cells.(26–29) ADCs engender many of the advantages of mAbs including cancer-specific targeting to lower toxicity in normal tissues, low immunogenicity, long plasma half-life and high stability, with the high cytotoxicity of small molecule chemotherapeutics.(30) After receptor binding, ADC is internalized via receptor-mediated endocytosis. The cytotoxic drug is then released into the cytoplasm of cancer cells via either lysosomal degradation or linker cleavage.(31, 32) As precedents, several antibodies carrying payloads, such as brentuximab vedotin (anti-CD30-MMAE), trastuzumab emtansine (anti-HER2-DM1), 131I-Tositumomab (I-131 labelled anti-CD20 mAb), 90Y-Ibritumomab tiuxetan (90Y labelled anti-CD20 mAb), and traztuzumab deruxtecan (anti-HER2 mAb-topoisomerase I inhibitor), have been developed to treat relapsed Hodgkin lymphoma, systemic anaplastic large cell lymphoma, relapsed, chemotherapy refractory or advanced HER2-positive breast cancer, non-hodgkin’s lymphoma (NHL), or NHL.(33) To our knowledge, neither mAb nor ADC has yet been developed for NET treatment. In addition to ADC, the SSTR2-targeted PEN-221 comprised a SSTR2 agonist [Tyr3, Cys8]octreotate amide linked to mertansine has been developed to treat small cell lung cancer.(34) The AN-238, a cytotoxic analogue of SST conjugated with peptide RC-121 and 2-pyrrolinodoxorubicin (2-pyrrolino-DOX), and two camptothecins (CPT)-potent somatostatin analog (SSA) conjugates, JF-10–71 and JF-10–81, have also been developed to treat choroidal neovascularization and lung cancer.(34–36)
The objective of this study was to develop an innovative targeted therapy to treat SSTR2-overexpressing NETs. A surface receptor analysis of multiple patient tissues and normal organ tissues showed that SSTR2 is highly expressed in most of NET patients. A new anti-SSTR2 mAb was developed to efficiently target NET and deliver an FDA approved potent cytotoxic payload, Monomethyl auristatin E (MMAE), which can effectively block microtubulin polymerization and inhibit NET cell growth. The specific targeting, tolerated dosage, pharmacokinetics, and anti-tumor efficacy of the anti-SSTR2 ADC were investigated using a NET xenograft mouse model. Our results demonstrate that the developed ADC was capable of specifically targeting and effectively reducing tumor growth, indicating promise for further development as a novel therapeutic for these tumors.
Materials and Methods
The animal studies conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85–23) and have been approved by the Institutional Biosafety Committee at the University of Alabama at Birmingham under the animal project number of IACUC-21929. The investigators were not blinded to the group allocating during the experiment and assessing the outcome.
NET patient tissue microarray
The TMA was prepared by Research Pathology Core to analyze the SSTR2 surface expression in NET. The patient tissues were obtained from the University Surgical Oncology Tumor Bank through an Institutional Review Board (IRB) approved protocol. The NET microarray consisted of 38 patient tissue cores, which contained 1 high-grade (G3) and 37 low-grade (G1) and intermediate-grade (G2) samples (2A to 9D), and 5 normal tissue cores of liver, spleen, placenta, prostate, and tonsil (negative controls, 1A-1E). The TMA slides of 33 normal human organs were purchased from US Biomax (Rockville, MD) to confirm the binding specificity of our anti-SSTR2 mAb using IHC staining with NET tissues as positive controls. The normal organs that we tested included cerebrum, cerebellum, peripheral nerve, adrenal gland, thyroid gland, spleen, thymus, bone marrow, lymph node, tonsil, pancreas, liver, esophagus, stomach, small intestine, colon, lung, salivary, pharynx, kidney, bladder, testis, prostate, penis, ovary, uterine tube, breast, endometrium, cervix, cardiac muscle, skeletal muscle, mesothelium, and skin.
Cell lines and media
Multiple human NET cell lines, including pancreatic NET BON-1 (kindly provided by Dr. Mark Hellmich from University of Texas, Galveston, TX) and QGP-1 (ACCEGEN Biotechnology, Fairfield, NJ), BON-Luc carrying a firefly luciferase reporter gene (generated in our lab by overexpressing FLuc in BON-1 cells), were used for in vitro or in vivo studies. BON-1 and OGP-1 cells were maintained in DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS) in T25 or T75 flasks. The non-neoplastic SSTR2-negative control cell lines, including pulmonary fibroblast WI-38 (ATCC, Manassas, VA) and foreskin fibroblast 917 (ATCC), were maintained in DMEM with 10% FBS, 1% non-essential amino acids, and 1% sodium pyruvate. Adherent mAb producing hybridoma cells were maintained in DMEM with 10% FBS in T flasks, while the adapted suspensive hybridoma was cultivated in Hybridoma-SFM with 4 mM L-glutamine and 1% anti-clumping agent (v/v) in shaker flasks with agitation of 130 rpm. All seed cultures were incubated at 37 °C and 5% CO2 in a humidified incubator (Caron, Marietta, OH). The cell growth, i.e. viable cell density (VCD) and viability, was measured using a Countess II automated cell counter or trypan blue (Fisher Scientific, Waltham, MA). All basal media, supplements, and reagents used in this study were purchased from Fisher Scientific or Life Technologies (Part of Fisher) unless otherwise specified.
Anti-SSTR2 mAb development
Both human SSTR2 (UniProtKB P30874) and mouse SSTR2 (UniProtKB P30875) are integral membrane glycoproteins with the same topology, including four extracellular topological domains, seven helical transmembrane, and four cytoplasmic topological domains. Protein BLAST analysis showed that their four extracellular domains had similarity of 81%, 100%, 100%, and 90%, respectively. We developed an SSTR2 mAb to target the 1st extracellular domain (cQTEPYYDLTSNA, aa 33–44) and the 2nd extracellular domain (cALVHWPFGKAICRVV, aa 104–118) using hybridoma technology (PCT patent, US2019/0055145). The immune splenocytes with the best anti-SSTR2 antibody expression were fused with myeloma cells (Sp2/0) to obtain 100 hybridoma subclones. The top 4 clones were screened using peptides (the 1st and the 2nd extracellular domains)-based ELISA and were adapted to serum-free suspension cultures to produce mAbs.(37) The tumor cell surface binding of these 4 mAbs was evaluated using flow cytometry and confocal microscopy imaging. These methods were used to define the lead clone which had strong and specific binding to NET (BON-1) cells but low binding to non-cancerous control cells. The isotype of the lead clone was determined using a mouse antibody isotyping kit (Sigma, St. Louis, MO).
Anti-SSTR2 mAb production and purification
The mAb production was performed in a 5-L stirred-tank bioreactor controlled at Temp 37 °C, pH 7.0, DO 50% and agitation 70 rpm. The bioreactor was seeded with VCD of 0.3–0.5 ×106 cells/mL in Hybridoma-SFM with 6 g/L glucose, 6 mM L-glutamine, 3.5 g/L Cell Boost #6, and 1% anti-clumping agent. The production cultures were sampled daily to monitor cell growth (i.e., VCD, viability, double time, and growth rate) using cell counter, glucose concentration using glucose analyser, and mAb production using NGC system (Bio-Rad, Hercules, CA). The anti-SSTR2 mAb was purified using our two-step antibody purification protocol by the NGC system (Bio-Rad, Hercules, CA) equipped with Protein A and ion exchange columns.(38, 39)
ADC construction
In this study, ADC was constructed following our published cysteine-based conjugation procedure. (38, 39) Briefly, the rebridging linker was synthesized by reacting 3.91 mmol 6-aminohexanoic acid with 3.91 mmol 3,4-dibromofuran-2,5-dione in 20 mL acetic acid at room temperature (RT) for 10 mins and purified with silica gel. Then the linker-MMAE payload was conjugated by mixing 33.85 μmol synthesized rebridging linker, 13.55 μmol N,N’-diisopropylcarbodiimide, 13.55 μmol N,N-diisopropylethylamine, and 13.55 μmol MMAE in 0.25 mL dichloromethane for 17 hrs and purified with HPLC. The 5 mg/mL anti-SSTR2 mAb was reduced with 1 mM dithiothreitol in 50 mM borate buffer at pH 8.0 at 37 °C for 1 hr and purified with buffer exchange. Finally the ADC was conjugated by mixing the linker-MMAE payloads with the reduced mAb with payload:mAb molar ratio of 4.4 and incubated at 4 °C for 1 hr. The generated ADC was purified with PD SpinTrap™ G25 column (GE Healthcare, Chicago, IL) or high-performance liquid chromatography (Waters, Milford, MA). The average drug-antibody ratio (DAR) was calculated as Ratio = (εAb248-RεAb280)/(RεD280-εD248), where R = A248/A280 = Absorbance ratio.(38)
In vitro anti-cancer cytotoxicity (IC50)
BON cells were utilized to evaluate the anti-NET cytotoxicity of the anti-SSTR2 ADC and MMAE (control) in 96-well plate following our published protocol.(38) Briefly, the BON cells were seeded with viable cell density of 50,000 cells/mL in 75 μL of DMEM/F12 medium complete medium, and incubated at 37 °C for 24 hrs. Then the anti-cancer cytotoxicity assay was initiated by adding 75 μL of medium containing ADCs and free drug with final concentrations of 0.5, 1, 2, 10 and 25 nM with triplication. After 72 hrs incubation, the toxicity was measured through CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, MI). The luminescent signal was proportional to the viable cell number and used to calculate the relative viability in all treatments. The IC50 value was calculated using ED50V10 Excel add-in.
SDS-PAGE and Western blotting
The Mem-PER plus membrane protein extraction kit was used to extract membrane proteins for surface receptor evaluation. The protein concentration was determined by the Pierce BCA assay. Non-reducing SDS-PAGE was run using NuPAGE™ 4–12% Bis-Tris protein gels. The primary rabbit anti-mouse antibody and HRP-conjugated secondary anti-rabbit antibody were purchased from Abcam (Cambridge, UK). The blotted membrane was treated with Luminata Forte Western HRP substrate (Millipore, Boston, MA), and imaged with MyECL imager with ImageJ software.
Flow cytometry
Flow cytometry was performed to quantify surface receptor binding of SSTR2 mAb using a BD LSRII flow cytometer (BD Biosciences, San Jose, CA). The mAb was labelled with an Alexa Fluor™ 647 labelling kit to generate AF647-mAb. The NET cell lines (BON and QGP-1) and negative control fibroblast cell line (917) were tested. Detailed methods are described elsewhere (38, 39) Briefly 1×106 cells were mixed with 1 μg AF647-mAb in 100 μL PBS and incubated at room temperature for 30 mins in dark. The labelled cells were washed with PBS for three times and resuspended in 500 μL buffer for flow cytometry analysis. The commercial anti-SSTR2 mAb (RD Systems, Minneapolis, MN, catalog# MAB4224) was used as control.
SSTR2 binding affinity and specificity analysis
The SSTR2 mAb-receptor binding affinity was measured following the previously reported procedure.(40) Specifically, 120 ng of somatostatin receptor 2 was coated on 96-well plates and incubated at 4 °C overnight. Plates were washed using PBS plus 0.05% Tween 20for 3 times and blocked with protein-free blocking buffer at 37 °C for 1 hr. Our anti-SSTR2 mAb was added following concentration gradient of 0, 5, 10, 50, 100, 500, 1,000, 10,000, 50,000, and 100,000 pM, incubated at 37 °C for 1 hr, and plates were washed for 5 times. Goat anti-mouse IgG HRP was added to each well at a concentration of 50 ng/mL, followed by 30-min incubation at 37 °C and 3 times of washing. Then 100 μL of TMB substrate solution was added to each well and incubated at RT for 30 mins, and 100 μL of 1M H2SO4 was added to stop color development. The absorbance was read using BioTek plate reader at a wavelength of 450 nm.
Confocal imaging
Confocal microscopy was used to observe the dynamic surface binding and internalization of mAb and ADC in NET cells following our established protocol.(38, 39) Specifically, BacMam GFP Transduction Control was used to stain the cytoplasm and nucleus, and the AF647-mAb or AF647-ADC was used to target cells. The stained cells were observed using an Olympus 1X-81 confocal microscope with a laser scanning head (Olympus IX81, Center Valley, PA). The MitoSox images were recorded and analyzed offline via ImageJ software.
Tolerated dose (TD) and pharmacokinetics (PK) study
To investigate the tolerated dose and metabolic rate of ADC, five different doses (4, 8, 12, 16, 20 mg/kg body weight [BW]) of ADC were administered intravenously (i.v.) to 5 groups of randomized 6-week nude (nu/nu) mice purchased from Jackson Labs (Bar Harbor, ME). The body weight was measured every two days for 23 days post injection. Blood samples were collected from tails at 2, 5, 24, 48, 72, 120 hrs post-injection (6 time points in total for each mouse). Blood was centrifuged at 2,000 g for 5 mins to precipitate cells and the supernatant was collected for ELISA. The previously developed PK model was used in this study.(41) Briefly, clearance , volume of distribution , half life , recommended dose , and dosing interval . The calculated D and τ were used in the anti-tumor efficacy animal study.
In vivo anti-NET efficacy study
The Mycoplasma-free BON-Luc (3×106 cells) were injected subcutaneously onto the flank of 4–6-week nude (nu/nu) mice (Jackson Labs, Bar Harbor, ME). The NET xenograft mice with tumor volume of 50–60 mm3 were randomized into 3 groups (n = 6): saline, anti-SSTR2 mAb, and mAb-MMAE conjugate. The mAb or ADC was administrated intravenously through tail vein following a dose of 8 mg/kg-BW (empirically determined from PK study) in 50 or 100 μL of saline. The same volume of mAb or saline was injected in control groups. Mice developed palpable nodules within 14 days and tumor volume and mouse body weight were measured every two days. Both electronic caliper and bioluminescence via In Vivo Imaging system (IVIS) were used to monitor tumor size. Four injections were conducted with average injection interval of 4.5 days during the entire treatment period (total of 24 days, our standard 3-week ADC treatment). At the end of the experiment, mice were euthanized to collect tumors and other organs (e.g. brain) for further analysis.
Hematoxylin and eosin (H&E) staining
Tissue samples were dehydrated in ethanol, cleared in xylene, embedded in paraffin, and sectioned at 5 μm with Leica microtome and mounted on frosted microscope slides (Fisher Scientific). Paraffin sectioned slides were dewaxed with xylene, and gradient hydrated with 100% ETOH, 95% ETOH, 70% ETOH, 50% ETOH, and dH2O. Slides were immersed in hematoxylin solution for 5 mins followed by tap water rinse for 2 mins, 1% HCl in 70% ETOH for 3 dips, 1% NH4OH until color turned blue and eosin Y solution for 30 secs. Finally, slides were dehydrated twice in absolute alcohols for 2 mins and cleared in xylene.
Immunohistochemistry (IHC) staining
The 5μm paraffin sections were used for IHC staining. Tissue microarray slides were rehydrated using xylene and ethanol, then immersed in citrate buffer (BioGenex, Fermont, CA) for a 10-min pressure cooker cycle to achieve antigen retrieval. Endogenous peroxidase activity was quenched by incubating slides in 3% hydrogen peroxide for 10 mins. Blocking was performed for 1 hr at RT using 3% goat serum in PBS. SSTR2 was detected with an overnight 4°C incubation using 1.8 mg/mL of anti-SSTR2 mAb (RD Systems mAb in the IHC of TMA and our mAb in other IHC experiments) with final concentration of 10 μg/mL, followed by an anti-mouse biotin-labeled secondary antibody and HRP streptavidin. Slides were stained with DAB kit (Vector, SK-4100) and counter stained with hematoxylin. Before being cover slipped and imaged, slides were dehydrated and cleared using ethanol and xylene.
Scoring of IHC
ImageJ was used for IHC quantitative scoring to analyze the SSTR2 expression or anti-SSTR2 mAb binding. The positive staining (red color) and negative staining (blue color) intensity as quantified using RGB Measure function (Plugins-Analyze-RGB Measure). The SSTR2 expression score was calculated as SSTR2 score = [(red intensity/blue intensity)NET tissues/average of (red intensity/blue intensity)nomal tissues-1]*10. The scoring criteria were defined as high expression (+++, 3): score >3.0, medium expression (++, 2): score of 1.5–2.0, and low expression (+, 1). The five normal tissues (liver, spleen, placenta, prostate and tonsil) are our negative control samples with SSTR2 score of 0.
Statistical analysis
All the data were presented as mean ± standard error of the mean (SEM). Two-tailed Student’s t tests were used to determine the probability of significance between two groups. Comparison among multiple groups was performed using a one-way ANOVA followed by post-hoc (Dunnett’s) analysis. The sample size of animal study followed the published ADC therapy study.(42) Statistical significance with ***P value of < 0.001 was considered for all tests.
Results
SSTR2 overexpression in NET
To assess SSTR2 expression, NET tissue microarray slides were first stained with H&E to confirm the presence and location of NET cells in each core (Fig. 1A), then was stained using SSTR2 mAb to evaluate the receptor surface expression. The IHC images were presented in Fig. 1B and the SSTR2 expression scores of each tissue were summarized in Supplemental Table 1. The IHC staining demonstrated that approximately 76% (29/38) of patient tissues cores (2A to 9D) had positive SSTR2 expression with strong cell membrane localization (Fig. 1B), while SSTR2 was not detectable in normal liver, spleen, placenta, prostate, and tonsil tissue cores (1A-1E). Of all the NET tissue samples, 50% (19/38) had very high SSTR2 expression (score: 3), 26% (10/38) showed intermediate expression (score: 2), and 24% (9/38) showed low expression (score: 1). All the SSTR2 scores of NET tissues were higher than the five normal tissues. The scores of 2 and 3 were defined as strong expression in this study. Moreover, the IHC staining of the 33 types of normal human tissues with our anti-SSTR2 mAb showed that there was no detectable SSTR2 surface expression in most of these normal organs except pancreas and skin which had weak signal (Fig. 2A). The Human Atlas Project database reported a high level of SSTR2 mRNA in brain, lung, liver, skin, placenta, prostate, tonsil, and pancreas. However, the high-resolution images of these normal organs showed minimal or undetectable surface SSTR2 (Fig. 2B).
Figure 1.
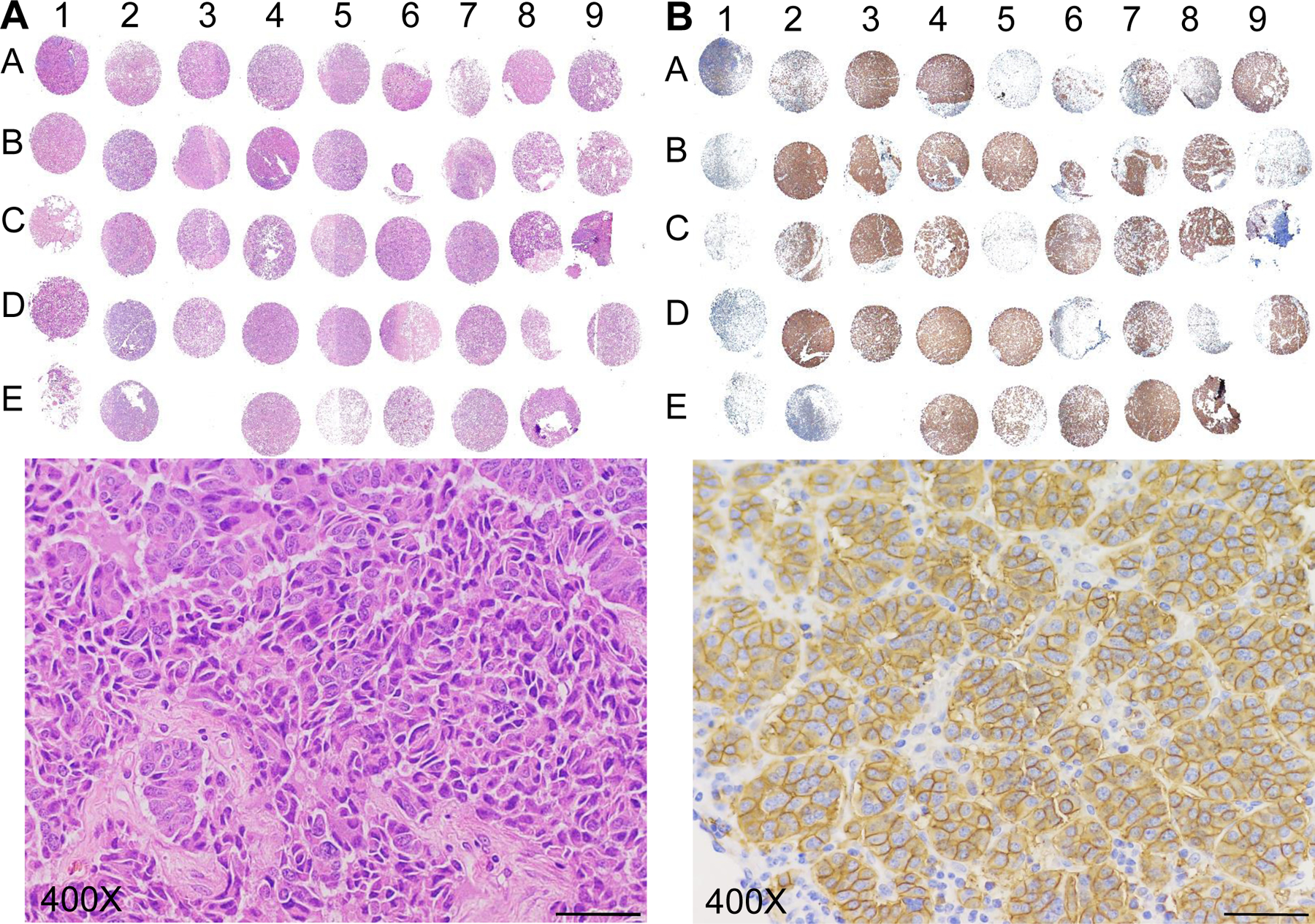
Tissue microarray (TMA) to detect SSTR2 expression in patients. A, H&E staining of the TMA including human pancreatic NET tissues (columns 2–9, n = 38) and normal tissues (control, column 1, n = 5). B, IHC analysis of SSTR2 in the TMA. Scale bar equals 20 μm.
Figure 2.
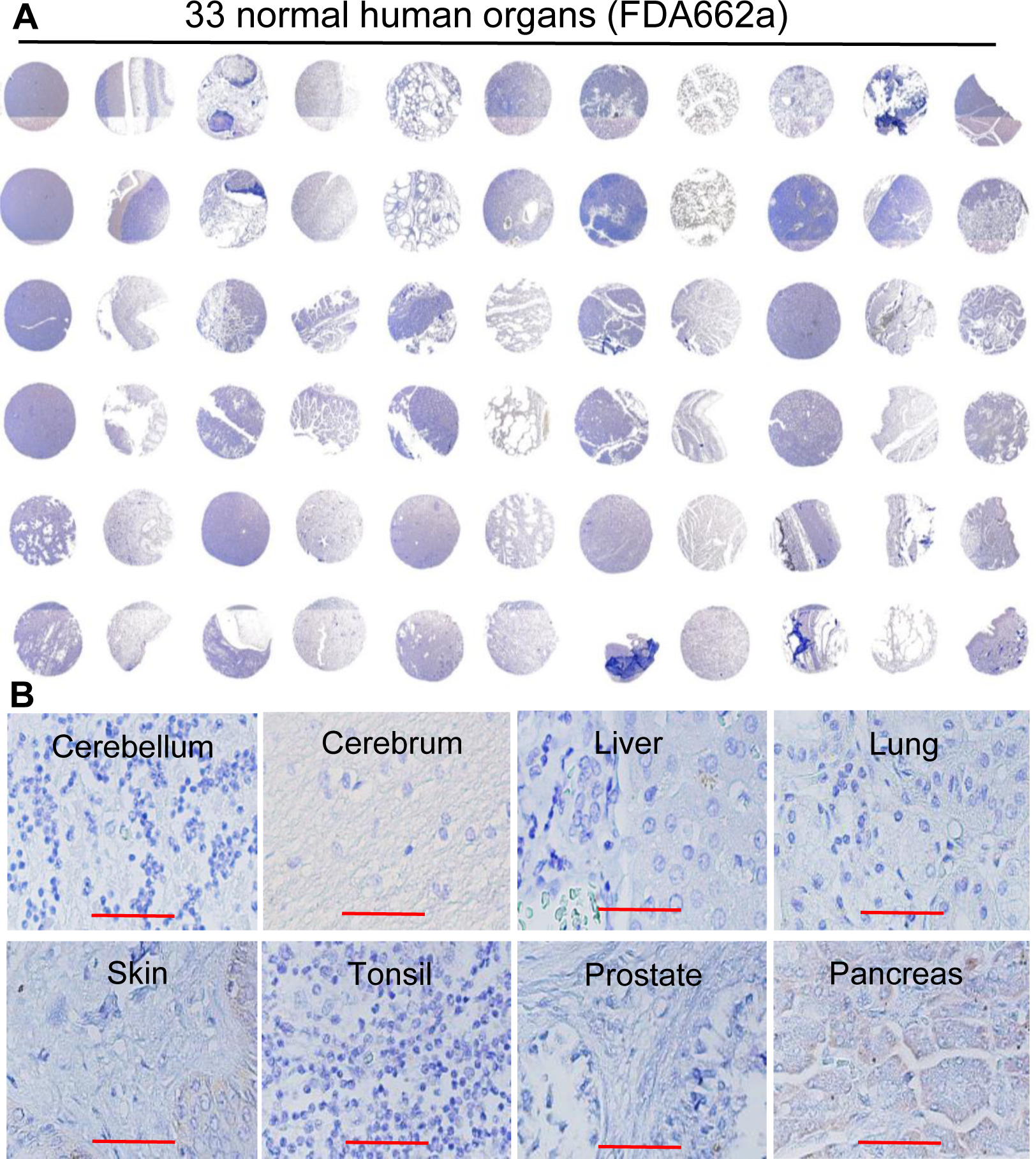
Evaluation of the NET-specific targeting of our anti-SSTR2 antibody using IHC of normal human organs. A, Surface SSTR2 staining in 33 normal human organs (US Biomax, FDA662a, n = 2), including cerebrum, cerebellum, peripheral nerve, adrenal gland, thyroid gland, spleen, thymus, bone marrow, lymph node, tonsil, pancreas, liver, esophagus, stomach, small intestine, colon, lung, salivary, pharynx, kidney, bladder, testis, prostate, penis, ovary, uterine tube, breast, endometrium, cervix, cardiac muscle, skeletal muscle, mesothelium, and skin. B, Representative high-resolution IHC imaging of cerebellum, cerebrum, liver, lung, skin, tonsil, prostate, and pancreas. Scale bar equals 50 μm.
Development of anti-SSTR2 mAb to target NET
The hybridoma clones secreting anti-SSTR2 mAb were screened using ELISA to identify the top mAb clones with strong binding to the 1st, the 2nd or both extracellular domains of SSTR2 (Fig. 3A). In flow cytometry analysis, the surface binding capacity of these four mAbs to BON-1 cells was 50%, 80%, 90% and 98%, respectively (Fig. 3B). Clone 4 was defined as the “lead clone”, fully characterized, and used throughout the remainder of the study. An isotype analysis revealed that the lead clone is IgG1 kappa, and SDS-PAGE analysis confirmed its molecular weight of 150 kDa (Fig. 3C). Further evaluation showed that the anti-SSTR2 mAb had high surface binding to NET cell lines BON-1 and QGP-1 (>90%) and low binding to fibroblast cell lines 917 and WI-38 (<7.5%) (Fig. 3D). Additionally, we cloned and sequenced the mAb, and confirmed the novelty of our anti-SSTR2 mAb (PCT patent TH Docket No. 222119–8030). To optimally produce mAb, we adapted the adherent hybridoma cells to suspension culture in stirred-tank bioreactors (Fig. 3E). The cultures in T-flask, spinner flask, and stirred-tank bioreactor generated 8.6, 39.8, and 53.3 mg/L of anti-SSTR2 mAb with a specific growth rate of 0.016, 0.024 and 0.035 hr−1, respectively.
Figure 3.
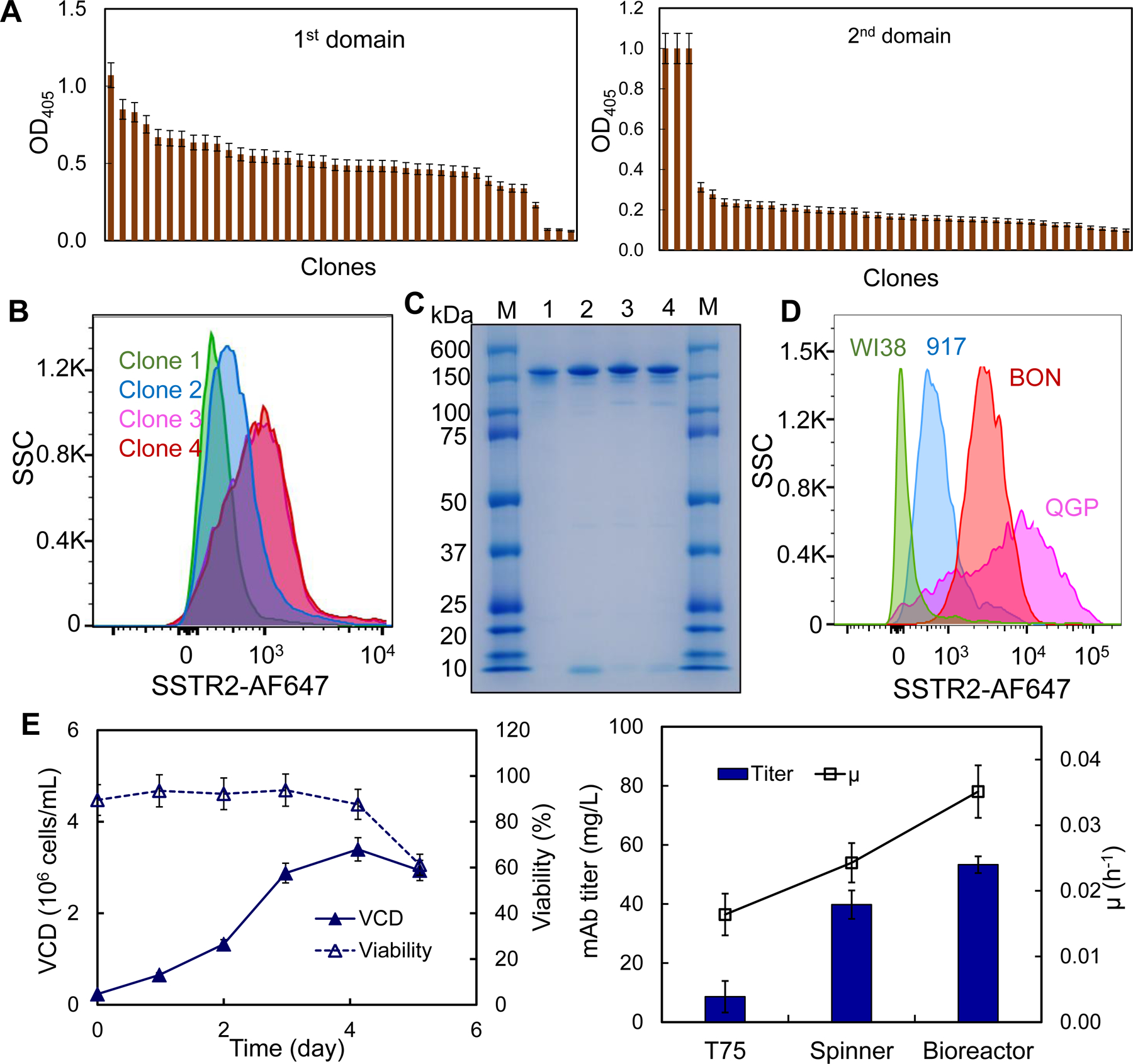
Anti-SSTR2 mAb development and production. A, Rank of top anti-SSTR2 mAb clones based on the titer in ELISA screening (data represent mean ± SEM, n = 3). B, Evaluation of top 4 clones using flow cytometry. C, SDS-PAGE to confirm the integrity and purity of mAb (M: marker; 1–4: Clones 1–4). D, Evaluation of SSTR2 binding of lead clone in control cell lines (WI38 and 917) and NET cell lines (BON and QGP). E, mAb production and hybridoma cell growth in fed-batch suspension cultures (data represent mean ± SEM, n = 3). Viable cell density (VCD):  , cell viability:
, cell viability:  , specific growth rate (μ):
, specific growth rate (μ):  .
.
High surface binding and high affinity to SSTR2
To assess the in vitro NET-specific targeting of our anti-SSTR2 mAb, we performed live-cell, dynamic CLSM imaging and flow cytometry analysis. The AF647-mAb accumulated on the BON-1 cell surface, displayed as a “red circle”, within 20 mins post incubation due to immunoaffinity (Fig. 4A). The mAb was then internalized through endocytosis and localized in cytoplasm (detected with BacMam GFP control) within 40 mins. Also our anti-SSTR2 mAb exhibited much stronger surface binding to BON-1 cells than the commercial mAb (R&D Systems), 95±3% vs. 38±2% (Fig. 4B). The UniProtKB database shows that the extracellular domains of human SSTR2 targeted by our mAb have no similarity with those in human SSTR1, 3, 4 and 5 (Supplemental Table 2), indicating that the cross reactivity among SSTRs is minimal. Moreover, human SSTR2 (UniProt P30874) and mouse SSTR2 (UniProt P30875) have the same topology, and the 1st and the 2nd extracellular domains of human SSTR2 that our mAb targets have 100% similarity with mouse SSTR2. Western blotting (Fig. 4C) confirmed that our anti-SSTR2 mAb can bind the SSTR2 present in human BON-1 xenografts, as consistent with our previous study which showed our mAb can specifically target the BON-Luc xenograft in mouse model (43), and bind the mouse SSTR2 in isolated medullary thyroid carcinoma (MTC, type of neuroendocrine thyroid cancer) tissue from a spontaneous MTC mouse model.(44, 45) Additionally, the antibody affinity experiment showed that our anti-SSTR2 mAb had equilibrium dissociation constant (KD) of 6.7 nM and 6.6 nM to human SSTR2 and mouse SSTR2, respectively. All these data revealed that the SSTR2 mAb developed in this study can bind both human and mouse SSTR2.
Figure 4.
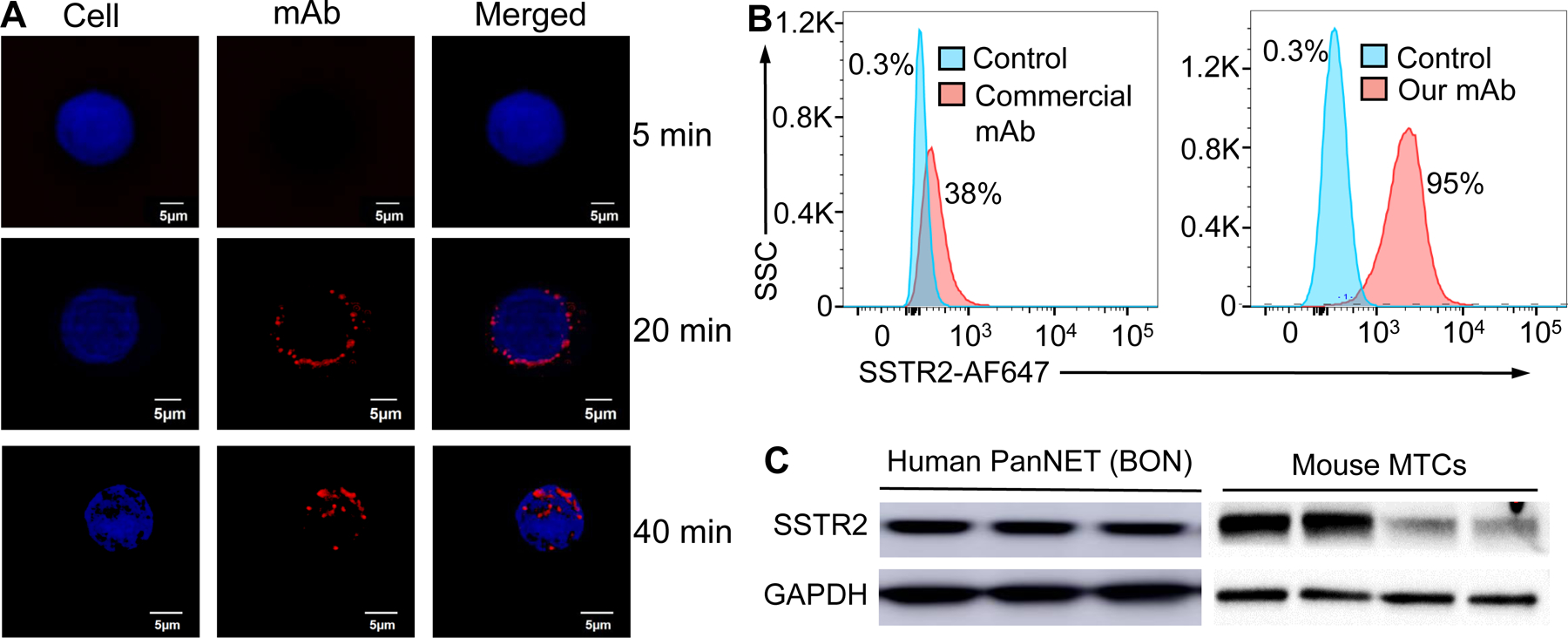
Evaluation of surface binding by anti-SSTR2 mAb. A, Live-cell CLSM dynamic imaging of anti-SSTR2 mAb. Two-color CLSM: whole cell labeled with GFP (displayed as blue) and SSTR2 mAb-MMAE labeled with AF647 (red). Scale bar equals 5 μm. B, Flow cytometry to analyze the surface binding of anti-SSTR2 mAb to NET cell (BON-1) and negative control cell (917). Stained with 1 μg of mAb-AF647/million cells on ice for 30 mins. C, Western blotting of human NET (BON) xenografted tissue and mouse MTC tissues (n = 3–4) using our mAb.
Anti-SSTR2 ADC construction
Our previously established cysteine-based conjugation procedure was used to construct ADC, where a rebridging peptide-based linker was synthesized to maintain mAb integrity during the MMAE conjugation (Fig. 5A).(38) The Agilent 6500 Q-TOF LC/MS confirmed the right structure of linker, and SDS-PAGE confirmed the high integrity of ADC structure (Fig. 5B). The drug-to-antibody ratio (DAR) of the constructed ADC was approximately 4.0.
Figure 5.
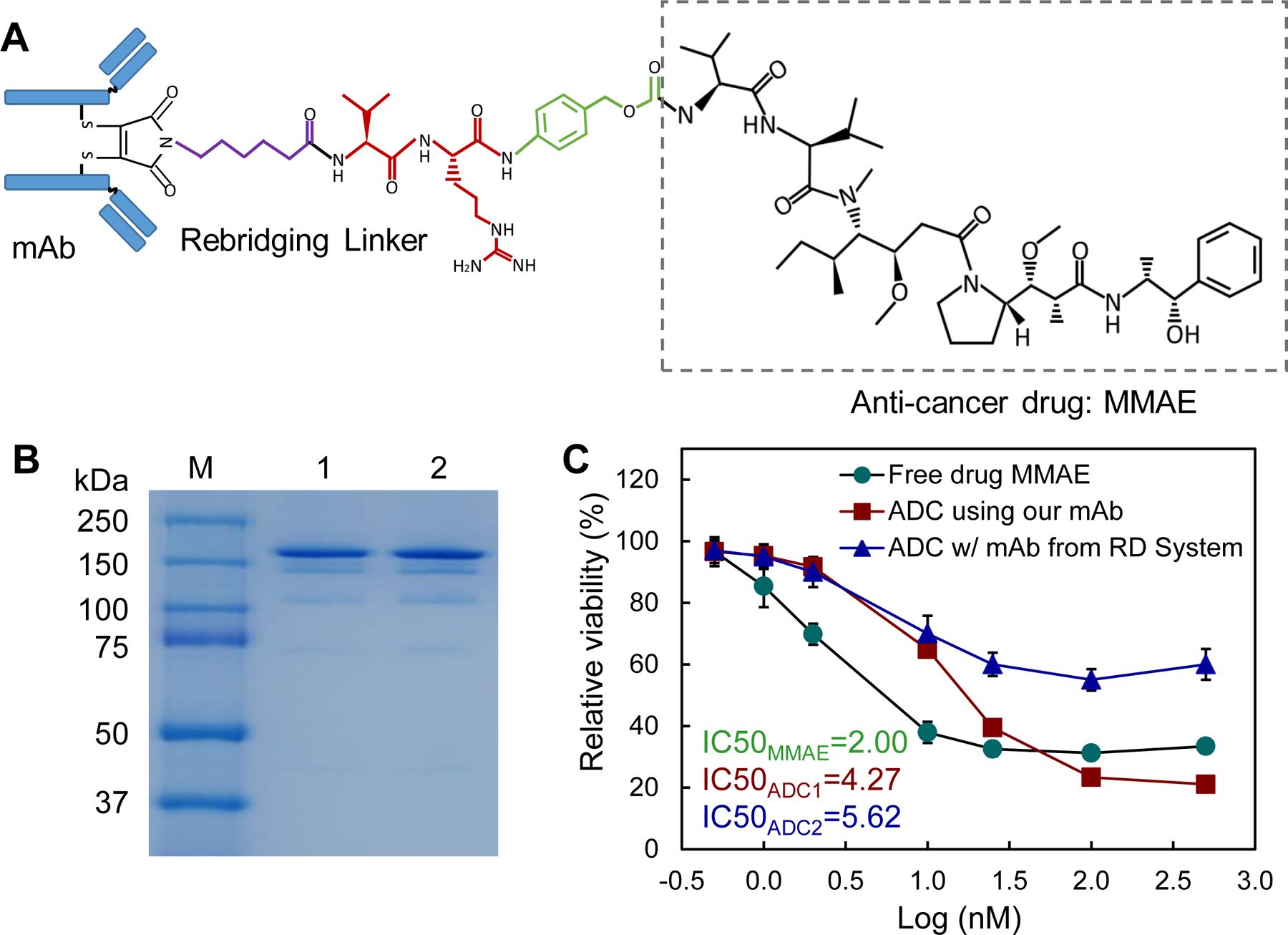
ADC construction and in vitro characterization. A, Molecule structure of anti-SSTR2 mAb-MMAE using re-bridging linker. B, SDS-PAGE to check the integrity of mAb-MMAE. C, The IC50 anti-cancer toxicity of free drug ( ), ADC constructed using commercial anti-SSTR2 mAb (R&D Systems,
), ADC constructed using commercial anti-SSTR2 mAb (R&D Systems,  ), and ADC constructed using our anti-SSTR2 mAb (
), and ADC constructed using our anti-SSTR2 mAb ( ) (data represent mean ± SEM, n = 3).
) (data represent mean ± SEM, n = 3).
In vitro ADC cytotoxicity
We evaluated the in vitro anti-cancer cytotoxicity of the ADC in BON-1 cells by comparing free drug (MMAE) and two ADCs constructed using the anti-SSTR2 mAb developed in this study or the R&D Systems mAb. MMAE is a highly potent cytotoxin that can block microtubulin polymerization.(46–49) The average final viabilities were 31.6%, 39.3%, and 19.8%, viable cell numbers were 15,800, 19,625, and 9,900 cells/mL, and IC50 values were 2.00, 4.27, and 5.62 nM post a 3-day treatment with MMAE, ADC constructed using our mAb, and ADC constructed using commercial mAb, respectively (Fig. 5C). Thus, the mAb-MMAE ADC and free drug had similar cytotoxic potency for NET cells.
Tolerated dose
To investigate the tolerated dose, 5 different doses of anti-SSTR2 ADC were injected into nude (nu/nu) mice (non-tumor bearing) via tail vein: 4, 8, 12, 16, and 20 mg/kg BW (n = 2). Mice were monitored twice daily for a total of 21 days and showed no overt changes in general health including water intake, breathing, and locomotion. ADC at a dose range of 4–20 mg/kg BW had no obvious effects on body weight or overall survival (Fig. 6A). At the end of the study, mice were sacrificed to collect major organs (brain, lung, heart, kidney and liver) for further studies. Since The Human Atlas Project reported the highest level of SSTR2 mRNA brain, so we performed H&E staining of brain tissue. To detect if normal brain is affected or damaged by any possible off-target, we performed H&E staining. As shown in Fig. 6B, brain tissue had no obvious morphology change or necrosis after ADC treatment, indicating that our anti-SSTR2 ADC did not cause cytotoxicity in normal brain. These results indicated that the anti-SSTR2 ADC therapy had no evident off-target effects in vivo.
Figure 6.
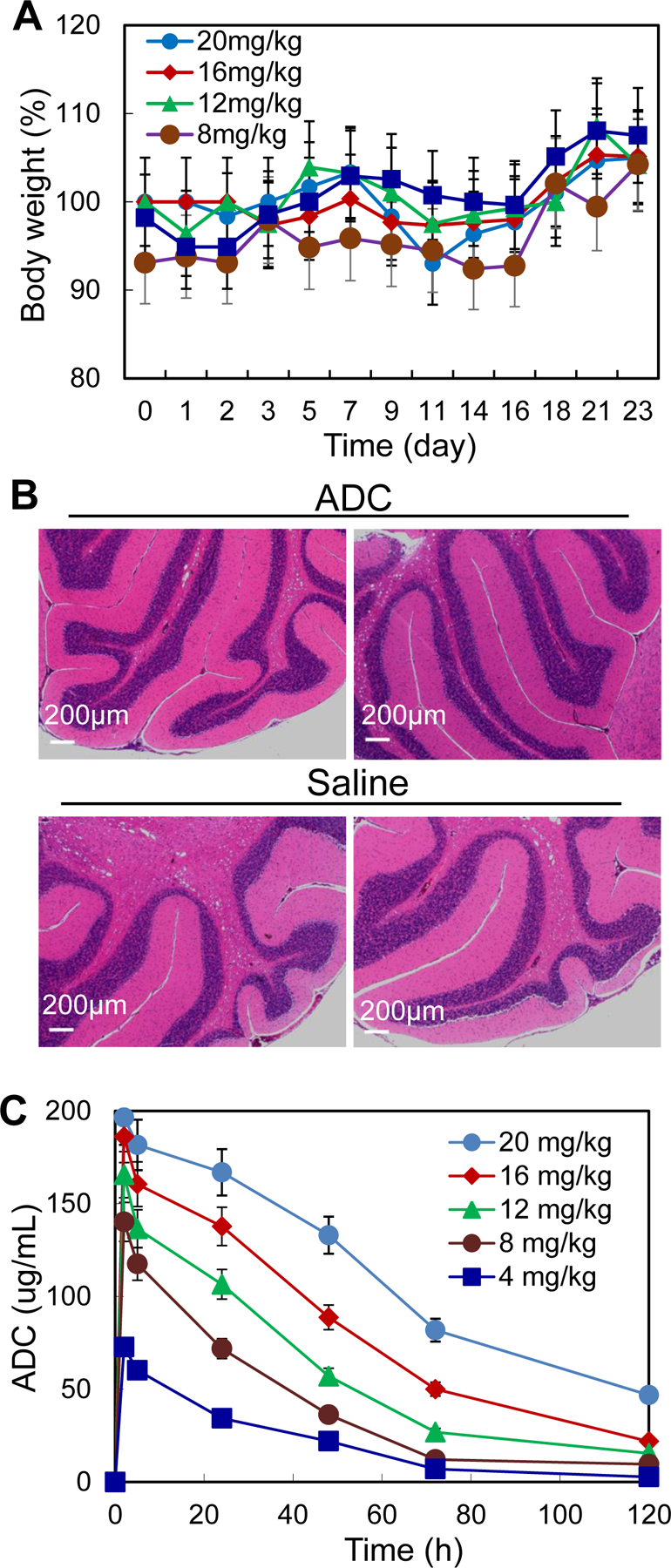
TD and PK studies of ADC. A, TD to test the effect of five ADC dosages including 4, 8, 12, 16 and 20 mg/kg-BW. B, H&E staining of brain tissues. Scale bar equals to 200 μm. C, PK to evaluate the stability and kinetics parameters of ADC (data represent mean ± SEM).
Pharmacokinetics (PK)
A PK study was conducted, where ADC intravenously injecting into nude mice at five doses of 4, 8, 12, 16, and 20 mg/kg. Plasma samples were collected (10–50 μL) from the tail at 0, 2, 8, and 16 hrs, and 1, 2, 3, 5, and 7 days post-ADC injection and titrated for ADC by ELISA (Fig. 6C). The PK modeling indicated that the serum half life (t1/2) = 1.38–2.33 days, Cmax = 72–196 μg/mL, recommended dose (D) = 3.78–14.30 mg/kg BW, and recommended dosing interval (τ) = 4.40–9.10 days. Therefore, based on the calculated D and τ, we selected a dose of 8 mg/kg with administration interval of 4–5 days for the remaining in vivo anti-NET study. Our standard ADC treatment period (3 weeks) was used to simulate the clinical ADC therapy. Moreover, we titrated the plasma samples that were collected from the anti-NET efficacy study using HPLC and detected no cleaved MMAE in plasma, indicating the high conjugation (linker) stability of ADC.
In vivo anti-tumor efficacy
The mice bearing BON-Luc xenografts were treated in a dosing interval of 4.5 days with either anti-SSTR2 ADC (8 mg/kg), anti-SSTR2 mAb (8 mg/kg, control), or saline (vehicle, control) in three groups (n = 6). Fig. 7A showed that tumor growth was significantly attenuated with a tumor volume of 62–67% in the ADC treatment group compared to controls (p≤ 0.001). The tumor fluorescence flux was measured with IVIS, showing 71–73% of signal in the treatment group compared to controls (Fig. 7B, p≤0.001). Terminal tumor weight measurement further confirmed the significant treatment efficacy of ADC (Figs. 7C–D). In order to evaluate the toxicity of ADC, we continued measuring body weight in tumor-bearing mice. As expected, there was no obvious difference among the three groups in overall body weight change, further supporting that the toxicity of ADC was limited or well tolerated (Fig. 7E). The SSTR2 expression in NET tumors during treatment was confirmed in Western blot analysis (Fig. 7F). The surface staining of SSTR2 in tumors from ADC treatment group appeared to be lower than the control group (Fig. 7G), likely due to the NET cell death caused by ADC which was confirmed through H&E staining (Fig. 7H). These findings support the hypothesis that our anti-SSTR2 ADC is an effective drug delivery vehicle with antitumor efficacy and a tolerable toxicity profile.
Figure 7.
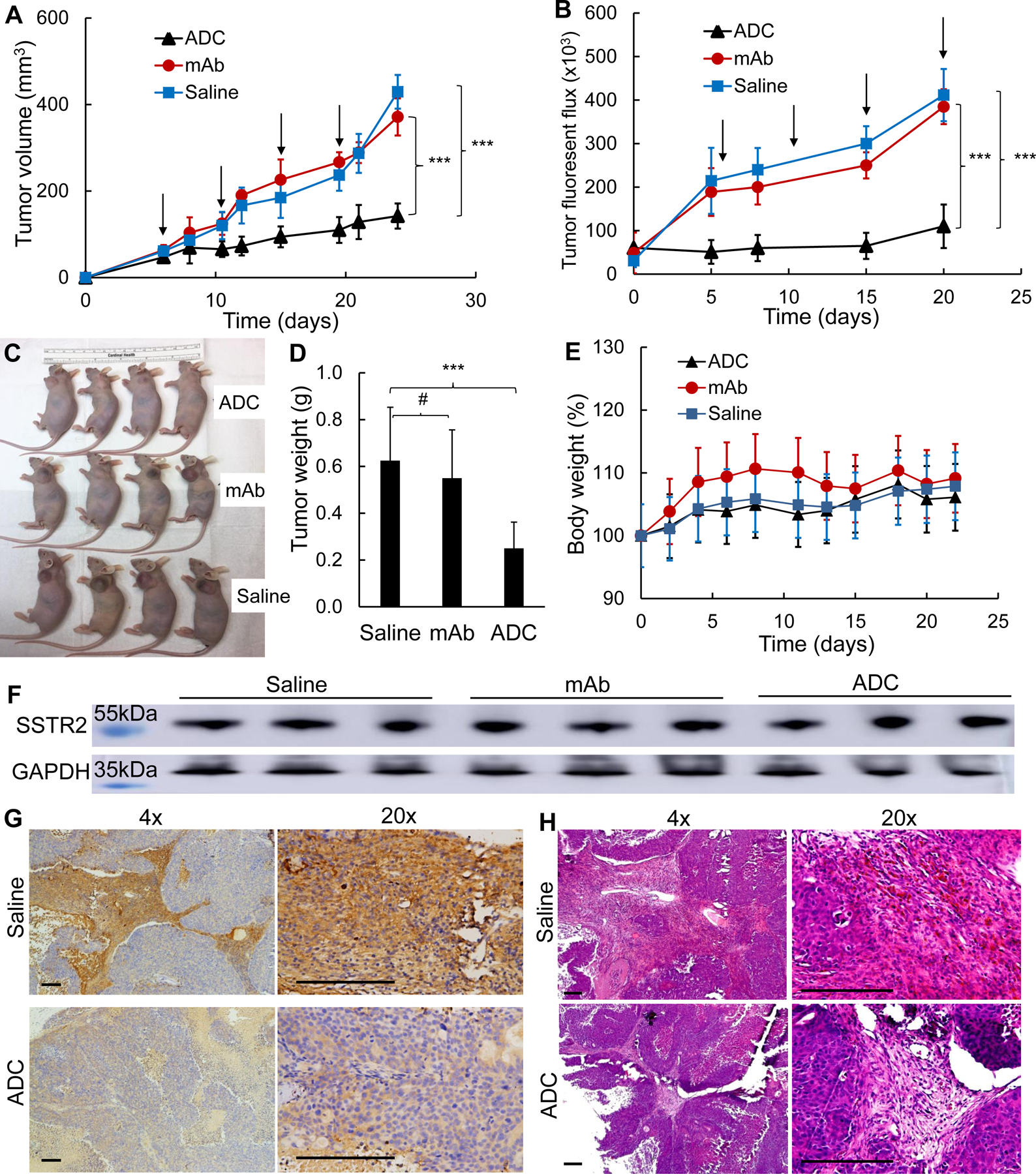
Anti-tumor efficacy study of ADC in NET (BON-Luc) xenografted mouse model. A, Tumor volume changes after BON-Luc cells injection and treatment (data represent mean ± SEM, n = 6). 3×106 mycoplasma-free BON-Luc cells were subcutaneously injected into nude mice. Saline, ADC, or mAb were administrated on Day 6, 10.5, 15, 19.5. Tumor size was measured with calipers and volume was calculated as ellipsoid. Tumor volumes between 3 groups were analyzed with mixed design ANOVA and multiple comparison. *** p<0.001. Black arrow indicating ADC (8 mg/kg BW) treatment date. B, Tumor fluorescence flux measurement with IVIS image system (data represent mean ± SEM, n = 6). C, Tumor bearing mice harvested. D, Wet weight of the tumors excised from harvested mice. Statistical analysis was done with one-way ANOVA, and multiple comparison. #, non-significant. *** p≤0.001. E, Body weight changes of 3 treatment groups. ▲: treatment group injected with ADC,  : control group injected with mAb, and
: control group injected with mAb, and  : control group injected with saline. F, Western blotting of tumors from represented mice (n = 3). G, Representative images of IHC of SSTR2 with tumor tissues in saline and ADC groups (n = 3). Scale bar equals to 50 μm. H, Representative images of H&E staining with tumor tissues in saline and ADC groups (n = 3). Scale bar equals 50 μm.
: control group injected with saline. F, Western blotting of tumors from represented mice (n = 3). G, Representative images of IHC of SSTR2 with tumor tissues in saline and ADC groups (n = 3). Scale bar equals to 50 μm. H, Representative images of H&E staining with tumor tissues in saline and ADC groups (n = 3). Scale bar equals 50 μm.
Discussion
To develop the effective and safe targeted cancer therapies, a unique biomarker that specifically defines the cancer cells from the non-cancerous cells must be identified and thoroughly characterized. The Human Atlas Project reports high mRNA expression of SSTR2 in several normal human tissues (such as brain), but our study showed that the surface protein expression in these tissues (and other normal tissues) is low or undetectable. Our IHC staining of 33 normal human organs showed that spleen and tonsil had no detectable surface SSTR2 expression. The published autoradiography and IHC revealed that SSTRs were mainly located in the red pulp of the spleen which contains diffusely distributed SSTRs.(50–53) The difference could be caused by the different spleen sections used in literature and our IHC staining (i.e. red pulp vs. white pulp) or different detection reagents (i.e. SST analog binding to various SSTR receptors in literature vs. high SSTR2-specific mAb in this study). Unlike our IHC staining of tonsil, the proteinatlas reported medium SSTR2 expression in squamous epithelial cells but no expression in germinal center cells and non-germinal center cells in tonsil. This difference could be caused by the section preparation or the detection reagent used in IHC staining. In future we need to further analyze the SSTR2 surface expression in squamous epithelial cells and compare our anti-SSTR2 mAb with other commercial staining reagents.
There are 5 SSTR subtypes (SSTR1–5) expressed in tumors and normal tissues. Leijon et al.’s previous IHC staining of 151 primary pheochromocytomas and paragangliomas. Their results showed that 74.8% NETs had strong or intermediate SSTR2 expression while tumors had variable individual SSTR3 profiles, and 71.4% metastasized NETs had strong expression of membrane SSTR2 but low expression of cytoplasmic and granular SSTR3.(25) In addition to overall expression, Fotouhi et al. reported that SSTR2 expression was downregulated when small intestinal NET cells progressed through the small intestinal layers. (54) These studies demonstrated that the SSTR2 expression in NETs is highly heterogenous. This study only investigated the overall SSTR2 expression in NET samples and the surface SSTR2 binding of our new mAb. In future we will analyze the SSTR2 expression heterogeneity in NET samples and also perform an in vivo biodistribution study using advanced positron emission tomography (PET).
Although several studies reported SSTR2 protein expression in central nervous system, gastrointestinal tract, and pancreas,(55) the surface expression of SSTR2 in NET tissues was confirmed >20-fold higher than that in normal tissues. Considering that ADC is a dose-dependent targeted therapy, the drastically high expression in NETs allows safe targeting of the SSTR2 with therapeutic drugs. Moreover, our study and other studies (22–24) demonstrate that more than 70% of NET patients abundantly express SSTR2. All the results collected from patient tumor tissues, normal organs, and cell lines demonstrate that SSTR2 is an ideal receptor for targeted cancer therapy.
Supporting our findings, not all patients with NETs overexpress SSTR2.(56, 57) For example, only 45–66% of pulmonary NET patients and 80–95% gastroenteropancreatic NET patients overexpress SSTR2.(56) To benefit the SSTR2 negative patients, we performed a comparative membrane proteomics study and found that the carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) has high expression in pancreatic NET cells (BON-1 and QGP-1) but not in non-NET cancerous pancreatic adenocarcinoma cells (PANC-1 and MiAPaCa-1) and non-cancerous fibroblast cell (WI-38). High CEACAM1 expression has been suggested in various cancers, including medullary thyroid cancer which represents a type of NET.(58, 59) Although further evaluation is needed, CEACAM1 might be an alternative receptor for ADC therapy in NET patients with minimal SSTR2 density.
In this study, we developed, characterized and confirmed a novel monoclonal antibody to target the identified SSTR2 receptor for NET therapy. Unlike a commercially available mAb developed using the whole SSTR2 membrane protein as immunogen, our anti-SSTR2 mAb was designed to selectively target two extracellular domains of SSTR2. As a result, our mAb showed a higher and more specific surface binding to NET cells than the commercial mAb. The new mAb was also designed to exhibit strong cross-reactivity to both human and mouse SSTR2, allowing the results presented here to be more translatable to test and use in human patients. Importantly, the maximum tolerated dose study we performed did not detect any adverse body weight or behavior changes at dose of up to 20 mg ADC/kg. For example, no histopathology was detected in murine brain tissue where the highest SSTR2 mRNA expression occurs. Furthermore, the constructed anti-SSTR2 monoclonal antibody-drug significantly reduced the tumor growth in s.c. xenograft mice. Taken together, the developed anti-SSTR2 mAb can specifically target the SSTR2-overexpressing NET cell lines, patient-derived tissues and xenografts, and the targeting delivered potent small molecule via ADC has high cytotoxicity to NET with minimal side effects.
Lutathera that combines the endoradiotherapy ([177Lu]Lu-DOTA-TATE) with its diagnostic partner [68Ga]Ga-DOTA-TATE or [68Ga]Ga-DOTA-TOC (DOTA-(D-Phe1, Tyr3)-octreotide has been approved to treat SSTR positive gastroenteropancreatic NET.(19, 20) The mechanism is to target SSTRs positive lesions using SST analogue and kill cells via the DNA damage provoked by 177Lu. The disadvantages of this therapy are: the radiopharmaceutical shelf life is short (i.e. 72 hrs from the time point of calibration at the end of production); the active concentration changes over time due to the decay of 177Lu; and the therapeutic impact on rapidly proliferating NETs is relatively poor. Moreover, the FDA approved SST analogue (e.g. octreotide LAR) with long-term stability has been used for diagnosis and imaging. Clinical trials show that octreotide effectively improves NET symptoms such as diarrhea, flushing, bronchoconstriction and carcinoid cardiac disease but does not demonstrate obviously improvement of the overall survival.(60, 61) As compared to Lutathera, our new anti-SSTR2 mAb-based ADC has the advantages of longer shelf life, higher cytotoxicity to treat NET, and potential to improve survival of patients. As compared to octreotide, our anti-SSTR2 mAb has higher specificity to target the membrane SSTR2, which can be used as diagnosis reagent and SSTR2-targeted delivery vehicle. We will evaluate and compare our ADC with Lutathera and octreotide in future.
Previous studies have reported that SSTR2 involves in apoptosis, regulation of cyclin-dependent kinase inhibitors, and inhibition of proliferation signaling.(62, 63) These findings indicated that the anti-SSTR2 mAb could regulate NET cell growth via SSTR2-mediated signaling cascades, but it needs a full investigation in future study. Moreover, the possible synergism of mAb and ADC-delivered cytotoxic payload, the optimal ADC dosage and treatment strategy, and a full MTD study will be performed in vivo. Also it is imperative to further evaluate the potential side effects, particular with regards to the patients demonstrating low-level SSTR2 expression.
In conclusion, our anti-SSTR2 ADC has a great potential to treat NET due to its capability or potential to target SSTR2 positive NET, reduce undesirable side effects, and effectively reduce NET growth.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. J. Bart Rose and Ms. Rachael Guenter and the Tissue-Based Translational Research Lab in the Department of Pathology at University of Alabama at Birmingham (UAB) for the design and construction of the tissue microarray.
Funding
This work was supported by SDHB Pheo Para Coalition (J.A.B.), National Institute of Health (NIH) R21HL 127599A1 (L.Z.), NIH R21CA226491-01A1 (R.J. and X.M.L.), NIH 1R01CA238273-01A1 (X.M.L.), and North American Neuroendocrine Tumor Society (NANETS) Basic/Translational Science Investigator award (R.J.).
Footnotes
Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–72. [DOI] [PubMed] [Google Scholar]
- 2.Kulke MH, Benson AB 3rd, Bergsland E, Berlin JD, Blaszkowsky LS, Choti MA, et al. Neuroendocrine tumors. J Natl Compr Canc Netw. 2012;10(6):724–64. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Hardacre JM, Uzar A, Cameron JL, Choti MA. Isolated liver metastases from neuroendocrine tumors: does resection prolong survival? J Am Coll Surg. 1998;187(1):88–92; discussion −3. [DOI] [PubMed] [Google Scholar]
- 4.Norton JA. Endocrine tumours of the gastrointestinal tract. Surgical treatment of neuroendocrine metastases. Best Pract Res Clin Gastroenterol. 2005;19(4):577–83. [DOI] [PubMed] [Google Scholar]
- 5.Mayo SC, de Jong MC, Pulitano C, Clary BM, Reddy SK, Gamblin TC, et al. Surgical management of hepatic neuroendocrine tumor metastasis: results from an international multi-institutional analysis. Ann Surg Oncol. 2010;17(12):3129–36. [DOI] [PubMed] [Google Scholar]
- 6.Adler JT, Meyer-Rochow GY, Chen H, Benn DE, Robinson BG, Sippel RS, et al. Pheochromocytoma: current approaches and future directions. Oncologist. 2008;13(7):779–93. [DOI] [PubMed] [Google Scholar]
- 7.Pinchot SN, Pitt SC, Sippel RS, Kunnimalaiyaan M, Chen H. Novel targets for the treatment and palliation of gastrointestinal neuroendocrine tumors. Curr Opin Investig Drugs. 2008;9(6):576–82. [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Pruitt A, Nicol TL, Gorgulu S, Choti MA. Complete hepatic resection of metastases from leiomyosarcoma prolongs survival. J Gastrointest Surg. 1998;2(2):151–5. [DOI] [PubMed] [Google Scholar]
- 9.Chen H Therapeutic options for patients with metastatic gastrointestinal carcinoid. J Surg Oncol. 2008;97(3):203–4. [DOI] [PubMed] [Google Scholar]
- 10.Shiba S, Morizane C, Hiraoka N, Sasaki M, Koga F, Sakamoto Y, et al. Pancreatic neuroendocrine tumors: A single-center 20-year experience with 100 patients. Pancreatology. 2016;16(1):99–105. [DOI] [PubMed] [Google Scholar]
- 11.Brown KT, Koh BY, Brody LA, Getrajdman GI, Susman J, Fong Y, et al. Particle embolization of hepatic neuroendocrine metastases for control of pain and hormonal symptoms. J Vasc Interv Radiol. 1999;10(4):397–403. [DOI] [PubMed] [Google Scholar]
- 12.Isozaki T, Kiba T, Numata K, Saito S, Shimamura T, Kitamura T, et al. Medullary thyroid carcinoma with multiple hepatic metastases: treatment with transcatheter arterial embolization and percutaneous ethanol injection. Intern Med. 1999;38(1):17–21. [DOI] [PubMed] [Google Scholar]
- 13.Lal A, Chen H. Treatment of advanced carcinoid tumors. Curr Opin Oncol. 2006;18(1):9–15. [DOI] [PubMed] [Google Scholar]
- 14.Lehnert T Liver transplantation for metastatic neuroendocrine carcinoma: an analysis of 103 patients. Transplantation. 1998;66(10):1307–12. [DOI] [PubMed] [Google Scholar]
- 15.Zhang R, Straus FH, DeGroot LJ. Effective genetic therapy of established medullary thyroid carcinomas with murine interleukin-2: dissemination and cytotoxicity studies in a rat tumor model. Endocrinology. 1999;140(5):2152–8. [DOI] [PubMed] [Google Scholar]
- 16.Boudreaux JP, Putty B, Frey DJ, Woltering E, Anthony L, Daly I, et al. Surgical treatment of advanced-stage carcinoid tumors: lessons learned. Ann Surg. 2005;241(6):839–45; discussion 45–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen C, Faraggi M, Giraudet AL, de Labriolle-Vaylet C, Aparicio T, Rouzet F, et al. Long-term efficacy of radionuclide therapy in patients with disseminated neuroendocrine tumors uncontrolled by conventional therapy. J Nucl Med. 2004;45(10):1660–8. [PubMed] [Google Scholar]
- 18.Fiorentini G, Rossi S, Bonechi F, Vaira M, De Simone M, Dentico P, et al. Intra-arterial hepatic chemoembolization in liver metastases from neuroendocrine tumors: a phase II study. J Chemother. 2004;16(3):293–7. [DOI] [PubMed] [Google Scholar]
- 19.Oberg K, Kvols L, Caplin M, Delle Fave G, de Herder W, Rindi G, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. 2004;15(6):966–73. [DOI] [PubMed] [Google Scholar]
- 20.Hennrich U, Kopka K. Lutathera((R)): The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals (Basel). 2019;12(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53(1):1–24. [PubMed] [Google Scholar]
- 22.Pinchot SN, Holen K, Sippel RS, Chen H. Carcinoid tumors. Oncologist. 2008;13(12):1255–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zatelli MC, Tagliati F, Taylor JE, Rossi R, Culler MD, degli Uberti EC. Somatostatin receptor subtypes 2 and 5 differentially affect proliferation in vitro of the human medullary thyroid carcinoma cell line tt. J Clin Endocrinol Metab. 2001;86(5):2161–9. [DOI] [PubMed] [Google Scholar]
- 24.Sun LC, Coy DH. Somatostatin receptor-targeted anti-cancer therapy. Curr Drug Deliv. 2011;8(1):2–10. [DOI] [PubMed] [Google Scholar]
- 25.Leijon H, Remes S, Hagstrom J, Louhimo J, Maenpaa H, Schalin-Jantti C, et al. Variable somatostatin receptor subtype expression in 151 primary pheochromocytomas and paragangliomas. Hum Pathol. 2019;86:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou L, Xu N, Sun Y, Liu XM. Targeted biopharmaceuticals for cancer treatment. Cancer Lett. 2014;352(2):145–51. [DOI] [PubMed] [Google Scholar]
- 27.Almasbak H, Aarvak T, Vemuri MC. CAR T Cell Therapy: A Game Changer in Cancer Treatment. J Immunol Res. 2016;2016:5474602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dai H, Wang Y, Lu X, Han W. Chimeric Antigen Receptors Modified T-Cells for Cancer Therapy. J Natl Cancer Inst. 2016;108(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. 2016;59(4):340–8. [DOI] [PubMed] [Google Scholar]
- 30.Little M, Kipriyanov SM, Le Gall F, Moldenhauer G. Of mice and men: hybridoma and recombinant antibodies. Immunology Today. 2000;21(8):364–70. [DOI] [PubMed] [Google Scholar]
- 31.Stump B, Steinmann J. Conjugation process development and scale-up. Antibody-Drug Conjugates: Springer; 2013. p. 235–48. [DOI] [PubMed] [Google Scholar]
- 32.Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med. 2015;7(302):302ra136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pereira DS, Guevara CI, Jin L, Mbong N, Verlinsky A, Hsu SJ, et al. AGS67E, an Anti-CD37 Monomethyl Auristatin E Antibody-Drug Conjugate as a Potential Therapeutic for B/T-Cell Malignancies and AML: A New Role for CD37 in AML. Mol Cancer Ther. 2015;14(7):1650–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whalen KA, White BH, Quinn JM, Kriksciukaite K, Alargova R, Au Yeung TP, et al. Targeting the Somatostatin Receptor 2 with the Miniaturized Drug Conjugate, PEN-221: A Potent and Novel Therapeutic for the Treatment of Small Cell Lung Cancer. Mol Cancer Ther. 2019;18(11):1926–36. [DOI] [PubMed] [Google Scholar]
- 35.Kiaris H, Schally AV, Nagy A, Szepeshazi K, Hebert F, Halmos G. A targeted cytotoxic somatostatin (SST) analogue, AN-238, inhibits the growth of H-69 small-cell lung carcinoma (SCLC) and H-157 non-SCLC in nude mice. European Journal of Cancer. 2001;37:620–8. [DOI] [PubMed] [Google Scholar]
- 36.Sun L, Fuselier JA, Coy DH. Effects of camptothecin conjugated to a somatostatin analog vector on growth of tumor cell lines in culture and related tumors in rodents. Drug Deliv. 2004;11(4):231–8. [DOI] [PubMed] [Google Scholar]
- 37.Xu N, Ou J, Gilani A-K, Zhou L, Liu M. High-level expression of recombinant IgG1 by CHO K1 platform. Frontiers of Chemical Science and Engineering. 2015;9(3):376–80. [Google Scholar]
- 38.Ou J, Si Y, Goh K, Yasui N, Guo Y, Song J, et al. Bioprocess development of antibody-drug conjugate production for cancer treatment. PLoS One. 2018;13(10):e0206246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu N, Ou J, Si Y, Goh KY, Flanigan DD, Han X, et al. Proteomics insight into the production of monoclonal antibody. Biochemical Engineering Journal. 2019;145:177–85. [Google Scholar]
- 40.Hasegawa K, Kudoh S, Ito T. Somatostatin receptor staining in FFPE sections using a ligand derivative dye as an alternative to immunostaining. PLoS One. 2017;12(2):e0172030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu N, Liu M, Liu M. Pharmacology, Pharmacokinetics, and Pharmacodynamics of Antibodies. Biosimilairs of Monoclonal Antibodies. 2016. [Google Scholar]
- 42.Sherbenou DW, Aftab BT, Su Y, Behrens CR, Wiita A, Logan AC, et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J Clin Invest. 2016;126(12):4640–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Si Y, Kim S, Zhang E, Tang Y, Jaskula-Sztul R, Markert JM, et al. Targeted Exosomes for Drug Delivery: Biomanufacturing, Surface Tagging, and Validation. Biotechnol J. 2019:e1900163. [DOI] [PubMed] [Google Scholar]
- 44.Pozo K, Castro-Rivera E, Tan C, Plattner F, Schwach G, Siegl V, et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell. 2013;24(4):499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pozo K, Hillmann A, Augustyn A, Plattner F, Hai T, Singh T, et al. Differential expression of cell cycle regulators in CDK5-dependent medullary thyroid carcinoma tumorigenesis. Oncotarget. 2015;6(14):12080–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102(4):1458–65. [DOI] [PubMed] [Google Scholar]
- 47.Yao H, Jiang F, Lu A, Zhang G. Methods to Design and Synthesize Antibody-Drug Conjugates (ADCs). Int J Mol Sci. 2016;17(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham D, Parajuli KR, Zhang C, Wang G, Mei J, Zhang Q, et al. Monomethyl Auristatin E Phosphate Inhibits Human Prostate Cancer Growth. Prostate. 2016;76(15):1420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Yu C, Jiang J, Huang C, Yao X, Xu Q, et al. An anti-HER2 antibody conjugated with monomethyl auristatin E is highly effective in HER2-positive human gastric cancer. Cancer Biol Ther. 2016;17(4):346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarikaya I, Sarikaya A, Alnafisi N, Alenezi S. Significance of splenic uptake on somatostatin receptor imaging studies. Nucl Med Rev Cent East Eur. 2018;21(1):66–70. [DOI] [PubMed] [Google Scholar]
- 51.Melis M, Kaemmerer D, de Swart J, Kulkarni HR, Lupp A, Sanger J, et al. Localization of Radiolabeled Somatostatin Analogs in the Spleen. Clin Nucl Med. 2016;41(2):e111–4. [DOI] [PubMed] [Google Scholar]
- 52.Reubi JC, Waser B, Horisberger U, Krenning E, Lamberts SW, Gebbers J-O, et al. In vitro autoradiographic and in vivo scintigraphic localization of somatostatin receptors in human lymphatic tissue. Blood. 1993;82:2143–51. [PubMed] [Google Scholar]
- 53.Reubi JC, Ursula H, Kappeler A, Laissue JA. Localization of receptors for vasoactive intestinal peptide, somatostatin, and substance P in distinct compartments of human lymphoid organs. Blood. 1998;92:191–7. [PubMed] [Google Scholar]
- 54.Fotouhi O, Zedenius J, Hoog A, Juhlin CC. Regional differences in somatostatin receptor 2 (SSTR2) immunoreactivity is coupled to level of bowel invasion in small intestinal neuroendocrine tumors. Neuroendocrinology Letters. 2018;39(4):305–9. [PubMed] [Google Scholar]
- 55.Cakir M, Dworakowska D, Grossman A. Somatostatin receptor biology in neuroendocrine and pituitary tumours: part 1--molecular pathways. J Cell Mol Med. 2010;14(11):2570–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Righi L, Volante M, Tavaglione V, Bille A, Daniele L, Angusti T, et al. Somatostatin receptor tissue distribution in lung neuroendocrine tumours: a clinicopathologic and immunohistochemical study of 218 ‘clinically aggressive’ cases. Ann Oncol. 2010;21(3):548–55. [DOI] [PubMed] [Google Scholar]
- 57.Sherman SK, Maxwell JE, Carr JC, Wang D, O’Dorisio MS, O’Dorisio TM, et al. GIPR expression in gastric and duodenal neuroendocrine tumors. J Surg Res. 2014;190(2):587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thies A, Moll I, Berger J, Wagener C, Brummer J, Schulze HJ, et al. CEACAM1 expression in cutaneous malignant melanoma predicts the development of metastatic disease. J Clin Oncol. 2002;20(10):2530–6. [DOI] [PubMed] [Google Scholar]
- 59.Tilki D, Irmak S, Oliveira-Ferrer L, Hauschild J, Miethe K, Atakaya H, et al. CEA-related cell adhesion molecule-1 is involved in angiogenic switch in prostate cancer. Oncogene. 2006;25(36):4965–74. [DOI] [PubMed] [Google Scholar]
- 60.Hejna M, Schmidinger M, Raderer M. The clinical role of somatostatin analogues as antineoplastic agents: much ado about nothing? Ann Oncol. 2002;13(5):653–68. [DOI] [PubMed] [Google Scholar]
- 61.Yau H, Kinaan M, Quinn SL, Moraitis AG. Octreotide long-acting repeatable in the treatment of neuroendocrine tumors: patient selection and perspectives. Biologics. 2017;11:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guillermet J, Saint-Laurent N, Rochaix P, Cuvillier O, Levade T, Schally AV, et al. Somatostatin receptor subtype 2 sensitizes human pancreatic cancer cells to death ligand-induced apoptosis. Proc Natl Acad Sci U S A. 2003;100(1):155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lahlou H, Guillermet J, Hortala M, Vernejoul F, Pyronnet S, Bousquet C, et al. Molecular signaling of somatostatin receptors. Ann N Y Acad Sci. 2004;1014:121–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.