

Abstract
Defining traits of platinum-tolerant cancer cells could expose new treatment vulnerabilities. Here, new markers associated with platinum-tolerant cells and tumors were identified using in vitro and in vivo ovarian cancer (OC) models treated repetitively with carboplatin and validated in human specimens. Platinum-tolerant cells and tumors were enriched in ALDH(+) cells, formed more spheroids, and expressed increased levels of stemness-related transcription factors compared to parental cells. Additionally, platinum-tolerant cells and tumors exhibited expression of the Wnt receptor Frizzled 7 (FZD7). Knockdown of FZD7 improved sensitivity to platinum, decreased spheroid formation, and delayed tumor initiation. The molecular signature distinguishing FZD7(+) from FZD7(−) cells included epithelial-to-mesenchymal (EMT), stemness, and oxidative phosphorylation-enriched gene sets. Overexpression of FZD7 activated the oncogenic factor Tp63, driving upregulation of glutathione metabolism pathways, including glutathione peroxidase 4 (GPX4), which protected cells from chemotherapy-induced oxidative stress. FZD7(+) platinum-tolerant OC cells were more sensitive and underwent ferroptosis after treatment with GPX4 inhibitors. FZD7, Tp63, and glutathione metabolism gene sets were strongly correlated in the OC Tumor Cancer Genome Atlas (TCGA) database and in residual human OC specimens after chemotherapy. These results support the existence of a platinum-tolerant cell population with partial cancer stem cell features, characterized by FZD7 expression and dependent on FZD7-β-catenin-Tp63-GPX4 pathway for survival. The findings reveal a novel therapeutic vulnerability of platinum-tolerant cancer cells and provide new insight into a potential “persister cancer cell” phenotype.
Keywords: Frizzled-7, platinum tolerance, platinum resistance, ovarian cancer, cancer stem cells, ferroptosis, glutathione peroxidase 4 (GPX4)
Introduction
Ovarian cancer (OC) is the leading cause of death from female gynecological cancers. Although initially a highly chemo-responsive tumor (1), most patients with OC experience tumor relapse and recurrent, resistant OC is fatal (2). “Persister” or drug-tolerant cells have been described as cells surviving cytotoxic drug exposure (3) and represent a reservoir for the outgrowth of drug-resistant clones. Recent studies in various cancers have reported the molecular signature of “persister” cells, including upregulation of stemness factors, mesenchymal-like gene expression, enrichment in glutathione peroxidase 4 (GPX4) and other genes related to lipid peroxidation, which antagonize ferroptosis allowing cells to survive after cytotoxic drug exposure (3–5). Small molecule inhibitors targeting GPX4 were shown to induce lipid peroxidation and eliminate tyrosine kinase receptor inhibitor tolerant cells through ferroptosis (3). It has been suggested that “persister” cells share characteristics with cancer stem cells (CSCs), but also have distinct traits. Specific markers to allow their identification and early targeting remain elusive. Here we sought to characterize the OC “persister” phenotype.
Our and other previous studies showed that while chemotherapy is effective at cytoreducing the mass of heterogeneous cancer cells, residual tumors persist and are enriched in CSCs (6, 7). Ovarian CSCs share some of the normal stem cells’ characteristics, including the ability to self-renew, differentiate, express specific stem cell surface markers (6, 8, 9), and exhibit enhanced tumor initiation capacity (TIC) (10). Importantly, ovarian CSCs possess a phenotype associated with drug resistance, including diminished apoptotic responses, increased efflux mechanisms and antioxidation defense (4, 8, 11, 12). The boundaries between stemness and platinum resistant (Pt-R) phenotypes remain blurry and, while an overlap exists, it is assumed that distinct pathways drive the two entities.
As platinum tolerant (Pt-T) cancer cells drive tumor relapse, we aimed to identify specific markers, by using in vitro and in vivo models of repeated exposure to the cytotoxic agent. We observed that Pt-T cells and tumors contained an increased ALDH+ cell population, expressing stemness related transcription factors (TFs), and able to form more spheroids compared to chemotherapy naïve cells. We identified the Frizzled 7 receptor (FZD7) as a novel cell surface marker significantly upregulated in the platinum tolerant cell population. FZD7 knock down increased sensitivity to Pt, decreased spheroid formation, and inhibited TIC. FZD7(+) cells harbored a “persister cell”-like signature, including down-regulated genes associated to DNA damage response, upregulated epithelial to mesenchymal (EMT) and stemness, and decreased expression of genes associated with oxidative phosphorylation. Expression of the antioxidant enzyme, GPX4 was increased in FZD7(+) Pt-T cells, rendering them sensitive to treatment with GPX4 inhibitors. Mechanistically, FZD7 caused activation of the transcriptional regulator, Tp63, which drove upregulation of glutathione metabolism genes, protecting cells from oxidative stress. In all, our results support the existence of a “persister” Pt-T cell population, sharing traits with CSCs, marked by upregulation of the receptor FZD7 and harboring a dependency on FZD7-β catenin-Tp63 mediated GPX4 expression and anti-oxidant activity.
Materials and methods.
Human specimens.
Deidentified high grade serous ovarian tumors (HGSOC) and associated malignant ascites were collected and processed fresh from patients who provided written informed consent. Tumor tissues were enzymatically disassociated into single cell suspensions and cultured as previously described (6, 8). A tissue microarray (TMA) was built from de-identified HGSOC specimens (n = 23) from patients who had undergone 3–6 cycles of platinum-taxane neoadjuvant chemotherapy (IRB approved CSR protocol #1247). Each specimen was entered in duplicate and fallopian tube epithelium (n=6) served as control. Patients’ characteristics are in Table S1. Human subject studies were conducted in accordance with the Declaration of Helsinki and approved the institutional review board (Northwestern University IRB#: STU00202468).
Cell lines and culture conditions.
SKOV3 and OVCAR3 cells were purchased from the American Type Culture Collection (ATCC). OVCAR5 cells were a generous gift from Dr. Marcus Peter, Northwestern University, COV362 cells were from Dr. Kenneth Nephew, Indiana University, immortalized human fallopian tube luminal epithelial cells (FT190) were from Dr. R. Drapkin of University of Pennsylvania (13). PEO1 and PEO4 cells were from Sigma Aldrich. Cell culture conditions are in Supplemental Material (SM). Low passage cells were used and all cell lines were tested to be pathogen and Mycoplasma negative (Charles River Research Animal Diagnostic Services).
Chemicals and reagents.
RSL3 was purchased from Fisher Scientific (Cat# 611810). ML210 (Cat# SML0521), cisplatin (Cat# 1134357), and carboplatin (Cat# C2538) were from Sigma-Aldrich. WNT3a was from Fisher Scientific (Cat# 5036WN010CF, R&D Systems), and IWR-1-endo was from Santa Cruz (sc-295215A).
In vitro development of Pt-R cells:
To generate Pt-T OC cells, SKOV3, OVCAR5, COV362, and OVCAR3 cells were treated with 3 or 4 repeated or increasing doses of cisplatin or carboplatin for 24 hours. Surviving cells were allowed to recover for 3 to 4 weeks before receiving the next treatment. Changes in resistance to platinum were estimated by calculating half maximal inhibitory concentration (IC50) values as described below.
In vivo experiments:
Animal studies were conducted according to a protocol (# IS00003060) approved by the Institutional Animal Care and Use Committee of Northwestern University and are described in SM. Experiments using PDX tumors were performed in the Developmental Therapeutics Core (DTC) of the Lurie Cancer Center, as previously described (14) and following a similar protocol (see SM).
Isolation of tumor cells.
Tumors from patients or xenografts were minced and enzymatically dissociated in Dulbecco’s modified Eagle’s medium/F12 (Thermo Fisher Scientific, Ref# 11320) containing collagenase (300 IU/ml, Sigma-Aldrich, Cat#C7657) and hyaluronidase (300 IU/ml, Sigma-Aldrich, Cat# H3506) for 2–4 hours at 37°C. The tissue digest was passed several times through a 16–18G needle using Cell Stripper (Corning, Cat# 25-056-CI) to dissociate remaining cell aggregates. Red blood cell lysis used RBC lysis buffer (BioLegend, Cat#420301), followed by DNaseI (Sigma Aldrich, Cat# DN25) treatment and filtering through a 40μm cell strainer (Fisher Scientific, Cat#NC0147038) to yield single cell suspension.
Aldefluor assay and flow cytometry.
Aldehyde dehydrogenase (ALDH) activity was measured using an Aldefluor assay kit (Stemcell Technologies, Cat#01700, Cambridge, MA, USA) following the manufacturer’s instructions and as described previously (6).
Cell survival assay.
Cell survival was measured with a Cell Counting Kit 8 (CCK8, Dojindo Molecular Technologies, Cat# CK04, Rockville, MD, USA), following the manufacturer’s protocol. Absorbances (450 nm) were measured with a microplate reader (BioTek ELX800, BioTeK, Winooski, VT).
Detailed protocols for spheroid formation assay; clonogenic assay, RNA extraction, quantitative Rt-PCR, western blotting and IHC are included in SM. Primers are included in Supplementary Table S2.
Extreme limited dilution assay.
A serial dilution of OVCAR5_shControl or OVCAR5_shFZD7 cells (5, 10, 50, 100, 500, 1000, and 5000 cells) were sorted by FACS directly into 96-well low-attached plates and cultured in MammoCult medium for 14 days as described above. Each dilution included 10 replicates. The total number of wells containing spheroids for each dilution were counted. The CSC frequency and statistical significance were determined using ELDA software at http://bioinf.wehi.edu.au/software/elda/(15).
Half maximal inhibitory concentration (IC50).
The IC50 values of the various treatment compounds were determined by the CCK8 assay as described in SM. IC50 values were determined by logarithm-normalized sigmoidal dose curve fitting using Prism 6 software (GraphPad Software Inc., San Diego, CA).
Lipid peroxidation assay:
Intracellular lipid peroxidation was determined by a Lipid Peroxidation Assay (Sigma-Aldrich, Cat# MAK085) following the manufacturer’s protocol (see SM).
Oxygen consumption rate.
Cells were seeded on 96-well plates at 100,000 cells/well and incubated overnight. 10μL of extracellular O2 consumption reagent (Oxygen Consumption Rate Assay kit, Abcam Cat#197243) were added to each well, and fluorescence was measured with a plate reader (SpectraMax i3X, Molecular Devices, San Jose, CA, USA) at 3 min intervals for 180 min at excitation/emission = 380/650 nm. Alternatively, oxygen consumption was measured using a Seahorse assay. Briefly, OVCAR5 shControl and shFZD7 cell lines were seeded in Seahorse 96-well microplate (Agilent, Cat#102416-100, Santa Clara, CA, USA) at a density of 10-80K per well. After incubation overnight, oxygen consumption was measured and calculated by Seahorse XFe96 Analyzer (Agilent, Santa Clara, CA, USA).
BODIPY staining for lipid peroxidation.
Cells were treated as described in SM. After treatment, cells were stained with BODIPY 581/591 C11 (5μM) for an hour at 37°C, washed with PBS, and fixed with 4% PFA on ice for 30 mins. The mean fluorescence intensity (minimum of 10,000 events per condition) was measured by FACS (LSR Fortessa, BD, Franklin lake, NJ). BODIPY emission was recorded on channels for FITC at 520nm and PE at 580nm. The data were displayed as histograms and mean fluorescence intensity of FITC was calculated.
Intracellular ROS levels were measured by monitoring the oxidation of cell permeable 2′,7′-dichlorofluorescein diacetate (DCFHDA, Sigma-Aldrich) at excitation and emission wavelengths of 480 and 535 nm. 150,000 cells cultured in 35 mm glass bottom dish were treated with 1 or 2μM ML210 alone or with 800nM DFOA for 24 hours. Cell cultures were then treated with 10μM DCFDA (Abcam, Cat#ab113851) for 15 minutes to detect ROS level through confocal fluorescence microscopy. ROS level was measured as integral fluorescence intensity normalized by the cellular area in a frame (n=15 frames) using ImageJ (https://imagej.nih.gov/ij).
RNA sequencing (RNA-seq) and data analysis.
The RNA-seq libraries (n=3 per experimental group) were prepared using the NEBNext Ultra II RNA library prep kit from Illumina (New England Biolabs Inc., Ipswich, MA, see SM). Trimmed reads were aligned to the ENSEMBL human genome version GRCh38 using STAR (2.5.2)(16) and SAMtools (17). Mapped reads were then counted using HTSeq (18). Differentially expressed genes were determined by exact test analysis followed by multiple hypothesis correction using false discovery rate (FDR) on the edgeR package (19). Genes with FDR ≤ 0.05 were considered differentially expressed. Normalized counts for all genes were ranked and subject to Gene Set Enrichment Analysis (20). Data are deposited in GEO (GSE148003).
Analysis of data from The Cancer Genome Atlas (TCGA) included correlation analysis between gene pairs and survival analysis, described in SM.
Statistical analyses of experimental data.
All data are presented as mean values ± SD of triplicate measurements. Two-tailed Student’s t-test or ANOVA (one-way or two-way) were used to determine effects of treatments. P < 0.05 were considered significant. All analyses were performed using Prism 6.0 software (GraphPad Software).
Results
Stemness and ferroptosis signatures are enriched in platinum-tolerant cancer cells.
Pt-T OC cells were generated through repeated in vitro exposure of OC cell lines (OVCAR3, OVCAR5, COV362 and SKOV3) to platinum at IC50 concentrations (Supplemental Figure S1A), while Pt-R xenografts were obtained by treating tumor harboring mice with carboplatin for 4–6 weekly cycles (Figure S1B). Repeated platinum exposure of OC cells induced a stable phenotype, with at least 2-fold increase in platinum IC50 (Supplementary Table S3) compared to parental chemotherapy naïve cells. Pt-T OC cells were enriched in ALDH(+) cells (Figure 1A and S2A), formed increased numbers of spheroids (Figure 1B and S2B), and contained cells expressing CSC-related TFs (Oct4, Nanog and Sox 2) (Figure 1C and S2C–D) compared to controls. RNA sequencing compared Pt-R vs. parental naive cells, with transcriptomic signatures revealing enrichment in stemness associated gene sets (Figure 1D and S2E). Similar observations were made in vivo, including in carboplatin treated OVCAR3 (Figures 1E–F) and SKOV3 (Figure S2F) xenografts. ALDH(+) cells were enriched (Figure 1E) and stemness associated genes (ALDH1A1, Oct4, Nanog and Sox 2) were upregulated (Figure 1F and S2F) in carboplatin-treated compared to PBS-treated xenografts, as we noted previously (6, 7).
Figure 1. Stemness and ferroptosis signatures are enriched in Pt-T OC cells.
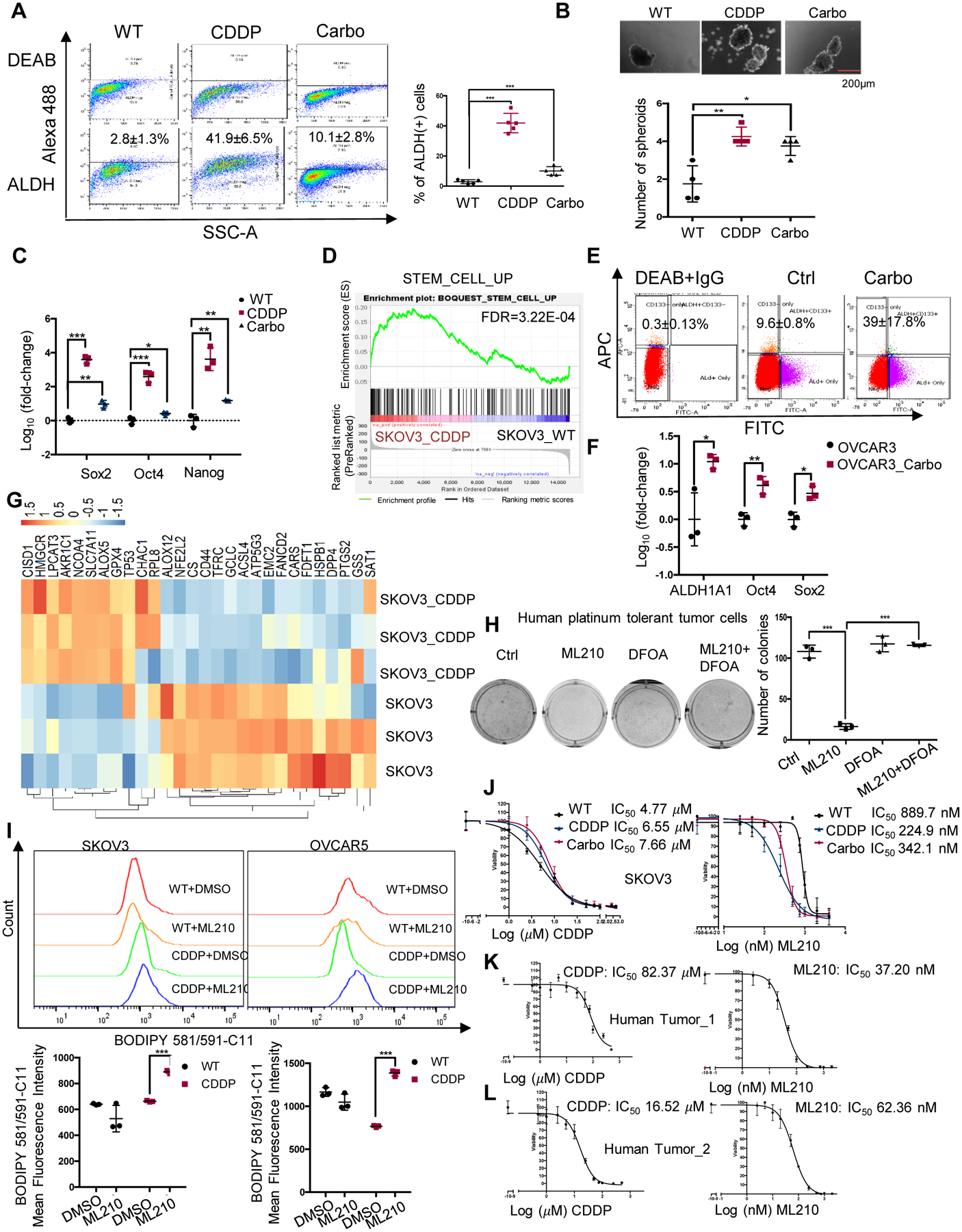
(A) Representative FACS side scatter analysis of the ALDH(+) population (left), and percentage (mean ± SD, n=5) of ALDH(+) cells (right) in parental (WT), cisplatin tolerant (CDDP), and carboplatin tolerant (Carbo) SKOV3 cells. (B) Representative images (top), and numbers (mean ± SD, n=4) of spheroids (bottom) formed by 1000 parental (WT), CDDP, and carboplatin tolerant (Carbo) SKOV3 cells after 7 days of culture under non-attachment conditions. (C) mRNA levels (fold-change ± SD, n=3) of stemness-related TFs (Sox2, Oct4 and Nanog) measured by real-time RT-PCR in CCDP and Carbo tolerant vs. parental SKOV3 cells (WT). (D) GSEA shows upregulated stemness pathway (Stem Cell_UP) in SKOV3 CDDP-tolerant vs. parental cells (FDR=3.22E-04). (E, F) FACS side scatter analysis of percentage of ALDH(+) cells (E), and fold-change (mean ± SD, n=3) ALDH1A1, Nanog, and Oct4 mRNA expression levels (F) in OVCAR3 xenografts treated with PBS (Ctrl) or carboplatin (Carbo). (G) Hierarchical clustering heatmap for DEG (FDR<0.05) ferroptosis-related genes in SKOV3_CDDP vs. control cells (n=3 replicates/group). (H) Representative pictures of a colony formation assay (left), and numbers (mean ± SD, n=3) of colonies (right) developed from 4000 cells isolated from Pt-R human tumors treated with DMSO (Ctrl), ML210 (500nM), DFOA (800nM), or ML210 plus DFOA for 24 hours. (I) Fluorescence histograms (top), and mean (± SD, n=3) fluorescence (bottom) of BODIPY 581/591-C11 staining show lipid peroxidation in SKOV3 and OVCAR5 parental (WT) cells and cisplatin tolerant (CDDP) cells treated with DMSO or ML210 (1 uM) for 20 hours. (J) Survival curves for WT, cisplatin tolerant (CDDP) and carboplatin tolerant (Carbo) SKOV3 cells in response to cisplatin (left) or GPX4 inhibitor ML210 (right). Cisplatin and ML210 IC50 values are shown. (K-L) Survival curves of cells from primary HGSOC tumors treated with cisplatin (left), or GPX4 inhibitor ML210 (right). IC50 for cisplatin and ML210 are shown. For all comparisons: *P<0.05, **P<0.01, ***P<0.001.
Given the possibility that CSCs would be more resistant to chemotherapy due to upregulated anti-redox mechanisms (11) and considering a recently proposed association between oxidative stress and ferroptosis, a new form of cell death triggered by oxidized lipids, we examined a gene set related to “ferroptosis” (21) in Pt-T compared to parental OC cells. Clear differences including upregulated genes involved in glutathione metabolism and anti-oxidant defense mechanisms were observed in Pt-T OC cells vs. chemotherapy-naïve cells (SKOV3, Fig. 1G and OVCAR5, Fig. S2G). Interestingly, this molecular signature was observed in HGSOC cells with higher baseline resistance to platinum (IC50 >5μM; COV362, OVCAR8, SNU119, and OVCAR4) compared with cells more sensitive to platinum (IC50 <5μM; TYKNU, IGROV1, OVCAR3; Fig S2H) (7, 22), suggesting the pathway is a common signature of Pt resistant cells. The selenoprotein glutathione peroxidase 4 (GPX4), a key protein regulating anti-oxidant response, was among the upregulated genes in Pt-T cells. GPX4 inhibitors impede anti-oxidant defense mechanisms and promote death of cells dependent on this pathway (3). Indeed, Pt-R OC cells were more sensitive to the GPX4 inhibitor, ML210, compared to control cells (SKOV3, Fig. S3A; OVCAR5, Fig. S3B; COV362, Fig. S3C). ML210 induced inhibition of colony formation was inhibited by the iron chelator deferoxamine (DFOA), consistent with induction of a ferroptosis phenotype (Figs. S3A–C). Furthermore, primary OC cells derived from malignant ascites from patients with Pt-R OC, were found to be dependent on GPX4, as ML210 potently reduced colony formation in these cells (Fig. 1H), this inhibition being rescued by DFOA.
ML210 caused increased oxidized membrane lipids levels, as measured by flow cytometry using the C11-BODIPY dye in Pt-T cells compared to naïve cells, supporting increased susceptibility to ferroptosis of Pt-R OC cells (SKOV3 and OVCAR5, Fig. 1I; COV362, Fig S3D). Additionally, Pt-T SKOV3 cells were more sensitive to ML210 compared to parental cells (IC50 of 224nM and 342nM vs. 889nM, Figures 1J, Fig S3E–F). Primary OC cells derived from malignant ascites associated with Pt-R OC, displayed resistance to platinum in vitro (Figures 1K–L, left panels; IC50 of 82μM and 16.52μM), and responded to low doses of the GPX4 inhibitors, ML210 (Figures 1K–L, right panels; IC50 of 37.20nM and 62.36nM) and RSL-3 (Figure S3G–H, IC50 of 14.20nM and 17.72nM). Trypan blue staining of parental and Pt-T SKOV3 and OVCAR5 cells treated with ML210 showed that the inhibitor induced more cell death in resistant OC cells (p <0.05, Figures S3I–J). These results derived from multiple in vitro and in vivo OC models, including primary human cancer cells, support the existence of a “persister cell” phenotype induced by Pt, sharing partial stemness characteristics, and highly susceptible to ferroptosis.
Frizzled 7 (FZD7) is upregulated in Pt-T OC cells and tumors.
To identify potential markers linked to the “persister” phenotype, an RT-PCR based platform representing 90 cancer stemness associated genes was used. A number of known CSC markers were found to be upregulated in the Pt-T cells (CD44, PROM1, SOX2), along with membrane transporters known to be associated with Pt resistance (ABCG2, ABCB5), and regulators of EMT (TGFBR1, SNAI1, BMP7, TWIST 1 and 2, SNAI 1 and 2, see Table S4). Among transcripts representing membrane proteins, which could potentially be used as novel markers, FZD7, a transmembrane receptor involved in canonical Wnt/β-catenin/TCF and non-canonical Wnt/planar cell polarity (PCP) signaling (23, 24), was one of the top highly expressed transcripts (> 8-fold) in Pt-R compared to control cells (Table S4 and Figure S4A). Increased FZD7 expression levels were confirmed in Pt-T models (SKOV3, OVCAR5, and OVCAR3) generated as described, compared to control cells at mRNA (Figure 2A) and protein level (Figure 2B), but also in Pt-R PEO4 cells compared to sensitive PEO1 OC cells, an isogenic cell line pair, derived from the same patient at different times during the disease course (25) (Figs. 2C–D). Likewise, FZD7 mRNA expression levels were upregulated in platinum treated SKOV3 (Fig. 2E) and OVCAR3 (Fig. 2F) xenografts compared to vehicle-treated tumors.
Figure 2. Frizzled 7 (FZD7) is upregulated in Pt-T OCs.
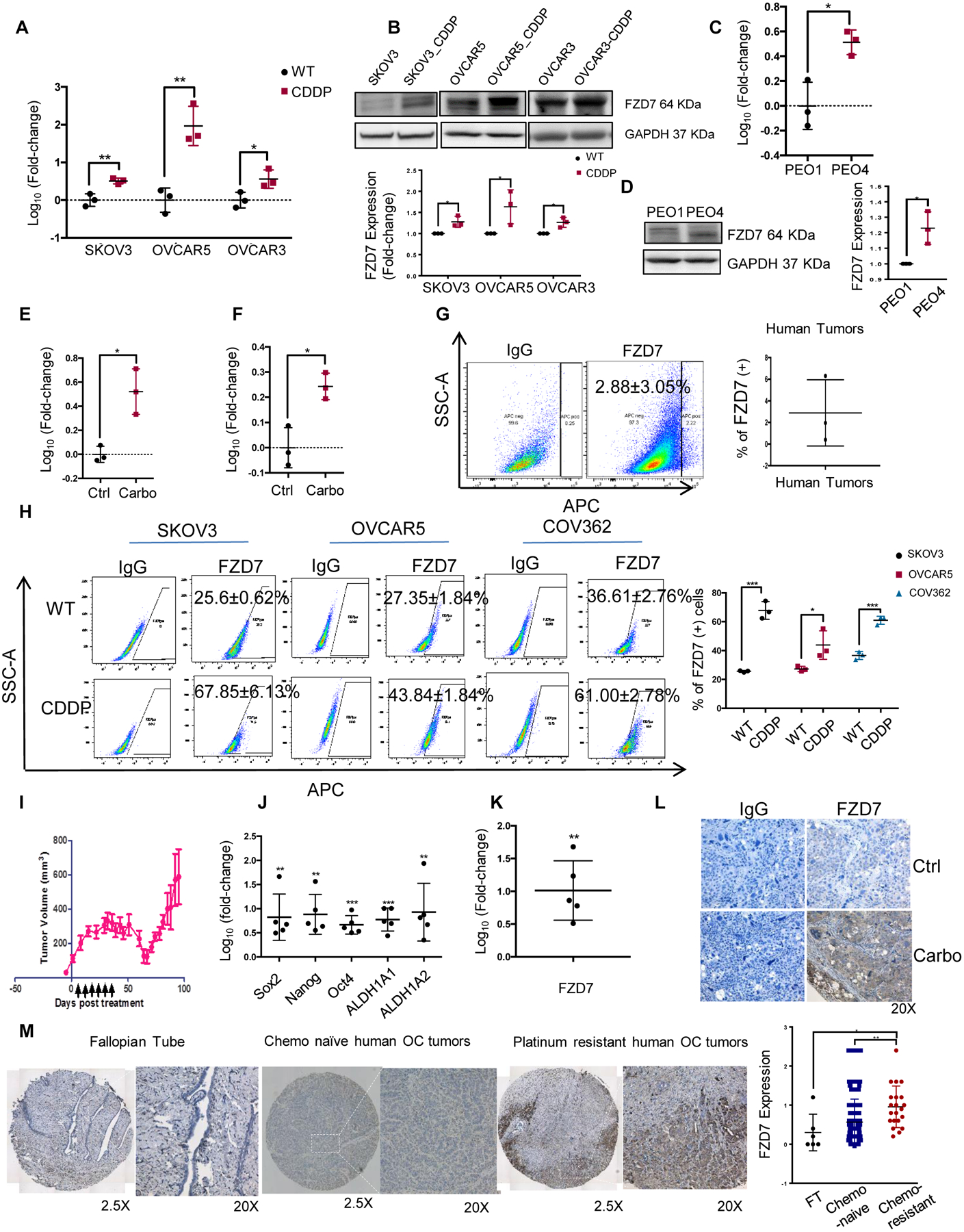
(A) Fold-change (mean ± SD, n=3) of FZD7 mRNA expression levels measured by real-time RT-PCR in cisplatin tolerant (CDDP) compared with parental OVCAR3, OVCAR5, SKOV3 cells. (B) Western blotting for FZD7 in parental and Pt-T SKOV3, OVCAR5 and OVCAR3 cells and Pt-T (-CDDP). Quantification shows fold change of FZD7 expression (n=3 experiments). (C-D) FZD7 mRNA expression levels (mean fold-change ± SD, n = 3) (C), and FZD7 protein levels measured by western blotting (D; including quantification in 3 experiments) in Pt-R PEO4 vs. PEO1 cells. (E) Fold-change (mean ± SD) of FZD7 mRNA levels in carboplatin-treated (Carbo) and control (Ctrl) SKOV3 (E) and OVCAR3 (F) xenografts (n = 3 per group). (G) FACS side scatter analysis (left) and average (±SD, n = 3) of FZD7(+) cells dissociated from HGSOC tumors. (H) FACS side scatter analysis of FZD7(+) cells (left), and percentage (mean ± SD, n=3) of FZD7(+) cells (right) in WT and Pt-T SKOV3, OVCAR5 and COV362 cells. (I) NSG mice carrying PDX received carboplatin (15mg/kg weekly) to induce platinum tolerance. Arrows indicate carboplatin treatment. Mean volumes (± SD) are shown (n=5). (J) Mean fold change (± SD, n=5) for Sox2, Nanog, Oct4, ALDH1A, and ALDH1A2 mRNA expression levels in Pt-T vs. control PDXs. (K, L) Mean fold-change of FZD7 mRNA levels (K), and representative images of FZD7 IHC staining (L) in Pt-T vs. control PDXs (Ctrl). (M) FZD7 IHC staining and H-scores (mean ± SD) (right) in sections of fallopian tube (n=6), chemo-naïve OC tumors (n=117) and Pt-T tumors (n=23) included in two tissue microarrays. For all comparisons: *P< 0.05, **P<0.01, and ***P<0.001.
Flow cytometry was used to determine whether a FZD7 high (FZD7+) cell population is detectable. FZD7(+) cells were detected among cells dissociated from primary OC, previously untreated with chemotherapy, and represented ~ 3% of all cells (Figure 2G). In OC cell lines, FZD7(+) cells were identified as a distinct sub-population representing ~25–35% of cells (SKOV3, OVCAR5 and COV362; Figure 2H). Additionally, the FZD7(+) cell population was detectable and was enriched in Pt-T compared to parental cells (SKOV3, OVCAR5, COV362; Figure 2H), suggesting that this cell membrane receptor may be a marker of “persister” cells, pre-existing in un-selected cell populations prior to Pt exposure, and enriched after exposure to the drug.
Further, we used patient derived xenografts (PDX) generated from newly diagnosed HGSOC (14), which were treated weekly with carboplatin. After initial response, recurrent tumors emerged (Figure 2I), which were enriched in ALDH (+) cells (Fig. S4B), CSC-related TFs (Sox 2, Nanog, Oct4), as well as ALDH1A1 and ALDH1A2 (Figure 2J). FZD7 expression levels were upregulated at mRNA (Figure 2K) and protein level (Figure 2L), as measured by immunohistochemistry (IHC) in Pt-T PDX vs. control. Next, IHC assessed FZD7 expression in primary HGSOC specimens and in tumors collected after 3–6 cycles of neo-adjuvant chemotherapy, containing surviving cells after standard platinum-taxane treatment, which are presumably Pt-T. Patients’ characteristics are included in Supplementary Table S1. Increased FZD7 staining intensity (measured as H-score) was observed in cancer cells residual after chemotherapy in these specimens (n = 23), when compared to chemotherapy-naive tumors (n = 117, p < 0.01) (26) and to fallopian tube epithelium (control n = 6, Figure 2M, p < 0.05).
Functional role of FZD7 in OC cells.
FZD7(+) and FZD7(−) cells were FACS sorted from cell lines and human tumors. Differences in mRNA expression levels between FZD7(+) and FZD7(−) cell are shown in Figure 3A. FZD7(+) OC cells were less sensitive to cisplatin (CDDP) (Figures 3B–D; p < 0.05), supporting that the receptor marks a Pt-T population. Additionally, FZD7(+) cells formed spheroids more efficiently compared to FZD7(−) cells (Figure 3E–F, p < 0.01), and expressed higher levels of stemness associated TFs (SKOV3, Fig. 3G, OVCAR5, Fig. 3H; COV362, Fig. 3I; p < 0.05), supporting that they share stemness-related features.
Figure 3. Functional role of FZD7 in OC cells.
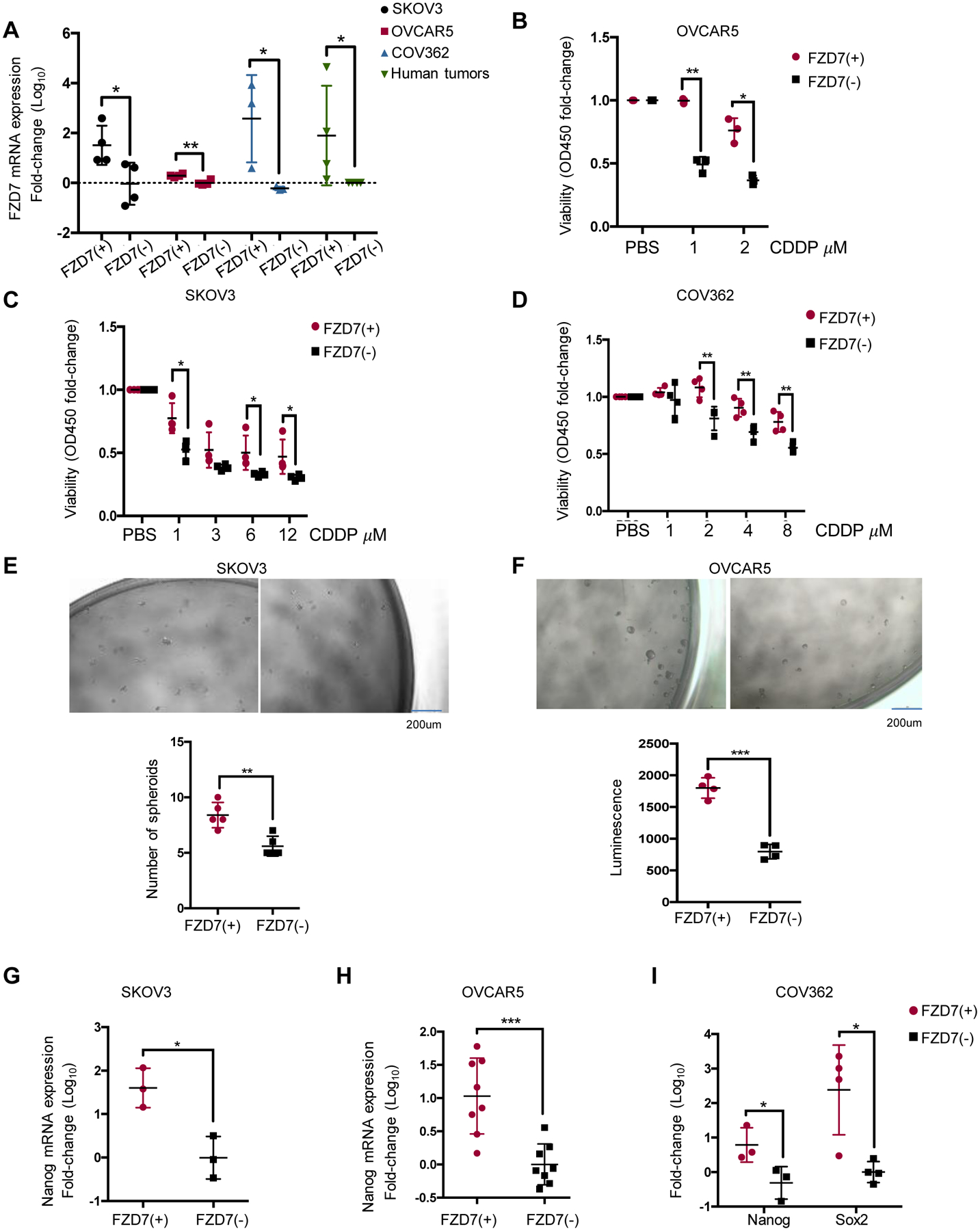
(A) FZD7 mRNA levels (mean fold-change ± SD, n=3–4) in FZD7 (+) and FZD7 (−) cells FACS selected from SKOV3, OVCAR5, and COV362 cells and HGSOC tumors. (B-D) Cell viability (mean fold-change ± SD, n=4) of FZD7(+) and FZD7(−) cells from OVCAR5 (n=3) (B), SKOV3 (n=3) (C) or COV362 (n=4) (D) cells, plated, treated with the indicated doses of CDDP for 24 hours, and cultured for additional three days. (E, F) Representative pictures and numbers (mean ± SD, n = 4–5) of spheroids formed after 7 days of culture by FZD7(+) and FZD7(−) cells FACS sorted from SKOV3 (E) and OVCAR5 (F) OC cells. Spheroids were counted or cell numbers were estimated by CellTiter-Glo 3D cell viability assay. (G-I) mRNA levels (fold-change ± SD) of Nanog in FZD7(+) compared with FZD7(−) cells from SKOV3 (G) (n=3), OVCAR5 (H) (n=8) and COV362 (I)(n=3) cells. For all comparisons: *P<0.05, **P<0.01, ***P<0.001.
To further examine its functions, the receptor was knocked-down (KD) by stable transduction of shRNA or was transiently overexpressed. Decreased FZD7 mRNA expression was confirmed by Q-RT-PCR in SKOV3 and OVCAR5 cells transduced with two shRNA sequences targeting the receptor (Figs. 4A–B). FZD7 KD decreased spheroid formation (Figs 4A–B) and expression of stemness associated TFs (Figure 4C and S5A). In vitro serial limited dilution assay showed that receptor KD decreased stem cell frequency, as calculated by the ELDA software (27) in FZD7 KD vs. control cells, (p = 0.034, Fig. S5B). Likewise, FZD7 KD in Pt-T SKOV3_CDDP cells (Fig. S5C) decreased sphere formation (Fig. S5D). Stable FZD7 KD in primary Pt-R tumor cells caused decreased expression levels of the stemness-associated gene ALDH1A1 (Figures 4D–E). Conversely, transient overexpression of FZD7 (Figs. 4E–F) promoted proliferation of SKOV3 and OVCAR5 cells as spheres (Fig. 4G) and increased expression of stemness associated TFs (Figure S5E–F). FZD7 KD decreased IC50 to cisplatin by ~ 2-fold (SKOV3, Fig. 4H; SKOV3_CDDP, Fig. 4I; Fig. S5G–H); while the receptor’s overexpression increased Pt resistance (Fig. 4J; Fig. S5I).
Figure 4. FZD7 regulates stemness characteristics.
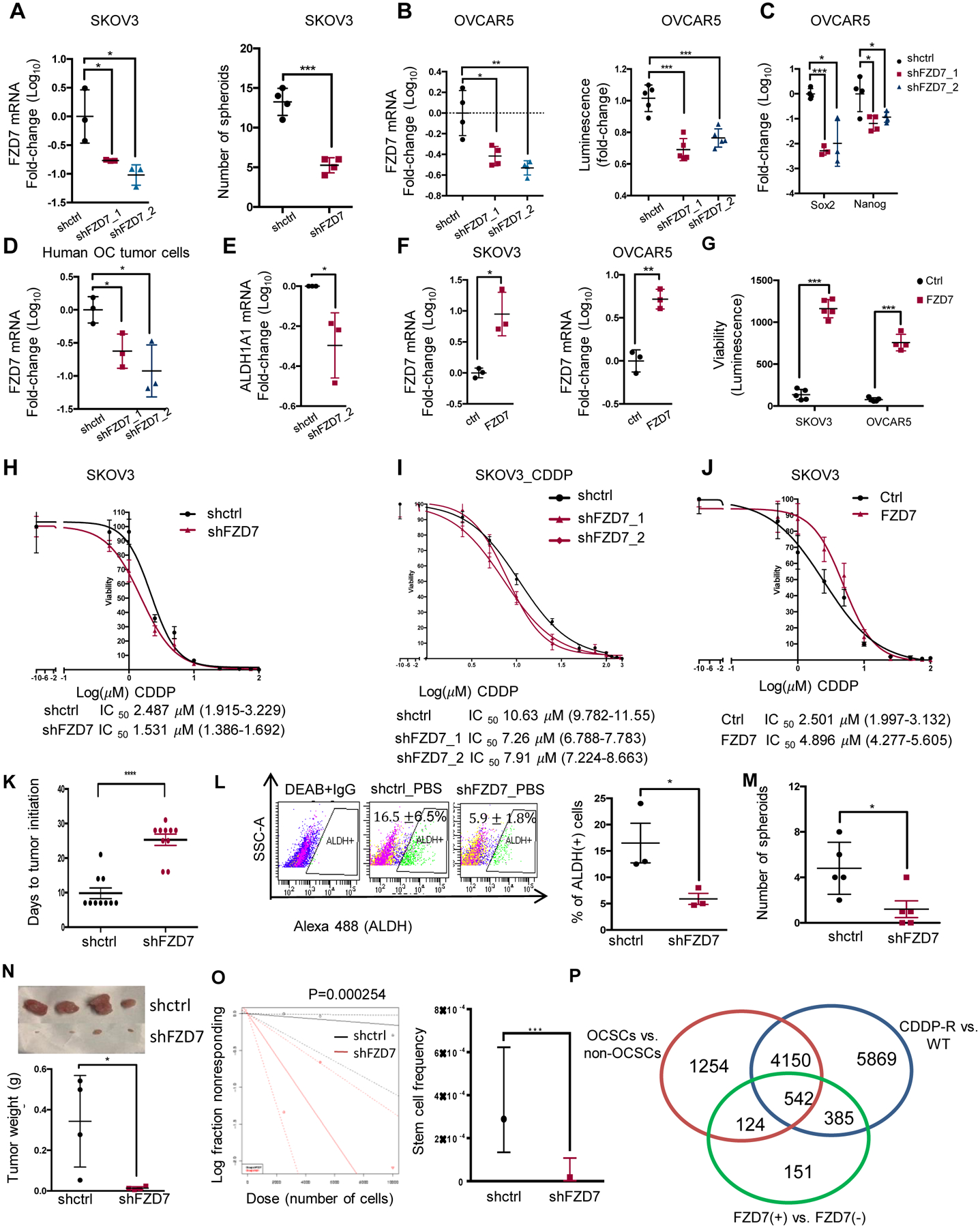
(A) (Left) FZD7 mRNA levels (mean fold-change ± SD, n=3) in SKOV3 cells transduced with shRNAs targeting FZD7 (shFZD7) vs. control shRNAs (shctrl). (Right) Numbers of spheroids (mean ± SD, n=4) formed by 2,000 shFZD7 or shctrl SKOV3 cells cultured for 14 days and counted under a microscope. (B) FZD7 mRNA expression levels (mean fold-change ± SD, n=4; left) and numbers of spheroids (n=5; right) in OVCAR5 cells transduced with shRNAs directed at FZD7 (shFZD7) vs. control shRNAs (shctrl). Cell viability assessed numbers of cells growing as spheroids using the CellTiter-Glo kit. (C) Sox2 and Nanog mRNA levels (mean fold-change ± SD, n=3–4) in OVCAR5 cells transduced with shFZD7 vs. shctrl. (D) FZD7 mRNA levels (mean fold-change ± SD, n=3) in Pt-R primary HGSOC cells transduced with shRNAs targeting FZD7 (shFZD7) vs. control shRNA (shctrl). (E) ALDH1A1 mRNA levels (mean fold-change ± SD, n = 3) in primary tumor cells transduced with shFZD7 cells vs. shctrl. (F) Average fold-change (± SD, n = 3) of FZD7 mRNA in SKOV3 (left) and OVCAR5 (right) cells transfected with FZD7-pcDNA3.1 vs. empty vector (ctrl). (G) Spheroid formation estimated with a CellTiter-Glo viability kit (bottom) from 1,000 ctrl and FZD7 expressing SKOV3 or OVCAR5 cells (described in F) and cultured for 7 days (n =4–5 per group). (H-J) Effects of CDDP on cell survival measured by CCK8 assays in SKOV3_shctrl and SKOV3_shFZD7 (H), SKOV3 cisplatin tolerant cells (SKOV3_CDDP) transduced with shRNAs targeting FZD7 (shFZD7_1, _2) or control shRNA (I), and SKOV3 cells transfected with FZD7-pcDNA3.1 (FZD7) or empty vector (Ctrl) (J). Cells were treated with cisplatin for 24 hours and cultured for additional 3 days (n=3–4). Cisplatin IC50 value is shown. (K) Days to tumor initiation (mean ± SD, n = 10) of sq xenografts induced by 2×106 shctrl and shFZD7 transduced OVCAR5 cells. (L) FACS side scatter analysis of ALDH(+) cells (top), and percentage (mean ± SD, n=3) of ALDH(+) cells (bottom) from cell suspensions generated from OVCAR5_shctrl and OVCAR5_shFZD7 xenografts. (M) Numbers (mean ± SD, n=5) of spheroids (bottom) formed during 14 days by 1000 cells derived from cell suspensions from OVCAR5_shctrl or OVCAR5_FZD7 xenografts. (N-O). In vivo limited dilution assay used serially diluted numbers (2,500, 5,000, and 10,000) of OVCAR5_shctrl and shFZD7 cells injected sq into nude mice (n=4 replicates per group). (N) Average tumor weights (± SD) are shown (for the 10,000 cells group). (O) Stem cell frequencies were calculated by using the Extreme Limiting Dilution Analysis (http://bioinf.wehi.edu.au/software/elda/; p = 0.000254). (P) A Venn diagram shows the number overlapping and unique DEGs between OVCAR5-derived OCSCs (ALDH+CD133+) versus non-OCSCs (ALDH-CD133-), OVCAR5 cisplatin tolerant (CDDP-R) vs. parental (WT), and FZD7(+) versus FZD7(−) OVCAR5 cells (FDR < 0.05). ALDH+CD133/ALDH−CD133- and FZD7+/FZD7− cells were sorted by FACS. For all comparisons: *P<0.05, **P<0.01, ***P<0.001.
To test the effects of FZD7 to tumor growth, a subcutaneous (s.c.) xenograft model was used. FZD7 KD in OVCAR5 cells delayed tumor initiation (sh-control 9.8±4 days. vs. sh-FZD7 25.3±4.9 days, Figure 4K, p < 0.0001) and decreased tumor size (Fig. S5J) and tumor weight (0.80±0.41 g vs. 0.14±0.15 g, p = 0.04, Fig S5K, n = 4/group). FZD7 KD in xenografts was confirmed by IHC (Figure S5L). ALDH (+) cells (16.5±6.5% vs. 5.9±1.8%, p= 0.05; Fig. 4L, n = 3) and spheroid forming ability (Fig. 4M, p = 0.02) were decreased in cells dissociated from FZD7 KD vs. control xenografts. Further, to test the effects of FZD7 KD to tumor initiation, an in vivo serial limited dilution assay was carried out by using 2,500; 5,000; and 10,000 FZD7 KD and control cells. FZD7 KD significantly inhibited TIC (Figure 4N) and reduced stem cell frequency, as calculated by the ELDA software (Figure 4O). Combined, these results support that FZD7 is linked to stemness and chemo-resistance.
Molecular signatures of FZD7(+) cancer cells:
Gene signatures distinguishing FZD7(+) vs. FZD7(−) cells; ovarian CSCs (ALDH+CD133+) vs. non-CSCs (ALDH−CD133−), and Pt-T vs. platinum-naïve OVCAR5 cells were examined and integrated (Figure 4P). FZD7(+) and (–) were sorted by FACS (Fig. S5J). Ovarian CSCs and non-CSCs were FACS-sorted by using dual stem cell markers, CD133 and Aldefluor activity (Fig. S5M–N). There were 666 differentially expressed genes (DEG) shared between FZD7(+)/FZD7(−) and CSCs/non-CSCs datasets, 5404 DEGs being unique to CSCs and 536 DEGs unique to FZD7(+) cells (Figure 4P). Additionally, there were 927 DEGs overlapping between FZD7+/FZD7− and resistant/parental cells, 10019 DEGs unique to the Pt-T cells and 275 genes uniquely associated with FZD7(+) cells (Figure 4P). Overlapping DEGs between FZD7+/FZD7− and OCSC/non-OCSCs were enriched in Cancer Stem Cell, but enriched DNA Repair signatures between FZD7+/− and resistant/parental cells (Fig. S6A–B). Additionally, FZD7(+) vs. FZD7(−) cells displayed signatures enriched in stemness (Fig. S6C), EMT (Fig. S6D) and downregulated DNA damage response genes (Fig. S6E). Together, these data suggest that FZD7(+) cells possess both shared, but also distinct features, relative to stemness and chemo-resistance, consistent with the phenotypes described above. Importantly, mitochondrial and oxidative phosphorylation gene sets were enriched among DEGs distinguishing FZD7(+) vs. FZD7(−) cells (Fig. S6F–H) and Ingenuity Pathway Analysis (IPA) identified Oxidative Phosphorylation and Mitochondria Dysfunction as the top enriched pathways in FZD7(+) cells, suggesting that the receptor marks a cell population harboring altered oxidative stress responses.
FZD7 marks a cell population enriched in GPX4.
Given that GPX4, an antioxidant enzyme which reduces reactive oxygen species (ROS), preventing formation of toxic lipid peroxides (28, 29), has been implicated in maintenance of normal mitochondrial function and oxidative phosphorylation (3, 28), we examined whether FZD7 expression impacted GPX4 expression and function in Pt-T OC cells. GPX4 levels were significantly increased in FACS sorted FZD7(+) vs. FZD7(−) cells derived from SKOV3, OVCAR5, COV362 cells or cancer cells dissociated from human tumors (Fig. 5A, p < 0.05). Furthermore, FZD7 KD by shRNA in OVCAR5 and SKOV3 cells resulted in repressed GPX4 mRNA (Fig. 5B, p < 0.001, and Fig S7A, p < 0.05) and protein expression levels (Fig. 5C). GPX4 expression was decreased in Pt-T SKOV3 cells (Fig. 5D) and in primary Pt-R OC cells (Fig. 5E) transduced with shRNA targeting FZD7. Conversely, FZD7 overexpression induced increased GPX4 mRNA and protein expression levels (Figs. 5F–H).
Figure 5. FZD7 regulates GPX4 and intracellular redox states.
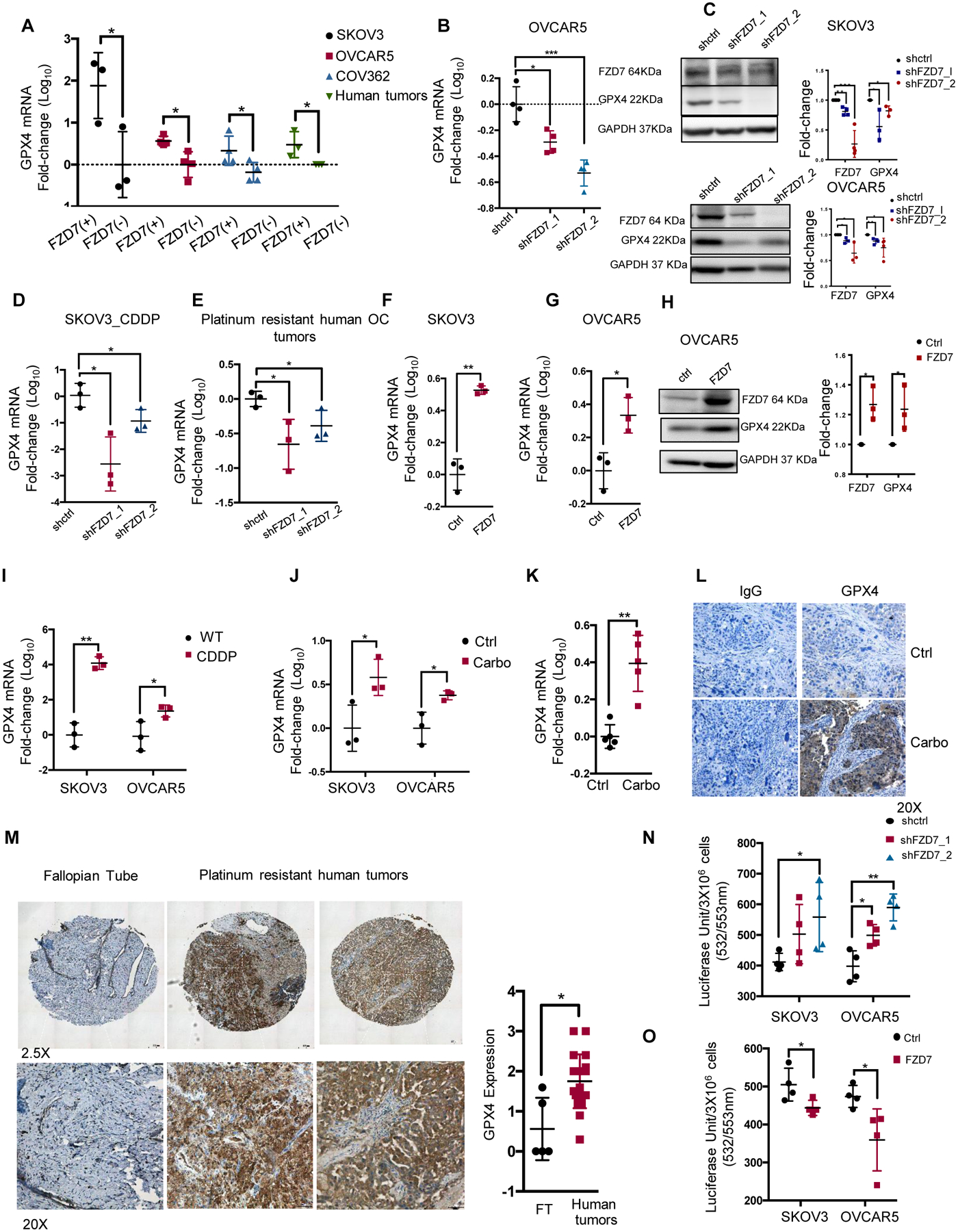
(A) Fold-change (mean ± SD, n=3–4) of GPX4 mRNA levels in FZD7(+) vs. FZD7(−) cells FACS sorted from SKOV3, OVCAR5, COV362 cells and cell suspensions from HGSOC tumors. (B) Fold-change (mean ± SD, n=4) of GPX4 mRNA expression levels in OVCAR5 cells transduced with shRNAs targeting FZD7 (shFZD7) vs. control shRNAs (shctrl). (C) Western blotting for FZD7, GPX4 and GAPDH in SKOV3 and OVCAR5 cells stably transduced with shctrl and shFZD7. Quantification shows fold change of FZD7 and GPX4 expression across 3 experiments. (D-E) GPX4 mRNA expression levels (fold-change ± SD, n = 3) in SKOV3 Pt-T cells (CDDP) (D), and Pt-R primary human HGSOC cells (E) transduced with shRNAs directed at FZD7 (shFZD7) vs. control shRNAs (shctrl). (F, G) Average fold-change (± SD, n=3) of GPX4 mRNA levels in SKOV3 (F) and OVCAR5 (G) cells transfected with FZD7-pcDNA3.1 vs. empty vector (ctrl). (H) Western blotting for FZD7, GPX4 and GAPDH in OVCAR5 cells transfected with FZD7-pcDNA3.1 vs. empty vector (ctrl). Quantification shows fold change of FZD7 and GPX4 expression across 3 experiments. (I-K) GPX4 mRNA expression (mean fold-change ± SD) in SKOV3 and OVCAR5 Pt-T (CDDP) vs. parental cells (WT) (I; n = 3/group). (J) SKOV3 and OVCAR5 xenografts (n=3 per group; J) and PDX tumors (K; n=5 per group) treated with PBS (Ctrl) or carboplatin. (L) Representative images of GPX4 IHC staining in sections of control (Ctrl) and Pt-T PDXs. (M) Representative pictures of GPX4 IHC (left) and H-scores (mean ± SD) (right) in sections of fallopian tube (n = 6) and Pt-R HGSOC tumors (n = 23). (N) Lipid peroxidation in SKOV3 and OVCAR5 cells transduced with scrambled shRNA (shctrl) or shRNAs targeting FZD7 (shFZD7) and expressed as average luciferase units/3×106 cells (± SD, n=4) (O) Intracellular lipid peroxidation measured in SKOV3 and OVCAR5 cells transfected with FZD7 or control vector (Ctrl) and expressed as average luciferase units/3×106 cells ± SD (n=4). For all comparisons: *P<0.5, **P<0.01, ***P<0.001.
Furthermore, GPX4 and FZD7 mRNA expression levels were coordinately upregulated in Pt-T models, including Pt-T vs. parental cells (SKOV3, OVCAR5, Fig. 5I, p < 0.05), Pt-T xenografts (Fig. 5J, p < 0.05) and PDXs (Fig. 5K, p < 0.01). Increased GPX4 expression in Pt-T PDX vs. controls was confirmed by IHC (Fig. 5L). IHC examined GPX4 in HGSOC specimens collected after neo-adjuvant chemotherapy, noting increased GPX4 staining in these residual tumors (n = 23) when compared to fallopian tube epithelium, Fig. 5M, p = 0.02). Together, the results confirm a positive correlation between FZD7 and GPX4 expression in OC cells and in Pt-T models, supporting that FZD7(+) cells have increased anti-oxidant capacity. To confirm the functional relevance, GPX4 enzymatic activity was measured by using the malondialdehyde (MDA) assay, which quantifies intracellular lipid peroxide levels. Lipid peroxides were found to be increased in FZD7 KD vs. control cells (Fig. 5N, p < 0.05) and decreased in OC cells overexpressing FZD7 (Fig. 5O, p < 0.05) supporting that FZD7(+) cells clear these toxic products more effectively, due to higher levels of GPX4.
GPX4 participates in regulation of intra-cellular redox states by utilizing glutathione (GSH) as the critical antioxidant (21). GSH is synthesized from glutamate-cysteine under the action of glutamate-cysteine ligase (GCL) (21). The cycling of reduced GSH to oxidized glutathione disulfide (GSSG) removes ROS derived from hydrogen peroxide and lipid hydroperoxides through various glutathione peroxidases (GPXs), including GPX4 (21). GSH recycling from GSSH is catalyzed by glutathione reductase (GSR), using NADPH, whose synthesis is regulated by isocitrate dehydrogenase 2 (IDH2) (21). FZD7 KD decreased the expression levels of multiple genes in this pathway, including GPX2, GSS, IDH2, GSR, GCL and SLC7A11 (OVCAR5, Fig. 6A and SKOV3, Fig. S7B). Conversely, FZD7 overexpression caused increased expression levels of GSS, GSR, GCL and SLC7A11 (OVCAR5, Fig. 6B and SKOV3, Fig S7C), suggesting a significant direct correlation between FZD7 and glutathione metabolism related genes.
Figure 6. FZD7 marks a cell population susceptible to GPX4 inhibitors.
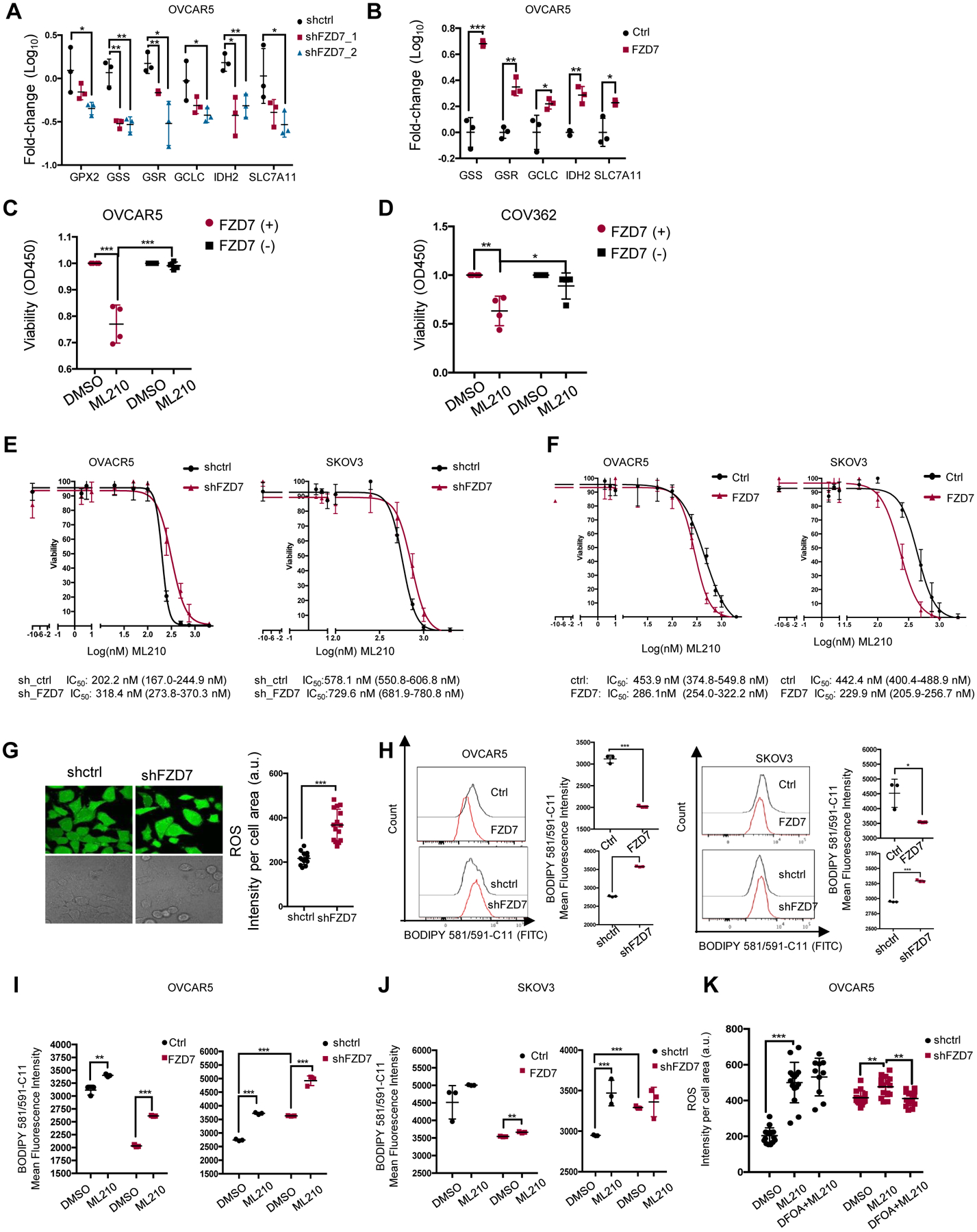
(A) Average fold-change (±SD, n = 3) in mRNA expression levels of selected glutathione metabolism genes in OVCAR5 cells transduced with shRNAs targeting FZD7(shFZD7) vs. control shRNA (A), and in OVCAR5 cells transfected with FZD7-pcDNA3.1 (FZD7) vs. control vector (Ctrl) (B) (C-D) Viability of FZD7(+) and FZD7(−) cells sorted from OVCAR5 and COV362 (D) cells and treated with DMSO or ML210 (OVCAR5, 2μM; COV362, 1μM) for 72 hours. Data are presented as average fold-change (± SD, n = 4) of absorbance values relative to control. (E) Survival curves of OVCAR5 (left) and SKOV3 (right) cells transduced with control shRNAs (shctrl) or shRNAs targeting FZD7 (shFZD7) and treated with ML210 for 3 days (n=3–4). ML210 IC50 values are shown below. (F) Survival curves of OVCAR5 (left) and SKOV3 (right) cells transfected with FZD7-pcDNA3.1 or control vector and treated with ML210 for 3 days. ML210 IC50 values are shown below (n=3–4). (G) Images (left) and quantification of intracellular ROS levels (right) in OVCAR5 cells transfected with control shRNA (shctrl) or shRNA targeting FZD7 (shFZD7). Data are presented as means (± SD) of DCF fluorescence intensity per cell area (n = 15). (H) Histograms of fluorescence intensity (left) and mean (± SD, n = 3) (right) of BODIPY 581/591-C11 in OVCAR5 (left) and SKOV3 (right) cells transfected with vector (ctrl), FZD7-pCDNA3.1 (FZD7), control shRNA (shctrl), or shRNAs targeting FZD7 (shFZD7). (I, J) Mean (± SD, n=3) fluorescence intensity of BODIPY 581/591-C11 show effects of ML210 (1 μM for 20 hours) on lipid peroxidation levels in SKOV3 (I) and OVCAR5 (J) cells transfected with empty vector (ctrl), FZD7-pcDNA3.1 (FZD7), control shRNA (shctrl), or shRNA against FZD7 (shFZD7). (K) Intracellular ROS levels in OVCAR5 cells transfected with shctrl and shFZD7 treated with DMSO, ML210 (2μM, 24 hours) and ML210 + DFOA (800nM, 24 hours) measured by assessing DCFHDA oxidation. Average intensity per cell area (± SD) is shown (n=15). For all comparisons: *P<0.05, **P<0.01, ***P<0.001.
FZD7 Marks a Cell Population Susceptible to GPX4 Inhibitors:
Given the correlations between FZD7, upregulated in Pt-T cells, and GPX4-mediated cellular redox maintenance, we hypothesized that inhibition of this axis will eliminate resistant cells. Small molecule inhibitors of GPX4 have been shown to increase cellular oxidative stress and induce ferroptosis (3, 30–32). Thus, we examined the sensitivity of OC cells with high vs. low FZD7 expression levels to GPX4 inhibitors, ML210 and RSL3 (3, 32, 33). FZD7(+) sorted cells were more sensitive to the GPX4 inhibitor ML210 compared to FZD7(−) cells (OVCAR5, Fig. 6C; COV362, Fig. 6D, SKOV3, Fig. S7D). FZD7 KD in OVCAR5 and SKOV3 cells also slightly reduced sensitivity to GPX4 inhibitors (Fig. 6E; Fig. S7E), while FZD7 overexpression slightly increased sensitivity to ML210 compared to vector-transduced cells (Fig. 6F; Fig. S7F).
Through its anti-oxidant function, GPX4 protects mitochondria from damage, maintaining normal oxidative phosphorylation. To test whether these processes were altered in FZD7(+) vs. FZD7(−) cells, as a consequence of differential GPX4 expression, oxygen utilization was measured by using fluorescence labeled oxygen uptake assay and the seahorse assays. FZD7 KD resulted in decreased oxygen uptake (Fig. S7G) and consumption rate (Fig. S7H), supporting the role of this pathway maintaining normal mitochondrial function. Additionally, intracellular ROS levels, quantified by DCFHDA staining were decreased in FZD7 KD cells compared to controls (Fig. 6G). As increased ROS levels contribute to oxidation of polyunsaturated lipids, leading to ferroptosis, C11-BODIPY staining was used in cells expressing different levels of FZD7 and/or exposed to GPX4 inhibitors. Oxidation of the polyunsaturated butadienyl portion of the dye in the presence of ROS is reflected in a shift of the fluorescence emission peaks from red to green, a hallmark of ferroptosis. The mean green (FITC) fluorescence intensity caused by oxidized lipids was decreased in cells overexpressing FZD7 and increased in FZD7 KD cells (Fig. 6H), consistent with increased susceptibility to ferroptosis of FZD7(+) cells. ML210-induced increase in fluorescence was higher in SKOV3 and OVCAR5 cells overexpressing FZD7 compared to controls and decreased in FZD7 KD cells (Fig. 6I–J). Likewise, baseline and ML210 induced intracellular ROS levels (rescued by DFOA) were higher in control vs. FZD7 KD OVCAR5 cells (Fig. 6K). Collectively, the data suggest that cells marked by FZD7 are more susceptible to ferroptosis and could be eliminated by targeting GPX4.
FZD7 regulates GPX4 expression and glutathione metabolism by activating canonical β catenin/p63 pathway.
As a classical Wnt receptor, FZD7 participates in both canonical β-catenin and non-canonical signaling. One of the known β-catenin targets is the transcription factor p63, directly transactivated by the TCF/LEF complex (34). Its most common isoform, ΔNTp63, lacking its N-terminus domain, has been implicated in maintaining intracellular redox homeostasis by regulating genes involved in glutathione metabolism, including GPX4 (21). We therefore hypothesized that in Pt-T OC cells, FZD7 could alter glutathione metabolism and protect OC cells from oxidative stress, through activation of β-catenin/TP63 signaling. Tp63 expression was upregulated in Pt-R PDX (Fig. 7A) and OC cells compared to controls (Fig. S8A). Tp63 expression was higher in FZD7(+) vs. FZD7(−) cells derived from SKOV3, OVCAR5, COV362 and human HGSOC (Fig. 7B) and FZD7 KD caused decreased Tp63 expression in OVCAR5 (Fig. 7C–D) and SKOV3 cells (Fig. S8B and Fig. 7D), while FZD7 overexpression led to increased Tp63 expression (Figs. 7E–G) at mRNA and protein levels. To demonstrate that the correlations between FZD7, TP63, and GPX4 were dependent upon the engagement of β-catenin, we used Wnt3A stimulation and the β-catenin inhibitor IWR-1-endo. Stimulation with Wnt3A induced p63 and GPX4 expression in control, but not in FZD7 KD cells, while treatment with IWR-1-endo inhibited the expression of both p63 and GPX4 (Figure 7H, OVCAR5 and S8F, SKOV3). Similar observations were made when FZD7 was overexpressed (Figure S8F), supporting that GPX4 upregulation is directly regulated by β-catenin.
Figure 7. FZD7 regulates GPX4 expression and gluthatione metabolism by activating the canonical β catenin/p63 pathway.
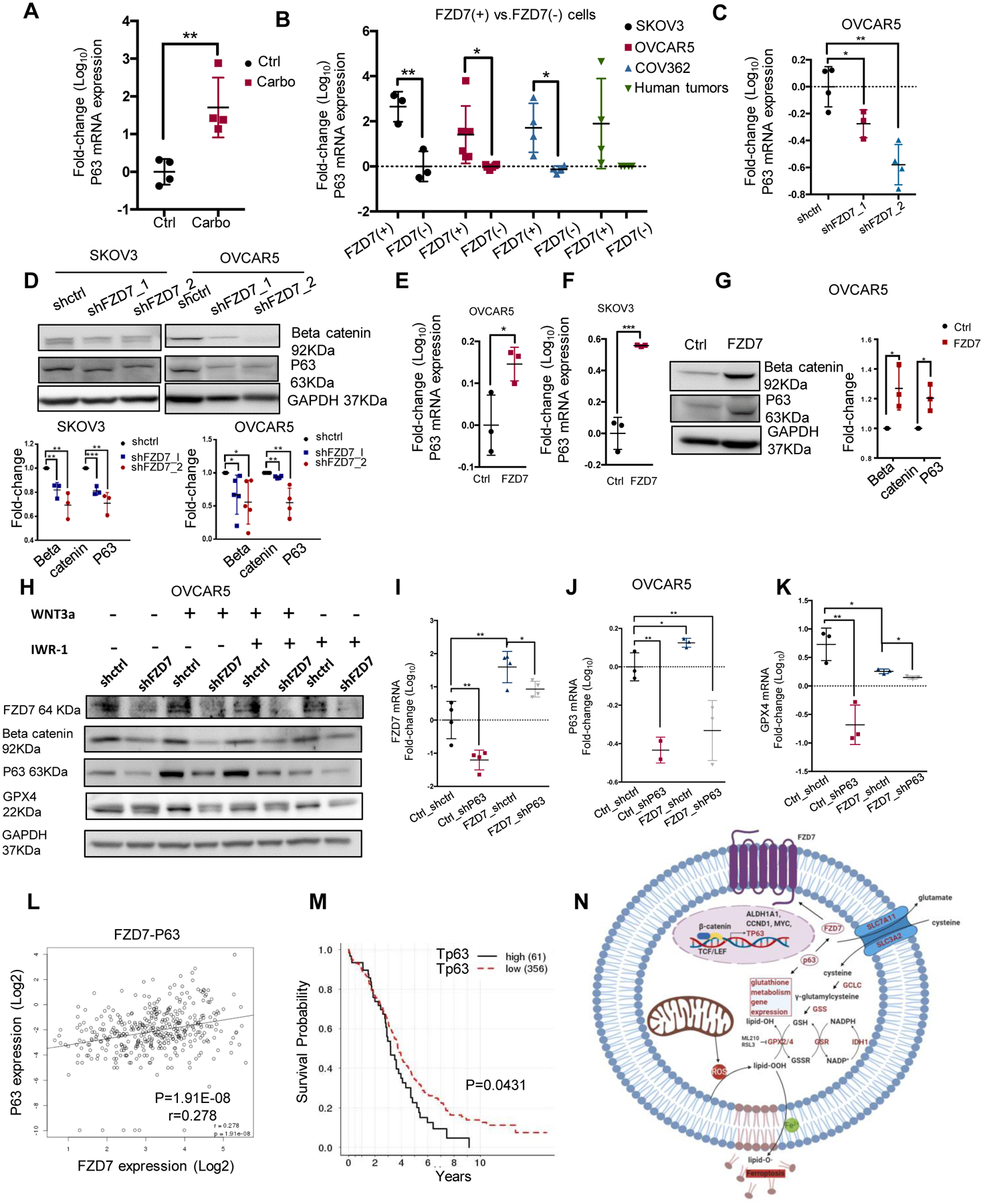
(A) P63 mRNA levels (fold-change ± SD, n=4) in Pt-T PDXs (carbo) vs. controls (Ctrl). (B) P63 mRNA levels (fold-change ± SD, n=3–6) in FZD7(+) versus FZD7(−)cells sorted from SKOV3, OVCAR5, and COV362 cell lines, and cell suspensions from human tumors. (C) P63 mRNA expression levels (fold-change ± SD, n = 4) in OVCAR5 cells transduced with shRNAs targeting FZD7 (shFZD7) vs. control shRNA (shctrl). (D) Western blot for β-catenin, P63, and GAPDH in SKOV3 and OVCAR5 cells transduced with shRNAs targeting FZD7 (shFZD7) or control shRNA (shctrl). Quantification shows fold change of β-catenin and P63 expression across 3 experiments. (E, F) P63 mRNA levels (fold-change ± SD, n=3) in OVCAR5 (E) and SKOV3 (F) cells transfected with FZD7 vs. control vector (Ctrl). (G) Western blot for β-catenin, P63, and GAPDH in OVCAR5 cells transfected with control (ctrl) or FZD7-pcDNA3.1 (FZD7). Quantification shows fold change of β-catenin and P63 expression across 3 experiments. (H) Western blot for FZD7, β-catenin, P63, GPX4 and GAPDH in OVCAR5 cells transfected with shctrl, shFZD7 (J) treated with WNT3a (150ng/ul) and/or IWR-1-endo (1 μM) for 24 hours (n = 2). (I-K) FZD7(I), P63 (J), and GPX4 (K) mRNA expression levels (fold-change ± SD, n=3–4) in OVCAR5 cells transduced with control shRNAs (Ctrl_shctrl, Ctrl_shP63, FZD7_shctrl) or transfected with FZD7 expression vector and subsequently transduced with shRNA targeting P63 (FZD7_shP63). For all comparisons: *P<0.05, **P<0.01, ***P<0.001. (L) Scatter plot shows the correlation between P63 and FZD7 mRNA expression levels in HGSOC tumors (n=419) profiled in the TCGA database. Pearson correlation coefficients and P-values are shown. (M) Kaplan-Meier survival curves for HGSOC patients profiled in the TCGA having high (n=61) or low (n=318) P63 (TP63-012) mRNA expression levels. High or low levels were defined based on statistically determined cutoff point that maximizes absolute value of the standardized two-sample linear rank statistic. (N) Model demonstrates the proposed mechanism by which FZD7 engages the anti-oxidant pathway governed by GPX4.
Lastly, to determine whether FZD7 regulates GPX4 expression by altering Tp63 function, the effects of Tp63 knockdown on GPX4 expression in cells expressing high vs. low levels of FZD7 were tested. Overexpression of FZD7 and KD of p63 in OVCAR5 cells were confirmed at mRNA level (Fig. 7I–K; SKOV3, Fig S8C–E). GPX4 upregulation induced by FZD7 overexpression was abrogated in cells in which p63 was KD (Fig. 7K; SKOV3, Fig S8E), supporting that this TF, engaged by β-catenin, downstream of FZD7, is an important regulator. Interestingly, Tp63 KD caused reduced expression of FZD7 in OC cells transfected with either control vector or FZD7, indicating a feedback regulatory role of Tp63 on FZD7. These experimental results were validated by examining the TCGA HGSOC database (35). FZD7 and Tp63 expression levels were positively correlated (Fig. 7L, r = 0.278, p < 0.0001) and Tp63 expression was positively associated with expression levels of genes related to glutathione metabolism, including GSS, GCLC and SLC7A11 (Fig. S8G–I, p < 0.01). Higher Tp63 expression levels were also significantly associated with poor overall survival in this patient cohort (Fig. 7M, p = 0.0431). In all, our results support the existence of a “persister” cell population, marked by FZD7, metabolically characterized by increased glutathione dependent anti-oxidant circuits and susceptible to ferroptosis. A potential mechanism leading to upregulation of GPX4 in platinum-tolerant OC cells marked by FZD7 is engagement of Tp63, trans-activated by β-catenin downstream of this receptor (Figure 7N).
Discussion:
Our data support that a Pt-T (“persister”) cancer cell population serving as a reservoir for resistant tumors, shares common features with CSCs and is characterized by FZD7 expression. We demonstrate that the survival of these cells is dependent on an active FZD7-β-catenin-Tp63-GPX4 pathway, which renders these cells susceptible to inducers of ferroptosis. Our findings have several implications.
First, we identified FZD7 as a receptor enriched in Pt-T cancer cells and tumors. FZD7 is a transmembrane receptor which transduces signals involved in both the canonical and non-canonical Wnt pathways (36). Previous data indicated that FZD7 plays essential roles in stem cell biology and cancer (37). In breast and hepatocellular carcinoma, FZD7 has oncogenic functions, promoting cell proliferation, migration, and invasion (24, 38–41). Here we show that FZD7 marks a population representing ~2–25% cells in Pt-T cell lines or tumors. The receptor’s KD sensitized OC cells to platinum and its overexpression rendered cells resistant. Our group previously identified FZD7 as a receptor facilitating interaction of ovarian CSCs with the tumor niche (42) and the current findings corroborate the link between this receptor and cancer stemness. FZD7(+) cells were shown to proliferate more robustly as spheres, to express higher levels of stemness associated TFs and display enhanced TIC. Interestingly, a related receptor, FZD10, was recently linked to PARP inhibitor resistance (43). Thus, it is likely that activation of the Wnt pathway through activation of one or more FZD receptors contributes to emergence of resistance to DNA-damaging agents.
Secondly, we report that FZD7 marks a population of cells highly susceptible to ferroptosis. Ferroptosis is a newly described type of cell death distinct from apoptosis and necrosis (44) characterized by iron-dependent accumulation of ROS resulting in increased lipid peroxidation and eventually leading to cell death (33). Ferroptosis is dependent on NADPH/H(+), polyunsaturated fatty acid metabolism, and the mevalonate and glutaminolysis metabolic pathways (31). Class 1 (system Xc(−) inhibitors) and class 2 (GPX4) inhibitors are small-molecules that induce ferroptosis (44). Here we observed that FZD7(+) platinum tolerant cells were highly sensitive to ferroptosis induced by small molecules targeting GPX4. Tyrosine kinase inhibitor-tolerant cells have been reported to be sensitive to ferroptosis (3); however, no markers to identify cells prone to ferroptosis have been described.
Thirdly, we found GPX4 to be significantly upregulated in Pt-T cells, xenografts, PDXs, and ovarian tumors residual after neoadjuvant chemotherapy. GPX4 detoxifies lipid peroxides (L-OOHs) by converting them to corresponding alcohols (L-OH), preventing the buildup of toxic, membrane oriented, lipid reactive oxygen species (L-ROS) (45). Aside from GPX4, other glutathione metabolism related genes, such as GSH and GSR are also involved in anti-oxidation defense. Cancer cells with acquired drug resistance were reported to have increased cellular GSH levels (46). Lower levels of endogenous ROS and higher levels of antioxidants and GSH were found in temozolomide (TMZ)-resistant glioblastoma cells (47) and silencing the GSH biosynthesis pathway triggered ferroptosis in clear cell carcinoma (48). Thus, modulation of redox homeostasis by GSH/GSR appears to be an important key modulating sensitivity of cancer cells to chemotherapy (47). Interestingly, in our study, the expression of glutathione metabolism-related genes GSS, GSR, GPX2, IDH were directly correlated with expression of FZD7.
Lastly, our results shed light on a potential mechanism explaining the connection between FZD7 and activation of the glutathione regulatory machinery. A recent study reported that expression of GPX4 and of other genes involved in glutathione metabolism are regulated transcriptionally by Tp63 (21). Tp63 (along with p53 and p73) belongs to the Tp53 family (49). TP63 regulates the self-renewal of progenitor cells in epithelial tissues through its by-product ΔNp63, which has dominant-negative effects on other p53 family isoforms and exerts tumorigenic functions (34). Unlike Tp53, inactivated in a majority of human cancers, including OC, p63 is rarely mutated or inactivated. Overexpression of Tp63 was associated with poor survival in OC (49, 50), similar to our findings exploring the TCGA database. Tp63 was shown to regulate the expression of FZD7 and enhance Wnt signaling in mammary tissue (49). A putative crosstalk between Wnt/β catenin pathway and ΔNp63, the TP63 isoform lacking the N-terminus domain, was reported in skin, hair follicles, mammary glands and limb buds during development (41). We observed a similar direct and strong correlation between TP63 and FZD7 expression in OC cells, supporting that expression of this receptor is regulated by this TF. Furthermore, GPX4 expression, increased in FZD7 overexpressing cells, was downregulated by Tp63 knockdown, supporting this mechanism.
In all, our results propose FZD7 as a new marker for cancer cell populations likely to survive exposure to platinum, enriched in anti-oxidant response mechanisms. These rare cells responsible for disease relapse after chemotherapy, share stemness features and are susceptible to eradication through ferroptosis. Our data provide compelling evidence that targeting Pt-T FZD7(+) cells by inducing ferroptosis is effective and could represents a new strategy in a disease of high unmet need.
Supplementary Material
Significance:
Frizzled-7 marks platinum-tolerant cancer cells harboring stemness features and altered glutathione metabolism that depend on GPX4 for survival and are highly susceptible to ferroptosis.
Acknowledgments:
This research was supported by funding from the Ovarian Cancer Research Alliance, the National Cancer Institute (R01-CA224275), and the Diana Princess of Wales endowed Professorship from the Lurie Cancer Center to DM. Tumor specimens were procured through the Tissue Pathology Core and sequencing was performed in the NUSeq Core supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. Flow cytometry analyses were performed in the Northwestern University – Flow Cytometry Core Facility supported by Cancer Center Support Grant NCI CA060553. This research was supported in part through the computational resources and staff contributions provided for the Quest high performance computing facility at Northwestern University which is jointly supported by the Office of the Provost, the Office for Research, and Northwestern University Information Technology.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References:
- 1.Berek JS, Bertelsen K, du Bois A, Brady MF, Carmichael J, Eisenhauer EA, et al. Advanced epithelial ovarian cancer: 1998 consensus statements. Ann Oncol. 1999;10 Suppl 1:87–92. [DOI] [PubMed] [Google Scholar]
- 2.Tummala MK, and McGuire WP. Recurrent ovarian cancer. Clin Adv Hematol Oncol. 2005;3(9):723–36. [PubMed] [Google Scholar]
- 3.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell. 2017;20(2):233–46 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014;74(17):4922–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Zong X, Mitra S, Mitra AK, Matei D, and Nephew KP. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight. 2018;3(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer research. 2011;71(11):3991–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen LV, Vanner R, Dirks P, and Eaves CJ. Cancer stem cells: an evolving concept. Nature reviews Cancer. 2012;12(2):133–43. [DOI] [PubMed] [Google Scholar]
- 11.Nwani NG, Condello S, Wang Y, Swetzig WM, Barber E, Hurley T, et al. A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer. Cancers (Basel). 2019;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shi X, Zhang Y, Zheng J, and Pan J. Reactive oxygen species in cancer stem cells. Antioxid Redox Signal. 2012;16(11):1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell. 2013;24(6):751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong R, Qiang W, Guo H, Xu X, Kim JJ, Mazar A, et al. Histologic and molecular analysis of patient derived xenografts of high-grade serous ovarian carcinoma. J Hematol Oncol. 2016;9(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Y, and Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70–8. [DOI] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anders S, Pyl PT, and Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson MD, McCarthy DJ, and Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang GX, Tu HC, Dong Y, Skanderup AJ, Wang Y, Takeda S, et al. DeltaNp63 Inhibits Oxidative Stress-Induced Cell Death, Including Ferroptosis, and Cooperates with the BCL-2 Family to Promote Clonogenic Survival. Cell Rep. 2017;21(10):2926–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bicaku E, Xiong Y, Marchion DC, Chon HS, Stickles XB, Chen N, et al. In vitro analysis of ovarian cancer response to cisplatin, carboplatin, and paclitaxel identifies common pathways that are also associated with overall patient survival. Br J Cancer. 2012;106(12):1967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asad M, Wong MK, Tan TZ, Choolani M, Low J, Mori S, et al. FZD7 drives in vitro aggressiveness in Stem-A subtype of ovarian cancer via regulation of non-canonical Wnt/PCP pathway. Cell Death Dis. 2014;5:e1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merle P, Kim M, Herrmann M, Gupte A, Lefrancois L, Califano S, et al. Oncogenic role of the frizzled-7/beta-catenin pathway in hepatocellular carcinoma. J Hepatol. 2005;43(5):854–62. [DOI] [PubMed] [Google Scholar]
- 25.Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer research. 2009;69(16):6381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMillen BD, Aponte MM, Liu Z, Helenowski IB, Scholtens DM, Buttin BM, et al. Expression analysis of MIR182 and its associated target genes in advanced ovarian carcinoma. Mod Pathol. 2012;25(12):1644–53. [DOI] [PubMed] [Google Scholar]
- 27.Gedye C, Sirskyj D, Lobo NC, Meens J, Hyatt E, Robinette M, et al. Cancer stem cells are underestimated by standard experimental methods in clear cell renal cell carcinoma. Sci Rep. 2016;6:25220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang H, Van Remmen H, Frohlich V, Lechleiter J, Richardson A, and Ran Q. Gpx4 protects mitochondrial ATP generation against oxidative damage. Biochem Biophys Res Commun. 2007;356(4):893–8. [DOI] [PubMed] [Google Scholar]
- 29.Maiorino M, Conrad M, and Ursini F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid Redox Signal. 2018;29(1):61–74. [DOI] [PubMed] [Google Scholar]
- 30.Pan X, Lin Z, Jiang D, Yu Y, Yang D, Zhou H, et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett. 2019;17(3):3001–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seibt TM, Proneth B, and Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. 2019;133:144–52. [DOI] [PubMed] [Google Scholar]
- 32.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol. 2018;9:1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruptier C, De Gasperis A, Ansieau S, Granjon A, Taniere P, Lafosse I, et al. TP63 P2 promoter functional analysis identifies beta-catenin as a key regulator of DeltaNp63 expression. Oncogene. 2011;30(46):4656–65. [DOI] [PubMed] [Google Scholar]
- 35.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phesse T, Flanagan D, and Vincan E. Frizzled7: A Promising Achilles’ Heel for Targeting the Wnt Receptor Complex to Treat Cancer. Cancers (Basel). 2016;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King TD, Zhang W, Suto MJ, and Li YH. Frizzled7 as an emerging target for cancer therapy. Cell Signal. 2012;24(4):846–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC, et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene. 2011;30(43):4437–46. [DOI] [PubMed] [Google Scholar]
- 39.Wu W, Dang S, Feng Q, Liang J, Wang Y, and Fan N. MicroRNA-542–3p inhibits the growth of hepatocellular carcinoma cells by targeting FZD7/Wnt signaling pathway. Biochem Biophys Res Commun. 2017;482(1):100–5. [DOI] [PubMed] [Google Scholar]
- 40.Ueno K, Hiura M, Suehiro Y, Hazama S, Hirata H, Oka M, et al. Frizzled-7 as a potential therapeutic target in colorectal cancer. Neoplasia. 2008;10(7):697–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons GE, Pandey S, Nedeljkovic-Kurepa A, Saxena M, Wang A, and Pruitt K. Frizzled 7 Expression Is Positively Regulated by SIRT1 and beta-Catenin in Breast Cancer Cells. Plos One. 2014;9(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Condello S, Sima L, Ivan C, Cardenas H, Schiltz G, Mishra RK, et al. Tissue Tranglutaminase Regulates Interactions between Ovarian Cancer Stem Cells and the Tumor Niche. Cancer Research. 2018;78(11):2990–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, et al. N(6)-Methylation of Adenosine of FZD10 mRNA Contributes to PARP Inhibitor Resistance. Cancer research. 2019;79(11):2812–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu H, Guo P, Xie X, Wang Y, and Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21(4):648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forcina GC, and Dixon SJ. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics. 2019;19(18):e1800311. [DOI] [PubMed] [Google Scholar]
- 46.Chen HH, and Kuo MT. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met Based Drugs. 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Z, Du S, Du Y, Ren J, Ying G, and Yan Z. Glutathione reductase mediates drug resistance in glioblastoma cells by regulating redox homeostasis. J Neurochem. 2018;144(1):93–104. [DOI] [PubMed] [Google Scholar]
- 48.Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. 2018;37(40):5435–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, et al. DeltaNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol. 2014;16(10):1004–15, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marchini S, Marabese M, Marrazzo E, Mariani P, Cattaneo D, Fossati R, et al. Delta Np63 expression is associated with poor survival in ovarian cancer. Annals of Oncology. 2008;19(3):501–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.