

Abstract
Antithrombin (AT) is a major plasma glycoprotein of the serpin superfamily which regulates the proteolytic activity of the procoagulant proteases of both intrinsic and extrinsic pathways. Two important structural features that participate in the regulatory function of AT include a mobile reactive center loop (RCL) that binds to active-site of coagulation proteases, trapping them in the form of inactive covalent complexes and a basic D-helix that binds to therapeutic heparins and heparan sulfate proteoglycans (HSPGs) on vascular endothelial cells. The binding of D-helix of AT by therapeutic heparins promotes the reactivity of the serpin with coagulation proteases by several orders of magnitude by both a conformational activation of the serpin and a template (bridging) mechanism. In addition to its essential anticoagulant function, AT elicits a potent anti-inflammatory signaling response when it binds to distinct vascular endothelial cell HSPGs, thereby inducing prostacyclin synthesis. Syndecans-4 has been found as a specific membrane-bound HSPG receptor on endothelial cells that relays the signaling effect of AT to the relevant second messenger molecules in the signal transduction pathways inside the cell. However, following cleavage by coagulation proteases and/or by spontaneous conversion to a latent form, AT loses both its anti-inflammatory activity and high affinity interaction with heparin and HSPGs. Interestingly, these low-affinity heparin conformers of AT elicit potent proapoptotic and antiangiogenic activities by also binding to specific HSPGs by unknown mechanisms. This review article will summarize current knowledge about mechanisms through which different conformers of AT exert their serine protease inhibitory and intracellular signaling functions in these biological pathways.
Keywords: antithrombin, anticoagulant, anti-inflammatory, heparin, heparan sulfate
Introduction to antithrombin structure and function
Antithrombin (AT, also called AT III) is a serine protease inhibitor of the serpin superfamily which regulates the proteolytic activity of procoagulant proteases of both intrinsic and extrinsic pathways [1-4]. It is synthesized in the liver as a single-chain glycoprotein with 432 amino acids, three disulfide bonds and four potential glycosylation sites [5,6]. It circulates in plasma with a concentration of 0.125 mg/ml (~2.5 μM) and its major form in the circulation referred to as α-AT carries carbohydrates at all four glycosylation sites, however, there is a minor fraction (5-10%) which does not have the carbohydrate chain at Asn-135 and is referred to as β-AT [7-9]. The initial x-ray crystal structure of AT in the dimeric form with an inactive latent or a cleaved form of the serpin was determined by two groups [10,11]. Similar to other serpins, the structure of AT is comprised of three β-sheets (A to C) and nine α-helices (A to I) [10,11]. The reactive center loop (RCL) extends from P15 to P5’ (α1-proteinase inhibitor (α1-PI) numbering system [12]) and connects the larger 5-stranded β-sheet A to the smaller 3-stranded β-sheet C [10,11]. Similar to other inhibitory serpins, AT has a metastable structure that is not folded in its thermodynamically most stable conformation but kinetically trapped in a state of higher free energy. This structural feature in AT and other inhibitory serpins is required for the inhibitory mechanism and its structural stability is achieved only when the RCL is inserted as a 4th strand in β-sheet A of the serpin upon binding to the active-site of a target coagulation protease and/or by spontaneous conversion to a latent conformation [10,11].
AT regulation of coagulation proteases
AT can inhibit all procoagulant proteases of the blood clotting cascade, but its primary targets appear to be thrombin and factors Xa (FXa) and IXa (FIXa) [1,2]. Similar to other serpins, AT inhibits its target proteases by a branched pathway, suicide substrate inhibition mechanism in which a Michaelis-type enzyme-inhibitor complex, formed in the first reaction step, is converted to a covalent acyl-enzyme intermediate complex in the second step of the reaction [1,2,13]. Thus, the RCL of AT initially interacts with the catalytic pocket of coagulation proteases by a manner that is similar to interaction of activation peptides of true substrates with proteases. However, unlike true substrates, upon attack of the P1-Arg (Arg-393) of the RCL by the catalytic Ser-195 of the protease, the RCL undergoes a large-scale conformational change that leads to its insertion into β-sheet A as a central fourth strand, thereby the RCL dragging the acylated target protease to the opposite pole of the serpin some ~70 Å away from its original position (Figure 1) [1,14]. AT is a heparin-binding serpin and its reactivity with coagulation proteases is markedly improved when heparin binds to several basic residues of the serpin located on a basic solvent exposed loop referred to as D-helix [1,2,15-17]. In the absence of heparin, AT exhibits relatively low reactivity with coagulation proteases. The basis for the slow reactivity of AT with coagulation proteases has been demonstrated to be due to a cryptic protease-interactive exosite on AT not being available for productive interaction with proteases in the absence of heparin (18,19). Binding of heparin and/or vascular glycosaminoglycans to D-helix conformationally activates AT by altering this cryptic exosite, making it available for interaction with target coagulation proteases of the serpin (see below).
Figure 1.
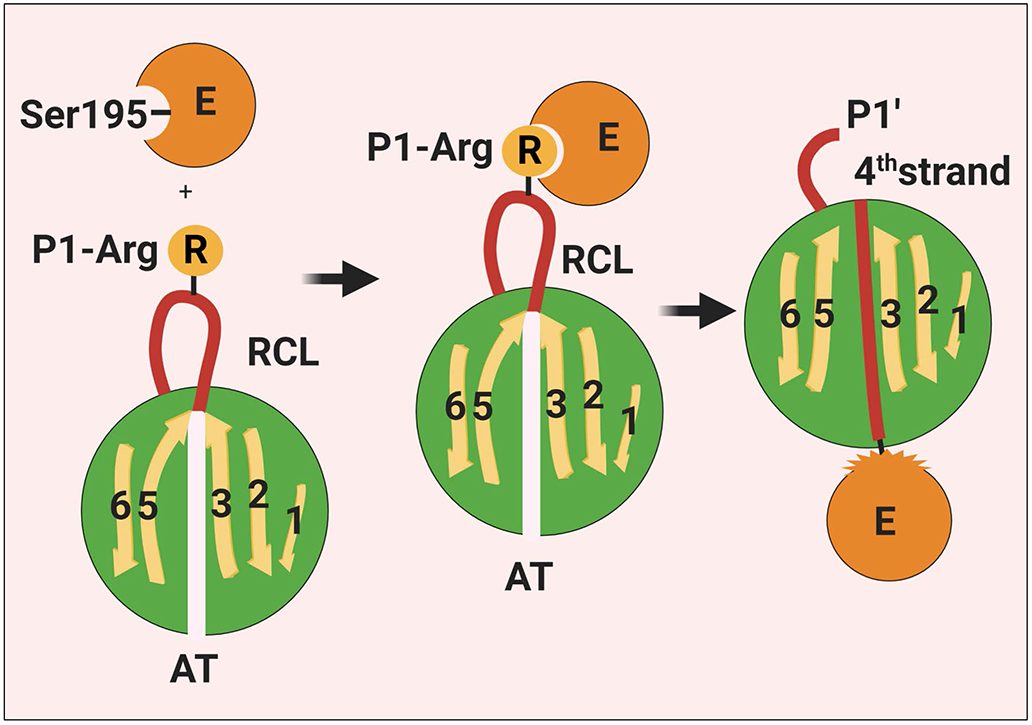
Schematic representation of the interaction of a coagulation protease with AT. The binding of the P1-Arg residue on RCL of AT to the active-site pocket of a coagulation protease induces acylation/cleavage of the P1-P1’ bond which then triggers a large scale conformational change in the serpin that leads to the insertion of the RCL into β-sheet A as a central 4th strand, thereby the RCL dragging the covalently-bound protease to the opposite pole of the serpin. The conformational change also results in disruption of the catalytic machinery of the protease. Thus, the protease gets trapped as an acylated complex with no catalytic function. See the text for more details. RCL, reactive center loop; E, enzyme; AT, antithrombin. Figure was prepared by software provided by Biorender.com.
Interaction with heparin
The basic D-helix of AT constitutes the heparin-binding site of the serpin. Binding of a distinct pentasaccharide sequence, which is present on approximately 1/3 of unfractionated heparins, to D-helix of AT induces a conformational change in the serpin that results in its activation [15,20]. The activated conformer of AT inhibits FXa and FIXa several hundredfold faster than native serpin [21-23]. It is however interesting to note unlike rapid inhibition of FXa and FIXa, conformational activation of AT makes an insignificant contribution to inhibition of thrombin [24]. For acceleration of AT inhibition of thrombin, a high molecular weight heparin with sufficient chain length is required in order to bridge both thrombin and AT in a ternary complex, thereby promoting the reaction by a template mechanism [17]. The mechanism by which pentasaccharide activates AT to promote its reactivity with FXa and FIXa but not with thrombin has been extensively studied. Comparisons of structures of AT in the absence and presence of synthetic pentasaccharide fragments of heparin have indicated that P14 and P15 residues of AT at the N-terminal end of the RCL are pre-inserted between strands 3 and 5 of β-sheet A in the absence but not in the presence of pentasaccharide [16,20]. Moreover, it has been noted D-helix of AT is shorter by two-turns in native AT and binding of pentasaccharide induces structural changes in the serpin that lead to elongation of D-helix that appears to be linked to expulsion of pre-inserted RCL residues from β-sheet A [16]. The mechanism by which pentasaccharide-mediated structural changes leads to activation of AT was unequivocally revealed by mutagenesis studies in which it was demonstrated binding of pentasaccharide on D-helix of AT leads to alteration of a cryptic protease-interactive exosite, located on strand 3 of β-sheet C (s3C), that specifically interacts with a complementary exosite (autolysis loop) on FXa and FIXa [18,19,25-28] as illustrated in Figure 2. Previous studies suggested that the P1-Arg-393 in native AT was limited in its ability to interact with proteases due to an intramolecular interaction with the serpin and that a pentasaccharide-mediated conformational activation of AT may involve breaking this interaction [29,30]. However, this mechanism of activation makes a minimum contribution (<2-fold) to activation of AT since mutagenesis of any one of the P6-P3’ residues of the RCL in AT had minimal effect on the extent of the rate accelerating effect of pentasaccharide in FXa inhibition by mutant serpins [18]. Furthermore, studies with the P1-Arg-393 to Trp mutant suggested that this residue is similarly accessible in both native and heparin-activated states of AT and contributes similarly to the specificity of the serpin interaction with thrombin and factor Xa in the two conformational states [31]. We have identified basic residues of the autolysis loop of both FIXa and FXa constituting an exosite that specifically and productively interacts with the cryptic exosite of AT only when AT is activated by pentasaccharide [26-28].
Figure 2.
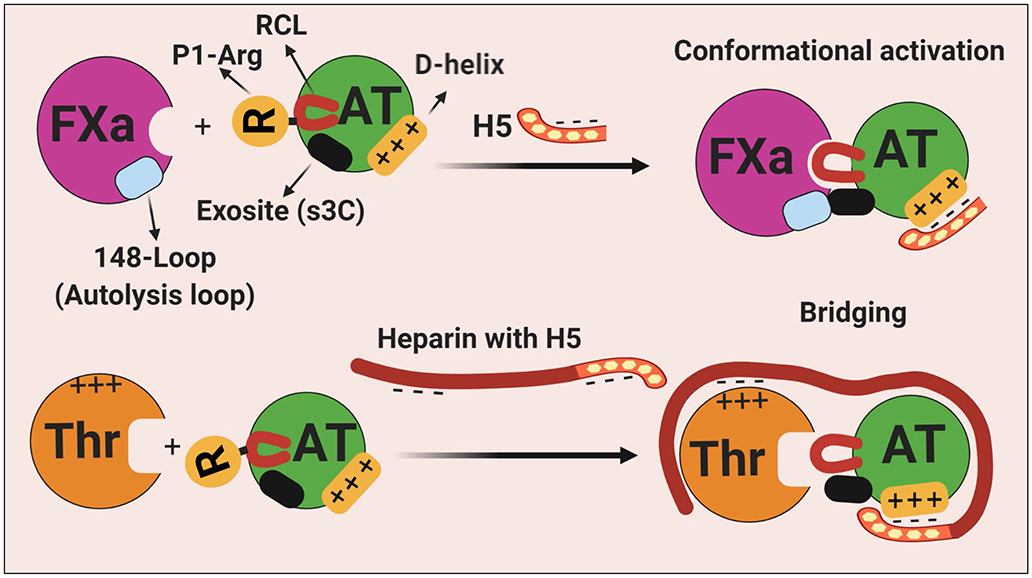
Schematic representation of heparin-mediated activation and promotion of protease inhibition by AT. (Top) In the native conformation of AT, the RCL is less exposed and a cryptic exosite (on s3C) is not available for productive interaction with the protease. The binding of the pentasaccharide fragment of heparin on D-helix results in the expulsion of the RCL that is coupled to the alteration of the cryptic exosite outside of the RCL. The conformationally altered exosite on AT interacts with a complementary exosite (the basic 148-loop, also called autolysis loop) on FXa (also on FIXa). (Bottom) Long-chain high molecular weight heparin bind both thrombin (on basic exosite-2) and D-helix of AT to promote the inhibition of the protease by a template (bridging mechanism). Thrombin is incapable of recognizing the activated conformer of AT because it lacks the complementary exosite site (the autolysis loop is negatively charged in thrombin) to interact with the heparin-exposed cryptic site on AT. See the text for more details. H5, pentasaccharide; s3C, strand 3 of β-sheet C; RCL, reactive center loop; AT, antithrombin; Thr, thrombin; FXa, factor Xa. Figure was prepared by software provided by Biorender.com.
Interaction with heparan sulfate proteoglycans
AT can bind specific HSPG receptors on endothelial cells (ECs) via basic residues of D-helix by the same mechanism it interacts with therapeutic heparins [32-34]. It has been reported a small population of vascular HSPGs (1-5%) possesses glycosaminoglycans (GAGs) which contain similar sequences as in pentasaccharide with the same characteristic 3-O-sulfate (3-OS) modification that is essential for high-affinity interaction of heparin with D-helix of AT [32]. This observation together with extensive in vitro data on the mechanism of the cofactor function of heparin has led to the hypothesis that vascular GAGs may function as heparin-like cofactors to promote the reactivity of AT with coagulation proteases under different pathophysiological conditions. In light of recent results showing that interaction of D-helix of AT with vascular GAGs induces signaling in ECs [34-36], determining the contribution of the GAG-AT interaction on the vasculature to physiological function of AT in two pathways has begged further investigation. It is known the 3-OS modification of GAGs on HSPGs is primarily mediated by heparan sulfate 3-O-sulfotransferase-1 (3-OST-1) in vascular ECs [37,38]. A possible physiological significance of the AT interaction with 3-OS containing vascular GAGs was provided by studies employing 3-OST-1 deficient mice in prothrombotic and pro-inflammatory models [37,38]. It was found that these mice have normal hemostasis and exhibit comparable phenotypes as wild-type mice in response to prothrombotic challenges [37,38]. However, 3-OST-1 deficient mice exhibited severe pro-inflammatory phenotypes in response to lipopolysaccharide (LPS) challenge, raising the possibility that interaction of AT with 3-OS containing vascular GAGs may primarily be responsible for the anti-inflammatory rather than the protease inhibitory function of the serpin [37,38]. However, low-affinity heparins lacking 3-OS modification are also known to accelerate AT inhibition of coagulation proteases albeit to a lesser extent. Thus, possible role of more abundant HSPG GAGs lacking 3-OS modification in accelerating AT inhibition of coagulation proteases cannot be discounted at this time.
Anti-inflammatory signaling function of AT
AT, similar to activated protein C [39], exhibits both anticoagulant and anti-inflammatory signaling activities. The signaling activity of AT is mediated through its D-helix-dependent interaction with vascular HSPGs. The interaction induces prostacyclin (PGI2) synthesis by ECs, leading to elaboration of a protective signaling pathway that inhibits NF-κB and expression of proinflammatory cytokines and cell adhesion molecules [34-36,40,41]. PGI2 is also a potent vasodilator and inhibitor of platelet aggregation [42], thus indirectly contributing to the anticoagulant function of AT. A PGI2-dependent anti-inflammatory function for AT has also been established in different animal models including severe sepsis [40,41,43]. However, in a randomized, placebo-controlled human clinical trial (KyberSept), AT did not show a beneficial effect on the mortality rate in patients with severe sepsis [44,45]. A potential problem with this clinical study was that it also used a low-dose of heparin concomitant with AT. Since interaction of AT with vascular HSPGs is necessary for the anti-inflammatory effect of AT, it is possible heparin has antagonized this effect in the septic setting [45,46]. In a couple of recent studies, we observed a potent cardioprotective effect for AT in an ischemia-reperfusion (I-R) injury model through the serpin activating adenosine monophosphate kinase (AMPK) signaling pathway [47,48]. The cardioprotective effect of AT was mediated through its D-helix-dependent interaction with vascular HSPGs since the heparan sulfate antagonist surfen inhibited AMPK activation [48]. AMPK-dependent cardioprotective effect of AT was markedly higher with the Asn-135 to Gln mutant of AT which is analogous to β-AT and known to bind 3-OS containing GAGs with 5-10-fold higher affinity [48]. This isoform of AT which constitutes 5-10% of total AT in the circulation also exhibits a markedly higher barrier-protective effect in response to LPS in cultured ECs [8,9]. AT inhibited the inflammatory c-Jun N-terminal protein kinase (JNK) signaling pathway in the I-R injury model [47,48]. D-helix-dependent interaction of AT with HSPGs is solely responsible for its anti-inflammatory signaling function since engineering D-helix of AT in α1-PI, a non-heparin-binding serpin with an acidic D-helix, transferred all HSPG-dependent anti-inflammatory properties of AT to the chimeric serpin [49].
Results of a recent study investigating anti-inflammatory properties of AT on leukocytes have identified three membrane spanning receptor proteins; CD13 (also known as aminopeptidase N), CD300f (also known as LMIR3, CLM-1, IgSF13, IREM-1 or CMRF35-like molecule 1), and LRP-1 (lipoprotein receptor-related protein 1) that are involved in AT-dependent signaling in monocytes [50]. These receptors are known to be negative regulators of inflammatory signaling on monocytes with CD13 playing a role in internalization of Toll-like receptor 4 (TLR4) and CD300f being involved in blocking both MyD88 and TRIF-mediated TLR signaling through activation of Src homology region 2 domain-containing phosphatase 1 (SHP-1) [51-53]. Results of this previous study have indicated these signaling effects of AT on monocytes are primarily specific for β-AT. It is worth noting that, similar to ECs, D-helix-dependent interaction of AT with 3-OS containing GAGs (i.e., syndecan-4, Synd-4) is known to elicit anti-inflammatory responses on monocytes [34,36,54,55]. Thus, further studies will be required to understand whether these receptors, similar to Synd-4, contain high affinity AT-binding GAGs that transmit signaling function of AT in monocytes [48,54]. Further investigation will also be required to determine whether this mechanism of AT-mediated CD13 and CD300f signaling is unique for monocytes or if it can occur in ECs [50,56].
Signaling mechanism of AT
The anti-inflammatory signaling function of AT in ECs requires its D-helix-dependent interaction with Synd-4 and siRNA knockdown of the receptor is shown to inhibit signaling function of AT [34,55]. The siRNA knockdown of 3-OST-1 also inhibits anti-inflammatory signaling of AT [48], suggesting that Synd-4 GAGs contain 3-OS modifications to support high-affinity interaction of the receptor with AT. Synd-4 also functions as a co-receptor for receptor tyrosine kinases (in particular receptor for basic fibroblast growth factor FGF2) [57]. The mechanism by which Synd-4 functions as a co-receptor for growth factor signaling has been extensively studied. It has been demonstrated protein kinase C (PKC)-delta (PKC-δ)-mediated phosphorylation of Ser-179 (Ser-183 in rat Synd-4) of the cytoplasmic-domain of Synd-4 regulates specificity of receptor signaling [57]. It appears that when Ser-179 of Synd-4 cytoplasmic domain is phosphorylated, it cannot function as a co-receptor for growth factor signaling [57]. Since it is essential for vascular ECs to maintain their basal quiescent phenotype under normal physiological conditions, we recently hypothesized AT may exert its protective effect by binding to Synd-4 GAGs to stabilize the cytoplasmic-domain of the receptor at Ser-179 in phosphorylated form (Figure 3), thereby inducing production of PGI2 that culminates in inhibition of NF-κB activation and downregulation of expression of vascular cell adhesion molecules [58]. In support of this hypothesis, we demonstrated AT-mediated membrane localization of PKC-δ and its phosphorylation of Synd-4 Ser-179 is required for the serpin to exert its anti-inflammatory effect [58]. In further support of this hypothesis, we found AT can neither induce PGI2 secretion nor phosphorylation of Synd-4 cytoplasmic-domain in ECs expressing a dominant-negative form of PKC-δ (PKC-δ-DN) [58]. The anti-inflammatory signaling function of AT was also lost in ECs expressing PKC-δ-DN, suggesting PKC-δ activity is required for protective signaling function of AT [58]. Based on these results, we propose the following model for the mechanism of AT signaling. We hypothesize phosphorylation state of the Synd-4 cytoplasmic-domain dictates signaling specificity of the receptor. Thus, upon binding to 3-OS containing Synd-4, AT recruits PKC-δ, thereby leading to phosphorylation of Ser-179 of the receptor (Figure 3). We further hypothesize AT activates phospholipase A2 (PLA2), most likely the Ca2+-independent isoform of it (iPLA2), thereby selectively hydrolyzing arachidonylated plasmolegen phospholipids to produce arachidonic acid followed by its metabolism to PGI2 by cyclooxygenase-2 (Figure 3). It is known iPLA2 may also be activated by thrombin in ECs [59-61]. The released PGI2 binds to its Gs-protein coupled receptor (IP), thereby activating adenylyl cyclase (AC) and mediating synthesis of cAMP and activation of protein kinase A (PKA) in both vascular (autocrine signaling) and smooth muscle cells (paracrine signaling). AT-mediated cAMP signaling leads to phosphorylation of cAMP responsive element binding-protein (CREB) and its transport to the nucleus, thereby modulating gene expression, including NF-κB inhibition (Figure 3) [62]. Indeed, an iPLA2-dependent cAMP generation, PKA activation and CREB phosphorylation has been shown in ECs [62]. Interestingly, it has been reported iPLA2 is activated by a novel PKC [63]. It is possible that PKC-δ is the novel PKC responsible for activating iPLA2. It has been discovered in addition to PKA, cAMP can also signal through Epac-1 (exchange protein directly activated by cAMP), thereby activating Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) which serves as an upstream AMPK kinase (CaMKKβ-AMPK) [64]. Further studies will be required to validate this model, determine which one of these pathways is involved in AT-mediated AMPK signaling, and whether in addition to Synd-4, other 3-OS containing HSPGs are also involved in transmitting protective signaling function of AT through similar or other unknown mechanisms.
Figure 3.
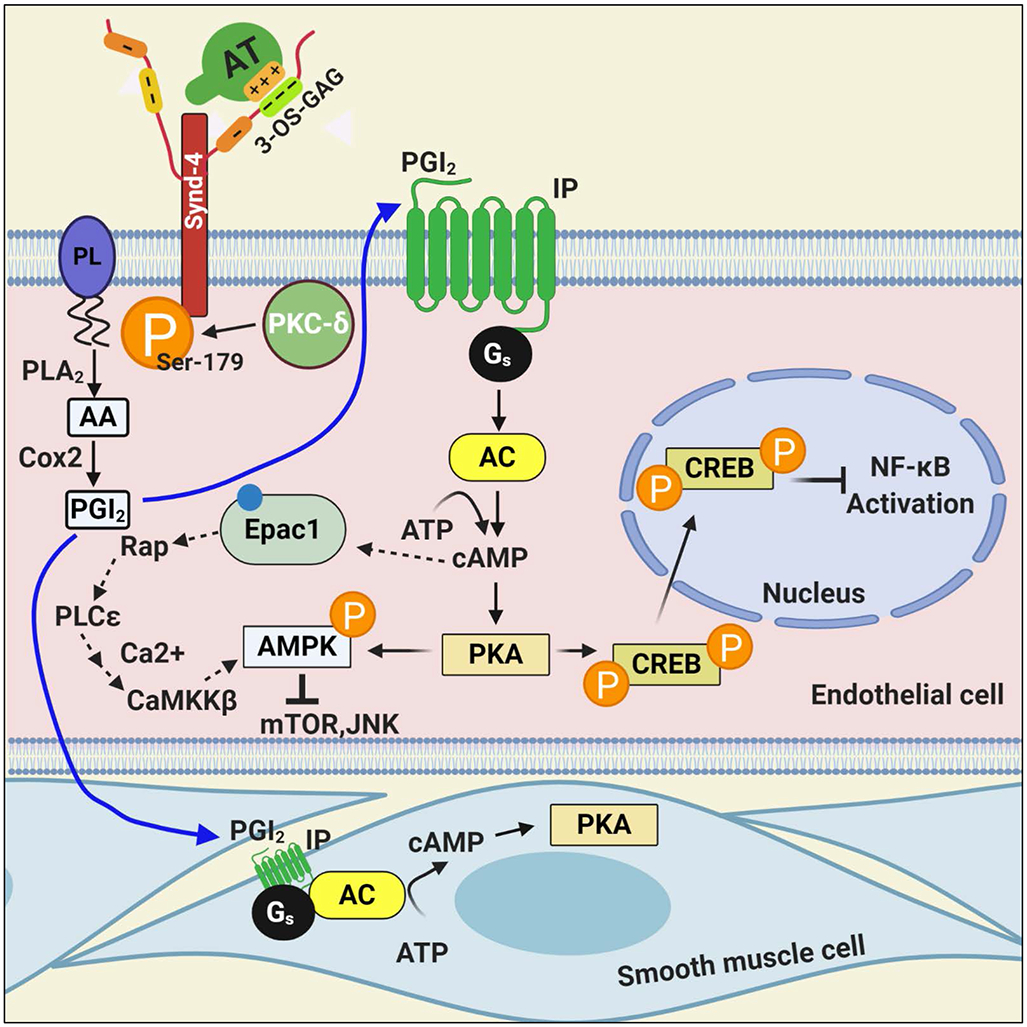
Hypothetical model of the anti-inflammatory signaling mechanism of AT. The binding of AT via its D-helix on 3-OS containing GAGs, covalently attached to Synd-4, recruits PKC-δ to the membrane, thereby leading to phosphorylation of the cytoplasmic domain of the Synd-4 at Ser-179. This process is linked to induction of PGI2 by a PLA2 hydrolyzing arachidonylated phospholipids to produce arachidonic acid followed by its metabolism to PGI2 by Cox-2. PGI2 binds to its Gs-protein coupled receptor, thereby activating adenylyl cyclase and mediating the synthesis of cAMP and activation of protein kinase A (PKA) in both vascular and smooth muscle cells. AT-mediated cAMP signaling leads to phosphorylation of cAMP responsive element binding-protein and its transport to the nucleus, thereby modulating gene expression, including NF-κB inhibition. In addition to PKA, cAMP can also signal through Epac-1, thereby activating CaMKKβ and AMPK in cardiomyocytes. It is not known which one the two pathways is involved in AT-mediated AMPK signaling. See the text for more details. PL, plasmolegen; PLA2, phospholipase A2; AA, arachidonic acid; Cox-2, cyclooxygenase-2; PGI2, prostacyclin; IP, PGI2 receptor; AC, adenylyl cyclase; PKA, protein kinase A; CREB, cAMP responsive element binding-protein; Epac-1, exchange protein directly activated by cAMP; AMPK, adenosine monophosphate kinase; CaMKKβ, Ca2+/calmodulin-dependent protein kinase kinase β; mTOR, mammalian target of rapamycin; JNK, c-Jun N-terminal protein kinase. Figure was prepared by software provided by Biorender.com.
Antiangiogenic function of AT
AT also possesses potent antiangiogenic activity [65-68]. However, this activity of AT is only observed when AT has been cleaved and/or the serpin has adopted a latent conformation (RCL is inserted into β-sheet A) [65-68]. Both cleaved and latent AT forms have very low-affinity for heparin [65-68]. These low-affinity conformers of AT have been shown to lack anti-inflammatory functions but both exhibit potent proapoptotic signaling activities in ECs [34]. Both anti-inflammatory and antiangiogenic/proapoptotic properties of AT are shown to be mediated through interaction of the same D-helix of the serpin with vessel wall HSPGs [34,65-68]. It has been hypothesized both native and latent/cleaved conformers of AT may utilize the same 3-OS containing pentasaccharide sequence to exert their biological activities and that the antiangiogenic epitope is cryptic in native AT but becomes available for interaction in the RCL-inserted conformers of the serpin [69]. In support of this hypothesis, we have demonstrated siRNA knockdown of Synd-4 not only reduces the anti-inflammatory function of native AT but also the proapoptotic activity of the latent serpin [34], Further studies will be required to understand the mechanism by which the interaction of high-affinity native and the low-affinity RCL-inserted conformers of AT with the same receptor elicit different signaling responses. One possible mechanism is that they engage different co-receptors for exerting their distinct signaling functions (see below).
Microarray analyses of global gene expression have indicated latent and cleaved conformers of AT significantly downregulate expression of a number of extracellular matrix-localized genes, known to play proangiogenic roles in ECs [70]. One of these proangiogenic genes, perlecan, is an HSPG that is known to function as a co-receptor for FGF2-induced angiogenesis [66,70]. Perlecan is a secreted multidomain basement protein with a key role in regulation of cell growth and angiogenesis [70]. The N-terminus of perlecan contains GAGs that bind growth factors, however, its C-terminal domain when cleaved by proteolysis exhibits a potent anti-proliferative effect by inhibiting proangiogenic integrins [70,71]. In this context, it is possible that latent and cleaved forms of AT by interaction with specific cell surface HSPGs induces a signaling pathway that results in proteolytic cleavage of the perlecan C-terminal domain in the basement membrane of vascular ECs. It is known nuclear localization of PKC-δ induces a potent proapoptotic effect in ECs [72]. In light of our recent results that Synd-4-dependent anti-inflammatory signaling effect of native AT requires the kinase function of PKC-δ [58], we postulated the RCL-inserted conformer of AT may exert their proapoptotic effects through nuclear localization of PKC-δ. Indeed, immunofluorescence analysis revealed that latent AT but not native AT enhances perinuclear/nuclear localization of PKC-δ in ECs [58], This function of latent AT which was also shared by TNFα, was not observed in cells over-expressing PKC-δ-DN, but was robust in cells over-expressing wild-type PKC-δ [58]. This conclusion was further supported by the TUNEL assay and analysis of isolated membrane and membrane-associated proteins of AT-treated ECs by Western-blotting [58]. The hypothetical model presented in Figure 4 depicts the outcome of the subcellular localization of PKC-δ by native and latent forms of AT. Further studies will be required to determine whether PKC-δ signaling plays a regulatory role toward perlecan in antiangiogenic functions of latent/cleaved forms of AT.
Figure 4.
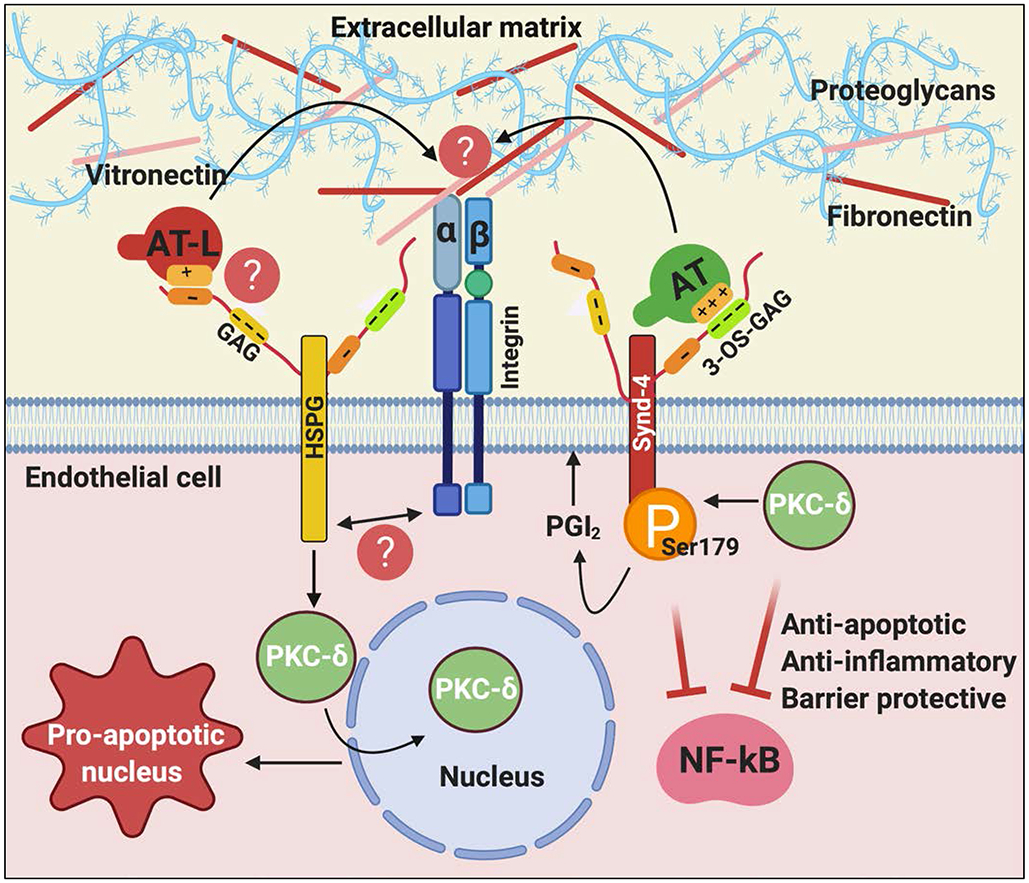
Hypothetical model of cytoprotective and pro-apoptotic signaling functions of native and latent forms of AT. The binding of native AT on 3-OS containing GAGs on Synd-4 recruits PKC-δ to the membrane, thereby phosphorylating the cytoplasmic domain of the receptor and inducing PGI2 synthesis. PGI2 signaling elicits anti-apoptotic, anti-inflammatory and barrier protective signaling responses. The binding of the low-affinity conformer of AT, latent AT, on vascular GAGs induces perinuclear/nuclear localization of PKC-δ, thereby eliciting pro-apoptotic/antiangiogenic signaling responses. We hypothesize that a crosstalk between HSPGs and different types of integrins modulates different mechanisms of AT function in these pathways by interacting with extracellular matrix proteins (i.e., vitronectin and fibronectin). See the text for more details. AT-L, latent AT; GAG, glycosaminoglycan; 3-OS, 3-O-sulfate. Figure was prepared by software provided by Biorender.com.
In experimental tumor models, latent and cleaved forms of AT have been shown to exhibit potent anti-proliferative and anti-tumorigenic effects [65]. However, the physiological significance of antiangiogenic AT forms, which constitute ~3% of plasma AT in healthy individuals (~4.8 μg/mL), is not known [73,74]. It is possible that these forms of AT have a role in regulating angiogenesis during wound healing after injury and/or inflammation where some amount of inactive loop-inserted AT becomes available as the result of the AT interaction with proteases of the clotting cascade. It is known a crosstalk between HSPGs and integrins is required for cell adhesion, survival, proliferation and migration [75,76]. Thus, we postulate interaction of different conformers of AT with distinct GAGs may be differentially linked to a receptor crosstalk involving the AT-bound receptor and different combinations of α and β integrins that leads to transmitting specific environmental cues from the extracellular matrix (ECM) to the actin cytoskeleton. It has been established a growth factor-mediated crosstalk between HSPGs and integrins (i.e., αvβ3, αvβδ and α5β1) modulate their specificity of function upon interaction with ECM proteins like vitronectin (VN) and fibronectin (FN) [75]. Thus, latent and cleaved conformers of AT can exert their antiangiogenic effect by interfering with binding of these integrins to ECM proteins (Figure 4). Plasminogen activator inhibitor 1 (PAI-1) functions by this mechanism to inhibit αvβ3 integrin binding to VN in ECs [77]. It is also known that inhibiting the interaction of αvβ3 and α5β1 integrins with VN and FN induces apoptosis and inhibits angiogenesis in response to FGF2 and VEGF [75]. Moreover, latent AT is shown to be antiangiogenic in wild-type but not in VN-null mice [78]. Further investigation is required to identify the nature of HSPG receptor(s) that transmit proapoptotic/antiangiogenic effects of latent AT and determine whether these effects are mediated by a PKC-δ-dependent crosstalk between different HSPGs and integrins.
Anti-bacterial and anti-viral effects of AT
A recent study found that β-AT, but not α-AT can bind to Gram-negative bacteria including E coli and to its purified cell wall component, LPS [50]. Circulating β-AT levels have been found to be significantly and preferentially consumed in infectious disease patients in intensive care units [50]. It has been hypothesized that binding of β-AT to bacterial cell wall can increase phagocytic activity of macrophages and subsequent bacterial lysis with their clearance from the circulation [50]. In support of this hypothesis, incubation of E. coli in the blood of mice overexpressing β-AT led to bacterial lysis and subsequent lower bacterial count [50], suggesting β-AT functions as an antimicrobial agent (Figure 5). By contrast, blood from mice overexpressing human α-AT showed higher number of bacterial counts when incubated with E. coli [50]. Furthermore, transgenic mice overexpressing β-AT, but not α-AT, were shown to exhibit significantly reduced LPS-induced pulmonary lesions and inflammatory cytokine levels and improved survival rates [50]. The antibacterial effect of AT has been shown to be mediated through D-helix of the serpin since a synthetic D-helix-derived peptide also exhibited a direct antimicrobial effect [79]. It is thought that neutrophil elastase or bacterial proteinases, released under in vivo conditions, may be responsible for the release of the D-helix peptide by fragmentation of AT and the antibacterial effect of the serpin [79]. Thus, β-AT has been hypothesized to have therapeutic utility as an antimicrobial drug in infectious disease settings [50], in particular in patients with sepsis where both coagulation and inflammatory pathways are highly upregulated. This question requires further investigation [56].
Figure 5.
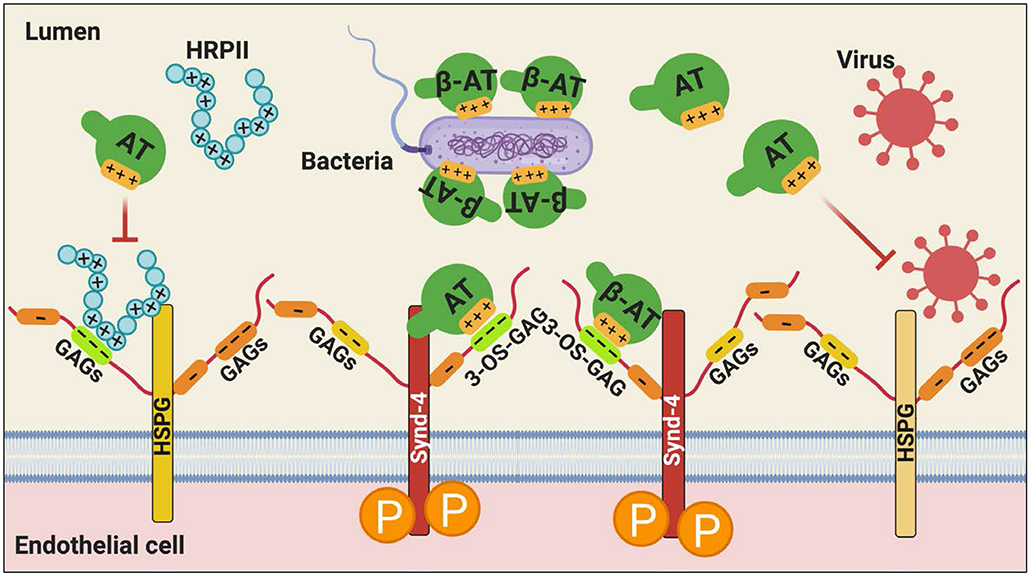
Hypothetical model of the protective effect of AT against infectious microorganisms. AT (particularly β-AT) can neutralize Gram-negative bacteria by binding via its D-helix to negatively charged molecules of the bacterial cell wall. AT can also inhibit the HSPG-dependent binding of certain family of viruses to cell surface GAGs, thereby preventing their entry into the host cell. AT can inhibit HRPII-dependent upregulation of pro-inflammatory and procoagulant responses, mediated by the Plasmodium falciparum-derived secretory protein, HRPII, in vascular endothelial cells not only by its anti-inflammatory signaling function but also by a competitive mechanism. See the text for more details. HSPG, heparan sulfate proteoglycan; Synd-4, syndecan-4; GAG, glycosaminoglycan; 3-OS, 3-O-sulfate; HRPII, histidine rich protein II. Figure was prepared by software provided by Biorender.com.
Similar to bacterial infection, viral infection has also been found to be associated with decreased AT levels and increased thrombosis [80]. A number of family of viruses including the coronavirus family containing membrane proteins with positively charged amino acids are reported to attach to HSPGs on cell surfaces (Figure 5) [81]. This interaction not only increases concentrations of the virus on cell surfaces but also increases chances of viral access into cells. The spike protein of severe acute respiratory syndrome coronavirus (SARS-CoV) was reported to interact with angiotensin-converting enzyme 2 (ACE2) [82]. Interestingly, however, masking cell surface HSPGs with lactoferrin was shown to inhibit binding of SARS-CoV on HEK-293E cells overexpressing ACE2 receptor [83]. Thus, it has been concluded that, in addition to ACE2, HSPGs are also essential cell-surface molecules involved in mediating the SARS-CoV entry. Similarly, incubation of human coronavirus NL63 with heparan sulfate reduced the replication of the virus in a dose dependent manner [84], and removing cell surface HSPGs with heparinase-I on HeLa cells decreased the entry of murine coronavirus by 50-60% [85]. AT therapy has been reported to elicit a potent anti-viral response against HIV-1, HCV, HSV-1 and HSV-2 by increasing the gene expression of prostaglandin synthetase-2 (PTGS2) in a dose dependent manner [86]. PTGS2 has been reported to downregulate NF-κB, an essential signaling pathway for transcription of genes involved in viral replication [86]. Finally, we have demonstrated that a histidine rich protein (HRPII), secreted by the malaria parasite, Plasmodium falciparum, binds to AT-specific HSPGs and inhibits HSPG-dependent anti-inflammatory function of the serpin by a competitive manner [87]. AT was shown to also inhibit the proinflammatory activity of HRPII by a concentration-dependent manner [87]. It was found HRPII elicits its proinflammatory effects by Src-dependent phosphorylation of vascular endothelial (VE)-cadherin and AT counteracted this effect by competitively inhibiting interaction of the parasite protein with vascular cell HSPGs [87]. Thus, in addition to its direct protective signaling function, AT may play a role in blocking the interaction of microorganisms with HSPGs and their entry into cells (Figure 5). In light of the observation that in addition to their HSPG-binding properties as a mechanism of virulence, infections with these pathogenic agents are also associated with increased hyper-coagulopathy, we believe therapeutic benefits of AT in these settings, in particular β-AT, which exhibits a normal anticoagulant activity but much higher affinity for HSPGs, warrants further investigation.
AT-deficiency caused by D-helix mutation
Acquired or congenital deficiency of AT is associated with increased risk of venous thrombosis and pulmonary embolism [88]. More than 300 natural variants of AT have been reported and based on circulating antigen and functional activity levels of the serpin, these variants are classified into two types with distinct characteristics [89,90]. The type-I AT deficiency is quantitative and is characterized by the mutation causing equally low antigen and activity levels [89,90]. By contrast, type-II deficiency is qualitative and is characterized by the AT variant exhibiting only a lower activity level [89,90]. Type-II AT deficiency is further divided into three subtypes based on the type of mutations: those affecting the reactive-site loop of AT and causing RCL-dependent impairment in the protease-inhibitory function for AT are called type-IIRS, those affecting the heparin-binding site are called type-IIHBS and those having pleiotropic effects on both sites of the serpin are referred to as type-IIPE [89,90]. Several interesting review articles have been published that describe the complete list and phenotypes of AT deficiencies [88-95], thus this subject will not be further discussed here. However, related to the main focus of this article, it is appropriate to discuss selected natural variants of HBS in which affinity of heparin for D-helix of the serpin has been adversely affected. At least 12 distinct HBS mutations of AT have been identified which are associated with higher incidence of venous and/or arterial thrombosis [88]. The underlying basis of thrombosis in carriers of these AT mutations, which exhibit normal progressive inhibitory activities toward coagulation proteases, has been primarily attributed to their loss of heparin cofactor-dependent protease inhibitory function. In light of the physiologically significant signaling function of AT as described above, we decided to characterize the anti-inflammatory functions of a couple of HBS mutants. The variants we picked were Ile-7 to Asn (I7N, Rouen III) and Leu-99 to Phe (L99F, Budapest III) variants, in neither of which the mutation is actually located on D-helix, nevertheless, both variants exhibit a lower-affinity for heparin [96-98]. Characterization of recombinant forms of these mutants in in vitro and in vivo assay systems revealed both mutants have lost their anti-inflammatory signaling activities in response to proinflammatory stimuli [99]. In another recent study, we identified a novel AT-deficient thrombosis patient who was a heterozygous carrier of Thr-90 to Ser substitution in AT. Characterization of a recombinant form of this mutant revealed the variant exhibits 4-5-fold lower anticoagulant activity due to the mutant acting as a substrate in its reaction with coagulation proteases [100]. Further studies revealed the mutation adversely affects conformations of both the RCL and D-helix of the serpin, thereby decreasing the rate of the RCL loop insertion of AT in the inhibitory pathway as well as exhibiting lower HSPG-dependent anti-inflammatory activity, thus qualifying it to be classified as a type-IIPE AT deficiency [100]. These results suggest that further studies are required to determine to what extent the loss of D-helix-dependent anti-inflammatory signaling function of HBS variants contribute to enhanced thrombosis in these patients.
Conclusions and perspectives
AT plays key roles in coagulation and inflammation by modulating proteolytic activities of coagulation proteases in plasma and also binding to 3-OS containing HSPGs on vascular ECs and eliciting anti-inflammatory responses by inducing prostacyclin synthesis. The Synd-4 HSPG has been identified as a 3-OS containing receptor for AT that is involved in transmitting the anti-inflammatory signaling function of the serpin inside the cell. Whether other HSPGs and/or non-HSPG membrane receptors also contribute to the signaling function of AT remains unknown. Cleaved and latent conformers of AT, which have much lower affinity for heparin, exhibit potent proapoptotic/antiangiogenic activities by binding to distinct vascular HSPGs sites that are not competed by the native AT. Subcellular localization of PKC-δ by high and low affinity conformers of AT appears to contribute to determining signaling specificity of the serpin: binding of native AT on Synd-4 GAGs recruits PKC-δ to the cytoplasmic membrane of ECs, thereby leading to phosphorylation of the cytoplasmic domain of Synd-4 and induction of prostacyclin synthesis. By contrast, binding of cleaved/latent forms of AT to low affinity HSPGs is associated with perinuclear/nuclear localization of PKC-δ in ECs and activation of a proapoptotic pathway, a property that is shared by TNF-α. Noting that cell-ECM interactions are highly critical for many physiological processes including cell survival, proliferation, angiogenesis and migration, which are all controlled by synergistic signaling by HSPGs and integrin family of receptors, a full understanding of the mechanism through which AT can exert its different intracellular functions will require deciphering as to how the AT-binding HSPGs communicate with different combination of α and β integrins.
Another important question that requires further investigation is determining to what extent interaction of D-helix of AT with 3-OS containing vascular HSPGs contributes to protease inhibitory function vs. the protective anti-inflammatory function of the serpin. Unlike the ample evidence for its anti-inflammatory function, there is sparse evidence for the hypothesis that AT interaction with HSPGs contributes to its protease inhibitory function and that most of the data in the literature is based on therapeutic heparins. A future challenge is to determine whether observations with low and high molecular weight heparins obtained by in vitro assays can occur under in vivo conditions (Figure 6). Finally, the possibility that β-AT is primarily responsible for the anti-inflammatory signaling function and α-AT is mainly involved in the protease inhibitory function of the serpin warrants further investigation. To this end, innovative studies with homogenous forms of the two AT isoforms, prepared by recombinant DNA methods, need to be developed in both cellular and animal models. In light of a markedly superior anti-inflammatory effect for β-AT in several recent studies, further investigation toward understanding the therapeutic utility of this isoform of AT in specific inflammatory disorders, may be warranted. This is particularly significant for the AT-deficient patients who are infected with pathogenic organisms which utilize HSPGs to promote procoagulant and proinflammatory pathways.
Figure 6.
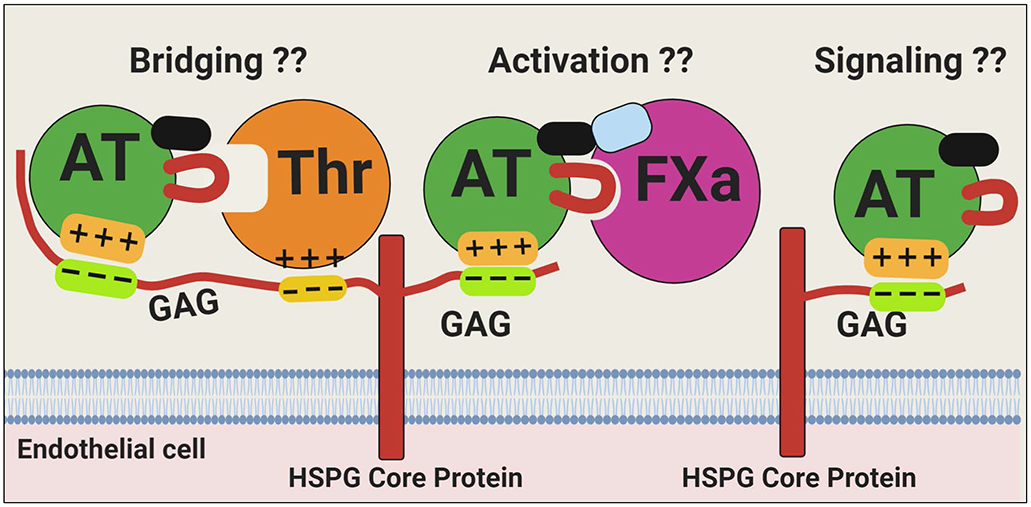
Hypothetical model of the interaction of AT with different vascular GAGs. Vascular HSPGs containing GAGs with different chain-lengths can bind D-helix of AT to elicit intracellular signaling responses and/or promote the serpin inhibition of coagulation proteases by a conformational activation (FXa) or by a template (thrombin) mechanism. These mechanistic concepts have been firmly established in cellular and in in vitro assays using therapeutic heparins. However, the relevance/significance of AT interaction with vascular GAGs to physiological functions (signaling or protease inhibitory function) of AT remains unknown (??) and requires further investigation. Figure was prepared by software provided by Biorender.com.
Acknowledgements
We thank thank Audrey Rezaie for editorial work on the manuscript.
Funding Sources
This study was supported by grants awarded by the National Heart, Lung, and Blood Institute of the National Institutes of Health HL101917 and HL062565 to ARR.
Footnotes
Disclosure of Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Olson ST, Richard B, Izaguirre G, Schedin-Weiss S, Gettins PG. Molecular mechanism of antithrombin regulation of blood clotting proteinases. A paradigm for understanding proteinase regulation by serpin family protein proteinase inhibitors. Biochimie. 2010; 92:1587–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gettins PG. Serpins structure, mechanism, and function. Chem Rev. 2002; 102: 4751–4803. [DOI] [PubMed] [Google Scholar]
- 3.Damus PS, Hicks M, Rosenberg RD. Anticoagulant action of heparin. Nature. 1973; 246: 355–357. [DOI] [PubMed] [Google Scholar]
- 4.Carrell RW, Skinner R, Jin L, Abrahams JP. Structural Mobility of Antithrombin and its Modulation by Heparin. Thromb Haemost. 1997; 78: 516–519. [PubMed] [Google Scholar]
- 5.Leon M, Aiach M, Coezy E, Gunennec JY, Feissinger JN. Antithrombin III synthesis in rat liver parenchymal cells. Thromb Res. 1983; 30: 369–375. [DOI] [PubMed] [Google Scholar]
- 6.Bock SC, Wion KL, Vehar GA, Lawn RM. Cloning and expression of the cDNA clone for human antithrombin III. Nucleic Acids Res. 1982; 10: 8113–8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ersdal-Badju E, Lu A, Peng X, Picard V, Zendehrouh P, Turk B, Björk I, Olson ST, Bock SC. Elimination of glycosylation heterogeneity affecting heparin affinity of recombinant human antithrombin III by expression of a β-like variant in baculovirus-infected insect cells. Biochem J. 1995; 310: 323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW. Structure of beta-antithrombin and the effect of glycosylation on antithrombin's heparin affinity and activity. J Mol Biol. 2003; 326: 823–833. [DOI] [PubMed] [Google Scholar]
- 9.Yamada T, Kanda Y, Takayama M, Hashimoto A, Sugihara T, Satoh-Kubota A, Suzuki-Takanami E, Yano K, Iida S, Satoh M. Comparison of biological activities of human antithrombins with high-mannose or complex-type nonfucosylated N-linked oligosaccharides. Glycobiology. 2016; 26: 482–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skinner R, Abrahams JP, Whisstock JC, Lesk AM, Carrell RW, Wardell MR. The 2.6 A Structure of Antithrombin Indicates a Conformational Change at the Heparin Binding Site. J Mol Biol. 1997; 266: 601–609. [DOI] [PubMed] [Google Scholar]
- 11.Schreuder HA, de Boer B, Dijkema R, Mulders J, Theunissen HJ, Grootenhuis PD, Hol WG. The intact and cleaved human antithrombin III complex as a model for serpin-proteinase interactions. Nat Struct Biol. 1994; 1: 48–54. [DOI] [PubMed] [Google Scholar]
- 12.Huber R, Carrell RW. Implications of the three-dimensional structure of alpha1-antitrypsin for structure and function of serpins. Biochemistry. 1989; 28: 8951–8966. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence DA, Olson ST, Muhammad S, Day DE, Kvassman JO, Ginsburg D, Shore JD. Partitioning of serpin-proteinase reactions between stable inhibition and substrate cleavage is regulated by the rate of serpin reactive center loop insertion into beta-sheet A. J Biol Chem. 2000; 275: 5839–5844. [DOI] [PubMed] [Google Scholar]
- 14.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000; 407: 923–926. [DOI] [PubMed] [Google Scholar]
- 15.Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci USA. 1997; 94: 14683–14688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belzar KJ, Zhou A, Carrell RW, Gettins PG, Huntington JA. Helix D elongation and allosteric activation of antithrombin. J Biol Chem. 2002; 277: 8551–8558. [DOI] [PubMed] [Google Scholar]
- 17.Danielsson A, Raub E, Lindahl U, Björk I. Role of ternary complexes, in which heparin binds both antithrombin and proteinase, in the acceleration of the reactions between antithrombin and thrombin or factor Xa. J Biol Chem. 1986; 261: 15467–15473. [PubMed] [Google Scholar]
- 18.Chuang YJ, Swanson R, Raja SM, Olson ST. Heparin Enhances the Specificity of Antithrombin for Thrombin and Factor Xa Independent of the Reactive Center Loop Sequence. Evidence for an Exosite Determinant of Factor Xa Specificity in Heparin-Activated Antithrombin. J Biol Chem. 2001; 276: 14961–14971. [DOI] [PubMed] [Google Scholar]
- 19.Gettins PG, Olson ST. Inhibitory serpins. New insights into their folding, polymerization, regulation and clearance. Biochem J. 2016; 473: 2273–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huntington JA, McCoy A, Belzar KJ, Pei XY, Gettins PGW, Carrell RW. The conformational activation of antithrombin. J Biol Chem. 2000; 275: 15377–15383. [DOI] [PubMed] [Google Scholar]
- 21.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Hérault JP, Herbert JM, Björk I. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb Haemost. 2004; 92: 929–939. [DOI] [PubMed] [Google Scholar]
- 22.Yang L, Manithody C, Rezaie AR. Localization of the heparin binding exosite of factor IXa. J Biol Chem. 2002; 277: 50756–50760. [DOI] [PubMed] [Google Scholar]
- 23.Wiebe EM, Stafford AR, Fredenburgh JC, Weitz JI. Mechanism of catalysis of inhibition of factor IXa by antithrombin in the presence of heparin or pentasaccharide. J Biol Chem. 2003; 278: 35767–35774. [DOI] [PubMed] [Google Scholar]
- 24.Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J Biol Chem. 1992; 267: 12528–12538. [PubMed] [Google Scholar]
- 25.Izaguirre G, Swanson R, Raja SM, Rezaie AR, Olson ST. Mechanism by which exosites promote the inhibition of blood coagulation proteases by heparin-activated antithrombin. J Biol Chem. 2007; 282: 33609–33622. [DOI] [PubMed] [Google Scholar]
- 26.Manithody C, Yang L, Rezaie AR. Role of basic residues of the autolysis loop in the catalytic function of factor Xa. Biochemistry. 2002; 41: 6780–6788. [DOI] [PubMed] [Google Scholar]
- 27.Yang L, Manithody C, Olson ST, Rezaie AR. Contribution of basic residues of the autolysis loop to the substrate and inhibitor specificity of factor IXa. J Biol Chem. 2003; 278: 25032–25038. [DOI] [PubMed] [Google Scholar]
- 28.Izaguirre G, Aguila S, Qi L, Swanson R, Roth R, Rezaie AR, Gettins PG, Olson ST. Conformational activation of antithrombin by heparin involves an altered exosite interaction with protease. J Biol Chem. 2014; 289: 34049–34064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jairajpuri MA, Lu A, Bock SC. Elimination of P1 arginine 393 interaction with underlying glutamic acid 255 partially activates antithrombin III for thrombin inhibition but not factor Xa inhibition. J Biol Chem. 2002; 277: 24460–24465. [DOI] [PubMed] [Google Scholar]
- 30.Johnson DJ, Langdown J, Li W, Luis SA, Baglin TP, Huntington JA. Crystal structure of monomeric native antithrombin reveals a novel reactive center loop conformation. J Biol Chem. 2006; 281: 35478–35486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chuang YJ, Swanson R, Raja SM, Bock SC, Olson ST. The antithrombin P1 residue is important for target proteinase specificity but not for heparin activation of the serpin. Characterization of P1 antithrombin variants with altered proteinase specificity but normal heparin activation. Biochemistry. 2001; 40: 6670–6679. [DOI] [PubMed] [Google Scholar]
- 32.de Agostini AI, Watkins SC, Slayter HS, Youssoufian H, Rosenberg RD. Localization of anticoagulantly active heparan sulfate proteoglycans in vascular endothelium: antithrombin binding on cultured endothelial cells and perfused rat aorta. J Cell Biol. 1990; 111: 1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Streusand VJ, Björk I, Gettins PG, Petitou M, Olson ST. Mechanism of acceleration of antithrombin-proteinase reactions by low affinity heparin. Role of the antithrombin binding pentasaccharide in heparin rate enhancement. J Biol Chem. 1995; 270: 9043–9051. [DOI] [PubMed] [Google Scholar]
- 34.Bae JS, Rezaie AR. Mutagenesis studies toward understanding the intracellular signaling mechanism of antithrombin. J Thromb Haemost. 2009; 7: 803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamauchi T, Umeda F, Inoguchi T, Nawata H. Antithrombin III stimulates prostacyclin production by cultured aortic endothelial cells. Biochem Biophys Res Commun. 1989; 163: 1404–1411. [DOI] [PubMed] [Google Scholar]
- 36.Opal SM. Therapeutic rationale for antithrombin III in sepsis. Crit Care Med. 2000; 28: S34–37. [DOI] [PubMed] [Google Scholar]
- 37.HajMohammadi S, Enjyoji K, Princivalle M, Christi P, Lech M, Beeler D, Rayburn H, Schwartz JJ, Barzegar S, de Agostini AI, Post MJ, Rosenberg RD, Shworak NW. Normal levels of anticoagulant heparan sulfate are not essential for normal hemostasis. J Clin Invest. 2003; 111: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shworak NW, Kobayashi T, De Agostini A, Smits NC. Anticoagulant heparan sulfate: To not clot-or not? Prog. Mol Biol Transl Sci. 2010; 93: 153–178. [DOI] [PubMed] [Google Scholar]
- 39.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007; 109: 3161–3172. [DOI] [PubMed] [Google Scholar]
- 40.Minnema MC, Chang ACK, Jansen PM, Lubbers YTP, Pratt BM, Whittaker BG, Taylor FB, Hack CE, Friedman B. Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood. 2000; 95: 1117–1123. [PubMed] [Google Scholar]
- 41.Harada N, Okajima K, Kushimoto S, Isobe H, Tanaka K. Antithrombin reduces ischemia/reperfusion injury of rat liver by increasing the hepatic level of prostacyclin. Blood. 1999; 93: 157–164. [PubMed] [Google Scholar]
- 42.Coughlin SR, Moskowitz MA, Zetter BR, Antoniades HN, Levine L. Platelet-dependent stimulation of prostacyclin synthesis by platelet-derived growth factor. Nature. 1980; 288: 600–602. [DOI] [PubMed] [Google Scholar]
- 43.Loubele S, Ten Cate H. Local administration of recombinant human antithrombin in a mouse model of peritoneal sepsis. J Thromb Haemost. 2006; 4: 2340–2342. [DOI] [PubMed] [Google Scholar]
- 44.Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Penzes I, Kubler A, Knaub S, Keinecke HO, Heinrichs H, Schindel F, Juers M, Bone RC, Opal SM, for the KyberSept Trial Study Group. High-dose antithrombin III in severe sepsis: A randomized controlled trial. JAMA. 2001; 286: 1869–1878. [DOI] [PubMed] [Google Scholar]
- 45.Wiedermann CJ, Hofmann JN, Juers M, Ostermann H, Kienast J, Briegel J, Strauss R, Keinecke HO, Warren BL, Opal SM, for the KyberSept Investigators. High-dose antithrombin III in the treatment of severe sepsis in patients with a high risk of death: Efficacy and safety. Crit Care Med. 2006; 34: 285–292. [DOI] [PubMed] [Google Scholar]
- 46.Roemisch J, Gray E, Hoffmann JN, Wiedermann CJ. Antithrombin: a new look at the actions of a serine protease inhibitor. Blood Coagul Fibrinolysis. 2002; 13: 657–670. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Wang Y, Wang J, Gao J, Tong C, Manithody C, Li J, Rezaie AR. Antithrombin is protective against myocardial ischemia and reperfusion injury. J Thromb Haemost. 2013; 11: 1020–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma Y, Wang J, Gao J, Yang H, Wang Y, Manithody C, Li J, Rezaie AR. Antithrombin up-regulates AMP-activated protein kinase signaling during myocardial ischaemia/reperfusion injury. Thromb Haemost. 2015; 113: 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L, Dinarvand P, Qureshi SH, Rezaie AR. Engineering D-helix of antithrombin in alpha-1-proteinase inhibitor confers antiinflammatory properties on the chimeric serpin. Thromb Haemost. 2014; 112: 164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Papareddy P, Rossnagel M, Doreen Hollwedel F, Kilic G, Veerla S, Naudin C, Smeds E, Westman J, Martinez-Martinez I, Egesten A, de la Morena-Barrio ME, Corral J, Linder A, Artoni A, Abbattista M, Novembrino C, Herbert Brakebusch C, Martinelli I, Kasetty G, Herwald H. A human antithrombin isoform dampens inflammatory responses and protects from organ damage during bacterial infection. Nat Microbiol. 2019; 4: 2442–2455. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh M, Subramani J, Rahman MM, Shapiro LH. CD13 restricts TLR4 endocytic signal transduction in inflammation. J Immunol. 2015; 194: 4466–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee SM, Kim EJ, Suk K, Lee WH. CD300F blocks both MyD88 and TRIF-mediated TLR signaling through activation of Src homology region 2 domain-containing phosphatase 1. J Immunol. 2011; 186: 6296–6303. [DOI] [PubMed] [Google Scholar]
- 53.Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 2000; 12: 361–378. [DOI] [PubMed] [Google Scholar]
- 54.Oelschläger C, Römisch J, Staubitz A, Stauss H, Leithäuser B, Tillmanns H, Hölschermann H. Antithrombin III inhibits nuclear factor kappaB activation in human monocytes and vascular endothelial cells. Blood. 2002; 99: 4015–4020. [DOI] [PubMed] [Google Scholar]
- 55.Kaneider NC, Förster E, Mosheimer B, Sturn DH, Wiedermann CJ. Syndecan-4-dependent signaling in the inhibition of endotoxin-induced endothelial adherence of neutrophils by antithrombin. Thromb Haemost. 2003; 90: 1150–1157. [DOI] [PubMed] [Google Scholar]
- 56.Rezaie AR, Giri H. Antithrombin: An anticoagulant, anti-inflammatory and antibacterial serpin. J Thromb Haemost. 2020; 18: 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elfenbein A, Simons M. Syndecan-4 signaling at a glance. J Cell Sci. 2013; 126: 3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Panicker SR, Biswas I, Giri H, Cai X, Rezaie AR. PKC (Protein Kinase C)-δ Modulates AT (Antithrombin) Signaling in Vascular Endothelial Cells. Arterioscler Thromb Vasc Biol. 2020; 40: 1748–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sharma J, Turk J, McHowat J. Endothelial cell prostaglandin I(2) and platelet-activating factor production are markedly attenuated in the calcium-independent phospholipase A(2)beta knockout mouse. Biochemistry. 2010; 49: 5473–5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharma J, Turk J, Mancuso DJ, Sims HF, Gross RW, McHowat J. Activation of group VI phospholipase A2 isoforms in cardiac endothelial cells. Am J Physiol Cell Physiol. 2011; 300: C872–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rickard A, Portell C, Kell PJ, Vinson SM, McHowat J. Protease-activated receptor stimulation activates a Ca2+-independent phospholipase A2 in bladder microvascular endothelial cells. Am J Physiol Renal Physiol. 2005; 288: F714–721. [DOI] [PubMed] [Google Scholar]
- 62.Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, Pimental D, van der Loo B. Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann Med. 2013; 45: 17–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meyer MC, Kell PJ, Creer MH, McHowat J. Calcium-independent phospholipase A2 is regulated by a novel protein kinase C in human coronary artery endothelial cells. Am J Physiol Cell Physiol. 2005; 288: C475–482. [DOI] [PubMed] [Google Scholar]
- 64.Park SJ, Ahmad F, Philp A, Baar K, Williams T, Luo H, Ke H, Rehmann H, Taussig R, Brown AL, Kim MK, Beaven MA, Burgin AB, Manganiello V, Chung JH. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012; 148: 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O'Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999; 285: 1926–1928. [DOI] [PubMed] [Google Scholar]
- 66.Zhang W, Chuang YJ, Swanson R, Li J, Seo K, Leung L, Lau LF, Olson ST. Antiangiogenic antithrombin down-regulates the expression of the proangiogenic heparan sulfate proteoglycan, perlecan, in endothelial cells. Blood. 2004; 103: 1185–1191. [DOI] [PubMed] [Google Scholar]
- 67.Zhang W, Swanson R, Izaguirre G, Xiong Y, Lau LF, Olson ST. The heparin-binding site of antithrombin is crucial for antiangiogenic activity. Blood. 2005; 106: 1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang W, Chuang YJ, Jin T, Swanson R, Xiong Y, Leung L, Olson ST. Antiangiogenic antithrombin induces global changes in the gene expression profile of endothelial cells. Cancer Res. 2006; 66: 5047–5055. [DOI] [PubMed] [Google Scholar]
- 69.Schedin-Weiss S, Richard B, Hjelm R, Olson ST. Antiangiogenic forms of antithrombin specifically bind to the anticoagulant heparin sequence. Biochemistry. 2008; 47: 13610–13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gubbiotti MA, Neill T, Iozzo RV. A current view of perlecan in physiology and pathology: A mosaic of functions. Matrix Biol. 2017; 57-58: 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bix G, Fu J, Gonzalez EM, Macro L, Barker A, Campbell S, Zutter MM, Santoro SA, Kim JK, Höök M, Reed CC, Iozzo RV. Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through alpha2beta1 integrin. J Cell Biol. 2004; 166: 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DeVries TA, Neville MC, Reyland ME. Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J. 2002; 21: 6050–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De la Morena-Barrio M, Sandoval E, Llamas P, Wypasek E, Toderici M, Navarro-Fernández J, Rodríguez-Alen A, Revilla N, López-Galvez R, Miñano A, Padilla J, de la Morena-Barrio B, Cuesta J, Corral J, Vicente V. High levels of latent antithrombin in plasma from patients with antithrombin deficiency. Thromb Haemost. 2017; 117: 880–888. [DOI] [PubMed] [Google Scholar]
- 74.Kjellberg M, Rimac B, Stenflo J. An immunochemical method for quantitative determination of latent antithrombin, the reactive center loop-inserted uncleaved form of antithrombin. J Thromb Haemost. 2007; 5: 127–132. [DOI] [PubMed] [Google Scholar]
- 75.Morgan MR, Humphries MJ, Bass MD. Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol. 2007; 8: 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science. 1994; 264: 569–571. [DOI] [PubMed] [Google Scholar]
- 77.Wu J, Strawn TL, Luo M, et al. Plasminogen activator inhibitor-1 inhibits angiogenic signaling by uncoupling vascular endothelial growth factor receptor-2-αVβ3 integrin cross talk. Arterioscler Thromb Vasc Biol. 2015; 35: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yi M, Sakai T, Fassler R, Ruoslahti E. Antiangiogenic proteins require plasma fibronectin or vitronectin for in vivo activity. Proc Natl Acad Sci USA. 2003; 100: 11435–11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Papareddy P, Kalle M, Bhongir RK, Morgelin M, Malmsten M, Schmidtchen A. Antimicrobial effects of helix D-derived peptides of human antithrombin III. J Biol Chem. 2014; 289: 29790–29800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saif MW, Greenberg B. HIV and thrombosis: a review. AIDS Patient Care STDS. 2001; 15: 15–24. [DOI] [PubMed] [Google Scholar]
- 81.Cagno V, Tseligka ED, Jones ST, Tapparel C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses. 2019; 11: 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020; 181: 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lang J, Yang N, Deng J, Liu K, Yang P, Zhang G, Jiang C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS One. 2011; 6: e23710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Milewska A, Zarebski M, Nowak P, Stozek K, Potempa J, Pyrc K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J Virol. 2014; 88: 13221–13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.de Haan CA, Li Z, te Lintelo E, Bosch BJ, Haijema BJ, Rottier PJ. Murine coronavirus with an extended host range uses heparan sulfate as an entry receptor. J Virol. 2005; 79: 14451–14456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Whitney JB, Asmal M, Geiben-Lynn R. Serpin induced antiviral activity of prostaglandin synthetase-2 against HIV-1 replication. PLoS One. 2011; 6: e18589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dinarvand P, Yang L, Biswas I, Giri H, Rezaie AR. Plasmodium falciparum histidine rich protein HRPII inhibits the anti-inflammatory function of antithrombin. J Thromb Haemost. 2019; December 19. doi: 10.1111/jth.14713. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lane DA, Bayston T, Olds RJ, Fitches AC, Cooper DN, Millar DS, Jochmans K, Perry DJ, Okajima K, Thein SL, Emmerich J. Antithrombin mutation database: 2nd (1997) update. For the Plasma Coagulation Inhibitors Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1997; 77: 197–211. [PubMed] [Google Scholar]
- 89.Perry DJ. Antithrombin and its inherited deficiencies. Blood Rev. 1994; 8: 37–55. [DOI] [PubMed] [Google Scholar]
- 90.Luxembourg B, Pavlova A, Geisen C, Spannagl M, Bergmann F, Krause M, Alesci S, Seifried E, Lindhoff-Last E. Impact of the type of SERPINC1 mutation and subtype of antithrombin deficiency on the thrombotic phenotype in hereditary antithrombin deficiency. Thromb Haemost. 2014; 111: 249–257. [DOI] [PubMed] [Google Scholar]
- 91.Rodgers GM. Role of antithrombin concentrate in treatment of hereditary antithrombin deficiency. An update. Thromb Haemost. 2009; 101: 806–812. [PubMed] [Google Scholar]
- 92.Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. 2008; 14: 1229–1239. [DOI] [PubMed] [Google Scholar]
- 93.Beresford CH. Antithrombin III deficiency. Blood Rev. 1988; 2: 239–250. [DOI] [PubMed] [Google Scholar]
- 94.Huntington JA. Serpin structure, function and dysfunction. J Thromb Haemost. 2011; 9: 26–34. [DOI] [PubMed] [Google Scholar]
- 95.Croles FN, Borjas-Howard J, Nasserinejad K, Leebeek FWG, Meijer K. Risk of Venous Thrombosis in Antithrombin Deficiency: A Systematic Review and Bayesian Meta-analysis. Semin Thromb Hemost. 2018; 44: 315–326. [DOI] [PubMed] [Google Scholar]
- 96.Gindele R, Oláh Z, Ilonczai P, Speker M, Udvari Á, Selmeczi A, Pfliegler G, Marján E, Kovács B, Boda Z, Muszbek L, Bereczky Z. Founder effect is responsible for the p.Leu131Phe heparin-binding-site antithrombin mutation common in Hungary: phenotype analysis in a large cohort. J Thromb Haemost. 2016; 14: 704–715. [DOI] [PubMed] [Google Scholar]
- 97.Olds RJ, Lane DA, Boisclair M, Sas G, Bock SC, Thein SL. Antithrombin Budapest 3. An antithrombin variant with reduced heparin affinity resulting from the substitution L99F. FEBS Lett. 1992; 300: 241–246. [DOI] [PubMed] [Google Scholar]
- 98.Brennan SO, Borg JY, George PM, Soria C, Soria J, Caen J, Carrell RW. New carbohydrate site in mutant antithrombin (7 Ile---Asn) with decreased heparin affinity. FEBS Lett. 1988; 237: 118–122. [DOI] [PubMed] [Google Scholar]
- 99.Dinarvand P, Yang L, Villoutreix BO, Rezaie AR. Expression and functional characterization of two natural heparin-binding site variants of antithrombin. J Thromb Haemost. 2018; 16: 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lu Y, Villoutreix BO, Biswas I, Ding Q, Wang X, Rezaie AR. Thr90Ser mutation in antithrombin is associated with recurrent thrombosis in a heterozygous carrier. Thromb Haemost. 2020; May 18. doi: 10.1055/s-0040-1710590. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]