

Abstract
Nine hemophilia A dogs were treated with adeno-associated viral (AAV) gene therapy and followed for up to 10 years. Administration of AAV8 or AAV9 vectors expressing canine factor VIII (AAV-cFVIII) corrected the FVIII deficiency to 1.9%−11.3% of normal FVIII levels. In two of nine dogs, FVIII activity increased gradually starting about four years after treatment. None of the dogs showed evidence of tumors or altered liver function. Analysis of integration sites in liver samples from six treated dogs identified 1,741 unique AAV integration events in genomic DNA and expanded cell clones in five dogs, with 44% of these integrations near genes involved in cell growth. All recovered integrated vectors were partially deleted and/or rearranged. Our data suggest that the increase in FVIII protein expression in two dogs may have been due to clonal expansion of cells harboring integrated vectors. These results support the clinical development of liver-directed AAV gene therapy for hemophilia A while emphasizing the importance of long-term monitoring for potential genotoxicity.
Editorial summary:
An AAV gene therapy study in hemophilia A dogs finds clonal expansions of transduced cells.
Gene therapy for hemophilia A has moved from preclinical studies in mice and dogs to successful trials in humans1–4. AAV vectors to deliver FVIII were first evaluated in hemophilia A dogs in the early 2000s5–7. While AAV gene therapy has demonstrated promising results in clinical studies for hemophilia B for more than eight years8,9, studies for hemophilia A are at an earlier stage1–4. Since recombinant (r)AAV commonly persists in an episomal form10, rAAV may not be maintained in tissues undergoing cell division. However, as hepatocytes in healthy dogs are typically quiescent11, the dog model is well suited to long-term studies designed to understand the durability and safety of AAV gene therapy and to evaluate tissues not accessible with human subjects.
One of the safety concerns for AAV gene therapy is the potential for genotoxic integration events. After introduction into cells, rAAV primarily remains episomal10 but integration into the host genome has been observed in mice12–16, non-human primates17 and humans17,18. To date there have been no serious adverse events involving genotoxicity by rAAV vector integration in humans or large animals. Other studies of wild type AAV in humans or rAAV in mouse models have detected tumors associated with AAV. Studies of wild-type AAV reported AAV integration events in human hepatocellular carcinoma (HCC), suggesting a possible role of AAV integration in transformation19–21. However, AAV copy number was much less than one per cell, so the idea that integration events caused HCC seems unlikely19–21. Another study showed that some AAV2 vectors contain a sequence in the 3’ untranslated region, adjacent to the AAV inverted terminal repeat (ITR), that has transcriptional enhancer in liver cells22. This element has been identified in HCC in humans and may be a mechanism of transformation in these tumors20. While HCC has not been observed in rAAV-treated adult mice or other mammals12,17,18,23, the development of rAAV-associated HCC has been reported after delivery to neonatal mice14,24–26 and to young adults of mouse strains that have a high incidence of spontaneous liver tumors27,28. This suggests the possibility that active cell division such as that occurring during development can be associated with genotoxic integration events14,24,25. In neonatal mice, rAAV integration events were identified recurrently in specific genomic regions in HCC tissue, suggesting possible insertional mutagenesis15. The above findings warrant further research to assess the safety of rAAV gene therapy.
In this study, we treated nine hemophilia A dogs with AAV-cFVIII and followed them for as long as ten years. Levels of cFVIII activity were maintained throughout the study, representing the longest sustained therapeutic levels of FVIII expression observed in a hemophilia A large animal model associated with a significant reduction in spontaneous bleeding events. Two dogs showed a gradual rise in FVIII expression. Upon termination of the study, AAV integration analysis on the liver tissue in these two dogs and four other dogs revealed clonal expansion of cells, with insertions near genes that are cancer-associated in humans, but not overt nodule formation or transformation.
Results
Sustained FVIII expression after AAV delivery
We treated hemophilia A dogs with two approaches, using either the two cFVIII chains (TC) encoded separately, or a single chain (SC) encoded in one vector6 (Fig. 1a and Table 1). The TC approach takes advantage of the normal intracellular processing of FVIII, in which FVIII undergoes proteolytic cleavage into two polypeptides that form a heterodimer that is subsequently secreted. In this approach, an AAV vector (AAV8 or AAV9) encoding the FVIII heavy chain (HC) was co-delivered with an AAV vector encoding the FVIII light chain (LC)6. The SC approach used an AAV8 vector to deliver B-domain deleted cFVIII6.
Fig. 1. Long term expression of AAV-cFVIII in hemophilia A dogs.
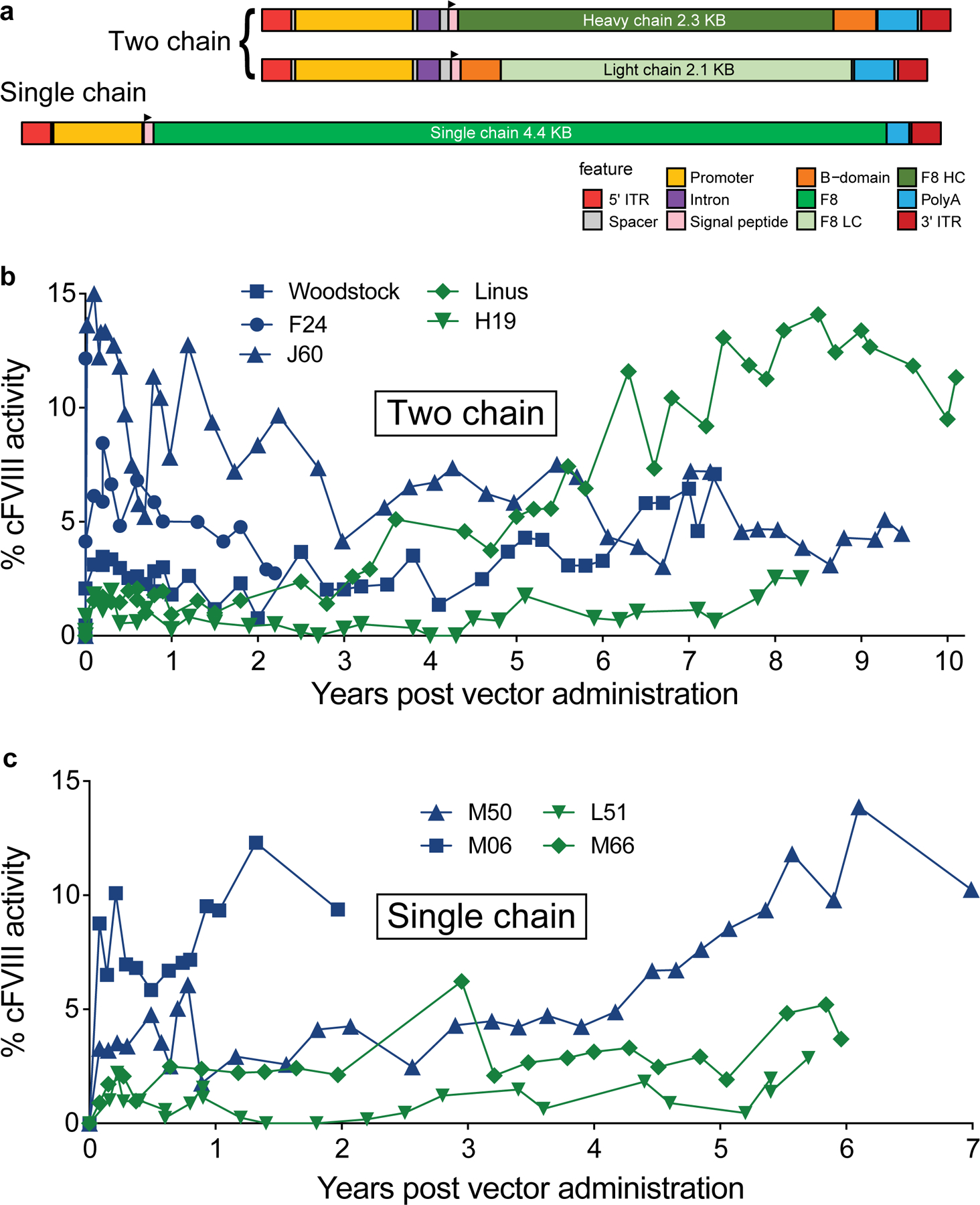
(a) Genetic structures of the vectors used, illustrating the two chain (TC) (top) and single chain (SC) (bottom) designs. The features of these constructs are indicated by colors shown in the legend. The TC delivery approach co-delivered the 2.3 Kb cF8 heavy chain in one AAV vector and the 2.1 Kb cF8 light chain in a second AAV vector. These vectors used the thyroxine-binding globulin (TBG) promoter and two copies of a1-microglobulin/bikunin enhancer sequences (695 bp) with an intron (175 bp) for liver-directed gene expression. The construct utilized a 263 bp SV40 polyadenylation signal (poly A). The SC delivery approach delivered the 4.4 Kb B-domain deleted cF8 driven by a minimal hepatic control region (192 bp) and human a-1 antitrypsin promoter (266 bp) (HCR-hAAT). This construct includes a 134 bp SV40 poly A. The grey regions indicate spacers or cloning sites. The TC vector was delivered by portal vein infusion while the SC vector was delivered by peripheral vein infusion. (b) Longitudinal quantification of FVIII levels after the TC delivery approach. In the two chain approach the total AAV dose was 2.5 × 1013 vg/kg dose for F24 (AAV8), Woodstock and J60 (AAV9)(blue). Linus (AAV8) and H19 (AAV9) were treated at the low vector dose (total vector dose of 1.2 × 1013 vg/kg)(green). (c) FVIII levels after the SC delivery approach. AAV8 delivery of the SC approach in hemophilia A dogs was at the vector dose of 4 × 1013 vg/kg (M50, M06)(blue) and at a vector dose of 2 × 1013 vg/kg (L51, M66)(green).
Table 1.
Summary of AAV administration, FVIII activity and DNA analysis of hemophilia A dogs.
| AAV Delivery Approach | Total AAV Vector Dose (vg/kg)1 | AAV serotype | Hem A Dog | Years Evaluated2 | % cFVIII activity | # of liver samples3 | DNA analysis of liver samples | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Final | Mean vector copy number per diploid genome ± SD | Copy number range | Number of genomic integration sites recovered (n=3 samples/dog)4 | ||||||
| Two Chain | 2.5 × 1013 | AAV8 | F24 | 2.2 | 5.7 ± 2.3 | 2.7 | nd | nd | nd | nd |
| AAV9 | Woodstock | 8.2 | 3.1 ± 1.5 | 7.1 | 5 | 0.28 ± 0.10 | 0.19–0.42 | nd | ||
| AAV9 | J60 | 9.5 | 8.1 ± 4.0 | 4.5 | 29 | 3.43 ± 2.34 | 0.76–7.82 | 271 | ||
| 1.2 × 1013 | AAV8 | Linus | 10.1 | 5.9 ± 4.6 | 11.3 | 15 | 0.28 ± 0.21 | 0.01–0.62 | 131 | |
| AAV9 | H19 | 8.3 | 0.9 ± 0.7 | 2.5 | 11 | 0.17 ± 0.09 | 0.07–0.33 | 160 | ||
| Single Chain | 4 × 1013 | AAV8 | M06 | 2.3 | 7.4 ± 2.6 | 9.4 | 8 | 1.44 ± 0.69 | 0.37–2.26 | 764 |
| M50 | 7.3 | 5.2 ± 3.6 | 10.3 | 29 | 0.35 ± 0.34 | 0.00–1.14 | 258 | |||
| 2 × 1013 | L51 | 6.1 | 0.8 ± 0.6 | 1.9 | 5 | 0.01 ± 0.02 | 0.00–0.04 | nd | ||
| M66 | 6.0 | 2.6 ± 1.3 | 3.7 | 13 | 0.32 ± 0.18 | 0.00–0.60 | 161 | |||
In the two chain delivery approach the vector dose was 1.25 × 1013 vector genomes per vector per kg (vg/v/kg) at the high dose and 6 × 1012 vg/v/kg at the low dose.
The number of years after AAV administration.
The number of liver samples analyzed in the vector copy number analysis.
The integration studies were performed on three liver samples per dog.
All the AAV-treated hemophilia A dogs maintained cFVIII expression for the entire study (2.2 to 10.4 years; Fig. 1b, 1c, Table 1). These hemophilia A dogs have an intron 22 inversion that is analogous to the most common mutation in humans, and have a severe bleeding phenotype (<1% cFVIII activity)29. After AAV administration, the mean levels of cFVIII activity for the dogs treated with the TC approach were 5.6% of normal for the dogs at the high vector dose and 3.4% for the dogs at the low vector dose (Table 1). One dog (J60) treated at the high vector dose required a splenectomy at the time of vector infusion and had a higher level of cFVIII expression (8.1% ± 4.0) than the other dogs treated at this dose (see Methods for additional details). For the SC treated dogs, the mean levels of cFVIII expression were 6.2% for the high vector dose and 1.7% for the low vector dose. At the final time points, all dogs had levels of cFVIII activity that were close to or greater than the mean cFVIII expression (Table 1). FVIII activity was also demonstrated by the shortening of the whole blood clotting time (WBCT) (Supplementary Fig. 1).
Two dogs (Linus and M50) showed a gradual increase in cFVIII expression that began more than four years after vector administration. Linus had 4% cFVIII activity at four years post-vector delivery, gradually increasing to 11% activity at 10 years after AAV administration (Fig. 1b). M50 had 4% cFVIII activity at four years post-AAV delivery that gradually increased to 10% activity at 7 years after vector administration (Fig. 1c). Two other dogs, Woodstock and M06, also had possible modest FVIII increases.
Clinical observations after AAV gene therapy
A cohort of naïve hemophilia A dogs (n=11) was followed prospectively for four years to determine the frequency of bleeding events. All bleeding events required treatment with recombinant cFVIII 30 or normal canine blood products. The annualized bleeding rate (ABR) was 12.7 ± 6.0 (see also 31; Supplementary Fig. 2), similar to the ABR for hemophilia patients treated episodically, who have 18.5 bleeds/patient/year32. In contrast, the nine AAV-treated hemophilia A dogs had a total of seven bleeding episodes during the combined total of 60 years of follow-up after AAV administration. The ABR per AAV-treated hemophilia A dog was between 0.00 and 0.45 with a mean of 0.13 ± 0.16, lower than reported for people with hemophilia on prophylaxis (ABRs of 4.8–6.0 bleeds/patient/year)32.
Clinical blood chemistries were obtained two to four times per year in AAV-treated dogs to monitor for adverse reactions. The alanine aminotransferase (ALT) level was within the normal limits for six of the nine dogs at most of the time points analyzed (Fig. 2a,b). Woodstock and Linus had an elevation (<2 times ULN) in ALT that began four years after AAV administration. These dogs were older (four years old) at the time of vector administration than the other dogs studied. L51 also had ALT levels that were >2 times the normal limits at multiple time points that are considered mildly elevated33. The aspartate aminotransferase (AST) levels were within normal limits for all dogs for the duration of the study (Fig 2c,d). These mild, asymptomatic liver enzyme elevations are not consistent with a specific liver disease pattern. The serum alpha-fetoprotein (AFP) is a biomarker for hepatic disease, including HCC in some patients34–36. The AFP values for all dogs were within the normal range at all times analyzed (Fig. 2e,f). Other blood chemistries were monitored throughout the study with no clinical concerns.
Fig. 2. Assessment of liver function after AAV-cFVIII administration.
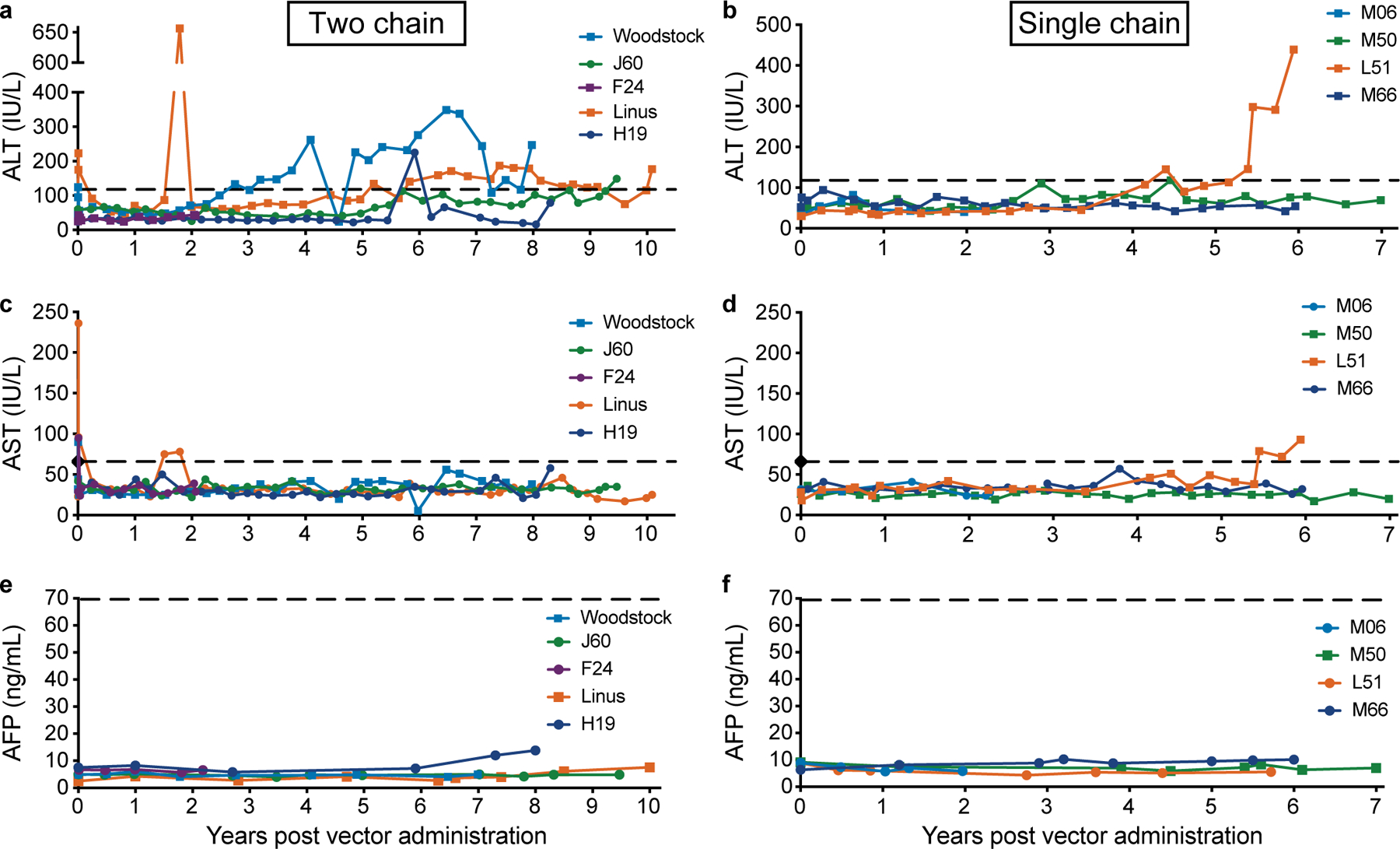
ALT (a and b), AST (c and d) and AFP (e and f) were monitored after AAV administration and throughout the study. In each case the x-axis shows time in years, the y-axis shows the liver analyte measured in blood. a), c) and e) Dogs treated with the two chain strategy. b), d), and f) Dogs treated with the single chain strategy. The upper limit of normal for ALT (120 IU/L), AST (65 IU/L) and AFP (70 ng/ml) are shown as dashed lines. Clinically, increase in liver enzyme levels in dogs are defined by veterinary clinical pathologists as mild (<5 times the upper limit of normal), moderate (5–10 times the upper limit of normal) and marked (>10 times the upper limit of normal) 33.
The liver pathology was evaluated at the final time point in the AAV-treated hemophilia A dogs and compared to naïve (to gene therapy) hemophilia A (n=10), hemophilia B (n=9) and normal (n=9) dogs of similar age from the same colony (Supplementary Tables 1 and 2). Some of the findings were consistent with features commonly observed in older dogs and were seen in both AAV treated and untreated (i.e., naïve) dogs. No pathologies definitively related to the treatment were identified. Three of nine AAV-treated hemophilia A dogs had clinical symptoms that led to ending the study early, but none were believed to be related to AAV gene therapy (Methods).
The immune response to cFVIII (Supplementary Fig. 3) and neutralizing antibodies (NAb) to the AAV capsid (Supplementary Table 3) were also followed during the study.
Investigation of the increase in FVIII expression
We investigated the mechanism of the rise in FVIII expression in Linus and M50 (Supplementary Table 4). At 6 years post vector administration, an ultrasound and liver biopsy were performed on Linus; no tumors, nodules, or notable inflammation in the liver were detected. The rise in FVIII levels was not associated with changes in levels of serum alpha-fetoprotein (AFP), fibrinogen, von Willebrand Factor (vWF) and clearance receptor in liver (Fig. 2e,f; Supplementary Fig. 4).
We next assessed AAV vector copy number (VCN) (Table 1). For dogs that had multiple liver samples per lobe available for analysis, the VCNs showed that the vector was distributed throughout the liver (Supplementary Table 5). Higher VCN levels were not detected in Linus and M50. J60 had a higher VCN than the other dogs; we speculate that the splenectomy at the time of vector administration may have increased liver transduction.
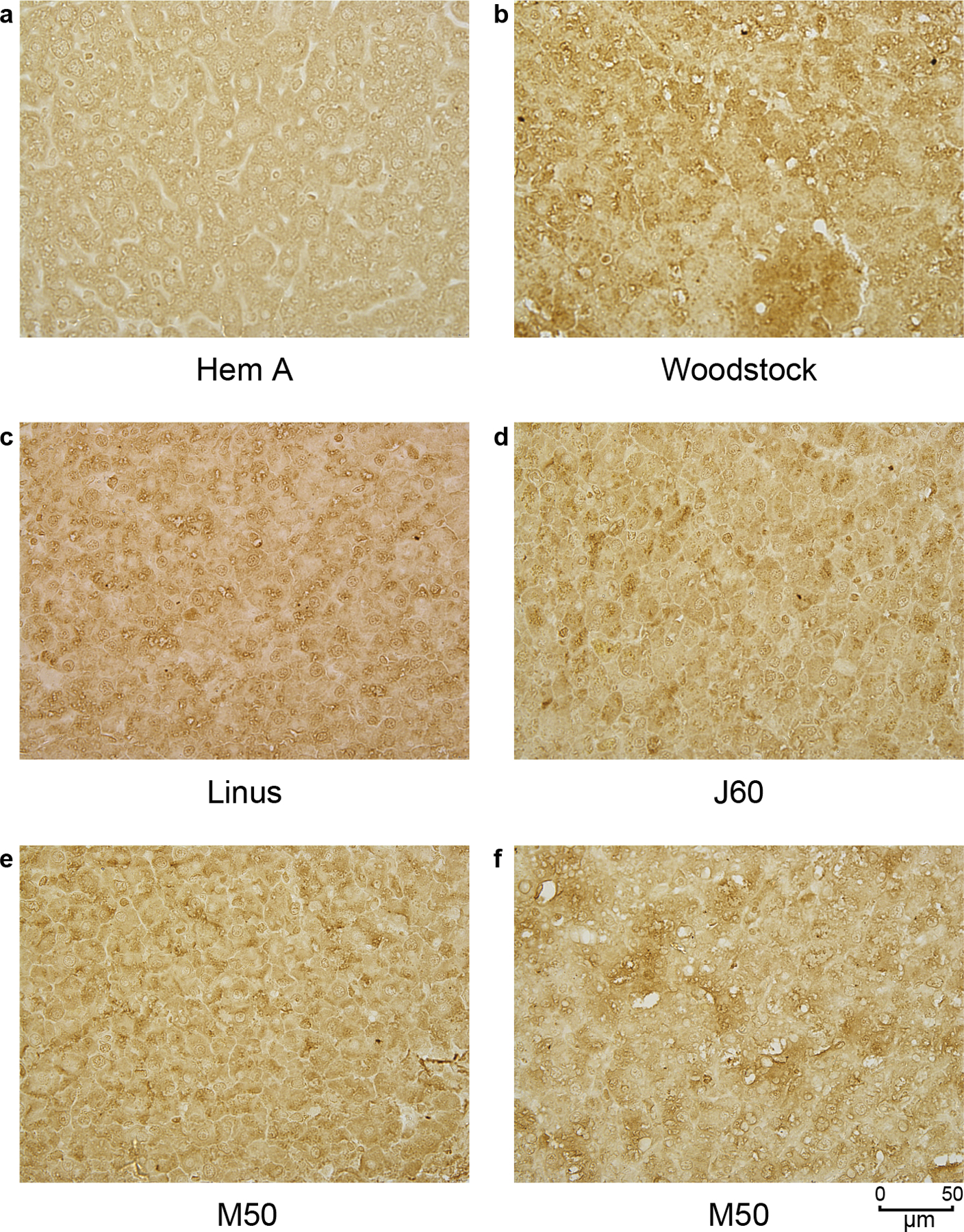
The pattern of cFVIII expression in the liver after AAV delivery was assessed using immunohistochemistry. Most of the tissue analyzed had cFVIII expression that was dispersed throughout the tissue, although some areas of the liver had what appeared to be clonal populations of cells expressing cFVIII (Extended Data File 1). No major differences were noted between the dogs.
Analysis of integration site distributions in livers of AAV-treated dogs
AAV vector genomes are expected to be present in multiple forms, with some integrated in the canine genome and some persisting as episomes. As integration can influence gene function and cell growth, we analyzed the locations of vector integration sites in the dog genome and the behavior of cell clones harboring these sites.
We analyzed 20 samples of liver tissue taken at necropsy from six AAV-treated dogs and two untreated hemophilia A dogs as controls. Liver DNA samples were selected to represent high, medium and low VCN ranges. Integration sites were recovered from genomic DNA samples by shearing DNA using sonication, ligating DNA linkers to the broken DNA ends, then carrying out nested PCR amplification using primers that bound to the ligated linker and the vector ITR sequences. (Supplementary Fig. 5). The resulting DNAs containing junctions between dog genomic DNA and vector DNA were then sequenced using the Illumina platform, and reads were mapped onto the dog genome (CanFam3).
From these data we can also estimate clonal abundance of cells harboring each unique integrated vector. For a tissue sample containing DNA with expanded clones, there will be multiple DNA chains from each expanded clone that contain an identical integration site. Each chain will usually be broken by sonication at a different location in the flanking dog DNA. As a result, there will be different locations of adaptor ligation in the cellular DNA flanking the integration site. These points of adaptor ligation can then be quantified by sequencing from both ends of amplification products, providing a measure of the number of cells sampled37.
We identified a total of 1,741 putative unique integration sites, corresponding to 3,263 inferred cells sampled (Supplementary Table 6). Statistical analysis revealed modest but significant homology between AAV and canine sequences at vector-host junctions (Supplementary Fig. 6), suggesting that annealing of complementary DNA sequences promoted integration in at least some cases. Numbers of cells sampled per integration site ranged from 1 to 130; the high values indicate extensive clonal proliferation following the integration event. These expanded clones were scarce—unique integration sites recovered from at least three cells corresponded to only 4.8% of the cell population. No integration events were identified in the Rian/Dlk/Dio region (identified as canine chromosome 8, ~68,900,000 to 69,800,000), previously associated with AAV integration and transformation in mice 14,25. The number of integration sites recovered per sample were positively correlated with VCN (Fig. 3a; p=3.0×10−4). Most ITRs that were associated with integration were truncated (Supplementary Fig. 7), as seen in previous studies13,38.
Fig. 3. Distributions of sites of AAV vector integration in the treated dogs.
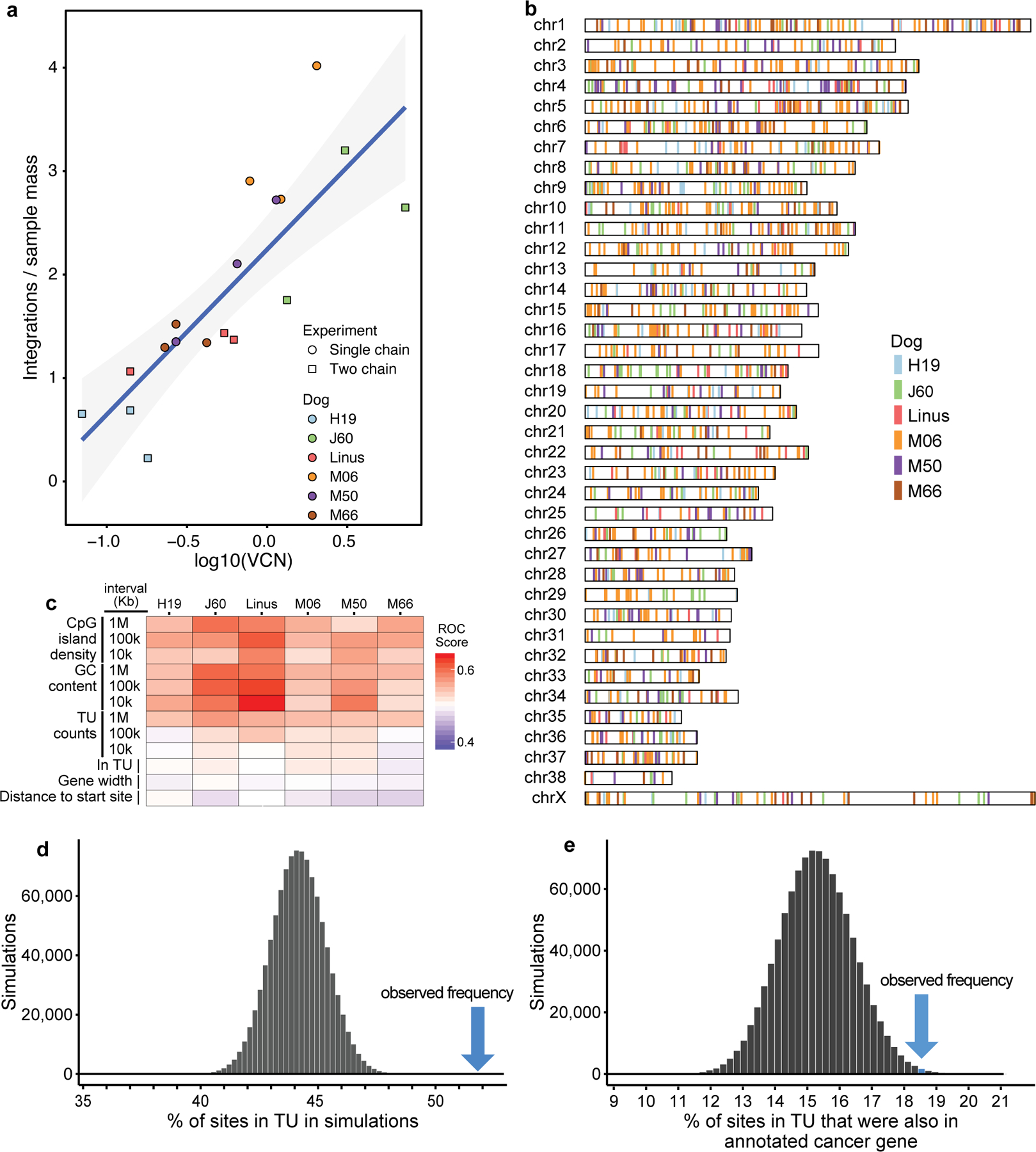
a) Correlation of the VCN value and the numbers of integration sites recovered from dog liver DNA specimens. Liver DNA samples (n=3 biologically independent samples) analyzed for each dog are shown as individual points. The gray envelope shows a 95% confidence interval. b) Distribution of AAV vector integration sites in the dog chromosomes. c) Distribution of AAV vector integration sites in the dog genome relative to genomic annotation. Associations are calculated using the ROC area method30. Values of the ROC area vary between 0 (negatively associated; blue) and 1 (positively associated; red). TU indicates transcription unit. Several length chromosomal intervals were used for comparison. d) Enrichment of integration sites in dog transcription units. The dark bars show the frequency of appearance of randomly selected sites in transcription units (TU), summarized over 106 random simulations. The blue arrow shows the observed frequency of AAV vector integration sites in TUs. e) Enrichment of integration sites within dog transcription units that are also cancer-associated genes. A list of human cancer-associated genes51 was used to annotate the dog genome.
We selected 18 integration sites for validation by targeted PCR and Sanger sequencing, focusing on expanded clones. We succeeded in validating 13 (Supplementary Fig. 5 and Supplementary Table 7). Two sites found in negative controls could not be validated, indicating probable laboratory contamination.
Global analysis of integration site distributions showed that integration events were distributed throughout the dog chromosomes (Fig. 3b). Integration was significantly favored in transcription units (Fig. 3c and d), and near CpG islands, which are associated with active transcription (Fig. 3c).
One possible contributor to cell persistence is insertional mutagenesis of genes involved in growth control. Comparison of our results to catalogs of cancer-associated genes in humans, mapped to canine homologs, showed that integration events were found modestly more frequently in cancer-associated genes than expected by chance (Fig. 3e), and gene ontology analysis disclosed modestly higher integration frequency in genes from several pathways regulating cellular growth (Supplementary Table 9).
To assess possible effects of insertional mutagenesis on cellular proliferation, we studied the vector integration sites in the most expanded clones. The most expanded clones (≥5 cells; n=54) are shown in Fig. 4a; all clones detected at least twice are in Supplementary Table 12. A challenge is that vector integration may mark a cell clone that expanded for a reason independent of insertional mutagenesis. Therefore, we looked for integrations that occurred multiple times independently in expanded clones from any of the dogs. Five genes were found at integration sites that expanded in multiple dogs (Fig. 4a): EGR2 (4 dogs), EGR3 (2 dogs), CCND1 (2 dogs), LTO1 (2 dogs) and ZNF365 (2 dogs). All these genes have been associated with transformation in humans. Two clones expanded beyond 100 cells, with vectors integrated at the genes “deleted in leukemia 2” (DLEU2) and “phosphatidylethanolamine binding protein 4” (PEBP4). In humans, DLEU2 is a commonly deleted gene in leukemia39. PEBP4 is implicated as a modulator of signaling pathways in multiple cancer cell types40,41.
Fig. 4. Clonal expansion of cells harboring AAV vectors.
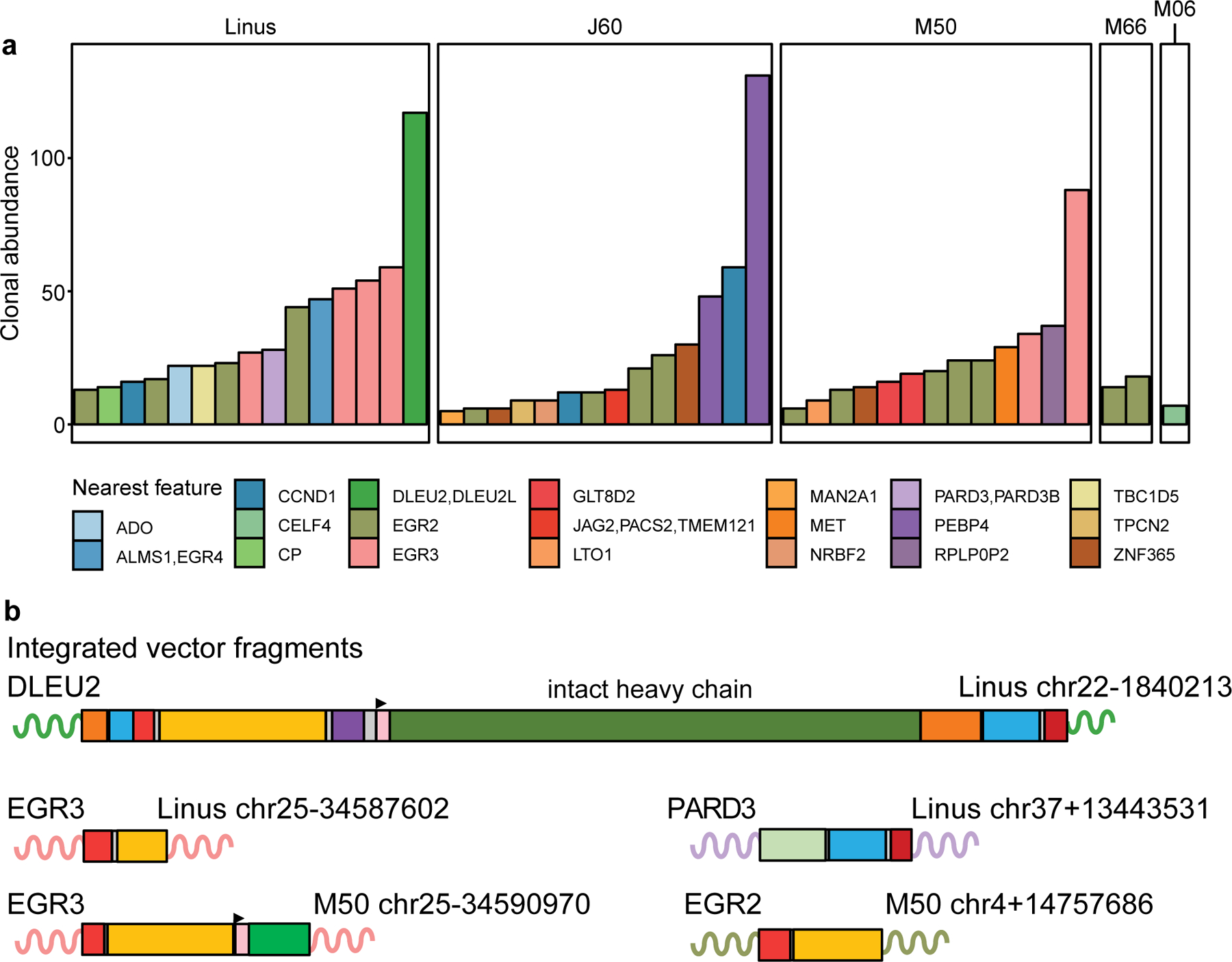
a) Summary of notable expanded clones in five of the six dogs studied. No expanded clones were detected in H19. Shown are most abundant 15 clones per dog represented by five or more cells sampled. The genes affected are shown by the color code. Data on each integration site studied is in Supplementary Table 12. b) Genetic map of vector sequences integrated in several of the dogs. The color code for vector segments as in Fig. 1a. The schematics illustrate truncations and rearrangements that may not contain the entire vector sequence. Sequences are provided (NIH SRA BioProject ID: PRJNA606282). The numbers indicate the chromosome location and coordinate of a junction between AAV vector sequences and the dog genome. The gene at the site of integration is labelled. The curved lines represent genomic DNA at the site of integration with the color matching the gene in panel a.
Clustering of integration sites in the canine genome.
Vector integration sites recovered in treated subjects can cluster on the target genome if integration at specific locations leads to increased cell growth or survival, so that at later times cells with such integration events are present at increased frequency. We used model-independent scan statistics 42 to assess possible clustering, and identified five clusters (limiting cluster sizes to less than 0.5 Megabases). These corresponded to apparent clusters at EGR2, EGR3, CCND1, ALB, and DUSP1 (Supplementary Table 10). Genetic maps of clusters at EGR2, EGR3 and CCND1 are shown in Fig. 5. All three of these genes also hosted integration events in expanded clones, providing two different indications that integration at these genes is associated with preferential cell proliferation or persistence.
Fig. 5.

Three examples of clusters of AAV vector integration sites found in the dog genome. Clusters are shown schematically at a) ADO-EGR2, b) CCND1, and c) EGR3. Transcription units are indicated by the grey rectangles. Integration sites are indicated by the shapes, with the inferred number of cells recovered harboring each site indicated by the nature of the shape (circle: 1–4 cells; square: 5–15 cells; and triangle >15 cells). The colors on the shapes indicate the dog of origin. The scales are shown by the bar at the top.
Structures of integrated vector DNAs
As the structures of AAV vector sequences in cells can be complex12,16,26, we investigated the structures of integrated vectors in expanded clones. We designed PCR primers that bound to the dog genome on either side of seven loci in expanded clones inferred to contain integrated vectors, amplified the full vector sequence, and sequenced them by Illumina sequencing, successfully validating five (Figure 4b; Supplementary Fig. 5).
The most expanded clone from Linus (integrated at DLEU2) contained an intact copy of the promoter and heavy chain portion of the factor VIII transgene (F8) (Fig. 4b). The ITRs were truncated at both ends, and in addition a portion of the polyA site was duplicated. After extensive attempts, four additional integrants were also characterized, integrated in two cases at EGR3, and one each at PARD3 and EGR2. In all these cases the vectors were greatly truncated, so that the transgenes were deleted, and the ITRs significantly shortened. Our reconstruction studies showed that intact ITRs inhibit PCR amplification, and internal repeat structures and longer concatemers are likely also recovered inefficiently, all potentially biasing the types of products recovered.
Rearrangements of the AAV vector
Our data provides additional evidence of extensive vector rearrangement. An average of 82% of integration sites showed apparent integration of AAV into F8 itself (Supplementary Fig. 5, Supplementary Fig. 8). These could be confidently ascribed to vector rearrangements (i.e. apparent integration of the vector into itself), and not integration in the genomic F8 copy, based on three observations: 1) No integration was seen in introns of the cellular F8 locus; introns are absent in the F8 cDNA in the vector (Supplementary Fig. 9 and 10). 2) The B-domain coding region of F8, which is absent in the vector but not the genomic locus, lacked apparent integration sites (Supplementary Fig. 10). 3) Numerous sequence reads were seen where F8 sequence crossed exon boundaries; such junctions are present in the F8 cDNA but not the genomic locus (Supplementary Fig. 9). This documents extensive formation of rearranged concatemeric AAV vector forms, which persisted in dog liver for the full duration of the study. For most of these concatemers, it is unknown whether they are integrated or episomal, nor whether these rearrangements occurred during vector production or in the target cells. Examples of rearranged sequences are shown in Supplementary Fig. 11. A previous report also identified joining of AAV vector sequences with each other as a prominent output from integration site analysis43
DISCUSSION
After years of preclinical studies, AAV gene therapy for hemophilia A has yielded therapeutic levels of FVIII in human patients. Although the durability and safety of the approach remain mostly unstudied in humans, long-term outcomes can be readily evaluated in dogs. We have demonstrated that expression of FVIII can persist in dogs for as long as 10 years. Unexpectedly, two of the nine treated dogs had gradual rises in FVIII expression, reaching levels that were four-fold higher than the those observed in the first four years. There were no clinical adverse events or evidence of malignancy related to AAV administration in any dog. Analysis of integration site distributions showed expansion of clones that harbored vector integration in genes potentially associated with growth control.
The clinical phenotype correlated with the levels of FVIII expression. After AAV administration, there was a >97% reduction in the ABR (mean = 0.13) even in dogs that expressed very low levels of FVIII. ABR of <1 is lower than that reported for human patients on prophylaxis32. Because gene therapy provides continuous expression of FVIII, it has the advantage that the outcome does not depend on the maintenance of trough FVIII levels and compliance to a treatment regimen. Our data confirm that even low levels of FVIII achieved by gene therapy substantially improve the disease phenotype.
We considered several hypotheses that might explain the increase in FVIII expression in two of the dogs. The increase was independent of the delivery approach (TC vs. SC), vector dose, and promoter element (Supplementary Table 4). The TC or SC vectors had different promoter elements, TBG and hAAT, respectively. The TBG promoter has been linked to HCC in mouse models14,28 while no studies have associated the hAAT promoter with HCC12,14,44. Both promoters are in clincial development4,45,46. The increase in FVIII expression was also not associated with any pathological findings, such as inflammation or biomarkers of hepatic disease.
The most plausible explanation emerged from the analysis of clonal behavior based on integration site analysis. Integration was favored in transcription units, and, in the most expanded clones, 44% were found near genes that in humans are associated with cell growth control and cancer. However, we could document only one intact transgene in our sequence data (an intact heavy chain at the DLEU2 site in Linus), suggesting a potential role for unsampled expanded clones elsewhere in the liver in rising FVIII levels.
In previous work, a 46 nt liver-specific enhancer-promoter element adjacent to the wild-type AAV2 ITR was associated with cancer in humans 22,21. In our analysis of integration sites, by chance we used the 46 nt element as the primer landing site for our AAV primer, so this sequence is inferred to be present in all the AAV sequences reported here. The 46 nt region is present in early AAV vectors47 but not in many of the current clinical AAV vectors 22.
Structures of AAV sequences in the dog livers were complex. Mapping of AAV sequences at five sites of integration yielded only one vector with an intact transgene, and even this vector was truncated and partially rearranged. Rearrangements were identified in both TC- and SC-treated dogs, indicating that the large size of the transgene in the SC construct did not make it more prone to rearrangements. Joining of vector ITRs to internal vector sequences was seen in 82% of all apparent integration sites. These events were clearly integrations into F8 in the vector and not the genomic F8 locus, because numerous reads spanned exon boundaries, the B domain (deleted in the vector) was not targeted, and no integration events were detected in F8 introns. One implication of these data is that the vector copy number as usually measured in patient samples may greatly over-estimate the number of functional copies of the encoded therapeutic gene. It will be of interest to investigate whether modification of the vector structure or the transduction procedure can increase the proportion of intact transgenes ultimately present in target cells. Another question for future investigation is whether the structure of loci containing vector DNAs changes over time after AAV infusion, for example, through iterative recombination that yields the greatly truncated vector derivatives observed at some integration sites (Fig. 4b).
This study has several limitations. The sample size is small, as is common in gene therapy studies of large vertebrates. Integration site analysis recovers some but not all integration events, so inferences are based on incomplete data. Because of the sampling scheme used, we were unable to pair integration site analysis with an analysis of expression of nearby host genes. In the integration site analysis, integration events associated with large deletions of the host chromosome could have been mis-identified as two independent integration events. For the vector-into-vector integration events, it is unknown whether the the sequences are integrated or episomal. Reconstruction studies showed that the DNA folding in the full ITR sequence interferes with efficient PCR, creating a possible recovery bias in favor of molecules with deleted ITRs. Lastly, hepatocytes can become polyploid during normal liver homeostasis, which could have been scored in our assays as low-level clonal expansion 48.
Our findings pose questions for ongoing clinical hemophilia gene therapy trials and other AAV gene therapy studies. To our knowledge, no previous study has documented a rise in FVIII expression after AAV-FVIII gene therapy in small or large animals. The FVIII levels in the dogs we studied remained within the range of a mild human hemophilia A phenotype; however, supra-physiological FVIII levels have been associated with thrombosis and would be concerning49,50. Although AAV clinical studies for hemophilia A, with three years of follow-up4, and for hemophilia B, with about 10 years of follow-up10, have not reported increases in transgene expression or vector-mediated serious adverse events, our data emphasize the importance of long term monitoring after AAV gene therapy.
Methods
AAV administration in hemophilia A dogs
The hemophilia A dogs were maintained at the University of North Carolina, Chapel Hill. All procedures in the dogs were approved by the Institutional Animal Care and Use Committee at the University of North Carolina. Vector administration and cFVIII transgene constructs were previously described. The two chain approach was delivered by the portal vein which requires exteriorization of the spleen6,52. While performing this procedure, one dog (J60) developed a small fracture that was unable to be repaired and obtain hemostasis. A decision was made to perfom a splenectomy from which the dog recovered without sequelae. J60 had a decline in FVIII expression in the absence of any rise in liver enzymes (Fig. 2a,c) or evidence of an immune response (Supplementary Figure 3). The single chain approach was delivered by intravenous infusion using a peripheral vein.
cFVIII activity, antigen and antibody assays
cFVIII activity was determined using the Chromogenix Coatest SP4 FVIII (Diapharma, Lexington, MA) using normal canine plasma as a standard. WBCT was performed as previously described6. cFVIII antigen levels were determined by ELISA as previously described6. To confirm the observation that the levels of cFVIII activity and antigen levels increased over time in Linus and M50, the activity and antigen levels were each measured in a single assay. Anti-cFVIII antibodies were detected by cFVIII-specific IgG1 and IgG2 antibodies by ELISA6. In each IgG ELISA assay, the pre-treatment baseline sample for each dog was used as a control and subtracted as the background for each dog for the duration of the study to account for any variation in the background for each assay. The Bethesda assay was used to measure anti-cFVIII neutralizing antibodies as previously described6.
Serological studies of antibody responses to the AAV vector
Heat-inactivated canine plasma samples were diluted (1:1 to 1:3155) in heat-inactivated fetal bovine serum before incubation with AAV8-Chicken-Beta-Actin-Renilla Luciferase for one hour at 37°C. Plasma samples incubated with the AAV8 vector were transferred onto human embryonic kidney cells stably expressing Ad-E4 (ATCC Number: CRL-2784) and incubated overnight at 37°C. Using the Renilla Luciferase Assay System (Promega, Madison, WI) and a luminometer (Veritas Microplate Luminometer, Turner BioSystems), the luciferase expression was assayed. The NAb titer is defined as the first dilution of the dog plasma at which there is 50% or greater inhibition of the reporter gene expression compared to the untreated HA canine plasma control sample.
Annualized bleeding rate
Naïve hemophilia A dogs (n=11) from two litters were followed prospectively for four years to obtain a detailed bleeding history for the dogs. The AAV treated dogs were monitored for bleeding events for the duration of the study. The annualized bleeding rate (ABR) was calculated based on the total number of bleeding events/number of years on the study.
Quantitative PCR for DNA copy number analysis
DNA from liver samples from different liver lobes of treated dogs was isolated using DNeasy Blood & Tissue Kit (Qiagen, Germantown, MD). When multiple liver samples from each liver lobe were available, an analysis of the vector copy number per liver lobe was performed (Supplementary Table 5). Gene copy numbers from each sample was determined using real-time quantitative polymerase chain reaction (qPCR) with TaqMan (Thermo Fisher Scientific, Waltham, MA). Primers were designed to recognize either the cF8 light chain (Forward: 5’ – AAGTGGCACAGTTACCGAGGGAAT – 3’; Reverse: 5’ – GCAACTGTTGAAGTCACAGCCCAA – 3’; Probe with a 5’ fluorescein derivative (6-FAM), Iowa Black FQ and Zen quenchers 5’ – AGTACATCCGTTTGCACCCAACCCAT −3’ or the cF8 heavy chain region (Forward: 5’ - AAGGGAGTCTGGCCAAAGAAAGGA – 3’; Reverse: 5’ - CATTGATGGTGTGCAGCTCATGCT – 3’; Probe with a 5’ fluorescein derivative (6-FAM), Iowa Black FQ and Zen quenchers: 5’ – AAATGCGTCTTTGACACAGGCTGAGG – 3’). These primers bind within the the cF8 cDNA sequence while the primers for the integration analysis bind outside of the cF8 sequence (see Integration Site Analysis; Supplementary Table 11). The level of cF8 copies was standardized against a series of dilutions of linearized pAAV plasmid containing the cFVIII-BDD transgene.
Assays to assess fibrinogen, alpha fetoprotein and vWF
Fibrinogen levels were assayed using the Canine Fibrinogen ELISA kit (ab205083, abcam, Cambridge, MA). Canine alpha fetoprotein was assayed using a Dog AFP ELISA kit (Kamiya Biomedical Company, Seattle, WA). vWF was detected by an ELISA assay. Canine vWF in citrated plasma samples was captured using Rabbit Anti-human vWF (Dako, Santa Clara 95051) and detected using the same antibodies HRP conjugated using the Lighting Link HRP kit (Novus Biologicals, LLC, Centennial CO).
Western blot analysis of LRP1
For western blot analysis of LRP1, whole cell lysate extraction was carried out from frozen liver tissue using RIPA lysis buffer (Cell Signaling Technology, Danvers, MA) with Complete Protease Inhibitor Cocktail (Roche, Mannheim, Germany). Protein concentrations were determined using Coomassie Bradford Protein Assay Kit (Thermo Scientific, Waltham, MA) and similar amount of proteins were loaded onto a NuPAGE 4–12% Bis-Tris gel (Thermo Fisher Scientific, Waltham, MA) under reducing conditions. Samples were then blotted onto a nitrocellulose membrane, and detected using Rabbit Monoclonal Anti-LRP1 antibodies (ab92544, Abcam, Cambridge, MA) and Goat Anti-rabbit IgG (H+L) IRDye 800 CW (925–32211, Li-COR, Lincoln, NE). After LRP staining, the membrane was stripped using Li-COR Stripping Buffer (Li-COR, Lincoln, NE) and stained for GAPDH as a loading control (Mouse recombinant Anti-GAPDH, 6C5, Abcam, Cambridge, MA; Goat Anti-mouse IgG (H+L) IRDye 680RD, Li-COR, Lincoln, NE). After densitometry the levels of protein for each dog was adjusted based on GAPDH and normalized to the untreated hemophilia A dog. Uncropped image of Western blot is provided (Source Data Supp Fig. 4).
Immunohistochemistry
Liver samples were fixed in formalin before paraffin-embedded. 5-μm thick slides were incubated in boiling 1mM EDTA for antigen retrieval in a microwave oven. Endogenous peroxidase was blocked using 2.13% sodium meta periodate buffer, followed by further blocking in buffer containing 5% goat serum (Cell Signaling Technology, Danvers, MA). FVIII was detected using mouse monoclonal anti-cFVIII or rabbit polyclonal anti-cFVIII (Green Mountain Antibodies, Burlington, VT) followed by biotinylated goat anti-rabbit IgG Antibody (Vector Laboratories, Burlingame, CA). Slides were incubated with VECTASTAIN ABC Kit (Vector Laboratories, Burlingame, CA) and immunohistochemical reactions were carried out using SignalStain DAB Substrate Kit (Cell Signaling Technology, Danvers, MA).
Analysis of anti-FVIII and anti-AAV antibodies
Although the dogs in this study had been exposed to plasma-derived cFVIII protein to treat bleeding episodes prior to vector delivery, they did not have any evidence of an immune response to cFVIII prior to AAV administration. Eight of the nine dogs had no evidence of anti-cFVIII antibodies throughout the study (Supplementary Fig. 2) 6.
Neutralizing antibodies (NAb) to the AAV capsid can prevent the transduction of AAV at the time of vector administration or interfere with readministration. Prior to vector administration, the anti-AAV8 Nab titers were <1:1 in all dogs except for one that showed possible low titers (1:1 to 1:3.16) (Supplementary Table 1). After AAV administration, titers rose to >1:3000 and remained between 1:1000 and 1:3000 at the terminal time points. These data indicate that the anti-AAV antibody titers persisted for years at high levels following AAV administration.
Liver Pathology
The liver was removed within minutes after euthanasia in eight of the nine dogs. The one exception was Woodstock who died from a fatal hemorrhage on the kidney. All five lobes were identified, palpated and examined for macroscopic abnormalities. All lobes were then cut into ~1 cm serial sections and the cut surfaces were inspected for abnormalities. Samples were then taken from all five lobes that were snap frozen in liquid nitrogen, frozen in Optimal Cutting Temperature (Fisher Scientific) or placed in 10% buffered formaldehyde.
The liver histopathology was assessed by analysis of hematoxylin and eosin stained liver sections (Supplementary Table 2). The veterinary pathologist reading these slides was blinded to the treatment groups. Two of the dogs had liver biopsies taken prior to AAV gene therapy (Woodstock and Linus) and one had a liver biopsy 6 years after gene therapy due to asymptomatic elevations of the liver enzymes and then was followed for an additional 4 years (Linus). As a control for the potential effect of AAV-gene therapy, liver sections from naïve hemophilia A (n=10), hemophilia B (n=9) and wild type dogs (n=9) of similar age from the same colony were also analyzed. No definitive treatment related findings were seen in any of the dogs examined. Most of the AAV-treated and control naïve dogs in this study exhibited at least one of the following findings that are compatible with age-related or background changes seen in dogs. These changes included a slight increase in connective tissue within the portal regions and around central veins, mixed inflammatory cell infiltrates in the same regions, bile duct hyperplasia, variable numbers of inflammatory cell aggregates and lipogranulomas, subcapsular post-necrotic scars and multifocal nodular hyperplasia. The connective tissue in some cases minimally extended away from the portal and centrilobular regions, without definitive evidence of bridging between these regions.
Acute hepatocellular necrosis was observed in one AAV-treated dog, H19, and mainly involved the centrilobular region. While an effect of the AAV treatment cannot be entirely ruled out, it was considered less likely given the distribution and time since the treatment administration (8.3 years). The history of severe diarrhea requiring the euthanasia of this dog likely played a role in this phenomenon (i.e. hypovolemia and/or endotoxin translocation from the gastrointestinal tract).
Although more commonly seen in association with hyperadrenocorticism (i.e. Cushing’s Disease or exogenous steroid administration), the hepatocellular swelling and clearing seen in most dogs can likely be considered as a background finding. This change may be associated with cytoplasmic glycogen accumulation and osmotic fluid intake. Alternatively, prolonged fixation of the tissues in buffered 10% formaldehyde may have contributed to these changes which would then be artifacts due to tissue processing. These findings are likely not significant given that the liver enzymes were not significantly elevated. Of the nine pathological variables assessed (Supplementary Tables 2 and 3), only hepatocellular swelling and clearing showed a significant difference between the AAV-treated and naïve hemophilia A dogs (exact Wilcoxon-Mann-Whitney test,53 p=0.004). However, the AAV-treated hemophilia A dogs were also significantly older than the naïve dogs at time of necropsy (8.9 vs. 5.5 years) and when the AAV-treated animals were compared to older naïve wild type dogs, there was no significant difference in hepatocellular swelling. Thus, further studies are needed to determine whether this pathology is associated with age or with treatment.
Of the three AAV-treated dogs having increased ALT, Woodstock and Linus exhibited slightly more swelling and clearing than other dogs, while L51 did not have any evidence of hepatocyte swelling and clearing. The latter dog presented more pronounced hemosiderin-laden macrophages, which likely represents sequelae to small hemorrhages. In contrast, dog M06 presented similar levels of hepatocyte swelling and clearing as Woodstock and Linus, without associated increased liver enzymes. A definitive histopathologic explanation for the increased ALT in these three dogs was difficult to ascertain.
Liver stained with hematoxylin and eosin from naïve (to gene therapy) hemophilia A, hemophilia B and wild type dogs were also analyzed (Supplementary Table 3). These dogs were similar in age range to the AAV-treated hemophilia A dogs in this study. The mean age of the dogs at the time of necropsy were 5.5 ± 2.2 years for the hemophilia A dogs, 5.6 ± 3.1 years for the hemophilia B dogs and 11.7 ± 2.4 years for the normal dogs. The changes seen in these naive dogs were very similar to the age-related changes seen in the AAV-treated hemophilia A dogs, both in the lesion type and degree of severity. Three naïve control dogs had elevations in their liver enzymes (alkaline phosphatase)(P19, X13, I32). The liver capsule in P19, a naïve hemophilia A dog, was regionally thickened and fibrotic and was elevated by a large accumulation of hemosiderin-laden macrophages and fibrous connective tissue. This likely represents a focus of chronic hemorrhage. Two additional naïve dogs (G35, hemophilia B; Diamond, hemophilia A) showed vascular changes that may be related to either amyloid deposition or arteriosclerosis. Three naive dogs had malignant neoplasms in their liver. Based on the hematoxylin and eosin staining, the neoplasms were diagnosed as of epithelial origin (Elton, wild type), metastatic osteosarcoma (I34, wild type) and lymphoma (O94, hemophilia A).
Clinical reasons for early termination
While six of the dogs were followed until the end of the study without clinical concerns, three dogs had clinical issues that led to the termination of the study. H19 had severe diarrhea and weight loss for unknown reasons and was euthanized 8.3 years after AAV administration. M06 had recurring rectal bleeds due to mucosal polyps and was euthanized 2.3 years after vector administration. Woodstock was found deceased 8.8 years after AAV delivery and upon autopsy the cause of death was determined to be a hemorrhage on the kidney. None of these clinical observations were related to the AAV administration.
Analysis of AAV integration sites
Six dogs were selected for AAV integration analysis: the two dogs that had an increase in FVIII expression, Linus (two chain approach) and M50 (single chain approach) and the dogs that received the same AAV dose of the two-chain vector (H19) or the single chain vector (M06) were included as a control to analyze alongside Linus and M50. In addition, one of the high dose two chain delivery dogs and one of the low dose single chain treated dogs were included for comparison (see Table 1). Integration site isolation and sequencing was carried out using ligation mediated PCR,54 under the following conditions. DNA for each sample (60–250ng) was sheared using Covaris M220 ultrasonicator under the following conditions—Peak Power: 50W, Duty Factor: 2%, Cycles: 200—for 95 seconds to achieve a fragment size of roughly 1000 bp. Two negative controls were added where molecular grade water was used instead of DNA. Bead purification of sheared DNA and linker ligation were performed as previously described54. PCR1 reaction was performed in triplicate to suppress effects of PCR jackpotting, with 25 microliters total volume per reaction, with 300 nM PCR1 linker primer, 300 nM ITR primer 1, 1X Clontech Advantage 2 PCR Buffer (Takara Bio), 200 μM dNTPs, and 1X Clontech Advantage 2 Polymerase Mix (Takara Bio). PCR1 thermocycling parameters were as follows: 1 minute initial denaturation at 95°C, then 5 linear amplification cycles of 95°C for 30 seconds, anneal at 80°C for 30 seconds, extend at 72°C for 1 minute 30 seconds, followed by 20 exponential amplification cycles of 95°C for 30 seconds, anneal at 80°C for 30 seconds, extend at 70°C 1 minute 30 seconds, followed by a final extension at 72°C for 4 minutes and an infinite hold at 4°C. Two microliters of PCR1 product were diluted into PCR2 reactions performed in triplicate, with 25 microliters total volume per reaction, with 300 nM PCR2 linker primer, 300 nM ITR primer 2, 1X Clontech Advantage 2 PCR Buffer (Takara Bio), 200 μM dNTPs, and 1X Clontech Advantage 2 Polymerase Mix (Takara Bio). PCR2 thermocycling parameters were as follows: 1 minute initial denaturation at 95°C, then 5 linear amplification cycles of 95°C for 30 seconds, anneal at 80°C for 30 seconds, extend at 72°C for 1 minute 30 seconds, followed by 15 exponential amplification cycles of 95°C for 30 seconds, anneal at 80°C for 30 seconds, extend at 70°C 1 minute 30 seconds, followed by a final extension at 72°C for 4 minutes and an infinite hold at 4°C. Finally, samples were pooled, purified, and quantified as previously described and sequenced on an Illumina MiSeq with a library loading concentration of 10 pM with a 15% PhiX spike-in.
Computational analysis of sites of AAV integration
AAV integrations were identified with the AAVenger software pipeline which identifies AAV integrations from paired-end sequencing data where sequenced fragments begin with the internal edge of integrated AAV ITR sequences and end with flanking genomic sequences. ITR sequence remnants were recorded after-which viral and adapter sequences were removed from sequencing reads before aligning to the host genome with the BLAT aligner. Aligned reads were then assembled into genomic fragments where the number of unique fragment lengths associated with integration positions were used as a lower-end estimate for clonal abundance55.
The canine genome was annotated using a sequence homology xenoRef human annotation track (https://genome.ucsc.edu/cgibin/hgTrackUi?hgsid=866058135_C33ztc5VtUOsQYEJhmgFwr4Xh4sH&c=chr26&g=xenoRefGene). This track was produced at UCSC from RNA sequence data generated by scientists worldwide and curated by the NCBI RefSeq project56,57. The RNAs were aligned against the dog genome using BLAT58; those with an alignment of less than 15% were discarded. When a single RNA aligned in multiple places, the alignment having the highest base identity was identified. Only alignments having a base identity level within 0.5% of the best and at least 25% base identity with the genomic sequence were kept.
The AAVenger software, raw sequencing data, and analysis software supporting this study is available at the Zenodo data server (DOI 10.5281/zenodo.3666122) while demultiplexed sample reads generated during the analysis are available at the NIH SRA (BioProject ID: PRJNA606282).
Numbers of AAV integration site junctions expected to be present in assay reactions.
Following recovery of integration sites using Illumina sequencing, we sought to validate several of the sites using targeted amplification and Sanger sequencing. As a first step, we calculated the expected numbers of target sequences in available samples. We approximated the number of integration site junctions from vector copy number (VCN) and relative sonic abundance data according to equation (1):
| (1), |
where 1000 canine genomes per 5.5 nanograms DNA is the size of the canine genome, copies AAV per genome is the VCN of the DNA sample, and abundance of integration per copy AAV is the relative sonic abundance of the integration site of interest (Supplementary Tables 7, 8). For each AAV ITR to genomic PCR amplification, we maximized the number of integration site junctions in the PCR template input while never using more than ten percent of sample per PCR reaction (Supplementary Tables 7, 8). For the long amplification of the high abundance integration into DLEU2, we input approximately 400 integration site junctions per PCR reaction, performing reactions in triplicate and using a total of 26% of DNA sample.
Validation of AAV integration site data
In reconstruction experiments we found that amplification across intact AAV inverted terminal repeat structures (ITRs) was inefficient, presumably due to extensive DNA secondary structure. In our sequence data, the great majority of ITRs were found to be truncated (Supplementary Fig 6). Based on previous literature14,38, we suggest that these probably provide a representative sample of the authentic population of integrants, but it remains possible that the data is biased in favor of recovery of integrants with truncated ITRs.
AAV integration results in heterogenous junctions, so that it can be challenging to distinguish between authentic AAV integration events in the dog genome, and artifactual molecules containing AAV and dog sequences generated during the PCR steps used in making sequence libraries. For this reason, we validated a set of the observed junctions using targeted PCR and Sanger sequencing.
Twenty integration sites were chosen for validation. These included high abundance sites in inferred expanded clones near cancer-associated genes, sites from less prominent clones, and two from negative control dogs that were expected to be artifacts. Primers were chosen so that one matched the expected sequence of the AAV ITR near the AAV-dog DNA junction, and the second was chosen to lie in flanking dog DNA. Of the 18 sites from AAV-treated dogs, 13 could be validated by target PCR (Supplementary Tables 7 and 8). Of the two sites from negative controls, neither could be validated (Supplementary Tables 7 and 8). Thus, we were successful in validating integration sites in expanded clones, and partially successful in validating sites in rare clones.
Primer designs are in Supplementary Table 11. We designed forward and reverse genomic primers flanking each integration site, and an additional primer to land on both the 5’ and 3’ AAV ITR regions (Supplementary Table 11). Prior to amplifying samples from livers of treated dogs we verified that dog genomic primers flanking the expected integration site, when tested in PCR reactions together, yielded dog genomic DNA products of the expected size. We then performed forty PCR reactions using the dog genomic forward primer or reverse primer paired with an AAV ITR primer binding to the A region of the ITR that is common to both ends of the genome. Dog liver DNA samples were amplified in 25 microliters total volume with 2.5 units LongAmp Taq DNA Polymerase (New England Biolabs), 1X LongAmp Taq Reaction Buffer (New England Biolabs), 300 μM dNTPs, 0.4 μM forward primer, and 0.4 μM reverse primer. PCR thermocycling parameters were as follows: 30 seconds initial denaturation at 94°C, then 35 cycles of 94°C for 30 seconds, anneal at 60°C for 30 seconds, extend at 65°C for 30 seconds, followed by infinite hold at 4°C. Each primer combination was also tested on negative control canine DNA in PCR reactions with the same chemical and thermocycling conditions in order to identify PCR products resulting from mispriming events.
PCR products were separated by electrophoresis on 1% agarose-TAE gels and visualized after EtBr staining. Significant bands in experimental or negative canine control samples were cut from gels, and DNA was extracted from the bands with a Monarch DNA Gel Extraction Kit (New England Biolabs). Concentration of extracted DNA was measured with a Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen). Extracted DNA was sequenced on a 3730XL sequencer (Applied Biosystems) in the Penn Genomics Analysis Core at the University of Pennsylvania. Fragments were sequenced from the genomic direction, or from the genomic and the AAV ITR directions when enough DNA product was available. Sequence results were tested for alignment to the canine genome using the BLAT search tool from the University of California Santa Cruz. Results of validation attempts are summarized in Supplementary Table 7. Of 20 attempts at validation, 13 validated. Two of the validation failures were apparent integration sites in untreated dogs, which we ascribe to laboratory contamination. Over all the integration site data and validation data, we identified both ends of 17 integrated AAV vectors (assuming that junctions with dog DNA are within 100 bp in the expected orientations). Failures were likely due to deletions in the vector, target site, or both15, 24,25,38.
The high abundance integration at the gene DLEU2 in Linus was further investigated by use of long-range PCR amplification. A complication is that reconstruction studies showed that amplification even from a plasmid template containing two full ITRs was undetectable. Conditions for this test were as follows: full-length FVIII AAV plasmid DNA was amplified with the two primer pairs designed to land on the plasmid, 350 nucleotides or 2 KB away from the ITR fragments. In each reaction, 10 nanograms full-length FVIII AAV plasmid were amplified in 25 microliters total volume with the following components: 2.5 units LongAmp Taq DNA Polymerase (New England Biolabs), 1X LongAmp Taq Reaction Buffer (New England Biolabs), 600 μM dNTPs, 0.4 μM forward primer, and 0.4 μM reverse primer. PCR thermocycling parameters were as follows: 4 minutes initial denaturation at 94°C, then 40 cycles of 94°C for 30 seconds, anneal at 60°C for 1 minute, extend at 65°C for 41 minutes 40 seconds, followed by a final extension at 65°C for 10 minutes and an infinite hold at 4°C. These conditions did not produce fragments of expected sizes. Further reconstruction studies under similar conditions showed that amplification could be accomplished through a single intact ITR. However, the Linus DLEU2 event is associated with deleted ITRs, so amplification was attempted across the full integration AAV vector at this locus. The forward and reverse dog genomic primers at position 1840213 in chromosome 22 were used to amplify the template DNA of interest in a PCR reaction with the following components: 2.5 units LongAmp Taq DNA Polymerase (New England Biolabs), 1X LongAmp Taq Reaction Buffer (New England Biolabs), 600 μM dNTPs, 0.4 μM forward primer, and 0.4 μM reverse primer. PCR thermocycling parameters were as follows: initial denaturation at 94°C for 30 seconds, then 30 cycles of 94°C for 30 seconds, anneal at 60°C for one minute, and extend at 65°C for 18 minutes 20 seconds, followed by a final extension at 65°C for 10 minutes and an infinite hold at 4°C. Extension time was calculated from the polymerase’s capacity to amplify one kilobase every 50 seconds, targeting a maximum amplification length of 22 kilobases.
PCR products were isolated after electrophoresis on a 1% agarose-TAE gel. Bands of length 4.9 kilobases were excised, and DNA was extracted from the bands with a Monarch DNA Gel Extraction Kit (New England Biolabs). Concentration of extracted DNA was measured with a Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen).
Samples were prepared for metagenomic sequencing using the Nextera XT DNA Library Prep Kit (Illumina), as per manufacturer guidelines. Samples were quantified by Quant-iT PicoGreen dsDNA Assay kit and pooled based on concentration. The sequencing libraries were quantified by Kapa qPCR (Kapa Biosystems) and Qubit dsDNA High Sensitivity assay (ThermoFisher Scientific), and fragment lengths were determined by Tape Station (Agilent). 8–10pM libraries were sequenced on the Illumina MiSeq with dual-indexed barcodes and 2 × 250 base read lengths.
Extended Data
Extended Data Figure 1.
Supplementary Material
Acknowledgements
We are grateful to members of the Sabatino and Bushman laboratories for help and suggestions. We acknowledge the Research Vector Core at The Children’s Hospital of Philadelphia for production of the single chain AAV vectors and the Penn Vector Core at The University of Pennsylvania for preparing the two-chain AAV vectors. We thank Megan Keiser for assistance with the immunohistochemistry and Amanda Messer for assisting with the analysis of the canine samples. We also thank Natalie Hoepp for discussions on canine liver clinical pathology. We thank Scott Sherrill-Mix for help with statistical analysis. This work was supported by National Institutes of Health grants RO1HL083017 (H.H.K.), R24HL63098 and N0175N92019D00041 (T.C.N.), RO1HL126850 (D.E.S.) and RO1AI082020, RO1AI045008 and RO1AI117950 (F.D.B). We also acknowledge support from the Penn Center for AIDS Research (P30AI045008) and the PennCHOP Microbiome Program (F.D.B.).
Footnotes
Competing Interests Statement
D.E.S. receives royalites from a licensing agreement with Spark Therapeutics. D.E.S. and G.N.N. are inventors on a patent on factor VIII and hemophilia A gene therapy.
Code Availability Statement
The AAVenger software and analysis software supporting this study is available at the Zenodo data server (DOI 10.5281/zenodo.3666122).
Data Availability Statement
The raw sequencing data supporting this study is available at the Zenodo data server (DOI 10.5281/zenodo.3666122) while demultiplexed sample reads generated during the analysis are available at the NIH SRA (BioProject ID: PRJNA606282).
REFERENCES
- 1.Rangarajan S et al. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med 377, 2519–2530 (2017). [DOI] [PubMed] [Google Scholar]
- 2.High KA et al. A Phase 1/2 Trial of Investigational Spk-8011 in Hemophilia A Demonstrates Durable Expression and Prevention of Bleeds. in (ed. Blood) 132, 487 (2018). [Google Scholar]
- 3.Nathwani AC et al. GO-8: Preliminary Results of a Phase I/II Dose Escalation Trial of Gene Therapy for Haemophilia A Using a Novel Human Factor VIII Variant. in (ed. Blood) 132, 489 (2018). [Google Scholar]
- 4.Pasi KJ et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. N Engl J Med 382, 29–40 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Jiang H et al. Multiyear therapeutic benefit of AAV serotypes 2, 6, and 8 delivering factor VIII to hemophilia A mice and dogs. Blood 108, 107–115 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Sabatino DE et al. Efficacy and safety of long-term prophylaxis in severe hemophilia A dogs following liver gene therapy using AAV vectors. Mol. Ther 19, 442–449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar R et al. Long-term efficacy of adeno-associated virus serotypes 8 and 9 in hemophilia A dogs and mice. Human Gene Therapy 17, 427–439 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Nathwani AC et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med 371, 1994–2004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nathwani AC et al. Adeno-Associated Mediated Gene Transfer for Hemophilia B: 8 Year Follow Up and Impact of Removing ‘Empty Viral Particles’ on Safety and Efficacy Gene Transfer. in (ed. Blood) 132, 491 (2018). [Google Scholar]
- 10.Nakai H et al. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. Journal of Virology 75, 6969–6976 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schotanus BA, Penning LC & Spee B Potential of regenerative medicine techniques in canine hepatology. Vet Q 33, 207–216 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Li H et al. Assessing the potential for AAV vector genotoxicity in a murine model. Blood 117, 3311–3319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakai H et al. Large-scale molecular characterization of adeno-associated virus vector integration in mouse liver. Journal of Virology 79, 3606–3614 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandler RJ et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. Journal of Clinical Investigation 125, 870–880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandler RJ, Sands MS & Venditti CP Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Human Gene Therapy 28, 314–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong L et al. Recombinant adeno-associated virus integration sites in murine liver after ornithine transcarbamylase gene correction. Human Gene Therapy 24, 520–525 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gil-Farina I et al. Recombinant AAV Integration Is Not Associated With Hepatic Genotoxicity in Nonhuman Primates and Patients. Mol. Ther 24, 1100–1105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaeppel C et al. A largely random AAV integration profile after LPLD gene therapy. Nat. Med 19, 889–891 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Nault J-C et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet 47, 1187–1193 (2015). [DOI] [PubMed] [Google Scholar]
- 20.La Bella T et al. Adeno-associated virus in the liver: natural history and consequences in tumour development. Gut 69, 737–747 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Büning H & Schmidt M Adeno-associated Vector Toxicity-To Be or Not to Be? Mol. Ther 23, 1673–1675 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Logan GJ et al. Identification of liver-specific enhancer-promoter activity in the 3’ untranslated region of the wild-type AAV2 genome. Nat. Genet 49, 1267–1273 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Bell P et al. No Evidence for Tumorigenesis of AAV Vectors in a Large-Scale Study in Mice. Molecular Therapy 12, 299–306 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Donsante A et al. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Therapy 8, 1343–1346 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Donsante A et al. AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma. Science 317, 477–477 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Walia JS et al. Long-Term Correction of Sandhoff Disease Following Intravenous Delivery of rAAV9 to Mouse Neonates. Molecular Therapy 23, 414–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosas LE et al. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol. Ther 20, 2098–2110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bell P et al. Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver. Molecular Therapy 14, 34–44 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Lozier JN et al. The Chapel Hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc. Natl. Acad. Sci. U.S.A 99, 12991–12996 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sabatino DE et al. Recombinant canine B-domain-deleted FVIII exhibits high specific activity and is safe in the canine hemophilia A model. Blood 114, 4562–4565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCormack WM et al. Helper-dependent adenoviral gene therapy mediates long-term correction of the clotting defect in the canine hemophilia A model. J Thromb Haemost 4, 1218–1225 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berntorp E, Spotts G, Patrone L & Ewenstein BM Advancing personalized care in hemophilia A: ten years’ experience with an advanced category antihemophilic factor prepared using a plasma/albumin-free method. Biologics 8, 115–127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Center SA Interpretation of liver enzymes. Vet. Clin. North Am. Small Anim. Pract 37, 297–333 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Galle PR et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int 39, 2214–2229 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Kitao S et al. Alpha-fetoprotein in serum and tumor tissues in dogs with hepatocellular carcinoma. J. Vet. Diagn. Invest 18, 291–295 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Yamada T et al. Serum alpha-fetoprotein values in dogs with various hepatic diseases. J. Vet. Med. Sci 61, 657–659 (1999). [DOI] [PubMed] [Google Scholar]
- 37.Berry CC et al. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics 28, 755–762 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang CC et al. Cellular recombination pathways and viral terminal repeat hairpin structures are sufficient for adeno-associated virus integration in vivo and in vitro. Journal of Virology 71, 9231–9247 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaidano G, Foà R & Dalla-Favera R Molecular pathogenesis of chronic lymphocytic leukemia. J. Clin. Invest 122, 3432–3438 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang RQ et al. Knockdown of PEBP4 inhibits human glioma cell growth and invasive potential via ERK1/2 signaling pathway. Mol. Carcinog 58, 135–143 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Zhang D et al. PEBP4 promoted the growth and migration of cancer cells in pancreatic ductal adenocarcinoma. Tumour Biol 37, 1699–1705 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Berry CC, Ocwieja KE, Malani N & Bushman FD Comparing DNA integration site clusters with scan statistics. Bioinformatics 30, 1493–1500 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cogné B et al. NGS library preparation may generate artifactual integration sites of AAV vectors. Nat. Med 20, 577–578 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Kao C-Y et al. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb Haemost 110, 244–256 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Nathwani AC et al. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med 371, 1994–2004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gene Therapy in Patients with Mucopolysaccharidosis Disease.
- 47.Samulski RJ, Chang LS & Shenk T A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. Journal of Virology 61, 3096–3101 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donne R, Saroul-Aïnama M, Cordier P, Celton-Morizur S & Desdouets C Polyploidy in liver development, homeostasis and disease. Nat Rev Gastroenterol Hepatol 131, 452 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Kyrle PA et al. High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med 343, 457–462 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Rietveld IM et al. High levels of coagulation factors and venous thrombosis risk: strongest association for factor VIIIand von Willebrand factor. J Thromb Haemost 17, 99–109 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Sadelain M, Papapetrou EP & Bushman FD Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 12, 51–58 (2011). [DOI] [PubMed] [Google Scholar]
Methods-only References
- 52.Sherman A et al. Portal Vein Delviery of Viral Vectors for Gene Therapy for Hemophilia in Gene Correction: Methods and Protocols. (Humana Press, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hothorn T, Hornik K, van de Wiel M & Zeileis A Implementing a Class of Permutation Tests: The coin package. Journal of Statistical Software 28, 1–23 (2008).27774042 [Google Scholar]
- 54.Sherman E et al. INSPIIRED: A Pipeline for Quantitative Analysis of Sites of New DNA Integration in Cellular Genomes. Mol Ther Methods Clin Dev 4, 39–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berry CC et al. INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions. Mol Ther Methods Clin Dev 4, 17–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pruitt KD, Tatusova T & Maglott DR NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 33, D501–4 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pruitt KD et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 42, D756–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kent WJ BLAT--the BLAST-like alignment tool. Genome Res 12, 656–664 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data supporting this study is available at the Zenodo data server (DOI 10.5281/zenodo.3666122) while demultiplexed sample reads generated during the analysis are available at the NIH SRA (BioProject ID: PRJNA606282).