

Abstract
SWI/SNF chromatin remodelers modify the position and spacing of nucleosomes and, in humans, are linked to cancer. To provide insights into the assembly and regulation of this protein family, we focused on a subcomplex of S. cerevisiae RSC comprising its ATPase (Sth1), the essential actin-related proteins (ARPs) Arp7 and Arp9, and the ARP-binding protein Rtt102. Cryo-EM and biochemical analysis of this subcomplex shows that ARP binding induces a helical conformation in the HSA domain of Sth1. Surprisingly, the ARP module is rotated 120° relative to full RSC, about a pivot point previously identified as a regulatory hub in Sth1, suggesting that large conformational changes are part of Sth1 regulation and RSC assembly. We also show that a conserved interaction between Sth1 and the nucleosome acidic patch enhances remodeling. As some cancer-associated mutations dysregulate rather than inactivate SWI/SNF remodelers, our insights into RSC complex regulation advance a mechanistic understanding of chromatin remodeling in disease states.
Introduction
The position and composition of nucleosomes throughout the genome are tightly regulated. Key to this are the so-called chromatin remodelers, which non-covalently modify nucleosome architecture using a conserved ATPase subunit1. Chromatin remodeling is involved in nearly every aspect of transcriptional regulation and must therefore be controlled in a cell-cycle, cell-type, and developmentally specific manner2. To achieve this high degree of regulation, cells have evolved multiple remodeler families, with each catalyzing distinct remodeling events, including sliding, ejection, and histone variant exchange. At the heart of each remodeler is an ATPase subunit with a conserved RecA domain, which couples ATP hydrolysis to DNA translocation. Domains flanking the core ATPase as well as accessory subunits allow each family to adapt the same biochemical RecA function to produce diverse remodelling outcomes. It is not yet fully understood how this functional diversity is achieved.
The SWI/SNF family of chromatin remodelers, which in yeast includes the eponymous SWI/SNF complex and the RSC complex, modify the position and spacing of nucleosomes at promoter regions throughout the genome and are capable of ejecting histone octamers from the DNA3,4. In yeast, RSC is a 17-subunit complex with a MW of > 1 MDa5,6 and its subunit architecture and biological function are broadly conserved from yeast to humans. Meta-genomic analysis has revealed that SWI/SNF-family genes in humans are highly mutated in cancer7–9, with mutation rates of all genes in aggregate (19%) rivaling those of TP53 (26%)9,10. Most of the mutations lie in accessory subunits, yet oncogenesis has in at least one case been shown to require an active ATPase subunit11. This suggests that that misregulation of remodeling is a hallmark of human disease.
Significant genetic and biochemical insight has been gained by focusing on the essential actin-related proteins (ARPs) Arp7 and Arp9, which are conserved in all SWI/SNF remodelers from yeast to humans12,13. The “ARP module”, which consists of Arp7, Arp9, and the fungal-specific protein Rtt102, binds to the HSA domain in Sth1, a helical segment N-terminal to the ATPase domain14–16. Binding of the ARP module lowers the intrinsic rate of ATPase turnover of Sth1, yet paradoxically increases remodeling rates, suggesting that it acts to convert ATP turnover into productive remodelling events17. Additionally, two other SWI/SNF-specific domains, the postHSA and Protrusion 1 (P1), seem to interact both physically16,18 and genetically15 with the HSA domain, indicating that they act as a regulatory hub to control ATPase turnover and remodelling17. Removal of the ARPs from the full RSC complex results in basal remodelling roughly equivalent to that of Sth1 alone, suggesting that they perform a core function in activating remodeling by RSC17. A mechanistic understanding of these regulatory interactions is still missing as the recent cryo-EM structures of the yeast RSC19–21 and SWI/SNF22 complexes and the BAF complex23, the human ortholog of SWI/SNF, did not reach high enough resolution in the ARP module or the HSA–postHSA–P1 regulatory hub. To bridge this gap, we focused on the well-characterized Sth1–Arp7–Arp9–Rtt102 RSC subcomplex (Fig.1a) that has been extensively used in biochemical studies to gain much of our current understanding of the regulation of remodeling in RSC16,17,24–26.
Fig.1 |. Cryo-EM structure of the Sth1-Arp7-Arp9-Rtt102 RSC subcomplex (RSCSAR) bound to a nucleosome.
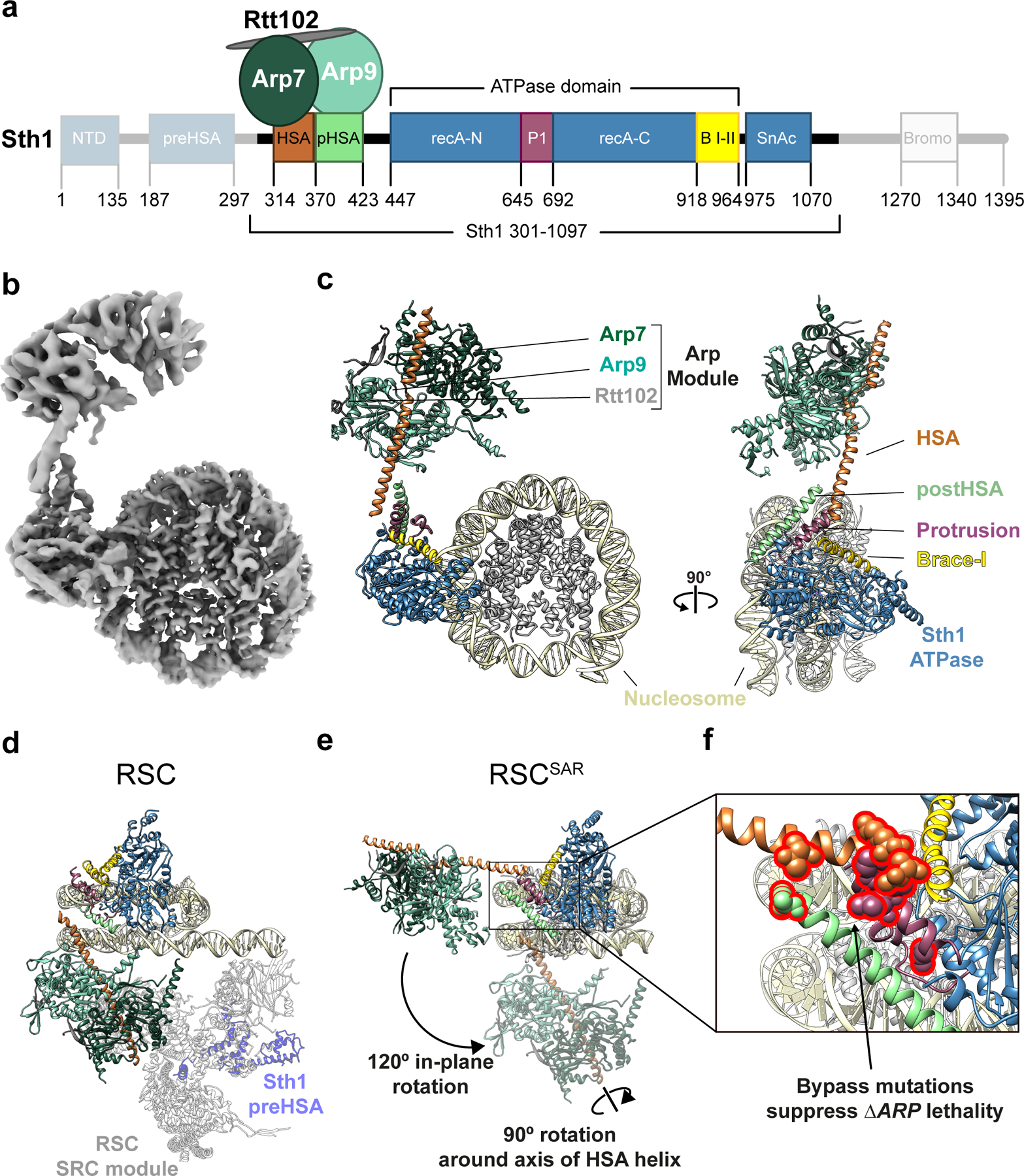
a, Schematic representation of RSCSAR, with Sth1 domains and their boundaries indicated. The same color scheme is used in all figures. b, 3.9 Å Cryo-EM map of RSCSAR bound to a nucleosome. c, Molecular model of the RSCSAR:nucleosome complex. d, e, Large rotations relate the position of the ARP module in RSCSAR and RSC. d, Structure of a RSC:nucleosome complex (PDB 6KW3) with the portion corresponding to RSCSAR shown with the same colors introduced above. The N-terminal portion of Sth1 preceding the HSA, which is absent in the Sth1 construct used in RSCSAR, is shown in dim purple. The Substrate Recognition Complex (SRC) of RSC is shown in grey. e, RSCSAR is shown with the nucleosome and Sth1 in the same orientation as that in (d). The position occupied by the ARP module in RSC (d) is shown in dim colors. The movements required to convert the ARP module from its position in RSCSAR to that in RSC—a 120° in-plane rotation and a 90° rotation about the HSA helix—are indicated. f, Close up of the regulatory hub located at the base of the HSA helix, consisting of the C-terminal end of the HSA helix, the postHSA and P1.
We present here a cryo-EM reconstruction of the Sth1–Arp7–Arp9–Rtt102 complex (which we will refer to as RSCSAR) bound to a nucleosome in the presence of the ATP analog ADP beryllium fluoride (ADP-BeF3). Using circular dichroism (CD) spectroscopy, we show that binding of the ARP module induces a folding transition of the HSA domain of Sth1 from a disordered loop into an α-helix. Our structure revealed that the folded HSA domain physically interacts with the postHSA and P1 domains, forming a pseudo-four helix bundle. Importantly, mutations that bypass ΔARP lethality in yeast cluster to this helical bundle. Surprisingly, we also observed a 120° rotation of the ARP module in RSCSAR compared to the full RSC complex, with the regulatory hub serving as its pivot point. We propose that binding of the ARP subunits creates a rigid “rod” that communicates conformational changes between the HSA–postHSA–P1 regulatory hub of the Sth1 ATPase and other subunits of the RSC complex. Our cryo-EM analysis also identified a conserved domain in SWI/SNF ATPases that interacts directly with the nucleosome acidic patch and helix 1 on histone H2B. Functional assays show that disrupting this interaction significantly reduces remodeling. Finally, our structure showed that the RSCSAR complex can peel away DNA at the nucleosomal exit site, a function previously attributed to the full complex.
Results
Cryo-EM structure of the RSCSAR subcomplex
Cryo-EM reconstructions of the RSC19–21, SWI/SNF22 and BAF23 complexes revealed, for the first time, the overall architecture of these important remodelers. However, they did not provide a mechanistic basis for how accessory subunits, in particular the ARPs, regulate the core ATPase domain in Sth1. The BAF-nucleosome structure, which shows the strongest ARP density among the different structures, has a resolution of 9–15 Å in this region23; focused refinement was used to improve the density of the ARPs and the HSA helix, but it excluded the interface of the HSA domain with the ATPase. To address how accessory subunits regulate the catalytic ATPase domain, we turned to a RSC subcomplex that shows regulation of its remodeling activity by the ARPs16,17,24–26. This complex includes a truncated form of Sth1 (AA 301–1097) and three subunits—Arp7, Arp9 and Rtt102—that constitute the “ARP module”, accounting for ~25% of the total mass of RSC (Fig. 1a). We reasoned that this simpler system might allow us to achieve higher resolution at the ATPase–ARP interface and thus shed light on the regulation of Sth1 by the ARPs. We refer to this complex throughout the manuscript as RSCSAR (SAR = Sth1 + ARPs + Rtt102).
To understand the structural consequences of ARP-module binding, we determined cryo-EM structures of RSCSAR both alone and bound to the nucleosome. We obtained a 4.2 Å structure of RSCSAR by itself, showing well-ordered density for the ARP module (Sth1HSA–Arp7–Arp9–Rtt102), but lacking density for the ATPase domain of Sth1 (Extended Data Fig. 1). We used this map to build a model of Sth1HSA–Arp7–Arp9–Rtt102 (Extended Data Fig. 1b,c) which shows differences in the HSA domain when compared to the Snf2HSA–Arp7–Arp9–Rtt102 crystal structure14 (Extended Data Fig. 1d). In the crystal structure, the Snf2 HSA domain binds to Arp7–Arp9 as a single α-helix, whereas in the cryo-EM structures HSA forms two helices connected by a short loop; the significance of this difference is unclear.
We next determined a cryo-EM structure of RSCSAR bound to the nucleosome in the presence of Mg2+ and ADP-BeF3 at a nominal resolution of 3.9 Å (Fig. 1b,c and Extended Data Figs. 2–4). Initial reconstructions showed strong density for the Sth1 ATPase domain and the nucleosome, and clear, albeit fragmented, density near the N-terminus of Sth1 (Extended Data Fig. 2). Using 3D classification, followed by multi-body refinement, we were able to improve the resolution of the map (Nucleosome: 3.7 Å; ATPase domain: 4.0 Å; ARP module: 6.5 Å) and unambiguously assign all components of the RSCSAR complex (Fig. 1b,c, Extended Data Fig. 2–4). Overall, our RSCSAR-nucleosome structure shows the ATPase bound to the nucleosome at superhelical location 2 (SHL2), consistent with previous cryo-EM structures18,27,28. Surprisingly, the location of the ARP module (Fig. 1b,c) differs dramatically from that observed in RSC, SWI/SNF, and BAF, as we discuss below (Fig. 1d,e and Extended Data Fig. 5)19–23. The C-terminal end of the HSA helix contacts the ATPase and engages the postHSA and P1 domains, as well as the Brace Helices (Fig. 1e,f and Fig. 2). The importance of these domains, which act as a regulatory hub within the ATPase domain, is also discussed below.
Fig.2 |. The structure of the HSA–postHSA–P1 regulatory hub changes in the presence of the ARPs.
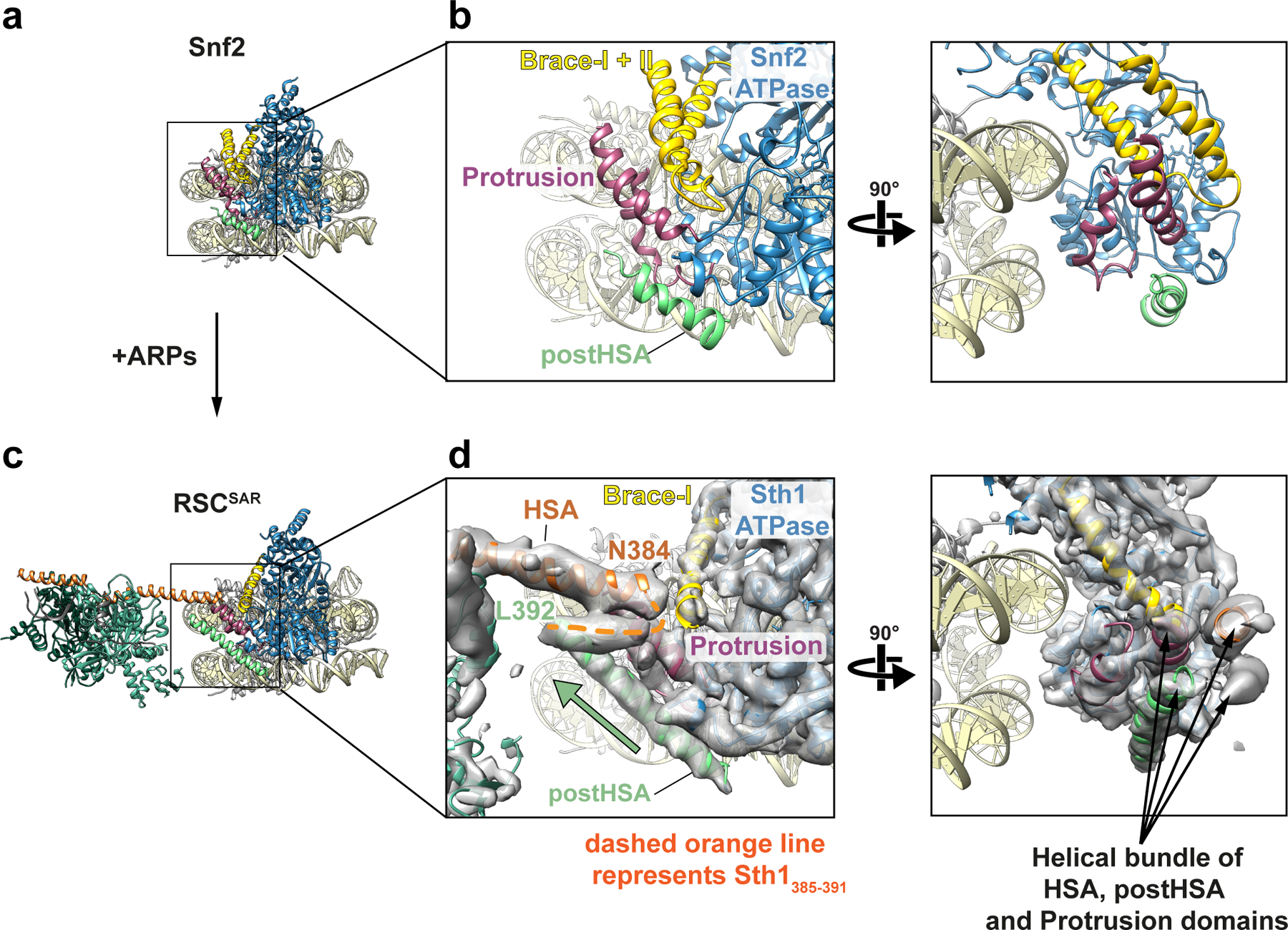
a, Cryo-EM structure of Snf2 bound to the nucleosome in the presence of ADP-BeF3 (PDB 5Z3U). Tan: nucleosome; Blue: ATPase domain; Yellow: Brace Helices; Dark pink: P1 domain; Green: postHSA domain. b, Two views showing the interaction of postHSA–P1 domains and the Brace Helices. c, Structure of RSCSAR bound to the nucleosome in the presence of ADP-BeF3 (this work). Coloring is the same as in panel (a) plus Orange: HSA domain; Dark Green: Arp7; Light Green: Arp9; Grey: Rtt102. d, Equivalent views as those in (b), showing the interaction of HSA–postHSA–P1 domains and the Brace Helices. The cryo-EM density for RSCSAR is included in this panel. A pseudo four-helix bundle is formed by the HSA–postHSA–P1 domains. The green arrow highlights the longer postHSA helix present in RSCSAR vs. Snf2 (compare (b) and (d)).
Assembly of the full RSC-nucleosome complex requires large conformational changes in the ARP module
In each of the reported structures of SWI/SNF remodelers19–23, the ATPase is bound at SHL2 on the nucleosome while the rest of the complex occupies a position distal to the nucleosome (Fig. 1d and Extended Data Fig. 5). The portion of RSC not included in our complex is named the Substrate Recognition Complex (SRC)21 and contains several DNA- and histone-binding domains that likely aid in determining RSC localization throughout the genome. The main connection between the Sth1 ATPase domain and the SRC is the ARP module, with the HSA helix acting as the bridge. Arp7–Arp9–Rtt102 bind to the HSA helix, but otherwise make no other contacts with the complex or the nucleosome (Extended Data Fig. 5b). Notably, the ARP module itself occupies very different positions in RSCSAR and the full RSC complexes: going from its orientation in RSCSAR to that in full RSC involves a 120° rotation away from the plane of the nucleosome and a 90° rotation about the axis of the HSA helix (Fig. 1d,e). This results in a ~80 Å displacement of the center of mass of the ARP module between the two structures. When engaged in the full RSC complex, the ARP module extends perpendicularly from the plane of the nucleosome and serves as a rigid connection between the ATPase (Sth1) and the SRC module, which together engage both faces of the nucleosome (Fig. 1d and Extended Data Fig. 5b).
Although we were able to build a full model of the RSCSAR complex, we did observe significant conformational heterogeneity in our dataset. Our biggest improvement in resolution came from using multi-body refinement29 (Extended Data Fig. 3), which refines user-defined portions of the model as independent bodies. This analysis showed that the ARP module undergoes conformational changes of upwards of ~70 Å in RSCSAR, with the structures representing this motion forming a cone of conformations anchored at the N-terminus of the ATPase domain of Sth1, where the HSA domain engages two structural elements of the regulatory hub: the postHSA and P1 domains (Extended Data Fig. 3d). Importantly, focused 3D classification did not identify particles where the ARP module occupied the same position as that seen in the full RSC complex. While we cannot rule out the possibility that a small subset of RSCSAR particles representing that conformation do exist in our dataset, the predominant conformation of the ARP module in the RSCSAR complex is clearly distinct from that seen in full RSC. Thus, formation of the full RSC complex likely requires a large conformational change in the ARP module during assembly, upon engagement with the nucleosome, or both.
The HSA helix interacts with a regulatory hub in the ATPase domain of Sth1
Previous data have shown that three accessory domains are essential to the function of Sth1–the HSA, postHSA, and P1 domains15. The first cryo-EM structures of Snf2 bound to the nucleosome revealed that the postHSA and P1 domains physically interact with each other (Fig. 2b). Our cryo-EM structure of RSCSAR bound to a nucleosome showed that in the presence of the ARP module, the HSA helix interacts with the postHSA and P1 domains, creating a pseudo four-helix bundle (Fig. 2d and Extended Data Fig. 4e). Interestingly, mutations that bypass the need for the ARP9 gene in yeast all cluster in this region15 and 8 out of 9 point mutants are found in ordered regions of the RSCSAR structure (Fig. 1f). Additionally, a truncation of AA 385–392 in Sth1 that was also found to suppress loss of ARP9 falls within this pseudo four-helix bundle. These residues lie in a region of Sth1 (residues 385–391) that was omitted from our model building due to the moderate resolution of this portion of the map (Fig. 2d, orange dashed line; Extended Data Fig. 4e, right panel). However, we are confident in the assignment of this region based on structural constraints from the termini of the HSA and postHSA domains, which are based on the high-resolution crystal structures18,28 used as reference for model building. To understand the consequences of ARP binding and RSC assembly on this important regulatory region, we compared cryo-EM structures of Snf2 27, RSCSAR, and RSC21 (Fig. 2 and Extended Data Fig. 6). Because all of these structures are engaged with the nucleosome and in the ADP-BeF3 nucleotide state, we expect the major differences between them to be due to the presence or absence of accessory subunits. Compared with Snf2 and RSC, we see an extension of the postHSA helix by 5 helical turns in RSCSAR (Fig. 2b,d and Extended Data Fig. 6b) and a displacement of the P1 domain by ~7 Å (Extended Data Fig. 6c). These changes are likely caused by the presence of an ordered HSA domain engaging the regulatory hub, which is only observed in RSCSAR.
In addition to the HSA–postHSA–P1 regulatory hub, SWI/SNF remodelers also contain a set of helices that link the two lobes of the ATPase, termed Brace Helix I and II28. The Brace Helices interact with the P1 domain and are also in close proximity to the ordered HSA domain in the RSCSAR structure (Fig. 2d). While Brace Helix I shows strong density in our cryo-EM map, Brace Helix II is disordered (Extended Data Fig. 4b,c). The conformation of the Brace Helices is likely intimately associated with the nucleotide state of the ATPase, as both are ordered in the Snf2-apo and Snf2-ADP structures27 (Extended Data Fig. 7). Although Brace Helix II was modeled in the Sfn2-ADP-BeF3 structure (Extended Data Figs. 4b and 7)27, there is no ordered density in the corresponding part of the cryo-EM map27 (Extended Data Fig. 7). As Brace Helix II is disordered in the Snf2-ADP-BeF3 (1 subunit) and the RSCSAR-ADP-BeF3 (4 subunits) structures, it is likely that unfolding of this domain is primarily a consequence of the nucleotide state of the ATPase domain and not the presence or absence of accessory subunits. To better understand the consequences of the nucleotide hydrolysis cycle on the conformation of the ATPase and the HSA–postHSA–P1 regulatory hub, we compared the structures of Snf2 in various nucleotide states (apo, ADP, ADP-BeF3) and RSCSAR-ADP-BeF3 (Extended Data Fig. 8). From this analysis, it is clear that the overall conformations of the ATPase in the apo and ADP states are the most similar. Binding of ADP-BeF3 causes the C-lobe to shift closer to the nucleosome and towards the N-lobe (Extended Data Fig. 8b, blue arrows), the P1 and postHSA domains towards the nucleosome (Extended Data Fig. 8b, magenta arrows), and Brace-Helix II to become disordered (Extended Data Fig. 8b, yellow arrows). Additionally, while Brace-Helix I is ordered in all structures, the angle of the helix changes by ~15° in the ADP-BeF3 state in both the Snf2 and RSCSAR structures (Extended Data Fig. 8c). Since the RSCSAR structure shows that the HSA domain interacts with the postHSA and P1 domains and Brace-Helix I, all of which change conformation throughout the nucleotide hydrolysis cycle, it suggests that the HSA domain serves as a conduit to relay the conformation of the ATPase lobes to the rest of the RSC complex, and vice versa.
Binding of the ARP module induces folding of the HSA and postHSA domains
The HSA and postHSA domains in RSCSAR showed extended helical structures compared with those seen in the cryo-EM structures of the related Snf2 enzyme18,27 (Fig. 2b,d and Extended Data Figs. 4c and 6b). Given that a major difference between our structure of RSCSAR and those of the Snf2-nucleosome complex is the presence of the ARPs, and that the ARPs have no binding partners outside the HSA domain and Rtt102 in either RSC or RSCSAR, we hypothesized that a key function of the ARPs might be to induce folding of the HSA domain into an alpha helix.
To determine if the HSA domain could adopt a helical conformation on its own, we performed Circular Dichroism (CD) spectroscopy on a peptide including the region of the HSA domain (residues 316–377) that was ordered in our cryo-EM reconstructions and in previous X-ray structures of the ARP module14,16. This analysis showed that the HSA peptide exhibits a random coil conformation in the absence of the ARP module (Fig. 3a, blue line). In contrast, comparing spectra of the ARP module with and without the HSA peptide (Fig. 3b) showed that addition of the ARP module induced a helical fold in the HSA domain (Fig. 3a, orange line).
Fig.3 |. Binding of the ARP module induces folding of the HSA helix.
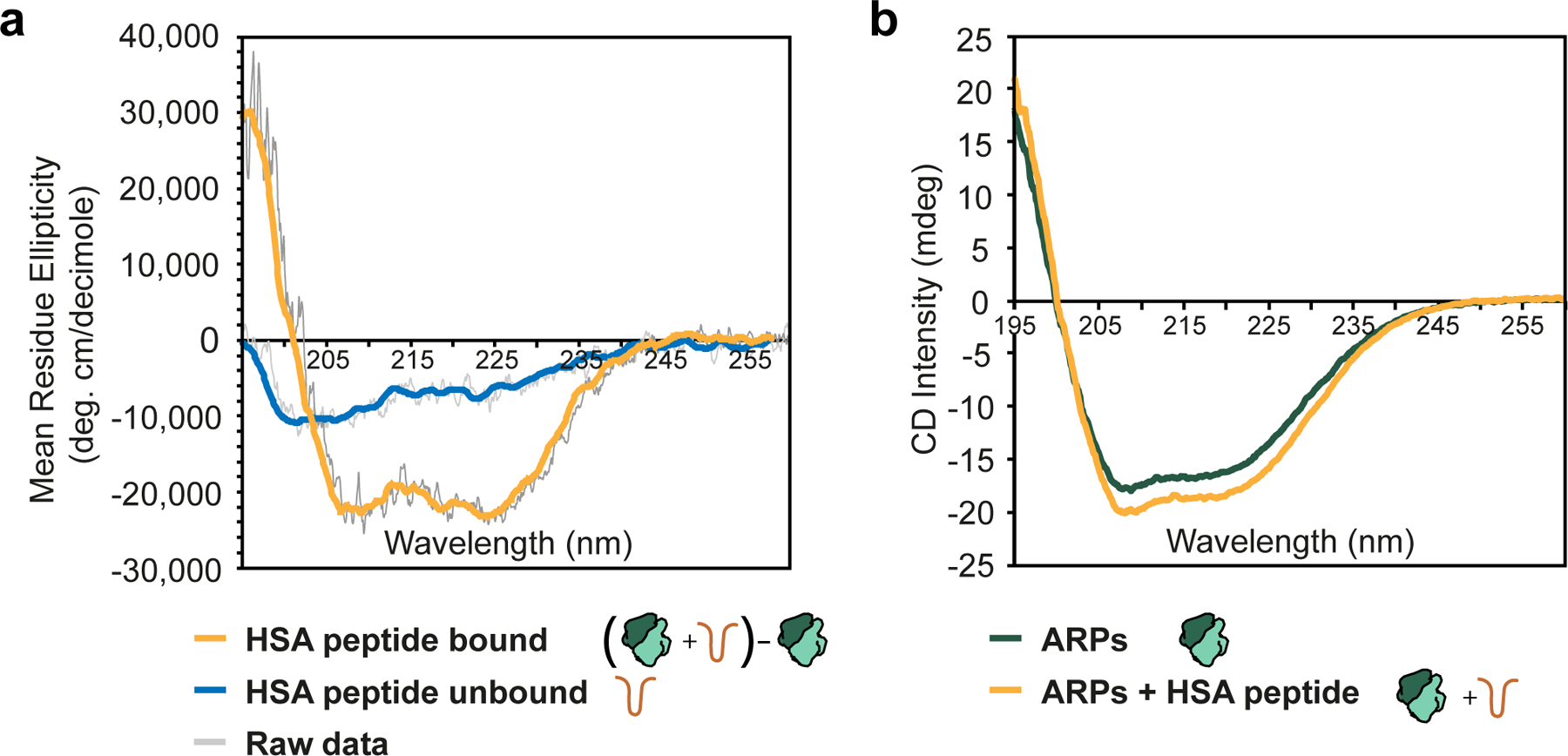
a, Molar ellipticity plot for the HSA peptide in the presence (orange) and absence (blue) of the ARP module. Raw data are shown in grey, with the colored line representing a smoothed trace. The HSA+ARPs data was calculated by subtracting the plots shown in (b) and calculating molar ellipticity based on the HSA peptide (see Methods). b, Circular dichroism plot for the ARP module (green) and the ARP module plus HSA peptide (orange). Raw, unsmoothed data are shown. All spectra were collected with identical protein concentrations and with complexes at equimolar ratios.
RSCSAR can break DNA:histone contacts at the exit site
The mechanism of step-wise DNA translocation by SNF2-family ATPase enzymes was recently elucidated using cryo-EM27. To understand how RSCSAR (4 subunits) might distort the nucleosomal DNA relative to Snf2 (1 subunit), we compared our RSCSAR structure with that of Snf2-nucleosome in the ADP-BeF3 bound state (Fig. 4, Extended Data Fig. 9). The structures reveal a nearly identical conformation of the nucleosomal DNA at SHL2 and at other superhelical sites throughout the nucleosome (Extended Data Fig. 9a). The conformation of the ATPase domain of Sth1 in the RSCSAR model is in good agreement with that of Snf2-ADP-BeF3 (Extended Data Fig. 6), except for the changes in the HSA, postHSA, and P1 domains discussed above. In contrast, we observe a large difference in the conformation of the DNA at the nucleosome exit site compared to that seen with Snf2. While all reported Snf2 cryo-EM datasets show a canonical nucleosomal DNA18,27, 26.5% of particles in our dataset have an exit site DNA peeled by approximately 40°, as was reported in structures of both RSC and SWI/SNF bound to the nucleosome20,21 (Fig. 4 and Extended Data Figs. 1 and 9). This observation suggests that peeling of the exit site DNA and breaking of multiple DNA-histone contacts is a feature of the RSCSAR complex and does not require a full complement of RSC subunits, as proposed21. Peeling appears to implicate several basic residues on the N-lobe of the ATPase that lie in close proximity to the exit site DNA (Extended Data Fig. 9c). Previous work has shown that mutation of three basic residues in the N-lobe of Snf2 reduces remodeling rates18. A comparison of the canonical and peeled DNA conformations in the RSCSAR dataset (Extended Data Fig. 9d) shows that these residues are ~19–36 Å from the exit site DNA in the unpeeled conformation but could make direct contact with the DNA in the peeled conformation; however, due to the low resolution of the Sth1-DNA contact in this cryo-EM map, the model shown in Extended Data Fig. 9 remains speculative. As mutation of these basic residues reduces remodeling, and we show that they could directly contact the DNA, it follows that peeling of the exit site DNA is likely an important component of the remodeling reaction and is mediated, at least in part, by the ATPase itself.
Fig.4 |. RSCSAR peels off nucleosomal DNA at the exit site.
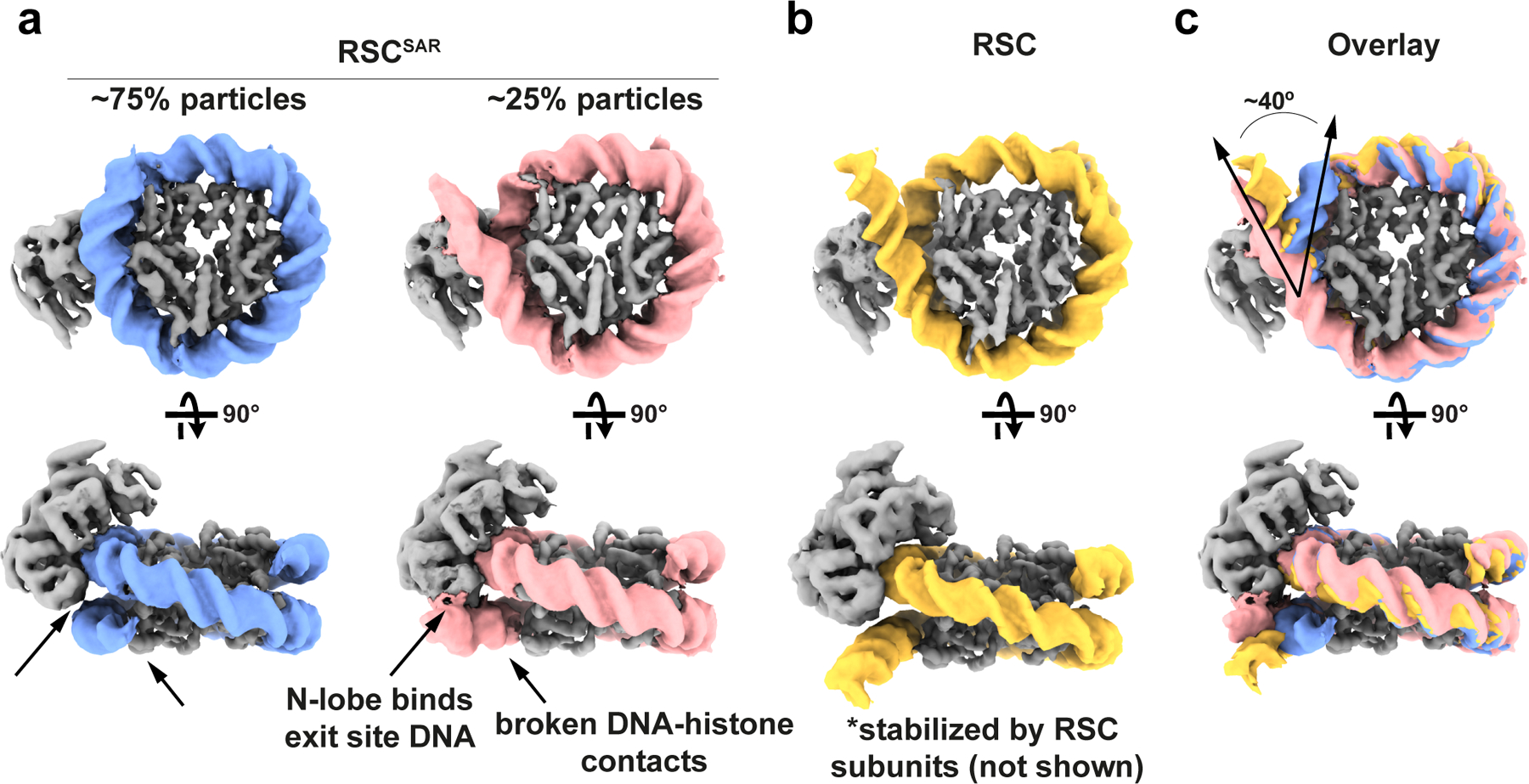
a, 3D classification of RSCSAR particles showed that ~25% have a peeled DNA conformation at the DNA exit site. Two cryo-EM maps are shown representing these populations. Blue: nucleosomal DNA with unpeeled exit site, Pink: nucleosomal DNA with peeled exit site. b, The exit site DNA in the published RSC-nucleosome cryo-EM structure showed a peeled conformation that was stabilized by accessory subunits in RSC not present in RSCSAR. This panel shows a density generated from a molecular model (PDB 6KW3); only the ATPase domain of Sth1 and the nucleosome were included to simplify the comparison with the structures in (a). Yellow: DNA from RSC-nucleosome model. c, An overlay of the three structures (RSCSAR peeled; RSCSAR unpeeled, and RSC) shows that RSCSAR is able to induce a DNA conformation at the exit site comparable to that of full RSC in the absence of the accessory subunits present in the latter. Colors equivalent to panels a and b.
Sth1 uses a conserved domain to bind the nucleosome’s acidic patch and promote remodeling
During cryo-EM processing we noticed that the C-terminal region of Sth1 extends from the ATPase domain and appears to make contact with the surface of the histone core (Fig. 5a, red dashed circle, and Extended Data Fig. 10a,b). Due to the lower resolution of this part of the map, this density is more visible in maps that are not sharpened or are filtered by local resolution (Extended Data Fig. 10a,b). This Sth1 density seems to directly interact with a region of the nucleosome known as the acidic patch, in agreement with a similar density observed in cryo-EM reconstructions of the full RSC complex20.
Fig.5 |. A basic stretch in Sth1 binds to the nucleosome acidic patch.
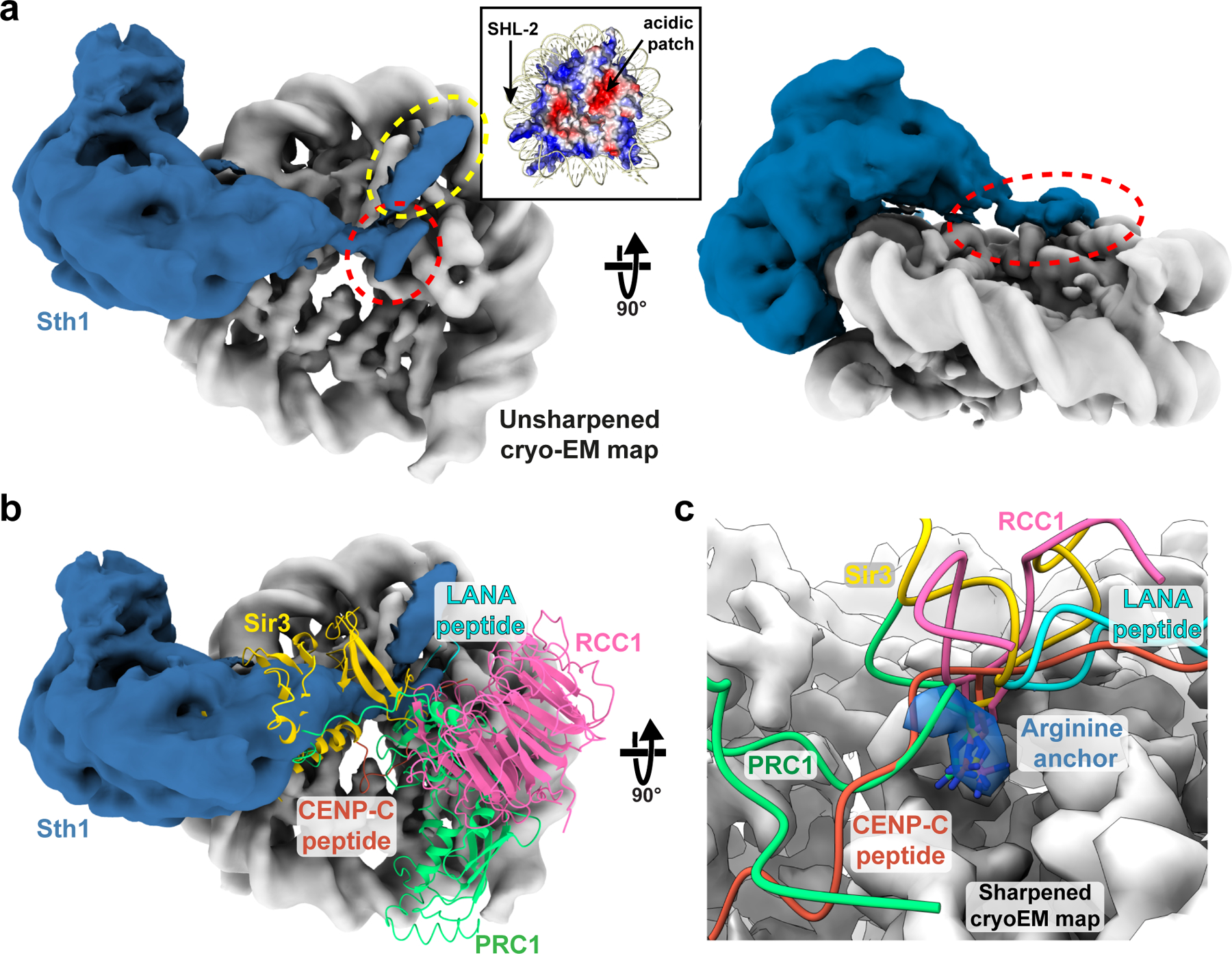
a, The RSCSAR map was manually segmented and colored grey for the nucleosome and blue for Sth1. Inset: the nucleosome is shown colored by electrostatic potential, red (acidic) and blue (basic). The region of Sth1 that sits on top of the nucleosome’s acidic patch is highlighted with a red dashed ellipsoid and a separate interaction with helix 1 of H2B is highlighted with a yellow ellipsoid. b, Same as panel (a) but with several nucleosome-binding proteins shown in ribbon representation: Sir3 (PDB 3TU4); RCC1 (PDB 3MVD); LANA peptide (PDB 1ZLA); CENP-C peptide (PDB 4INM); and PRC1 (PDB 4R8P). c, Close up of the acidic patch and the “arginine anchor” motif. Cryo-EM density from a sharpened map of RSCSAR is shown, with density for the arginine anchor colored blue.
The nucleosome acidic patch comprises 8 acidic residues found on histones H2A and H2B and is used by many proteins to interact with the nucleosome30–34 (Fig. 5b). The characteristic binding motif for the acidic patch is an arginine anchor that makes hydrogen bonds with H2A residues E61, D90, and D92 (Fig. 5c). We observed a pocket of unassigned density directly above the acidic patch that corresponds with the binding site for the arginine anchor (Fig. 5a, red circle, and 5c). Following the SnAc domain, Sth1 contains an arginine/lysine-rich area, which we will refer to as the “basic patch”, (Fig. 6a), and the distance from the last modeled residue of the SnAc domain would place this basic patch in reasonable proximity to the acidic patch (31 residues to span ~40 Å distance). Furthermore, this C-terminal basic patch (Sth1 residues 1084–1096) is conserved in SWI/SNF remodelers, but not in other remodeler families (Fig. 6a).
Fig.6 |. Mutations in a basic region of Sth1 disrupt nucleosome remodeling.
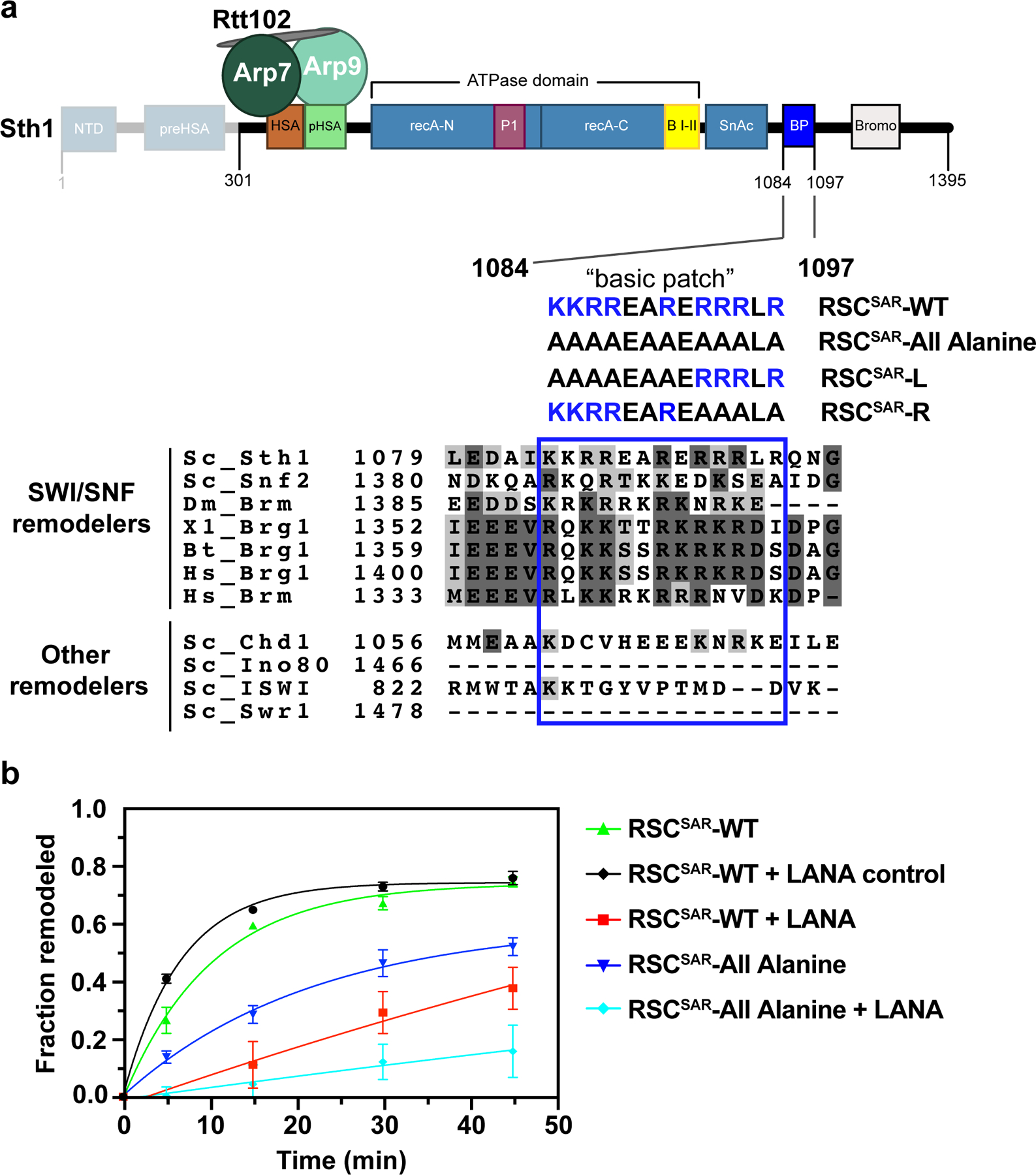
a, Schematic of the RSCSAR construct used in nucleosome remodeling assays. Sth1301–1097 was used for cryoEM analysis whereas Sth1301–1395 was used for activity assays. A “basic patch” that follows the ATPase and SnAc domains is highlighted, with a sequence alignment shown below. Sequences of Sth1 mutants used for remodeling assays are shown below the wild type sequence. b, Restriction enzyme accessibility assay of wildtype and mutant RSCSAR. The LANA peptide binds the acidic patch and has been shown to reduce remodeling rates in other systems. Error is reported as standard deviation of the mean of n=3 technical replicates and curves were fit to a one-phase association non-linear regression curve.
We hypothesized that SWI/SNF remodelers use the basic patch to promote remodeling. To test this idea, we generated a RSCSAR mutant where all arginines and lysines in the basic patch were mutated to alanine (RSCSAR-All Alanine) (Fig. 6a). Remodeling was monitored using a restriction enzyme accessibility assay. Comparison of RSCSAR-WT and RSCSAR-All Alanine remodeling rates showed that removing the basic patch reduces the rate of remodeling (Fig. 6b, green vs blue traces, and Extended Data Fig. 10c–f). To further test whether the reduction in remodeling activity was due to a compromised interaction between the nucleosome’s acidic patch and Sth1’s basic patch, we performed a competition assay using the LANA peptide from Kaposi’s sarcoma herpesvirus31, a known acidic patch binder. Both wildtype and mutant RSCSAR complexes showed a significant decrease in remodeling in the presence of the LANA peptide, but were unaffected by a LANA control in which basic residues are mutated to Ala (Fig. 6b, black, red, and cyan traces). To try and identify the most critical residues in the basic patch, we generated RSCSAR mutants where all arginines and lysines in either its first half (RSCSAR-L) or second half (RSCSAR-R) were mutated to alanine (Fig. 6a) and performed remodeling assays with them (Extended Data Fig. 10c–f). Compared with RSCSAR-WT, RSCSAR-L and RSCSAR-R both showed reduced remodeling activity (Extended Data Fig. 10c–f), with RSCSAR-L having a more modest effect and RSCSAR-R being comparable to RSCSAR-All Alanine (Extended Data Fig. 10d–f). As was the case for RSCSAR-All Alanine, RSCSAR-L and -R showed a significant decrease in remodeling in the presence of the LANA peptide (Extended Data Fig. 10e,f).
In addition to the cryo-EM density observed immediately above the acidic patch, we noticed another, stronger density along the surface of helix 1 of histone H2B (Fig. 5a, yellow dashed circle and Extended Data Fig. 10a,b). This interaction with H2B seems to be unique to SWI/SNF remodelers, as other known acidic patch interactors (LANA peptide, RCC1, Sir3, CENP-C, PRC1) do not localize to this region (Fig. 5b,c).
Discussion
In this work, we have used a well-studied RSC subcomplex to understand, mechanistically, how the essential ARP module regulates remodeling activity. We determined cryo-EM reconstructions of the RSCSAR complex, defining its overall architecture (Fig. 1) and the conformation of a regulatory hub within the ATPase domain (Fig. 2). Our structure and biochemical analysis showed that binding of the ARPs induces a helical fold in the HSA domain of Sth1 (Figs. 3 and 7a), and comparisons between our structure of RSCSAR and those of the related Snf2 ATPase (without ARPs) revealed that the folded HSA domain forms a pseudo-helical bundle with two structural elements of the regulatory hub: the postHSA domain and P1 domains (Figs. 2 and 7a). The most striking overall feature of the RSCSAR structure is the flexibility and position of the ARP module, which is rotated by ~120° in RSC about a pivot point located where the HSA helix meets the postHSA–P1 regulatory hub (Fig. 1c and Extended Data Fig. 3d). In RSC, the ARP module bridges the ATPase to the SRC, which interacts with the opposite face of the nucleosome (Figs. 1d, e, and 7b, c). Despite these large conformational differences, the ARP module appears to behave as a rigid body and the ARPs themselves do not interact with anything other than the HSA helix in RSCSAR, full RSC, or SWI/SNF. Therefore, we propose that the connection mediated by the ARP module, whose rigidity (i.e. helical nature) depends on the ARPs, is responsible for coupling the nucleotide state and conformation of the ATPase, via its regulatory hub, to the SRC.
Fig. 7 |. The ARP module: assembly and regulatory role in the RSC complex.
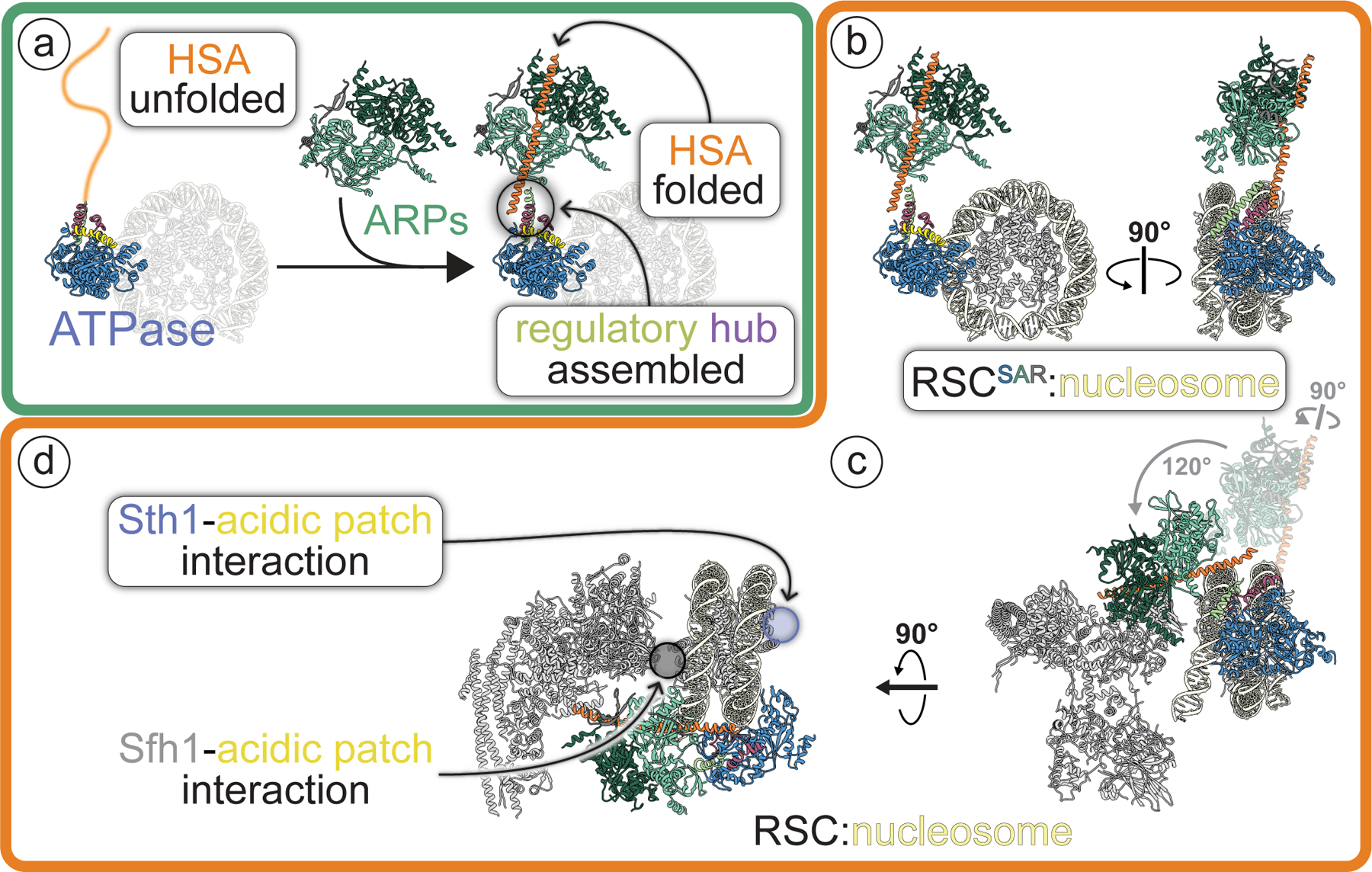
This figure places the findings of this work in the context of the full RSC complex (highlighted by rounded white rectangles). a, Arp7–Arp9–Rtt102 (“ARPs”) mediate the folding of Sth1’s HSA helix and formation of the ARP module (Arp7–Arp9–Rtt102 + Sth1HSA). In the presence of a folded HSA helix, a regulatory hub containing the postHSA and P1 segments is assembled where the HSA connects to the ATPase domain of Sth1. All subunits and motifs are colored following the schematic shown in Fig. 1a. A dimmed nucleosome is included in this panel as an orientation reference. b, The structure of RSCSAR bound to a nucleosome. c, The structure of RSC bound to a nucleosome (PDB 6KW3) shown in the same orientation as RSCSAR in (b). The ARP module from RSCSAR is shown in dim colors to highlight the different positions it adopts in RSCSAR and RSC. The rotations relating its conformation in RSCSAR to that in RSC, which have their pivot point in the regulatory hub, are indicated in grey. d, Same structure as in (c), rotated to highlight that RSC can interact with the acidic patches on both sites of the nucleosome using its Sth1 and Sfh1 subunits.
The striking difference in the position of the ARP module in RSCSAR vs. RSC may have important implications for RSC assembly. Previous studies have proposed that human SWI/SNF complexes assemble in a hierarchical fashion35, with the so-called “ATPase module”, roughly equivalent to the RSCSAR complex used in our study, operating as an independent subcomplex that becomes loaded onto the bulk of the SWI/SNF complex in the final stages of assembly. Our data suggests that not only does the assembly of SWI/SNF complexes require an ordered addition of subunits as suggested35, but that the position of the ARP regulatory module must undergo a rearrangement for the complex to reach its final, most active form (Fig. 7b, c). As SWI/SNF complexes are both conformationally and compositionally heterogeneous, there are likely additional stages along the assembly pathway. Further analysis, particularly of assembly intermediates, will be necessary to evaluate this possibility.
Although our structure of RSCSAR and those of yeast SWI/SNF and RSC showed the ARPs interacting only with the HSA helix, a cryo-EM structure of the BAF complex, the human ortholog of SWI/SNF, suggested that the ARP module might associate with the N-lobe of the ATPase23. While this may reflect organismal differences, it should be noted that the BAF structure was determined in the absence of nucleotide while the SWI/SNF and RSC and RSCSAR structures were determined in the presence of ADP-BeF3; it is therefore possible that the conformation and interaction network of the ARP module changes throughout the nucleotide hydrolysis cycle. A complete, high-resolution structural characterization of a SWI/SNF complex in all nucleotide states remains essential to fully understand the remodeling reaction.
During data processing we found a subset of particles showing a “peeled” DNA conformation at the nucleosome exit site (Fig. 4a). This conformation requires breaking of several DNA-histone contacts. Previous Snf2-nucleosome structures did not report this peeled DNA conformation18,27. A major difference between this study and previous studies is the inclusion of the ARP module in our work, suggesting that the ARP module may aid the ATPase in peeling the exit site DNA. Since other RSC subunits have also been shown to interact with the exit site DNA20,21, it is likely that multiple components of the full complex contribute to this process.
Lastly, our cryo-EM analysis of RSCSAR showed that the ATPase interacts with the surface of the nucleosome, in agreement with a recent RSC-nucleosome structure20. We identified two distinct interactions: an interaction along the H2B surface (Fig. 5a) and a highly basic region of Sth1 that interacts with the nucleosome’s acidic patch (Fig. 7d). Based on sequence constraints, we propose that a conserved stretch of basic residues C-terminal to the ATPase domain directly binds to the acidic patch (Fig. 6a). Unfortunately, the resolution of our cryo-EM reconstruction was insufficient to build a molecular model of this interaction, although we do observe clear density for a single residue corresponding to an “Arginine Anchor”, a well-characterized motif that engages the acidic patch. Since the nucleosome has the highest resolution in the map, yet the basic patch is not well resolved, this difference may be due to non-specific interaction of the Sth1 basic patch with the nucleosome acidic patch. It is likely that more than one of the 9 basic residues in the Sth1 basic patch can mediate binding. Further analysis is required to better understand the molecular details of this interaction.
It is becoming increasingly clear that the acidic patch is a near universal feature of nucleosome engagement, and several remodeler families have been previously shown to bind this motif. Consistent with this work, the human ortholog of Sth1, Brg1, was shown to have decreased remodeling of nucleosomes carrying acidic patch mutations36. CHD1 binds the acidic patch37, but lacks the C-terminal basic motif identified in this work, suggesting that it uses a different mechanism. ISWI also contains a small basic stretch C-terminal to the ATPase domain that mediates binding to the acidic patch38. This motif seems to have a strong interplay with accessory domains in ISWI that do not exist in SWI/SNF remodelers, so further analysis of the acidic patch interaction in the context of each remodeler is necessary to understand if and how it contributes to the functional diversity of remodelers. Several studies have reported that SWI/SNF remodelers engage the acidic patch on the opposite face of the nucleosome using their Sfh1 (RSC) or Snf5 (SWI/SNF) subunit (Fig. 7e)19–21,23. This interaction is vital to the function of the enzyme, as mutations in a conserved basic domain in SMARCB1 (Snf5 homolog) disrupt binding to the nucleosome, reduce remodeling rates, and have defects in cell-type differentiation39. It therefore appears that SWI/SNF remodelers are able to simultaneously engage the acidic patch on both faces of the nucleosome (Fig. 7e). This two-sided engagement with both acidic patches would allow SWI/SNF remodelers to maintain hold of the histone octamer as it threads DNA around the nucleosome.
Methods
Sample preparation and biochemical analysis
Protein expression and purification
Sth1 constructs were cloned as described16. Sth1 constructs were expressed in BL21 RIPL cells and grown in TB supplemented with kanamycin and chloramphenicol until reaching an OD600 of ~0.8. Cells were induced with 0.5 m M isopropyl β-D-1-thiogalactopyranoside (IPTG) at 16 °C and grown overnight, followed by centrifugation. Cell pellets were resuspended in lysis buffer (20 mM Tris pH 8, 300 mM NaCl, 2 mM imidazole, 1 mM DTT, and 10% glycerol) supplemented with protease inhibitors and DNase, lysed by sonication, and clarified by centrifugation. Lysates were applied to tandem 1 mL HiTrap FF columns (Ge Healthcare) charged with Ni2+, and washed with 20 mM imidazole. Proteins were eluted with a gradient of 20–250 mM imidazole. Proteins were diluted to 75 mM NaCl and loaded onto a MonoQ 5/50 GL (GE Healthcare) equilibrated in QO buffer (20 mM Tris pH 8, 50 mM NaCl, 1 mM DTT, 5% glycerol). Proteins were eluted with a 50–600 mM gradient, and then run over a final S200 10/300 Increase (Ge Healthcare) size exclusion column equilibrated in 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM DTT, and 5 % glycerol. For assembly of ATPase module complexes, Sth1 was mixed with Arp7/Arp9/Rtt102 in a 1:2 ratio and incubated on ice for a minimum of 30 minutes prior to use.
Xenopus laevis histones were expressed in BL21-Rosetta cells and purified from inclusion bodies, essentially as described in40. DNA containing the Widom 601 positioning sequence was purified using restriction enzyme digestion of a plasmid containing 16 copies of the 601 sequence36. Nucleosomes were assembled using the salt-gradient dialysis method, essentially as described40.
GraFix crosslinking
Samples were crosslinked essentially as described41. Briefly, nucleosomes (185 bp DNA, X. laevis histones) and yeast RSCSAR (Sth1301–1097, Arp7, Arp9, Rtt102) were combined at a final concentration of 3 μM and 9 μM, respectively, in a base buffer containing 20 mM HEPES, pH 7.5, 50 mM KOAc, 3 mM MgCl2, 1 mM ADP, 1 mM BeF3. Glycerol gradients were made by layering 10 mL of light solution (base buffer + 10% glycerol) on top of 2 mL of heavy solution (base buffer + 30% glycerol + 0.10% glutaraldehyde) and mixing the solution using a BioComp Gradient Master 107 set to rotate for 90 seconds with an 83° tilt. Gradients were allowed to settle at 4°C for at least 1 hour before centrifugation. 100 μL of protein solution was layered on top of the glycerol gradient and spun using an SW-60 rotor at 100,000 g for 18 hours at 4°C. Samples were fractionated and analyzed using native PAGE. Fractions containing cross-linked RSCSAR-nucleosome samples were pooled, dialyzed in base buffer to remove glycerol, and concentrated to ~5 μM.
Remodeling assays
In a 40 μL reaction, proteins at 10 nM were incubated for 5 minutes with 20 nM 225 bp nucleosomes (EpiCypher) at 30 °C in 20 mM Tris pH 7.5, 50 mM potassium acetate, 2 mM MgCl2, 0.1 mg/mL BSA, 1 mM DTT, 2 units MfeI, and 2 units PmlI. The remodeling reaction was started with the addition of 1 mM ATP. 5 uL aliquots were removed at each time point and added to 2 uL 5 mg/mL Proteinase K (NEB) in 5% SDS, and incubated at 50°C for 30 minutes. Samples were analyzed in quadruplicate on 8–16% TGX gels (BioRad) that had previously been equilibrated in 0.5X TBE for 30 minutes at 150V. Gels were stained with Sypro ruby (Thermofisher) and quantified by densitometry using the program ImageLab (BioRad). Each sample, the 225 bp DNA band was quantified and normalized relative to the no-protein control to obtain the fraction of remodeled nucleosome. Unedited gels are included in Supplementary Figure 1.
Cryo-EM structure determination and model building
Sample preparation and data collection
All grids were made following the same general protocol. UltrAuFoil R1.2/1.3 300 mesh grids (Quantifoil GmbH) grids were glow-discharged for 30 seconds at 25 mAmp and used within 15 minutes. 4 μl of sample was applied to a charged grid and blotted and plunge-frozen in nitrogen-cooled liquid ethane using a Vitrobot Mark IV robot (Thermo Fisher) set to blot force 20, blot time 4 s, 100% humidity, 4° C. Grids were made using 1 μM - 5 μM sample, with concentrations increased 2–5 fold when supplemented with 0.05% n-Octyl-β-D-Glucopyranoside (β-OG).
Data were collected using a Talos Arctica 200 keV TEM (Thermo Fisher) operating in nanoprobe mode and equipped with a K2 Summit Direct Detector (Gatan) operating in counting mode. For the RSCSAR apo sample (Arp module), a Volta Phase Plate was used. The VPP was advanced every 30 minutes to ensure phase shifts < 135°. Magnification was set to 36,000 for a final pixel size of 1.16 Å and defocus was set to −0.6 μm to −2.5 μm. Exposure rate was 5–7 e−/pixel/s, depending on dataset, with 200 ms frames, and a total exposure of 50–55 e−/ Å2. New camera gain references were collected before each dataset and the hardware dark reference was updated at least every 24 hours. Parallel illumination of the microscope was performed according to42. Holes containing thin ice were manually queued for automatic data collection using the Leginon software suite. Data were processed on-the-fly using the Appion software suite43 and each micrograph was visually inspected. Subpar micrographs (ethane contamination, crystalline ice, empty holes, etc.) were manually excluded from the dataset.
Arp module cryo-EM structure determination
RSCSAR was purified by recombinant expression in E. coli (see above). Grids were prepared as described, with a range of protein and detergent concentrations. A total of 5 datasets were combined and processed as a single dataset. All datasets were collected with similar exposure rates, total exposure, frame rate, and magnification, and processed as a single dataset. A total of 5365 movies were collected. MRC stacks were compressed to TIF using the mrc2tif script in IMOD44. Gain correction, motion correction, dose weighting, and CTF estimation were performed using MotionCor245 and CTFFIND446 in Relion-347. Micrographs with defocus values > −2.5 μm and CTF fits worse than 5 Å were removed from the dataset. Particles were picked from motion-corrected micrographs using crYOLO48. A total of 1,986,341 particles were picked and subjected to multiple rounds of 2D classification. An initial model was determined using cryoSPARC49, yielding a structure that contains clear density for the Arp module (Sth1 HSA helix, full-length Arp7/Arp9/Rtt102). The Sth1 ATPase domain is disordered in our map, and extensive effort to find a well-ordered population did not yield a structure. 3D classification in Relion-3 was used to find a subset of 415,957 particles that classified into models with well-defined secondary structure. 3D refinement yielded a structure with a GSFSC resolution of ~4.6 Å resolution (unmasked). The resolution improved to ~4.2 Å after masking and post-processing in Relion-3. The unsharpened, sharpened, and half maps were deposited in the EM Data Bank as EMD-21489.
Arp module model building and validation
Our cryo-EM map shows clear density for the RSC Arp module, including the Arp7/Arp9/Rtt102 heterotrimer, and the HSA helix of Sth1. To build a molecular model of the RSC Arp module, the crystal structure of Snf2HSA/Arp7/Arp9/Rtt102 (PDB 4I6M14) was docked into the map. The Snf2 HSA helix was mutated to Sth1 identity and numbering and manually rebuilt in certain regions using Coot50. After docking, we realized that there was density in the ATP-binding pocket of Arp7, despite no ATP being included in our buffers (Extended Data Fig. 1e). This site is enzymatically dead and the ligand is likely from the recombinant expression. To model ATP in this binding site, the ATP ligand from the Arp7ATP/Arp9/Rtt102 crystal structure (PDB 5TGC16) was docked into the map. To refine our model, we used a cloud-based Rosetta pipeline51. Initially, ~1000 models were generated using RosettaCM52. The top 10% based on Rosetta energy score were scored using MolProbity53. The ten best models based on MolProbity score were refined using Rosetta Relax, which improved the MolProbity and Clashscore for all models. A final model consisting of 10 models was deposited as 6VZG.
RSCSAR-nucleosome cryoEM structure determination
RSCSAR -nucleosome was purified and crosslinked as described above. Multiple data sets were collected on grids with various protein and detergent concentrations. All datasets were collected with similar exposure rates, total exposure, frame rate, and magnification, and processed as a single dataset. A total of 9292 movies were collected. MRC stacks were compressed to TIF using the mrc2tif script in IMOD 44. Gain correction, motion correction, dose weighting, and CTF estimation were performed using MotionCor245 and CTFFIND446 in Relion-347. Micrographs with defocus values > −2.5 μm and CTF fits worse than 4 Å were removed from the dataset. Particles were picked from motion-corrected micrographs using crYOLO48. A total of 2,020,734 particles were picked and initial 2D classification showed structures that were clearly nucleosomes with density attached to SHL2 and SHL-2. An initial model was determined using cryoSPARC49, yielding a 3D structure with clear density for the nucleosome and either one or two copies of Sth1. Additional fragmented density was seen at the N-terminus of Sth1. Instead of using 2D classification to ‘clean’ our dataset, we used multiple rounds of 3D classification to isolate particles that partitioned into clear nucleosome-bound structures. This yielded a ‘clean’ dataset of 747,408 particles.
Refinement of the ‘clean’ dataset yielded a reconstruction at 3.7 Å resolution after masking and post-processing. The full dataset clearly has a mixture of particles with either 1 or 2 copies of Sth1 bound, yielding a structure with full occupancy at SHL2 and partial occupancy at SHL-2. To improve the density of the Arp module, which shows fragmented density in this reconstruction, we used 3D classification. A total of 293,940 particles partition into 3D classes with improved Arp density, and refine to a final resolution of ~3.9 Å after masking and post-processing. To further improve the Arp density, multi-body refinement29 was performed using masks for 3 bodies (nucleosome, Sth1, Arp module). Masks were partially overlapping and included > 100 kDa of mass for each body. This dramatically improved the density of the Arp module, and yielded moderate improvements in resolution for the nucleosome and Sth1 ATPase domain. Unsharpened, sharpened, and half-maps for the consensus refinement and the three bodies of the multi-body refinement were deposited in the EMDB as EMD-21484.
RSCSAR -nucleosome model building and validation
After multi-body refinement, our cryo-EM map shows clear density for the nucleosome, the ATPase domain of Sth1 bound to SHL2, and the Arp module bound to the HSA domain of Sth1. To build the molecular model of RSCSAR bound to the nucleosome, we first docked in the model of a related structure, yeast Snf2 bound to the nucleosome in the presence of Mg-ADP-BeF3 (PDB 5Z3U27). The Snf2 model was mutated to Sth1 residue number and sequence identity in Coot. In addition to the ATPase domain, clear density is seen for the HSA domain and the two lobes of the Arp module, allowing for un-ambiguous docking of our RSC Arp module structure (Sth1 HSA, Arp7/Arp9/Rtt102) into the map. After rigid-body docking these two models, the main structural differences were in the HSA, postHSA, and protrusion domains of Sth1. Notably, the HSA and postHSA domains are more ordered in our map compared to the starting models. The helices were extended with ideal geometry in Coot. However, at this point the termini of these two domains were incompatible (the HSA domain ended at residue 384 and the postHSA domain began at 381). To relieve this clash, we instead modeled the postHSA and protrusion domains from another Snf2 structure into our map (PDB 5HZR). This yielded a postHSA conformation with much better agreement relative to the position of the HSA domain. To mitigate bias in our modeling, we created multiple hybrid models using a variety of conformations of the postHSA and protrusion domains of 5HZR. The position of the postHSA and protrusion domains can vary significantly depending on how the automatic or manual docking is performed, so we made a variety of models and used these as inputs for RosettaCM52. Approximately 1000 models were generated and ranked using Rosetta energy score54 and MolProbity score53. The top ten models from this analysis had good agreement in the postHSA domain. The nucleosome was refined using phenix.real_space_refine. The top ten Sth1 models were combined with the real space refined nucleosome model and further refined using Rosetta Relax, which improved geometry and clashscores for all models. The nucleosome DNA was replaced with the refined coordinates from phenix.real_space_refine55. The Arp module was not refined in the map, but rather rigid-body docked and slight modifications to Sth1 residues 362–383 were made in Coot50. Lastly, B-factors were refined in Phenix56. Residues 385–391 of Sth1 were not modeled despite density of moderate resolution connecting the HSA domain (residues 316–384) and the postHSA domain (residues 392–424). A final model consisting of 10 models was deposited in the PDB as 6VZ4.
RSCSAR-nucleosome “peeled DNA exit site” cryoEM structure determination
During processing we noticed that 3D classification often showed heterogeneity in the DNA exit site of the nucleosome. To better classify this heterogeneity, we used signal subtraction and 3D classification without alignment to sort the 293,940 particles from our consensus refinement based on the conformation of the DNA exit site (Extended Data Fig. 1). We found the ~25% of particles segregated into classes that showed a “peeled” DNA conformation at the exit site. A subset of these particles, corresponding to two 3D classes containing 112,364 particles, were re-centered and re-extracted and subjected to gold-standard 3D refinement in Relion3. This yielded a final reconstruction with a resolution of 4.3 Å after masking and post-processing. The unsharpened, sharpened, and half-maps were deposited in the EMDB as EMD-21493.
Reporting Summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1. Cryo-EM structure determination of the Sth1-Arp7-Arp9-Rtt102 RSC subcomplex (RSCSAR).
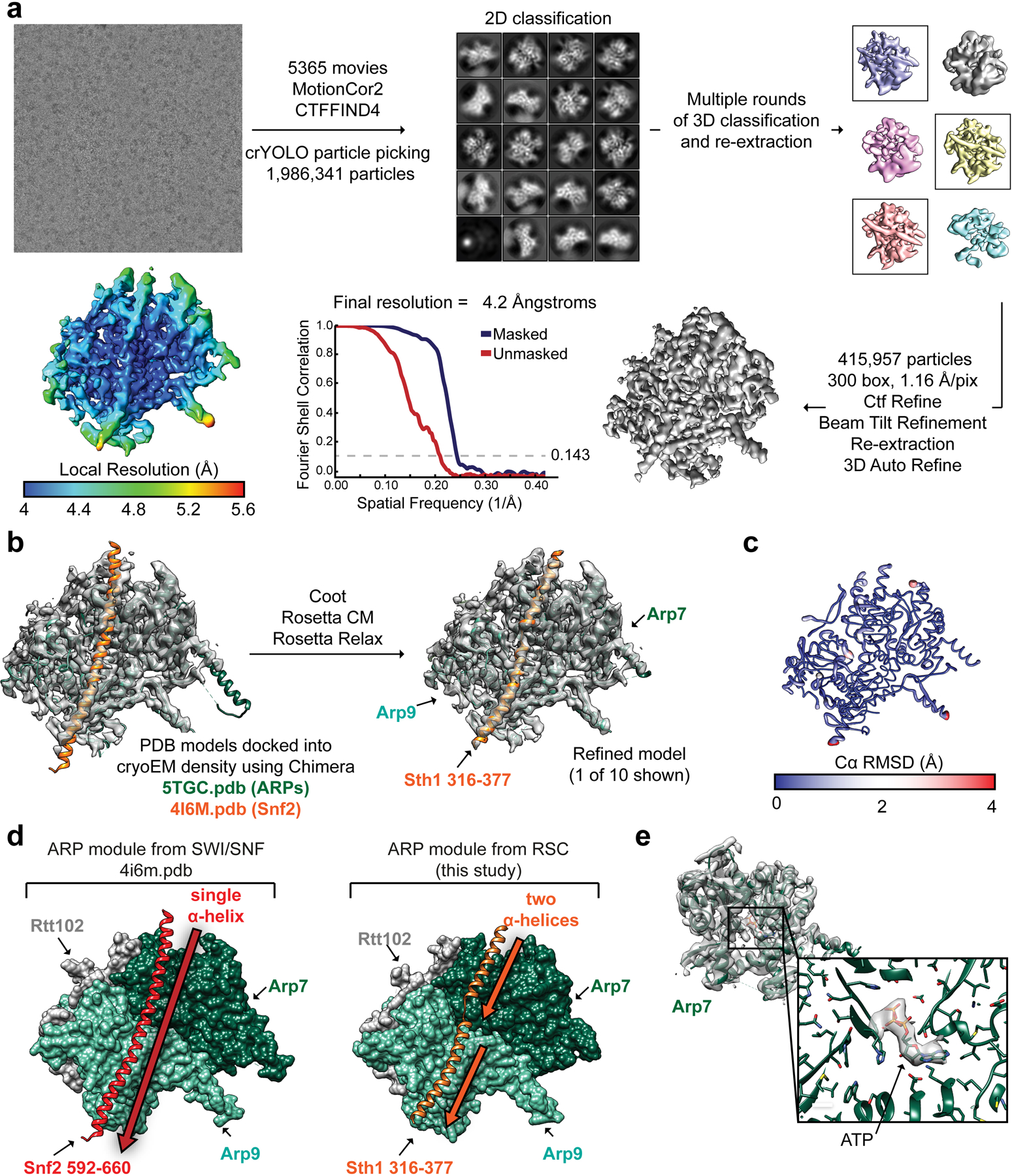
a, Workflow for cryo-EM data acquisition and structure determination. Our processing only revealed density for the ARP module. b, Schematic for model building using Coot and Rosetta. Chains from PDB 5TGC and PDB 4I6M were used as a starting model. c, The top 10 Rosetta models were deposited as PDB 6VZG and the top model is shown with its ribbon thickness and color indicating the RMSD among all 10 models. d, The ARP module form SWI/SNF (left) has a single, unbroken α-helix for the HSA domain while that of RSC (right) comprises two α-helices separated by a loop. e, Our cryo-EM has density for a known ATP binding site in Arp7. This site is catalytically dead and the ATP is likely from endogenous ATP pools.
Extended Data Fig. 2. Cryo-EM structure determination of RSCSAR bound to the nucleosome.
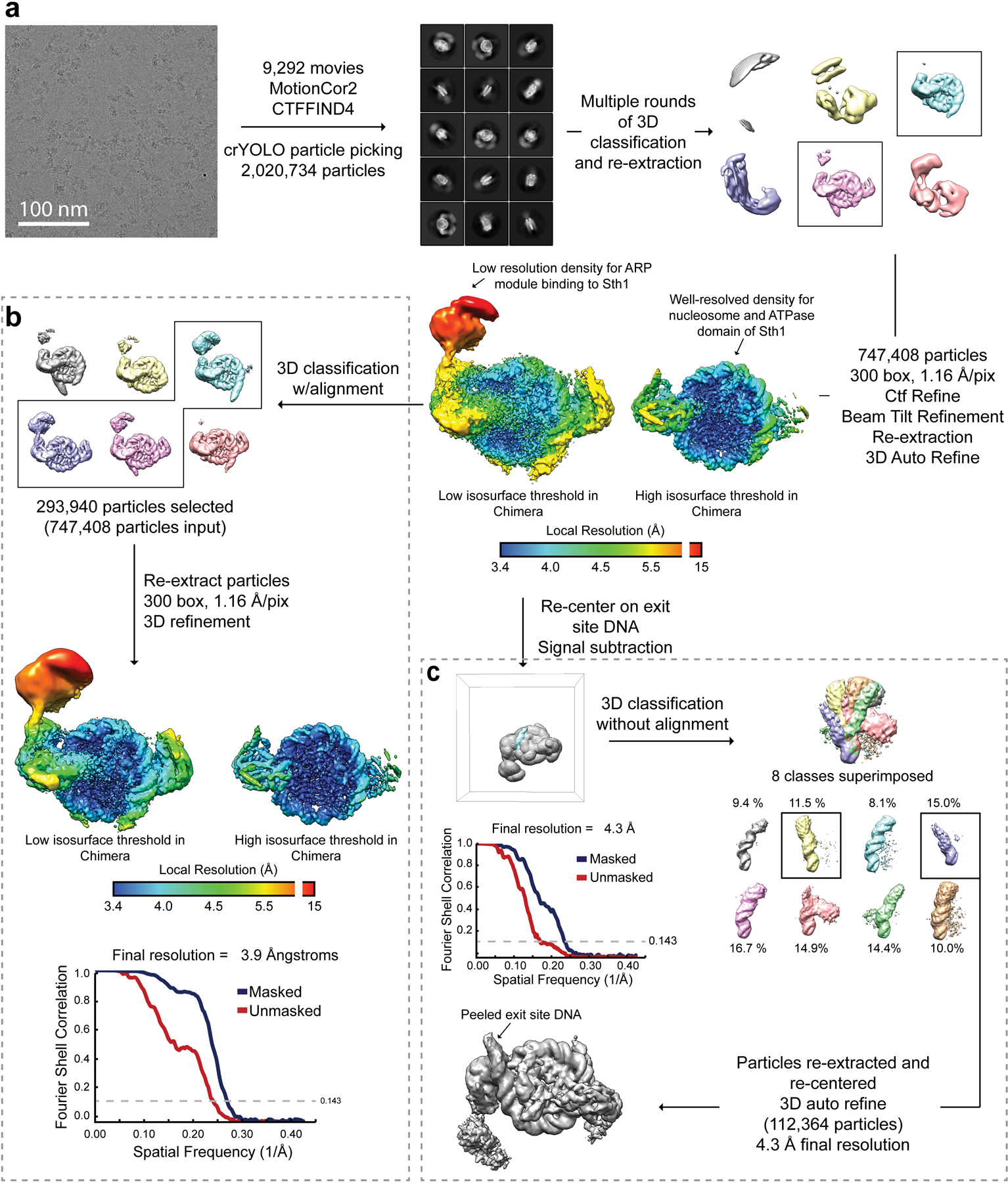
a, Workflow for cryo-EM data acquisition and structure determination. b, 3D classification with alignment was used to find ~300,000 particles with better ARP module density. This dataset reached a final global FSC resolution of ~3.9 Å with a local resolution range of 3.4–15 Å. c, Partial signal subtraction followed by 3D classification without alignment was used to identify a sub-set of particles with an alternate DNA conformation at the nucleosome DNA exit site.
Extended Data Fig. 3. Multi-body refinement of RSCSAR bound to the nucleosome in the ADP-BeF3 state.
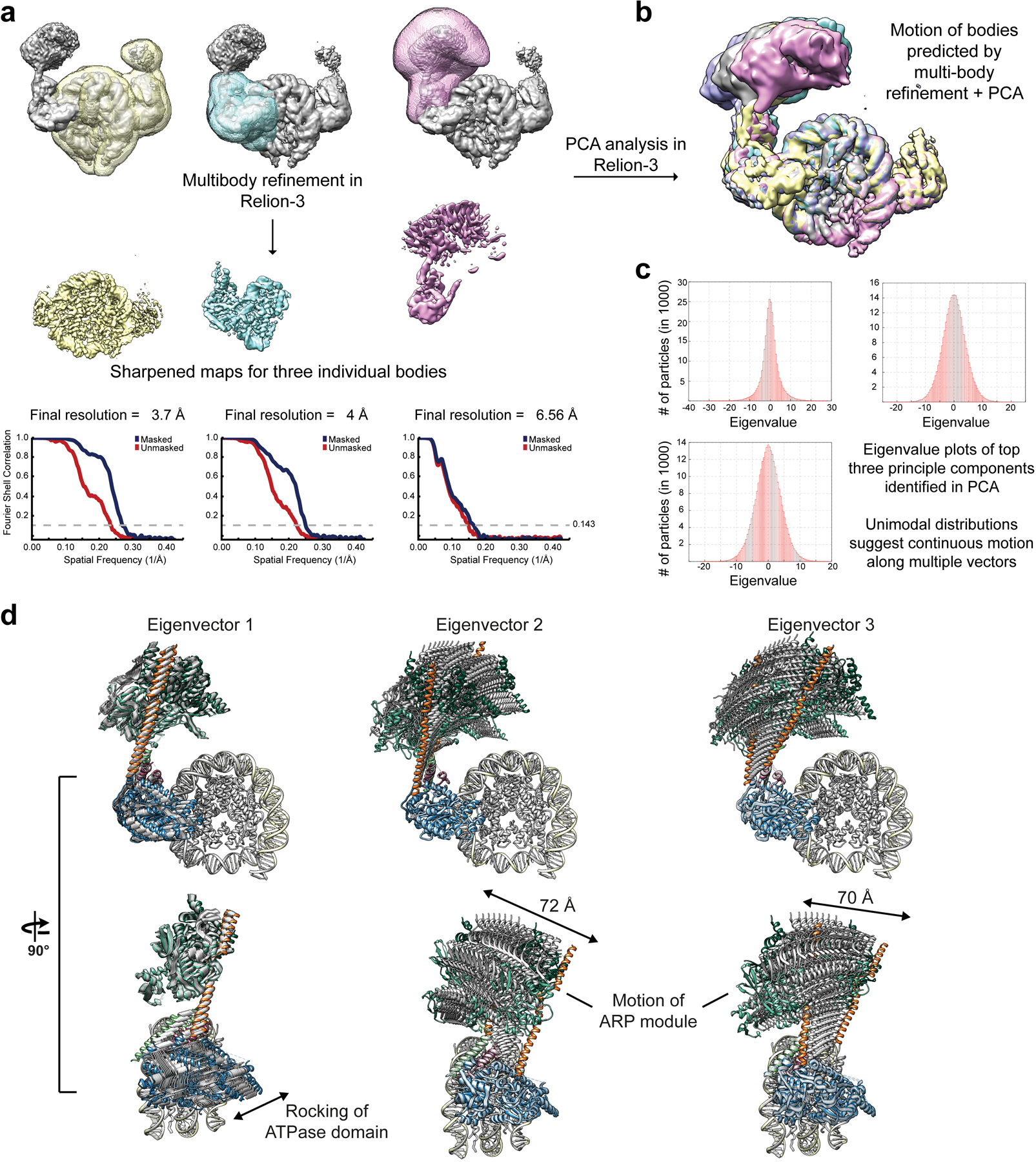
a, The consensus refinement of RSCSAR-nucleosome from Extended Data Fig. 2b was used to generate 3 masks and as a starting model for multi-body refinement. b, PCA was performed in Relion-3 and used to generate maps that show the conformational heterogeneity present in our data set. Several of these maps are shown overlaid. c, Eigenvalue plots for the top three eigenvectors. d, The maps generated in panel (b) were used to build PDB models. 10 models for each eigenvector were generated and are shown superimposed. The main sources of heterogeneity in our data set are a rocking of the ATPase domain (eigenvector 1) and rocking of the ARP module along two different axes (eigenvector 2 and 3).
Extended Data Fig. 4. Model building of RSCSAR-nucleosome (ADP-BeF3) and comparison with Snf2-nucleosome (ADP-BeF3).
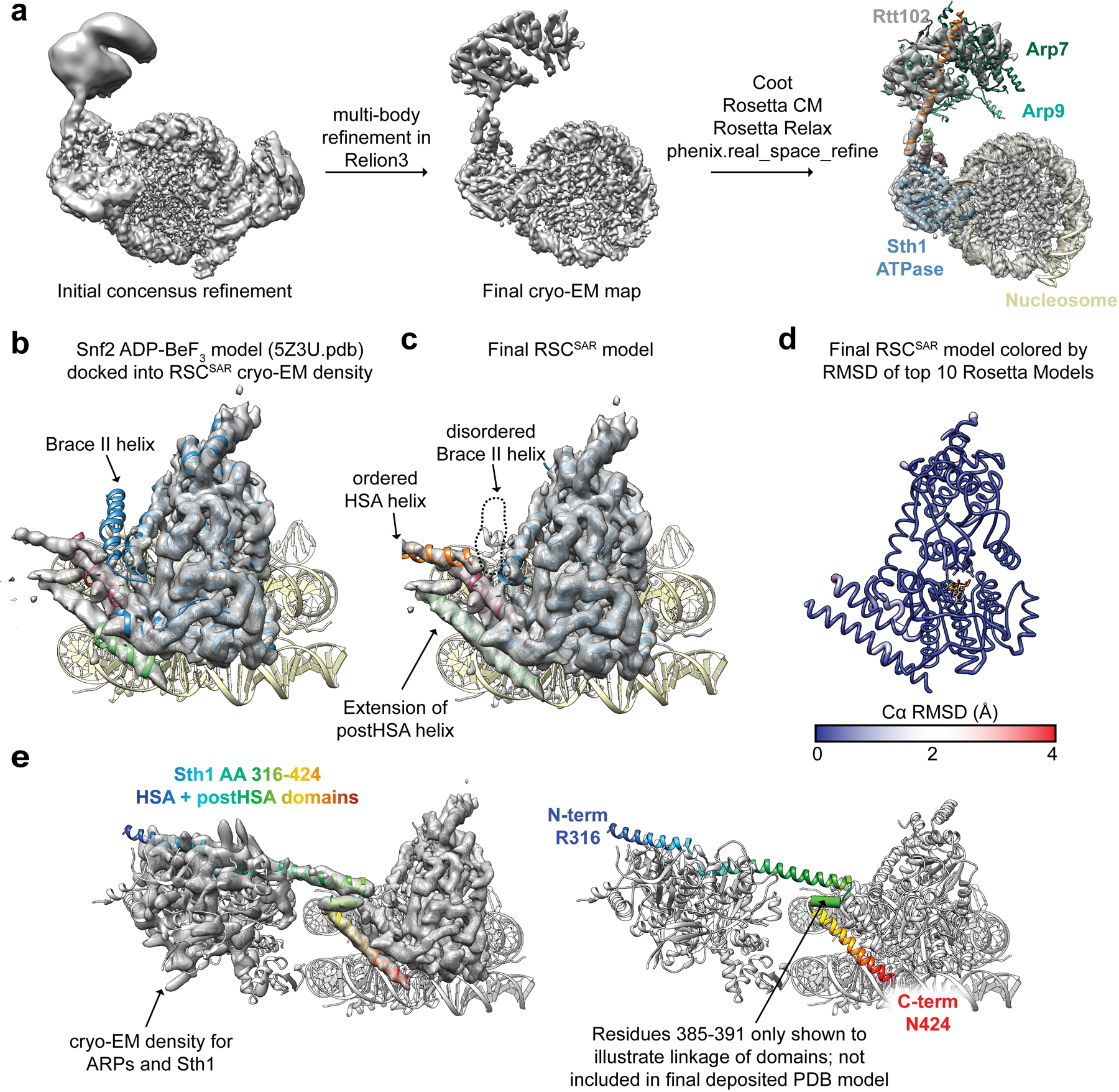
a, Model building schematic for RSCSAR-nucleosome (ADP-BeF3). Snf2-nucleosome (PDB 5Z3U) and the T. thermophilus Snf2 crystal structure (PDB 5HZR) were used as a starting point for model building and refinement using Coot, Rosetta, and phenix.real_space_refine. The ARP module built in Extended Data Fig.1 was rigid-body docked into the map, yielding a near-complete model for all components in our sample. b, PDB 5Z3U is shown docked into our cryo-EM map. c, The same view as in panel (b) but showing Sth1-nucleosome (ADP-BeF3). d, The top Rosetta model is shown with its ribbon thickness and color indicating the RMSD among the top 10 models. e, The RSCSAR-nucleosome structure is shown with the HSA and postHSA domains of Sth1 colored in a rainbow from N to C terminus. Residues 385–391 are shown as a cylinder to signal the linkage between the two domains and indicate that the density is compatible with an alpha helix, although this region is not included in the deposited.
Extended Data Fig. 5. The ARP module of RSC only interacts with the HSA domain of Sth1.
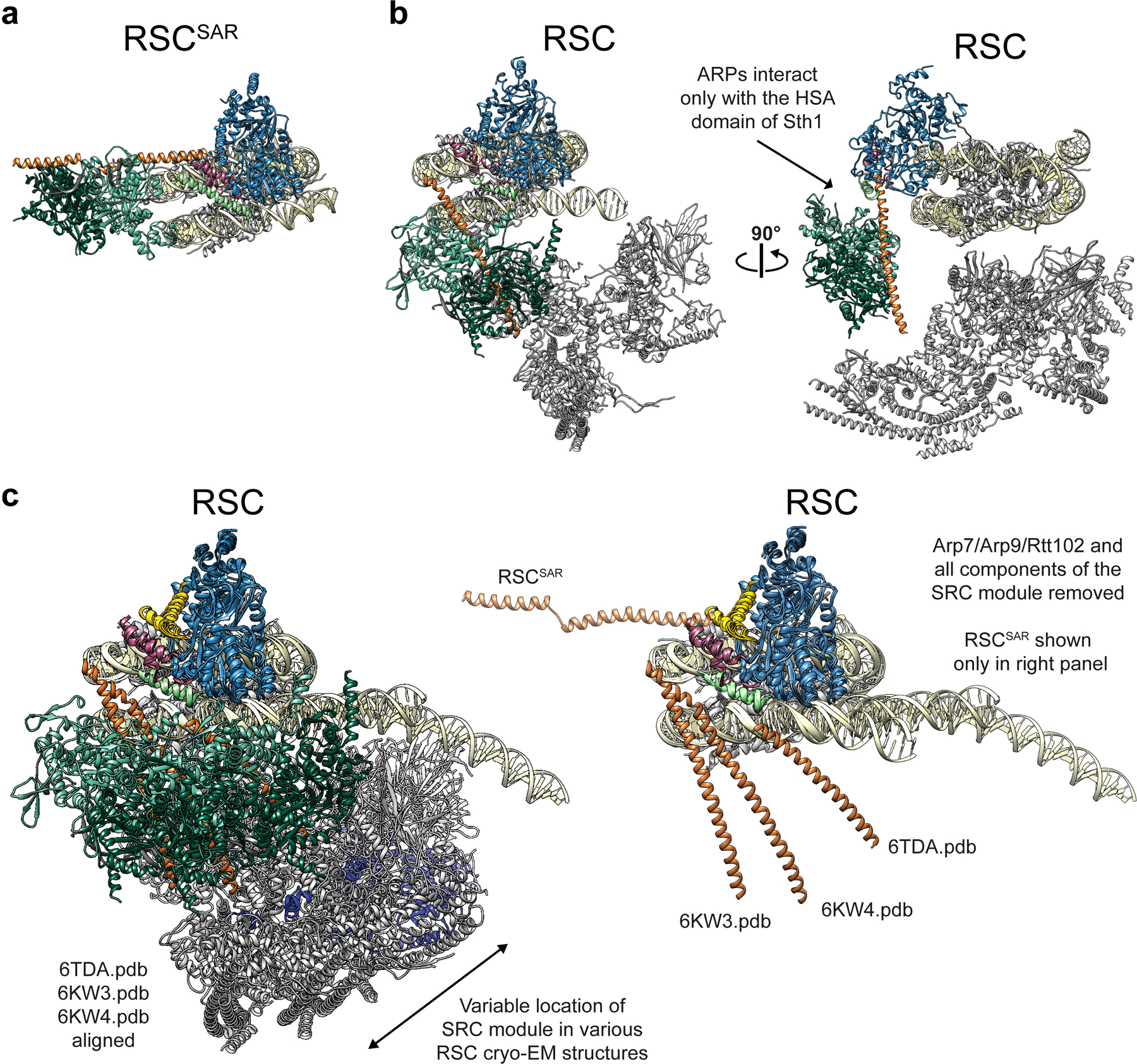
a, Our model of RSCSAR-nucleosome (ADP-BeF3). b, Model of RSC-nucleosome (PDB 6KW3) shown in the same orientation as panel (a) (left) and rotated 90° (right). The ARP module only interacts with the HSA domain in both RSC and RSCSAR. c, Multiple RSC-nucleosome (ADP-BeF3) cryo-EM structures were aligned. Left, all components shown and colored using the same convention. Right, Arp7–Arp9–Rtt102 and all components of the SRC are omitted to highlight the conformational flexibility of the Sth1 HSA domain (orange) among the three structures. The HSA domain from RSCSAR is included in this panel to indicate that, despite their conformational flexibility, the HSA domains from the three RSC-nucleosome structures have a similar general orientation relative to the HSA from RSCSAR.
Extended Data Fig. 6. Comparison of the nucleosome-bound ATPase domain of Snf2, RSC, and RSCSAR in the ADP-BeF3 nucleotide state.
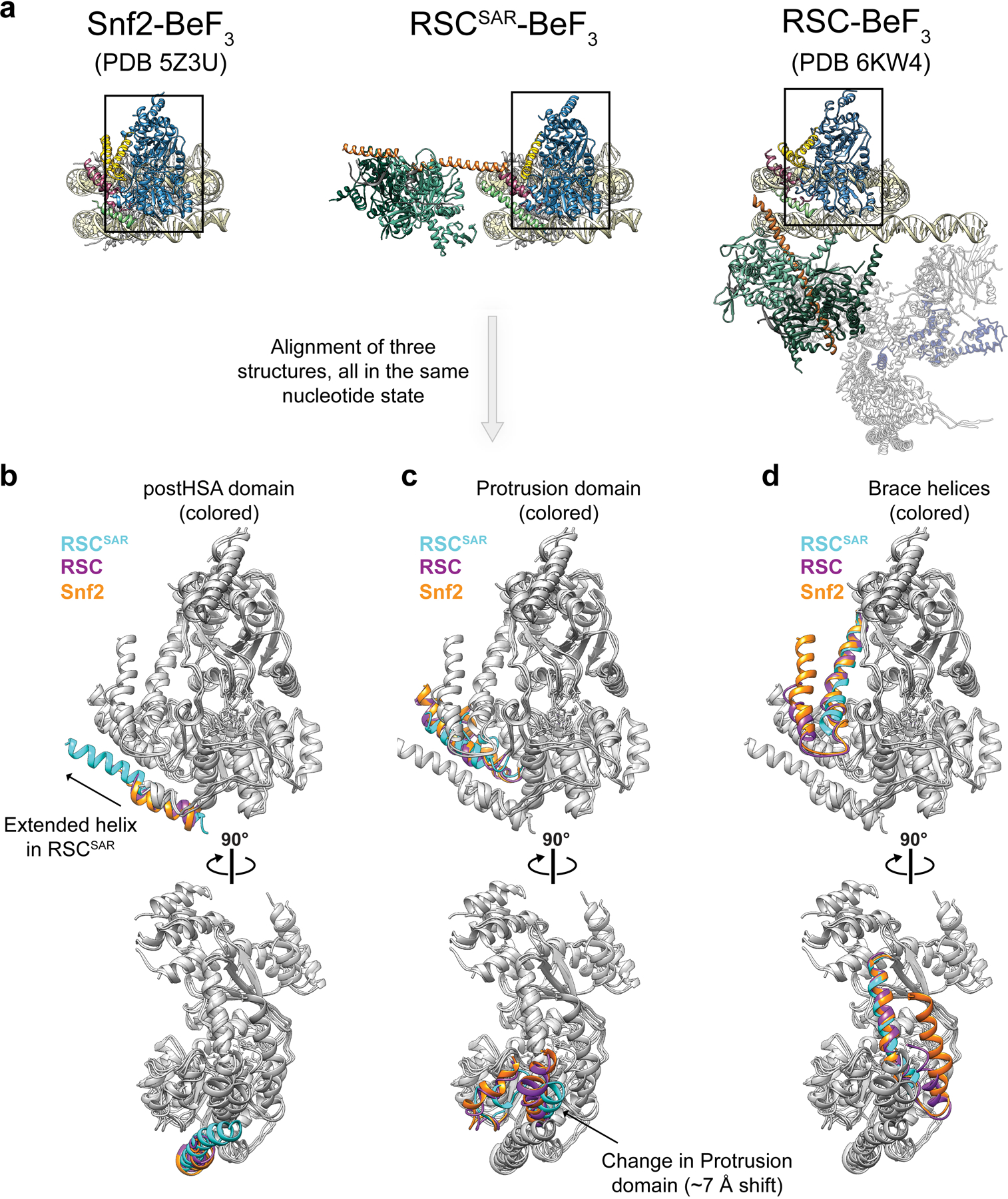
a, The PDB models for Snf2, RSCSAR, and RSC are shown with the same orientation and domain coloring. All three structures are from S.cerevisae, bound to the nucleosome and in the ADP-BeF3 nucleotide state. b-d, The ATPase domain of each structure from (a) was docked into the cryo-EM map of RSCSAR. The postHSA domain (b), Protrusion 1 domain (c), and Brace Helices (d) were colored according to their respective PDB model in each panel. Cyan: RSCSAR, Magenta: RSC (PDB 6KW3), Orange: Snf2 (PDB 5Z3U).
Extended Data Fig. 7. Comparison of cryo-EM maps of Snf2-nucleosome in the apo, ADP, and ADP-BeF3 states and RSCSAR-nucleosome (ADP-BeF3).
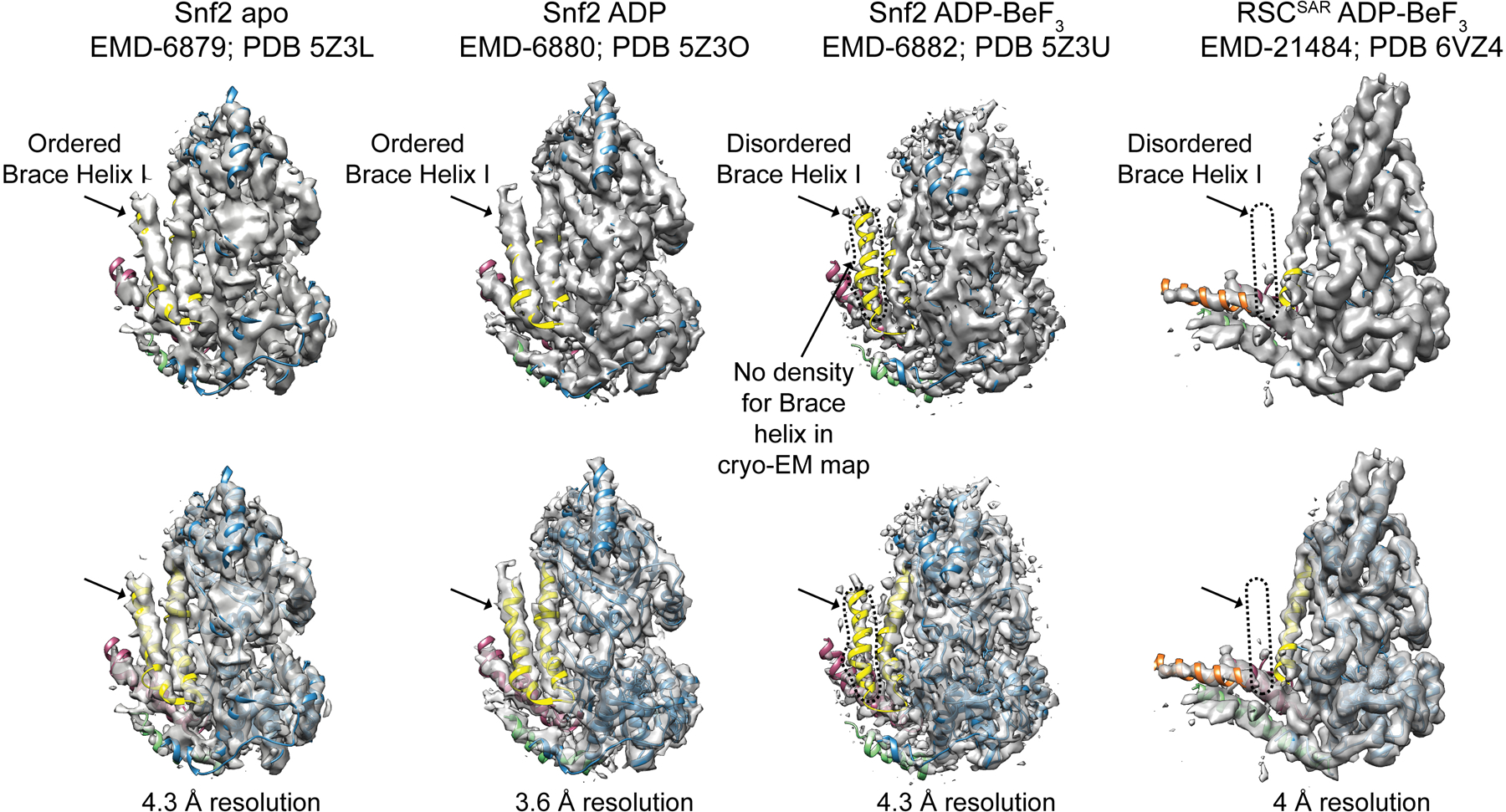
Each structure is shown with cryo-EM map and corresponding PDB model. All structures are bound to the nucleosome but this region is omitted for clarity. The same map at the same contour level is shown opaque (top) and transparent (bottom). Note that density for the Brace Helix I is absent in the ADP-BeF3 state but present in the apo and ADP-bound states.
Extended Data Fig. 8. Structural comparison of the ATPase domain of SWI/SNF remodelers in the apo, ADP, and ADP-BeF3 nucleotide states.
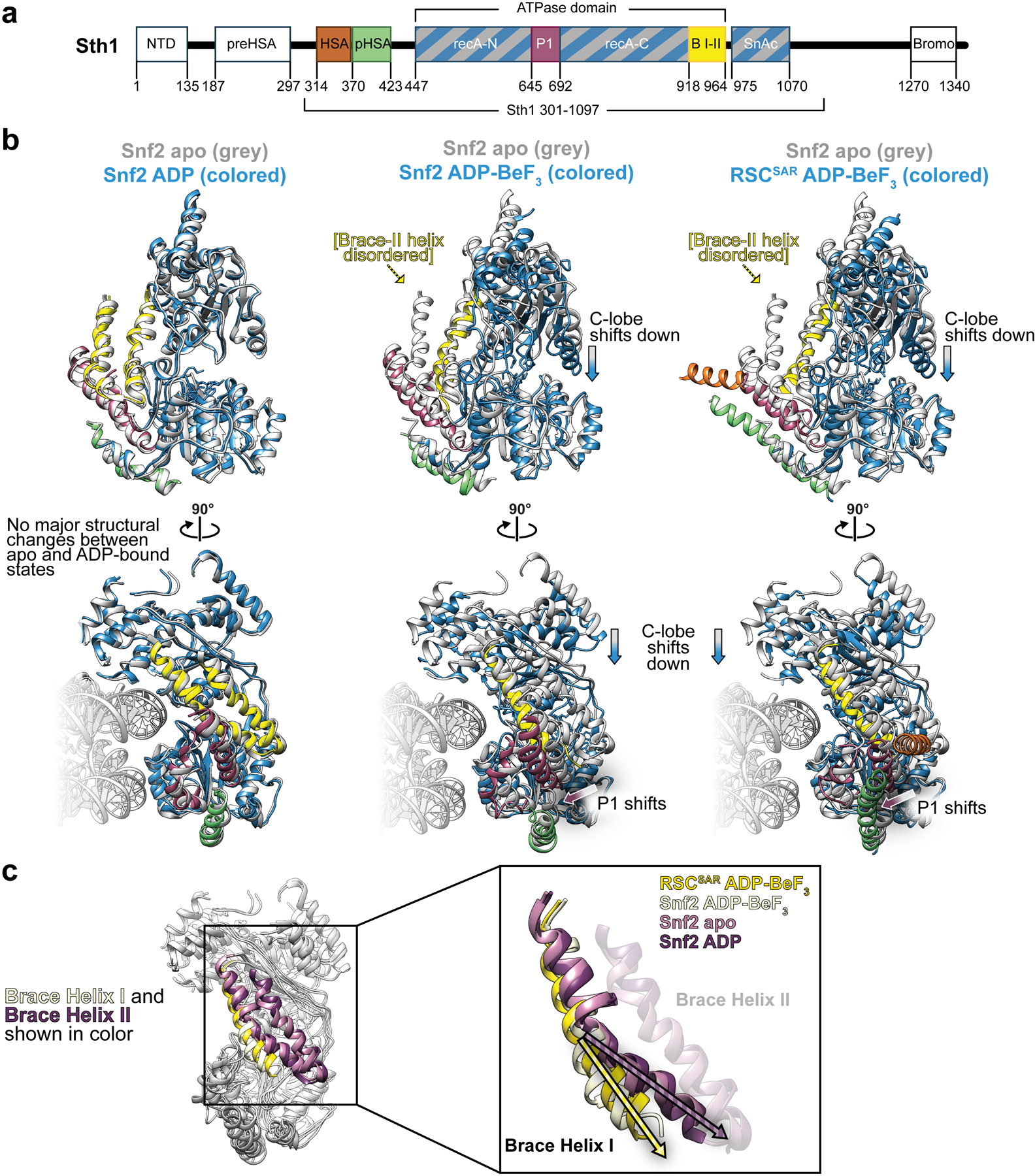
a, Domain schematic of Sth1 from RSCSAR. b, Structural alignment of the ATPase domain of Snf2 apo with Snf2 - ADP (left), Snf2-ADP-BeF3 (middle), and RSCSAR- BeF3 (right). All structures are colored using the domain convention shown in (a). c, All structures from (b) are aligned and colored grey, except for the Brace Helices which are colored by model: Snf2 apo, pink; Snf2 ADP, purple; Snf2 ADP-BeF3, tan; RSCSAR BeF3, yellow. PDB codes for structures shown: Snf2 apo (5X0Y), ADP (5Z3O), ADP-BeF3 (5Z3U).
Extended Data Fig. 9. Conformation of the nucleosomal DNA in the cryo-EM structure of RSCSAR-nucleosome (ADP-BeF3).
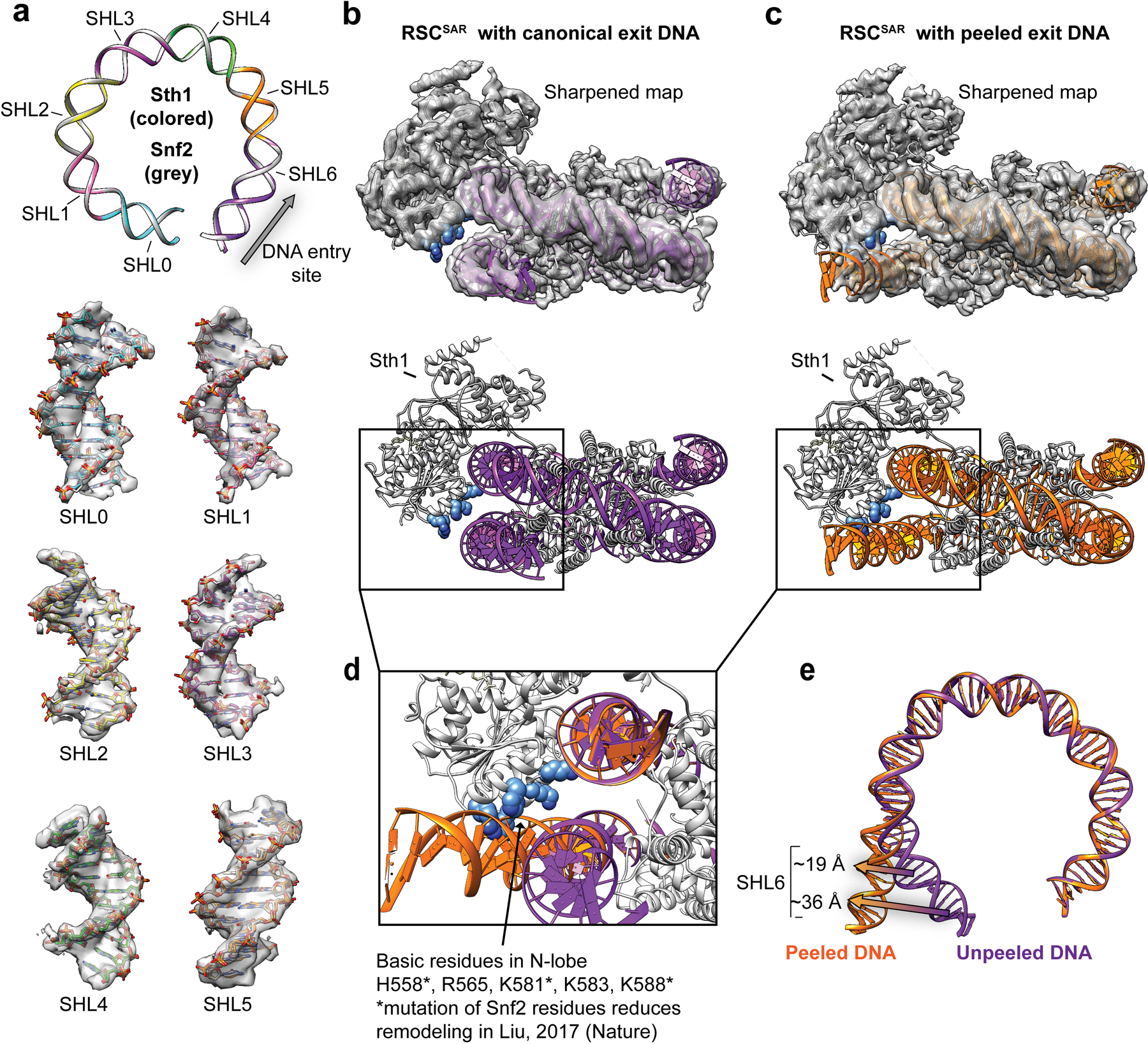
a, The models of Snf2 (PDB 5Z3U) and RSCSAR were aligned. A ribbon representation of the nucleosomal DNA is shown for Snf2 (grey) and Sth1 (colored by SHL). Super helical Locations for the aligned models are shown inside the cryo-EM density of RSCSAR. b, RSCSAR with a canonical conformation of the nucleosome. Top panel shows map and model from the initial 3.9 Å consensus refinement (Extended Data Fig. 2b). The nucleosomal DNA is colored purple and basic residues in the N-lobe are colored blue. c, RSCSAR with a peeled conformation of the nucleosomal DNA exit site. Top panel shows map and model from the 4.3 Å refinement of a subset of particles with a peeled DNA conformation (Extended Data. Fig. 2c). The nucleosomal DNA is colored orange and basic residues in the N-lobe are colored blue. c, Alignment of canonical and peeled DNA structures. Note that basic residues in the N-lobe appear to make contact with the exit site DNA in the peeled conformation. d, SHL6 shifts between the canonical and peeled DNA conformations.
Extended Data Fig. 10. A basic patch in RSCSAR binds to the nucleosome and is important for remodeling.
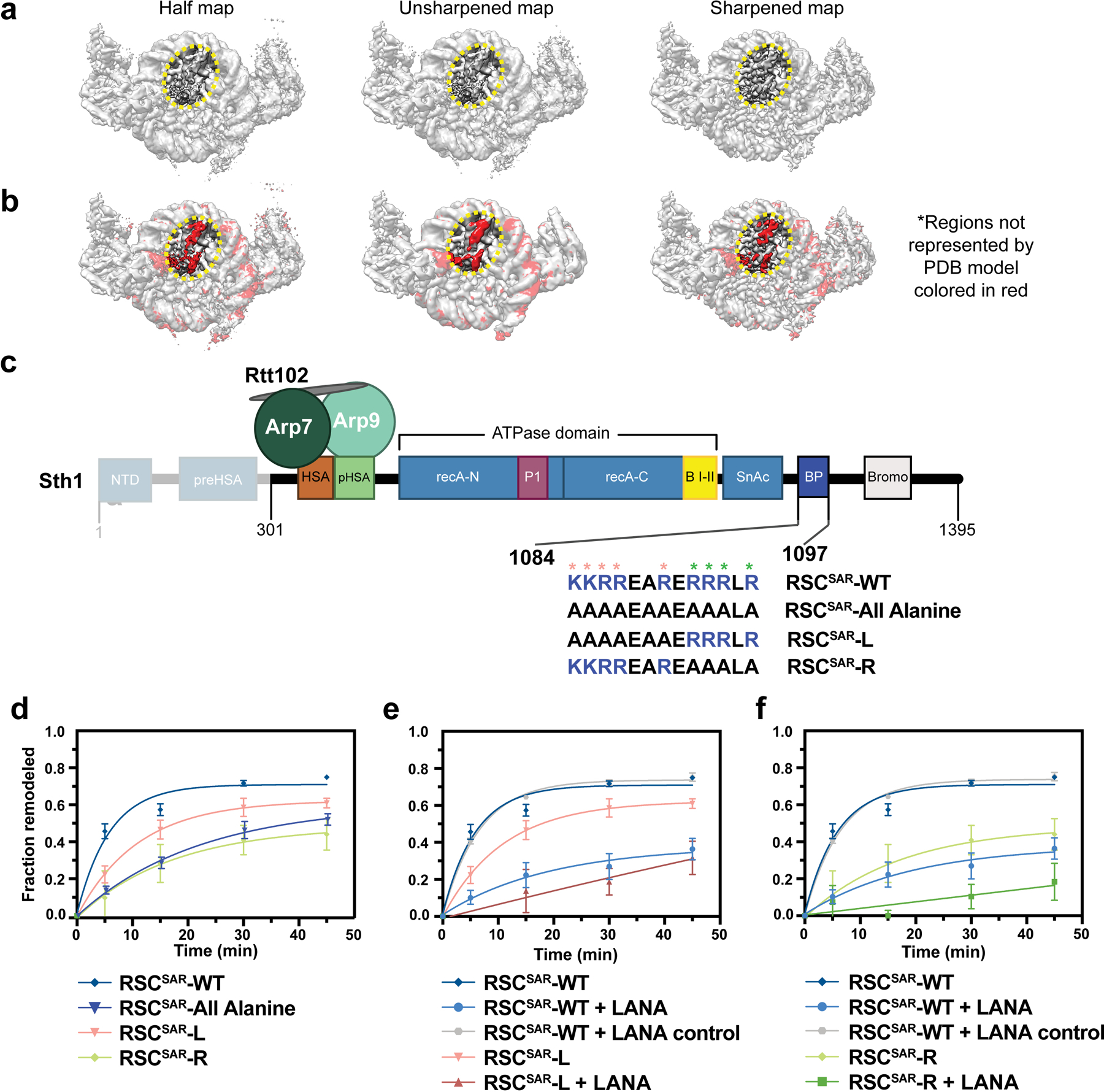
a, Different maps from the RSCSAR consensus refinement are shown in the same orientation. A region of unassigned density is circled in yellow b, The same maps shown in panel (a) with red indicating regions of the maps that are not accounted for by the PDB model of the nucleosome. c, Schematic of the RSCSAR construct used in nucleosome remodeling assays. Sequences of Sth1 mutants used for remodeling assays are shown below the wild type sequence. Residues mutated in the RSCSAR-L and RSCSAR-R variants are noted (salmon, L; green, R). d, Remodeling assay of wildtype, RSCSAR-All Alanine, RSCSAR-L, and RSCSAR-R mutants. WT data are the same as in Fig. 6b. e, Remodeling assay of wildtype and RSCSAR-R mutant. f, Remodeling assay of wildtype and RSCSAR-L mutant. Error is reported as standard deviation where n=3 replicates.
Supplementary Material
Table 1.
Cryo-EM data collection, refinement and validation statistics
| RSCSAR apo (EMD-21489, PDB 6VZG) |
RSCSAR nucleosome (BeF3) (EMD-21484, PDB 6VZ4) |
RSCSAR nucleosome (BeF3) Peeled conformation (EMD-21493) |
|
|---|---|---|---|
| Data collection and processing | |||
| Magnification | 36,000 | 36,000 | 36,000 |
| Voltage (kV) | 200 | 200 | 200 |
| Camera mode | Super-resolution | Counting | Counting |
| Electron exposure (e–/Å2) | 50–55 | 50–55 | 50–55 |
| Defocus range (μm) | 0.6–2.5 | 0.6–2.5 | 0.6–2.5 |
| Pixel size (Å) | 0.58 | 1.16 | 1.16 |
| Symmetry imposed | C1 | C1 | C1 |
| Initial particle images (no.) | 1,986,341 | 2,020,734 | 2,020,734 |
| Final particle images (no.) | 415,957 | 293,940 | 112,364 |
| Map resolution (Å) | 4.2 | 3.9 | 4.3 |
| FSC threshold | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 4.0–5.8 | 3.5–15 | |
| Refinement | |||
| Initial model used | PDB 4I6M, 5TGC | PDB 5Z3U, 5HZR | |
| Model resolution (Å) | 4.2 | 3.9 | |
| FSC threshold | 0.143 | 0.143 | |
| Model resolution range (Å) | 4.0–5.8 | 3.5–15 | |
| Map sharpening B factor (Å2) | −162 | −118 | −114 |
| Model composition | |||
| Number of models | 10 | 10 | |
| Non-hydrogen atoms (per model) | 7420 | 23950 | |
| Protein residues (per model) | 906 | 2509 | |
| Ligands (per model) | 1 ATP | 1 ADP; 1 Mg2+; 1 BeF3, 1 ATP |
|
| B factors (Å2) | |||
| Protein | 101.4 | 116.64 | |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.014 | 0.013 | |
| Bond angles (°) | 1.35 | 1.13 | |
| Validation | |||
| MolProbity score | 1.23 | 1.27 | |
| Clashscore | 1.89 | 2.08 | |
| EM Ringer Score | 1.37 | 1.77 | |
| Map CC (CC mask) | 0.791 | 0.832 | |
| Poor rotamers (%) | 0.01 | 0.26 | |
| Ramachandran plot | |||
| Favored (%) | 95.91 | 95.85 | |
| Allowed (%) | 3.18 | 3.21 | |
| Disallowed (%) | 0.91 | 0.94 |
Acknowledgements
We thank the UC San Diego Cryo-Electron Microscopy Facility, which was supported in part by NIH grants to T. Baker and a gift from the Agouron Institute to UCSD and the UC San Diego Physics Computing Facility for IT support. RWB is a Damon Runyon fellow supported by the Damon Runyon Cancer Research Foundation (DRG-2285-16). JMR is a Merck fellow of the Damon Runyon Cancer Research Foundation (DRG-2370-19). BT is an American Heart Association pre-doctoral fellow (14PRE19970011). This work was funded by National Institutes of Health Grants R01 GM092895 (AEL), R01 GM073791 (RD), and T32 AR053461 (PJC).
Footnotes
Competing Interests
The authors declare no competing interests.
Data Availability
The cryo-EM maps for apo RSCSAR (Sth1HSA-Arp7-Arp9-Rtt102 model) are deposited in the EMDB as EMD-21489 and molecular models are deposited in the wwPDB as PDB 6VZG. The cryo-EM maps for RSCSAR bound to the nucleosome in the presence of ADP BeF3 are deposited in the EMDB as EMD-21484 and molecular models are deposited in the wwPDB as PDB 6VZ4. The cryo-EM maps of RSCSAR bound to the nucleosome in the presence of ADP BeF3 with a peeled DNA conformation are deposited in the EMDB as EMD-21493. Unsharpened, sharpened, and half maps were deposited for each EMDB entry.
References
- 1.Bartholomew B Regulating the chromatin landscape: structural and mechanistic perspectives. Annual review of biochemistry 83, 671–696 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clapier CR & Cairns BR The Biology of Chromatin Remodeling Complexes. Annual Review of Biochemistry 78, 273–304 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Kwon H, Imbalzano AN, Khavari PA, Kingston RE & Green MR Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 370, 477–481 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Parnell TJ, Huff JT & Cairns BR RSC regulates nucleosome positioning at Pol II genes and density at Pol III genes. The EMBO journal 27, 100–110 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cairns BR, Levinson RS, Yamamoto KR & Kornberg RD Essential role of Swp73p in the function of yeast Swi/Snf complex. Genes & Development 10, 2131–2144 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Stern M, Jensen R & Herskowitz I Five SWI genes are required for expression of the HO gene in yeast. Journal of Molecular Biology 178, 853–868 (1984). [DOI] [PubMed] [Google Scholar]
- 7.Kadoch C & Crabtree GR Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Science advances 1, e1500447–e1500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reisman D, Glaros S & Thompson EA The SWI/SNF complex and cancer. Oncogene 28, 1653–1668 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Shain AH & Pollack JR The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PloS one 8, e55119–e55119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadoch C et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature genetics 45, 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X et al. Oncogenesis Caused by Loss of the SNF5 Tumor Suppressor Is Dependent on Activity of BRG1, the ATPase of the SWI/SNF Chromatin Remodeling Complex. Cancer Res 69, 8094 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peterson CL, Zhao Y & Chait BT Subunits of the Yeast SWI/SNF Complex Are Members of the Actin-related Protein (ARP) Family. J. Biol. Chem 273, 23641–23644 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Zhao K et al. Rapid and Phosphoinositol-Dependent Binding of the SWI/SNF-like BAF Complex to Chromatin after T Lymphocyte Receptor Signaling. Cell 95, 625–636 (1998). [DOI] [PubMed] [Google Scholar]
- 14.Schubert HL et al. Structure of an actin-related subcomplex of the SWI/SNF chromatin remodeler. Proceedings of the National Academy of Sciences 110, 3345–3350 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szerlong H et al. The HSA domain binds nuclear actin-related proteins to regulate chromatin-remodeling ATPases. Nature structural & molecular biology 15, 469–476 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turegun B, Baker RW, Leschziner AE & Dominguez R Actin-related proteins regulate the RSC chromatin remodeler by weakening intramolecular interactions of the Sth1 ATPase. Communications biology 1, 1–1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clapier CR et al. Regulation of DNA Translocation Efficiency within the Chromatin Remodeler RSC/Sth1 Potentiates Nucleosome Sliding and Ejection. Molecular cell 62, 453–461 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Li M, Xia X, Li X & Chen Z Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 544, 440 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Patel AB et al. Architecture of the chromatin remodeler RSC and insights into its nucleosome engagement. eLife 8, e54449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wagner FR et al. Structure of SWI/SNF chromatin remodeller RSC bound to a nucleosome. Nature (2020) doi: 10.1038/s41586-020-2088-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye Y et al. Structure of the RSC complex bound to the nucleosome. Science eaay0033 (2019) doi: 10.1126/science.aay0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han Y, Reyes AA, Malik S & He Y Cryo-EM structure of SWI/SNF complex bound to a nucleosome. Nature (2020) doi: 10.1038/s41586-020-2087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He S et al. Structure of nucleosome-bound human BAF complex. Science 367, 875 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Eastlund A, Malik SS & Fischer CJ Kinetic mechanism of DNA translocation by the RSC molecular motor. Archives of Biochemistry and Biophysics 532, 73–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malik SS, Rich E, Viswanathan R, Cairns BR & Fischer CJ Allosteric Interactions of DNA and Nucleotides with S. cerevisiae RSC. Biochemistry 50, 7881–7890 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sirinakis G et al. The RSC chromatin remodelling ATPase translocates DNA with high force and small step size. The EMBO Journal 30, 2364–2372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li M et al. Mechanism of DNA translocation underlying chromatin remodelling by Snf2. Nature 567, 409–413 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Xia X, Liu X, Li T, Fang X & Chen Z Structure of chromatin remodeler Swi2/Snf2 in the resting state. Nature Structural and Molecular Biology 23, nsmb.3259 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Nakane T, Kimanius D, Lindahl E, Scheres SH & Brunger AT Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. eLife 7, e36861 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Armache K-J, Garlick JD, Canzio D, Narlikar GJ & Kingston RE Structural Basis of Silencing: Sir3 BAH Domain in Complex with a Nucleosome at 3.0 A Resolution. Science 334, 977–982 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barbera AJ et al. The Nucleosomal Surface as a Docking Station for Kaposi's Sarcoma Herpesvirus LANA. Science 311, 856 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Kato H et al. A Conserved Mechanism for Centromeric Nucleosome Recognition by Centromere Protein CENP-C. Science 340, 1110–1113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Makde RD, England JR, Yennawar HP & Tan S Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature 467, 562–566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGinty RK, Henrici RC & Tan S Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature 514, 591–596 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mashtalir N et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 175, 1272–1288.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dann GP et al. ISWI chromatin remodellers sense nucleosome modifications to determine substrate preference. Nature 548, 607 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levendosky RF & Bowman GD Asymmetry between the two acidic patches dictates the direction of nucleosome sliding by the ISWI chromatin remodeler. eLife 8, e45472 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dao HT, Dul BE, Dann GP, Liszczak GP & Muir TW A basic motif anchoring ISWI to nucleosome acidic patch regulates nucleosome spacing. Nature Chemical Biology 16, 134–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Valencia AM et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell 179, 1342–1356.e23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods References
- 40.Dyer PN et al. Reconstitution of Nucleosome Core Particles from Recombinant Histones and DNA in vol. 375 23–44 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Stark H Chapter Five GraFix: Stabilization of Fragile Macromolecular Complexes for Single Particle Cryo-EM. Methods in Enzymology 481, 109–126 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Herzik JMA, Wu M & Lander GC Achieving better-than-3-Å resolution by single-particle cryo-EM at 200 keV. Nature methods 14, 1075–1078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. Journal of structural biology 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kremer JR, Mastronarde DN & McIntosh JR Computer Visualization of Three-Dimensional Image Data Using IMOD. Journal of Structural Biology 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of Structural Biology 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner T et al. SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Communications Biology 2, 218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallographica Section D: Biological Crystallography 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Cianfrocco MA, Lahiri I, DiMaio F & Leschziner AE cryoem-cloud-tools: A software platform to deploy and manage cryo-EM jobs in the cloud. Journal of Structural Biology 203, 230–235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song Y et al. High-resolution comparative modeling with RosettaCM. Structure (London, England : 1993) 21, 1735–1742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica. Section D, Biological crystallography 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alford RF et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. Journal of chemical theory and computation 13, 3031–3048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Afonine PV et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta crystallographica. Section D, Structural biology 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liebschner D et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallographica Section D 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps for apo RSCSAR (Sth1HSA-Arp7-Arp9-Rtt102 model) are deposited in the EMDB as EMD-21489 and molecular models are deposited in the wwPDB as PDB 6VZG. The cryo-EM maps for RSCSAR bound to the nucleosome in the presence of ADP BeF3 are deposited in the EMDB as EMD-21484 and molecular models are deposited in the wwPDB as PDB 6VZ4. The cryo-EM maps of RSCSAR bound to the nucleosome in the presence of ADP BeF3 with a peeled DNA conformation are deposited in the EMDB as EMD-21493. Unsharpened, sharpened, and half maps were deposited for each EMDB entry.