

Abstract
Many intracellular proteins are modified by N-acetylglucosamine, a posttranslational modification termed O-GlcNAc. This modification is found on serine and threonine side-chains and has the potential to regulate signaling pathways through interplay with phosphorylation. Here, we discover and characterize one such example. We find that O-GlcNAc levels control the sensitivity of fibroblasts to actin contraction induced by the signaling lipid sphingosine-1-phosphate (S1P), culminating in the phosphorylation of myosin light chain (MLC) and cellular contraction. Specifically, O-GlcNAc modification of the phosphatase subunit MYPT1 inhibits this pathway by blocking MYPT1 phosphorylation, maintaining its activity and causing the dephosphorylation of MLC. Finally, we demonstrate that O-GlcNAc levels alter the sensitivity of primary human dermal fibroblasts in a collagen-matrix model of wound healing. Our findings have important implications for the role of O-GlcNAc in fibroblast motility and differentiation, particularly in diabetic wound healing.
INTRODUCTION
O-GlcNAc is a form of protein glycosylation that is uniquely suited to regulate cellular signaling pathways (Figure 1a). This intracellular posttranslational modification involves the transfer of the monosaccharide N-acetylglucosamine to serine and threonine side chains of proteins1,2. Unlike most forms of glycosylation, this GlcNAc moiety is not elaborated by additional carbohydrates and is dynamically regulated by two enzymes3. O-GlcNAc transferase (OGT) uses the donor sugar UDP-GlcNAc to install O-GlcNAc, while O-GlcNAcase (OGA) removes it, setting up a scenario potentially similar to the cycling of protein phosphorylation. The amounts of O-GlcNAc change upon alterations in cellular environment through the sensing of different biological inputs. For example, one of the main roles of O-GlcNAc is to act as a nutrient sensor to control various cellular pathways4. In particular, flux of glucose into the cell, which is transformed by the hexosamine biosynthetic pathway (HBP) into increased amounts of UDP-GlcNAc5, results in a dynamic increase in O-GlcNAc levels. Interestingly, the substrate selection and activity of OGT are highly dependent on the concentration of UDP-GlcNAc6, allowing OGT to “read-out” extracellular glucose levels. Therefore, it is not surprising that hyperglycemia in diabetes results in elevated O-GlcNAc in comparison to healthy individuals and indicates that O-GlcNAc should modulate various proteins and pathways and that misregulation of these events contributes to disease. An attractive hypothesis has been a direct relationship between the O-GlcNAc modification and phosphorylation (Figure 1b) 7. For example, O-GlcNAc modification of the kinase Akt at Thr305 and Thr312 prevents phosphorylation at nearby Thr308 and impairs Akt signaling8. Similarly, O-GlcNAc modification of casein kinase II (CK2) at Ser347 antagonizes phosphorylation at nearby Thr344, thereby reducing its stability/activity9. However, despite these and some other examples, and the theoretical attractiveness of O-GlcNAc as a regulator of cell signaling, the cases where we understand how this modification controls specific cascades remains limited. Here, we add another example of O-GlcNAc/phosphorylation interplay. Specifically, we used chemical tools to investigate our serendipitous discovery linking the signaling lipid sphingosine-1-phosphate (S1P) to actin cytoskeletal remodeling through the O-GlcNAc modification of the phosphatase regulatory subunit MYPT1.
Figure 1. O-GlcNAc controls the sensitivity of fibroblasts to sphingosine-1-phosphate (S1P) mediated cell contraction.
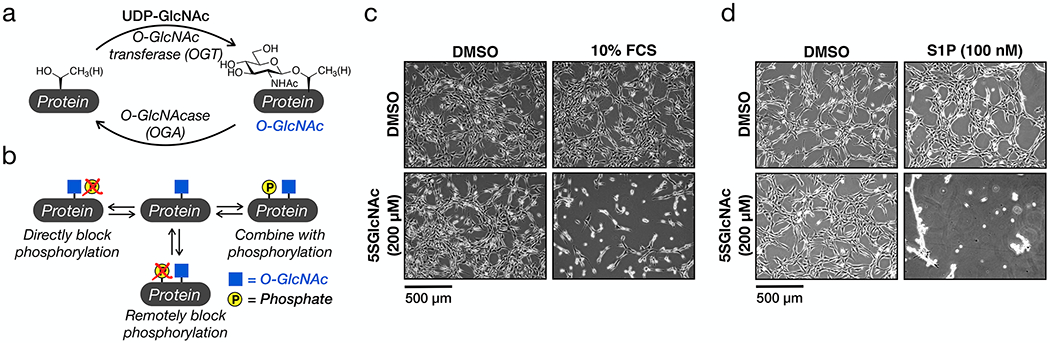
a) O-GlcNAcylation is the dynamic addition to N-acetylglucosamine to serine and threonine residues of intracellular proteins. b) O-GlcNAcylation has its own functions but can also antagonize phosphorylation, both directly and remotely, as well as combine with phosphorylation to illicit biological outcomes. c) NIH3T3 cells were treated with the indicated combinations of DMSO, the OGT inhibitor 5SGlcNAc, and/or 10% FCS. The contraction phenotype was then visualized using bright-field microscopy. d) NIH3T3 cells were treated with the indicated combinations of DMSO, 5SGlcNAc, and/or S1P. The contraction phenotype was then visualized using bright-field microscopy. The data in c and d is representative of at least two biological replicates.
RESULTS
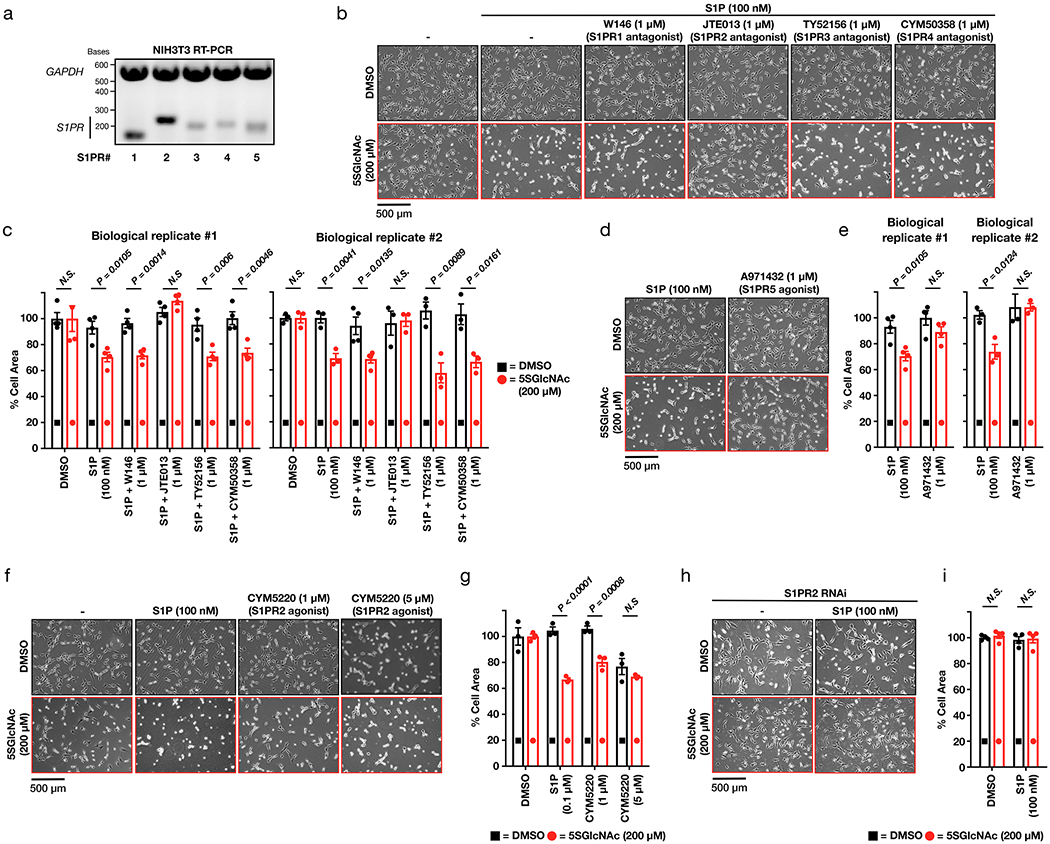
O-GlcNAc controls S1P mediated fibroblast contraction.
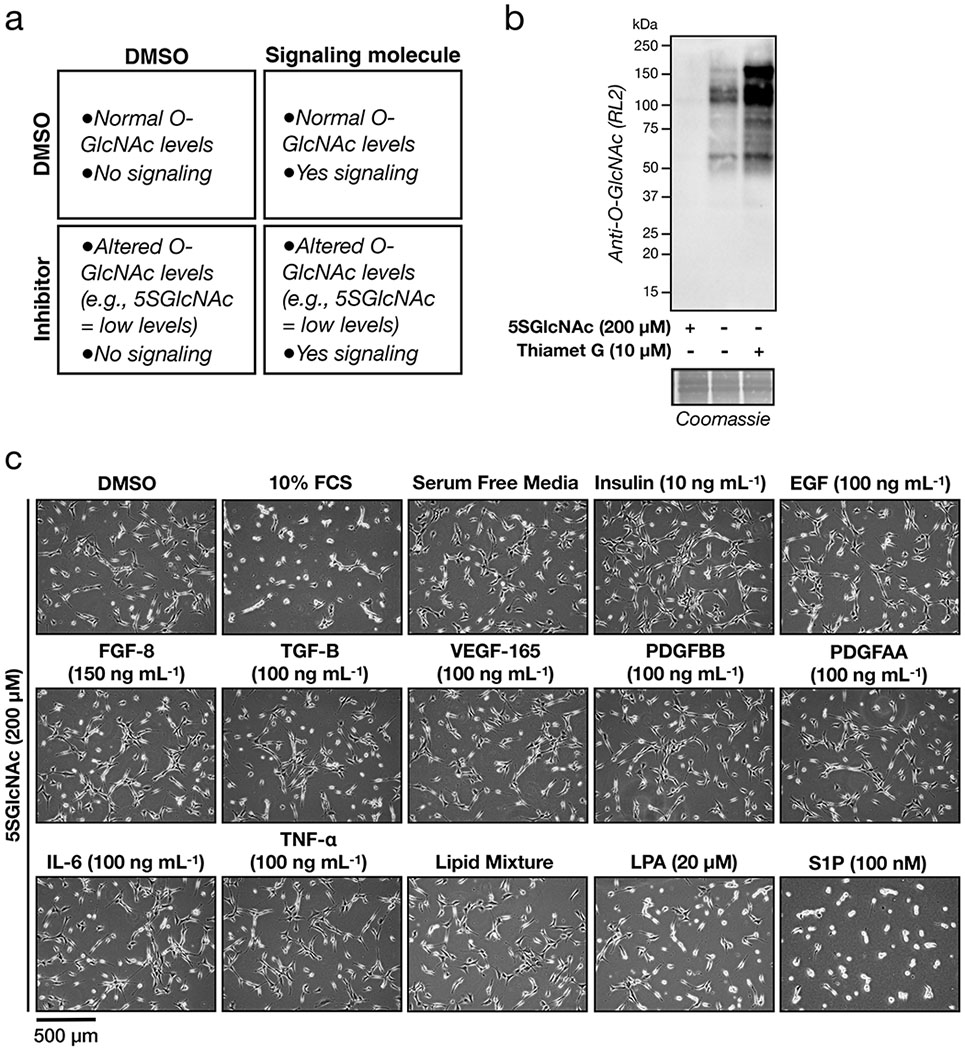
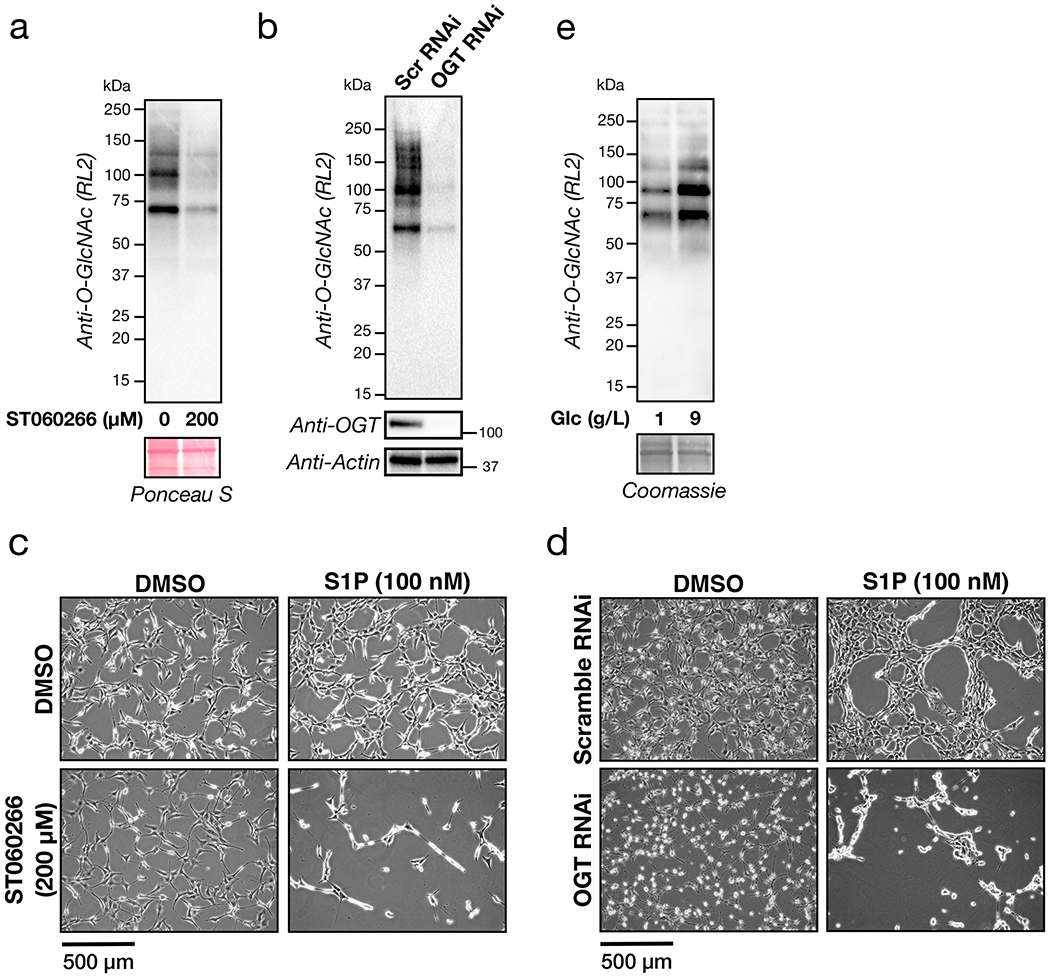
We encountered a phenotypic change in NIH3T3 fibroblasts upon reduction of O-GlcNAc levels followed by treatment with fetal calf serum (FCS). As schematized in Extended Data Figure 1a, these fibroblasts were treated with the OGT inhibitor 5SGlcNAc10 (200 μM) for 16 h to give a downregulation of O-GlcNAc (Extended Data Figure 1b). Upon addition of 10% FCS, the cells underwent a rapid (10 - 30 min) morphological change, resulting in their contraction (Figure 1c and Supplementary Video 1). Intrigued, we then screened protein factors, cytokines, and lipids commonly found in serum. Strikingly, we only detected this phenotype upon S1P treatment (100 nM), and to a lesser extent lysophosphatidic acid (LPA) treatment (20 μM) (Figure 1d, Extended Data Figure 1c and Supplementary Video 2), indicating that signaling initiated by these lipids is responsible for our observation. Here, we chose to explore S1P in greater detail. To confirm that this phenotype is indeed due to a reduction in O-GlcNAc, we next treated fibroblasts with either an orthogonal OGT inhibitor (ST060266, 200 μM) or RNAi against OGT (Extended Data Figures 2a & b). In both cases, we found a similar morphological change upon addition of S1P (Extended Data Figures 2c & d), confirming the O-GlcNAc dependence of the phenotype. To test whether this was apoptosis, we used Western blotting to visualize caspase-3 cleavage during 5SGlcNAc and/or S1P treatment but did not observe any active protease (Supplementary Figure 1). Next, we examined whether the cells would return to a relaxed state. We treated NIH3T3 cells with with DMSO or 5SGlcNAc (200 μM) followed by S1P (100 nM) for an extended time course (Supplementary Figure 2). The cells with decreased O-GlcNAc levels quickly contracted but returned to a relaxed state over the course of the assay. Together, these data suggest that O-GlcNAc controls the sensitivity of fibroblasts to a dynamic, S1P-mediated signaling pathway.
Published studies previously demonstrated that μM concentrations of S1P cause fibroblast contraction in collagen matrices11. To test if we are observing the same pathway, we treated NIH3T3 cells with either DMSO vehicle or 5SGlcNAc (200 μM) for 16 h followed by a range of S1P concentrations (0.05 to 5 μM). Fibroblasts with “normal” O-GlcNAc levels underwent contraction at the higher concentrations of S1P, but cells treated with the OGT inhibitor displayed increased sensitivity to lower amounts of S1P (Figure 2a). We then quantified the amounts of cellular contraction by measuring the surface area occupied by the cells and found the difference in contraction to be statistically significant (Figure 2b). Importantly, this result was observed in multiple biological replicates (Supplementary Figure 3a). We next asked whether raising the modification levels would decrease their sensitivity to the signaling lipid. Accordingly, we treated these fibroblasts with either DMSO vehicle or the OGA inhibitor Thiamet-G12 (10 μM), resulting in increased O-GlcNAc (Extended Data Figure 1b). We found the Thiamet-G treated cells to be significantly more resistant to S1P concentrations compared to the control (Figures 2c & d), and again this difference was recapitulated in multiple biological replicates (Supplementary Figure 3b).
Figure 2. O-GlcNAc controls the sensitivity of cells to S1P-mediated contraction.
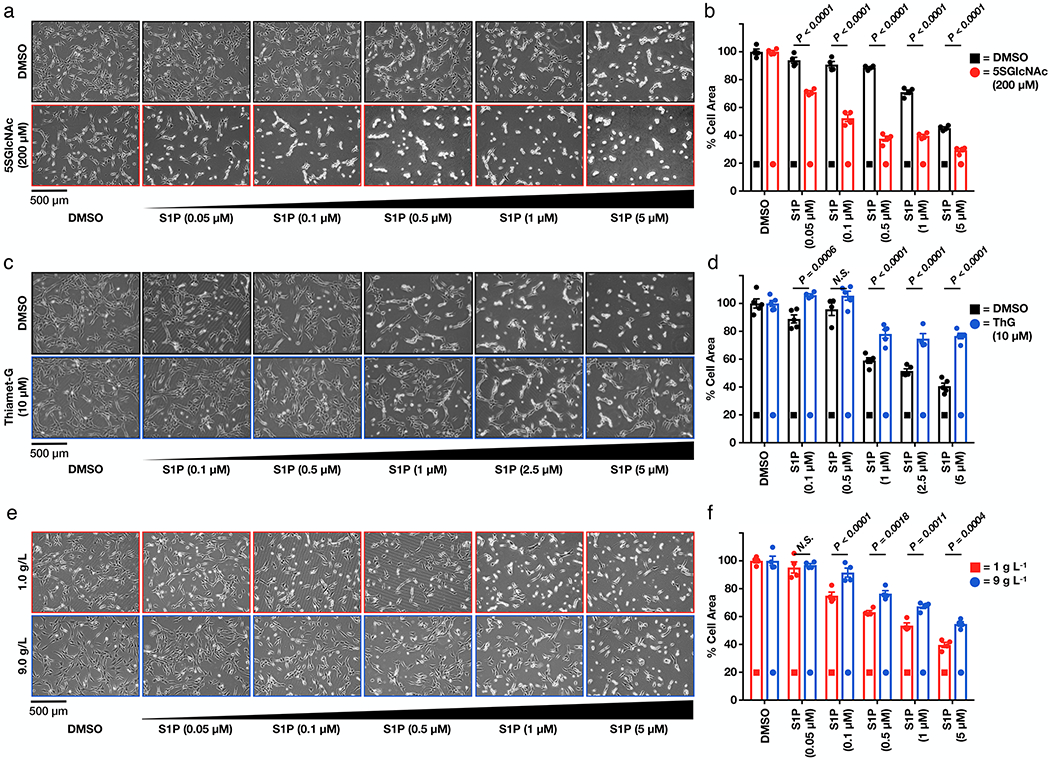
a) Lowering O-GlcNAc levels increases the sensitivity of NIH3T3 cells to S1P induced cell contraction. NIH3T3 cells were pre-treated with either DMSO or 5SGlcNAc before addition of the indicated concentrations of S1P. The contraction phenotype was then visualized using bright-field microscopy. b) Quantitation of the data in (a). Results are the mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test. c) Raising O-GlcNAc levels decreases the sensitivity of NIH3T3 cells to S1P induced cell contraction. NIH3T3 cells that had been treated with either DMSO or Thiamet-G before addition of the indicated concentrations of S1P. The contraction phenotype was then visualized using bright-field microscopy. d) Quantitation of the data in (c). Results are the mean ± SEM of the relative culture plate area taken-up by cells in at least four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test. e) Glucose concentration controls the sensitivity of NIH3T3 cells to S1P induced cell contraction. NIH3T3 cells were cultured in the indicated amounts of glucose before addition of the indicated concentrations of S1P. The contraction phenotype was then visualized using bright-field microscopy. f) Quantitation of the data in (e). Results are the mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test.
As mentioned in the introduction, O-GlcNAc modifications rise due to higher levels of circulating glucose, with important consequences in diabetes. This observation and others raised the possibility that the modification could act as a nutrient sensor to control S1P-dependent signaling. We therefore cultured NIH3T3 cells in media containing either 1 or 9 g L−1 glucose for 48 h, resulting in the expected differences in O-GlcNAc levels (Extended Data Figure 2e). We found that the cells cultured in low glucose were indeed more sensitive to S1P-mediated contraction compared to those grown in high glucose (Figures 2e & f and Supplementary Figures 3c). Together these results indicate that O-GlcNAc acts as a molecular buffer to control S1P signaling that culminates in fibroblast contraction.
O-GlcNAc controls signaling through the S1PR2 receptor.
S1P is an endogenous signaling lipid13 produced intracellularly, and it can then be transported outside of the cell and signal through a family of five G-protein coupled receptors (GPCRs) (S1PR1 through S1PR5, Supplementary Figure 4). Coupling of these GPCRs to different classes of G-proteins results in the activation of several associated downstream signaling pathways. S1P plays roles in many biological events, including cell survival, proliferation, motility, and adherence. Therefore, it is not surprising that it has been shown to contribute to diverse areas of human health and disease, such as cardiovascular development and control of blood pressure, regulation of immune cell migration, and wound healing14–17.
We next used RT-PCR to determine which of the S1PRs were actively transcribed in our fibroblasts and found mRNA for all five receptors (Extended Data Figure 3a). Given that this approach was unable to narrow down the potential S1PR responsible, we turned to small molecule pharmacology. Specifically, we first treated NIH3T3 cells with either DMSO or 5SGlcNAc (200 μM) to modulate O-GlcNAc levels as above. We then added individual selective antagonists of S1PR1 through S1PR4 (1 μM) for 10 min immediately followed by S1P (100 nM). Visualization of the cellular contraction phenotype demonstrated that only blockage of S1PR2 signaling could prevent contraction (Extended Data Figures 3b & c), indicating that the other three receptors (S1PR1, R3, and R4) are not involved. S1PR5 does not yet have a selective antagonist, and therefore we could not rule it out with these data. However, when we treated NIH3T3 cells with a selective agonist of this receptor, we observed no contraction regardless of O-GlcNAc levels (Extended Data Figures 3d & e). Next, we tested whether S1PR2 agonism alone is sufficient to activate the pathway by exposing DMSO- or 5SGlcNAc-treated cells to different concentrations of a selective S1PR2 agonist. We found that low concentrations of this agonist (1 μM) resulted in cell contraction under low O-GlcNAc levels while a higher concentration (5 μM) induced the phenotype universally (Extended Data Figures 3f & g). Finally, we knocked down S1PR2 using RNAi and found that S1P (100 nM) did not induce cell contraction under either normal or reduced O-GlcNAc levels (Extended Data Figures 3h & i). Together these results demonstrate that O-GlcNAc controls a signaling pathway that is likely downstream of S1PR2.
O-GlcNAc controls a canonical actin-contraction pathway.
Previous work has shown that stressed collagen-matrix contraction by fibroblasts is dependent on Rho kinase18. Therefore, we decided to focus on S1P signaling through Rho GTPase, where S1PR agonism results in activation of Rho and Rho kinase (ROCK1/2) 19, ultimately giving actin contraction (Figure 3a and Supplementary Figure 4) 20. More specifically, activated ROCK1/2 will phosphorylate myosin light chain (MLC) at two serines (Thr18 & Ser19) resulting in the formation of an active myosin complex and actin filament contraction21. To avoid unwanted activation and terminate this pathway, cells are equipped with molecular brakes in the form of the phosphatase regulatory subunit MYPT1 that mediates the dephosphorylation MLC. Previous work has shown that ROCK1/2 can also phosphorylate MYTP1 at Thr696 and Thr85322-24. This results in the deactivation of MYPT125, effectively cutting the breaks on the pathway and ensuring myosin activation.
Figure 3. O-GlcNAc levels control S1P-mediated phosphorylation of MLC and MYPT1.
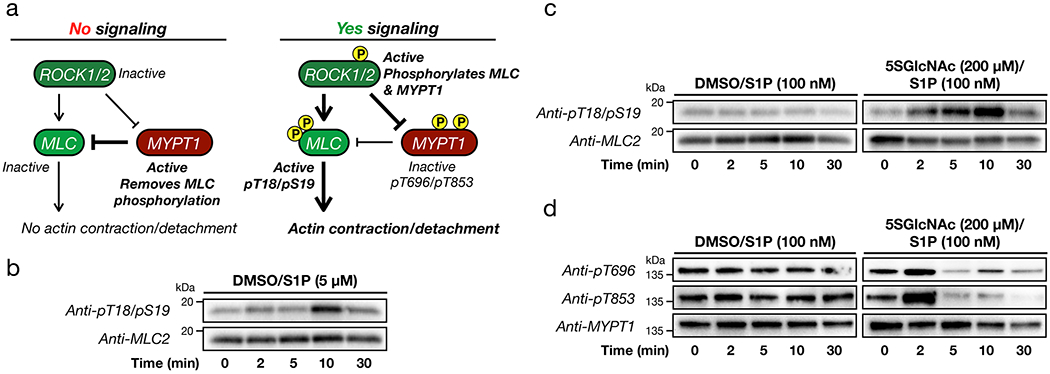
a) Schematic of the ROCK-signaling pathway that can result in cellular contraction. Under no or low signaling conditions ROCK does not phosphorylate MLC or MYPT1, and MYPT1 remains active to dephosphorylate MLC. Upon induction of signaling, ROCK phosphorylates both MLC to activate contraction and MYPT1 to inactivate dephosphorylation, effectively pushing on the gas and cutting the brake. b) MLC is phosphorylated under high S1P signaling. NIH3T3 cells were treated with S1P for the indicated lengths of time. MLC phosphorylation, and thus activation, was visualized using Western blotting. c) MLC is phosphorylated under low S1P signaling when O-GlcNAc levels are reduced. NIH3T3 cells were treated with either DMSO or 5SGlcNAc followed by S1P for the indicated lengths of time. MLC phosphorylation, and thus its activation, was visualized using Western blotting. d) MYPT1 is phosphorylated under low S1P signaling when O-GlcNAc levels are reduced. NIH3T3 cells were treated with either DMSO or 5SGlcNAc followed by S1P for the indicated lengths of time. MYPT1 phosphorylation, and thus its deactivation, was visualized using Western blotting. The data in b-d is representative of 2 biological replicates.
To test whether ROCK1/2 activity was required for our observed contraction phenotype, we treated NIH3T3 cells with either DMSO or 5SGlcNAc (200 μM) for 16 h, followed by the ROCK1/2 inhibitor Y27632 (10 μM) for an additional hour before addition of S1P (0.05 - 5 μM). We observed no contraction of cells at any concentration of S1P (Extended Data Figure 4). This result confirms the importance of ROCK1/2, so we moved on to explore the phosphorylation status of its targets MLC26 and MYPT127,28. We first treated NIH3T3 cells with normal O-GlcNAc levels to a high concentration of S1P (5 μM), which results in their contraction. After different lengths of time, we analyzed the lysates by Western blotting (Figure 3b). As expected, we observed increasing phosphorylation of MLC at Ser18/Thr19 that corresponded well with the overall timing of cell contraction. Next, we performed a similar analysis of NIH3T3 cells that had been pre-treated with either DMSO or 5SGlcNAc (200 μM) for 16 h before addition of a low concentration of S1P (100 nM) (Figure 3c & d). In cells treated with DMSO, we found no detectable increase in MLC or MYTP1 phosphorylation. However, in cells treated with 5SGlcNAc, we detected a rapid increase in phosphorylation of both MLC and MYPT1. Importantly, the observed oscillation of phosphorylation on MYTP1 is consistent with previous reports and relates to the function of MYTP1 as its own phosphatase and therefore a biological timer of this signaling pathway29.
MYPT1 is heavily and dynamically O-GlcNAc modified.
We next set out to identify which protein in the Rho/ROCK signaling pathway is potentially regulated by O-GlcNAc. We chose to focus on MLC and MYPT1 first, as they are known to be O-GlcNAc modified30,31. To ascertain the O-GlcNAc modification status of these proteins, we took advantage of chemoenzymatic labeling (Supplementary Figure 5) 32. We first used this method to install a cleavable biotin-tag onto all endogenous O-GlcNAc modifications in NIH3T3 cell lysates. After incubation with streptavidin beads and washing, the O-GlcNAc modified proteins were eluted and analyzed by Western blotting, and we identified both MLC and MYPT1 to be O-GlcNAc modified (Figure 4a). Notably, while both MLC and MYPT1 are O-GlcNAc modified, when we normalized the amount of input to our O-GlcNAc pulldown, we found that MYPT1 was modified at dramatically higher levels compared to MLC and similar to the constitutively O-GlcNAc-modified Nup62 (Figure 4b). Therefore, we chose to move forward with MYPT1 as the most likely candidate to be regulated in the pathway.
Figure 4. MYPT1 is heavily and dynamically O-GlcNAc modified near its ROCK-binding domain.
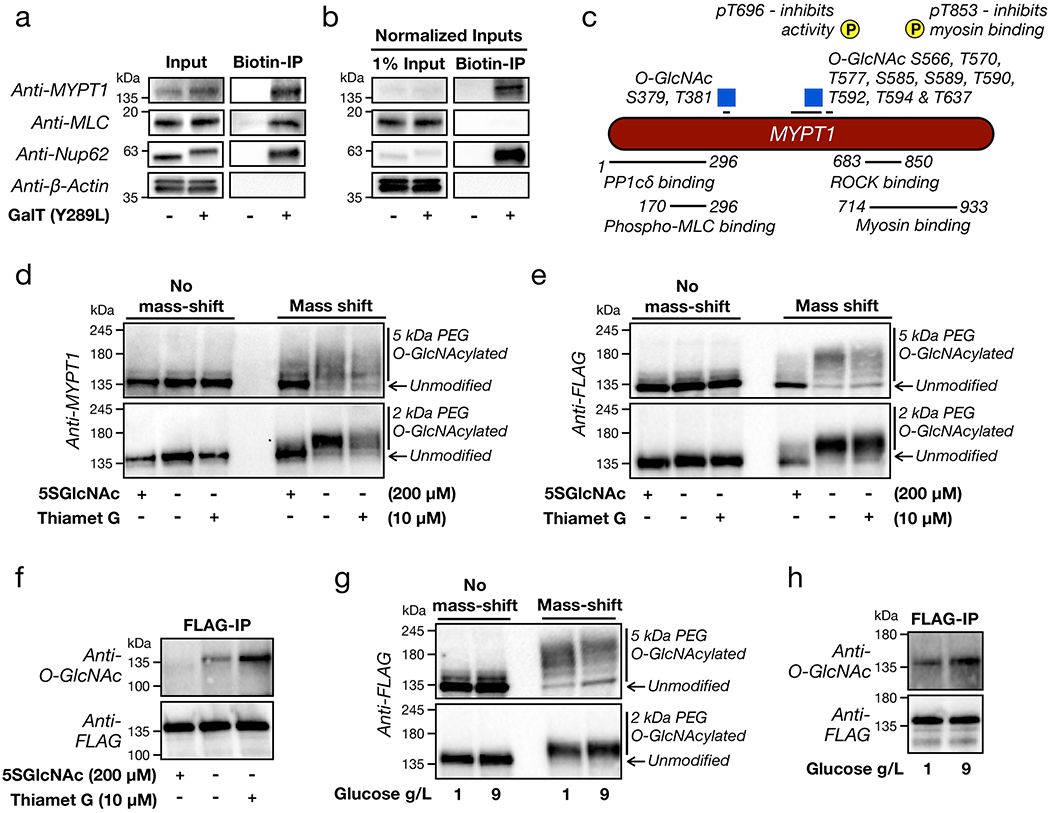
a) MLC and MYPT1 are both O-GlcNAc modified. O-GlcNAc modified proteins from NIH3T3 cells were enriched using chemoenzymatic labelling and analyzed by Western blotting. Nucleoporin 62 (Nup62) is a known, heavily O-GlcNAc modified protein, and β-actin serves as a negative control. b) MYPT1 is highly O-GlcNAc modified, while MLC is not. the samples in (a) were normalized for inputs and analyzed by Western blotting. c) Schematic of identified MYPT1 O-GlcNAc sites, as well as the two deactivating phosphorylation sites and regions of MYPT1 responsible for critical protein-protein interactions. d) Endogenous MYPT1 is heavily and dynamically O-GlcNAc modified. NIH3T3 cells were treated with either 5SGlcNAc, Thiamet-G, or DMSO vehicle. The O-GlcNAc modified proteins were then subjected to chemoenzymatic modification and then “mass-shifted” by PEGylation causing the modified fraction of proteins to run at higher molecular weights when analyzed by Western blotting. e-h) NIH3T3 cells stably expressing FLAG-tagged MYTP1 were treated under the indicated conditions before analysis by either mass-shifting or IP-Western blot. The data in a, b, & d-i are representative of at least 2 biological replicates.
Recent proteomics experiments using the same chemoenzymatic strategy have identified many different endogenous MYPT1 O-GlcNAc sites33–35 localized around two different regions of the protein (Figure 4c). Modification at Ser379 and Thr381 is located near the portion of MYPT1 responsible for binding the phosphatase catalytic subunit PP1cδ and phosphorylated MLC36–38. The second region contains eight identified O-GlcNAc sites (Ser566, Thr570, Thr577, Ser585, Ser589, Thr590, Thr592 & Thr594) clustered within a serine/threonine rich region, as well as ninth site more distant at Thr637. These modification sites are located closer to the inhibitory phosphorylation sites and the portions of MYPT1 responsible for interacting with ROCK1/236–39. Despite the fact that MYPT1 is heavily O-GlcNAc modified, the exact stoichiometry of the modification had not been previously measured to our knowledge. To accomplish this, we took advantage of the chemoenzymatic methodology (Supplementary Figure 5), but instead of installing an enrichment tag, we modified each O-GlcNAc moiety with a polyethylene glycol (PEG) chain of either 2 or 5 kDa in molecular weight. This mass-shifting approach causes the O-GlcNAc modified fraction of a protein to run higher on an SDS-PAGE gel for analysis by Western blotting40,41. We applied this technique to NIH3T3 cells under three sets of conditions: 5SGlcNAc treatment (200 μM), DMSO, or Thiamet-G treatment (10 μM). We then visualized MYTP1 by Western blotting (Figure 4d). Under basal O-GlcNAc levels (i.e., DMSO treatment), we found that MYTP1 is indeed highly modified with essentially 100% of the protein running at mass-shifted molecular weights, and these modifications are largely removed upon treatment with 5SGlcNAc. With Thiamet-G treatment, we observed some potential further upward shift of the MYPT1 bands, suggesting an increase in O-GlcNAc levels. However, the most obvious change was an overall decrease in our ability to detect MYPT1. We believe that this is due to the reduced affinity of some antibodies to highly PEGylated proteins, which we previously found for Nup6241. To overcome this issue, we next generated NIH3T3 cells that stably express a FLAG-tagged version of MYPT1 (Supplementary Figure 6a) and performed the same mass-shifting analysis (Figure 4e). Again, we saw near total O-GlcNAc modification of MYPT1, and we confirmed that the modification stoichiometry could be dialed down or up upon 5SGlcNAc or Thiamet-G treatment. Unfortunately, we still detected some loss of anti-FLAG signal upon Thiamet-G treatment. Therefore, we next used an anti-FLAG immunoprecipitation (IP) to enrich MYPT1 and blotted against O-GlcNAc, confirming, that the protein’s modification status can be both down- and upregulated by inhibitor treatment (Figure 4f). Finally, we used both mass-shifting and anti-FLAG IP to demonstrate that MYTP1 O-GlcNAc levels will change in response to alterations in glucose concentration (1 vs. 9 g L−1) in the media (Figures 4g & h). These glucose-responsive differences in O-GlcNAc were smaller than those created by inhibitor treatment, in agreement with their more subtle effect on the cellular contraction phenotype (Figures 2e & f).
MYTP1 O-GlcNAc modification blocks its phosphorylation.
Previous experiments demonstrated that the serine/threonine region of MYPT1 slightly inhibited its binding to ROCK39, and we hypothesized that O-GlcNAcylation in this location would increase this inhibition. To test this possibility, we generated mutant MYPT1 constructs (Extended Data Figure 5a) using human MYPT1, which is 93% identical to the mouse protein and enabled us to use RNA interference to selectively knockdown the endogenous MYPT1 in NIH3T3 cells (Supplementary Figure 6b). The first mutant, MYPT1(S/TtoA), contained serine and threonine mutations at each of the 8 sites in this region previously identified as O-GlcNAc modified by mass spectrometry33–35. Because OGT has been found to modify multiple residues in serine/threonine rich regions that cannot be readily differentiated by mass spectrometry, we also generated three deletion mutants: MYPT1(d564-578) and MYPT1(d588-602), each lacking one of the two stretches of serines/threonines, and MYPT1Δ where the entire serine/threonine-rich region (residues 550-600) was deleted. All of these mutants displayed similar expression to stably expressed MYPT1 (Supplementary Figure 6a). With these cell lines in hand, we used RNAi to knock down the endogenous copy of MYPT1, treated them with S1P (50 or 100 nM), and measured cellular contraction (Extended Data Figure 5b). We observed essentially no contraction in MYPT1, MYPT1(S/TtoA), or MYPT1(d564-578) expressing cells. In contrast, we found MYPT1Δ cells to have significant levels of contraction and MYPT1(d588-602) to have an intermediate phenotype. These results suggested that the entire domain contains O-GlcNAc modifications that are important for controlling cell contraction. We then used the mass-shifting assay and IP-Western blotting to examine the O-GlcNAc stoichiometry of MYPT1Δ and found the levels noticeably but not completely reduced (Extended Data Figure 6), consistent with a reasonable fraction of O-GlcNAc in this region.
Next, we subjected MYPT1- or MYPT1Δ-expressing cells to the full range of S1P concentrations (0.05 - 5 μM) and measured the amounts of cell contraction (Figures 5a & b). We found that MYPT1Δ cells were significantly more sensitive to S1P-mediated contraction. Again, this phenotype was observed in multiple biological replicates (Supplementary Figure 7). We then visualized the phosphorylation status of MLC and MYPT1Δ after treatment with a low concentration of S1P (100 nM) and observed less phosphorylation of both proteins in the cells expressing MYPT1 versus those expressing MYPT1Δ (Figure 5c). These results are consistent with a model where O-GlcNAc in the serine/threonine region inhibits MYPT1 phosphorylation and is lost in the MYTP1Δ mutant (Figure 5d).
Figure 5. MYPT1 O-GlcNAc modification prevents its phosphorylation and deactivation, thereby inhibiting S1P-mediated contraction.
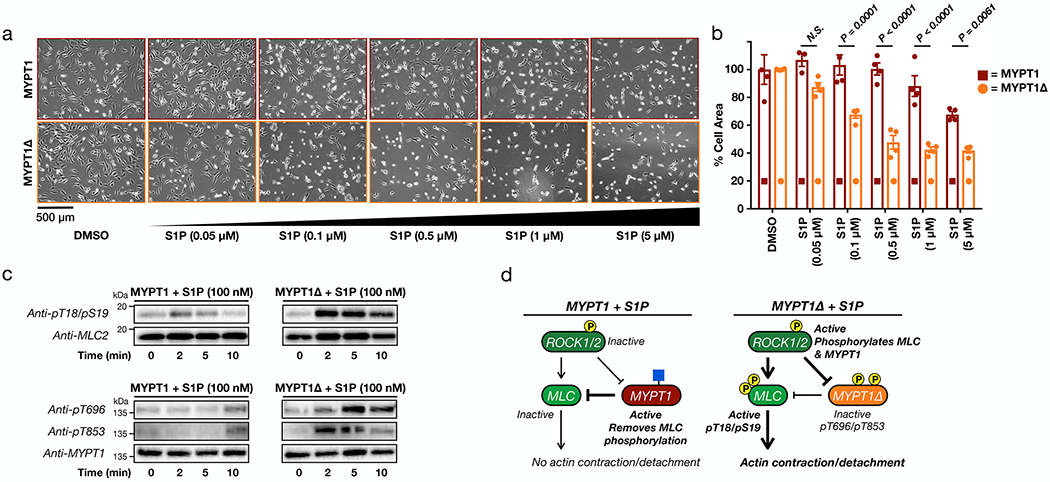
a) MYTP1Δ expression sensitizes cells to S1P-mediated contraction. NIH3T3 cells stably expressing either MYPT1 or MYTP1Δ were transfected with RNAi to downregulate endogenous MYTP1. They were then treated with the indicated concentrations of S1P. The contraction phenotype was visualized using bright-field microscopy. b) Quantitation of the data in (a). Results are the mean ± SEM of the relative culture plate area taken-up by cells in at least three randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test. c) MYPT1Δ is more readily phosphorylated upon low S1P signaling. NIH3T3 cells stably expressing either MYPT1 or MYTP1Δ were transfected with RNAi to downregulate endogenous MYTP1. They were then treated with S1P for the indicated lengths of time. MLC and MYPT1/MYPT1Δ phosphorylation were analyzed by Western blotting. These data are representative of 2 biological replicates. d) Our model. When MYPT1 is O-GlcNAc modified it is more resistant to phosphorylation and deactivation, thus maintaining the brakes on contraction. When MYTP1 O-GlcNAc is lost, it is phosphorylated, resulting in MLC phosphorylation and contraction.
We were concerned that removal of 50 amino acids could affect the functions of MYPT1. For example, MYPT1 can form a complex with OGT and direct it to protein substrates31. However, we found no apparent changes in O-GlcNAc levels or OGT expression comparing MYPT1Δ to MYPT1 (Supplementary Figure 6c). We also tested whether MYPT1Δ was still an active phosphatase. We subjected MYPT1Δ cells to RNAi to remove endogenous MYPT1 and treated with S1P (100 nM) for a long time course of 180 min. We then examined the phosphorylation of MLC after 60 and 180 min. As expected, we observed phosphorylation of MLC at 60 min, but this modification was removed by 180 min (Extended Data Figure 7a), demonstrating that MYPT1Δ can still dephosphorylate MLC to turn off the S1P-signaling pathway. We also observed the cells return to a relaxed state on the culture plate after 120 min (Extended Data Figure 7b). Together these data demonstrate that MYPT1Δ is still an active phosphatase despite deletion of the serine/threonine rich region.
Next, we set out to test whether O-GlcNAc inhibits the interaction between MYPT1 and ROCK. We treated NIH3T3 cells expressing FLAG-tagged MYPT1 with either DMSO or 5SGlcNAc (200 μM) and performed an anti-FLAG co-immunoprecipitation using the Catch and Release system (Thermo). We then blotted against endogenous ROCK, normalized the level to overall protein enrichment by Coomassie staining, and found that loss of O-GlcNAc resulted in a significant increase in the amount of ROCK interacting with MYPT1 (Extended Data Figure 8). These results support a model where O-GlcNAc blocks the protein-protein interaction between MYPT1 and ROCK.
Finally, we wanted to rule out the possibility that deletion of the serine/threonine region of MYPT1 has an effect beyond inhibiting O-GlcNAc and/or that O-GlcNAc on another protein contributes to the observed contraction. If neither of these possibilities are true, we would predict that cells expressing MYPT1Δ would respond to S1P in the same way as cells expressing MYPT1 treated with 5SGlcNAc. Accordingly, we compared three different conditions: cells stably expressing MYPT1, cells stably expressing MYPT1 in the presence of 5SGlcNAc, and cells stably expressing MYPT1Δ. In all cases RNAi was used to remove the endogenous copy of MYPT1. Treatment of these populations of cells with S1P (0.05 - 5 μM) demonstrated that MYPT1Δ sensitized cells to the same extent as 5SGlcNAc treatment (Extended Data Figure 9a & b) and (Supplementary Figure 8a). We also performed the inverse experiment, where one would predict that increasing O-GlcNAc in MYPT1Δ cells would not rescue the contraction phenotype. We treated MYPT1Δ expressing cells with DMSO or Thiamet-G (10 μM) before subjecting them to S1P (0.05 - 5 μM) and quantitation of cell contraction (Extended Data Figure 9c & d) and (Supplementary Figure 8b). These data are consistent with our model that O-GlcNAc on MYPT1 is at least largely responsible for regulating cell contraction.
O-GlcNAc inhibits S1P-caused collagen-matrix contraction.
Wound healing involves several overlapping phases carried out by dermal cells, including fibroblasts42,43. Fibroblasts migrate and proliferate into the matrix formed by the fibrin clot, secrete extracellular matrix (ECM) components, and differentiate into myofibroblasts to generate granulation tissue. The fibroblasts also contract themselves and the ECM to facilitate wound closure and remodeling. Three-dimensional collagen matrices are in vitro models to study fibroblast behavior in tissue-like environments18,44, and a stressed collagen-matrix serves as a model for wound contraction (Supplementary Figure 9). In this model, dermal fibroblasts are mixed with collagen, allowed to polymerize, and then cultured for 16 - 48 h. During this time, the cells form actin stress fibers and focal adhesions. Subsequent release of the matrix and addition of signaling molecules results in matrix contraction. To determine if O-GlcNAc levels control this process, we first treated dermal fibroblasts in 2D culture with either DMSO or 5SGlcNAc (200 μM) (Supplementary Figure 10a) for 16 h followed by S1P (0.05 - 5 μM). Similar to the NIH3T3 cells, we observed increased sensitivity of fibroblasts with low O-GlcNAc levels to S1P (Figures 6a & b and Supplementary Figure 10b).
Figure 6. O-GlcNAc controls S1P-mediated collagen matrix contraction.
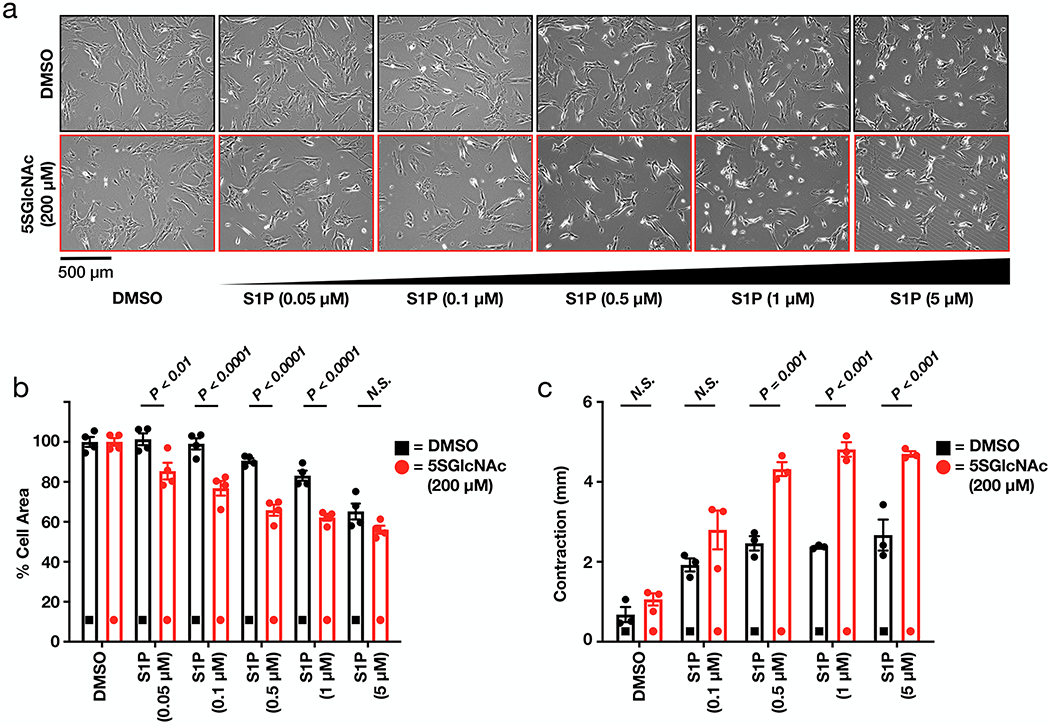
a) Lowering O-GlcNAc levels increases the sensitivity of human dermal fibroblasts to S1P induced cell contraction in 2D culture. Fibroblasts were pre-treated with either DMSO or 5SGlcNAc before addition of the indicated concentrations of S1P. The contraction phenotype was then visualized using bright-field microscopy. b) Quantitation of the data in (a) Results are the mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test. c) Lowering O-GlcNAc increased the contraction of stressed collagen matrices in response to S1P. Contraction in millimeters was quantified as change in each matrix diameter from the initiation (t = 0 min) to termination (t = 30 min) of the assay. Results are the mean ± SEM of the millimeters contracted from three separate experiments. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test.
We then suspended dermal fibroblasts in collagen and plated them in 24-well dishes. After 30 h, the cells were treated with either DMSO or 5SGlcNAc (200 μM) and incubated for an additional 16 h. The fibroblasts were then treated with S1P (0.1 - 5 μM) and contraction was initiated by releasing the matrices from the culture plate. Images were taken immediately (t = 0 min) and after 30 min (Supplementary Figure 11). The extent of contraction was quantified by comparing the diameter of a matrix at 0 min to the diameter of the same matrix after 30 min (Figure 6c). Consistent with previous reports11, S1P caused matrix contraction by dermal fibroblasts with normal O-GlcNAc levels, but we observed significantly more contraction by cells treated with 5SGlcNAc. Together, these results demonstrate that O-GlcNAc levels control the contraction of human dermal fibroblasts in response to S1P and that this observation extends to a physiologically relevant model of wound healing.
DISCUSSION
These results strongly suggest that that MYPT1 O-GlcNAc functions as a nutrient sensor to regulate the sensitivity of fibroblasts to S1P-mediated contraction (Extended Data Figure 10). At this time, we do not yet know the biological consequences of this regulation in more complex setting, but there is reason to believe that they could be numerous. In fibroblasts, S1P signaling has been shown to increase wound healing in mice45,46. We speculate that the elevated O-GlcNAc levels in diabetes may attenuate S1P signaling with detrimental consequences for fibroblast biology in wound healing. Our results in stressed-collagen matrices support this hypothesis, and we plan to continue this line of investigation. Furthermore, MYPT1 activity is critical for controlling smooth muscle contraction and therefore blood pressure. Several different GPCR agonists, angiotensin, etc., activate the same Rho/ROCK pathway leading to smooth muscle contraction47–49. Given that O-GlcNAc of MYTP1 acts downstream of ROCK, we hypothesize that this modification will control the sensitivity of smooth muscle cells, with potential implications in atherosclerosis and heart disease.
In summary, we present a biological model where O-GlcNAc modification of MYPT1 maintains its phosphatase activity by inhibiting phosphorylation marks by ROCK1/2. This O-GlcNAcylation can then function in its well-established role as a sensor of the cellular environment and state to control the responsiveness of cells to contractile stimuli. In the past, the documented interaction between OGT and MYPT1 was thought to play only an adapter function for OGT substrate selection31, but we demonstrate here that O-GlcNAcylation of MYPT1 has its own important direct functions. Very recently, O-GlcNAc on MYPT1 was also shown to inhibit its phosphorylation by CDK1 with important consequences for the cell cycle50. We do not yet know the precise molecular mechanism by which O-GlcNAc inhibits MYPT1 phosphorylation. For example, we do not know if there are specific O-GlcNAc modification sites in the MYPT1 serine/threonine rich region that are most consequential, but our data suggests that our observations are likely a consequence of multiple O-GlcNAc modifications spread heterogeneously across the stretches of consecutive serine/threonine resides in this region. Thus, we believe that any individual site responsible for our observed phenotype probably does not exist. Our Co-IP suggests that O-GlcNAc inhibits the ROCK/MYPT1 interaction. However, we cannot rule out the contribution of additional mechanisms. For example, O-GlcNAc might result in a conformation of MYPT1 that is refractory to phosphorylation. Finally, our experiments comparing the effects of 5SGlcNAc or Thiamet-G treatment to MYTP1Δ expression cannot completely rule out some contribution of other O-GlcNAc modified proteins. Unfortunately, this “loss-of-modification” experiment is a general limitation of O-GlcNAc studies in cells, as to-date there are no robust methods for the site- or even protein-selective introduction of O-GlcNAc. However, we believe that the breadth of our experiments strongly support both our conclusions and the future exploration of these modifications in a variety of biological contexts.
ONLINE METHODS
Synthesis of known small molecules.
Known compounds Thiamet-G121 and Ac45SGlcNAc102 were synthesized according to literature procedures. Both were dissolved as 1,000x stocks in DMSO.
Cell culture.
Mouse embryonic fibroblast cell-line NIH3T3 (ATCC) was propogated in DMEM medium (Genessee Scientific) supplemented with 10% fetal calf serum (FCS, Atlanta Biologics). NIH3T3 cell-lines stably expressing either FLAG-tagged MYPT1, MYPT1(S/TtoA), MYPT1(d564-578), MYPT1(d588-602) or MYPT1Δ were grown in DMEM + 10% FCS supplemented with 250 ng mL−1 puromycin (1 mg mL−1 stock in H2O). Human dermal fibroblast cell-line BJ-5ta (ATCC) was propagated in 4:1 DMEM:Medium 199 (ATCC) + 10% fetal bovine serum (FBS, Atlanta Biologics) supplemented with 10 ng mL−1 hygromycin B (10 mg mL−1 stock in H2O). All cell-lines were grown in a humidified incubator at 37 °C and 5% CO2 atmosphere.
Antibodies.
All antibodies were incubated in OneBlock™ Western-CL blocking buffer purchased from Genesee Scientific (20-313). Anti-FLAG-Tag (2368S), anti-MYPT1 (2634S), anti-MLC2 (3672S), anti-pT18/pS19 MLC (3674S), anti-pT696 MYPT1 (5163S), anti-pT853 MYPT1 (4563S), anti-OGT (24083S), anti-Caspase-3 (9762), and anti-Cleaved Caspase-3 (9664) were purchased from Cell Signaling Technology. Anti-Nup62 (610497) was purchased from BD Biosciences. Anti-RL2 (MA1-072) was purchased from ThermoFisher Scientific. Anti-β-actin (A5441) and Anti-Rock1/2 (07-1458) were purchased from MilliporeSigma. Horseradish peroxidase (HRP)-conjugated secondary antibodies were purchased from Jackson ImmunoResearch.
General procedure for Western blotting.
Post lysis, proteins were separated by SDS-PAGE (200 V, 45 min) before being transferred to a PDVF membrane (Bio-Rad) with a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad, 20 V, 1 h). Western blots were blocked in OneBlock™ Western-CL blocking buffer (Genesee) for 1 h rt. Blots were then incubated with primary antibody (1:1,000 dilution) in fresh blocking buffer at 4 °C overnight. Following overnight incubation, blots were washed in TBST (Cell Signaling, 3× 10 min) and incubated with (HRP)-conjugated secondary antibody (1:10,000 dilution) in fresh blocking buffer for 1 h at rt. Once complete, blots were again washed in TBST (3× 10 min). Blots were developed using ECL reagents (Bio-Rad) and the ChemiDoc XRS+ molecular imager (Bio-Rad, Bio-Rad Image Lab 4.1).
General procedure for 2D contraction assay.
For initial characterization of phenotype, NIH3T3 cells were seeded at 1 × 105 cells in 6-well dishes 8 h prior to treatment with either DMSO vehicle, 5SGlcNAc (200 μM), or Thiamet-G (10 μM). Following incubation for 16 or 20 h for 5SGlcNAc or Thiamet-G/DMSO respectively, cells were treated with FCS (Atlanta Biologics, 10% by volume) or fresh serum-free media for 30 min. Each well was analyzed using a Leica Microscope (Leica Acquire 3.4.1) to capture bright-field microscopy images at 20x magnification. Quantification of contraction phenotype was determined by taking the mean ± SEM of the relative culture plate area covered by cells in four randomly selected frames per well. Images were analyzed using Adobe Photoshop (Photoshop CC 2019). Background pixels were selected using the Magic Wand Tool in Adobe Photoshop’s toolbar and subtracted from total pixels. This value was then normalized using a control, untreated well allowing for quantification of the difference in area taken up by cells before and after contraction. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test performed in Prism (Graphpad Prism 7). Representative images for each treatment were selected from one of the four frames used for quantification.
Serum screen.
Seeded NIH3T3 cells were treated with 5SGlcNAc (200 μM) as stated above. After 16 h, wells were treated with one of the following; FCS (10% v/v), replacement with serum-free media, VEGF-165 (100 ng mL−1), EGF (100 ng mL−1), IGF (10 ng mL−1), TGF-β (100 ng mL−1), TNF-α (100 ng mL−1), FGF-8 (150 ng mL−1), PDGF-AA (100 ng mL−1), PDGF-BB (100 ng mL−1), IL-6 (100 ng mL−1), LPA (20 μM), Lipid Mixture (MilliporeSigma, L0288-100ML), or S1P (100 nM). After a 30 min incubation at 37 °C, contraction for each treatment was measured as indicated above.
OGT inhibition with ST060266.
Seeded NIH3T3 cells were treated with covalent OGT inhibitor ST060266 (MilliporeSigma, 200 μM) for 16 h before treatment with S1P (100 nM). Contraction was measured as indicated above. OGT inhibition was determined using the pan O-GlcNAc antibody anti-RL2.
OGT knockdown with RNAi.
Custom plasmids containing either mOGT siRNA (sequence: 5’-GAUUAAGCCUGUUGAAGUCTT-3’) or scramble siRNA were purchased from Genescript. NIH3T3 cells were transfected using Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher Scientific; 13778030) according to the manufacturer’s protocol. After 24 h, cells were seeded at 1 × 105 cells in 6-well dish for an additional 24 h before treatment with S1P (100 nM). Contraction was characterized as stated above. OGT knockdown was determined via Western blotting using anti-OGT to visualized OGT expression and anti-RL2 to visualized global O-GlcNAcylation.
5SGlcNAc/DMSO/Thiamet-G S1P concentration course.
Seeded NIH3T3 cells were treated with either DMSO vehicle, 5SGlcNAc (200 μM), or Thiamet-G (10 μM). After 20, 16, or 20 h respectively, wells were treated with S1P (0.05 - 5 μM) for 30 min. Contraction was characterized consistent with previously described experiments.
Relaxation assay S1P time course.
NIH3T3 cells were treated with either DMSO vehicle or 5SGlcNAc (200 μM) for 16 h before addition of S1P (100 nM). Images of each well were taken at 0, 30, 60, 120, and 180 min. Contraction was characterized as stated above.
High/low glucose S1P concentration course.
NIH3T3 cells were cultured in media containing either high (9 g L−1) or low (1 g L−1) concentrations of glucose for 24 h prior to being seeded as indicated above and incubated for an additional 24 h at which point cells were treated with S1P (0.05 - 5 μM) for 30 min. Contraction was characterized and changes in global O-GlcNAcylation were visualized with Western blotting using anti-RL2 as a pan O-GlcNAc antibody.
S1PR1-4 antagonist screen.
Seeded NIH3T3 cells were treated with 5SGlcNAc. After 16 h, cells were treated with one of the following; S1PR1 antagonist W146 (1 μM), S1PR2 antagonist JTE013 (1 μM), S1PR3 antagonist TY52156 (1 μM), S1PR4 antagonist CYM50358 (1 μM) for 10 min. Each well was then treated with S1P (100 nM) for 30 min and contraction was characterized as stated above.
S1PR2 and S1PR5 agonist screen.
Seeded NIH3T3 cells were treated with 5SGlcNAc. After 16 h, cells were treated with either S1PR2 agonist CYM5220 (1 or 5 μM) or S1PR5 agonist A971432 (1 μM) for 30 min. Contraction was characterized consistent with previously described experiments.
S1PR2 knockdown with RNAi.
NIH3T3 cells were seeded at 30% confluency in a 10 cm dish 24 h before transfection. 10 nmole of RNAi targeted against mouse S1PR2 or a scramble sequence was purchased from MilliporeSigma (siRNA ID: SASI_Mm01_00082880) and diluted in 1 mL in nuclease free H2O to make a stock concentration of 10 μM. Cells were transfected with 125 pmole (12.5 μL) of RNAi using Lipofectamine RNAiMAX transfection reagent (ThermoFisher Scientific) according to the manufacturer’s protocol. 24 h post-transfection, cells were seeded 1 × 105 cells in 6-well dishes. After 8 h, wells were treated with either DMSO vehicle or 5SGlcNAc (200 μM) for 16 h. Each well was then treated with S1P (100 nM) for 30 min and contraction was characterized as stated previously.
ROCK1/2 inhibition with Y27632.
NIH3T3 cells were seeded and treated with 5SGlcNAc (200 μM). After 15 h, cells were treated with ROCK1/2 inhibitor Y27632 (10 μM) for 1 h prior to treatment with S1P (0.05 - 5 μM) for 30 min. Contraction was characterized as stated above.
MYPT1 vs MYPT1 mutants S1P concentration course.
NIH3T3 cell-lines expressing human FLAG-tagged MYPT1, MYPT1(S/TtoA), MYPT1(d564-578), MYPT1(d588-602), or MYPT1Δ were transfected at 40% confluency with RNAi targeting mouse MYPT1 (transfection details described in subsequent section). After 24 h, cells were seeded 1 × 105 cells in 6-well dishes and grown for an additional 24 h. After 48 h total, cell-lines were treated with S1P (0.05 or 0.1 μM) for 30 min. Contraction was characterized as stated above.
MYPT1 vs MYPT1Δ S1P concentration course.
NIH3T3 cell-lines expressing human FLAG-tagged MYPT1 or MYPT1Δ were transfected at 40% confluency with RNAi targeting mouse MYPT1 (transfection details described in a subsequent section). After 24 h, cells were seeded 1 × 105 cells in 6-well dishes and grown for an additional 24 h. After 48 h total, cell-lines were treated with S1P (0.05 - 5 μM) for 30 min. Contraction was characterized as stated above.
MYPT1 vs MYPT1Δ relaxation assay S1P time course.
NIH3T3 cell-lines expressing human FLAG-tagged MYPT1 or MYPT1Δ were transfected at 40% confluency with RNAi targeting mouse MYPT1 (transfection details described in a subsequent section). After 24 h, cells were seeded 1 × 105 cells in 6-well dishes and grown for an additional 24 h. After 48 h total, cell-lines were treated with S1P (100 nM). Each well was imaged at 0, 30, 60, and 120 min post S1P treatment. Contraction was characterized as stated above.
MYPT1 vs MYPT1 + 5SGlcNAc vs MYPT1Δ S1P concentration course.
Cell-lines expressing human FLAG-tagged MYPT1 or MYPT1Δ were transfected at 40% confluency with RNAi targeting mouse MYPT1 (transfection details described in a subsequent section). 24 h post transfection, cells were seeded 1 × 105 cells in 6-well dishes. After 8 h, one plate of cells expressing FLAG-tagged MYPT1 was treated with 5SGlcNAc (200 μM) and one plate each of FLAG-tagged MYPT1 or MYPT1Δ were treated with DMSO vehicle. All three sets of cells were incubated for an additional 16 h before treatment with S1P (0.05 - 5 μM) for 30 min. Contraction was characterized as stated above.
MYPT1Δ vs MYPT1Δ + Thiamet-G S1P concentration course.
Cell-lines expressing human FLAG-tagged MYPT1Δ were transfected at 40% confluency with RNAi targeting mouse MYPT1 (transfection details described in a subsequent section). 24 h post transfection, cells were seeded 1 × 105 cells in 6-well dishes for 8 h before treatment with either DMSO vehicle or Thiamet-G (10 μM). Cells were incubated for an additional 20 h at which point cells were treated with S1P (0.05 - 5 μM) for 30 min. Contraction was characterized as stated above.
Human dermal fibroblast S1P concentration course.
Human dermal fibroblast cell-line, BJ-5ta, were seeded 1.5 × 105 cells in a 6-well dish. After 8 h, cells were treated with either DMSO vehicle or 5SGlcNAc (200 μM). Cells were incubated for an additional 16 h before treatment with S1P (0.05 - 5 μM) for 30 min. Contraction was characterized as stated above.
Video characterization of S1P-mediated contraction.
Seeded NIH3T3 cells were treated with either DMSO vehicle or 5SGlcNAc (200 μM) for 16 h. The following videos were taken using a Leica Microscope at 20x magnification: DMSO, DMSO + S1P (100 nM), DMSO + 10% FCS, 5SGlcNAc, 5SGlcNAc + S1P (100 nM), 5SGlcNAc + 10% FCS. For each null video, (DMSO or 5SGlcNAc), plates were placed under microscope and recorded for 10 min. For wells treated with S1P or 10% FCS, video recording started immediately following treatment and continued for 10 min.
Caspase-3 activation Western blot.
NIH3T3 cells were treated with either DMSO vehicle or 5SGlcNAc (200 μM) for 16 h before the addition of S1P (100 nM) for 0, 30, 60, or 120 min at which point cells were collected by trypsinization and washed two times with PBS (2 min, 2,000 x g, 4 °C). Cell pellets were then resuspended in 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) supplemented with 5 mg mL−1 c0mplete mini protease inhibitor cocktail (MilliporeSigma), 5 mg mL−1 PhosSTOP (MilliporeSigma), 1 mM phenylmethylsulfonyl fluoride. Cells were lysed via tip sonication (3× 5 sec on 5 sec off) and cell debris were pelleted (10 min, 10,000 x g, rt). Protein concentration was determined by BCA Assay (Pierce, ThermoScientific), and the lysate was diluted to 4 mg mL−1 in lysis buffer before an equivalent volume of 2x loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol, pH 6.8). 20 μL (40 μg) per sample was loaded per lane. Western blotting was performed according to the General Western Blot procedure described above. Caspase-3 cleavage was visualized using anti-Caspase-3 and anti-Cleaved Caspase-3.
RT-PCR confirming mRNA expression of S1PR1-5 in NIH3T3 cells.
NIH3T3 cell lysate was harvested via trypsinization and washed twice with 1 mL cold PBS (5 min, 2,000 x g, 4 °C). RNA extraction was performed using RNeasy Minikit (Qiagen) according to manufacturer’s protocol. RNA concentration was acquired using UV/Vis absorption and 1 μg RNA per reaction was subjected to RT-PCR. RT-PCR primers for each S1P receptor were designed with their mRNA sequences found using NCBI Gene Search tool (ncbi.nlm.nih.gov/gene). Sequences were copied into a RT-PCR primer design database (MIT Primer3). Primers were generated by accepting all design database default parameters and choosing primer pairs with the highest score. To verify sequence autonomy and avoid sequences that span mRNA introns, amplified region was copied into the UCSC genome browser and a BLAT search was performed. RT-PCR primers for S1PR1 (Forward: 5’-CTCCGGTTTCTTCTCACCCT-3’, Reverse: 5’-TCCCAACAACTTGACCCAGT-3’), S1PR2 (Forward: 5’-GGTGCTGTCTGACCTCTTCT-3’, Reverse: AAGGAGGACATCTGGGAAGC-3’), S1PR3 (Forward: 5’-ATCCAACCCTCACCCTGAAG-3’, Reverse: AGGGAGAGAAGCAAGACCAC-3’), S1PR4 (Forward: 5’-GAGGGCAACTTGACGTGTTT-3’, Reverse: 5’-ATTCAGAACAGGAAGGGCGA-3’), and S1PR5 (Forward: 5’-TGTGGGGAAGTCTTGTTGGT-3’, Reverse: 5’-ACATCACCTGGTTCTGAGCA-3’) were ordered along with primers for GAPDH mRNA (Forward: 5’-AGGCCGGTGCTGAGTATGTC-3’, Reverse: TGCCTGCTTCACCACCTTCT-3’) for use as loading controls (Integrated DNA Technologies). PCR reaction was carried out using Superscript IV One-Step RT-PCR System (Invitrogen) according to manufacturer’s protocol. 10 μL from each PCR reaction was removed from the PCR tube and added to fresh tubes containing 2 μL loading buffer (New England BioLabs). The entire 12 μL was loaded into a 1% agarose DNA gel (500 mg agarose, 5 μL ethidium bromide, 50 mL 1x Bio-Rad TAE buffer) and run for 45 min at 100 V. DNA bands were visualized using the ChemiDoc XRS+ molecular imager (Bio-Rad, Bio-Rad Image Lab 4.1).
MYPT1 and MLC phosphorylation Western blot time course.
General procedure.
NIH3T3 or stable cell-lines were seeded 5 × 105 cells per 10 cm dish. To each plate, S1P at indicated concentration was added and plates were incubated for 0, 2, 5, 10, or 30 min. At each time point, dishes were removed from the incubator, media was decanted, and plates were immediately placed on ice, rinsed once with ice cold PBS, and harvested by scraping into pre-cooled 15 mL falcon tubes. Cells were pelleted by centrifugation (5 min, 2,000 x g, 4 °C). Pellets were resuspended in 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) and lysed via tip sonication (3× 5 sec on 5 sec off). Lysate was centrifuged (10 min, 10,000 x g, rt) and supernatant was moved to a fresh tube. Protein concentration was determined by BCA Assay (Pierce, ThermoScientific) and gel samples were prepared at 2 mg mL−1 with appropriate volumes of 4% SDS buffer and 2x loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol, pH 6.8). 20 μL (40 μg) per sample was loaded per lane. Western blotting was performed according to the General Western Blot procedure described above. Induction of MYPT1 phosphorylation was visualized using anti-pT696 MYPT1 and anti-p853 MYPT1. Activation of MLC was visualized using anti-pT18/S19 MLC2. Expression of MYPT1 and MLC2 were used as loading controls.
MLC activation with high S1P signaling.
To validate S1P’s role in mediating MLC activation, NIH3T3 cells were treated with a high concentration S1P (5 μM) for 0, 2, 5, 10, and 30 min, and worked up as indicated in the previous section. Activation of the pathway was visualized with Western blotting with anti-pT18/S19 MLC and anti-MLC2.
O-GlcNAcylation sensitizes S1P-mediated phosphorylation of MLC and MYPT1.
To indicate the role O-GlcNAcylation plays in MLC activation, 8 h after plating, cells were treated with DMSO vehicle or 5SGlcNAc (200 μM) and incubated for an additional 16 h. To each plate per original treatment (DMSO or 5SGlcNAc) a low concentration S1P (100 nM) was added and plates were incubated for 0, 2, 5, 10, and 30 min and worked up as described previously. Induction of MYPT1 phosphorylation was visualized using anti-pT696 MYPT1 and anti-p853 MYPT1. Activation of MLC was visualized using anti-pT18/S19 MLC2. Expression of MYPT1 and MLC2 with anti-MYPT1 and anti-MLC2 respectively were used as loading controls.
MYPT1 vs MYPT1Δ phosphorylation Western blot time course.
Stable expressing human Wild-Type or Δ550-600 MYPT1 constructs were seeded at 30% confluency 24 h before transfection with RNAi targeting mouse MYPT1. After 24 h, cells were seeded 5 × 105 cells per 10 cm dish and grown for an additional 24 h at which point they were treated with a low concentration of S1P (100 nM) for 0, 2, 5, or 10 min. Cells were worked up as described above. Induction of MYPT1 phosphorylation was visualized using anti-pT696 MYPT1 and anti-p853 MYPT1. Activation of MLC was visualized using anti-pT18/S19 MLC2. Expression of MYPT1 and MLC2 with anti-MYPT1 and anti-MLC2 respectively were used as loading controls.
MYPT1Δ MLC activation time course.
To ensure that MYPT1Δ continues to perform its biological functions, NIH3T3 cells expressing FLAG-tagged MYPT1Δ were seeded at 30% confluency 24 h before transfection with RNAi targeting mouse MYPT1. After 24 h, cells were seeded 5 × 105 cells per 10 cm dish and grown for an additional 24 h at which point they were treated with S1P (100 nM) for 0, 60, and 180 min. Cells were worked up as described above. Activation of MLC was visualized using anti-pT18/S19 MLC2. Expression MLC2 with anti-MLC2 was used as a loading controls.
General procedure for chemoenzymatic labeling.
NIH3T3 cells were collected by trypsinization and washed two times with 1 mL cold PBS (2 min, 2,000 x g, 4 °C). Cell pellets were then resuspended in 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) supplemented with 5 mg mL−1 c0mplete mini protease inhibitor cocktail (MilliporeSigma), 5 mg mL−1 PhosSTOP (MilliporeSigma), 1 mM phenylmethylsulfonyl fluoride. Cells were lysed via tip sonication (3× 5 sec on 5 sec off) and any remaining cell debris was pelleted (10 min, 10,000 x g, rt) and supernatant was moved to fresh tubes. Protein concentration was determined by BCA Assay (Pierce, ThermoScientific), and the lysate was diluted to 1 mg mL−1 in 1% SDS chemoenzymatic buffer (1% SDS, 20 mM HEPES, pH 7.9). Proteins were precipitated by adding the following in order: 3x volume of MeOH, 0.75x volume of CHCl3, and 2x volume of H2O. Tubes were then briefly vortexed and centrifuged (5 min, 13,000 x g, rt). The aqueous phase (top layer) was discarded carefully to avoid disturbing the interface between layers. 2.5x volume of MeOH was added, the mixture was vortexed, and samples were centrifuged (5 min, 13,000 x g, rt). The supernatant was decanted and the protein pellet was allowed to air-dry for 5-10 min before being resuspended in 1% SDS chemoenzymatic buffer. Protein concentration was then re-normalized using the BCA Assay and diluted to 2.5 mg mL−1 in 1% SDS chemoenzymatic buffer. To start the chemoenzymatic transfer reaction, the following reagents were added in order for an 100 μg scale: 49 μL of H2O, 80 μL of labeling buffer (2.5x; 5% NP-40, 125 mM NaCl, 50 mM HEPES, pH 7.9), 55 μL of MnCl2 (100 mM in H2O), and 50 μL of UDP-GalNAz (0.5 mM in 10 mM HEPES, pH 7.9). This was mixed by pipetting gently. Finally, 7.5 μL of purified GalT(Y289L) (in 10 mM Tris, pH 8.0). The reaction mixture was incubated for 16 h at 4 °C without agitation. Following incubation, unreacted UDP-GalNAz was removed by MeOH/CHCl3/H2O precipitation. Air-dried protein pellets were resuspended in 1% SDS CuAAC buffer (1% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) and subjected to either immunoprecipitation or conjugation with DBCO-PEG mass-tags.
Endogenous MYPT1 and MLC chemoenzymatic/biotin IP sample preparation.
NIH3T3 cells at 80% confluency were harvested, resuspended in 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) supplemented with 5 mg mL−1 c0mplete mini protease inhibitor cocktail (MilliporeSigma), 5 mg mL−1 PhosSTOP (MilliporeSigma), 1 mM phenylmethylsulfonyl fluoride, and lysed via tip sonication (3× 5 sec on 5 sec off) before being subjected to chemoenzymatic transfer according to the protocol described above. Following chemoenzymatic transfer, proteins were precipitated by MeOH/CHCl3/H2O. The aqueous phase (top layer) was discarded carefully to avoid disturbing the interface. Next, 2.5x volume of MeOH was added, the mixture was vortexes briefly, and the tube was centrifuged (5 min, 13,000 x g, rt). The supernatant was decanted and the protein pellet was allowed to air-dry for 5-10 min before being resuspended in 1% SDS buffer (1% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4). Inputs were generated by adding 25 μL of the lysis buffer and 25 μL of 4x LB (200 mM Tris, 8% SDS, 40% glycerol, 0.4% bromophenol blue, 2.8% β- mercaptoethanol, pH 6.8) to 50 μL of lysate. IP sample preparation is continued in the next section.
CuAAC enrichment and Immunoprecipitation.
Cell lysates (1 mg at 1 mg mL−1) labeled chemoenzymatically were subjected to CuAAC enrichment performed using a freshly made master mix containing alkyne-azo-biotin (100 μM from 5 mM stock in DMSO, Click Chemistry Tools), TCEP (1 mM from 50 mM freshly prepared stock in H2O), TBTA (100 μM from 10 mM stock in DMSO), CuSO4:5H2O (1 mM from 50 mM freshly prepared stock in H2O) and incubated for 1 h in the dark. Proteins were precipitated by addition of 4x volume of ice-cold MeOH and incubated at −20 °C for 2 h. Proteins were collected by centrifugation (10 min, 5,000 x g, 4 °C) and washed 3x with ice-cold MeOH. Pellets were then air-dried for 15 min before being resuspended in 800 μL resuspension buffer (6 M urea, 2 M thiourea, 10 mM HEPES, pH 8.0). Streptavidin beads (25 μL of a 50% slurry per sample, ThermoFisher Scientific) were allocated into 2 mL dolphin-nosed tubes and prepared by washing 2x with 1 mL PBS and 1x with 1 mL resuspension buffer. Following each wash, samples were centrifuged (1 min, 2,000 x g, rt) and the supernatant was carefully discarded. Once prepared, beads were resuspended in 200 μL resuspension buffer. Protein samples were added to the dolphin-nosed tubes and incubated on a rotator (2 h, rt, full rotation). Beads were washed 2x with 1 mL resuspension buffer, 2x with 1 mL PBS, and 2x with 1 mL 1% SDS in PBS. Following each wash, samples were centrifuged (1 min, 2,000 x g, rt) and the supernatant was carefully discarded. Beads were then incubated in 25 μL of sodium dithionite solution (1% SDS, 25 mM sodium dithionite) for 30 min at rt to elute bound proteins. Beads were centrifuged (2 min, 2,000 x g, rt) and the eluent was collected. Elution was repeated and the two elution products were pooled. Proteins were precipitated in 1 mL ice-cold MeOH overnight at −20 °C. Proteins were collected by centrifugation (10 min, 10,000 x g, 4 °C) and the supernatant was carefully decanted. Pellets air-dried for 5 min before a final resuspension in 30 μL of 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4), and bath sonicated to ensure complete dissolution. Gel samples were prepared by adding 30 μL of 2× loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol) and boiling (5 min, 98 °C). Inputs and IP samples were separated by SDS-PAGE and Western blotted for Nup62 (positive control), β-actin (negative control), MYPT1, and MLC2 according to the procedure described above.
Mass-shift of MYPT1.
Mass-shifting was performed essentially as previously described413. Specifically, NIH3T3 cells were plated in 2× 150 mm dishes and grown to 80% confluency. Cells were harvested by trypsinization and washed two times with 1 mL cold PBS (2 min, 2,000 x g, 4 °C). Cell pellets were resuspended in 500 μL 4% SDS lysis buffer (4% SDS, 10 mM TEA pH 7.4, 150 mM NaCl) supplemented with 5 mg mL−1 c0mplete mini protease inhibitor cocktail (MilliporeSigma), 5 mg mL−1 PhosSTOP (MilliporeSigma), and 1 mM phenylmethylsulfonyl fluoride. The resulting suspension was lysed using a tip sonicator (3× 5 sec on 5 sec off). 5 μL of TCEP (500 mM stock dissolved in 1 M NaOH) was then added to each sample and boiled (10 min, 98 °C). Samples were cooled to room temperature before adding 40 μL iodoacetimade (600 mM stock dissolved in 4% SDS buffer) and incubated in the dark for 30 min. Samples were diluted to a total volume of 2 mL using 1% SDS buffer (1% SDS, 50 mM TEA pH 7.4, 150 mM NaCl). Proteins were precipitated by adding 3x volume of MeOH, 0.75x volume of CHCl3, and 2x volume of H2O and briefly vortexing followed by centrifugation (5 min, 5,000 x g, rt). The aqueous phase (top layer) was discarded carefully to prevent disturbing the pellet floating in the interface between layers. An additional 2.5x volume of MeOH was then added to the tube followed by vortexing, pelleting the protein (10 min, 5,000 x g, rt), and decanting the supernatant. The resulting pellet was allowed to air-dry for 5 min before being resuspended in 400 μL 1% SDS chemoenzymatic buffer (1% SDS, 20 mM HEPES, pH 7.9). Protein concentration was normalized using the BCA Assay and diluted to 2.5 mg mL−1 in 1% SDS chemoenzymatic buffer. UDP-GalNAz Enzymatic Labeling was set up and scaled as described in the General Chemoenzymatic Labeling section above. For each sample, 200 μg of protein lysate was incubated with both GalT and UDP-GalNAz. Negative controls for each sample, incubating with just GalT, were generated using 100 μg of protein lysate. After chemoenzymatic labeling, unreacted UDP-GalNAz was removed via MeOH/CHCl3/H2O precipitation. Air-dried protein pellets were resuspended in 90 μL (negative samples) or 180 μL (positive samples) of 1% SDS (1% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4). 10 μL (negative samples) or 20 μL (positive samples) of 10 mM DBCO-PEG5000 or DBCO-PEG2000 in DMSO were added and the mixture was boiled (5 min, 98 °C). Unreacted DBCO-PEG was removed by MeOH/CHCl3/H2O precipitation. The pellet was again to air-dried for 5-10 min. Finally, 25 μL 4% SDS buffer was added to each sample. The mixture was briefly sonicated in a bath sonicator to ensure complete resuspension before 25 μL of 2x loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol, pH 6.8) was added and the samples were boiled (5 min, 98 °C). 20 μL (40 μg) of each gel sample was loaded per lane. Western blots were performed according to the General Western Blot procedure described above with the following two changes: anti-MYPT1 primary antibody was incubated at a dilution of 1:500 and (HRP)-conjugated secondary antibodies were incubated at a dilution of 1:5,000.
FLAG-IP for RL2 detection of MYPT1.
FLAG-tagged MYPT1 expressing NIH3T3 grown to 80% confluency were harvested by trypsinization and washed two times with PBS (2 min, 2,000 x g, 4 °C) Cell pellets were then resuspended in 200 μL of lysis buffer (1% Triton-x 100, 50 mM Tris, 150 mM NaCl, 1 mM EDTA) supplemented with 5 mg mL−1 c0mplete mini protease inhibitor cocktail (MilliporeSigma), 5 mg mL−1 PhosSTOP (MilliporeSigma), 1 mM phenylmethylsulfonyl fluoride, and 100 μM Thiamet-G. This suspension was lysed using a tip sonicator (3× 5 sec on 5 sec off) on ice. To remove remaining cell debris, the suspension was centrifuged (5 min, 15,000 x g, 4 °C) and the supernatant was transferred to a fresh eppendorf tube. Protein concentration was determined by BCA Assay and diluted to 2 mg mL−1 in lysis buffer. Inputs were generated by adding 25 μL of the lysis buffer and 25 μL of 4x LB (200 mM Tris, 8% SDS, 40% glycerol, 0.4% bromophenol blue, 2.8% β- mercaptoethanol, pH 6.8) to 50 μL of lysate. 20 μL of Anti-FLAG-M2 magnetic beads (MilliporeSigma) were added to an eppendorf tube and washed with lysis buffer (3× 1 mL). Lysate was diluted to 1 mg mL−1 in lysis buffer. 1 mL of lysate was added to the beads followed by incubation for 1 h rotating at 4 °C. Beads were collected and washed (6× 1 mL lysis buffer). To elute from beads, 30 μL of lysis buffer and 10 μL of 4x LB were added to beads, vortexed briefly, and boiled (5 min, 98 °C). Beads were pelleted by centrifugation (1 min, 15,000 x g, rt). The resulting supernatant was transferred to a new eppendorf tube. For detection of MYPT1, anti-FLAG antibody was used and 5 μL of input and 10 μL of IP was loaded. For detection of RL2, anti-RL2 antibody was used and 15 μL of input and 30 μL of IP was loaded. Western blot was performed according to General Western Blot Procedure described above.
FLAG-IP for RL2 detection of MYPT1 vs MYPT1Δ.
FLAG-tagged MYPT1 or MYPT1Δ NIH3T3 cell-lines were harvested and worked-up as described in the previous section. Western blot was performed according to General Western Blot Procedure described above.
Co-Immunoprecipitation of FLAG-tagged MYPT1 or FLAG-tagged MYPT1Δ and ROCK1/2.
NIH3T3 cells were treated with DMSO vehicle or 5SGlcNAc (200 μM) for 16 h before being harvested by trypsinization. Cell lysate was washed twice with 1 mL cold PBS (5 min, 2,000 x g, 4 °C) and resuspended in 500 μL lysis buffer (1% IGPAL, 50 mM TEA, 150 mM NaCl, pH 7.4) supplemented with 5 mg mL-1 c0mplete mini protease inhibitor cocktail (MilliporeSigma), 5 mg mL-1 PhosSTOP (MilliporeSigma), 1 mM phenylmethylsulfonyl fluoride. The cell suspension was lysed using a tip sonicator (3× 5 sec on 5 sec off) and any remaining cell debris were pelleted (10 min, 10,000 x g, 4 °C). Lysate was moved to a pre-chilled tube and kept on ice. Protein concentration was determined by BCA Assay (Pierce, ThermoScientific) and diluted to 2 mg mL-1 in lysis buffer. Inputs were generated by adding 25 μL of the lysis buffer and 25 μL of 4x LB (200 mM Tris, 8% SDS, 40% glycerol, 0.4% bromophenol blue, 2.8% β- mercaptoethanol, pH 6.8) to 50 μL of lysate. An anti-flag co-immunoprecipitation was performed using the Catch and Release system (ThermoScientific) according to the manufacturers protocol. Briefly, spin columns were washed twice with 400 μL 1x Wash Buffer (30 sec, 2000 x g, 4 °C). 500 μg of cell lysate (250 μL) per sample was added to designated spin column followed by 5 μL PMSF (200 μM stock in IPA), 225 μL 1x Wash Buffer, 10 μL Affinity Ligand, and 10 μL of anti-FLAG antibody. Plugged spin columns were then incubated on a rotator (1 h, 4 °C, full rotation). Spin columns were next centrifuged (30 sec, 2000 x g, 4 °C) and washed 3x with 1x Wash Buffer (30 sec, 2000 x g, 4 °C) to remove unbound proteins. Spin columns were then placed in fresh capture tubes and bound proteins were eluted in 70 μL of 1x Denaturing Elution buffer containing βME to generating gel samples. Input and IP gel samples were boiled (5 min, 98 °C) and separated by SDS-PAGE. ROCK enrichment was detected by Western blotting against anti-ROCK1/2 and normalized to overall protein capture via Coomassie staining with ImageJ (ImageJ 1.52q). The results were quantified and presented as mean ± SEM of the normalized ROCK levels (n = 3 biological replicates). Statistical significance was determined using a 2-tailed, unpaired Student’s t-test in Prism (Graphpad Prism 7).
Generation of NIH3T3 cell-lines stably expressing FLAG-tagged MYPT1 mutants.
FLAG-tagged MYPT1, MYPT1(S/TtoA), MYPT1(d564-578), MYPT1(d588-602), and MYPT1Δ NIH3T3 cell-lines were generated using the PiggyBac™ Transposon Vector System (System Biosciences). Briefly, NIH3T3 cells at 30% confluency in 10 cm dishes were co-transfected with 5 μg PiggyBac transposase promoter (plasmid ID: PB531A-1) and 10 μg PiggyBac transposon plasmid pPB-CAG-IRES2-puro containing either human MYPT1, human MYPT1(S/TtoA), human MYPT1(d564-578), human MYPT1(d588-602), or human MYPT1Δ550-600 (MYPT1Δ). One plate was transfected with 10 μg of pcDNA3 to serve negative selection control. 24 h post-transfection, plates were split 1:2 into normal growth media (DMEM + 10% FCS) and supplemented with 1 μg mL−1 puromycin (1 mg mL−1 stock in H2O). Cells were selected for three days at which point puromycin concentration was reduced to 250 ng mL-1. FLAG-tagged MYPT1 expression was confirmed with analysis by Western blotting against anti-FLAG and anti-MYPT1. Effect of MYPT1 vs MYPT1Δ on global O-GlcNAcylation and OGT expression were analyzed by Western blotting against anti-RL2 and anti-OGT.
Collagen gel contraction assay.
3D collagen contraction assay was performed using a two-step cell contraction assay kit purchased from Cell Biolabs (CBA-201) according to manufacturers protocol. Briefly, BJ-5ta cells were suspended in fresh media at a confluency of 2 × 106 cells per mL. A 4:1 mixture of collagen stock solution to cell suspension was mixed in a 15 mL falcon tube and 500 μL per well was plated in a 24-well dish. The cell-collagen matrix was incubated at 37 °C for 1 h to allow collagen polymerization before adding 1 mL of media to each well. After 30 hours, wells were supplemented with either DMSO vehicle or 5SGlcNAc inhibitor (200 μM) and incubated for another 16 hours at which point the media was changed to serum-free media containing S1P (0.1 - 5 μM). Contraction was initiated by gently releasing the sides and bottom of the matrices from the plate. Images were taken immediately after media change (t=0) and after 30 min using the ChemiDoc XRS+ molecular imager (Bio-Rad, BioRad Image Lab 4.1). Contraction was measured using ImageJ (ImageJ 1.52q). Specifically, collagen matrix diameter was measured by taking the average of one horizontal (0 degrees) and one vertical (90 degrees) measurement of each matrix.
Endogenous mouse MYPT1 knockdown with RNAi.
NIH3T3 or stably expressing cell-lines were seeded at 30% confluency in 10 cm dishes 24 h before transfection. 5 nmole of RNAi targeted against mouse MYPT1 or a scramble sequence was purchased from ThermoFisher Scientific (siRNA ID: s70342) and diluted in 500 μL in nuclease free H2O to make a stock concentration of 10 μM. Cells were transfected with 125 pmole (12.5 μL) of RNAi using Lipofectamine RNAiMAX transfection reagent (ThermoFisher Scientific) according to the manufacturer’s protocol. Knockdown efficiency was measured via Western blotting against MYPT1. Western blot procedure is described in detail previously.
Extended Data
Extended Data Figure 1. Modulating O-GlcNAcylation levels and identification of S1P.
a) Schematic layout of our general experimental conditions. b) O-GlcNAc levels can be changed upon OGT or OGA inhibition. NIH3T3 cells were treated with either the OGT inhibitor 5SGlcNAc, the OGA inhibitor Thiamet G, or DMSO vehicle respectively before the O-GlcNAc levels were analyzed by Western blotting. c) Only serum- or S1P-treatment results in notable cell contraction when O-GlcNAc levels have been lowered. NIH3T3 cells were treated with DMSO or 5SGlcNAc. The indicated signaling molecules were then added, and the contraction phenotype was then visualized using bright-field microscopy. The data in b-c is representative of 2 biological replicates.
Extended Data Figure 2. O-GlcNAc controls the sensitivity of fibroblasts to sphingosine-1-phosphate (S1P) mediated cell contraction.
a) O-GlcNAc levels can be changed upon OGT inhibition. NIH3T3 cells were treated with either the OGT inhibitor ST060266 or DMSO vehicle respectively before the O-GlcNAc levels were analyzed by Western blotting. b) O-GlcNAc levels can be lowered by OGT RNAi. NIH3T3 cells were transfected with RNAi targeting OGT or a scrambled sequence before analysis by Western blotting. c) NIH3T3 cells were treated with the indicated combinations of DMSO, the OGT inhibitor ST060266 (200 μM) and/or S1P. The contraction phenotype was then visualized using bright-field microscopy. d) NIH3T3 cells were transfected with either scramble or OGT-targeted RNAi for 48 h before addition of either DMSO or S1P. The contraction phenotype was then visualized using bright-field microscopy. e) O-GlcNAc levels can be changed by culturing cells in different glucose concentrations. NIH3T3 cells were cultured in the indicated concentrations of glucose before analysis by Western blotting. The data in a-e is representative of at least 2 biological replicates.
Extended Data Figure 3. Signaling through the second S1P receptor, S1PR2, is responsible for the contraction phenotype.
a) NIH3T3 cells can express all five S1P GPCRs (S1PR1 to 5). mRNA was collected from NIH3T3 cells before being subjected to RT-PCR and visualization on an DNA-agarose gel. These data are representative of 2 biological replicates. b) Antagonizing S1PR2, but not the other receptors, inhibits S1P-mediated cell contraction. NIH3T3 cells were treated with either DMSO or 5SGlcNAc. The same cells were then treated with either additional DMSO or the indicated selective antagonists followed by S1P. The contraction phenotype was then visualized using bright-field microscopy. c) Quantitation of the data in (b). Results are the mean ± SEM of the relative culture plate area taken-up by cells in at least three randomly selected frames. Statistical significance was determined using a 2-tailed student’s t-test. d) S1PR5 agonism does not induce cell contraction. NIH3T3 cells were treated with either DMSO or 5SGlcNAc. The same cells were then treated with either S1P or the S1PR5-selective agonist A971432. The contraction phenotype was then visualized using bright-field microscopy. e) Quantitation of the data in (d). Results are the mean ± SEM of the relative culture plate area taken-up by cells in at least three randomly selected frames. Statistical significance was determined using a 2-tailed student’s t-test. f) Lowering O-GlcNAc levels increases the sensitivity of NIH3T3 cells to S1PR2 induced cell contraction. NIH3T3 cells were treated with either DMSO or 5SGlcNAc before addition of the indicated concentrations of the S1PR2-selective agonist CYM5220. The contraction phenotype was then visualized using bright-field microscopy. g) Quantitation of the data in (f). Results are the mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test. h) S1PR2 knockdown using siRNA blocks contraction phenotype. NIH3T3 cells were transfected with either scramble or S1Pr2-targeted RNAi before addition of DMSO or S1P. The contraction phenotype was then visualized using bright-field microscopy. i). Quantitation of the data in (h). Results are the mean ± SEM of the relative culture plate area taken-up by cells in three randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test.
Extended Data Figure 4. Inhibition of Rho kinase (ROCK1/2) blocks S1P-mediated cell contraction.
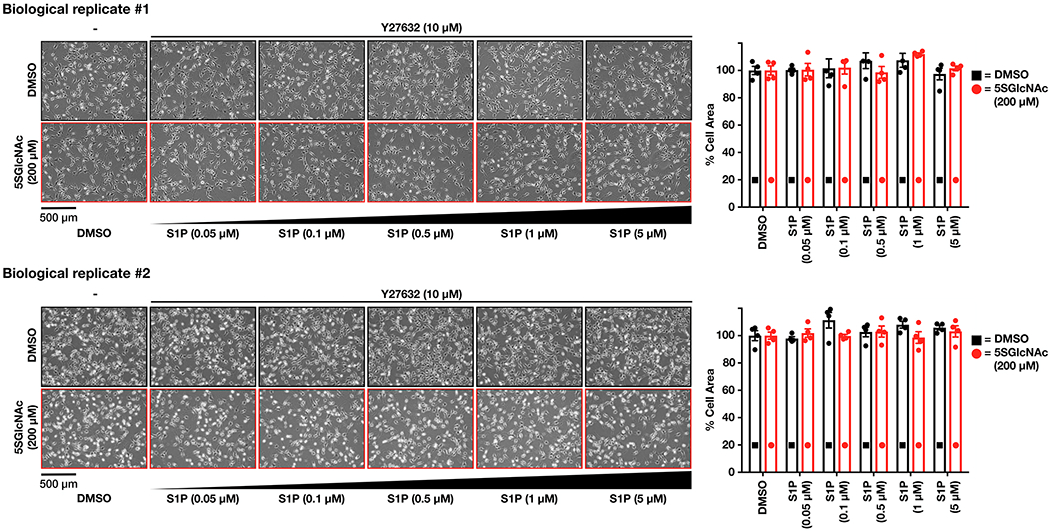
NIH3T3 cells were treated with either DMSO or 5SGlcNAc. The same cells were then treated with either additional DMSO or the ROCK1/2 inhibitor Y27632 followed by the indicated concentrations of S1P. The contraction phenotype was then visualized using bright-field microscopy. The results were then quantified and are presented as mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames.
Extended Data Figure 5. Analysis of MYPT1 mutants shows that deletion of the serine/threonine domain sensitizes cells to S1P-mediated contraction.
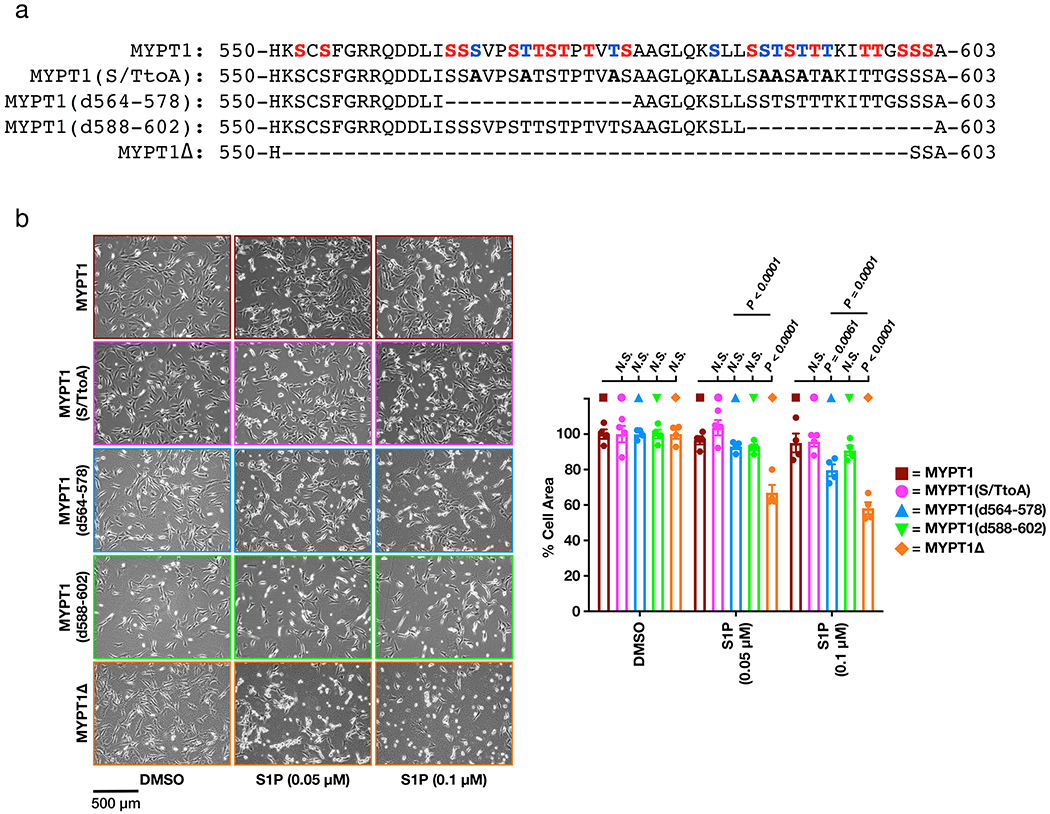
a) Sequence alignment of MYPT1 and the different mutants tested here. Red indicates potential O-GlcNAc modification sites (S or T), and blue indicates O-GlcNAc sites previously identified by mass spectrometry. MYTP1(S/TtoA) mutates all of the previously identified O-GlcNAc sites, MYPT1(d564-578) deletes the first serine/threonine rich region, MYPT1(d588-602) deletes the second serine/threonine rich region, and MYPT1Δ deletes the entire serine/threonine rich domain. b) MYPT1Δ, and to a lesser extent MYPT1(d564-578), sensitizes cells to S1P-mediated contraction. NIH3T3 cells expressing the indicated MYPT1 proteins and the endogenous copy was removed by RNAi. DMSO or S1P was then added and the contraction of the cells was measured. Results are the mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test.
Extended Data Figure 6. Deletion of the MYPT1 serine/threonine rich domain yields MYPT1Δ with reduced O-GlcNAc modification.
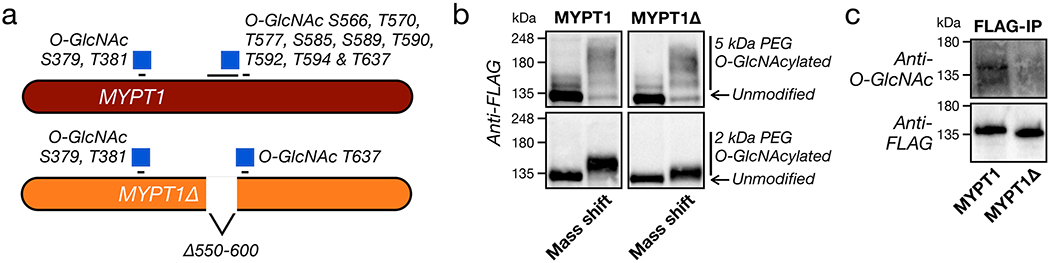
a) Schematic of the MYPT1Δ protein, which lacks the major O-GlcNAc region of the protein. b&c) MYPT1Δ loses a notable amount of O-GlcNAc. The O-GlcNAc levels of FLAG-tagged MYPT1 or MYPT1Δ were analyzed using mass-shifting or IP-Western blot. The data in are representative of at least 2 biological replicates.
Extended Data Figure 7. MYPT1Δ is an active phosphatase that can dephosphorylate MLC and return cells to a relaxed state.
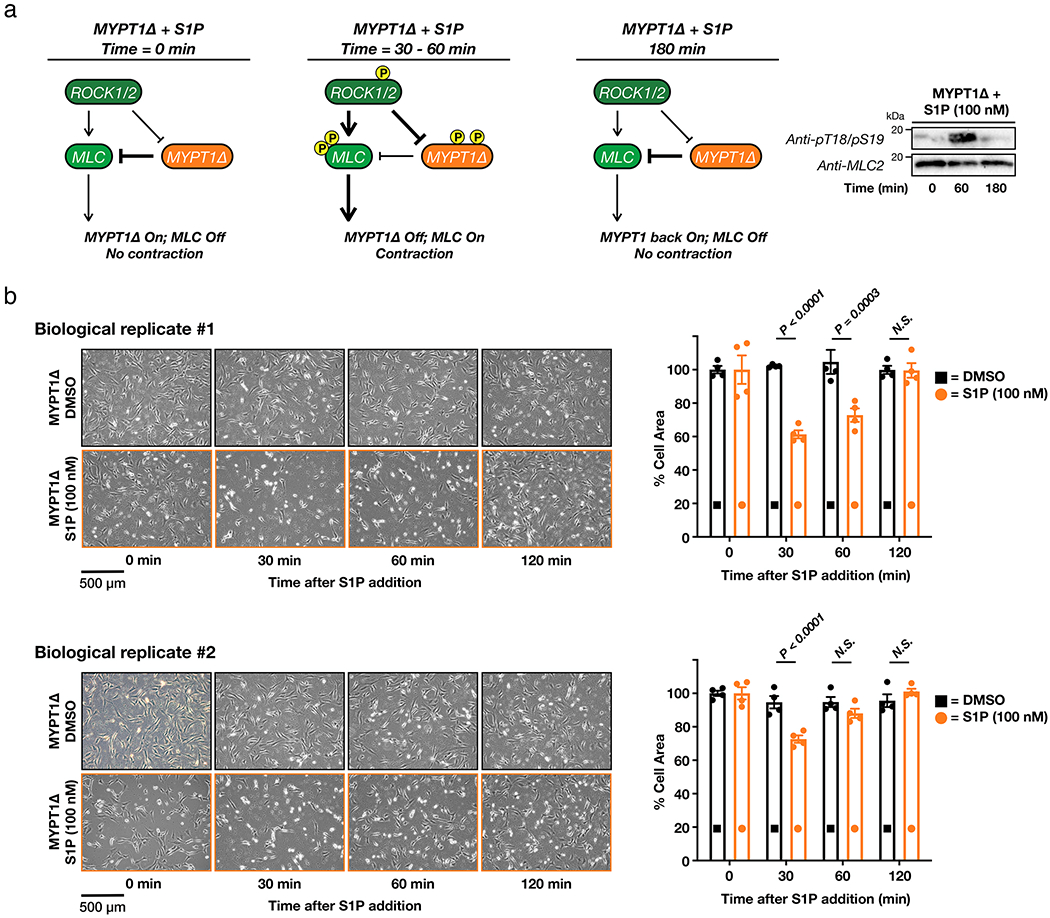
a) If MYPT1Δ is an active phosphatase, we expect that it will become phosphorylated and deactivated by ROCK after S1P treatment but will return to a desphosphorylated and active state after a longer period of time. This will result in MLC dephosphorylation and relaxation of the cells. This is exactly what we observed by Western blotting. These data are representative of 2 biological replicates. b) Cells expressing MYPT1Δ return to a relaxed state over 180 min. The contraction phenotype was visualized using bright-field microscopy. The results were then quantified and are presented as mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test.
Extended Data Figure 8. O-GlcNAc blocks the interaction between MYPT1 and ROCK.
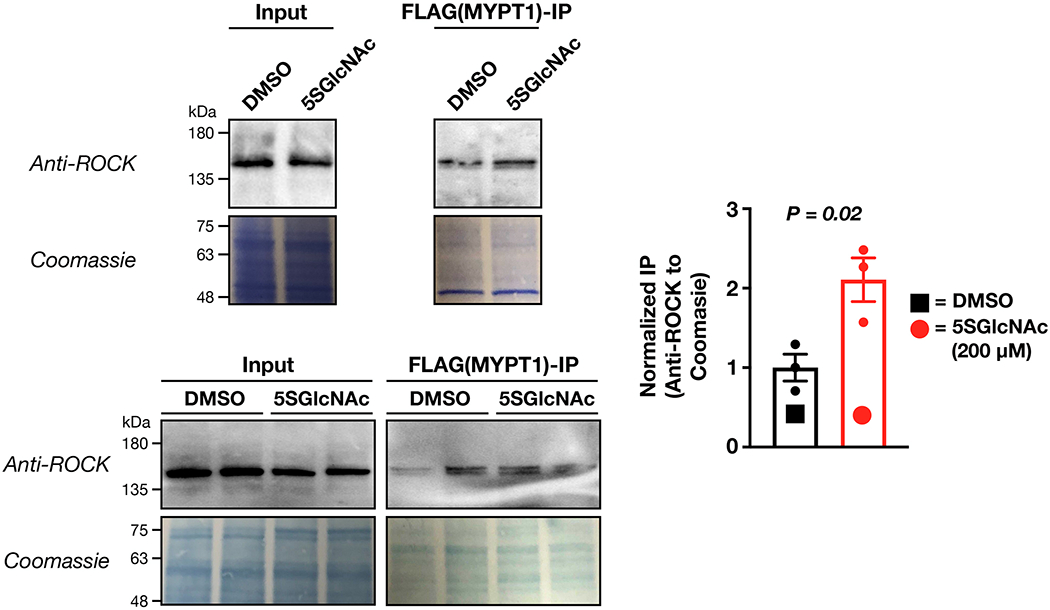
NIH3T3 cells expressing flag-tagged MYPT1 were treated with either DMSO or 5SGlcNAc (200 μM). An anti-flag co-immunoprecipitation was then performed using the Catch and Release system (Thermo). ROCK enrichment was detected by Western blotting and normalized to overall protein capture (Coomassie staining). The results were quantities and presented as mean ± SEM of the normalized ROCK levels (n = 3 biological replicates). Statistical significance was determined using a 2-tailed, unpaired Student’s t-test.
Extended Data Figure 9. Direct MYPT1 O-GlcNAcylation is largely responsible for the phenotype.
a) NIH3T3 cells stably expressing either MYPT1 or MYTP1Δ were transfected with RNAi to downregulate endogenous MYTP1. MYTP1-expressing cells were then treated with either DMSO or 5SGlcNAc. Cells under all three sets of conditions were then treated with the indicated concentrations of S1P. The contraction phenotype was visualized using bright-field microscopy. The results were then quantified and are presented as mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test. b) NIH3T3 cells stably expressing either MYPT1 or MYTP1Δ were transfected with RNAi to downregulate endogenous MYTP1. MYTP1-expressing cells were then treated with either DMSO or Thiamet-G. Cells under all three sets of conditions were then treated with the indicated concentrations of S1P. The contraction phenotype was visualized using bright-field microscopy. The results were then quantified and are presented as mean ± SEM of the relative culture plate area taken-up by cells in four randomly selected frames. Statistical significance was determined using a 2-way ANOVA test followed by Sidak’s multiple comparisons test.
Extended Data Figure 10. Our experimental model.
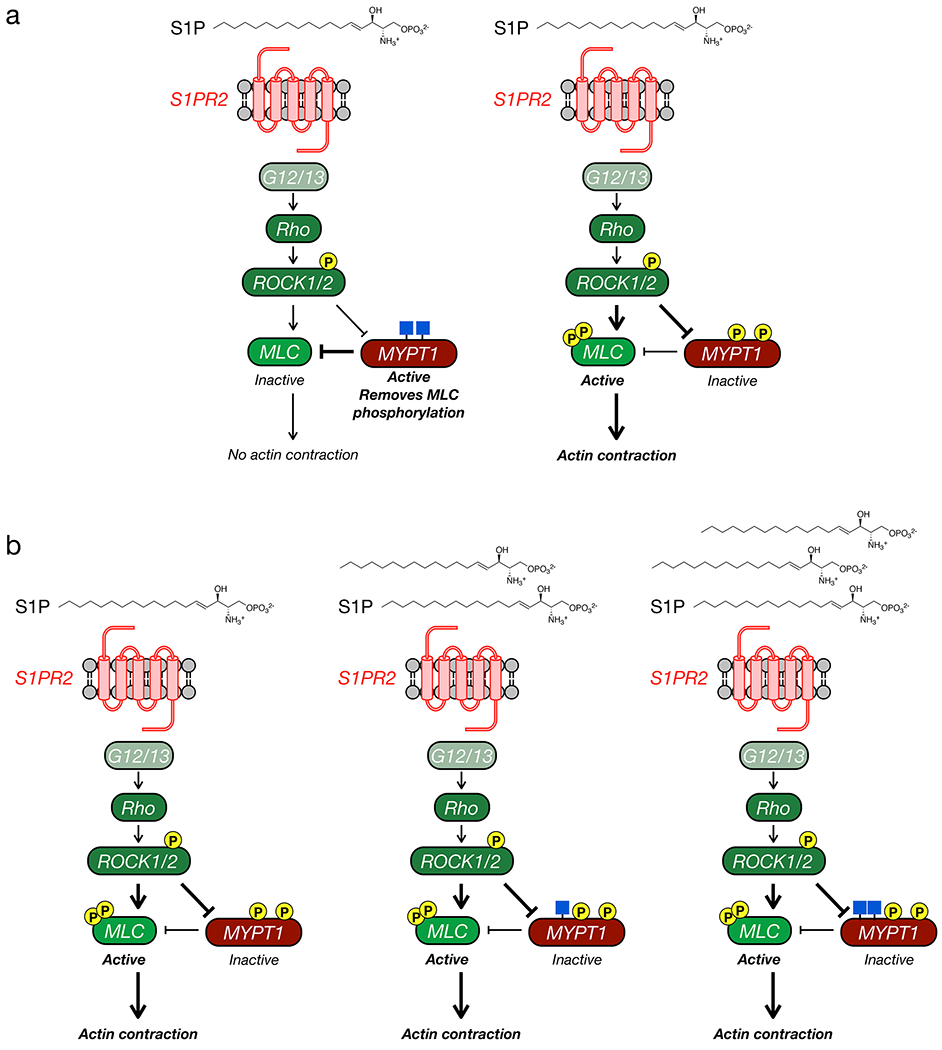
a) MYPT1 O-GlcNAcylation inhibits its phosphorylation by ROCK1/2. This maintains MYPT1 phosphatase activity, resulting in inactive MLC and no actin contraction. Loss of O-GlcNAc enables ROCK1/2 to phosphorylate and deactivate MYTP1. b) Therefore, MYPT1 O-GlcNAcylation levels control the sensitivity of cells to the concentration of S1P, where more MYTP1 O-GlcNAcylation requires more S1P to illicit actin contraction and cell detachment.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank F. Grinnell for consultations on collagen matrix contraction. This research was supported by the American Cancer Society Research Scholar Grant (RSG-14-225-01-CCG), the University of Southern California, the Anton Burg Foundation, and the National Institutes of Health (R01GM125939) to M.R.P.. N.J.P. is supported by NIGMS T32GM118289. The authors thank K. Moremen at the University of Georgia, who is supported by the National Institutes of Health (P41GM103390 and R01GM130915), for the generous gift of GalT(Y289L).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
DATA AVAILABILITY STATEMENT
RT-PCR primers were designed using the MIT Primer3 design database (http://biotools.nubic.northwestern.edu/Primer3.html) with sequences found using the NCBI Gene Search tool (ncbi.nlm.nih.gov/gene).
REFERENCES
- 1.Yang X & Qian K Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18, 452–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zachara NE Critical observations that shaped our understanding of the function(s) of intracellular glycosylation (O‐Glc NAc). FEBS Lett 592, 3950–3975 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King DT, Males A, Davies GJ & Vocadlo DJ Molecular mechanisms regulating O-linked N-acetylglucosamine (O-GlcNAc)–processing enzymes. Current Opinion in Chemical Biology 53, 131–144 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Hart GW Nutrient regulation of signaling and transcription. J Biol Chem 294, 2211–2231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marshall S, Bacote V & Traxinger RR Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system. Role of hexosamine biosynthesis in the induction of insulin resistance. 266, 4706–4712 (1991). [PubMed] [Google Scholar]
- 6.Shen DL, Gloster TM, Yuzwa SA & Vocadlo DJ Insights into O-linked N-acetylglucosamine (O-GlcNAc) processing and dynamics through kinetic analysis of O-GlcNAc transferase and O-GlcNAcase activity on protein substrates. J Biol Chem 287, 15395–15408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart GW, Slawson C, Ramirez-Correa G & Lagerlof O Cross Talk Between O-GlcNAcylation and Phosphorylation: Roles in Signaling, Transcription, and Chronic Disease. Annu Rev Biochem 80, 825–858 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang S et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS ONE 7, e37427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarrant MK et al. Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat Chem Biol 8, 262–269 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gloster TM et al. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol 7, 174–181 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang H, Rhee S, Ho C-H & Grinnell F Distinguishing fibroblast promigratory and procontractile growth factor environments in 3-D collagen matrices. FASEB J 22, 2151–2160 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Yuzwa SA et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol 4, 483–490 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Spiegel S & Milstien S Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4, 397–407 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Watterson KR, Lanning DA, Diegelmann RF & Spiegel S Regulation of fibroblast functions by lysophospholipid mediators: potential roles in wound healing. Wound Repair Regen 15, 607–616 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Takabe K, Paugh SW, Milstien S & Spiegel S ‘Inside-out’ signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol. Rev 60, 181–195 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendelson K, Evans T & Hla T Sphingosine 1-phosphate signalling. Development 141, 5–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eto M & Kitazawa T Diversity and plasticity in signaling pathways that regulate smooth muscle responsiveness: Paradigms and paradoxes for the myosin phosphatase, the master regulator of smooth muscle contraction. J. Smooth Muscle Res. 53, 1–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhee S & Grinnell F Fibroblast mechanics in 3D collagen matrices. Adv Drug Deliv Rev 59, 1299–1305 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riento K & Ridley AJ Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 4, 446–456 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Amano M, Nakayama M & Kaibuchi K Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) 67, 545–554 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Totsukawa G et al. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. The Journal of Cell Biology 150, 797–806 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng J et al. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. 274, 37385–37390 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Kawano Y et al. Phosphorylation of myosin-binding subunit (MBS) of myosin phosphatase by Rho-kinase in vivo. The Journal of Cell Biology 147, 1023–1038 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murányi A et al. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett 579, 6611–6615 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Khasnis M, Nakatomi A, Gumpper K & Eto M Reconstituted Human Myosin Light Chain Phosphatase Reveals Distinct Roles of Two Inhibitory Phosphorylation Sites of the Regulatory Subunit, MYPT1. Biochemistry 53, 2701–2709 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heissler SM & Sellers JR Myosin light chains: Teaching old dogs new tricks. Bioarchitecture 4, 169–188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito M, Nakano T, Erdodi F & Hartshorne DJ Myosin phosphatase: structure, regulation and function. Mol Cell Biochem 259, 197–209 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Grassie ME, Moffat LD, Walsh MP & MacDonald JA The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1δ. Arch Biochem Biophys 510, 147–159 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Takemoto K, Ishihara S, Mizutani T, Kawabata K & Haga H Compressive stress induces dephosphorylation of the myosin regulatory light chain via RhoA phosphorylation by the adenylyl cyclase/protein kinase A signaling pathway. PLoS ONE 10, e0117937 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hédou J et al. O-linked N-acetylglucosaminylation is involved in the Ca2+ activation properties of rat skeletal muscle. 282, 10360–10369 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Cheung WD, Sakabe K, Housley MP, Dias WB & Hart GW O-linked beta-N-acetylglucosaminyltransferase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. 283, 33935–33941 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clark PM et al. Direct In-Gel Fluorescence Detection and Cellular Imaging of O-GlcNAc-Modified Proteins. J Am Chem Soc 130, 11576–11577 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J Pathol 243, 78–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin K et al. Quantitative Profiling of Protein O-GlcNAcylation Sites by an Isotope-Tagged Cleavable Linker. ACS Chem Biol 13, 1983–1989 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Li J et al. An Isotope-Coded Photocleavable Probe for Quantitative Profiling of Protein O-GlcNAcylation. ACS Chem Biol 14, 4–10 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Ichikawa K et al. Interactions and properties of smooth muscle myosin phosphatase. Biochemistry 35, 6313–6320 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Hirano K, Phan BC & Hartshorne DJ Interactions of the subunits of smooth muscle myosin phosphatase. 272, 3683–3688 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Tanaka J et al. Interaction of myosin phosphatase target subunit 1 with the catalytic subunit of type 1 protein phosphatase. Biochemistry 37, 16697–16703 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Wang Y et al. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res 104, 531–540 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rexach JE et al. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat Chem Biol 6, 645–651 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darabedian N, Thompson JW, Chuh KN, Hsieh-Wilson LC & Pratt MR Optimization of Chemoenzymatic Mass Tagging by Strain-Promoted Cycloaddition (SPAAC) for the Determination of O-GlcNAc Stoichiometry by Western Blotting. Biochemistry 57, 5769–5774 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gosain A & DiPietro LA Aging and wound healing. World J Surg 28, 321–326 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Guo S & DiPietro LA Factors affecting wound healing. J. Dent. Res 89, 219–229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grinnell F & Petroll WM Cell motility and mechanics in three-dimensional collagen matrices. Annu Rev Cell Dev Biol 26, 335–361 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Kawanabe T, Kawakami T, Yatomi Y, Shimada S & Soma Y Sphingosine 1-phosphate accelerates wound healing in diabetic mice. J. Dermatol. Sci 48, 53–60 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Aoki M et al. Sphingosine-1-Phosphate Facilitates Skin Wound Healing by Increasing Angiogenesis and Inflammatory Cell Recruitment with Less Scar Formation. Int J Mol Med 20, E3381 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.SOMLYO AP & SOMLYO AV Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev 83, 1325–1358 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Grassie ME et al. Cross-talk between Rho-associated kinase and cyclic nucleotide-dependent kinase signaling pathways in the regulation of smooth muscle myosin light chain phosphatase. J Biol Chem 287, 36356–36369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiao Y-N et al. Myosin phosphatase target subunit 1 (MYPT1) regulates the contraction and relaxation of vascular smooth muscle and maintains blood pressure. J Biol Chem 289, 22512–22523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu C et al. O-GlcNAcylation of myosin phosphatase targeting subunit 1 (MYPT1) dictates timely disjunction of centrosomes. J Biol Chem 295, 7341–7349 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuzwa SA et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol 4, 483–490 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Gloster TM et al. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol 7, 174–181 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darabedian N, Thompson JW, Chuh KN, Hsieh-Wilson LC & Pratt MR Optimization of Chemoenzymatic Mass Tagging by Strain-Promoted Cycloaddition (SPAAC) for the Determination of O-GlcNAc Stoichiometry by Western Blotting. Biochemistry 57, 5769–5774 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.