

Abstract
Cell sorting is a powerful tool in basic research and therapeutic enrichment. However, common cell sorting methods, such as fluorescence-activated cell sorting (FACS) and magnetic-activated cell sorting (MACS) have significant limitations, such as generally low cell yields or restriction to binary separation, respectively. To address these limitations, we developed a two-step cell sorting method called mass-added density centrifugation (MADC) to enable nonbinary separation of large cell numbers based on surface protein levels. In the first MADC step (mass-adding), antibody-directed massive microparticles bind target surface proteins to modulate single-cell density proportionally to target protein level. Second, microparticle-laden cells are subjected to discontinuous density gradient centrifugation, whereby they separate into discrete density bands which can be isolated for downstream use. MADC will prove especially advantageous for obtaining sufficient cell numbers for protein analyses from large source populations, and it is a fast process that can facilitate live cell enrichment for therapies that require tens of millions of cells. Here, we demonstrate MADC’s utility for both live and fixed cell sorts of multiple cell types based on abundance of an example target protein, CD44. CD44 quantity in separated cell groups was assayed with western blots and correlated with modulated cell density. This novel sorting method enables rapid, nonbinary isolation of large quantities of cells based on surface protein levels and should prove useful in both basic science and therapeutic applications.
Key terms: cell separation, density gradient, density centrifugation, biomarker, enrichment, high-throughput
Cell sorting is a crucial tool for enrichment of specific cell types from a heterogeneous population, with applications in research and medicine (1). The goal of cell enrichment is to increase the prevalence of a cell type of interest from a sample that contains a mixture of other cell types. Current cell sorting methods, including differential centrifugation, fluorescence-based sorting, and immunomagnetic-based isolation, each have limitations that make it difficult to efficiently sort large numbers of cells based on surface protein levels without sacrificing cell viability and/or yield. Density gradient centrifugation, a technique developed in the 1950’s, has proven a valuable tool for isolating cell populations based on inherent density differences (2,3). Unfortunately, this method is not universally applicable, as cells of interest do not always have distinct densities from other cells in the sample. In these cases, researchers must use surface markers for the identification and enrichment of specific cell types.
Fluorescence-activated cell sorting (FACS) is the current, state-of-the-art method for isolating populations of cells based on surface marker proteins. This method is advantageous in that multiple markers can be assessed at once, and cells can be isolated based on protein level. FACS has facilitated the study of cell heterogeneity and has demonstrated fantastic utility for both research and therapeutic purposes (4–6). Despite its many advantages, FACS has a few major limitations. First, flow cytometers are costly and require frequent maintenance as well as a skilled operator. Additionally, FACS is relatively limited in its cell throughput, with recent studies reporting poor cell yields (~70% loss of cells) and long processing times when working with large cell numbers (7). Magnetic-activated cell sorting (MACS) is a more high-throughput alternative to FACS and is an attractive choice for applications requiring large cell numbers including spectrometry, proteomics, and therapeutic applications where millions-to-billions of cells are essential (8–11). In MACS, cells are labeled with magnetic microparticles targeting specific surface proteins and subsequently placed in a magnetic field. Labeled cells are retained in the field while unlabeled cells flow through (12). This method is advantageous in that it can sort many cells rapidly and does not require expensive machinery. However, MACS processing is limited to a binary output (i.e., target protein positive vs. negative subpopulations); therefore it cannot enrich for subpopulations based on specific target surface protein quantity.
To address the shortcomings of FACS and MACS, we developed a novel cell sorting method, mass-added density centrifugation (MADC), to rapidly isolate large amounts of cells for applications where protein levels need to be resolved. We hypothesized that we could modulate single-cell density proportionally to its target protein level to enable separation of cells on a density gradient by distributing the modified, single-cell densities into discrete bands. The MADC workflow involves two major steps, the first being mass-adding in which the density of single cells is altered according to protein abundance, followed by density centrifugation. In the mass-adding step, we achieve single-cell density modulation with a series of three incubations: (1) labeling the target surface protein with a primary antibody, (2) targeting primary antibodies with secondary, biotinylated anti-IgG antibodies to increase signal, and (3) binding tertiary, streptavidin-coated, massive microparticles. These small (0.4 μm diameter) microparticles effectively increase the mass of the cell without appreciably affecting the composite volume, thereby modulating composite density proportionally to surface protein abundance. The second step of MADC, density centrifugation, involves centrifuging the mass-added cells on a discrete density gradient to separate cells with like-densities. After centrifugation, groups of cells with similar target surface protein quantities are distributed into distinct density layers and can be isolated from the gradient for downstream applications.
To demonstrate its utility, we applied MADC to the enrichment of human melanoma cells (MeWo) and primary adipose-derived stem cells (ASCs) based on differential CD44 abundance. CD44 was selected as a representative protein of interest since it is a common cell surface protein with applications in many areas of research. After confirming robust density changes with the mass-adding procedure, we used western blots to confirm variable CD44 protein levels in cells isolated from distinct density layers. Good reproducibility and high, post-sort cell viabilities indicated MADC is a robust method for rapidly sorting millions of cells based on surface protein levels.
Materials and Methods
Cell Culture
Human melanoma (MeWo) cell line (ATCC® HTB-65™) was cultured in T182 tissue culture flasks (Genesee Scientific, San Diego, CA) at 37°C with 5% CO2. Medium consisted of Minimum Essential Media (GE Healthcare HyClone, Pittsburgh, PA) supplemented with 10% fetal bovine serum (FBS, Zen-Bio, Research Triangle, NC), 1% nonessential amino acids (GE Healthcare HyClone), 1% penicillin–streptomycin (GE Healthcare HyClone), and 1% l-glutamine (GE Healthcare HyClone). Medium was changed every other day, and cells were expanded until 90% confluent. Cells were detached using 0.25% trypsin (GE Healthcare HyClone) then either passaged at 1/10th density or fixed with 4% paraformaldehyde (Thermo Fisher Scientific, Waltham, MA) for 15 min for use in fixed-cell MADC trials.
For human adipose-derived stem cell (ASC) studies, primary human lipoaspirate was procured from the abdomen of one informed and consenting male donor (43y/o, prior diagnoses of panniculitis and lipodystrophy) in accordance with guidelines approved by the Institutional Review Board of Rhode Island Hospital. Stromal vascular fraction (SVF) cells were isolated according to established protocols and immediately placed in cryogenic storage (13). SVF cells were thawed and passaged three times to expand cell numbers and enrich for ASCs for use in cell-sorting studies (14). ASCs were maintained in control medium consisting of Dulbecco’s modified Eagle’s medium: nutrient mixture F-12 (GE Healthcare HyClone), 10% FBS, and 1% antibiotic/antimycotic (GE Healthcare HyClone) (15). Cells were maintained at 37°C with 5% CO2, and medium was changed every other day until cells were 90% confluent in T182 tissue culture flasks before splitting at 1/10th density into new flasks. ASCs in this study were used up to passage 8.
Mass-Adding Procedure
Fixed cells
4% paraformaldehyde-fixed cells were centrifuged (800g × 2 min), then resuspended at 5 million cells/ml in phosphate-buffered saline (PBS, Thermo Fisher Scientific) supplemented with 5% FBS to minimize nonspecific antibody binding and cell clumping. Mouse anti-human CD44 antibody (Cat. 555476BD; Bioscience, San Jose, CA) was added to the cell suspension at a 1:500 dilution and incubated at room temperature on a shaker for 30 min. Cells were pelleted (800g × 2 min) and washed once in PBS + 5% FBS. Cells were resuspended to 5 million cells/ml, and biotinylated goat anti-mouse IgG antibody (Cat. BA-9200; Vector labs, Burlingame, CA) was added at an optimized 1:500 dilution followed by a 30-min incubation. Cells were washed in PBS + 5% FBS twice, then resuspended to 2 million/ml. High iron, 0.4 μm, streptavidin-coated microbeads (Cat. SVM-05–5H; Spherotech, Lake Forest, IL,) were then added in a cell-number dependent fashion to yield greater than 1,200 beads/cell (210 μl beads at a concentration of 5.74 × 1010 particles/ml) per 10 million cells and incubated for 30 min on a shaker at room temperature. The labeled cells were then filtered through a 70 μm nylon mesh (Thermo Fisher Scientific) to remove clumps before pipetting on top of a Percoll gradient (Thermo Fisher Scientific) for density separation.
Live cells
Cells expanded in T182 flasks were detached using 0.25% trypsin and spun down at 400 g for 5 min. The pellet was resuspended in ice-cold Hank’s Balanced Salt Solution (HBSS, Thermo Fisher Scientific), supplemented with 10% FBS (HBSS + 10%FBS). Cell counts and viabilities were determined using trypan exclusion. Cells were diluted to 5 million cells/ml, and mouse anti-human CD44 antibody was added to the cell suspension at a 1:500 dilution. Cells were incubated in primary antibody for 10 min while shaking on ice. An equal volume of HBSS + 10% FBS was then added to dilute out the antibody before spinning down at 300 g for 2 min. Solution was removed and cells were resuspended to 5 million cells/ml before adding a 1:500 dilution of secondary antibody, biotinylated goat anti-mouse IgG antibody. Cells were incubated for 10 min while shaking on ice. An equal volume of HBSS + 10% FBS was then added before spinning down at 300g for 2 min. Cells were resuspended in HBSS + 10% FBS to a maximum volume that could be layered onto an Optiprep (Sigma-Aldrich, St. Louis, MO) gradient. Beads were added to the cell suspension in a cell-number dependent fashion to yield greater than 1,200 beads/cell (210 μl beads at a concentration of 5.74 × 1010 particles/ml) per 10 million cells, followed by a 10-min incubation shaking on ice. Cells were then passed through a 70 μm mesh (Thermo Fisher Scientific) to remove any clumps before layering the suspension on the Optiprep gradient for density centrifugation.
Density Gradient Formation and Centrifugation
Designing the discrete density gradient
Discrete gradient densities were determined experimentally. Literature about the densities of live vs. fixed cells is limited, so first we tested the cell density of non-mass-added live and fixed cells by centrifuging the cells on a discrete gradient ranging from 1.030 to 1.050 g/ml. Fixed and live cells were found to exhibit slightly different densities. For example, fixed MeWo densities were less than 1.040 g/ml and live MeWo were less than 1.060 g/ml. These pilot cell density tests determined the least-dense fraction on the MADC gradient. The spread of the density gradient (lowest density to highest density) also required initial optimization due to the differences in surface protein levels between the cell types tested. For example, MeWo cells required gradients with higher densities, up to 1.100 g/ml, compared to ASCs that inherently expressed less CD44, which only needed gradients that went up to 1.090 g/ml. Generally, we found that setting the most-dense fraction at ~0.05 g/ml higher than the starting density worked well to resolve cell groups with increased density due to mass-adding.
Density gradient for fixed cell separation
Percoll solution was diluted with 1.5 M sodium chloride (NaCl, Thermo Fisher Scientific) to make stock isotonic Percoll (SIP, density 1.125 g/ml). The SIP solution was further diluted with 0.15 M NaCl according to manufacturer’s instructions to generate desired density solutions (16). The 15 ml tubes (Genesee Scientific) were rinsed with 100% FBS to prevent cells from adhering to the sides. Then, Percoll solutions of decreasing density were pipetted into the tube, with care taken to gently layer solutions to prevent mixing at gradient interfaces.
Density gradient for live cell separation
Optiprep (iodixanol) solution was used for live-cell separations due to increased cell viability post-sort. Optiprep solution was diluted with HBSS, supplemented with 3% bovine serum albumin (BSA, Thermo Fisher Scientific) to generate desired density solutions according to manufacturer’s instructions (17). The 15 ml tubes were rinsed with 100% FBS to prevent cells from adhering to the sides. Then, Optiprep solutions of decreasing density were pipetted into the tube, with care taken to gently layer solutions to prevent mixing at gradient interfaces.
Density centrifugation
After mass-added cells had been filtered through a 70 μm mesh, cells were layered carefully on top of prepared Percoll (fixed cells) or Optiprep (live cells) density gradients. The gradients were then centrifuged at 600g for 10 min to separate cells into distinct density layers. Gradients were fractionated by pipetting off Percoll or Optiprep solution 1 ml at a time and collecting bands of cells of interest into new 15 ml tubes. The individual fractions were resuspended in 5 ml PBS + 10% FBS (fixed cells) or 5 ml HBSS + 10% FBS (live cells) to dilute out gradient solution before spinning down at 400g for 5 min to pellet the cells for use in downstream assays or plating.
Surface Protein Abundance
Western blot assays were conducted to determine CD44 protein levels of cells isolated in distinct density layers after MADC separation. For live-cell protein quantification, MADC-sorted cells from distinct density layers were lysed on ice for 30 min using radioimmunoprecipitation (RIPA, Santa Cruz Biotechnologies, Santa Cruz, CA) lysis buffer. For paraformaldehyde-fixed-cell protein tests, the previously described formaldehyde-fixed intracellular target-sorted antigen retrieval (FITSAR) method was used to isolate protein from MADC-sorted cells (18,19). Briefly, cells were suspended in lysis buffer consisting of 300 mM Tris hydrochloride (Sigma-Aldrich) with 2% sodium dodecyl sulfate (SDS; Thermo Fisher Scientific) and 2X protease/phosphatase inhibitor (Pierce™, Thermo Fisher Scientific) and boiled at 100°C for 35 min, followed by a 2-h incubation at 60°C. The samples were then spun down at 14,000g for 10 min before collecting the supernatant in a fresh 1.7 ml tube (Genesee Scientific). Protein concentrations in the lysates were analyzed using a bicinchoninic acid (BCA) Assay (Pierce™, Thermo Fisher Scientific) prior to gel loading. For each gradient, a fixed amount of protein across all cell fractions was loaded and separated on SDS-PAGE Any KD gels (Bio-Rad, Hercules, CA) and transferred onto Immobilon™-FL polyvinylidene fluoride membranes (Millipore, Burlington, MA). The membrane was rinsed in water then incubated in Revert Total Protein Stain (LiCor, Lincoln, NE) for 5 min. Total protein stain was imaged in the 700 nm channel using an Odyssey CLx near-infrared scanner (LiCOR). The membrane was then submerged in 0.1 M sodium hydroxide (Sigma-Aldrich) and 30% (v/v) methanol (Sigma-Aldrich) in water for 5 min to remove the stain before blocking in nonmammalian Odyssey® Blocking Buffer (LiCOR) for 1 h at room temperature to limit interference with the IRDye™ secondary antibodies. Following blocking, membranes were incubated with a mouse anti-human CD44 antibody (1:500 dilution, Cat. 3570S; Cell Signaling Technology, Danvers, MA) for 1 h at room temperature or at 4°C overnight. Membranes were washed three times at 15-min intervals in 1X Tris-buffered saline Tween (TBST, Thermo Fisher Scientific) and then labeled with a goat anti-mouse IRDye® 800CW (1:1000 dilution, Cat. 925–32210, LiCOR) secondary antibody for 1 h. Membranes were washed three more times at 15-min intervals in 1X TBST before imaging on the Odyssey CLx near-infrared scanner on the 800 nm channel. For all western blots, densitometry analyses were performed using ImageJ version 1.51d. Protein quantification data were normalized to total protein staining.
Reproducibility Assay
The entire MADC procedure was conducted in triplicate. Ten million paraformaldehyde-fixed MeWo cells were processed for each, independent run (30 million cells total). After initial antibody labeling steps, the cells were pipetted onto a Percoll density gradient with three layers: 1.07, 1.08, and 1.22 g/ml. After a 10-min centrifugation at 600g, cells from the top (<1.07 g/ml), middle (<1.22 g/ml), and bottom (>1.022 g/ml) fractions were isolated. Cells were then labeled with wheat germ agglutinin (WGA) at a 1:500 dilution for 10 min at room temperature. Cells from the top fraction were labeled with Alexa Fluor 647-conjugated WGA (WGA-647; Thermo Fischer Scientific). Cells from the middle fraction were labeled with Alexa Fluor 488-conjugated WGA (WGA-477; Thermo Fischer Scientific). Cells from the bottom fraction were left unstained. After thorough rinsing in PBS, cells were fixed in a second preparation of 4% paraformaldehyde to prevent WGA leaching in solution, which was observed in pilot work to lead to artifactually stained cells. Following this treatment, cells from the separate fractions were re-mixed and layered onto another density gradient (identical to the first: 1.07, 1.08, and 1.22 g/ml). After 10-min centrifugation at 600g, gradients were fractionated as previously described to obtain cells from the top (<1.07 g/ml), middle (<1.22 g/ml), and bottom (>1.022 g/ml). The three fractions were then spun down separately and resuspended in 1 ml PBS for analysis on a Guava easyCyte 8HT flow cytometer (Luminex, Austin, TX). Control samples for establishing gates consisted of unstained, WGA-647-stained, and WGA-488-stained paraformaldehyde-fixed MeWo cells. Cell distributions spanning across the second gradient separation were analyzed based on cell counts within defined gates around WGA-647, WGA-488, and unstained cells for each isolated fraction. A double-stained cell population with high red and green fluorescence appeared in the mixed-cell fractions. This group was excluded from analysis as it was attributed to the artifactual staining mentioned above. Data were analyzed with InCyte 3.3 software.
Proliferation Assay
To confirm cells were alive and capable of attaching, migrating, and proliferating in culture, post MADC-sorted cells were counted and plated into three six-well plates (Corning, Corning, NY) at 2,000 cells per well. Then at 2-day intervals a plate was removed, and its cells were fixed with 10% formalin (Thermo Fisher Scientific) for 10 min at room temperature. The cells were then stained with 4′,6-diamidino-2-phenylindole (DAPI, 0.1 μg/ml, Thermo Fisher Scientific) for 5 min at room temperature. DAPI-stained cells were then imaged on a Cytation3, and nuclei were counted using a custom MATLAB script. Nuclei per image were then extrapolated to total well area to assess the number of cells per well for each time point. Population doubling times were calculated for time spans encompassing Days 0–3, 3–6, and 6–9 for Adipose-derived stem cells, and for Days 0–2, 2–5, and 5–7 for MeWo cells.
Statistical Analysis
All statistical analyses were performed using SigmaPlot version 12.5. Western blot protein levels for the density fractions were compared using a one-way analysis of variance (ANOVA) with post hoc Holm–Sidak analysis for significance in pairwise comparisons. For the repeatability assay, we compared how cells from each fraction of the first gradient distributed into fractions of the second gradient with a one-way ANOVA. For example, we compared the distribution of AlexaFluor 647 labeled cells (from the top fraction of the first gradient) across the three fractions of the second gradient. A post hoc Holm–Sidak test was used to analyze significance in pairwise comparisons. Population doubling times for control (non-mass-added), mass-added unsorted cells, mass-added CD44high, and mass-added CD44low cells, were compared for each timepoint with a one-way ANOVA.
Results
Single-Cell Density Modulation is Achieved During the MADC Mass-Adding Step
To test whether cell density could be modified by microparticle labeling, fixed-MeWo cell distribution throughout a density gradient was compared between mass-added and control, unprocessed cells (Fig. 1A). The gradient on the right shows the clear distribution of mass-added cells throughout the density gradient, indicated by banding at density interfaces. The gradient on the left shows the distribution of control, non-mass-added cells, which are only found at the upper-most density layers (<1.040 g/ml). Cells from increasing density fractions displayed higher microparticle coverage when observed under a brightfield microscope (Supporting Information Fig. S1). Density bands from the gradients were isolated, and cells in each fraction were quantified to confirm that the distribution of mass-added cells ranged from <1.035 g/ml to upward of 1.070 g/ml (pellet) (Fig. 1B). These results indicated that the mass-adding step was able to increase cell density upward of 0.035 g/ml, a difference easily resolvable on a discrete density gradient. Secondary-only controls with MeWo cells incubated with an anti-biotin antibody and microparticles showed no discernable shift in densities. This gradient confirms that cell density is modulated by the mass-adding MADC step by anchoring dense microparticles to specific surface proteins.
Figure 1.
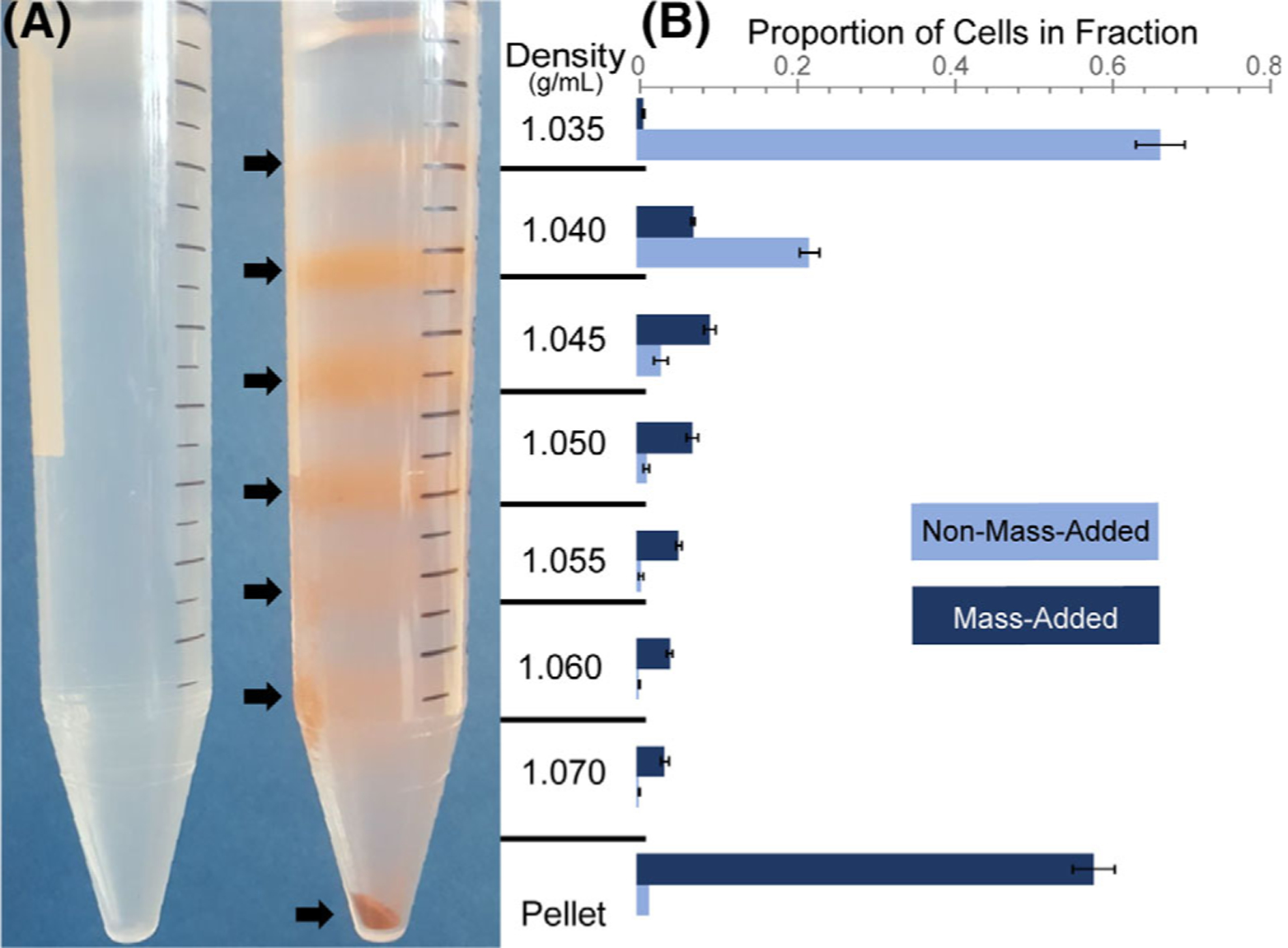
Cell density increased after MADC mass-adding step. (A) Individual cell densities could increase to over 1.070 g/ml in the mass-added cell density column (right) as compared to control, non-mass-added cells, which largely settled at densities less than 1.040 g/ml (left). Note: Brownish red color due to binding of the iron microparticles to cells. (B) The distribution of mass-added and control cells throughout the gradient was quantified using flow cytometry. Mass-added cells that did not end up in the pellet showed a clear shift in their densities, spanning the entire gradient.
Target Protein Levels Correlate with Cell Density
Western blots were used to determine whether MADC cell sorting is protein-level dependent. Live MeWo cells isolated from distinct density bands after MADC separation were lysed and analyzed for relative CD44 levels. Cells isolated from increasing-density fractions correlated with increased CD44 abundance (Fig. 2). Repeated iterations of MADC separations into four fractions of cells reflected similar protein concentration correlations to cell density. There was a significant difference in CD44 level between cells of the first and fourth (P = 0.005) fraction, as well as between the second and fourth fraction (P = 0.029).
Figure 2.
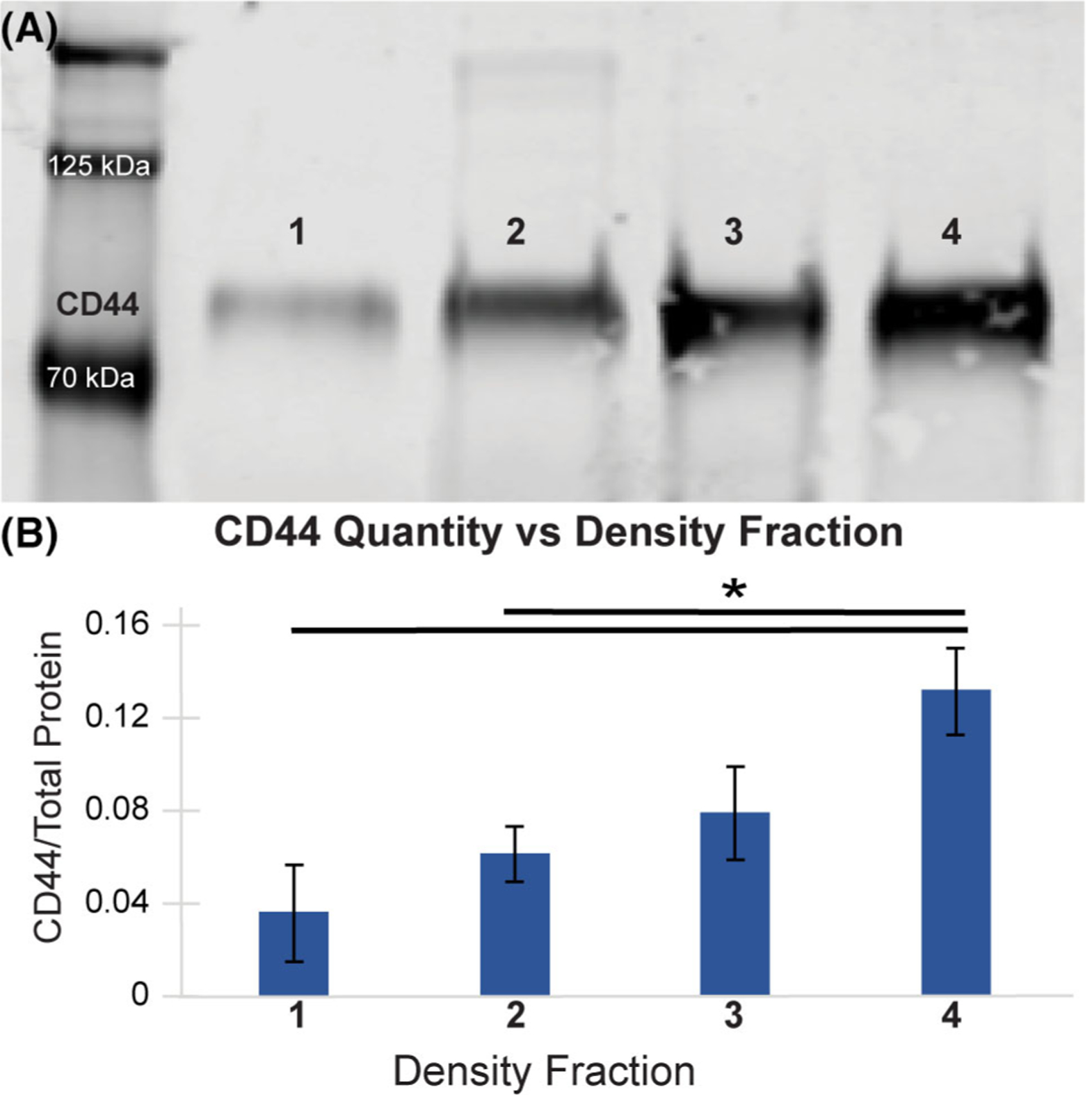
CD44 abundance was higher in cells isolated from more dense fractions. (A) Western blots probing for CD44 on cells sorted by MADC revealed a corresponding increase in CD44 levels with gradient fraction density. Note: Lysates were collected from a typical live-cell MADC separation trial. (B) Densitometry analysis of CD44 intensity, normalized by total protein levels for each lane, revealed the same trend of increasing CD44 abundance in cells deposited in higher density fractions (*P < 0.05). Note: Densitometry analysis combines western blots from three, separate, live-cell MeWo MADC separations.
Assessing Reproducibility of the MADC Process
We performed a reproducibility experiment to determine the reliability of cell distribution in each density layer if a sample was centrifuged sequentially on two identically constructed gradients. This test was also used to quantify cell loss using the MADC procedure. After a MADC sort of fixed MeWo cells based on CD44 abundance, cells from the top, middle and bottom were labeled with Alexa Fluor 647-conjugated wheat germ agglutinin (WGA-647) for the top fraction, Alexa Fluor 488-conjugated wheat germ agglutinin (WGA-477) for the middle fraction, and left unstained for the bottom fraction. These cells were then mixed and re-separated on a second gradient. Distribution of cells to the three density layers in the second density gradient was then analyzed by flow cytometry (Fig. 3, analysis example in the Supporting Information Fig. S2) For all fractions, a large majority of cells re-deposited in the same density band as in the original gradient (70–96%). Statistically, the number of cells re-depositing in their same gradient location between the first and second separations (WGA-647 to the top, WGA-488 to the middle, and unstained to the bottom), versus other density bands in the second separation, was significant for all fractions (P < 0.001). About 70% of cells from the top fraction of the first separation (WGA-647 stained) re-deposited in the top fraction of the second separation. Seventy percent of cells from the middle fraction (WGA-488 stained) re-deposited to the middle fraction. Ninety-six percent of cells from the bottom fraction (unstained) re-deposited to the bottom fraction. The entire process, from the mass-adding procedure to fractionation and cell isolation proved to be largely cell-retentive, with ~25% cell loss. For a standard MADC run, this cell loss would likely be lower, given that the reproducibility assay involves two gradient separations with considerable handling for the intermediate cell processing.
Figure 3.
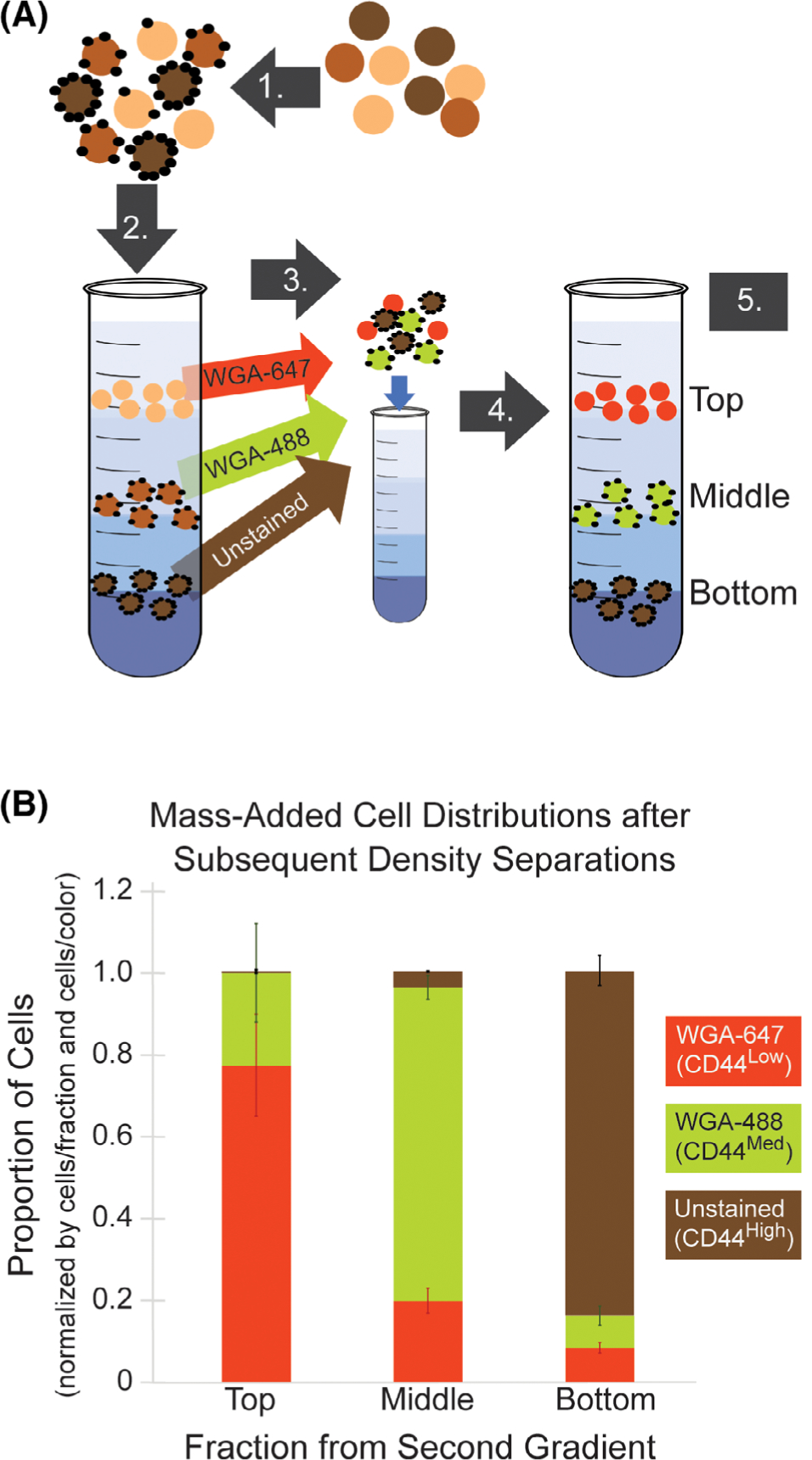
Cells re-deposited at ≥70% rates in the same density band upon subsequent separation trials. (A) Experimental schematic for the MADC reproducibility assay (as described in Methods Section). 1. Cells were labeled with mass-adding particles. 2. Mass-added cells were centrifuged on a density gradient (first gradient separation). 3. Cell fractions were isolated and WGA stained, then fractions were re-mixed. 4. WGA-stained cells were centrifuged on a second density gradient (second gradient separation). 5. Cell fractions were isolated and fluorescence tagging analyzed by flow cytometry to determine the distribution of cells in each fraction. (B) Cell distributions upon subsequent separation trials indicated that most cells re-distributed to the same density fraction, as compared to the number of cells depositing in different fractions. Note: Fixed MeWo cells were used in this experiment.
MADC Maintains High Cell Viabilities Post-Sort
Live-cell MADC sorts were conducted to examine feasibility for experiments where live cells are needed in downstream applications. Both MeWo and ASCs were separated on MADC gradients in duplicate. Both cell types maintained high viabilities (>90%) throughout the entire MADC process, with no significant difference between mass-added and unprocessed control cells incubated on ice. In these sorts, we were able to process upwards of 20 million cells on one 15 ml gradient column centrifuged for 10 min, attesting to the high-throughput possibilities of this method, particularly if conducted in an arrayed setup. To assess the potential effects of mass-adding particles on cells, we tracked proliferation rates of cells plated post-MADC sorting (Fig. 4). When comparing CD44high, CD44low, and unprocessed-control cells, there was no significant difference in proliferation rates when assessed over a week postsort (P > 0.05).
Figure 4.
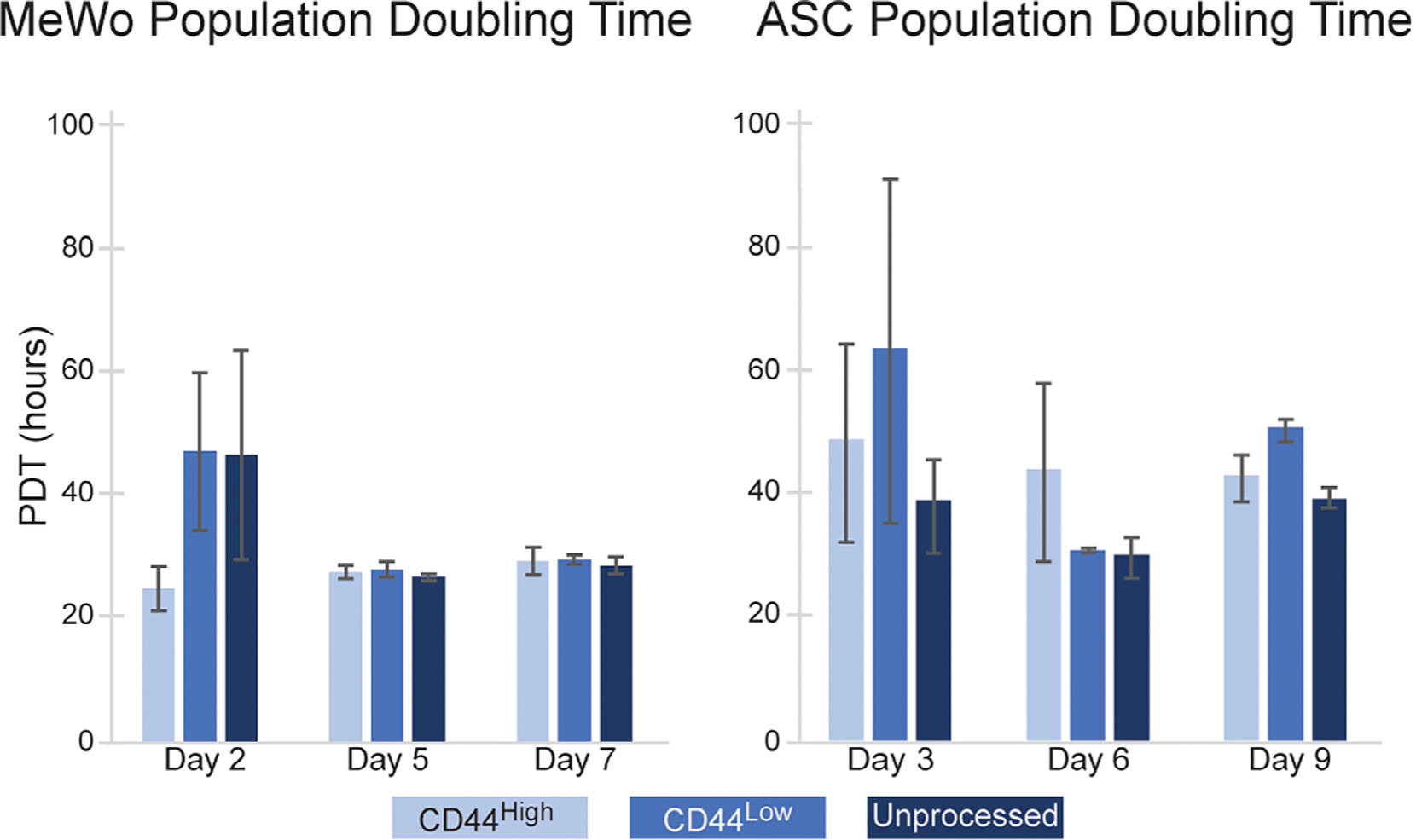
Cell proliferation rates for MeWo and ASCs were unaffected by MADC cell processing. There was no statistically significant difference in population doubling times between MADC processed and control, unprocessed cells (P > 0.05). Plating effects impacted all conditions, as indicated by longer PDTs through Days 2 and 3. However, this effect seemed to disappear by Days 5 and 6.
Discussion
The goal of our work was to develop a novel cell sorting method that allowed for rapid selection of large quantities of cells according to the abundance of a target surface protein. To do this, we first confirmed that single-cell density modulation with a mass-adding step was resolvable on a density gradient and that density shifts were proportional to protein abundance. Next, we characterized reproducibility and cell losses, confirming MADC as a reliable sorting method with high cell yields. Lastly, we applied MADC to live-cell sorts to demonstrate its utility in maintaining high viabilities (>90%) with minimal effects on basic cell behavior (e.g., proliferation rates). Our results demonstrate MADC as an attractive cell sorting alternative to FACS and MACS for high-throughput, protein quantity-dependent sorts.
The fundamental principle of MADC is for single-cell density modulation to correlate with surface protein levels. Western blot experiments revealed a trend existed for cells from increasing density fractions having increasing protein abundance, confirming our expected outcome. To date, studies looking at cell heterogeneity based on protein levels have been limited to FACs-sorted cells, a technology not readily available to all labs, particularly ones in low resource settings. MADC enables researchers to rapidly and inexpensively sort cells based on protein levels to enable closer examination of cell heterogeneity. For example, our CD44-sorted cells could be used for downstream cancer or stem cell research, as CD44 expression levels correlate with stem-like cell behavior and cancer malignancy (20–22). Beyond research use, MADC could be applied to sorting stem cells, and other beneficial cell types, to improve treatment outcomes. A major challenge for cell-based therapies is that often more than hundreds of millions of cells are needed (23–25). This requirement necessitates in vitro expansion of cells to achieve high enough numbers (24–26). However, vitro expansion can have adverse effects on cell behavior resulting in decreased stem cell potency and unpredictable treatment outcomes (27,28). In such cases, MADC can be applied to enrich expanded stem cells for those retaining the most optimal stem-like qualities, while simultaneously acquiring a more homogenous cell population. MADC is a versatile cell sorting technique, with the power to expand research into cell heterogeneity and improve cell-based therapies.
The MADC protocol, as detailed here, was found to be well suited for fixed-cell isolations as well as live-cell sorts. For the optimized live-cell MADC procedure, cell viability remained above 90%. Optiprep gradient solution was selected due to higher viabilities in live-cell sorts. The diluting medium for the iodixanol solution was HBSS supplemented with FBS to provide more nutrients to cells during processing (29). Alternatively, Percoll is diluted with an isotonic salt solution which provides little pH buffering action or toxin neutralization. It is also possible that Optiprep results in less shear during density centrifugation, as it is a small molecule-based density solution (iodixanol) rather than a polydisperse particle-based (silica) solution like Percoll. Post-sort there was no observable impact of MADC on cell health, as determined with a proliferation assay and general morphological observations. However, cell behavior is very complex. Therefore, future work will focus on characterization of the effect of microparticles on cell behaviors, such as impact on stem cell differentiation. We do not expect microparticle-decoration on cells to pose unresolvable problems for therapeutic applications. A study by David et al. found no negative consequences of injecting cells tagged with MACS microbeads in vivo (30). When stem cells tagged with MACS microbeads (compositionally similar to our microparticles) were injected into a mouse heart, the microbeads were quickly cleared and showed no adverse impact, thereby increasing our confidence that this method could be applied as a therapeutic cell enrichment method. Alternatively, MADC can be modified by substituting a cleavable biotin to then easily remove microparticles post-sort to minimize adverse effects (31). In addition to demonstrating utility for sorting both live and fixed cells, we found MADC to be a reliable method, with good reproducibility.
In our reproducibility assay, we tested whether cells would distribute to the same density fractions when centrifuged twice in a row on identical density gradients. For the most part, labeled cells re-distributed to the same fractions they originally separated into. However, this was not always the case. We found that a portion of cells from the top and middle fractions would redistribute to other fractions following the second separation process. Of the cells originally in the top fraction, 21% re-distributed to the middle fraction and 9% to the bottom fraction. Of the cells originally in the middle fraction, 22% re-distributed to the top fraction of the second gradient while 9% re-distributed to the bottom fraction. Of the cells originally in the bottom fraction, <1% re-distributed to the top fraction and 3% to the middle fraction. The general redistribution of cells to different layers could be caused by a combination of factors including: dislodging of microparticles during intermediate cell-processing steps (e.g., WGA staining), entrapment of more dense cells within the upper-buoyant cell bands, inadvertent mixing during gradient fractionization prior to flow cytometry, and/or imprecise gate placement during cell analysis. Given that in a normal MADC separation, cells are only mass-added, and density centrifuged once, microparticle shearing is not a problem for practical applications. Despite the drift of cells to less dense fractions in the second gradient, we found that the majority of cells from the first gradient settled into the same density-bands when centrifuged on an identical gradient a second time. These results indicate that distribution throughout the gradient is not random, but rather that individual cell density is indeed modulated in the mass-adding step. Altogether, the consistent reproducibility of cell distribution on subsequent density separations indicates that MADC can separate cells based on density in a manner dependent on microparticle decoration, and as such, protein quantity.
Here, we present MADC as a novel cell sorting method that complements gold-standard approaches like FACS and MACS. MADC is a suitable option for research, or therapeutic, purposes as it is rapid and inexpensive, requiring only a centrifuge and cell labeling materials. A limitation of MADC is that multiplexing to target multiple proteins in a single gradient centrifugation is not feasible. Instead, future work can explore the possibility of sequential MADC separations targeting different proteins by removing microparticles after each separation, by way of a cleavable biotin modification (31). In this manuscript, we present MADC in its basic form, which is best suited for separating cells based on a single target protein expressed at variable levels. Rigorous optimization of antibody combinations and gradient densities is required to achieve high-quality outcomes. A given cell type may differ in original starting density, and the magnitude of density shifts will depend on the absolute number of surface proteins available for antibody binding. Due to a lack of publications reporting cell densities and amount of surface proteins expressed, there is some experimental calibration required for each cell type and target protein, necessitating a high initial time investment before MADC sorts can be routinely applied. Despite these limitations, MADC addresses key shortcomings of both FACS and MACS and will enable scientists to rapidly sort high quantities of cells based on protein levels for downstream applications ranging from proteomic studies to therapeutics.
Supplementary Material
Acknowledgments
Financial support for this work was provided by National Institutes of Health, National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR063642). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Literature Cited
- 1.Jia Z, Liang Y, Xu X, Li X, Liu Q, Ou Y, Duan L, Zhu W, Lu W, Xiong J, et al. Isolation and characterization of human mesenchymal stem cells derived from synovial fluid by magnetic-activated cell sorting (MACS). Cell Biol Int 2018;42(3):262–271. [DOI] [PubMed] [Google Scholar]
- 2.Schneider C Cytochemical studies of mammalian tissues; the isolation of cell components by differential centrifugation: A review. Cancer Res 1951;11:23. [PubMed] [Google Scholar]
- 3.Ulmer AJ, Scholz W, Ernst M, Brandt E, Flad H-D. Isolation and subfractionation of human peripheral blood mononuclear cells (PBMC) by density gradient centrifugation on Percoll. Immunobiology 1984;166(3):238–250. [DOI] [PubMed] [Google Scholar]
- 4.Boumelhem BB, Assinder SJ, Bell-Anderson KS, Fraser ST. Flow cytometric single cell analysis reveals heterogeneity between adipose depots. Adipocyte 2017;6(2): 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herzenberg LA, Parks D, Sahaf B, Perez O, Roederer M, Herzenberg LA. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin Chem 2002;48(10):1819–1827. [PubMed] [Google Scholar]
- 6.Tytor M, Wingren S, Olofsson J. Heterogeneity of squamous cell carcinomas of the Oral cavity studied by flow cytometry. Pathology 1991;187(1):30–35. [DOI] [PubMed] [Google Scholar]
- 7.Sutermaster BA, Darling EM. Considerations for high-yield, high-throughput cell enrichment: fluorescence versus magnetic sorting. Sci Rep 2019;9(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.García-Olmo D, García-Arranz M, Herreros D, Pascual I, Peiro C, Rodríguez-Montes JA. A phase I clinical trial of the treatment of Crohn’s fistula by adipose mesenchymal stem cell transplantation. Dis Colon Rectum 2005;48(7):1416–1423. [DOI] [PubMed] [Google Scholar]
- 9.Pers Y-M, Rackwitz L, Ferreira R, Pullig O, Delfour C, Barry F, Sensebe L, Casteilla L, Fleury S, Bourin P, et al. Adipose mesenchymal stromal cell-based therapy for severe osteoarthritis of the knee: A phase I dose-escalation trial. Stem Cells Transl Med 2016;5(7):847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-Olmo D, García-Arranz M, García LG, Cuellar ES, Blanco IF, Prianes LA, Montes JAR, Pinto FL, Marcos DH, García-Sancho L. Autologous stem cell transplantation for treatment of rectovaginal fistula in perianal Crohn’s disease: A new cell-based therapy. Int J Colorectal Dis 2003;18(5):451–454. [DOI] [PubMed] [Google Scholar]
- 11.Bura A, Planat-Benard V, Bourin P, Silvestre J-S, Gross F, Grolleau J-L, Saint-Lebese B, Peyrafitte JA, Fleury S, Gadelorge M, et al. Phase I trial: The use of autologous cultured adipose-derived stroma/stem cells to treat patients with non-revascularizable critical limb ischemia. Cytotherapy 2014;16(2):245–257. [DOI] [PubMed] [Google Scholar]
- 12.Miltenyi S, Müller W, Weichel W, Radbruch A. High gradient magnetic cell separation with MACS. Cytometry 1990;11(2):231–238. [DOI] [PubMed] [Google Scholar]
- 13.Estes BT, Diekman BO, Gimble JM, Guilak F. Isolation of adipose-derived stem cells and their induction to a chondrogenic phenotype. Nat Protoc 2010;5(7):1294–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varma MJO, Breuls RGM, Schouten TE, Jurgens WJFM, Bontkes HJ, Schuurhuis GJ, Ham SMV, Milligen FJV. Phenotypical and functional characterization of freshly isolated adipose tissue-derived stem cells. Stem Cells Dev 2007;16(1): 91–104. [DOI] [PubMed] [Google Scholar]
- 15.Estes BT, Diekman BO, Guilak F. Monolayer cell expansion conditions affect the chondrogenic potential of adipose-derived stem cells. Biotechnol Bioeng 2008;99(4): 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Healthcare GE. How to make and use gradients of Percoll [Internet]. 2007. https://www.sigmaaldrich.com/technical-documents/protocols/biology/cell-separation-media/gradients-of-percoll.html [Google Scholar]
- 17.Ford T, Graham J, Rickwood D. Iodixanol: A nonionic Iso-osmotic centrifugation medium for the formation of self-generated gradients. Anal Biochem 1994;220(2): 360–366. [DOI] [PubMed] [Google Scholar]
- 18.Sadick JS, Darling EM. Processing fixed and stored adipose-derived stem cells for quantitative protein array assays. Biotechniques 2017;63(6):275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sadick JS, Boutin ME, Hoffman-Kim D, Darling EM. Protein characterization of intracellular target-sorted, formalin-fixed cell subpopulations. Sci Rep 2016;6 (1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dietrich A, Tanczos E, Vanscheidt W, Schöpf E, Simon JC. High CD44 surface expression on primary tumours of malignant melanoma correlates with increased metastatic risk and reduced survival. Eur J cancer Oxf Engl 1997;33(6):926–930. [DOI] [PubMed] [Google Scholar]
- 21.Johansson A-C, Fleur LL, Melissaridou S, Roberg K. The relationship between EMT, CD44high/EGFRlow phenotype, and treatment response in head and neck cancer cell lines. J Oral Pathol Med 2016;45(9):640–646. [DOI] [PubMed] [Google Scholar]
- 22.Thapa R, Wilson GD. The importance of CD44 as a stem cell biomarker and therapeutic target in cancer. Stem Cells Int 2016;2016:2087204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kharaziha P, Hellström PM, Noorinayer B, Farzaneh F, Aghajani K, Jafari F, Telkabadi M, Atashi A, Honardoost M, Zali MR, et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: A phase I–II clinical trial. Eur J Gastroenterol Hepatol 2009;21(10):1199–1205. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Olmo D, Herreros D, Pascual I, Pascual JA, Del-Valle E, Zorrilla J, De-La-Quintana P, Garcia-Arranz M, Pascual M. Expanded adipose-derived stem cells for the treatment of complex perianal fistula: A phase II clinical trial. Dis Colon Rectum 2009;52(1):79–86. [DOI] [PubMed] [Google Scholar]
- 25.Pérez-Simon JA, López-Villar O, Andreu EJ, Rifón J, Muntion S, Diez Campelo M, Sánchez-Guijo FM, Martinez C, Valcarcel D, Del Cañizo C. Mesenchymal stem cells expanded in vitro with human serum for the treatment of acute and chronic graft-versus-host disease: Results of a phase I/II clinical trial. Haematologica 2011;96(7): 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.García-Arranz M, Herreros MD, González-Gómez C, Quintana P d l, Guadalajara H, Georgiev-Hristov T, Trébol J, Garcia-Olmo D. Treatment of Crohn’s-related rectovaginal fistula with allogeneic expanded-adipose derived stem cells: A phase I–IIa clinical trial. Stem Cells Transl Med 2016;5(11):1441–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bogdanova A, Berzins U, Nikulshin S, Skrastina D, Ezerta A, Legzdina D, Kozlovska T. Characterization of human adipose-derived stem cells cultured in autologous serum after subsequent passaging and long term cryopreservation. J Stem Cells 2014;9(3):135–148. [PubMed] [Google Scholar]
- 28.Phinney DG. Functional heterogeneity of mesenchymal stem cells: Implications for cell therapy. J Cell Biochem 2012;113(9):2806–2812. [DOI] [PubMed] [Google Scholar]
- 29.Johnson M Fetal Bovine Serum Mater Methods [Internet] 2020. March 27 [cited 2020 Apr 27]; /method/Fetal-Bovine-Serum.html [Google Scholar]
- 30.Müller P, Gaebel R, Lemcke H, Wiekhorst F, Hausburg F, Lang C, Zarniko N, Westphal B, Steinhoff G, David R. Intramyocardial fate and effect of iron nanoparticles co-injected with MACS® purified stem cell products. Biomaterials 2017;135:74–84. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Verhelst SHL. Cleavable trifunctional biotin reagents for protein labelling, capture and release. Chem Commun 2013;49(47):5366–5368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.