

Abstract
MYC is a highly validated oncogenic transcription factor and cancer target. However, the disordered nature of this protein has made it a challenging target, with no clinical stage, direct small molecule MYC inhibitors available. Recent work leveraging a large in silico chemical library and a rapid in vivo screen has expanded the chemotypes of direct small molecule inhibitors (MYCi). Novel MYCi represent a class of improved MYC chemical probes that bind directly to MYC to inhibit its function and promote its degradation by enhancing GSK-3β-mediated phosphorylation. One of these compounds, MYCi975, has shown remarkable tolerability and efficacy in vivo and is associated with a selective effect on MYC target gene expression. Additional effects of MYCi on the tumor immune microenvironment including immune cell infiltration and upregulation of PD-L1 expression provide a rationale for combining MYCi with anti-PD1/PD-L1 therapy to enhance anti-tumor efficacy. Our strategy for developing MYCi demonstrates an efficient way to identify selective and well-tolerated MYC inhibitors. The new MYCi provide tools for probing MYC function and serve as starting points for the development of novel anti-MYC therapeutics.
Introduction
MYC is a master transcription factor responsible for regulating essential cellular processes including proliferation, metabolism, biosynthesis, and apoptosis, which when corrupted are recognized as hallmarks of cancer (1). Upregulation of MYC expression occurs with high frequency in cancer. A recent pan-cancer analysis of over 9000 human cancers revealed amplification of MYC genes in ~28% of all malignancies (2). In addition, tumors use multiple mechanisms to upregulate MYC mRNA and protein expression, knowledge of which has led to estimates that MYC proteins are functionally involved in up to 70% of all human cancers (3,4).
Despite being one of the most frequently altered and highly validated oncogenes, there are currently no direct MYC inhibitors available in the clinic. The lack of well-defined small molecule binding pockets on this intrinsically disordered protein and the absence of in vivo evidence supporting MYC inhibition as a safe and efficacious approach to treat cancer have been challenges. Studies using transgenic models of a dominant negative MYC peptide, Omomyc, have shown that MYC inhibition triggers rapid regression of tumors in vivo, with only mild, and fully reversible side effects on healthy tissues (5,6). These experiments, paired with emerging chemical biology approaches, have reignited the search for MYC inhibitors in cancer.
Substantial efforts to target MYC, both directly and indirectly, are underway. This review focuses on direct MYC inhibitors and the lessons they afford in further unraveling MYC tumor biology. Indirect strategies for targeting MYC e.g. reducing MYC transcription with inhibitors of BRD4 proteins or inhibitors of G-quadruplex DNA structures in the MYC promoter have also yielded important insights (7–9).
Multiple chemical series have been reported to directly bind MYC in biophysical assays and these serve as promising starting points for the development of chemical probes (10–18). Most of these compounds disrupt MYC/MAX complex formation, which is essential for DNA binding and regulation of gene expression. However, the limited potency and the poor pharmacokinetic properties of these compounds have limited their utility as probes or therapeutic leads.
Our group has recently reported a novel series of distinct small molecules capable of binding to and inhibiting MYC, in vitro and in vivo (19). Here we discuss the molecular mechanisms by which these compounds function to inhibit and degrade MYC and their role in remodeling the tumor microenvironment through immune regulatory proteins and effector cells (Fig. 1). We contextualize this work by reviewing the direct MYC inhibitors available at present and the potential therapeutic opportunities these agents offer, particularly in combination with immune checkpoint blockade. The development of direct MYC inhibitors, while beset by challenges, is a desirable goal that offers unique opportunities to probe MYC function and to comprehensively assess the therapeutic potential of targeting this notorious oncogene.
Figure 1:
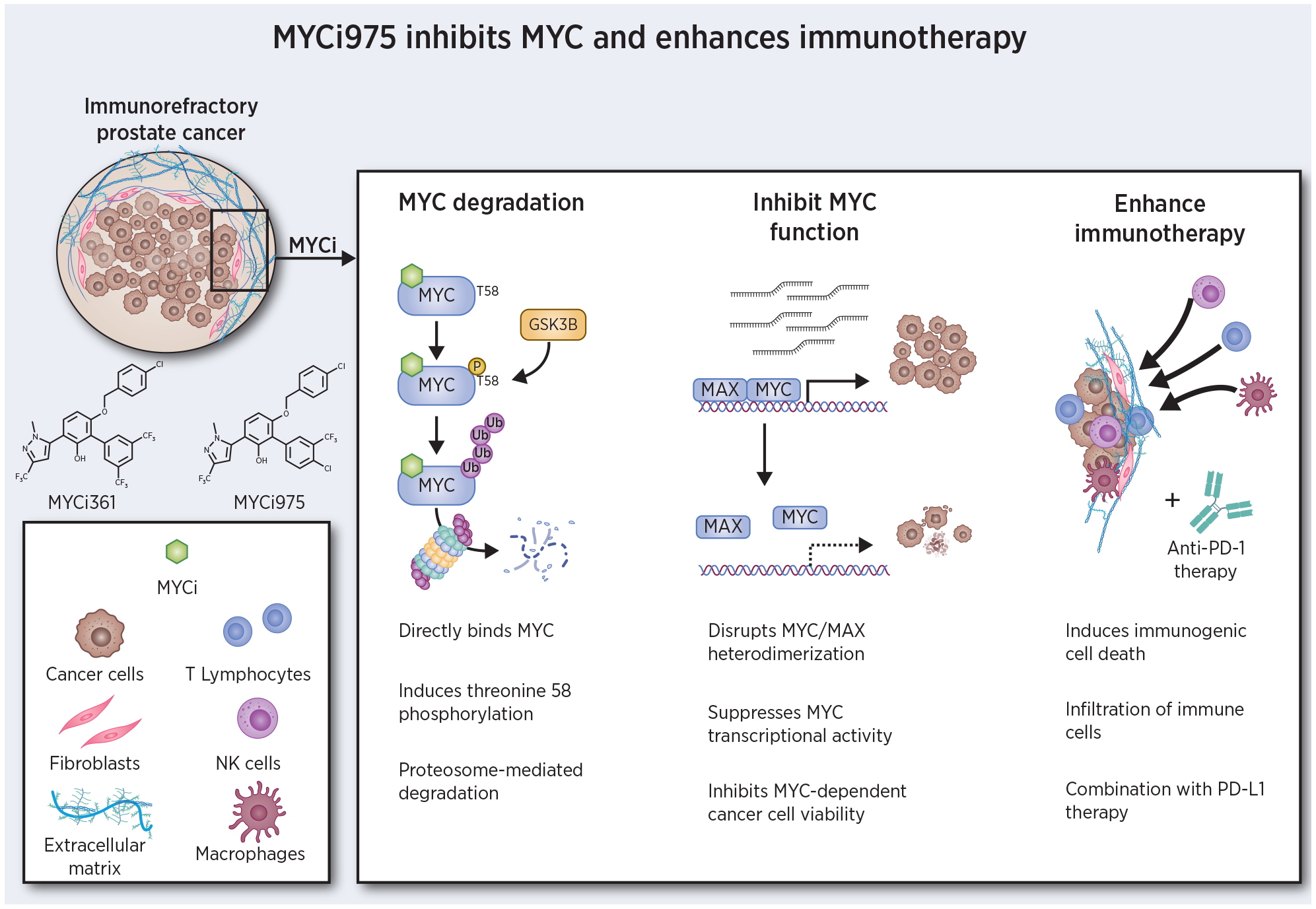
Mechanism of MYCi efficacy. MYCi directly binds to MYC, induces phosphorylation of threonine 58 leading to MYC degradation. MYCi inhibits MYC/MAX heterodimerization, blocking MYC-dependent transcription and proliferation. Following MYCi treatment, the tumor microenvironment is altered by infiltration of diverse immune effector cells and increased PD-L1 expression. Combination of MYCi with anti-PD-1/PD-L1 therapy results in improved efficacy.
The enduring challenge of drugging the “undruggable”
MYC is the poster child of so-called “undruggable” targets. The protein presents a featureless surface which lacks traditional binding pockets, a common characteristic of intrinsically disordered proteins. This challenge is further complicated by the lack of a resolved crystal structure of MYC as a monomer. No rational drug design approach exists for direct targeting of protein domains that present extensive regions of disorder. In the case of MYC and its obligate partner MAX, both present a disordered bHLHZip (basic region-helix-loop-helix-leucine zipper) domain as monomers. These two disordered monomeric leucine zipper motifs undergo coupled folding and binding to generate the ordered alpha-helical structure of the MYC-MAX heterodimer, with its characteristic left-handed coiled coil appearance. Resolution of the crystal structure of the MYC-MAX heterodimer spurred efforts to identify small molecules aimed at disrupting the ordered MYC-MAX heterodimer using a variety of high-throughput screening strategies (20). Although these efforts have not yielded a clinical MYC inhibitor thus far, they have nonetheless yielded important insights into MYC biology and provide a starting point for the development of newer probes.
As an example, Prochownik and colleagues have studied a series of inhibitors, including 10074-G5 and 10058-F4 which were shown to bind to MYC using a variety of biophysical assays(17,18). NMR experiments measuring chemical shift perturbations of amino acid backbone heteroatoms were used to explore how these small molecules bind MYC peptides. The experimental data are most consistent with a mode of binding described as “diffuse” binding, in which MYC remains disordered over the binding region. Consequently, it is apparent that there are multiple distinct conformations of the disordered primary amino acid region that can accommodate binding of the ligand. In turn, in silico modelling efforts have revealed that the binding of the ligand itself seems to be dispersed over the targeted disordered binding region, meaning different amino acids along the binding region interact differently with the ligand based on the conformation the disordered protein adopts. In short, MYC peptides that are bound by ligand remain in a dynamic ensemble (21,22). The in-depth study of the mechanism of binding of these inhibitors revealed that there are indeed regions on the disordered MYC protein that can accommodate small molecule binding and, more importantly, that direct targeting of the MYC monomer with small molecules is feasible.
We reasoned that these and other small molecule inhibitors shown to bind to MYC could be the starting point of a pharmacophore model that can be used to interrogate a large chemical library in silico (32 million compounds). Indeed several new chemotypes were identified with in vitro activity in MYC/MAX/DNA complex assays, MYC-driven E box reporter assays, and MYC-dependent proliferation assays. To facilitate the identification of compounds with in vivo activity, we used a rapid in vivo assay in which compounds are screened against an allograft bearing a MYC-driven luciferase reporter. This assay helps identify compounds that are stable in vivo and able to access tumor and modulate MYC activity. The assay also provides a gross idea of the in vivo tolerability of the test compounds. Integration of this key in vivo assay with established in vitro assays guided our SAR (structure-activity relationships) strategy. This approach resulted in the development of several active compounds, among which MYCi361 and MYCi975 were most extensively studied and reported (19). It is instructive to note that up to 85% of compounds with MYC-selective in vitro activity failed the in vivo screen, highlighting the importance of this step in identifying compounds with desirable features. In particular, MYCi975 showed favorable pharmacokinetics, in vivo tolerability and efficacy in mouse tumor models. Testing in vivo revealed improved tumor responses in the presence of an intact host immune system. MYCi treatment modulated the tumor immune microenvironment by increasing expression of the immune checkpoint protein PD-L1 and inducing immune cell infiltration. This provided the rationale for combination therapy with MYCi and anti-PD-1/PD-L1 immunotherapy.
Blocking and degrading MYC
Target engagement studies for MYCi361 and MYCi975 showed that the compounds engaged MYC in its native cellular environment by cellular thermal shift (CETSA) assay and could disrupt MYC/MAX complex formation. Additional biophysical assays including competitive fluorescence polarization and STD-NMR were used to show that MYCi’s bind to MYC but not MAX. Studies using biotinylated compounds indicated that MYCi361 and MYCi975 bind to amino acids 366–381 of MYC. This region of MYC has also been reported to bind to several other small molecules, identified by different groups using disparate experimental approaches, including 10074-G5, 10074-A4, JKY-2–169, and 7594–0035(11,12,18,23–27). This amino acid stretch within the C-terminal bHLHZip domain of the disordered MYC protein may represent a “hotspot” for binding to small molecules that we here call MYCHot1 (Fig. 2). This region, in addition to another “hotspot” region spanning amino acids 402–409 in MYC (MYCHot2) that binds 10058-F4 (18,24), consists of amino acid sequences that are enriched for hydrophobic residues in comparison to other regions of the bHLHZip domain (26). Additionally, these sequences are less disordered than surrounding regions and are predicted to display transient secondary structure (28). Notably, the MYCHot regions are poorly conserved in MAX, possibly explaining the specificity for binding MYC isoforms over MAX for many of these compounds (Fig. 2, sequence alignment). The binding of small molecule MYCi’s to the MYCHot1 region in the bHLH domain provides a structural rationale for their disruption of the MYC/MAX complex.
Figure 2:
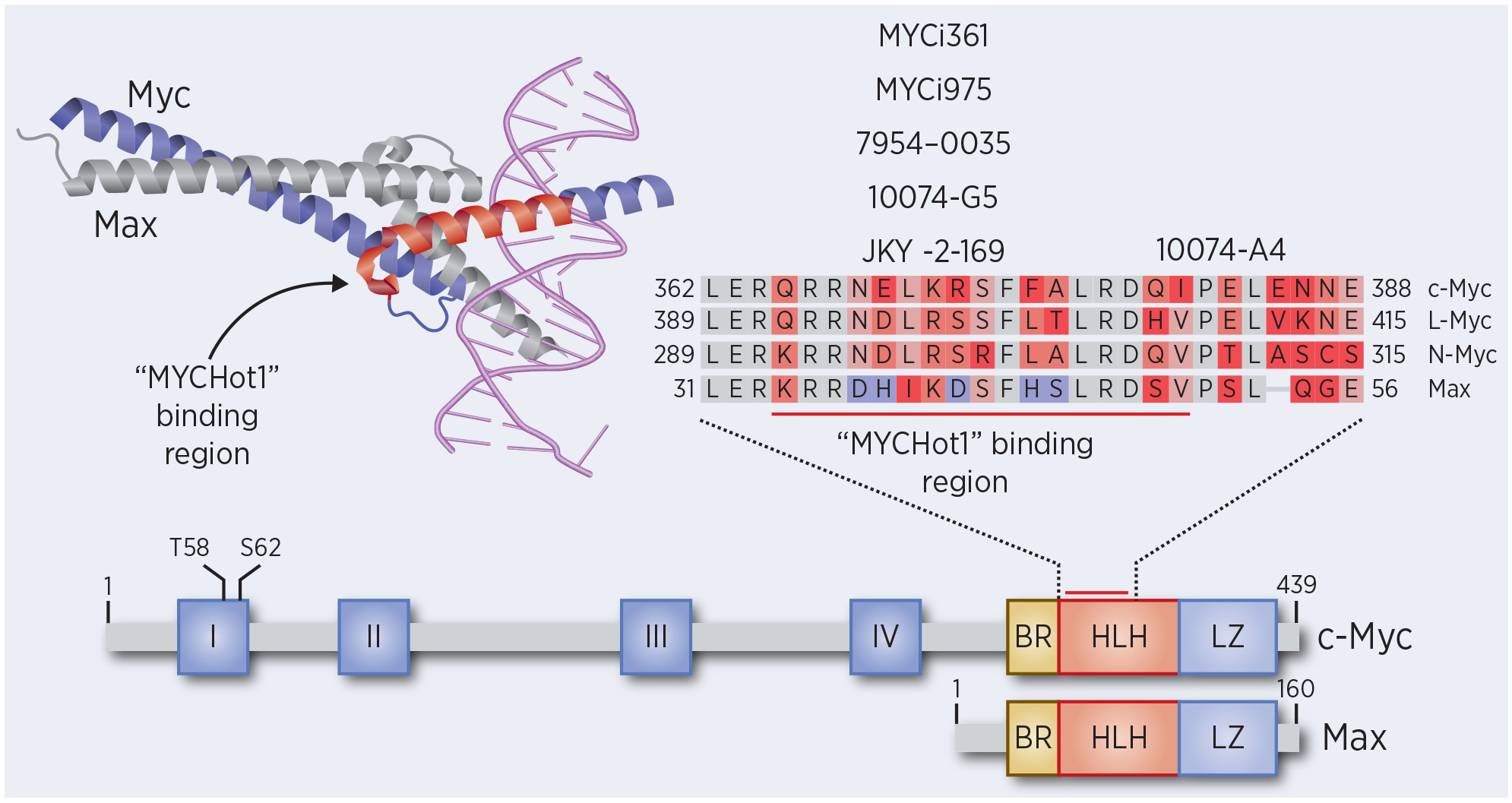
Crystal structure of the DNA-bound MYC/MAX heterodimer (PDB: 1NKP) highlighting the putative “MYCHot1” binding region. The functional domains on Myc and Max are shown with a zoomed in region of the HLH domain containing the “MYCHot1” binding region. Sequence alignment of the “MYCHot1” binding region for c-Myc, L-Myc, N-Myc, and Max. Mismatched residues are colored in red, with the notable differences between c-Myc and Max in purple. Corresponding MYC inhibitor binding locations are shown above the alignment.
Interaction of MYCi with MYC also led to enhanced degradation of MYC through the proteasome pathway (19). This could be a consequence of disruption of the MYC/MAX complex as other disruptors of the MYC/MAX complex also led to proteasome-mediated MYC degradation (12). However, examples of small molecules that disrupt MYC/MAX dimers without inducing MYC degradation have also been reported (13). Thus, it is more likely that specific interactions between some small molecules and MYC could affect MYC conformation to promote its degradation. We have investigated the potential mechanism for MYCi361- and MYCi975-induced degradation of MYC (19), focusing on the well-defined GSK-3β/MYCpT58/proteasome pathway that regulates MYC protein stability (29–31). Here, ordered phosphorylation of MYC on serine 62 (pS62) by kinases such as ERK, CDK and JNK primes MYC for subsequent isomerization and phosphorylation on threonine 58 (pT58) by GSK-3β(32). MYC pT58 is then recognized by E3 ubiquitin ligases and subsequently degraded by the 26S proteasome(31). MYCi co-opts this native mechanism to degrade MYC by selectively increasing phosphorylation of T58. Notably, MYCi enhanced MYC pT58 in an in vitro kinase assay with recombinant proteins (MYC, ERK, and GSK-3β), suggesting that the interaction of MYCi with MYC may modify the conformational ensemble the protein can sample to make it a better GSK-3β substrate. Importantly, MYCi did not enhance the phosphorylation of another established GSK-3β substrate β-catenin(19). Biophysical and structural data detailing how small molecule binding to the C-terminal region of MYC leads to phosphorylation of T58 located near the N-terminus are not yet available and merit further investigation.
A therapeutic window for MYC inhibition
Earlier studies with the MYC dominant negative peptide Omomyc yielded the important insight that MYC inhibition could be tolerated (5,6). Our recent work explored in detail the tolerability of MYCi small molecules. While MYCi361 showed evidence of in vivo toxicity, which may include on-target and off-target effects, MYCi 975 showed remarkable tolerability (19). Acute maximum tolerated dose finding studies indicate that MYCi975 could be tolerated in mice when given orally at up to 10X the anti-tumor efficacious dose of 100mg/kg, with testing of higher doses limited by solubility. A 2 week daily treatment regimen of MYCi975 at 100 mg/kg/d showed remarkably no histopathological or chemical pathological abnormalities. RNAseq data showed that MYCi975 regulated a smaller number of genes than MYCi361, which may explain its tolerability. Notably, in the tetracycline-controlled MYC lymphoma model P493–6, MYCi975 regulated genes largely overlapped with tetracycline-regulated genes, with the exception of a group of genes involved in metabolism of organic small molecule and ER stress that were uniquely induced by MYCi975. Thus, MYCi975 appears to selectively regulate some target genes and may not uniformly inhibit the expression of all MYC targets. Further studies of the selectivity of MYCi and other small-molecule MYC probes in regulating the MYC transcriptional program should prove informative in this regard, especially when coupled with in vivo tolerability and toxicity studies.
MYCi remodeling of the tumor immune microenvironment and therapeutic implications
MYC plays an important role in regulating the host anti-tumor immune response (33–35). MYC regulates the tumor microenvironment through effects on both immune regulatory proteins and immune effector cells. CD4+ T cells have been shown to be specifically required for the induction of cellular senescence, shutdown of angiogenesis, and chemokine expression that lead to sustained tumor regression upon MYC inactivation(36). MYC may also promote tumorigenesis by regulating expression of PD-L1 (37–39). Other mechanisms contributing to tumor regression following MYC inactivation may involve immunogenic cell death (ICD), innate immune responses by NK cells and B cells, as well as adaptive immune-dependent responses by CD4+ and CD8+T cells(34).
The critical role of the host immune response in the anti-tumor effects of MYC inactivation, coupled with the reported roles of MYC in immune cell function led us to focus on immunocompetent models for testing MYCi anti-tumor efficacy (35). MYCi361 and MYCi975 treatment of syngeneic prostate tumor bearing mice led to a remodeling of the tumor immune microenvironment by recruitment of innate and adaptive immune cells and increased expression of immunoregulatory proteins. Following MYCi treatment in this murine model of prostate cancer, an increase in intratumoral CD3+ T cells was observed. Further immunophenotyping showed a specific increase in CD3+CD4+ and CD3+CD8+ T cells, interferon ɣ (IFN-ɣ)-expressing CD4+ and CD8+ T cells, tumor necrosis factor alpha-expressing CD8+ cells, dendritic cells, as well as natural killer (NK) cells (19). Recruitment of these immune effector cells was accompanied by immunogenic cell death and an upregulation of tumor PD-L1 expression. These results provided the rationale for combination therapy with MYCi and anti-PD1 checkpoint blockade leading to improved anti-tumor efficacy in vivo.
The findings described with MYCi were primarily performed in a murine model of MYC-driven prostate cancer, MycCaP. In prostate cancer specifically, immune checkpoint blockade has had marginal benefit due to an immunosuppressive tumor microenvironment (40). The MycCap model recapitulates this immunosuppressive microenvironment as seen in patients, and this model has been shown to be resistant to anti-PD1 therapy alone (41). By increasing immune cell infiltration and tumor immunogenicity, MYCi circumvents the immunosuppressive nature of the prostate tumor environment and sensitizes an immunotherapy resistant cancer type to readily available, and well tolerated, immune checkpoint inhibitors. Further studies will be required to determine the applicability of this approach to other tumor types that respond poorly to immunotherapy, evaluate the efficacy of MYCi monotherapy in tumors with an immune-primed microenvironment, and delineate what immune effectors are essential to MYC-inhibitor induced tumor responses.
Perspective
In contrast to the major scientific advances in gene regulation, transcription, and tumorigenesis that MYC research has yielded, breakthroughs in therapeutically targeting MYC have been scant. In recent years, oncologic drug discovery has instead focused efforts on precision medicines aimed at more tractable targets, such as kinases. This approach suffers from diminishing returns as an ever more narrow patient population is targeted. It is important to continue to invest in key oncogenic drivers, such as MYC, that hold therapeutic promise across a diverse spectrum of cancers (42). While the process has been challenging, the discovery of direct binding small molecules targeting MYC has led to significant new insights into how MYC protein levels are regulated, how MYC transcriptional programs can be selectively altered, and how MYC functions to help tumors evade host immune detection. These insights have opened new avenues of research and below we highlight some of the key unanswered questions, and methodologies to consider, that may accelerate the discovery of the first clinically active MYC inhibitor.
The observed degradation of MYC by multiple distinct chemical series supports the hypothesis that MYC can be inhibited by small molecules that alter MYC protein stability for therapeutic purposes. The degradation phenotype should be evaluated systematically as a part of MYC drug discovery efforts, and the mechanistic basis of degradation should be investigated. The most well characterized pathway regulating MYC protein stability involves a stepwise phosphorylation of serine 62 followed by isomerization and phosphorylation of threonine 58 leading to proteasomal degradation (31). Small molecules may co-opt all or part of this pathway, as demonstrated by MYCi361 and MYCi975, which selectively alter phosphorylation at T58 (19). Finally, the possibility of MYC degradation by distinct mechanisms, whether involving native signaling pathways or engineered heterobifunctional approaches targeting MYC for degradation, should be considered or even actively sought. An increase in the number, and diversity, of chemical probes that directly bind to MYC will enable us to test whether the best approach to inhibit MYC is to prevent all of its functions via degradation, or to inhibit select functions while leaving others intact.
The increasing availability of chemical probes with suitable properties for in vivo testing described above enables the scientific community to test the effect of MYC inhibition on the tumor microenvironment and tumor-host immune interactions. MYC functions in a context-dependent manner to integrate multiple intracellular signals, process and interpret these inputs, and ultimately generate an output in the form of a transcriptional program that alters cell function and tumor behavior (43). Given the role of the immune system in the anti-tumor response to MYC inactivation and the widely varied efficacy of immunotherapy across tumor types, testing the consequence of inhibiting MYC should be done in model systems that best reflect human disease, namely immunocompetent in vivo models.
The MYCi’s highlighted in this review have some limitations as they are further developed for early human testing. The lipophilicity of the lead compound (logP value of 8.4) poses a challenge for formulating appropriate solutions for in vivo administration. The potency of these compounds could also be further improved.
Despite being considered one of the most highly validated oncogenes in cancer, MYC has remained an intractable target. While the challenge of targeting an intrinsically disordered protein is daunting, experimentally testing the full potential of MYC inhibition as a therapeutic approach to treat cancer necessitates the development of highly selective and well-tolerated systemic therapies. The work reviewed above lays the groundwork for this and identifies promising approaches to discover the elusive, clinically efficacious MYC inhibitor.
Acknowledgements
This work was supported, in part, by grants from the National Cancer Institute (F30 CA250196 to M.T. and P50 CA180995 to S.A.A.); by the Prostate Cancer Foundation Award (#19CHAL06 to S.A.A.); and by a grant from the Polsky Urologic Cancer Institute of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University at Northwestern Memorial Hospital.
Footnotes
Disclosure of Potential Conflicts of Interest: H.Han and S.A.Abdulkadir are co-inventors on patent applications covering the methods and assays to identify and characterize MYC inhibitors and derivatives. All other authors declare no competing interests.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 2.Schaub FX, Dhankani V, Berger AC, Trivedi M, Richardson AB, Shaw R, et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst 2018;6:282–300 e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dang CV. MYC on the path to cancer. Cell 2012;149:22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, et al. MYC Deregulation in Primary Human Cancers. Genes (Basel) 2017;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature 2008;455:679–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, et al. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 2013;27:504–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calabrese DR, Chen X, Leon EC, Gaikwad SM, Phyo Z, Hewitt WM, et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat Commun 2018;9:4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen-Petersen BL, Sears RC. Mission Possible: Advances in MYC Therapeutic Targeting in Cancer. BioDrugs 2019;33:539–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, et al. Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther 2007;6:2399–408 [DOI] [PubMed] [Google Scholar]
- 11.Fletcher S, Prochownik EV. Small-molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta 2015;1849:525–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi SH, Mahankali M, Lee SJ, Hull M, Petrassi HM, Chatterjee AK, et al. Targeted Disruption of Myc-Max Oncoprotein Complex by a Small Molecule. ACS Chem Biol 2017;12:2715–9 [DOI] [PubMed] [Google Scholar]
- 13.Castell A, Yan Q, Fawkner K, Hydbring P, Zhang F, Verschut V, et al. A selective high affinity MYC-binding compound inhibits MYC:MAX interaction and MYC-dependent tumor cell proliferation. Sci Rep 2018;8:10064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proc Natl Acad Sci U S A 2014;111:12556–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung KY, Wang H, Teriete P, Yap JL, Chen L, Lanning ME, et al. Perturbation of the c-Myc-Max protein-protein interaction via synthetic alpha-helix mimetics. J Med Chem 2015;58:3002–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C, Niu X, Jin F, Liu Z, Jin C, Lai L. Structure-based Inhibitor Design for the Intrinsically Disordered Protein c-Myc. Sci Rep 2016;6:22298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Follis AV, Hammoudeh DI, Wang H, Prochownik EV, Metallo SJ. Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol 2008;15:1149–55 [DOI] [PubMed] [Google Scholar]
- 18.Hammoudeh DI, Follis AV, Prochownik EV, Metallo SJ. Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc 2009;131:7390–401 [DOI] [PubMed] [Google Scholar]
- 19.Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019;36:483–97 e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003;112:193–205 [DOI] [PubMed] [Google Scholar]
- 21.Michel J, Cuchillo R. The impact of small molecule binding on the energy landscape of the intrinsically disordered protein C-myc. PLoS One 2012;7:e41070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin F, Yu C, Lai L, Liu Z. Ligand clouds around protein clouds: a scenario of ligand binding with intrinsically disordered proteins. PLoS Comput Biol 2013;9:e1003249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, et al. In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther 2010;335:715–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo J, Parise RA, Joseph E, Egorin MJ, Lazo JS, Prochownik EV, et al. Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol 2009;63:615–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Teriete P, Hu A, Raveendra-Panickar D, Pendelton K, Lazo JS, et al. Direct inhibition of c-Myc-Max heterodimers by celastrol and celastrol-inspired triterpenoids. Oncotarget 2015;6:32380–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beaulieu ME, Castillo F, Soucek L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carabet LA, Lallous N, Leblanc E, Ban F, Morin H, Lawn S, et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur J Med Chem 2018;160:108–19 [DOI] [PubMed] [Google Scholar]
- 28.Panova S, Cliff MJ, Macek P, Blackledge M, Jensen MR, Nissink JWM, et al. Mapping Hidden Residual Structure within the Myc bHLH-LZ Domain Using Chemical Denaturant Titration. Structure 2019;27:1537–46 e4 [DOI] [PubMed] [Google Scholar]
- 29.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol 2004;6:308–18 [DOI] [PubMed] [Google Scholar]
- 30.Arnold HK, Zhang X, Daniel CJ, Tibbitts D, Escamilla-Powers J, Farrell A, et al. The Axin1 scaffold protein promotes formation of a degradation complex for c-Myc. EMBO J 2009;28:500–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrell AS, Sears RC. MYC degradation. Cold Spring Harb Perspect Med 2014;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Z, He C, Wang J. Regulation mechanism of Fbxw7-related signaling pathways (Review). Oncol Rep 2015;34:2215–24 [DOI] [PubMed] [Google Scholar]
- 33.Felsher DW. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer 2010;1:597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Casey SC, Baylot V, Felsher DW. The MYC oncogene is a global regulator of the immune response. Blood 2018;131:2007–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, et al. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017;171:1301–15 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rakhra K, Bachireddy P, Zabuawala T, Zeiser R, Xu L, Kopelman A, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010;18:485–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016;352:227–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 2017;31:1633–7 [DOI] [PubMed] [Google Scholar]
- 39.Kim EY, Kim A, Kim SK, Chang YS. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 2017;110:63–7 [DOI] [PubMed] [Google Scholar]
- 40.Boettcher AN, Usman A, Morgans A, VanderWeele DJ, Sosman J, Wu JD. Past, Current, and Future of Immunotherapies for Prostate Cancer. Front Oncol 2019;9:884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anker JF, Naseem AF, Mok H, Schaeffer AJ, Abdulkadir SA, Thumbikat P. Multi-faceted immunomodulatory and tissue-tropic clinical bacterial isolate potentiates prostate cancer immunotherapy. Nat Commun 2018;9:1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thomas LR, Adams CM, Fesik SW, Eischen CM, Tansey WP. Targeting MYC through WDR5. Mol Cell Oncol 2020;7:1709388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer 2008;8:976–90 [DOI] [PubMed] [Google Scholar]