

Abstract
The highly reactive compound methylglyoxal (MG) can cause direct damage to cells and tissues by reacting with cellular macromolecules. MG has been identified as a biomarker associated with increased sepsis-induced mortality. Patients undergoing septic shock have significantly elevated circulating MG levels compared to post-operative patients and healthy controls. Furthermore, MG has been implicated in the development of Type II diabetes mellitus and Alzheimer’s Disease. Since MG is generated during glycolysis, we hypothesized that MG may be produced by classically activated (M1) macrophages, possibly contributing to the inflammatory response. LPS and IFN-γ-treated macrophages acquired an M1 phenotype (as evidenced by M1 markers and enhanced glycolysis) and formed MG-adducts, MG-H1, MG-H2, and MG-H3, that were detected using antibodies specific for MG-modified proteins (methylglyoxal 5-hydro-5-methylimidazolones). MG adducts were also increased in the lungs of LPS-treated mice. Macrophages treated with LPS and IFN-γ also exhibited decreased expression of Glyoxalase 1 (Glo1), an enzyme that metabolizes MG. Concentrations of exogenous, purified MG > 0.5 mM were toxic to macrophages; however, a nontoxic dose of 0.3 mM induced TNF-α and IL-1β, albeit to a lesser extent than LPS stimulation. Despite prior evidence that MG-adducts may signal through “Receptor for Advanced Glycation Endproducts” (RAGE), MG-mediated cell death and cytokine induction by exogenous MG was RAGE-independent in primary macrophages. Finally, RAGE-deficient mice did not exhibit a significant survival advantage following lethal LPS injection. Overall, our evidence suggests that MG may be produced by M1 macrophages during sepsis, following IFN-γ-dependent down regulation of Glo1, contributing to over-exuberant inflammation.
Keywords: Innate immunity, inflammation, cytokine regulation, metabolism
Graphical Abstract:
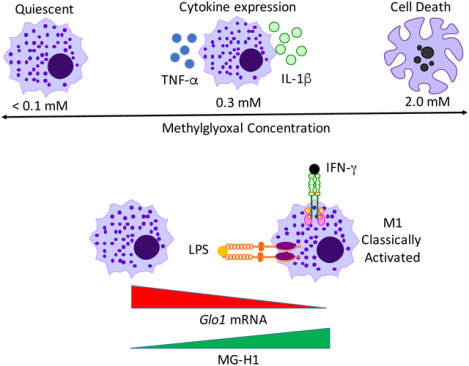
Summary:
Cellular and molecular effects of MG on macrophage function, including a role in promoting inflammatory cytokine induction independent of RAGE.
Introduction:
Even though sepsis has been a focus of biomedical research for decades, it remains one of the leading causes of death in intensive care units, with an estimated incidence of 1.5 million patients in the United States alone with a mortality rate of 20–50%. The incidence of sepsis has increased over the past 20 years, with costs associated with sepsis in the USA estimated at ~$20 billion [1]. These statistics indicate that there is an unmet need to find alternative treatments for sepsis. The early phase of sepsis has been characterized by changes in inflammation and metabolism. We propose that these two changes may be intertwined and that targeting the levels or activity of the alpha-carbonyl aldehyde, methylglyoxal (MG), a reactive carbonyl species (RCS) that is elevated compared to healthy controls during septic shock [2], may alleviate the inflammation and the associated morbidity and mortality of sepsis. In support of this hypothesis, MG has been identified as a biomarker associated with increased mortality in sepsis [2], although the mechanism(s) by which it exerts its effect(s) on sepsis is/are unclear. There are no specific scavengers of MG currently licensed for use to treat this disease.
One of the key pathways by which MG is formed is glycolysis, through the non-enzymatic loss of the phosphate group from glyceraldehyde phosphate and dihydroxyacetone phosphate [3]. These two substrates are isomers that are interconverted through the action of the enzyme triosephosphate isomerase (TPI). MG is produced by decomposition of the ene-diol triose intermediate that leaks from the active site of TPI [4]. Glycolysis is relevant to sepsis because the mitochondrial electron transport chain can be uncoupled or otherwise impaired during systemic inflammation through the activity of cytokines or nitric oxide (NO.) [5, 6], potentially forcing an overdependence on glycolysis for glucose catabolism and ATP synthesis. The need to mobilize glucose from stores to provide energy to failing organs and tissues may help explain why sepsis patients exhibit hyperglycemia as a stress response during disease [7]. The glyoxalase system, comprised of Glyoxalase 1 (Glo1) and Glyoxalase 2 (Glo2), normally detoxifies MG by converting it to lactate, using glutathione as a cofactor, in an effort to maintain homeostasis [8]. In relation to sepsis, Glo1 expression is impaired in patients with septic shock, while Glo2 expression was not analyzed [2]. This combination of systemic enhancement of glycolysis and decreased Glo1 activity likely contributes to the observation that septic patients have significantly elevated MG levels compared to post-operative patients and healthy controls [2]. Certain species of bacteria like Escherichia coli produce MG de novo [9], possibly contributing to its accumulation during bacterial sepsis.
Researchers have tried to determine the mechanistic role for MG in inflammation through stimulation of cells in tissue culture with purified MG or MG conjugated to bovine serum albumin (MG-BSA). As a member of the RCS family, MG can cause direct cellular damage by covalently modifying proteins, DNA, RNA, and phospholipids, potentially causing affected proteins to lose function [10] or DNA to undergo mutation [11]. Further, MG-treated cells can undergo apoptosis [12]. In the presence of oxygen, MG-mediated glycation of free amino groups in proteins can also lead to formation superoxide anion (O2−.) [13] that contributes to the apoptotic effect of MG [14]. However, apoptotic signaling is not the sole pathway triggered by MG. One common finding from tissue culture experiments is MG-induced activation of NF-κB and/or MAPK pathways [15–22]. In contrast, MG has also been shown to suppress NF-κB activation in response to TNF-α [23], highlighting the uncertainty of how MG truly regulates host inflammation. Overall, it is important to note that it is not clear whether MG-induced apoptosis or NF-κB activation requires formation of MG-protein adducts or whether MG alone is sufficient. Additionally, due to the fact that many prior studies have used cell lines, it is also difficult to ascertain if results will translate to primary cells of physiologic relevance.
MG-modified proteins like MG-BSA can undergo further reactions to form Advanced Glycation Endproducts (AGEs) that bind and signal through the Receptor for Advanced Glycation Endproducts (RAGE) to induce NF-κB-mediated inflammation, potentially amplifying tissue damage [24, 25]. Significantly, mice with a targeted mutation in Ager, the gene that encodes RAGE, have been reported to exhibit improved survival versus wild-type (WT) control mice following LPS-induced endotoxicity [26] and cecal ligation and puncture [27], two murine models of sepsis. In support of the latter study, treatment of mice with a RAGE-specific monoclonal antibody was protective in a model of sepsis induced by cecal ligation and puncture, but its efficacy against LPS-induced endotoxicity was not examined [28]. Although these studies highlight the potential importance of RAGE during sepsis, it should be noted that the role of RAGE in sepsis is not always clear-cut and may be context-dependent. For instance, RAGE plays no role in sepsis induced by bypassing the lung through intravenous injections of Streptococcus pneumoniae [29]. Although many different cell types and organ systems are affected in sepsis, macrophages figure centrally in this dysregulated immune response to infection (reviewed in [30]). The central role of macrophages in endotoxicity was first established conclusively by showing that LPS-induced lethality could be reconstituted in TLR4 signaling-deficient C3H/HeJ mice by adoptive transfer of LPS-sensitive WT macrophages [31, 32]. Therefore, it was the goal of our study to determine if classically activated, primary macrophages synthesize MG and how MG might regulate inflammation.
Materials and Methods:
Reagents.
Vitamin C (# A5960), Aminoguanidine (AG) (# 396494) Trypan Blue solution (# T8154) and Methylthiazolyldiphenyl tetrazolium bromide (MTT) (# 2128) were purchased from Sigma-Aldrich (St. Louis, MO). Protein-free E. coli K235 LPS was prepared as previously described [33]. Recombinant mouse IFN-γ (# 485-MI-100/CF) and recombinant mouse IL-4 (# 404-ML-010/CF) were both purchased from R & D systems. MG-BSA (# STA-306) was purchased from Cell Biolabs and purified prior to use in experiments by Pierce™ high capacity endotoxin removal columns (# 88273) according to the manufacturers’ instructions.
MG synthesis.
Methylglyoxal 1,1-dimethyl acetal (#170216) was purchased from Sigma-Aldrich (St. Louis, MO) and used to synthesize MG. Methylglyoxal 1,1-dimethyl acetal was redistilled using a ChemGlass 19 cm TS-14/20 Vigreux column and a Heidolph-Brinkman B169 electric water aspirator. The methylglyoxal 1,1-dimethyl acetal was in a 100 ml recovery flask containing Teflon boiling chips and submersed in a 55° C water bath. Fractions were collected in a radial distillation receiver. Redistilled methylglyoxal 1,1-dimethyl acetal (100 mmol) was hydrolyzed by adding it to 200 ml 2.5% (vol/vol) sulfuric acid in a 1 L round bottom flask fitted with a reflux condenser and was heated in a boiling water bath for 1 h. The MG was distilled out of the sulfuric acid solution now placed in a 37° C water bath using an all Teflon KnF Neuberger Laboport vacuum pump pulling a 2 mm vacuum through a 38 cm Hempel column filled with small glass Raschig rings in conjunction with an Argon bleed [34]. Fraction 3 from the distillation was shown to be free of methanol by using 1H NMR [35], and was assayed using aminoguanidine [36]. The process yielded about 12 ml of 49.8 mM pure MG.
Macrophage isolation, treatment, and infection.
The IACUC at University of Maryland, Baltimore approved all of our studies described below using animals. Ager−/− mice [27], backcrossed to C57BL/6 background, were the kind gift of Dr. Ann Marie Schmidt (NYU). Macrophages isolated from Nrf2−/− mice [37] were kindly provided by Dr. Thomas Kensler (Fred Hutchinson Cancer Research Center). TLR4−/− mice, also on a C57BL/6J background, were originally obtained from Dr. Shizuo Akira (Osaka University, Osaka, Japan) and were bred at the University of Maryland (Baltimore, MD, USA). Thioglycollate-elicted, mouse peritoneal macrophages were isolated from age- and sex-matched wild-type (WT) C57BL/6J (Jackson Laboratories), TLR4−/−, Ager−/−, and Nrf2−/− mice by peritoneal lavage and cultured as previously described [38]. Mouse macrophages were stimulated with LPS (10 ng/ml) and IFN-γ (20 ng/ml) or recombinant IL-4 (40 ng/ml) for 48 h to polarize the cells to an M1 or M2 phenotype, respectively. Bone marrow derived macrophages (BMDM) were obtained by lavaging the femurs of mice as previously reported [39].
Isolation and processing of lung tissue for in vitro cell culture.
Lungs were isolated from mice using a method adapted from a previously published protocol [40]. Mouse lungs were initially minced thoroughly with surgical scissors and then incubated with 5 mg/ml Collagenase D (#11088858001, Sigma-Aldrich, St. Louis, MO) for 30 min at 37° C in a water bath. The resulting tissue fragments were forced through a 70 μM nylon mesh filter into culture media, using a rubber stopper from a syringe. After centrifugation, the pelleted cells were resuspended in 10 ml of ACK lysis solution for 5 min at room temperature to lyse contaminating erythrocytes. After being washed again with culture medium and a second round of centrifugation, the cells were resuspended in RPMI 1640 (#15–040-CV) tissue culture medium supplemented with 2% Fetal Bovine Serum, Penicillin-Streptomycin (50 U/ml) and L-Glutamine (2 mM). Single cell suspensions were counted by hemocytometer, and plated in 6-well tissue culture dishes at a density of 2.5 – 3 × 106 cells/well.
Extracellular Flux analyses.
Macrophages were seeded at 1 – 1.5 × 105 cells/well in XF24 microplates (Agilent Technologies), and treated as described above, except that a P1000 pipette was used for aspiration for all washes and media changes. Standard mitochondrial and glycolytic stress tests were performed on an XF24 Extracellular Flux Analyzer (Agilent Technologies) with the following optimizations [41]: XF media was supplemented with 0.4% fatty-acid free bovine serum albumin (#A7030, Sigma-Aldrich, St. Louis, MO), then pH was adjusted to 7.4 with sodium hydroxide and the media was filter-sterilized (Steriflip, Millipore); compound delivery ports A, B, C were loaded with 75 μL of 10x, 11x, and 12x of the final drug concentrations, respectively; for the mitochondrial stress tests, a final concentration of 0.6 μM oligomycin, 4.0 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) + 10mM sodium pyruvate, and 1 μM antimycin A were found to be optimal; for the glycolytic stress test, the standard 10 mM L-glucose, 1 μM oligomycin, and 50 mM 2-deoxyglucose were used; after the standard temperature and pH equilibration, an additional 15 minute equilibration step was added to the XF run protocol, consisting of 3 cycles of 3-min mix and 2-min wait before baseline measurements were performed. Data were analyzed using the Wave for Desktop software suite (Agilent Technologies) and plotted in Prism 7.
Griess assay.
Griess assays to detect the nitric oxide byproduct nitrate in cell supernatants were performed using the Griess Reagent System (#G2930, Promega Corporation, Madison, WI) according to the manufacturer’s supplied protocol. Phosphoric acid, sulfanilamide, N-(1-naphthyl) ethylenediamine dihydrochloride and sodium nitrate (for standard curve) were purchased from Sigma and were prepared fresh on the day of the assay.
Mitochondrial reductase activity (MTT) assay and measurement of cell viability.
A colorimetric assay was used to measure mitochondrial succinic dehydrogenase activity, a surrogate measure for cell death. After 24 h of stimulation with the indicated concentration of MG or mock solvent, the cell supernatants were removed and replaced with filter sterilized MTT solution (1 mg/ml) dissolved in PBS (# 21–040-CV, Mediatech, Inc, Herndon, VA). The macrophage cultures were allowed to incubate for 1 h at 37° C and 5% CO2 in the presence of MTT. The MTT solution was removed and replaced with isopropanol to dissolve the water insoluble formazan products, yielding a purple solution whose absorbance was quantified at 595 or 630 nm using a Biotek microplate reader. The trypan blue exclusion assay to determine cell viability was performed as previously described [38].
Antibodies and Western blot (WB) analysis.
Cell lysates obtained from macrophages in tissue culture or lung tissue were prepared in cell lysis buffer (Cell Signaling Technology, Danvers, MA). The resulting lysates were centrifuged at 12,000 RPM for 10 min at 4° C to remove insoluble cell debris and frozen in single use aliquots at −80° C. Prior to immunoblotting, thawed lysates were mixed with an equal volume of 2x Laemmli sample buffer (Bio-Rad, Hercules, CA) and heated to 95° C for 5 min. Samples were separated by SDS-PAGE in a continuous 10% acrylamide pre-cast gel (Bio-Rad, Hercules, CA). Proteins were transferred from the gel to a polyvinyl difluoride membrane in transfer buffer for 1 h at 4° C. Afterwards, the membrane was dried to complete adherence of protein to the membrane. Prior to probing the membrane with specific anti-MG-H1, MG-H2, and MG-H3 antibodies [42], the membrane was washed with TBS and blocked with TBS containing 0.1% v/v Tween 20 buffer and 5% w/v blotting grade blocker as previously described [43]. A rabbit monoclonal antibody specific for Glo1 (ab137098, Abcam) and a rabbit polyclonal antibody specific to RAGE (ab37647) were both diluted 1:1000 in 5% w/v blotting grade blocker and incubated with the membrane overnight at 4° C.
Gene expression analysis by quantitative qRT-PCR.
Synthesis of cDNA from RNA has been previously described [38]. A 7900HT Fast Real-Time PCR system (Applied Biosystems, Carlsbad, CA) was used to monitor qRT-PCR using the 2x Power SYBR green mix PCR master mix (Life Technologies Corporation, Carlsbad, CA). Primers used to detect mouse Nrf2 and mouse Glo1 transcripts were as follows; Nrf2- forward- 5’-ACG ACA ACG GTC CTT TCC AG- 3’, Nrf2- reverse- 5’- AAT GAC GTT CAT GTC CCC GT- 3’, Glo1 forward- 5’- CCC TCG TGG ATT TGG TCA CA- 3’ Glo1- reverse- 5’- AGC CGT CAG GGT CTT GAA TG- 3’. All other individual genes were analyzed using cytokine gene-specific mouse primers that have previously been published [38, 44]. The total amount of mRNA was calculated using the Comparative ΔΔCt method [45] and expressed as fold-induction compared to untreated or uninfected cells.
In vivo LPS challenge model.
Age- and sex-matched C57BL/6J and Ager−/− mice were weighed one day prior to injection. Experimental groups were nearly evenly split between male and female subjects and were injected at 6 or 7 weeks of age. Each mouse was injected intraperitoneally with LPS (20 mg/kg) based on the weight from the previous day. Survival was monitored every twelve hours for the first 72 h and then daily for a total of 7 days.
Statistical analysis:
Experiments were performed in duplicate or triplicate for analysis of gene expression or cellular viability. Treatments containing two groups were tested for significance by a two-tailed Student’s t test. For analysis of cell death, the numerical values obtained from multiple assays were pooled and analyzed using GraphPad Prism 4 (GraphPad software Inc, La Jolla, CA) with a one-way analysis of variance (ANOVA). For analysis of gene expression, negative ΔCt values from the multiple experiments were pooled and analyzed with a one-way ANOVA. Significance was determined to be p < 0.05. For all one-way ANOVAs, a Tukey’s post-hoc test was used to test for significance between experimental groups within the data set. For survival studies, the log-rank test was used to determine significance between the two groups.
Online Supplemental Material:
Supplementary Figure 1 illustrates the protein expression of RAGE on different tissues in the mouse. It also shows the activity of MG on cells harvested from lung tissue that were cultured ex vivo.
Results:
Activation of macrophages by LPS and IFN-γ promotes generation of MG-modified proteins.
Although the principal source of MG during sepsis is unknown, we hypothesized that inflammatory macrophages may contribute to its production due, in part, to previous research that has shown that two mouse macrophage cell lines are capable of increased MG production when stimulated by LPS and IFN-γ [46]. Therefore, to test this in primary cells, we examined lysates from unstimulated or stimulated primary macrophages for the formation of MG-modified proteins using antibodies specific for MG-derived hydroimidazolone-1 (MG-H1), MG-H2, and MG-H3 [42]. These three isomers are formed in multi-step glycation reactions between MG and arginine side chains in proteins and represent a surrogate measurement of MG (Fig. 1A). Treatment of primary murine macrophages with LPS and IFN-γ produced increased levels of MG-H1, MG-H2, and MG-H3 compared to untreated controls at 24 h post-treatment (Fig. 1B), indicating that LPS and IFN-γ-induced activation of macrophages to an M1 phenotype correlates with MG production. The result in primary macrophages with M1 stimulation mirrors the time course of MG production previously reported in three macrophage cell lines of human and mouse origin that were treated with LPS and IFN-γ [46]. In contrast to the M1 priming regimen, treatment with IL-4 to induce M2 macrophages did not alter the level of MG-protein adducts over a 24 h span (Fig. 1C). To confirm that MG-adducts also formed in vivo, WT C57BL/6J mice were treated with a lethal dose of E. coli K235 LPS (600 μg i.p.). After 24 h, mice were euthanized and lungs excised and homogenized for Western blot analysis (WB) with anti-MG-H1 (Fig. 1D). LPS administration robustly induced MG-H1 adducts (Fig. 1D) compared to lung tissue from saline-treated control mice. Treatment of primary macrophages with highly purified, exogenous MG led to the rapid formation of MG-H1-specific bands with the peak band appearing at the predicted MW of albumin, ~65 kDa, within 4 hours of treatment (Fig. 1E). Consistent with the hypothesis that MG glycates serum proteins found in the media, MG treatment of cells cultured in the absence of serum exhibited significantly less MG-H1 formation across a spectrum of doses (Fig. 1F). The presence of bands at the highest dose of MG treatment at ~50–65 kDa, even in the absence of serum in the media, suggests that glycation is not exclusive to albumin (Fig. 1F). Thus, our data suggests that M1 macrophages exhibit increased production of MG-adducts in vitro and in vivo.
Figure 1: In vitro and in vivo generation of MG adducts by conditions that polarize macrophages to an M1 phenotype.
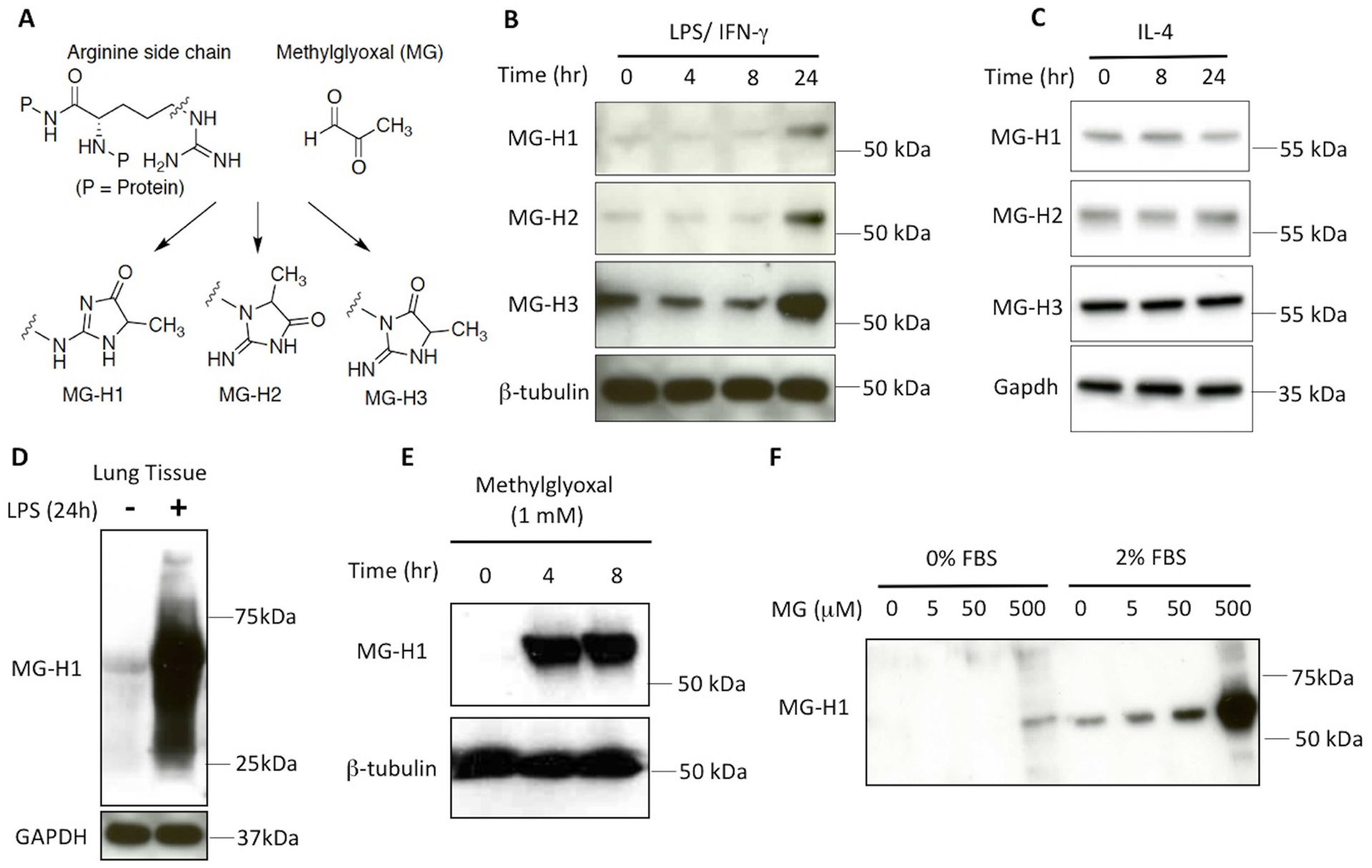
(A) MG reacts with arginine side chains on proteins to form three antigenically distinct isomers, MG-H1, MG-H2, or MG-H3, that can be detected by WB using specific antibodies[42]. (B) WT macrophages were stimulated with LPS (10 ng/ml) and IFN-γ (20 ng/ml) for the indicated times and SDS-PAGE separated lysates were probed with antibodies specific for MG-H1, MG-H2, MG-H3, and β-tubulin. (C) WT macrophages were stimulated with IL-4 (40 ng/ml) for the indicated times and SDS-PAGE separated lysates were probed with antibodies specific for MG-H1, MG-H2, MG-H3, and GAPDH. (D) Mice were untreated or injected with a lethal dose of LPS (600 μg) intraperitoneally. After 24 h, lung tissues were isolated and the resulting lysates were immunoblotted with anti-MG-H1 antibody or anti-GAPDH antibody (loading control). (E, F) WT macrophages were stimulated with MG (1 mM) for the indicated times (E), or with a broad range of MG concentrations (0–500 μM) for 4 h in the absence or presence of serum in the media (F). Cell lysates were separated by SDS-PAGE, transferred to membrane, and probed with the indicated antibodies. Individual panels in B-F portray a representative blot from experiments performed 3 times with similar outcomes.
Stimulation of primary macrophages with LPS and IFN-γ leads to enhanced glycolysis and decreased Glo1 expression.
Having established that MG was being produced by macrophages in vitro, we sought to better understand the mechanism underlying this increase. It has been estimated that only a very small proportion (0.089%) of all triosephosphates are converted to MG during glycolysis [47], so one possible explanation for the increase in MG is an increase in glycolytic metabolites such as triose phosphate isomers. LPS has been shown to alter host cellular metabolism [48–50] in addition to activating innate immune signaling pathways that lead to cytokine production. Therefore, we hypothesized that the proinflammatory conditions associated with early sepsis regulate MG production in primary macrophages. WT C57BL/6J peritoneal macrophages were differentiated in vitro into “classically activated” (M1) or “alternatively activated” (M2) macrophages by treatment with LPS and IFN-γ or IL-4, respectively. LPS and IFN-γ treatment is well-known to polarize cells into the highly inflammatory, classically activated M1 phenotype, while IL-4 stimulation of macrophages induces an anti-inflammatory response that has been termed alternatively activated or M2 (reviewed in [51]). These polarized populations acquired specific “markers” (M1: nitrite, a proxy for NO., and TNF-α mRNA; M2: arginase 1 and Ym1 mRNA) to denote their differentiation state (Fig. 2A). Importantly, these phenotypic changes are accompanied by a profound metabolic shift: M1 macrophages use glycolysis exclusively (the “Warburg effect” [52]), in contrast to the mixed use of mitochondrial respiration and glycolysis by M2 macrophages (Fig. 2B, 2C). These data illustrate that the primary macrophages used in our study confirm that glycolysis and MG production increase in parallel in response to LPS + IFN-γ stimulation.
Figure 2: Macrophages treated with LPS and IFN-γ acquire M1 markers and shift metabolism to aerobic glycolysis.
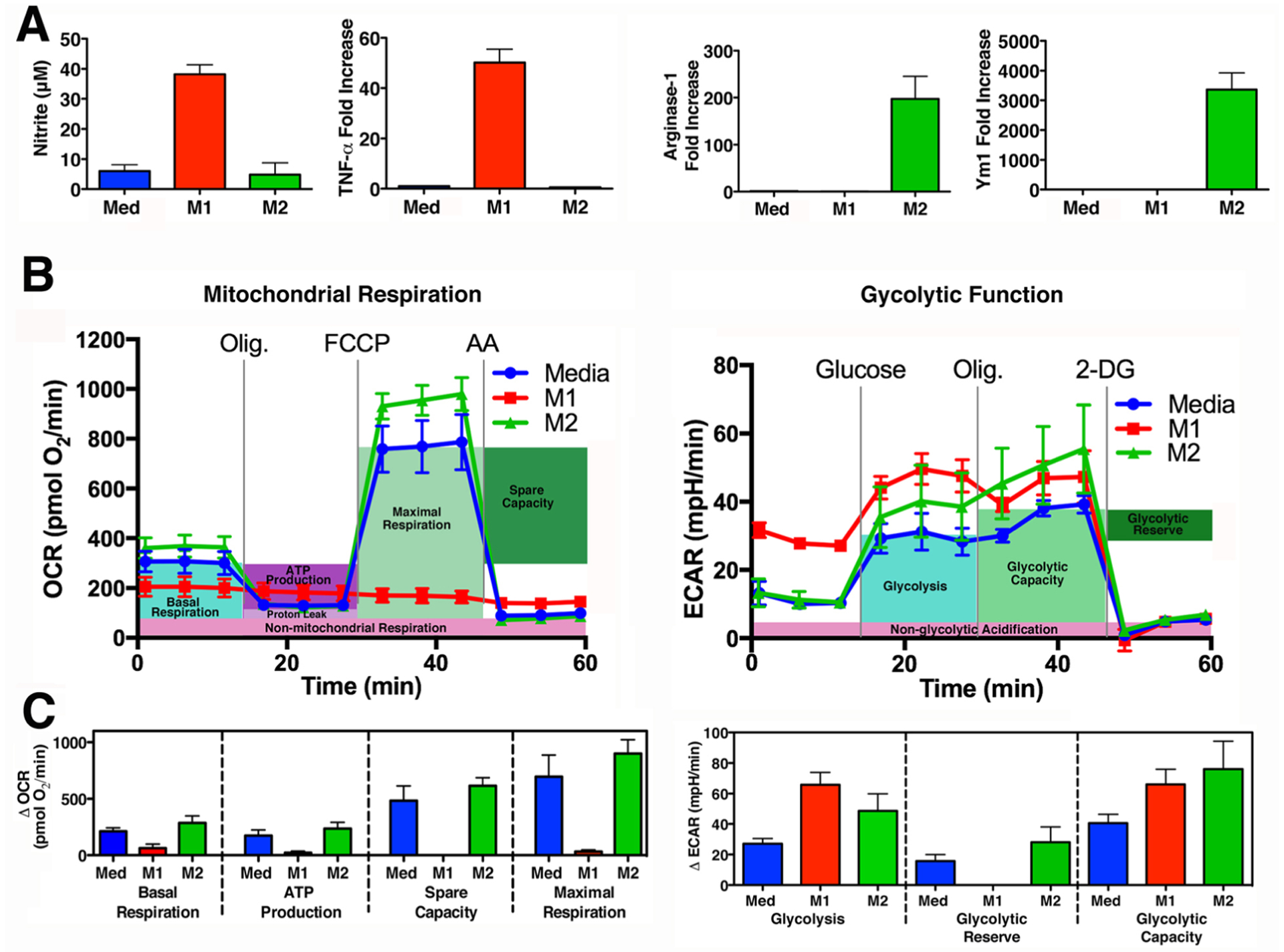
(A) Primary WT macrophages were treated with media alone (Med; blue), LPS (10 ng/ml) and IFN- γ (20 ng/ml) (M1; red) or IL-4 (40 ng/ml) (M2; green) for 48 h. M1 Activation was assessed by measuring the NO. byproduct, nitrite, using the Griess assay and by qRT-PCR to measure Tnf expression, by comparing inducible levels to levels in macrophages treated with medium alone after normalization to Hprt expression. M2 activation was assessed by measuring Arg1 and Ym1 expression. (B)(C) Glycolytic Stress Test and Mitochondrial Stress Test performed on macrophage cultures derived from 4 individual mice, treated as described above in (A) for 24 h. (B) Mean ± SEM shown as a Seahorse wave plot. ECAR (Extracellular Acidification Rate), OCR (Oxygen Consumption Rate), Olig (Oligomycin), 2-DG (2- deoxyglucose), FCCP (Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone), AA (Antimycin A). (C) Bar graphs of measurements to assess individual parameters of glycolysis and mitochondrial respiration.
Under normal physiologic conditions, Glo1, along with Glo2 and glutathione, protect the cell from excessive production of MG by converting it to lactate, thereby preventing the accumulation of intracellular MG triggered by increased extracellular glucose concentrations [53]. The transcription factor Nrf2 has been reported to be required for maximal Glo1 levels in liver and maximal Glo1 protein in multiple organs [54]. Since Nrf2 has also been shown to be activated by LPS [55], and Nrf2−/− mice exhibit greatly enhanced susceptibility in models of endotoxicity and sepsis [56], we hypothesized that Glo1 expression would be increased in M1 macrophages. Expression of the Nrf2-dependent gene Nqo1 [57], that encodes NAD(P)H dehydrogenase [quinone] 1, was increased at 24 h following treatment of macrophages with LPS or LPS and IFN-γ, but not by IFN-γ treatment alone (Fig. 3A). In contrast, Glo1 expression in macrophages was decreased following stimulation with LPS and IFN-γ, but not by LPS treatment alone (Fig. 3B). This suggests that IFN-γ signaling might be an important regulator of Glo1 expression. In line with this hypothesis, IFN-γ treatment of macrophages was sufficient to decrease Glo1 expression in the absence of LPS (Fig. 3B). Considering that this result runs counter to what we initially expected based on the literature [54], we sought to determine the Nrf2 contribution to Glo1 expression in macrophages. Mouse macrophages lacking Nrf2 showed a significant decrease in Nqo1, as expected, but identical levels of Glo1 (Fig. 3C). To verify that these changes translated to protein expression, we determined the level of Glo1 protein by Western analysis. We found that only LPS and IFN-γ treatment together, and not LPS alone, led to a decrease in Glo1 protein levels (Fig. 3D). Cumulatively, these data suggest that IFN-γ but not Nrf2, regulates expression of Glo1 in mouse macrophages.
Figure 3: Expression of the MG-detoxifying enzyme Glo1 is regulated by IFN-γ signaling, but not by the transcription factor Nrf2.
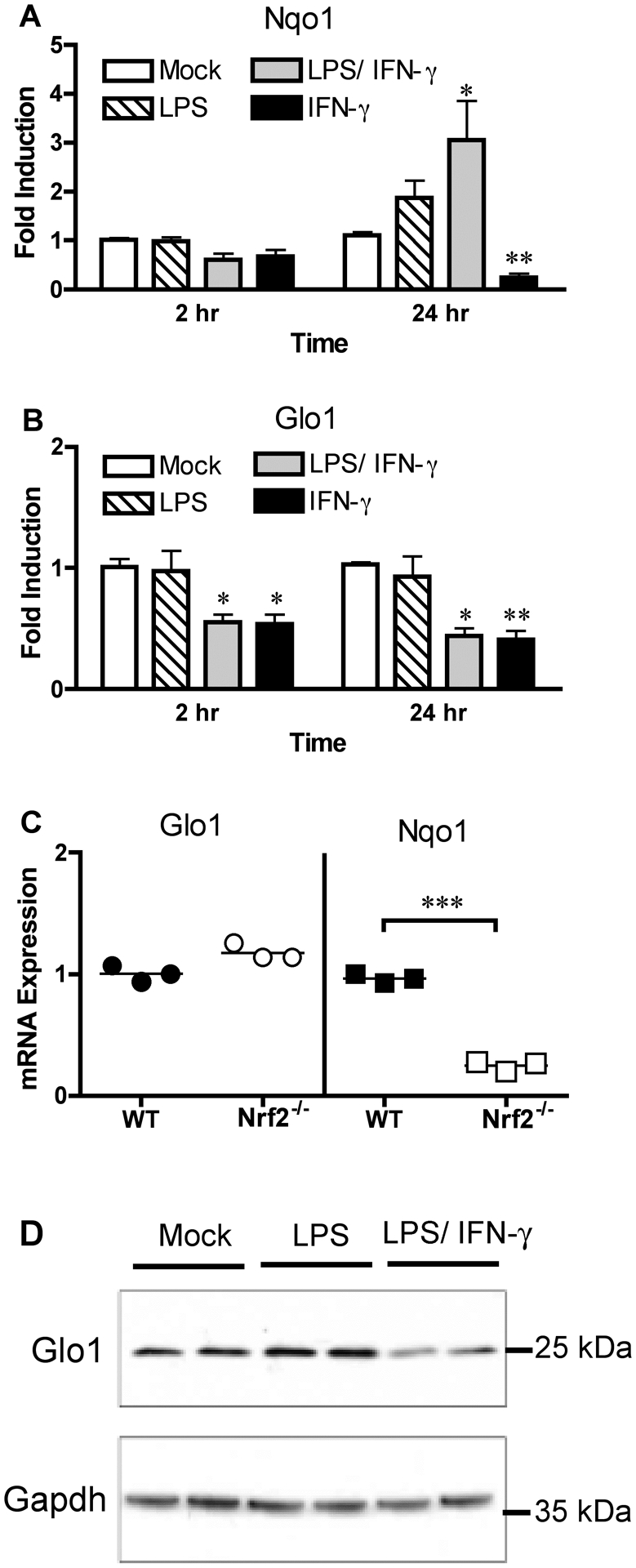
Mouse macrophages from WT mice were stimulated with LPS (10 ng/ml) and IFN-γ (50 ng/ml) individually or in tandem for the indicated time points. Expression levels of (A) Nqo1 and (B) Glo1 expression were determined by qRT-PCR. (C) Untreated WT and Nrf2−/− macrophages were examined for expression of Glo1 and Nqo1. (D) Glo1 protein levels were determined by Western blot of peritoneal macrophages left untreated, treated with LPS (100 ng/ml) alone, or treated with both LPS (10 ng/ml) and IFN-γ (50 ng/ml) for 24 h. Data in (A, B) represents the mean ± SD from 3 independent experiments. * Denotes p < 0.05. ** Denotes p < 0.01. Significance determined in comparison to mock treatment. Data in (C) represents the mean expression of Nqo1 or Glo1. Dots represent the values from the individual experiments that were used to calculate the mean. *** Denotes p < 0.001. Bands in (D) represent treatments from a single experiment performed in duplicate. The blot itself is representative of 2 independent experiments with similar outcomes.
Exogenous MG induces cell death in primary macrophages in a RAGE-independent manner.
Cell death has been a well-studied outcome of MG treatment, and has led researchers to hypothesize that targeting Glo1 to reduce the level of MG might be therapeutic in cancer [58]. We next sought to determine whether primary macrophages were sensitive to MG-induced cell death. Stimulation of WT macrophages with highly purified MG decreased cell viability in a dose-dependent manner (Fig. 4A) as assayed by redox-dependent MTT assay. Doses of MG up to 0.5 mM failed to decrease cellular redox potential significantly, but 2.0 mM completely blocked redox activities 24 h after treatment of macrophages. Cells treated with 2.0 mM MG were also 100% positive for trypan blue staining at this time point (Fig. 4B), indicating that loss of mitochondrial activity was associated with membrane permeability and cell death. Aminoguanidine (AG), an MG quencher [59], partially reversed MG-induced cellular toxicity (Fig. 4C). In contrast, pre-treatment of macrophages with the anti-oxidant Vitamin C failed to reverse MG-induced cytotoxicity (Fig. 4C), illustrating that de novo Reactive Oxygen Species (ROS) production does not contribute to cell death in this instance as had been previously suggested for MG-induced cell death in breast cancer cells [60]. The fact that AG partially reversed the cell death phenotype indicates that MG may be directly responsible for the observed cell death, but it was unknown if AG exerted its protection by preventing the formation of MG-adducts or through some other mechanism. Macrophages cultured in the absence of serum showed reduced mitochondrial activity compared to cells cultured with serum, but otherwise exhibited no protection against MG-induced cytotoxicity, suggesting that MG-adducts are not mediating the cell death phenotype (Fig. 4D). In support of this hypothesis, Ager−/− (RAGE-deficient) macrophages also exhibited nearly identical dose-dependent MG-induced cell death as WT controls over a broad spectrum of MG doses (Fig. 4E), indicating that RAGE is not required for MG-induced cell death.
Figure 4: Cell death induced by MG is reversible by pretreatment with AG, but is RAGE-independent.
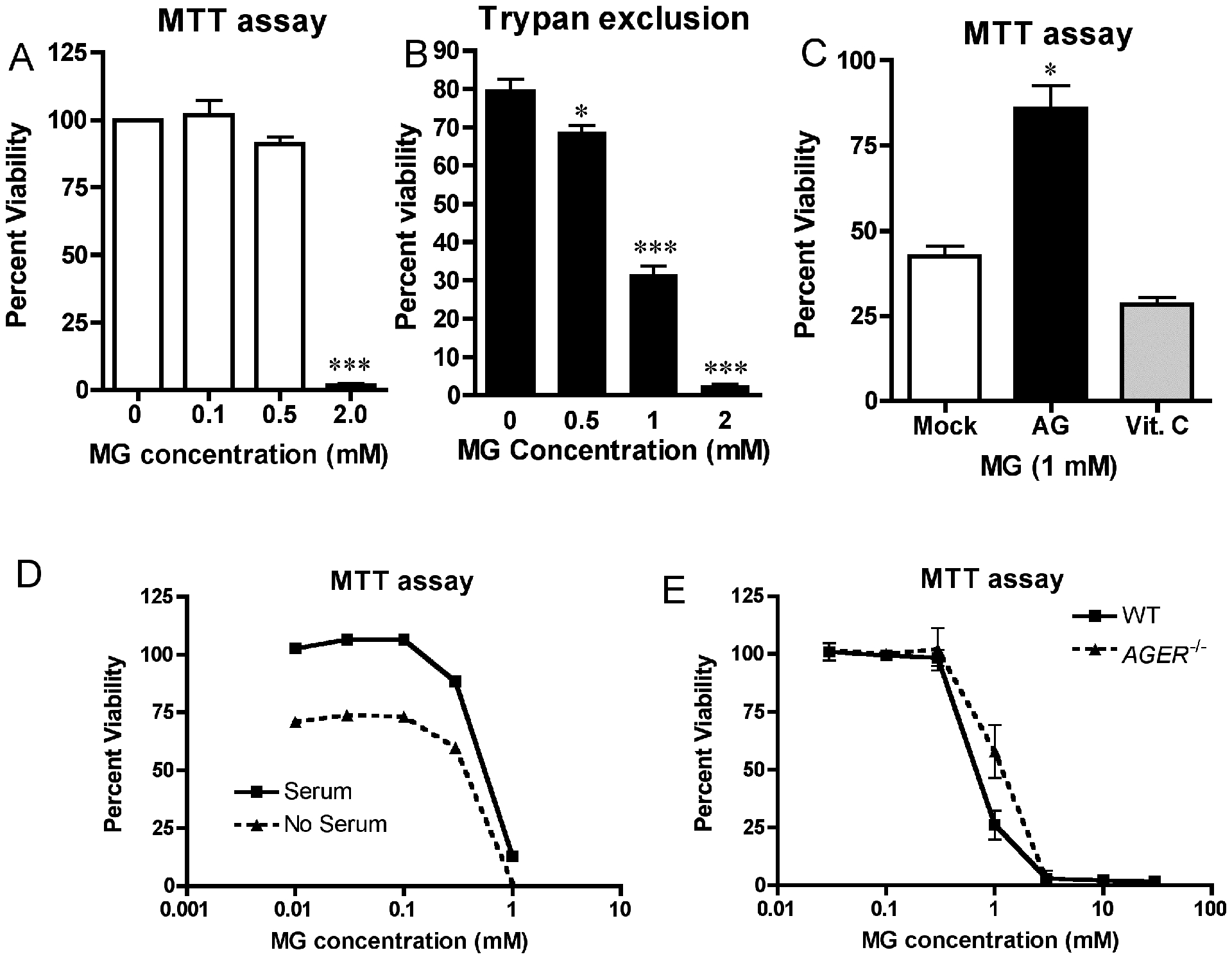
WT macrophages were stimulated in duplicate with the indicated doses of purified MG. Cellular viability was determined after 24 hours of incubation by Mitochondrial reductase (MTT) activity assay (A) or by trypan exclusion assay (B). (C) WT macrophages were treated as described in (A), except cells were incubated in the presence of the MG quencher, AG (1 mM), or the antioxidant, Vitamin C (5 mM), for 30 min prior to MG exposure. MTT activity was measured as in (A). (D) Macrophages were treated as in (A) except that serum was removed (or not) immediately prior to MG exposure. (E) WT and Ager−/− macrophages were stimulated with the indicated doses of MG. MTT activity was determined after 24 hours of incubation. Data for all panels represents the percent viability (mean ± SD) compared to untreated control cells and represent the results of 3 independent experiments. * Denotes p < 0.05. *** denotes p < 0.001.
Exogenous MG treatment of primary macrophages induces a modest upregulation of cytokine gene expression.
Due to the effect of MG on the cellular viability of macrophages at high doses, we sought to test the capacity of MG to activate innate immune signaling pathways at 0.3 mM, a dose that was consistently found to be non-toxic to macrophages, even after prolonged treatment of 24 h (Fig. 4). MG treatment of WT macrophages led to rapid, but modest, increases in the expression levels of Il1b, Tnf, and Cxcl1 mRNA after 1 h of stimulation, before returning to baseline at later time points (Fig. 5A, B, C). Macrophages from Ager−/− mice exhibited a similar enhancement of cytokine gene expression following MG treatment (Fig. 5A, B, C). These increases in cytokine gene expression were significantly lower than seen in response to LPS (Fig. 5A, B, C). Since plasma MG concentrations generally fall in the range of 100 – 200 nM [61], we sought to evaluate the effects of MG under more physiologically relevant conditions. When primary WT macrophages were treated with 0.1 mM or 0.03 mM MG, they failed to induce significant increases in Il1b, Tnf, or Cxcl1 gene expression in WT or Ager−/− macrophages, while higher (0.3 and 1.0 mM) concentrations of MG again induced gene expression in a RAGE-independent fashion (Fig. 5D, E, F). In order to determine if this phenomenon translated to other types of primary cells, we repeated our stimulations with mouse BMDM. Due to culturing in the presence of colony stimulating factor 1, these cells exhibit altered phenotypes to peritoneal macrophages, including production of auto-stimulatory signals like type I interferons [62]. In a similar fashion to our data from peritoneal macrophages, BMDMs also exhibited a slight increase in Il1b, Tnf, or Cxcl1 gene expression (Fig. 5G, H, I). In contrast to peritoneal macrophages, this increase in cytokine gene expression in the BMDMs persisted for a longer duration.
Figure 5: MG stimulation of primary mouse macrophages fails to induce robust cytokine expression compared to LPS.
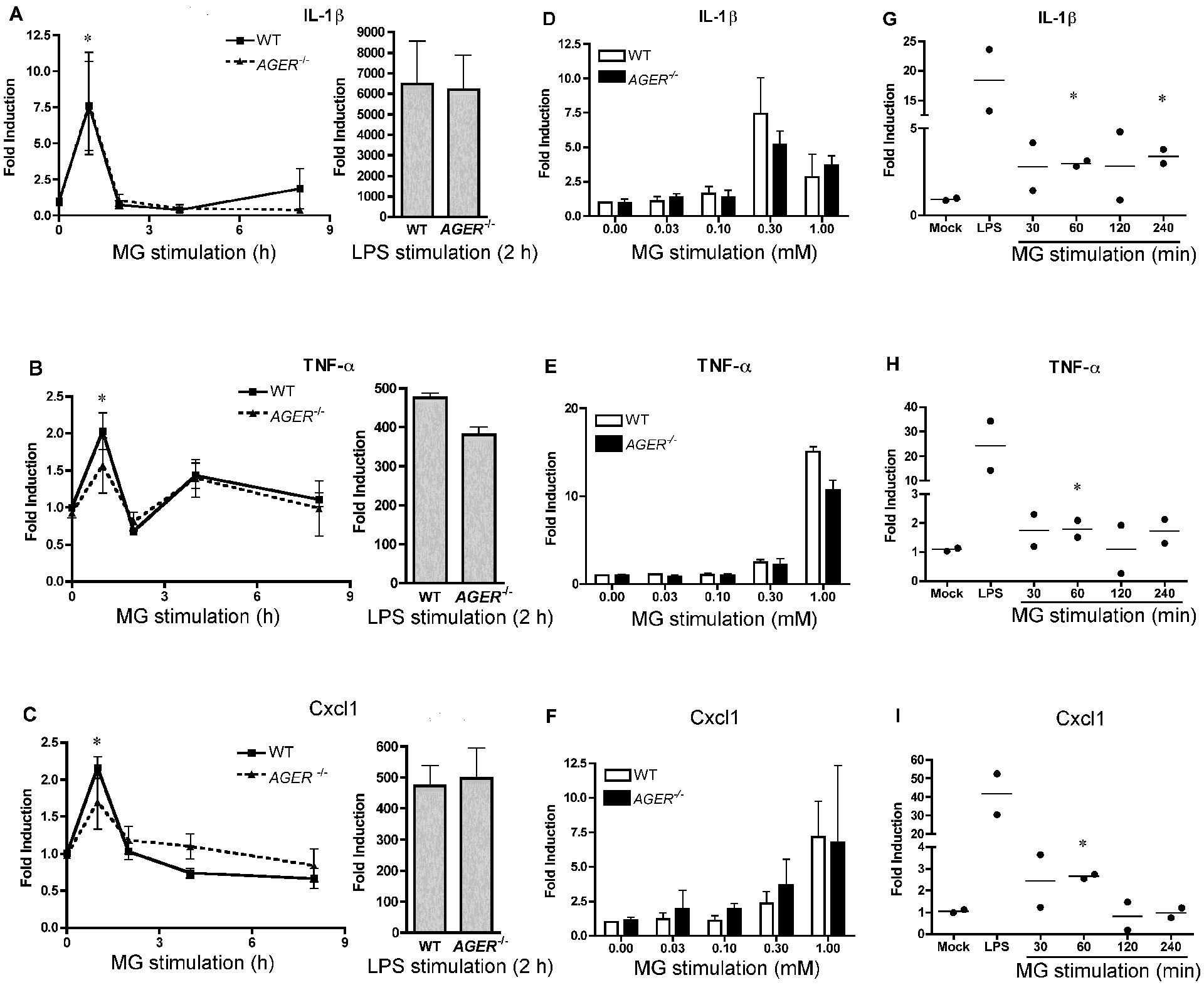
Mouse macrophages from WT or Ager−/− mice were stimulated with MG (0.3 mM) for the indicated time points (left graph) or LPS (100 ng/ml; right graph) for 2 h. Expression of (A) Il1b, (B) Tnf, (C) Cxcl1 were determined by qRT-PCR. Mouse macrophages from WT or Ager−/− mice were stimulated with varying doses of MG for 1 hr. Expression of (D) Il1b, (E) Tnf, or (F) Cxcl1 expression were determined by qRT-PCR. The data for panels (A- F) represents the mean ± SD of 3 independent experiments. * Denotes p < 0.05. Mouse BMDM from WT mice were stimulated with MG (0.3 mM) for the indicated time points or LPS (100 ng/ml) for 30 min. Expression of (G) Il1b, (H) Tnf, or (I) Cxcl1 expression were determined by qRT-PCR. Data in panels (G-I) represent the mean (lines) of 2 independent experiments. Each symbol represents the value from each experiment.
Due to the fact that MG has also been shown to regulate the activation of NF-κB by TNF-α [23], we tested whether MG regulated the gene expression induced by the TLR4 agonist LPS. Induction of Tnf, Il1b, and Il6 were unaffected by a 4 h pretreatment of cells with 0.3 mM MG followed by a 2 h exposure to a broad range of LPS concentrations (Fig. 6A, B, C); however, LPS-induced Cxcl1 gene expression was significantly increased by MG pretreatment (Fig. 6D).
Figure 6: MG regulates expression of the chemokine Cxcl1, but not proinflammatory genes, in response to concurrent LPS treatment.
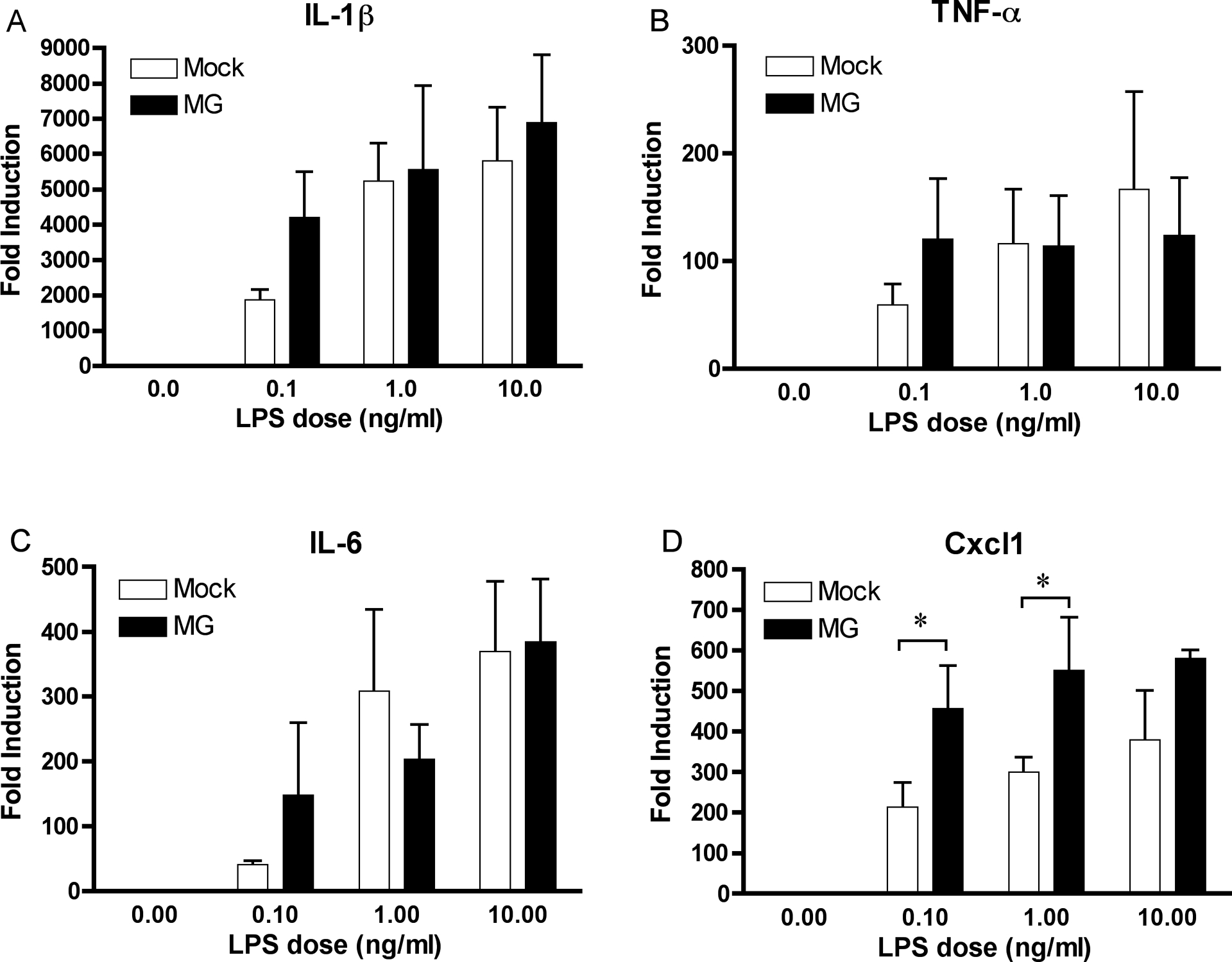
WT mouse macrophages were initially stimulated with MG (0.3 mM) or vehicle only (Mock) for 4 h. Macrophages were then stimulated for an additional 2 h with LPS (1– 100 ng/ml) in the continued presence of MG. Expression of (A) Il1b, (B) Tnf, (C) Il6, and (D) Cxcl1 were determined by qRT-PCR. The data represents the mean ± SD of 3 independent experiments. * Denotes a p value < 0.05.
Exogenous MG-BSA treatment of primary macrophages induces a modest upregulation of cytokine gene expression.
AGEs have been previously shown to activate NF-κB through an MyD88-dependent pathway downstream of RAGE [63]. This suggests that MG glycation of serum proteins that we observed (Fig. 1D) may be responsible for the low level of TNF-α and IL-1β gene induction seen in MG-treated macrophages. To investigate this hypothesis, we treated primary macrophages with serum proteins glycated by MG. Commercially available MG-BSA, formed by glycation of BSA by MG [64], induced Il1b and Tnf expression in WT macrophages (Fig. 7A–D) to levels similar to that observed upon treatment of macrophages with MG alone (Fig. 5). Unlike LPS-induced gene expression that is TLR4-dependent, upregulation of cytokine gene expression induced by MG-BSA was TLR4-independent (Fig. 7A, 7B). Neither LPS-induced nor MG-BSA-induced gene expression was affected by the absence of RAGE (Fig. 7C, 7D), consistent with our findings using purified MG alone. Although MG-BSA was much less stimulatory than LPS, these data highlight that the limited proinflammatory nature of MG-BSA is independent of both TLR4 and RAGE in primary macrophages.
Figure 7: MG-BSA activates innate signaling pathways in mouse macrophages independent of TLR4 and RAGE.
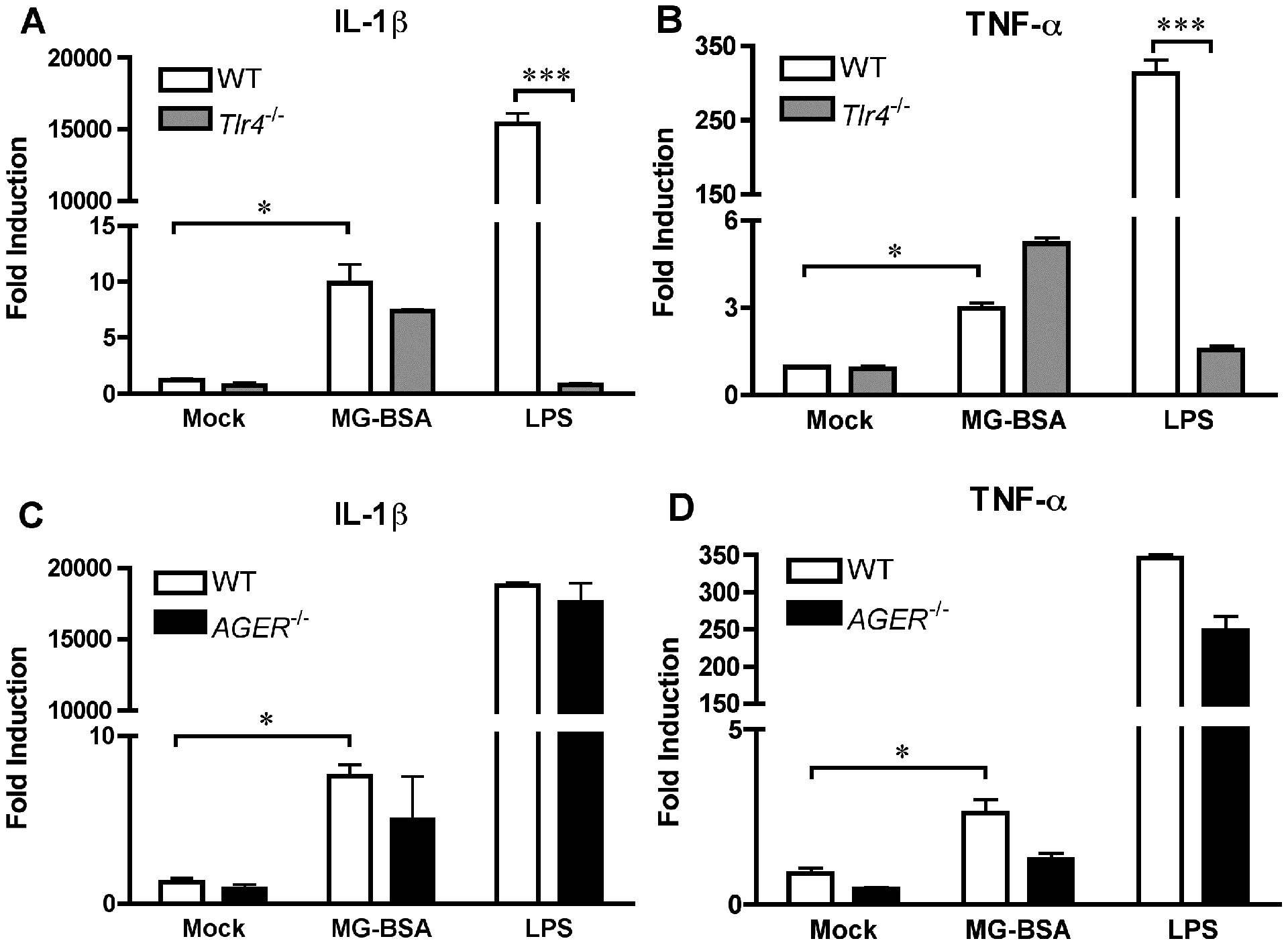
WT and Tlr4−/− macrophages (A, B) or WT and Ager−/− macrophages (C, D) were stimulated with MG-BSA (20 μg/ml) or LPS (100 ng/ml) for 2 h. Expression of Il1b and Tnf were determined by qRT-PCR. The data represents the mean ± SD of 3 independent experiments. * Denotes p < 0.05. *** denotes p < 0.001.
Lung cell cultures expressing RAGE show that RAGE is not sufficient for cytokine gene induction in response to MG.
Our examination of cytokine expression in primary mouse macrophages led us to speculate that the relatively muted cytokine response to MG was, perhaps, due to the lack of adequate RAGE expression in primary murine macrophages. Upon examination of multiple mouse tissues, we found the highest level of RAGE expression in lung tissue (Supplementary Fig. S1A), consistent with previously published work [65]. Upon longer exposure of the blot, we could detect low levels of RAGE in the spleen and kidney, but not in the liver (Supplementary Fig. S1A). The lower molecular weights observed below ~42 kDa could represent degradation products or soluble RAGE and might not reflect endogenous tissue expression. Since we were able to detect RAGE expression in the lung, we tested the ability of lung cell cultures to respond to MG in vitro. Whole lung cell cultures stimulated with LPS upregulated proinflammatory gene expression (Supplementary Fig. S1B–E). In contrast to the significant induction of Il1b, Tnf, Il6, and Cxcl1 mRNA by LPS, MG treatment did not significantly increase cytokine gene expression (Supplementary Fig. S1B–E). This indicates that lung cells, even with high levels of RAGE, are still not able to induce cytokines in response to MG.
RAGE deficiency is associated with increased Glo1 expression in RAGE−/− mice, but does not protect mice from LPS-induced lethality.
Recently, it was published that the same Ager−/− mice that we have used in our experiments have gene duplication of Glo1 [66]. To corroborate this finding, we isolated peritoneal macrophages and other tissues to analyze for Glo1 expression. Peritoneal macrophages from Ager−/− mice expressed significantly higher levels of Glo1 mRNA and Glo1 protein (Fig. 8A). Tissue harvested from the lung, liver, and spleen of Ager−/− mice all exhibited a similar statistically significant increase in Glo1 expression (Fig. 8B, 8C, 8D). However, this did not always translate to increased protein expression. Lungs from Ager−/− mice did not express increased Glo1 protein, while spleen tissue did exhibit increased protein expression (Fig. 8B, 8D). Glo1 expression was highest in liver tissue (Fig. 8C), and a second band of ~40 kDa was detectable in addition to the predicted 25 kDa band. This higher molecular weight band was increased in Ager−/− mice (Fig. 8C). Ager−/− mice have been previously reported to be protected from endotoxic shock compared to WT controls [26]; however, we did not see a differential response to LPS in primary macrophages lacking RAGE (Fig. 5). To address this inconsistency, we injected C57BL/6J (WT) and Ager−/− mice with LPS (20 mg/kg) and monitored survival for 7 days. This dose of LPS resulted in an ~LD60 in WT mice (Fig. 9). When analyzed for possible sex variations, there were no significant differences observed. For this reason, the data from male and female mice were pooled. Although WT animals succumbed to endotoxic shock slightly sooner than their Ager−/− counterparts, this difference was not statistically significant (Fig. 9). Overall, our data support the following model: we hypothesize that increased glycolytic triose phosphate isomers and decreased Glo1 expression contributes to increased MG in classically activated macrophages in vitro and subsequent formation of MG-AGEs. We believe that this may exacerbate inflammation during sepsis in a TLR4- and RAGE-independent fashion, leading to enhanced morbidity and mortality.
Figure 8: Macrophages, lung, liver, and spleen from Ager−/− mice all have elevated expression of Glo1.
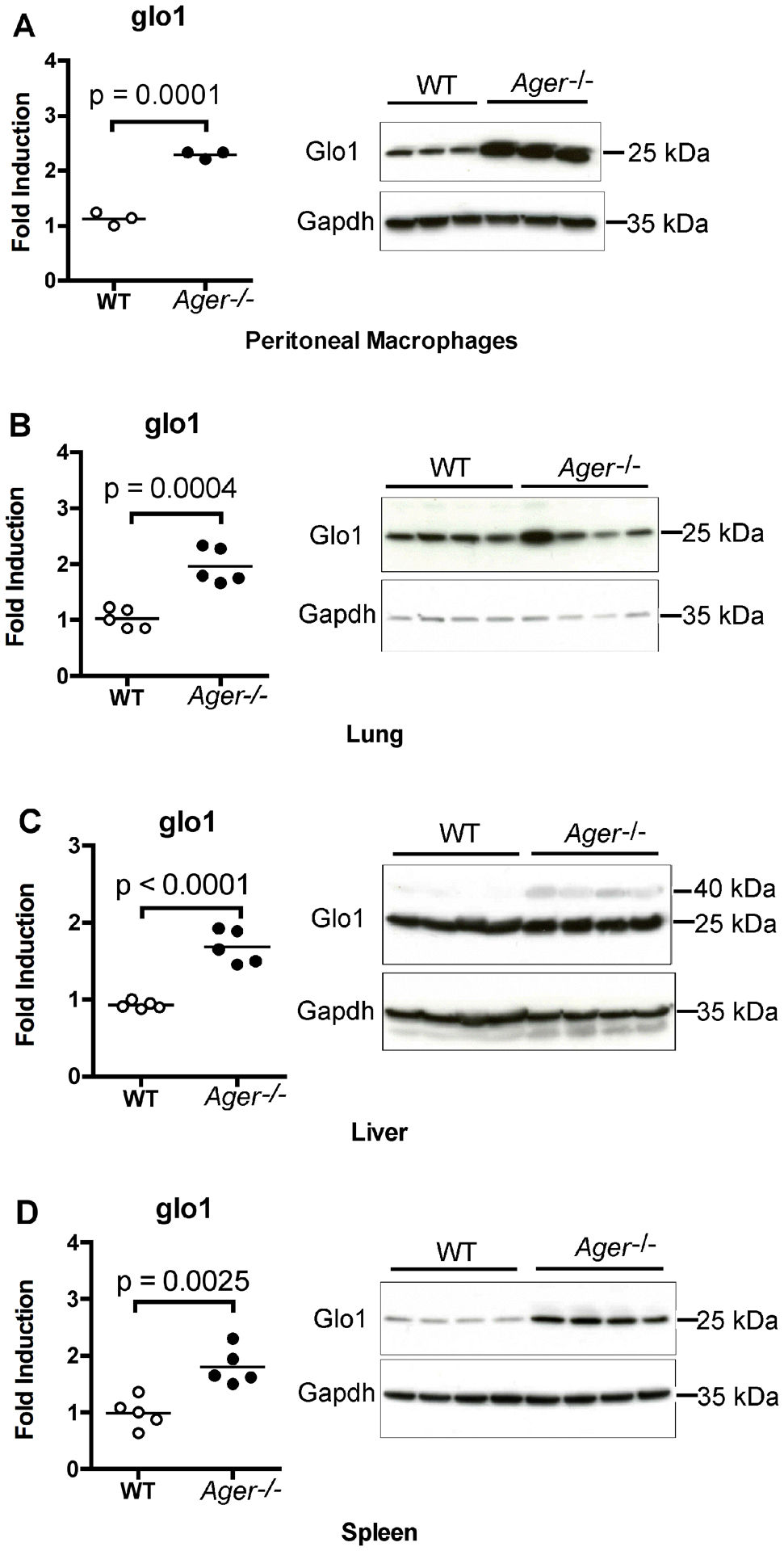
(A) Peritoneal macrophages were isolated from WT and Ager−/− mice. Levels of Glo1 mRNA and Glo1 protein were determined by qRT-PCR or Western blotting respectively. Each mark denotes the results from cells isolated from an individual mouse (N = 3 per group). (B- D) In a separate experiment, the spleens, lungs, and livers were removed from WT and Ager−/− mice. Levels of Glo1 mRNA and Glo1 protein were determined by qRT-PCR or Western blotting, respectively. Each mark denotes the results of tissue isolated from an individual mouse (N = 5 per group). A two-tailed Student’s t test was used to calculate the p values listed in the graphs. From these tissues, 4 per group were analyzed for protein expression.
Figure 9: Ager−/− mice exhibit survival equivalent to WT mice following LPS challenge.
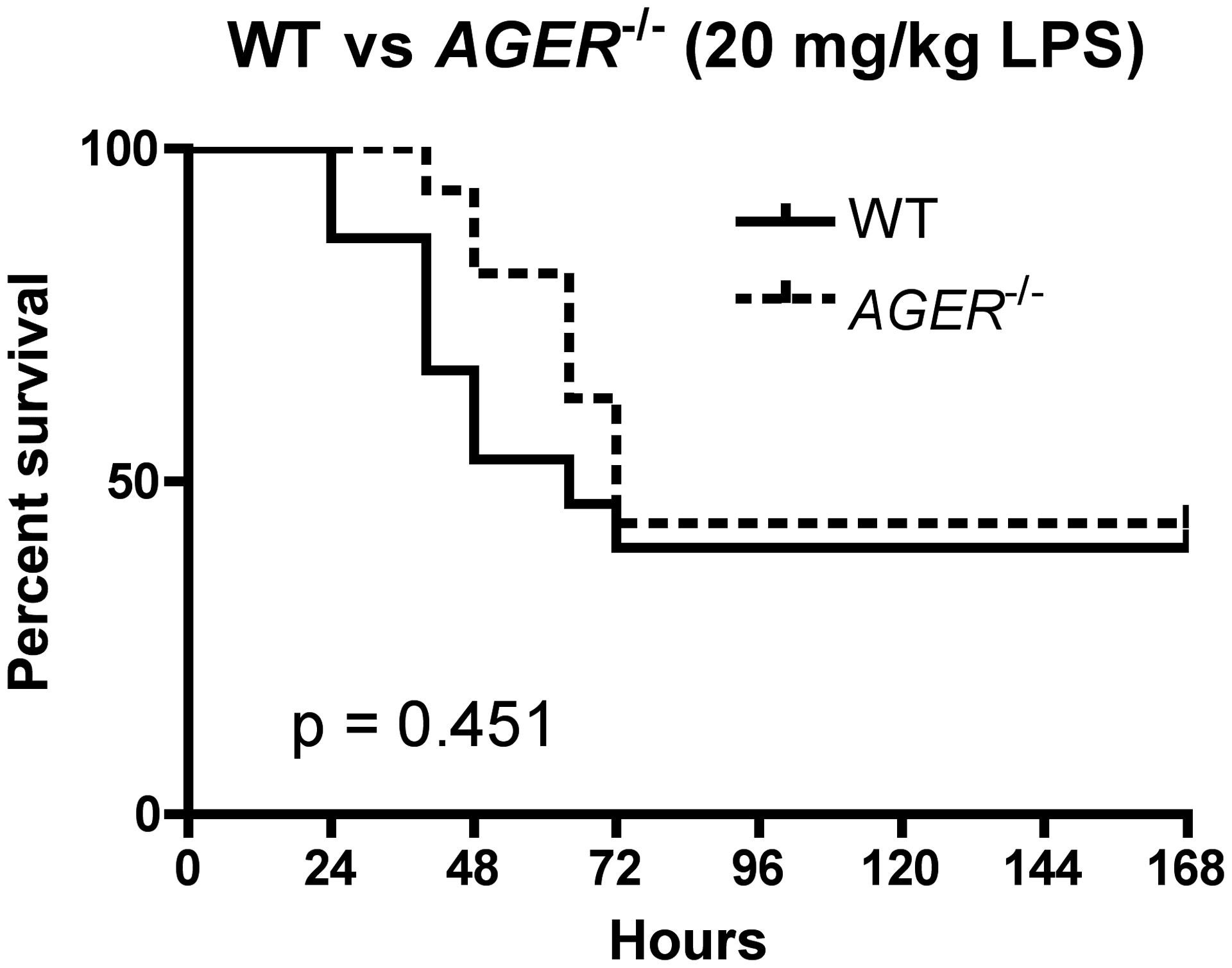
WT (N=15; 7 male, 8 female) and Ager−/− (N=16; 7 male, 9 female) were injected i.p. with 20 mg/kg of LPS. Survival was monitored every 12 h for the first 72 h and daily afterwards for a total of 7 days. P value calculated by log-rank test.
Discussion:
The highly reactive metabolite MG is elevated in the plasma of patients undergoing septic shock compared to the plasma levels in healthy volunteers [2]. Although this data has been used as support for the concept that MG is a biomarker for this disease, it is an intriguing possibility that MG could also be a proximal cause for this disease and could, therefore, be targeted therapeutically. We undertook this study to determine how MG might be formed under inflammatory conditions that might mimic those seen during sepsis or septic shock. While the source of plasma MG is unknown in sepsis, we found that classically activated, M1 macrophages differentiated in tissue culture with LPS and IFN-γ mimic the inflammatory response seen in sepsis, producing increased amounts of MG as monitored by the formation of MG-H1, MG-H2, and MG-H3. These MG adducts are a common glycation biomarker and can be found on human serum albumin [10] in the blood and also in tissues under physiological conditions, with MG-H1 being the most prominent species [67]. In our tissue culture model, MG-H1 peaked 24 h after stimulation and this timing correlated with a shift in the metabolism of the macrophages toward glycolysis as confirmed by our metabolic analyses. It is well known that TLR4 signaling can alter cellular metabolism. One example of this phenomenon is through increased activation of the first enzyme of the glycolytic pathway, hexokinase [50]. LPS also activates the transcription factor Hypoxia-inducible factor 1 alpha (HIF-1α) [49], which induces expression of hexokinase II and other glycolytic (iso)enzymes, presumably leading to greater glycolytic metabolite concentrations. The presence of whole microorganisms induces an increase in both glycolysis and oxidative phosphorylation in monocytes through engagement of TLRs [68]. Given that MG is formed from the glycolytic metabolites glyceraldehyde phosphate and dihydroxyacetone phosphate [47], we hypothesize that an increase in glycolysis would be expected to increase MG levels accordingly.
One question that arises is the identification of the respective MG-adduct. From our experiments, it is suggested that serum proteins taken into the cell or associated with the plasma membrane of the cell can be altered by exogenous or endogenous MG. But longer exposures of the Western blot carried out under serum-free conditions show a broader pattern of glycation, indicating that MG is not only targeting serum proteins, it is merely that these are easier to monitor in our current experimental setup due to homogeneity in size and due to relative abundance.
The increase in MG adducts was also associated with a concomitant decrease in Glo1 expression, which would be predicted to increase MG levels due to a decrease in its degradation. Although the overall timing of MG-H1 formation we observed in tissue culture and in mice injected with a lethal dose of LPS is consistent with MG-adducts peaking 24 h after onset of septic shock in septic patients [2], it is unclear how much macrophages contribute to the pool of MG during this disease. Once formed, we showed that MG and MG-adducts are capable of not only regulating inflammatory cytokines alone, but also can alter the inflammatory response to LPS, exacerbating the production of the chemokine Cxcl1. As Cxcl1 is known to attract neutrophils, this alteration in the cytokine microenvironment may contribute to the detrimental role of MG in sepsis that has been proposed.
It has also been proposed that reactive species like ROS and RCS are a self-perpetuating system, whereby one component enhances the production of the others in a vicious cycle of positive regulation [69]. This is supported by the finding that MG can induce mitochondrial damage leading to an increase of cellular ROS in osteoblasts [70]. This is relevant to our study because nitrotyrosinated TPI exhibits enhanced MG production compared to unmodified TPI in in vitro enzyme assays [71]. Additionally, global levels of nitrotyrosine were increased in a macrophage cell line treated with LPS and IFN-γ [46]. A prerequisite of protein nitrotyrosination is the interaction of proteins with nitrating agents such as peroxynitrite [72]. Peroxynitrite forms in the presence of the ROS, superoxide (O2.-), and the Reactive Nitrogen Species, NO.[73], all induced in M1 macrophages [51]. In professional phagocytic cells like macrophages, ROS can be generated during respiratory burst by NADPH oxidases of the NOX and DUOX families. In addition to glycolysis driving NOX to form O2.- in activated macrophages, MG also contributes to the O2.- formation in the presence of oxygen [13] by MG-induced glycation of mitochondrial Complex III [74]. Of the four NOX and 2 DUOX family members that contribute to the production of O2.-, primary macrophages express NOX2 only [38]. These findings from the literature suggest that MG might enhance TPI nitrotyrosination in a feed-forward manner once synthesized by the cell, leading to more MG being produced. While this is an intriguing hypothesis, the relative contribution of nitrotyrosinated TPI toward MG production in our in vitro system remains to be conclusively determined.
In addition to studying MG production, we also sought to determine whether MG contributed to the inflammatory response of macrophages, known to be physiologically relevant during sepsis. While it is possible that MG-induced cell death may lead to release of host-derived “danger-associated molecular patterns” (DAMPs) like High Mobility Group Box-1 (HMGB1) that could enhance inflammation in sepsis [75], we have shown that MG itself is not a potent activator of innate signaling pathways, in contrast to LPS. MG acts more like HMGB1 which is also much less potent TLR4 agonist than LPS for induction of inflammatory cytokines [43]. We also found that the ability of MG to activate proinflammatory gene expression required doses that were significantly higher than are found in plasma at homeostasis in healthy individuals. Whether the threshold concentration of MG in tissue to induce cytokines is ever met during sepsis has not been established. Recently, MG was identified as a biomarker for myeloid-derived suppressor cells and was shown in a co-culture model to regulate CD8+ T cells, depleting L- arginine levels, leading to paralysis of their effector functions [76]. This finding highlights a potential additional mechanism by which MG could exert its effects during sepsis and necessitates careful examination of this pathway moving forward.
MG-AGE has been identified as a ligand for RAGE [24] and we were able to detect rapid glycation of proteins by MG, but it is possible that more advanced crosslinking is necessary to call these nascent MG-adducts MG-AGEs. While there is also a wealth of literature that suggests that RAGE ligands activate proinflammatory cell signaling pathways [63, 77, 78], it is not apparent whether these studies have used ligand preparations that are free of endotoxins like LPS or other contaminating ligands that could potentially activate cells through TLRs and other innate immune receptors independent of RAGE. In this study, we purified our commercially acquired MG-BSA solutions through an endotoxin removal column and confirmed a loss of endotoxic activity in the recovered material with minimal loss of protein prior to treating macrophages (data not shown). This purified MG-BSA induced proinflammatory signaling independent of TLR4, indicating that LPS contamination was no longer a prime concern. But, surprisingly, this induction was also independent of RAGE. We are not the first to report a RAGE-independent effect for AGEs as it has been published that AGEs repress inflammasome activation when used at a 100-fold higher concentration than used in our study [79]. In our study, mixed lung cell cultures, that express high levels of RAGE, were also refractory to MG when stimulated ex vivo, failing to upregulate proinflammatory cytokines. This shows that even the higher levels of RAGE expression typically seen in the lung [65] are not sufficient to activate innate immune signaling in response to MG. These findings also may indicate that RAGE might need to be co-expressed with TLR4 in order for cells to activate MyD88-dependent signal transduction pathways as previously described [63].
In contrast to a prior study that reported a detrimental effect of RAGE during endotoxic shock [26], we observed only a minor delay in the time of death in LPS-treated Ager−/− mice that we determined not to be significantly different from WT mice (Fig. 9). This underscores that the relative importance of RAGE may be context-specific. Comparing the two studies, we used a lower dose (20 mg/kg versus 50 mg/kg) and mice on a different genetic background (C57BL/6J versus CD-1). Considering the importance of RAGE in the binding of DAMPs like HMGB1 during sepsis [80], it is conceivable that higher doses of LPS elicit more tissue damage and release of more immune activating DAMPs, making RAGE a more crucial component to pathology induced during endotoxicity when using higher doses of LPS. We also corroborated a study that reported that Glo1 was expressed at higher copy number in the Ager−/− mice we used in the study [66], by showing increased Glo1 expression in all tissues examined (Fig. 8). We would not expect that increased Glo1 expression would make the Ager−/− mice more susceptible to LPS counteracting a putatively protective role of RAGE, but it does indicate that increasing Glo1 expression would not likely exert a statistically protective effect during sepsis at least at the level of increase observed in the Ager−/− mice.
In summary, our data support a model in which systemic inflammation during sepsis leads to increased glycolysis and a concomitant decrease in Glo1 expression, leading to increased MG production and MG-adduct formation. We have shown that MG is sufficient to induce inflammatory cytokines. We propose that increased levels of MG-adducts aggregate to form AGEs, or cause release of DAMPs by inducing cell death, that combine to exacerbate the ongoing inflammatory response during sepsis, leading to tissue damage and organ failure. In support of our hypothesis, we detected an increase in MG-modified proteins in the lungs of LPS-treated mice, yet additional study will be required to measure MG levels directly in plasma and tissue during sepsis. Thus, targeting MG therapeutically may represent a novel approach to ameliorate the complications of sepsis. Although some researchers may feel that targeting Glo1 and RAGE is a reasonable strategy during sepsis to mitigate the effects of MG, it is important to note that our data suggests that neither targeting RAGE, nor increasing Glo1, are likely to be the most effective strategies as we have shown many phenotypes of MG that are RAGE-independent and that Glo1 overexpression is not protective in an in vivo model of sepsis. What remains is to more accurately determine the source of physiological MG and to devise ways to block its accumulation.
Supplementary Material
Acknowledgements
This study was supported by grant support from the NIH AI123371, AI125215, and S10 OD025101 (SNV). Metabolic studies were performed at the Center for Innovative Biomedical Resources (CIBR) Genomics Core Facility at the University of Maryland, School of Medicine (UMSOM).
Abbreviations:
- ANOVA
Analysis of variance
- BSA
Bovine Serum Albumin
- DAMP
Damage-Associated Molecular Patterns
- Glo
Glyoxalase
- HMGB1
High Mobility Group Box-1
- LPS
Lipopolysaccharide
- MG
Methylglyoxal
- NO.
Nitric Oxide
- RAGE
Receptor for Advanced Glycation Endproducts
- RCS
Reactive carbonyl species
- ROS
reactive oxygen species
- TPI
triosephosphate isomerase
Footnotes
Conflict of Interest Disclosure:
The authors declare no conflict of interest. The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Institute of Allergy and Infectious Diseases.
References:
- 1.Torio CM, Andrews RM (2011) National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, Rockville, MD. [PubMed] [Google Scholar]
- 2.Brenner T, Fleming T, Uhle F, Silaff S, Schmitt F, Salgado E, Ulrich A, Zimmermann S, Bruckner T, Martin E, Bierhaus A, Nawroth PP, Weigand MA, Hofer S (2014) Methylglyoxal as a new biomarker in patients with septic shock: an observational clinical study. Crit Care 18(6), 014–0683-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thornalley PJ, Langborg A, Minhas HS (1999) Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 344 Pt 1, 109–16. [PMC free article] [PubMed] [Google Scholar]
- 4.Pompliano DL, Peyman A, Knowles JR (1990) Stabilization of a reaction intermediate as a catalytic device: definition of the functional role of the flexible loop in triosephosphate isomerase. Biochemistry 29(13), 3186–94. [DOI] [PubMed] [Google Scholar]
- 5.Zell R, Geck P, Werdan P, Boekstegers P (1997) TNF-alpha and IL-1alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyoctes: evidence for primary impairment of mitochondrial function. Mol Cell Biochem 177, 61–67. [DOI] [PubMed] [Google Scholar]
- 6.Maneiro E, Lopez-Armada MJ, de Andres MC, Carames B, Martin MA, Bonilla A, Del Hoyo P, Galdo F, Arenas J, Blanco FJ (2005) Effect of nitric oxide on mitochondrial respiratory activity of human articular chondrocytes. 2005 64, 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marik PE, Raghavan M (2004) Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med. 30(5), 748–56. [DOI] [PubMed] [Google Scholar]
- 8.Thornalley PJ (2003) Glyoxalase I--structure, function and a critical role in the enzymatic defence against glycation.. Biochem Soc Trans. 31(Pt 6), 1343–8. [DOI] [PubMed] [Google Scholar]
- 9.Baskaran S, Rajan DP, Balasubramanian KA (1989) Formation of methylglyoxal by bacteria isolated from human faeces. J Med Microbiol. 28(3), 211–5. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed N, Dobler D, Dean M, Thornalley PJ (2005) Peptide mapping identifies hotspot site of modification in human serum albumin by methylglyoxal involved in ligand binding and esterase activity. J Biol Chem. 280(7), 5724–32. [DOI] [PubMed] [Google Scholar]
- 11.Murata-Kamiya N, Kamiya H, Kaji H, Kasai H (2000) Methylglyoxal induces G:C to C:G and G:C to T:A transversions in the supF gene on a shuttle vector plasmid replicated in mammalian cells. Mutat Res. 468(2), 173–82. [DOI] [PubMed] [Google Scholar]
- 12.Okado A, Kawasaki Y, Hasuike Y, Takahashi M, Teshima T, Fujii J, N. T (1996) Induction of apoptotic cell death by methylglyoxal and 3-deoxyglucosone in macrophage-derived cell lines. Biochem Biophys Res Commun 225(1), 219–24. [DOI] [PubMed] [Google Scholar]
- 13.Yim HS, Kang SO, Hah YC, Chock PB, Yim MB (1995) Free radicals generated during the glycation reaction of amino acids by methylglyoxal. A model study of protein-cross-linked free radicals. J Biol Chem. 270(47), 28228–33. [DOI] [PubMed] [Google Scholar]
- 14.Du J, Suzuki H, Nagase F, Akhand AA, Ma XY, Yokoyama T, Miyata T, Nakashima I (2001) Superoxide-mediated early oxidation and activation of ASK1 are important for initiating methylglyoxal-induced apoptosis process. Free Radic Biol Med. 31(4), 469–78. [DOI] [PubMed] [Google Scholar]
- 15.Yang CT, Meng FH, Chen L, Li X, Cen LJ, Wen YH, Li CC, Zhang H (2017) Inhibition of Methylglyoxal-induced AGEs/RAGE expression contributes to dermal protection by N-acetyl-L-Cysteine. Cell Physiol Biochem. 41(2), 742–754. [DOI] [PubMed] [Google Scholar]
- 16.Chu P, Han G, Ahsan A, Sun Z, Liu S, Zhang Z, Sun B, Song Y, Lin Y, Peng J, Tang Z (2017) Phosphocreatine protects endothelial cells from Methylglyoxal induced oxidative stress and apoptosis via the regulation of PI3k/Akt/eNOS and NF-kB pathway. Vascul Pharmacol. 91, 26–35. [DOI] [PubMed] [Google Scholar]
- 17.Lin CC, Chan CM, Huang YP, Hsu SH, Huang CL, Tsai SJ (2016) Methylglyoxal activates NF-kB nuclear translocation and induces COX-2 expression via a p38- dependent pathway in synovial cells. Life Sci. 149, 25–33. [DOI] [PubMed] [Google Scholar]
- 18.Su Y, Qadri SM, Cayabyab FS, Wu L, Liu L (2014) Regulation of methylglyoxal-elicited leukocyte recruitment by endothelial SGK1/GSK3 signaling. Biochim Biophys Acta. 1843(11), 2481–91. [DOI] [PubMed] [Google Scholar]
- 19.Wang YH, Yu HT, Pu XP, Du GH (2014) Myricitrin alleviates methylglyoxal-induced mitochondrial dysfunction and AGEs/RAGE/NF-κB pathway activation in SH-SY5Y cells. J Mol Neurosci. 53(4), 562–70. [DOI] [PubMed] [Google Scholar]
- 20.Yamawaki H, Saito K, Okada M, Hara Y (2008) Methylglyoxal mediates vascular inflammation via JNK and p38 in human endothelial cells. Am J Physiol Cell Physiol. 295(6), C1510–7. [DOI] [PubMed] [Google Scholar]
- 21.Pal A, Bhattacharya I, Bhattacharya K, Mandal C, Ray M (2009) Methylglyoxal induced activation of murine peritoneal macrophages and surface markers of T lymphocytes in sarcoma-180 bearing mice: involvement of MAP kinase, NF-kappa beta signal transduction pathway. Mol Immunol. 46(10), 2039–44. [DOI] [PubMed] [Google Scholar]
- 22.Kuntz S, Kunz C, Rudloff S (2010) Carbonyl compounds methylglyoxal and glyoxal affect interleukin-8 secretion in intestinal cells by superoxide anion generation and activation of MAPK p38. Mol Nutr Food Res. 54(10), 1458–67. [DOI] [PubMed] [Google Scholar]
- 23.Laga M, Cottyn A, Van Herreweghe F, Vanden Berghe W, Haegeman G, Van Oostveldt P, Vandekerckhove J, Vancompernolle K (2007) Methylglyoxal suppresses TNF-alpha-induced NF-kappaB activation by inhibiting NF-kappaB DNA binding. Biochem Pharmacol. 74(4), 579–89. [DOI] [PubMed] [Google Scholar]
- 24.Xue J, Ray R, Singer D, Bohme D, Burz DS, Rai V, Hoffmann R, Shekhtman A (2014) The receptor for advanced glycation end products (RAGE) specifically recognizes methylglyoxal-derived AGEs. Biochemistry 53, 3327–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan X, Subramaniam R, Weiss MF, Monnier VM (2003) Methylglyoxal-bovine serum albumin stimulates tumor necrosis factor alpha secretion in RAW 264.7 cells through activation of mitogen-activating protein kinase, nuclear factor kappaB and intracellular reactive oxygen species formation. 409(2), 274–86. [DOI] [PubMed] [Google Scholar]
- 26.Yamamoto Y, Harashima A, Saito H, Tsuneyama K, Munesue S, Motoyoshi S, Han D, Watanabe T, Asano M, Takasawa S, Okamoto H, Shimura S, Karasawa T, Yonekura H, Yamamoto H (2011) Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J Immunol 186(5), 3248–57. [DOI] [PubMed] [Google Scholar]
- 27.Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Gröne HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hämmerling GG, Nawroth PP, Arnold B (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 113(11), 1641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lutterloh EC, Opal SM, Pittman DD, Keith JC Jr, Tan XY, Clancy BM, Palmer H, Milarski K, Sun Y, Palardy JE, Parejo NA, Kessiman N (2007) Inhibition of the RAGE products increases survival in experimental models of severe sepsis and systemic infection. Crit Care 11, R122–R122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Achouiti A, de Vos AF, de Beer R, Florquin S, van ‘t Veer C, van der Poll T (2013) Limited role of the receptor for advanced glycation end products during Streptococcus pneumoniae bacteremia. J Innate Immun 5, 603–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG (2017) The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol 17, 407–420. [DOI] [PubMed] [Google Scholar]
- 31.Freudenberg MA, Galanos C (1988) Induction o tolerance to lipopolysaccharide (LPS)-D-galactosamine lethality by pretreatment with LPS is mediated by macrophages. Infect Immun 56, 1352–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freudenberg MA, Keppler D, Galanos C (1986) Requirement for lipopolysaccharide-responsive macrophages in galactosamine-induced sensitization to endotoxin. Infect Immun 51, 891–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIntire FC, Sievert HW, Barlow GH, Finley RA, Lee AY (1967) Chemical, physical, biological properties of a lipopolysaccharide from Escherichia coli K-235. Biochemistry 6, 2363–72. [DOI] [PubMed] [Google Scholar]
- 34.Rabbani N, Thornalley PJ (2014) Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat Protoc 9, 1969–79. [DOI] [PubMed] [Google Scholar]
- 35.Nemet I, Vikic-Topic D, Varga-Defterdarovic L (2004) Spectroscopic studies of methylglyoxal in water and dimethylsulfoxide. Bioorg Chem 32, 560–70. [DOI] [PubMed] [Google Scholar]
- 36.Thornalley PJ, Yurek-George A, Argirov OK (2000) Kinetics and mechanism of the reaction of aminoguanidine with the alpha-oxoaldehydes glyoxal, methylglyoxal, and 3-deoxyglucosone under physiological conditions. Biochem Pharmacol 60, 55–65. [DOI] [PubMed] [Google Scholar]
- 37.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayma I, Yamamoto M, Nabeshima Y (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236, 313–22. [DOI] [PubMed] [Google Scholar]
- 38.Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN (2012) 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem 287, 39776–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pennini ME, Perkins DJ, Salazar AM, Lipsky M, Vogel SN (2013) Complete dependence on IRAK4 kinase activity in TLR2, but not TLR4, signaling pathways underlies decreased cytokine production and increased susceptiblity to Streptococcus infection in IRAK4 kinase-inactive mice. J Immunol 190, 307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prantner D, Sikes JD, Hennings L, Savenka AV, Basnakian AG, Nagarajan UM (2011) Interferon regulatory transcription factor 3 protects mice from uterine horn pathology during Chlamydia muridarum genital infection. Infect Immun 79, 3922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, Cherok E, Khalil A, Yadava N, Ge SX, Francis TC, Kennedy NW, Picton LK, Kumar T, Uppuluri S, Miller AM, Itoh K, Karbowski M, Sesaki H, Hill RB, Polster BM (2017) The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev Cell 40, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang T, Streeter MD, Spiegel DA (2015) Generation and characterization of antibodies against arginine-derived advanced glycation endproducts. Bioorg Med Chem Lett. 25(21), 4881–6. [DOI] [PubMed] [Google Scholar]
- 43.Prantner D, Shirey KA, Lai W, Lu W, Cole AM, Vogel SN, Garzino-Demo A (2017) The Theta-defensin retrocyclin 101 inhibits TLR4- and TLR2- dependent signaling and protects mice against influenza infection. J Leukoc Biol 102, 1103–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirey KA, Cole LE, Keegan AD, Vogel SN (2008) Francisella tularensis live vaccine strain induces macrophage alternative activation as a survival mechanism. J Immunol 181, 4159–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–8. [DOI] [PubMed] [Google Scholar]
- 46.Dhananjayan K, Gunawardena D, Hearn N, Sonntag T, Moran C, Gyengesi E, Srikanth V, Münch G (2017) Activation of Macrophages and Microglia by Interferon-gamma and Lipopolysaccharide Increases Methylglyoxal Production: A New Mechanism in the Development of Vascular Complications and Cognitive Decline in Type 2 Diabetes Mellitus? J Alzheimers Dis. 59(2), 467–79. [DOI] [PubMed] [Google Scholar]
- 47.Thornalley PJ (1988) Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem J 254, 751–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huynh A, DuPage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, Townamchai N, Gerriets VA, Rathmell JC, Sharpe AH, Bluestone JA, Turka LA (2015) Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. 16(2), 188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496(7444), 238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith JA, Stallons LJ, Schnellmann RG (2014) Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am J Physiol Renal Physiol. 307(4), F435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA (2014) Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Warburg O (1925) The metabolism of carcinoma cells. The Journal of Cancer Research 9(1), 148–163. [Google Scholar]
- 53.Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, Brownlee M (1998) Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 101(5), 1142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xue M, Rabbani N, Momiji H, Imbasi P, Anwar MM, Kitteringham N, Park BK, Souma T, Moriguchi T, Yamamoto M, Thornalley PJ (2012) Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem J 443, 213–222. [DOI] [PubMed] [Google Scholar]
- 55.Yin S, Cao W (2015) Toll-like Receptor signaling induces Nrf2 pathway activation through p62- triggered Keap1 degradation. Mol Cell Biol. 35(15), 2673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S (2006) Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 116, 984–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Venugopal R, Jaiswal AK (1996) Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element- mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 93, 14960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baunacke M, Horn LC, Tretter S, Engel KM, Hemdan NY, Weichmann V, Stolzenburg JU, Bigl M, Birkenmeier G (2014) Exploring glyoxalase 1 expression in prostate cancer tissues: targeting the enzyme by ethyl pyruvate defangs some malignancy-associated properties. Prostate 74(1), 48–60. [DOI] [PubMed] [Google Scholar]
- 59.Lo TW, Selwood T, Thornalley PJ (1994) The reaction of methylglyoxal with aminoguanidine under physiological conditions and prevention of methylglyoxal binding to plasma proteins. Biochem Pharmacol. 48(10), 1865–70. [DOI] [PubMed] [Google Scholar]
- 60.Roy A, Ahir M, Bhattacharya S, Parida PK, Adhikary A, Jana K, Ray M (2017) Induction of mitochondrial apoptotic pathway in triple negative breast carcinoma cells by methylglyoxal via generation of reactive oxygen species. Mol Carcinogen. 56(9), 2086–2103. [DOI] [PubMed] [Google Scholar]
- 61.Lu J, Randell E, Han Y, Adeli K, Krahn J, Meng QH (2011) Increased plasma methylglyoxal level, inflammation, and vascular endothelial dysfunction in diabetic nephropathy. Clin Biochem. 44(4), 307–11. [DOI] [PubMed] [Google Scholar]
- 62.Warren MK, Vogel SN (1985) Bone marrow-derived macrophages: development and regulation of differentiation markers by colony-stimulating factor and interferons. J Immunol 134, 982–9. [PubMed] [Google Scholar]
- 63.Sakaguchi M, Murata H, Yamamoto K, Ono T, Sakaguchi Y, Motoyama A, Hibino T, Kataoka K, Huh NH (2011) TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS One. 6(8), e23132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lo TW, Westwood ME, McLellan AC, Selwood T, Thornalley PJ (1994) Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J Biol Chem 269, 32299–305. [PubMed] [Google Scholar]
- 65.Brett J, Schmidt AM, Yan SD, Zou YS, Pinsky E,W, Nowygrod D, Neeper R, Przysiecki M, Shaw C, Migheli A, Stern, D. A (1993) Survey of the Distribution of a Newly Characterized Receptor for Advanced Glycation End Products in Tissues. Am J Pathol. 143(6), 1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 66.Bartling B, Zunkel K, Al-Robaiy S, Dehghani F, Simm A (2020) Gene doubling increases glyoxalase 1 expression in RAGE knockout mice. Biochem Biophys Acta Gen Subj 1864, 129438. [DOI] [PubMed] [Google Scholar]
- 67.Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A (2003) Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J. 375(Pt 3), 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lachmandas E, Boutens L, Ratter JM, Hijmans A, Hooiveld GJ, Joosten LA, Rodenburg RJ, Fransen JA, Houtkooper RH, van Crevel R, Netea MG, Stienstra R (2016) Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat Microbiol. 2, 16246. [DOI] [PubMed] [Google Scholar]
- 69.Kalapos MP (2008) The tandem of free radicals and methylglyoxal. Chem Biol Interact. 171(3), 251–271. [DOI] [PubMed] [Google Scholar]
- 70.Chan WH, Wu HJ, Shiao NH (2007) Apoptotic signaling in methylglyoxal-treated human osteoblasts involves oxidative stress, c-Jun N-terminal kinase, caspase-3, and p21-activated kinase 2. J Cell Biochem. 100(4), 1056–69. [DOI] [PubMed] [Google Scholar]
- 71.Guix FX, Ill-Raga G, Bravo R, Nakaya T, de Fabritiis G, Coma M, Miscione GP, Villà-Freixa J, Suzuki T, Fernàndez-Busquets X, Valverde MA, de Strooper B, Muñoz FJ (2009) Amyloid-dependent triosephosphate isomerase nitrotyrosination induces glycation and tau fibrillation. Brain 132(Pt 5), 1335–45. [DOI] [PubMed] [Google Scholar]
- 72.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS (1992) Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 298(2), 431–7. [DOI] [PubMed] [Google Scholar]
- 73.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS (1992) Peroxynitrite, a cloaked oxidant formed by nitric oxide and superoxide. Chem Res Toxicol. 5(6), 834–42. [DOI] [PubMed] [Google Scholar]
- 74.Rosca MG, Mustata TG, Kinter MT, Ozdemir AM, Kern TS, Szweda LI, Brownlee M, Monnier VM, Weiss MF (2005) Glycation of mitochondrial proteins from diabetic rat kidney is associated with excess superoxide formation. Am J Physiol Renal Physiol 289, F420–430. [DOI] [PubMed] [Google Scholar]
- 75.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–251. [DOI] [PubMed] [Google Scholar]
- 76.Baumann T, Dunkel A, Schmid C, Schmitt S, Hiltensperger M, Lohr K, Laketa V, Donakonda S, Ahting U, Lorenz-Depiereux B, Heil JE, Schredelseker J, Simeoni L, Fecher C, Körber N, Bauer T, Hüser N, Hartman D, Laschinger M, Eyerich K, Eyerich S, Anton M, Streeter M, Wang T, Schraven B, Spiegel D, Assaad F, Misgeld T, Zischka H, Murray PJ, Heine A, Heikenwalder M, Korn T, Dawid C, Hofmann T, Knolle PA, Höchst B (2020) Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol. 21, 555–566. [DOI] [PubMed] [Google Scholar]
- 77.Ohtsu A, Shibutani Y, Seno K, Iwata H, Kuwayama T, Shirasuna K (2017) Advanced glycation end products and lipopolysaccharides stimulate interleukin-6 secretion via the RAGE/TLR4-NF-kappaB-ROS pathways and resveratrol attenuates these inflammatory responses in mouse macrophages. Exp Ther Med. 14(5), 4363–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen B, Miller AL, Rebelatto M, Brewah Y, Rowe DC, Clarke L, Czapiga M, Rosenthal K, Imamichi T, Chen Y, Chang CS, Chowdhury PS, Naiman B, Wang Y, Yang D, Humbles AA, Herbst R, Sims GP (2015) S100A9 induced inflammatory responses are mediated by distinct damage associated molecular patterns (DAMP) receptors in vitro and in vivo. PLoS One. 10(2), e0115828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Son S, Hwang I, Han SH, Shin JS, Shin OS, Yu JW (2017) Advanced glycation end products impair NLRP3 inflammasome-mediated innate immune responses in macrophages. J Biol Chem. 292(50), 20437–20448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, Huang Y, Peng H, Xiao L, Tang D, Scott MJ, Wang Q, Liu J, Xiao X, Watkins S, Li J, Yang H, Wang H, Chen F, Tracey KJ, Billiar TR, Lu B (2018) The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49(4), 740–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.