

Abstract
This study aimed to evaluate the effect of a single administration of IB-MECA, an A3 adenosine receptor agonist, upon the nociceptive response and central biomarkers of rats submitted to chronic pain models. A total of 136 adult male Wistar rats were divided into two protocols: (1) chronic inflammatory pain (CIP) using complete Freund’s adjuvant and (2) neuropathic pain (NP) by chronic constriction injury of the sciatic nerve. Thermal and mechanical hyperalgesia was measured using von Frey (VF), Randal-Selitto (RS), and hot plate (HP) tests. Rats were treated with a single dose of IB-MECA (0.5 μmol/kg i.p.), a vehicle (dimethyl sulfoxide—DMSO), or positive control (morphine, 5 mg/kg i.p.). Interleukin 1β (IL-1β), brain-derived neurotrophic factor (BDNF), and nerve growth factor (NGF) levels were measured in the brainstem and spinal cord using enzyme-linked immunosorbent assay (ELISA). The establishment of the chronic pain (CIP or NP) model was observed 14 days after induction by a decreased nociceptive threshold in all three tests (GEE, P < 0.05). The antinociceptive effect of a single dose of IB-MECA was observed in both chronic pain models, but this was more effective in NP model. There was an increase in IL-1β levels promoted by CIP. NP model promoted increase in the brainstem BDNF levels, which was reversed by IB-MECA
Keywords: Inflammatory pain, Neuropathic pain, Adenosine receptor, Rats, Biomarkers
Introduction
Pain is an essential mechanism for survival, acting as a warning sign of actual or potential tissue damage. While chronic pain has a high prevalence rate, there is a lack of effective treatments, principally due to the inability of pharmacological therapy to target the precise mechanism [1]. The adenosinergic signaling system has been investigated as a therapeutic target for its role in neurotransmission and nociception [2–4]. Adenosine receptors are subdivided into A1, A2A, A2B, and A3, and each receptor presents a specific distribution in the body and different functionalities. The A3 adenosine receptor (A3AR) is found in both the peripheral and central nervous systems, including glial cells, and has been linked to antinociceptive and anti-inflammatory roles [5–7].
Previous studies using an inflammatory pain model induced by complete Freund’s adjuvant (CFA) showed overexpression of A3AR in the synovia, peripheral blood mononuclear cells, and drain lymph nodes of female rats. The same authors showed downregulation of A3AR after chronic oral treatment with N6-(3-iodobenzyl)adenosine-5′-methyluronamide (IB-MECA), a selective agonist for A3AR [8]. In acute models of pain, intrathecal administration of IB-MECA can suppress the nociceptive response in the late phase of the formalin test in mice, supporting the involvement of A3AR in the nociception process [9]. Preclinical studies have demonstrated that IB-MECA is effective in the prevention and treatment of neuropathic pain induced by different chemotherapeutic agents [10, 11]. Although recent studies have shown that A3AR agonists can produce antinociceptive effects, the mechanisms triggered by this activation are still not clear [5, 10].
Different neurotransmitter systems are involved in pain transmission and modulation. Excessive and sustained inflammatory responses in the peripheral and central nervous systems have been associated with the initiation and maintenance of persistent pain [12]. The cytokine cascade is implicated in the peripheral and central sensitization of persistent pain, with an interaction between glial, immune, and neuronal cells [13, 14]. The role of IL-1β in pain conditions has been studied from different perspectives, mainly because it is involved in the modulation of the supraspinal circuitry of pain as well as a potential target for pain relief [15–17].
In addition, neurotrophins seem to contribute to the pathogenesis of chronic pain through brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), which play key roles in peripheral and central sensitization [18]. NGF is reported as a peripheral pain mediator, and it is upregulated in inflammatory states, while BDNF acts as a modulator of pain in central sites, and is released when nociceptors are activated [19]. In this way, both are implicated in the pathogenesis and maintenance of chronic pain conditions.
Considering the high prevalence of chronic pain in the general population and the lack of effective pharmacological treatments, this study aimed to investigate the effect of IB-MECA on the nociceptive and neurochemical responses of rats subjected to two chronic pain models.
Material and methods
Animals
A total of 136 adult male Wistar rats (55–65 days old; weight 200–250 g) were used. The number of animals required for the behavioral experiments and biochemical analysis was calculated as 8 rats per group, considering a difference of 1.5 standard deviations between the variables and α = 0.05 [20–22]. The rats were housed in groups of three in a polypropylene cage (49 cm × 34 cm × 16 cm) with sawdust-covered flooring. The rats were maintained in a controlled environment (22 ± 2 °C) under a standard light-dark cycle (lights on at 7 a.m. and lights-off at 7 p.m.), with ad libitum water and chow (Nuvital, Porto Alegre, Brazil). All experiments and procedures were approved by the Institutional Animal Care and Use Committee (GPPG-HCPA protocol no. 150530 and no. 2018-0377), and performed in accordance with the Guide for the Care and Use of Laboratory Animals, 8th ed. The experimental protocols complied with the ethical and methodological standards of the ARRIVE guidelines [23].
Experimental design
The rats were acclimated to the maintenance room for 2 weeks before the experiments. The rats were randomized by weight and paw withdrawal latency. Paw withdrawal latency was measured using the hot plate test to ensure that all rats had similar nociceptive behavior. The rats were divided into two experimental designs according to the pain model:
Protocol 1. Rats were subjected to the chronic inflammatory pain (CIP) model and were subdivided into eight groups (eight animals per group): control, control + vehicle, control + morphine, control + IB-MECA treatment, pain, pain + vehicle, pain + morphine, and pain + IB-MECA treatment.
Protocol 2. Rats were subjected to the chronic neuropathic pain (NP) model and were divided into nine groups: control, sham, sham + vehicle, sham + morphine, sham + IB-MECA treatment, pain, pain + vehicle pain, pain + morphine, and pain + IB-MECA treatment.
For both protocols, Randall-Selitto, von Frey, and hot plate tests were performed at baseline and 14 days after the induction of the CIP or NP models and 30 min after treatment with a single dose of the vehicle, morphine, or IB-MECA. The hot plate test was repeated 30, 60, and 90 min after treatment. The rats were killed 6 h after the treatment. The spinal cord and brainstem were collected for further analysis of Interleukin 1β (IL-1β), Brain-Derived Neurotrophic Factor (BDNF), and Neuronal Growth Factor (NGF) levels. For all procedures (nociceptive and neurochemical assays), investigators were blinded to avoid and prevent bias (Fig. 1).
Fig. 1.

Timeline of experimental design
Chronic inflammation pain model
Chronic inflammation was induced as previously described by Laste et al. [24], using complete Freund’ s adjuvant (CFA), which consists of heat-killed Mycobacterium tuberculosis in non-metabolizable oils (paraffin oil and mannide monooleate). In short, rats were anesthetized with isoflurane (5% for induction, 2.5% for maintenance), and placed in a dorsal position. A single intradermal injection of 100 μl of CFA diluted in 1 mg/mL of saline solution was administered to the left footpad [24]. All animals that underwent CFA injection received tramadol (5 mg/kg) intraperitoneally for immediate analgesia as well as every 12 h for 2 days [25]. The control group did not undergo any intervention.
Neuropathic pain model
Neuropathic pain was induced as previously described by Bennett and Xie [26] and adapted by Cioato et al. [27] using chronic constriction injury (CCI) of the sciatic nerve. Rats were anesthetized with isoflurane (5% for induction, 2.5% maintenance) and placed in a dorsal position for the trichotomy of the left thigh and skin antisepsis with 2% iodine alcohol. After skin incision of the left hind limb, the common sciatic nerve was exposed, and three ligatures were tied (Vycril 4.0) separated by an interval of 1 mm. The length of the affected nerve was approximately 5.0 mm, and the ligatures reduced the diameter of the nerve but did not interrupt the epineural circulation. The same investigator performed the ligatures in all rats to ensure an equal level of constriction. The skin was sutured using a Mononylon 4.0 thread. The sham groups were anesthetized and exposed to the sciatic nerve. After surgery and anesthetic recovery, the animals were allowed in their home cages where they remained until they were killed. All rats subjected to CCI received tramadol 5 mg/kg intraperitoneally for immediate analgesia as well as every 12 h for 2 days [25]. The control group did not undergo anesthesia or any surgical procedure.
Pharmacological treatment
The rats in the treatment groups received a selective adenosine A3 receptor agonist, N6-(3-iodobenzyl) adenosine-5′-methyluronamide (IB-MECA) dissolved in 3% dimethyl sulfoxide (DMSO) and applied by intraperitoneal (i.p.) injection. The treatment consisted of a single dose of 0.5 μmol/kg of IB-MECA (i.p.) [10]. Rats in the vehicle groups received a single dose of DMSO dissolved 3% in saline solution (i.p.). The positive control group received one dose of morphine 5 mg/kg (i.p.) as the gold standard for analgesia [28].
Von Frey test
Mechanical allodynia was assessed using von Frey aesthesiometer (Insight, São Paulo, Brazil) as described by Cioato et al. (2016) at baseline and 14 days after pain induction, and 30 min after the treatment with IB-MECA. Rats were habituated to cages for 10 min, 24 h prior to the test, and 5 min daily before tests to prevent novelty-induced analgesia by the apparatus. For testing, a polypropylene tip was inserted perpendicularly from underneath the floor grid and applied to the plantar side of the left hind paw with a gradually increasing pressure. The intensity of the stimulus supported up to the paw withdrawal in grams was automatically recorded. Three successive readings were measured between interval periods of 5 s and averaged. The average was used as the final measurement, and the paw withdrawal threshold was expressed in grams [29].
Randall-Selitto test
The Randall-Selitto test was performed at baseline and 14 days after pain induction, and 30 min after the treatment with IB-MECA. The rats were subjected to mechanical stimuli to determine the paw withdrawal threshold [30]. For mechanical stimulation, an Analgesymeter (type 7200, Ugo-Basile Biological Research, Comerio-Varese, Italy), which gradually increases the pressure, was applied to the dorsal surface of the left rat paw. The nociceptive threshold was defined as the force in grams that resulted in the withdrawal of the hind paw. A cutoff value of 100 g was used.
Hot plate test
The hot plate (HP) test was carried out at baseline and 14 days after pain induction to confirm the effectiveness of the pain model, and 30, 60, and 90 min after treatment with IB-MECA to assess the effects on the thermal nociceptive threshold. The hot plate test determines changes in the latency of behaviors such as jumping and hind paw-licking as indicators of modifications of the supraspinal pain process, considering the results of supraspinal sensory integration [31–34]. All rats were exposed to the HP for 5 min, 24 h prior to testing in order to avoid analgesia induced by the novelty of the apparatus [35]. The surface of the HP was pre-heated and maintained at a constant temperature of 55 ± 0.1 °C. As described previously by Cioato et al., rats were placed inside glass funnels on the heated surface and the time in seconds between the placement of the rat and the first response (foot-licking, jumping, or rapidly removing paws) was recorded as the latency of nociceptive response. The cutoff time was 20 s to prevent tissue damage [27].
Tissue collection
Six hours after treatment with IB-MECA, the rats were killed by decapitation. Their brainstems and spinal cords were collected, and stored at − 80 °C until the assays were performed.
Neurochemical assays
For ELISA assays, the structures were homogenized with protein inhibitor cocktail (#P8340, Sigma-Aldrich) in a volume of 1 mL for each structure. The tissue homogenization was made using tissue homogenization equipment (Jetta-G50, LabHouse) at 4000 rpm. The sample was centrifuged at 10,000 rpm in a refrigerated centrifuge, and the supernatant was aliquoted for further ELISA analysis. The brainstem and spinal cord IL-1β, BDNF, and NGF levels were determined by sandwich enzyme-linked immunosorbent assay (ELISA) using monoclonal antibodies for IL-1β, BDNF, and NGF (R&D Systems, Minneapolis, USA). Procedures were performed in accordance with the manufacturer’s protocol. Optical density was measured using a spectrophotometer at a wavelength of 450 nm. Data were expressed in picograms per milligram of protein. Total protein was measured using the Bradford assay and bovine serum albumin as a standard curve [36].
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). A P value of less than 0.05 was considered statistically significant. The statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS) version 20.0 for Windows. Generalized estimated equations followed by the Bonferroni was used to analyze nociceptive repeated-measures data with one within- subjects factor (time-point), and two between-subjects factors (pain model and treatment) [37]. A one-way analysis of variance (ANOVA) followed by Bonferroni was used to compare the neurochemical data across groups after treatment.
Results
Protocol 1: CFA inflammatory chronic pain model
Effects of a single administration of IB-MECA in mechanical and thermal hyperalgesia in rats subjected to the CFA inflammatory chronic pain model
To confirm the establishment of the chronic inflammatory pain model, the nociceptive response was evaluated using the GEE analysis for all three nociceptive tests. There was no difference between the groups at baseline in terms of mechanical and thermal latency withdrawal evaluated by the von Frey, Randall-Selitto, and hot plate tests (Figs. 2, 3, and 4). However, on the fourteenth day after the CFA injection, these animals presented decreased latency withdrawal in all three tests performed when compared to the control group (GEE/Bonferroni, Wald χ2 = 182.363, Wald χ2 = 362.634, Wald χ2 = 444.092, respectively, and P < 0.001 for all).
Fig. 2.

Mechanical hyperalgesia by von Frey test in rats subjected to CIP (n = 7–8 per group). Data presented as mean ± S.E.M of paw withdrawal threshold in grams. (a) Significant difference between pain, pain + vehicle, pain + morphine, and pain + IB-MECA from all other groups (GEE, P < 0.05) and (b) significant difference between pain and pain + vehicle from all other groups (GEE, P < 0.05)
Fig. 3.

Mechanical hyperalgesia by Randall-Selitto test in rats subjected to CIP (n = 7–8 per group). Data presented as mean ± S.E.M of paw withdrawal threshold in grams. (a) Significant difference between pain, pain + vehicle, pain + morphine, and pain + IB-MECA groups from all other groups (GEE, P < 0.05); (b) significant difference between pain, pain + vehicle, pain + morphine, and IB-MECA from all other groups (GEE, P < 0.05); and (c) significant difference between pain and pain + vehicle from other groups (GEE, P < 0.05)
Fig. 4.

Thermal hyperalgesia by hot plate test in rats subjected to CIP (n = 7–8 per group). Data presented as mean ± S.E.M of latency in seconds. (a) Significant difference between pain, pain + vehicle, pain + morphine and pain + IB-MECA from all other groups (GEE, P < 0.05); (b) significant difference between pain, pain + vehicle and pain + IB-MECA from control + morphine groups (GEE, P < 0.05); (c) significant difference between control, control + vehicle, control + IB-MECA from control + morphine, pain, and pain + vehicle groups (GEE, P < 0.05); (d) significant difference between pain and pain + vehicle from pain + IB-MECA group (GEE, P < 0.05); (e) significant difference between control + morphine from all other groups (GEE, P < 0.05); (f) significant difference between pain, pain + vehicle, pain + morphine and pain + IB-MECA from control, control + vehicle and control + IB-MECA groups (GEE, P < 0.05); and (g) significant difference between pain, pain + vehicle, pain + morphine and pain + IB-MECA from all other groups (GEE, P < 0.05)
When assessed 30 min after the IB-MECA injection, the mechanical hyperalgesia induced by chronic inflammatory pain evaluated through the von Frey test was reversed by a single dose of IB-MECA and an injection of morphine (Fig. 2). Otherwise, when evaluated using the Randall-Selitto test, mechanical hyperalgesia was partially reversed by IB-MECA or morphine injection (Fig. 3).
The thermal latency withdrawal was evaluated 30, 60, and 90 min after the IB-MECA injection. A complete reversal of hyperalgesia induced by the chronic inflammatory model using IB-MECA or morphine was observed at 30 min. This effect was not maintained in the other two time points evaluated. In addition, the control + morphine group presented increased latency 30 and 60 min after injection (Fig. 4).
Effects of a single administration of IB-MECA on biomarker levels in the spinal cord and brainstem of rats subjected to the CFA inflammatory chronic pain model
In the brainstem, there was an increase in the IL-1β levels in the control + IB-MECA group when compared to all other groups (one-way ANOVA, F(7, 44) = 4.348, P = 0.001) (Fig. 5a). However, in the spinal cord, a decrease in the IL-1β levels was observed in pain, pain plus vehicle, pain plus morphine, and pain plus IB-MECA groups when compared with the control group (one-way ANOVA, F(7, 45) = 19.770, P = 0.001) (Fig. 5b).
Fig. 5.
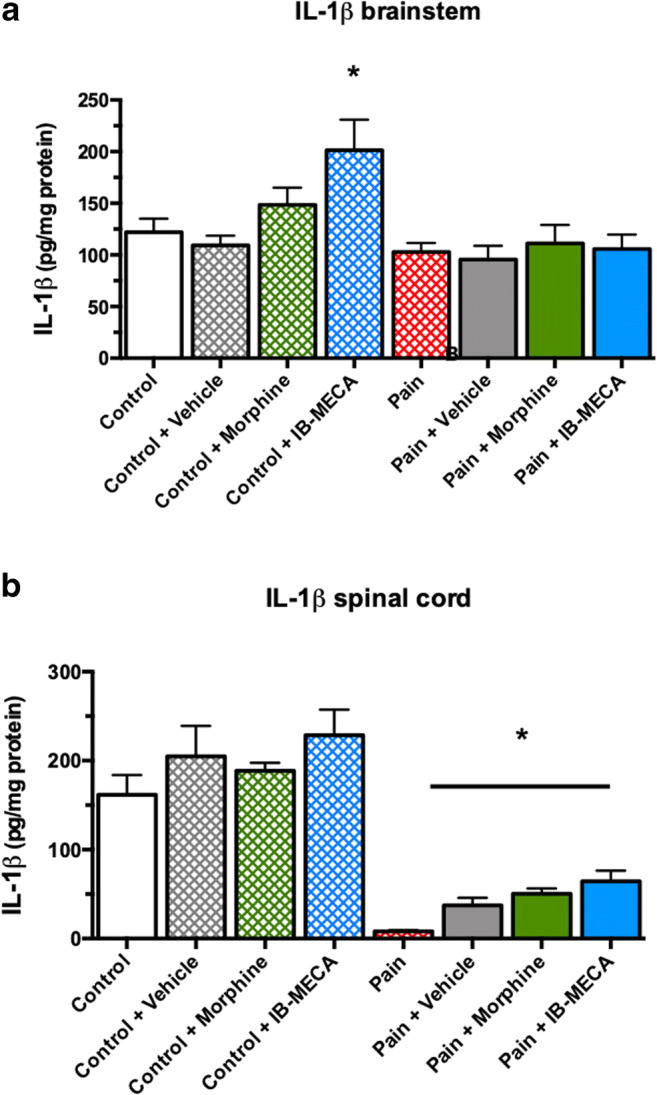
Effects of single administration of IB-MECA on IL-1β levels in rats subjected to CIP. a IL-1β levels in brainstem. Asterisk indicates significant difference between control + IB-MECA from all other groups (one-way ANOVA/Bonferroni, P < 0.05). b IL-1β levels in spinal cord. Asterisk indicates significant difference between pain, pain + vehicle, pain + morphine and pain + IB-MECA from all other groups (one-way ANOVA/Bonferroni, P < 0.05)
There was no difference in the BDNF levels in the brainstem (one-way ANOVA, F(7, 43) = 0.949, P > 0.05) and spinal cord (one-way ANOVA, F(7, 45) = 1.561, P > 0.05) (Fig. 6a, b). There was no difference in the NGF levels in the brainstem (one-way ANOVA, F(7, 44) = 0.871, P > 0.05) and spinal cord (one-way ANOVA, F(7, 45) = 0.702, P > 0.05) (Fig. 7a, b).
Fig. 6.
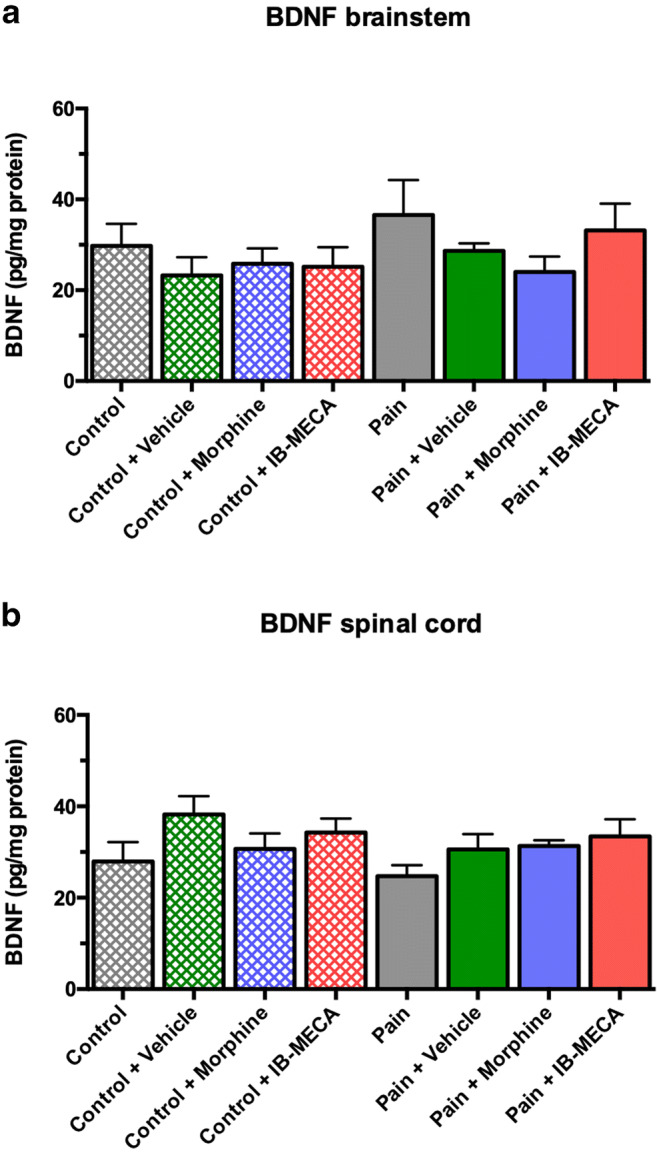
Effects of single administration of IB-MECA on BDNF levels in rats subjected to CIP. a BDNF levels in brainstem. No significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05). b BDNF levels in spinal cord. No significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05)
Fig. 7.

Effects of single administration of IB-MECA on NGF levels in rats subjected to CIP. a NGF levels in brainstem. No significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05). b NGF levels in spinal cord. No significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05)
Protocol 2: Chronic neuropathic pain model
Effect of a single administration of IB-MECA in mechanical and thermal hyperalgesia in rats subjected to the neuropathic chronic pain model
To confirm the establishment of the neuropathic pain model, the nociceptive response was evaluated using the GEE analysis for all three nociceptive tests. There was no difference between the groups at the baseline time of mechanical and thermal latency withdrawal evaluated in the von Frey, Randall-Selitto, and hot plate tests (Figs. 8, 9, and 10). However, on the fourteenth day after the CCI model, these animals presented decreased latency withdrawal in all three tests performed when compared to the control group (GEE/Bonferroni, Wald χ2 = 356,832, Wald χ2 = 193,656, Wald χ2 = 73,064, respectively, and P < 0.001 for all).
Fig. 8.

Mechanical hyperalgesia by von Frey test in rats subjected to NP (n = 7–8 per group). Data presented as mean ± S.E.M of paw withdrawal threshold in grams. (a) Significant difference between pain, pain + vehicle, pain + morphine, and pain + IB-MECA from all other groups (GEE, P < 0.05). (b) Significant difference between pain + IB-MECA from all other groups (GEE, P < 0.05). (c) Significant difference between pain and pain + vehicle from all other groups (GEE, P < 0.05)
Fig. 9.

Mechanical hyperalgesia by Randall-Selitto test in rats subjected to NP (n = 7–8 per group). Data presented as mean ± S.E.M of paw withdrawal threshold in grams. (a) Significant difference between pain, pain + vehicle, pain + morphine, and pain + IB-MECA from all other groups (GEE, P < 0.05). (b) Significant difference between pain and pain + vehicle from other groups (GEE, P < 0.05)
Fig. 10.

Thermal hyperalgesia by hot plate test in rats subjected to NP (n = 7–8 per group). Data presented as mean ± S.E.M of latency in seconds. (a) Significant difference between: control, sham, sham + vehicle, and sham + morphine groups from all other groups (GEE,P < 0.05). (b) Significant difference between pain + morphine and pain + IB-MECA from all other groups group (GEE, P < 0.05). (c) Significant difference of pain + IB-MECA group from other groups (GEE, P < 0.05). (d) Significant difference of pain and pain + vehicle from other groups (GEE, P < 0.05). (e) Significant difference between sham + morphine and sham from sham + vehicle, sham + IB-MECA, pain, pain + vehicle, and pain + IB-MECA groups (GEE, P < 0.05). (f) Significant difference between pain, pain + vehicle, and pain + IB-MECA from other groups (GEE, P < 0.05). (g) Significant difference between pain and pain + vehicle from all other groups (GEE, P < 0.05)
When the mechanical hyperalgesia in the Randall-Selitto test was analyzed, IB-MECA was able to completely reverse the nociceptive behavior induced by chronic constriction of the sciatic nerve 30 min after its administration. In addition, as expected, the sham group that received morphine showed an analgesic response in the Randall-Selitto test in comparison with other groups, including the morphine treated group (Fig. 9). A similar result was found upon mechanical hyperalgesia assessed by the von Frey test. However, IB-MECA partially reversed the hyperalgesia induced by CCI when compared with the pain groups untreated or treated with a vehicle (Fig. 8).
In relation to thermal hyperalgesia, our results showed that IB-MECA was able to partially reverse the decrease in latency to the nociceptive response induced neuropathic pain conditions 30 min after injection. This effect persisted until 60 and 90 min after the administration. We also showed that morphine was able to induce analgesia in the sham and pain groups, and its effects were observed 30 and 60 min after administration (Fig. 10).
Effect of a single administration of IB-MECA on the biomarker levels in the brainstem and spinal cord of rats subjected to the neuropathic chronic pain model
There was no difference in the IL-1β brainstem and spinal cord levels (one-way ANOVA, F(8, 62) = 1.245 and F(8, 62) = 1.215, respectively, P > 0.05) (Fig. 11). Regarding the BDNF levels, there was no difference in the spinal cord (one-way ANOVA, F(8, 62) = 0.647, respectively, P > 0.05) (Fig. 12a). In the brainstem, an increased level of this neurotrophin was observed in the sham group treated with IB-MECA and the pain group and pain groups treated with a vehicle and morphine when compared with the sham group (one-way ANOVA, F(8, 62) = 2.060, P < 0.05) (Fig. 12b). We did not observe any statistical difference in the NGF levels in the brainstem or spinal cord (one-way ANOVA, F(8 ,62) = 1.458 and F(8, 62) = 1.045, respectively, P > 0.05) (Fig. 13a and b).
Fig. 11.

Effects of single administration of IB-MECA on IL-1β levels in rats subjected to NP. a IL-1β levels in brainstem. There was not significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05). b IL-1β levels in spinal cord. There was not significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05)
Fig. 12.

Effect of single administration of IB-MECA on BDNF levels in rats subjected to NP. a BDNF levels in brainstem. Asterisk indicates significant difference between sham + IB-MECA, pain, pain + vehicle and pain + morphine from sham group (one-way ANOVA/Bonferroni, P < 0.05). b BDNF levels in spinal cord. There was not significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05)
Fig. 13.
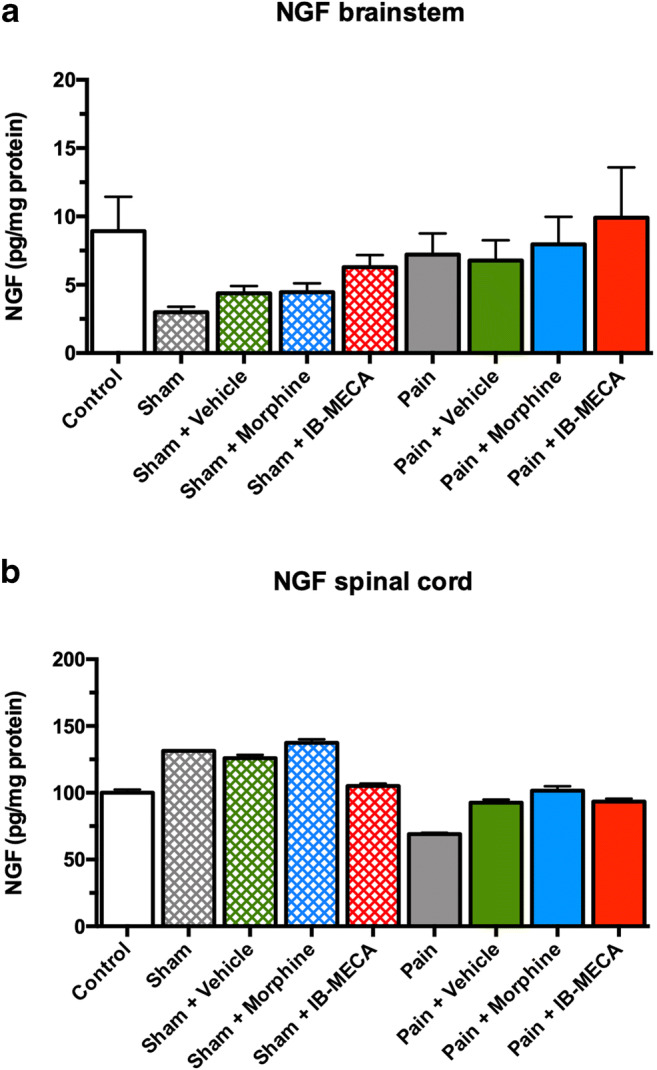
Effect of single administration of IB-MECA on NGF levels in rats subjected to NP. a NGF levels in brainstem. There was not significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05). b NGF levels in spinal cord. There was not significant difference between groups (one-way ANOVA/Bonferroni, P > 0.05)
Discussion
In this study, we showed the antinociceptive effect of a single administration of IB-MECA in well-established chronic pain models (inflammatory and neuropathic pain). However, this was more effective in the NP model. Interestingly, only the CIP model promoted a decrease in the spinal cord IL-1β levels. On the other hand, only NP increased the brainstem BDNF levels, which was reversed by IB-MECA. The antinociceptive effect linked to A3AR presented supraspinal and spinal components, despite the authors describing the involvement of A3AR in a pronociceptive response [38, 39]. Thus, efforts were made to comprehend the involvement of A3AR in the transmission or modulation of nociception.
Our findings regarding the use of IB-MECA for treating chronic pain corroborate previous preclinical studies that showed that it reduced pain in a CCI model and chemotherapy-related pain in rats [10]. However, our results in the inflammatory pain model contrast with a previous study that showed that A3AR activation did not alter nociceptive thresholds in non-neuropathic animal models, and resulted in the selective relief of persistent neuropathic pain [40]. Adenosine A3 receptors seem to have complex effects on the central nervous system, with both pro-inflammatory and anti-inflammatory roles [41]. Diverse mechanisms have been investigated from A3 activation for its effects on the inflammatory process [42–44].
In this way, IB-MECA has been studied as a potential pharmacological treatment for neuropathic pain [10, 11, 40, 45, 46] and inflammatory conditions [47–49]. It has been shown to be a more selective agonist for the A3 adenosine receptor rather than for A1 and A2 adenosine receptors [50]. Evidence demonstrates the influence of A3 receptors in rheumatoid arthritis, Crohn’s disease, and psoriasis, with a direct role in inflammation [51]. Different mechanisms have been proposed. For example, it attenuates neuropathic pain by suppressing microglial activation in tibial nerve injury [45]. In addition, IB-MECA alleviated mechanical hyperalgesia and thermal hypoalgesia in mice with diabetes induced by streptozotocin injection by inhibiting the activation of nuclear factor-κB, thereby decreasing the generation of tumor necrosis factor-α (TNF-α) [52]. In addition, it prevented the establishment of neuropathic pain induced by CCI in rodents [10]. Previous studies have suggested that A3AR is expressed in astrocytes and microglia [53]. It is known that these cells are closely involved in the initiation or perpetuation of neuropathic pain [45, 53, 54]. In this context, we have suggested that the analgesic effect of IB-MECA in the hypernociceptive behavior of rats may be due to BDNF in the NP model.
It is well reported that BDNF acts by modulating spinal and supraspinal levels through fast excitatory and inhibitory signals mediated by the glutamatergic and GABAergic systems, respectively, [55]. A study by Coull and colleagues showed the crucial involvement of BDNF as a signaling molecule between microglia and neurons, and the blockade of this pathway can be a new strategy to treat neuropathic pain [56]. In addition, the activation of A3 receptors could attenuate microglial activation, which involves a decrease in BDNF release and contributes to the restoration of GABA signaling in the spinal dorsal horn [45]. This theory can explain the decreased levels of BDNF induced by IB-MECA found in this study. However, it is necessary to assess the GABA levels and other markers involved in this signaling cascade to confirm this hypothesis.
Preclinical studies have shown that IB-MECA exerts its analgesic effects upon neuropathic pain induced by paclitaxel, inhibiting nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in the spinal cord [10, 11]. This process prevents the activation of NF-κB, mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinase (ERK), and p38, which are associated with decreased production of pro-inflammatory cytokines, such as TNF-α and IL-1β, while also increasing the production of anti-inflammatory cytokines, such as IL-10 [11, 57, 58]. However, in the present study, we showed that treatment with IB-MECA increased IL-1β levels in the control group when compared to all other groups but had no effect in the pain group. It is important to highlight that an injection of CFA in the hind paw of rats has been reported to increase local levels of IL-1β when evaluated acutely after induction of this pain model as well as a study by our research group [59–61]. Interestingly, in the current study, a CIP model decreased the spinal cord levels of IL-1β. A preclinical study described pro-inflammatory cytokine production induced by a CFA model in the central structures of rats [62]. Another study reported that IL-1 receptors are overexpressed in neurons in the spinal dorsal horn of rats subjected to the same inflammatory pain model [63]. Furthermore, this result agrees with the theory of glial involvement in the establishment and maintenance of pain conditions. However, more studies are needed to clarify the involvement of central cytokines and glial structures in inflammatory pain.
The current study has several limitations. First, we tested the analgesic effect of the acute administration of IB-MECA in chronic pain. However, we consider this to be the first step before repeated administrations. Second, we could not evaluate hormonal interference because our results are only from male adult rats. Third, it is possible that the rats habituated to the Hot Plates test, as the rats from the control group presented decreased latency during the time assessment [64].
Conclusions
In summary, IB-MECA represents a potential therapeutic target in chronic pain conditions, independent of its pathophysiology, once it has relieved the hypernociceptive behavior induced by the CCI and CFA models in rats. The analgesic effect of IB-MECA can be linked with the modulation of the brainstem BDNF levels only in the NP model. Further studies are needed for a better understanding of the mechanisms related to the antinociceptive effects of IB-MECA in chronic pain, especially in relation to inflammatory and neuropathic pain.
Stefania Giotti Cioato
Ph.D. in Biological Sciences: Pharmacology and Therapeutics at Federal University of Rio Grande do Sul. M.Sc. in Medicine: Medical Sciences at Federal University of Rio Grande do Sul. B.Sc. in Nursing at Federal University of Rio Grande do Sul.
Compliance with ethical standards
Conflicts of interest
Stefania Giotti Cioato declares that she has no conflict of interest. Liciane Fernandes Medeiros declares that she has no conflict of interest. Bettega Costa Lopes declares that he has no conflict of interest. Andressa de Souza declares that she has no conflict of interest. Helouise Richardt Medeiros declares that she has no conflict of interest. José Antônio Fagundes Assumpção declares that he has no conflict of interest. Wolnei Caumo declares that he has no conflict of interest. Rafael Roesler declares that he has no conflict of interest. Iraci L. S. Torres declares that she has no conflict of interest.
Ethical approval
This study was approved by the Institutional Animal Care and Use Committee (GPPG-HCPA protocol no. 150530 and no.2018-0377).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cohen SP, Mao J. Neuropathic pain : mechanisms and their clinical implications. Br J Med. 2014;348:1–12. doi: 10.1136/bmj.f7656. [DOI] [PubMed] [Google Scholar]
- 2.Zylka MJ. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol Med. 2011;17:188–196. doi: 10.1016/j.molmed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burnstock G. An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacology. 2015;104:4–17. doi: 10.1016/j.neuropharm.2015.05.031. [DOI] [PubMed] [Google Scholar]
- 4.Burnstock G. Purinergic mechanisms and pain. Adv Pharmacol. 2016;75:91–137. doi: 10.1016/bs.apha.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Sawynok J. Adenosine receptor targets for pain. Neuroscience. 2016;338:1–18. doi: 10.1016/j.neuroscience.2015.10.031. [DOI] [PubMed] [Google Scholar]
- 6.Jacobson KA, Gao Z-G. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borea PA, Varani K, Vincenzi F, et al. The A3 adenosine receptor: history and perspectives. Pharmacol Rev. 2014;67:74–102. doi: 10.1124/pr.113.008540. [DOI] [PubMed] [Google Scholar]
- 8.Cohen S, Barer F, Bar-Yehuda S, IJzerman AP, Jacobson KA, Fishman P. A3 adenosine receptor allosteric modulator induces an anti-inflammatory effect: in vivo studies and molecular mechanism of action. Mediat Inflamm. 2014;2014:1–8. doi: 10.1155/2014/708746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon MH, Bae HB, Choi J., II Antinociception of intrathecal adenosine receptor subtype agonists in rat formalin test. Anesth Analg. 2005;101:1417–1421. doi: 10.1213/01.ANE.0000180994.10087.6F. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Janes K, Chen C, Doyle T, Bryant L, Tosh DK, Jacobson KA, Salvemini D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012;26:1855–1865. doi: 10.1096/fj.11-201541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janes K, Esposito E, Doyle T, Cuzzocrea S, Tosh DK, Jacobson KA, Salvemini D. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain. 2014;155:2560–2567. doi: 10.1016/j.pain.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellis A, Bennett DLH. Neuroinflammation and the generation of neuropathic pain. Br J Anaesth. 2013;111:26–37. doi: 10.1093/bja/aet128. [DOI] [PubMed] [Google Scholar]
- 13.Clark AK, Old E, Malcangio M. Neuropathic pain and cytokines: current perspectives. J Pain Res. 2013;6:803–814. doi: 10.2147/JPR.S53660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huh Y, Ji R-R, Chen G (2017) Neuroinflammation, bone marrow stem cells, and chronic pain. Front Immunol 8 [DOI] [PMC free article] [PubMed]
- 15.Apkarian AV, Lavarello S, Randolf A, et al. Expression of IL-1beta in supraspinal brain regions in rats with neuropathic pain. Neurosci Lett. 2006;407:176–181. doi: 10.1016/j.neulet.2006.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.del Rey A, Yau H-J, Randolf A, Centeno MV, Wildmann J, Martina M, Besedovsky HO, Apkarian AV. Chronic neuropathic pain-like behavior correlates with IL-1β expression and disrupts cytokine interactions in the hippocampus. Pain. 2011;152:2827–2835. doi: 10.1016/j.pain.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.del Rey A, Apkarian AV, Martina M, Besedovsky HO. Chronic neuropathic pain-like behavior and brain-borne IL-1β. Ann N Y Acad Sci. 2012;1262:101–107. doi: 10.1111/j.1749-6632.2012.06621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan N, Smith MT. Neurotrophins and neuropathic pain: role in pathobiology. Molecules. 2015;20:10657–10688. doi: 10.3390/molecules200610657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- 20.Torres I, Buffon A, Silveira PP, et al. Effect of chronic and acute stress on ectonucleotidase activities in spinal cord. Physiol Behav. 2002;75:1–5. doi: 10.1016/S0031-9384(01)00605-9. [DOI] [PubMed] [Google Scholar]
- 21.Medeiros LF, Rozisky JR, de Souza A, Hidalgo MP, Netto CA, Caumo W, Battastini AMO, Torres ILS. Lifetime behavioural changes after exposure to anaesthetics in infant rats. Behav Brain Res. 2011;218:51–56. doi: 10.1016/j.bbr.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 22.Torres I, Cucco SNS, Bassani M, et al. Long-lasting delayed hyperalgesia after chronic restraint stress in rats—effect of morphine administration. Neurosci Res. 2003;45:277–283. doi: 10.1016/S0168-0102(02)00232-8. [DOI] [PubMed] [Google Scholar]
- 23.Kilkenny C, Browne WJ, Cuthill IC, et al. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:1–6. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laste G, Macedo IC, Rozisky JR, et al. Melatonin administration reduces inflammatory pain in rats. J Pain Res. 2012;5:359–362. doi: 10.2147/JPR.S34019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guzman-silva MA, Pollastri CE, Augusto J, et al. Tramadol minimizes potential pain during post-oophorectomy in Wistar rats. AATEX. 2008;14:91–92. [Google Scholar]
- 26.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 27.Cioato SG, Medeiros LF, Marques Filho PR, Vercelino R, de Souza A, Scarabelot VL, de Oliveira C, Adachi LNS, Fregni F, Caumo W, Torres ILS. Long-lasting effect of transcranial direct current stimulation in the reversal of hyperalgesia and cytokine alterations induced by the neuropathic pain model. Brain Stimul. 2016;9:209–217. doi: 10.1016/j.brs.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Rozisky JR, Dantas G, Adachi LS, Alves VS, Ferreira MBC, Sarkis JJF, Torres ILDS. Long-term effect of morphine administration in young rats on the analgesic opioid response in adult life. Int J Dev Neurosci. 2008;26:561–565. doi: 10.1016/j.ijdevneu.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Vivancos GG, Verri WA, Cunha TM, et al. An electronic pressure-meter nociception paw test for rats. Brazilian J Med Biol Res. 2004;37:39139–39139. doi: 10.1590/S0100-879X2004000300017. [DOI] [PubMed] [Google Scholar]
- 30.Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957;111:409–419. [PubMed] [Google Scholar]
- 31.Woolfe G, Macdonald A. The evaluation of the analgesic action of pethidine hydrochloride. J Pharmacol Exp Ther. 1944;80:300–307. [Google Scholar]
- 32.Ossipov MH, Kovelowski CJ, Nichols ML, Hruby VJ, Porreca F. Characterization of supraspinal antinociceptive actions of opioid delta agonists in the rat. Pain. 1995;62:287–293. doi: 10.1016/0304-3959(94)00231-3. [DOI] [PubMed] [Google Scholar]
- 33.Caggiula AR, Epstein LH, Perkins KA, Saylor S. Different methods of assessing nicotine-induced antinociception may engage different neural mechanisms. Psychopharmacology. 1995;122:301–306. doi: 10.1007/BF02246552. [DOI] [PubMed] [Google Scholar]
- 34.Rubinstein M, Mogil JS, Japón M, et al. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc Natl Acad Sci U S A. 1996;93:3995–4000. doi: 10.1073/pnas.93.9.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Netto CA, Siegfried B, Izquierdo I. Analgesia induced by exposure to a novel environment in rats: effect of concurrent and post-training stressful stimulation. Behav Neural Biol. 1987;48:304–309. doi: 10.1016/S0163-1047(87)90850-8. [DOI] [PubMed] [Google Scholar]
- 36.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 37.Ballinger GA. Using generalized estimating equations for longitudinal data analysis. Organ Res Methods. 2004;7:127–150. doi: 10.1177/1094428104263672. [DOI] [Google Scholar]
- 38.Sawynok J, Liu XJ. Adenosine in the spinal cord and periphery: release and regulation of pain. Prog Neurobiol. 2003;69:313–340. doi: 10.1016/S0301-0082(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 39.Sawynok J, Zarrindast MR, Reid AR, Doak GJ. Adenosine A3 receptor activation produces nociceptive behaviour and edema by release of histamine and 5-hydroxytryptamine. Eur J Pharmacol. 1997;333:1–7. doi: 10.1016/S0014-2999(97)01110-2. [DOI] [PubMed] [Google Scholar]
- 40.Little JW, Ford A, Symons-Liguori AM, Chen Z, Janes K, Doyle T, Xie J, Luongo L, Tosh DK, Maione S, Bannister K, Dickenson AH, Vanderah TW, Porreca F, Jacobson KA, Salvemini D. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain. 2015;138:28–35. doi: 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janes K, Symons-Liguori AM, Jacobson KA, Salvemini D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br J Pharmacol. 2016;173:1253–1267. doi: 10.1111/bph.13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sachdeva S, Gupta M. Adenosine and its receptors as therapeutic targets: an overview. Saudi Pharm J. 2013;21:245–253. doi: 10.1016/j.jsps.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gessi S, Merighi S, Varani K, Leung E, Mac Lennan S, Borea PA. The A3 adenosine receptor: an enigmatic player in cell biology. Pharmacol Ther. 2008;117:123–140. doi: 10.1016/j.pharmthera.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 44.Borea PA, Gessi S, Merighi S, Vincenzi F, Varani K. Pharmacology of adenosine receptors: the state of the art. Physiol Rev. 2018;98:1591–1625. doi: 10.1152/physrev.00049.2017. [DOI] [PubMed] [Google Scholar]
- 45.Terayama R, Tabata M, Maruhama K, Iida S. A3 adenosine receptor agonist attenuates neuropathic pain by suppressing activation of microglia and convergence of nociceptive inputs in the spinal dorsal horn. Exp Brain Res. 2018;236:3203–3213. doi: 10.1007/s00221-018-5377-1. [DOI] [PubMed] [Google Scholar]
- 46.Janes K, Wahlman C, Little JW, et al. Spinal neuroimmune activation is independent of T-cell infiltration and attenuated by A3 adenosine receptor agonists in a model of oxaliplatin-induced peripheral neuropathy. Brain Behav Immun. 2015;44:91–99. doi: 10.1016/j.bbi.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren T, Tian T, Feng X, et al. An adenosine A3 receptor agonist inhibits DSS-induced colitis in mice through modulation of the NF-κB signaling pathway. Sci Rep. 2015;5:9047. doi: 10.1038/srep09047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian Hua R, Min Min L, Xiao Meng A, et al. Activation of adenosine A3 receptor inhibits inflammatory cytokine production in colonic mucosa of patients with ulcerative colitis by down-regulating the nuclear factor-kappa B signaling. J Dig Dis. 2019;21:38–45. doi: 10.1111/1751-2980.12831. [DOI] [PubMed] [Google Scholar]
- 49.Ren T, Qiu Y, Wu W, et al. Activation of adenosine A3 receptor alleviates TNF- alpha -induced inflammation through inhibition of the NF-kB signaling pathway in human colonic epithelial cells. Mediat Inflamm. 2014;2014:1–11. doi: 10.1155/2014/818251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacobson KA, Merighi S, Varani K, et al. A3 adenosine receptors as modulators of inflammation: from medicinal chemistry to therapy. Med Res Rev. 2018;38:1031–1072. doi: 10.1002/med.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fishman P, Bar-Yehuda S. Pharmacology and therapeutic applications of A3 receptor subtype. Curr Top Med Chem. 2003;3:463–469. doi: 10.2174/1568026033392147. [DOI] [PubMed] [Google Scholar]
- 52.Fishman P, Bar-Yehuda S, Madi L, et al. The PI3K-NF-κB signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res Ther. 2006;8:1–9. doi: 10.1186/ar1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohsawa K, Sanagi T, Nakamura Y, Suzuki E, Inoue K, Kohsaka S. Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. J Neurochem. 2012;121:217–227. doi: 10.1111/j.1471-4159.2012.07693.x. [DOI] [PubMed] [Google Scholar]
- 54.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merighi A, Salio C, Ghirri A, et al. BDNF as a pain modulator. Prog Neurobiol. 2008;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 56.Coull JAM, Beggs S, Boudreau D, et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 57.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haskó G, Szabó C, Németh ZH, et al. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol. 1996;157:4634–4640. [PubMed] [Google Scholar]
- 59.Woolf CJ, Allchorne A, Poole S. Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor a 1. Br J Pharmacol. 1997;121:417–424. doi: 10.1038/sj.bjp.0701148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma QP, Woolf CJ. Progressive tactile hypersensitivity: an inflammation-induced incremental increase in the excitability of the spinal cord. Pain. 1996;67:97–106. doi: 10.1016/0304-3959(96)03105-3. [DOI] [PubMed] [Google Scholar]
- 61.Laste G, de Souza ICC, dos Santos VS, et al. Histopathological changes in three variations of Wistar rat adjuvant-induced arthritis model. Int J Pharm Res Sch. 2014;3:780–90. [Google Scholar]
- 62.Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- 63.Holló K, Ducza L, Hegyi Z, et al. Interleukin-1 receptor type 1 is overexpressed in neurons but not in glial cells within the rat superficial spinal dorsal horn in complete Freund adjuvant-induced inflammatory pain. J Neuroinflammation. 2017;14:1–18. doi: 10.1186/s12974-017-0902-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gunn A, Bobeck EN, Weber C, Morgan MM. The influence of non-nociceptive factors on hot plate latency in rats. J Pain. 2012;12:222–227. doi: 10.1016/j.jpain.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]