

Abstract
Purpose:
Triple-negative breast cancer (TNBC) is an aggressive subtype of breast cancer with poor survival outcomes. Metformin has been shown to have anti-tumor effects by lowering serum levels of the mitogen insulin and having pleiotropic effects on cancer cell signaling pathways. BMS-754807 is a potent and reversible inhibitor of both insulin-like growth factor 1 receptor (IGF-1R) and insulin receptor (IR). Both drugs have been reported to have some efficacy in TNBC. However, it is unclear whether the combination of the two drugs is more effective than single drug treatment in TNBC.
Methods:
We treated a panel of TNBC cell lines with metformin and BMS-754807 alone and in combination and tested cell viability using MTS assays. We used the CompuSyn software to analyze for additivity, synergism, or antagonism. We also examined the molecular mechanism by performing reverse phase protein assay (RPPA) to detect the candidate pathways altered by single drugs and the drug combination and used Western blotting to verify and expand the findings.
Results:
The combination of metformin and BMS-754807 showed synergy in 11 out of 13 TNBC cell lines tested (85%). RPPA analysis detected significant alterations by the drug combination of multiple proteins known to regulate cell cycle and tumor growth. In particular, the drug combination significantly increased levels of total and phosphorylated forms of the cell cycle inhibitor p27Kip1 and decreased the level of the p27Kip1 E3 ligase SCFSkp2.
Conclusions:
We conclude that the combination of metformin and BMS-754807 is more effective than either drug alone in inhibiting cell proliferation in the majority of TNBC cell lines, and that one important mechanism may be suppression of SCFSkp2 and subsequent stabilization of the cell cycle inhibitor p27Kip1. This combination treatment may represent an effective targeted therapy for a significant subset of TNBC cases and should be further evaluated.
Keywords: Triple-negative breast cancer (TNBC), Metformin, BMS-754807, IGF-1R inhibitor, insulin receptor inhibitor
Introduction
Breast cancer is the most prevalent invasive cancer in women [1]. Triple-negative breast cancer (TNBC) is an aggressive subtype that lacks expression of estrogen receptor (ER) and progesterone receptor (PR) and does not show amplification of the gene encoding human epidermal growth factor receptor 2 (HER2) [2]. Compared with other subtypes of breast cancer, TNBC has a worse prognosis and lacks effective targeted therapy. At present, chemotherapy is the mainstay of treatment for early stage and advanced TNBC. Developing efficacious targeted therapeutics is crucial for improving survival in patients with TNBC.
Insulin-like growth factor-1 receptor (IGF-1R) is a transmembrane tyrosine kinase growth factor receptor that plays a key role in the establishment and maintenance of the transformed phenotype [3]. Suppression of IGF-1R activity can prevent and treat breast cancer [3,4]. Insulin receptor (IR) is closely related to IGF-1R and is also increased in breast cancer [5]. High expression levels of total IR as well as insulin are correlated with recurrence and poor survival in breast cancer patients [6–8]. Activation of IGF-1R or IR activates PI3-Kinase, Akt, and Ras, potent oncoproteins that are deregulated in many cancers [9,10]. BMS-754807, a pyrrolotriazine that is a reversible dual inhibitor of IGF-1R/IR [11,12], has antitumor activity in a broad range of tumor types and enhances the antitumor response of other therapeutic agents [13–16]. BMS-754807 has been reported to suppress TNBC in vitro and in tumor xenografts [17]. It has been investigated in several phase I and phase II clinical trials as an anti-cancer drug [18,19] including in ER+ breast cancer patients and HER2+ patients, but it has not been tested in TNBC patients[20,21].
Metformin is a biguanide that is widely used as a front-line therapeutic for type 2 diabetes since it can lower blood glucose levels and alleviate insulin resistance in these patients [22,23]. The primary mechanism is the inhibition of complex I of mitochondrial electron transport, resulting in decreased ATP levels, increased AMP levels and subsequent activation of the AMP-activated protein kinase (AMPK) [24]. AMPK suppresses hepatic gluconeogenesis and improves glucose uptake in muscle and liver [25]. Activation of AMPK inhibits the growth of cancer cells [26]. Epidemiological studies have found that metformin decreases the incidence of cancer in diabetic patients [27,28] although others did not [24]. Some but not all studies have suggested that metformin can enhance the response to neoadjuvant treatment in breast cancer patients [29,30]. Metformin has been shown to inhibit TNBC cell proliferation/survival in vitro and in mouse models [31–34]. Anticancer molecular mechanisms of metformin have been reported to include systemic effects in controlling hyperinsulinemia and blood glucose levels, indirect effects on inflammation and body weight, AMPK-dependent effects in cancer cells (such as mTOR and mRNA translation suppression [35,36]), and AMPK-independent impact in cancer cells (such as inhibition of RAG GTPase and reactive oxygen species as a result of inhibition of mitochondrial complex I [24]).
Though several studies have investigated the response of TNBC cells to metformin or BMS754807 as single agents, no studies have examined the combination of these two drugs. In our work, we found that combining BMS-754807 with metformin in TNBC cell lines led to better therapeutic efficacy in TNBC cell lines than either drug alone. We also utilized reverse phase protein array (RPPA), a platform that allows for the broad-scale and quantitative measurement of the level and activation/phosphorylation state of hundreds of proteins [37], to investigate potential molecular mechanisms underlying the synergistic effects of BMS-754807 and metformin.
Materials and methods
Cell lines and culture conditions
Breast cancer cell lines as listed in Table 1 were obtained from American Type Culture Collection (ATCC) and cultured using the recommended culture conditions. Briefly, BT-549, HCC1937, HCC38, HCC1806, HCC70 and HCC1395 were cultured in RPMI-1640 medium (ATCC) supplemented with 10% fetal bovine serum (FBS). MDA-MB-231, MDA-MB-436, BT-20, MDA-MB-453, Hs 578T, MDA-MB-157 and MDA-MB-468 were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS). Cultures were maintained at 37°C in a 5% CO2 atmosphere using a tissue culture incubator. Cells were carried not more than 10 passages and not more than 3 months before using new frozen stocks.
Table 1.
The majority of TNBC cell lines are sensitive to combined treatment with metformin and BMS-754807
| Cell line | TNBC subtype | ED50 BMS (μM) | ED50 Metformin (mM) | CI Value | Effect |
|---|---|---|---|---|---|
| BT-20 | LAR Unclassified | 5.41 | 1.08 | 0.43 | Synergism |
| MDA-MB-453 | LAR LAR | 45.9 | 0.9 | 0.43 | Synergism |
| HCC1806 | Basal-like BL2 | 7.69 | 2.56 | 0.49 | Synergism |
| MDA-MB-436 | Mesenchymal-like MSL | 2.34 | 0.23 | 0.56 | Synergism |
| HCC70 | Basal-like BL2 | 1.16 | 5.82 | 0.59 | Synergism |
| MDA-MB-231 | Mesenchymal-like MSL | 3 | 15.33 | 0.63 | Synergism |
| MDA-MB-468 | Basal-like BL1 | 0.39 | 0.16 | 0.63 | Synergism |
| HCC1937 | Basal-like BL1 | 10.42 | 2.08 | 0.75 | Synergism |
| HCC38 | Basal-like BL1 | 0.95 | 1.9 | 0.75 | Synergism |
| MDA-MB-157 | Mesenchymal-like MSL | 3.29 | 0.55 | 0.92 | Synergism |
| BT-549 | Mesenchymal-like M | 5.09 | 12.73 | 0.93 | Synergism |
| Hs 578T | Mesenchymal-like MSL | 4.32 | 11.52 | 1.03 | Additive |
| HCC1395 | LAR Unclassified | 29.5 | 31.1 | 1.28 | Antagonism |
All the cells were cultured in regular growth medium for 24 hours and then treated with increasing concentrations of either a single agent or both agents for 72 hours. CI values were calculated by software CompuSyn to determine the anticancer effect of drug combination. The concentration of each drug at ED50 were given by the report from the software CompuSyn.
Drugs
BMS-754807 was purchased form Selleck Chemicals (Houston, TX), dissolved in DMSO (Sigma-Aldrich, Oakville, ON), and stored at −20°C. Metformin (1,1-Dimethylbiguanide-hydrochloride) was obtained from Sigma-Aldrich, dissolved in water, and stored at −20°C.
Cell viability and synergy assay
Cells were seeded in 96-well tissue culture plates at 2,000–4,000 cells/well depending on the growth characteristic of the cell line so that each was in growth phase at the time of assay (60%−80% confluency). After 24 hours seeding, cells were treated in triplicate with single drugs individually, as well as their constant-ratio combination over a 5-point range centered on the single agent concentrations that inhibited viability by 50% (IC50). Cells were further incubated at 37°C for 72 hours, and cell proliferation was measured using a methanethiosulfonate-based viability assay (CellTiter96 Aqueous One Solution Reagent, Promega Corporation, Madison, WI) according to the manufacturer’s instructions and then read by microplate reader (Biotek, SYNERGY HTX, Vermont, USA). Results were expressed as percentage of vehicle DMSO-treated cells. Results presented are mean ± SD from three separate experiments done in triplicate. The CI (combination index) value was calculated by software CompuSyn following the manufacturer’s guidance. A CI of <1, =1, or >1 indicates synergism, an additive effect, and antagonism, respectively [38].
Reverse phase protein lysate microarray (RPPA)
HCC1806 cells were seeded for 24 hours and then treated with vehicle (DMSO), metformin (5 mM), BMS-754807(15 μM), and the combination of two drugs (Metformin 5mM and BMS-754807 15μM) for 48 hours. Protein lysates were prepared from cultured cells with modified Tissue Protein Extraction Reagent (TPER; Pierce) and a cocktail of protease and phosphatase inhibitors (Roche Life Science). Lysates were spun at 14,000xg for 15min at 4°C, and the supernatants were transferred to fresh tubes. The centrifugation was repeated until the supernatants were clear. Protein concentration was determined by BCA assay (Pierce™). Lysates of 0.5mg/ml were denatured in 2x SDS sample buffer with 2.5% 2-mercaptoethanol at 100°C for 8 min. RPPA was performed by the Antibody-based Proteomics Core Facility at Baylor College of Medicine and analyzed as previously described [39]. Samples were probed with 216 antibodies.
Immunoblot analyses
HCC1806 cells were treated with vehicle (DMSO), BMS-754807 (15 μM), metformin (5 mM) and the combination of the two drugs for 48 hours and cells were lysed in RIPA buffer containing protease inhibitors and phosphatase inhibitors. Cell lysates were boiled at 95°C for 5 mins and loaded on 15% polyacrylamide gels. Proteins were separated by electrophoresis, transferred to nitrocellulose membranes. The membranes were then blocked in 5% non–fat milk in TBST (w/v) for 1 h at room temperature, incubated with a primary antibody, washed 4 times in TBST, incubated with a secondary antibody for 1 h, washed with TBST, and imaged by the Odyssey System (Li-Cor Biosciences, Lincoln, NE, USA). The relative intensity of a band to the β-actin control was measured using ImageJ software (NIH). One-way ANOVA and multiple comparisons were performed using Prism6. Antibody against SKP2 (2652, 1:1000) was purchased from Cell Signaling Technology (Danvers, MA). Antibody against Phospho-p27Kip1 (Thr187) (71–7700, 1:1000) was from Invitrogen (Grand Island, NY). Anti-p27 KIP1 antibody [Y236] (ab32034, 1:1000) was from Abcam (Cambridge, UK).
Statistical analysis
Error bars represent standard deviations from the mean of at least three replicates, unless otherwise indicated. The normalized data (Norm) were used for RPPA data analysis. The normalized data were log2 transformed for hypothesis testing. There are 4 groups (untreated, metformin treatment, BMS-754807 treatment and the drug combination treatment) and each group has 3 biological replicates. For each sample, the median value of the three technical replicates was used for statistical analysis. Two-way ANOVA was used to test main effect of different treatment and their interaction and then diverse contrasts were performed. p value < 0.05 was considered statistically significant.
Results
The majority of TNBC cell lines are sensitive to combined treatment with metformin and BMS-754807
Antiproliferative effects of metformin and BMS-754807 were tested via an MTS assay in a panel of TNBC cell lines [17]. The combination index (CI) was calculated using CompuSyn software to determine whether the drug combination demonstrated synergism, additivity, or antagonism (CI <1, =1, and >1, respectively) (Table 1). We found that 11 out of 13 cell lines (85%) showed synergy towards the combination of metformin and BMS-754807. These cell lines represent 6 of the 7 known TNBC subtypes [40] (Table 1). Among these synergistic cell lines, BT-20, HCC1806, MDA-MB-436, HCC70 and MDA-MB-468 were most sensitive to the drug combination.
Representative examples of the synergistic effect of metformin and BMS-754807 are shown in Fig 1. The cell viability of HCC1806 and BT-20 after treatment with increasing concentrations of the two drugs for 72 hours was measured by the MTS assay. At all concentrations tested, the combination of the two drugs enhanced the inhibition of cell proliferation compared with single agent treatment. Overall, our MTS assay results revealed the synergistic effects of metformin and BMS-754807 in the great majority of TNBC cell lines.
Fig. 1. The majority of TNBC cell lines are sensitive to combined treatment with metformin and BMS-754807.
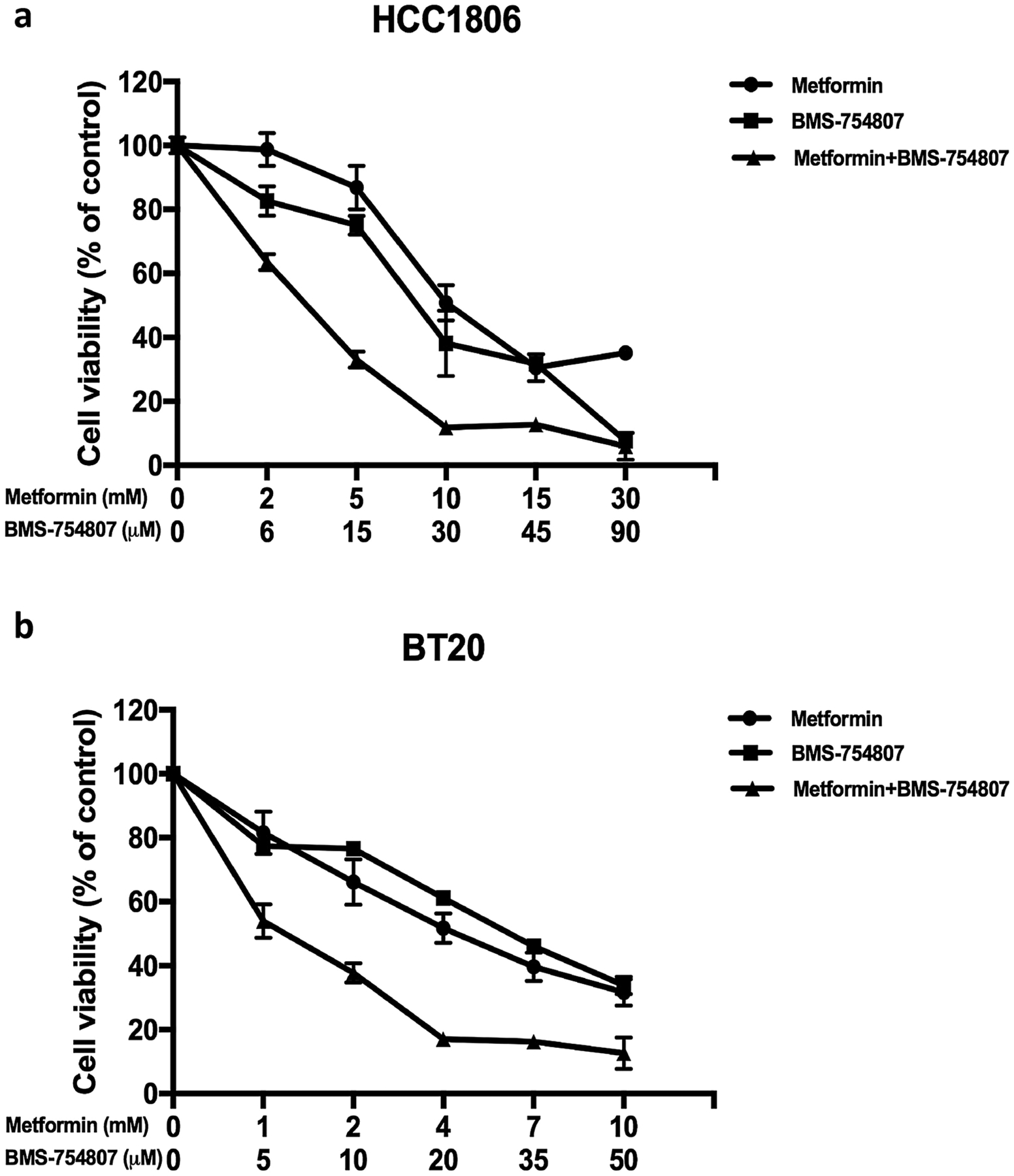
Proliferation assay of HCC1806 and BT-20 cells treated with metformin, BMS-754807 and their combination with different concentrations. Both HCC1806 and BT-20 cells were plated onto a 96-well plate (2000 – 5000 cells per well) in regular growth medium. After 24 hours of incubation, cells were treated with single agent or combined agents for 72 hours and then assayed for percent proliferation using MTS and normalized to control treatment. Experiments are representative of triplicate assays. Data are representative of averages of triplicate determinations ± SE.
Reverse-phase protein array identifies proteins differentially regulated by metformin and BMS-754807 alone and in combination
To discover protein profile changes caused by metformin or BMS-754807 or their combination, we performed a reverse-phase protein array (RPPA) analysis on HCC1806 cell lysates using 216 different cancer-related antibodies. These lysates were prepared from cells treated with vehicle (DMSO), metformin (5 mM), BMS-754807 (15μM), and the combination of the two drugs for 48 hours. From our analysis, we considered the significantly changed proteins between experimental groups by employing Tukey’s pairwise T-test (significant for p value < 0.05). We first compared each of the single drug treatment groups to the vehicle group to detect protein changes caused by treatment with metformin and BMS-754807 individually. We found that metformin altered 35 proteins while BMS-754807 changed 118 proteins in HCC1806 cells (Supplementary Table 1). Among those proteins significantly changed by single drug treatment, 19 were shared (Fig 2a). Examples include decreased levels of p-EGFR(Y845; Y1045), p-FAK(Y576/577; Y397), p-ALK(Y1604), p-HER2/ErbB2(Y877), c-Myc, Ezh2, RRM2, HIF-1α, and LRP6 (Table 2) and increased levels of p-p27(T187). These results confirm that these two agents individually have impacts on a number of cancer-related signaling pathways in HCC1806, reflecting our observation that HCC1806 was sensitive to each drug individually.
Fig. 2. Protein changes as a result of treatment with BMS-754807 or metformin as a single agent.
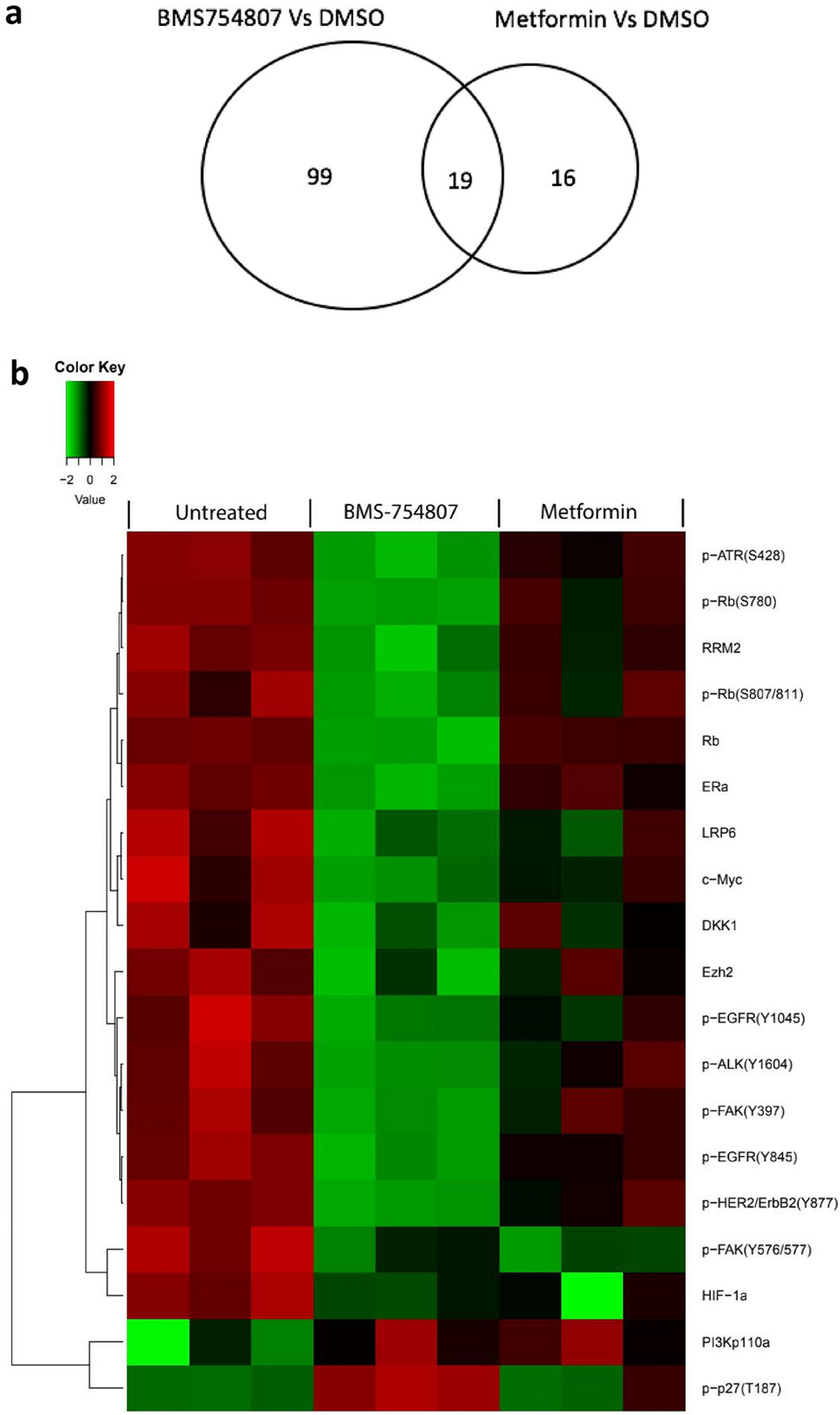
(a) Venn diagram showing the numbers of proteins in the listed comparisons. (b) Heatmap from RPPA analysis showing proteins/phosphoproteins differentially expressed in HCC1806 following treatment with DMSO, metformin alone, and BMS-754807 alone. The normalized data were log2 transformed. Each group has 3 biological replicates. For each sample, the median value of three technical replicates was used for statistical analysis. Two-way ANOVA was used to test the main effects of the different treatments and their interactions. A p value < 0.05 was considered statistically significant. Red indicates higher intensity while green indicates lower intensity.
Table 2.
Proteins whose levels are altered by single agent treatment with metformin and with BMS-754807
| Proteins | BMS-754807 vs. DMSO | Metformin vs. DMSO | ||
|---|---|---|---|---|
| p value | Fold Change | p value | Fold Change | |
| p-p27(T187) | <0.0001 | 2.96 | 0.0404 | 1.32 |
| PI3Kp110a | 0.0318 | 1.14 | 0.0239 | 1.15 |
| Rb | <0.0001 | −3.61 | 0.0118 | −1.20 |
| p-Rb(S807/811) | <0.0001 | −3.48 | 0.0261 | −1.40 |
| p-Rb(S780) | <0.0001 | −3.17 | 0.0002 | −1.42 |
| p-EGFR(Y845) | <0.0001 | −2.71 | 0.0062 | −1.42 |
| ERa | <0.0001 | −2.44 | 0.0387 | −1.23 |
| DKK1 | <0.0001 | −2.28 | 0.0188 | −1.40 |
| p-FAK(Y397) | <0.0001 | −2.12 | 0.0236 | −1.23 |
| p-ALK(Y1604) | <0.0001 | −2.09 | 0.0358 | −1.32 |
| Ezh2 | <0.0001 | −2.04 | 0.0321 | −1.30 |
| p-HER2/ErbB2(Y877) | <0.0001 | −2.01 | 0.0181 | −1.25 |
| RRM2 | <0.0001 | −1.97 | 0.0063 | −1.29 |
| c-Myc | <0.0001 | −1.84 | 0.0013 | −1.36 |
| HIF-1a | 0.0004 | −1.80 | 0.0001 | −1.80 |
| LRP6 | 0.0007 | −1.67 | 0.0330 | −1.35 |
| p-EGFR(Y1045) | 0.0002 | −1.62 | 0.0330 | −1.30 |
| p-ATR(S428) | <0.0001 | −1.53 | 0.0159 | −1.13 |
| p-FAK(Y576/577) | <0.0001 | −1.32 | <0.0001 | −1.38 |
For each sample, the median value of three technical replicates was used for statistical analysis. Two-way ANOVA was used to test the main effects of the different treatments and their interactions. p value < 0.05 was considered statistically significant.
To detect the biological pathways altered by the drug combination, we compared protein expression of cells receiving combination treatment vs. single drug treatment. Adding metformin to BMS-754807 increased the effects of BMS-754807 alone by 51 proteins, while adding BMS-754807 to metformin enhanced the effects of metformin by 121 proteins (Fig 3a, Supplementary Table 2). 35 proteins are shared in these two comparisons (Fig 3 and Table 3). One of these proteins, the cell proliferation marker Ki-67, was dramatically reduced by the combination compared to either drug alone, consistent with the synergistic inhibition of cell proliferation seen in the MTS assays. Other shared proteins may include targets that are crucial to the synergistic inhibition of tumor cell growth. For example, the expression level of cleaved caspase-7 was raised by the combination compared to either drug alone (Table 3 vs. Supplementary Table 1), implying that metformin and BMS-754807 together but not as single agents induce apoptosis in HCC1806. Aurora A, a mitotic serine/threonine kinase that plays a crucial role in mitosis, was significantly suppressed by the combination compared with BMS-754807 only (p < 0.0001, fold change = −2.13) or metformin only (p < 0.0001, fold change = −4.56). Other notable proteins that were downregulated include p-EGFR(Y1068), p-SHC(Y317), BRCA1, ATM, ILK1, Notch1, and STAT3 (Table 3). These proteins are known to promote cell proliferation and tumor growth. The phosphorylated (inactive) form of Rb was reduced by approximately 53% in the combination treatment vs. BMS-754807 alone, whereas the total Rb was reduced by approximately 14% (Table 3), indicating that the fraction of unphosphorylated (active) Rb, which can bind E2F and thereby inhibit cell cycle progression, was increased by the combination treatment.
Fig. 3. Protein changes as a result of treatment with BMS-754807 and metformin combination.
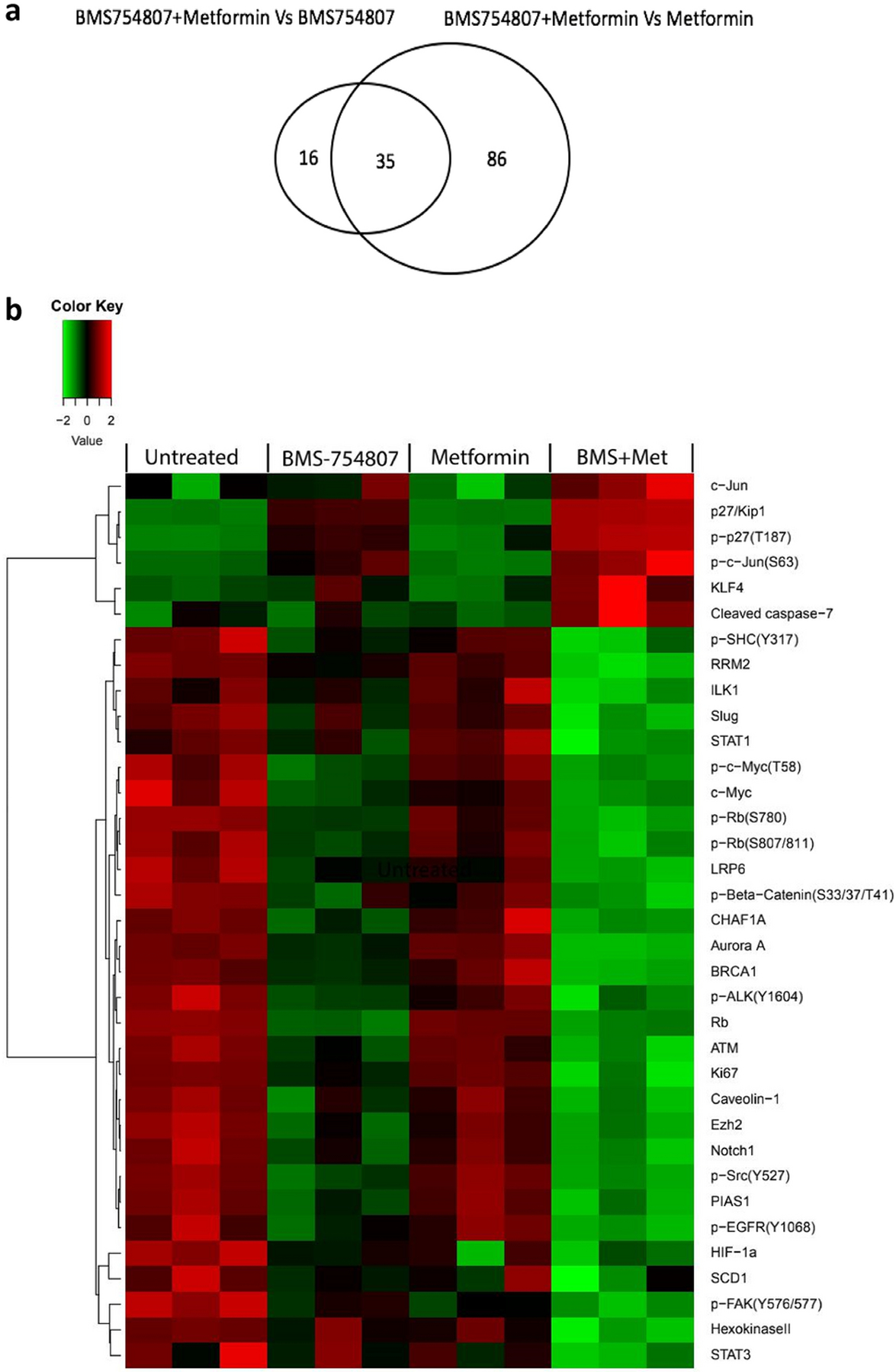
(a) Venn diagram showing the numbers of proteins in the listed comparison. (b) Heatmap from RPPA analysis showing proteins/phosphoproteins differentially expressed in HCC1806 following treatment with DMSO, metformin alone, BMS-754807 alone, and the drug combination. The normalized data were log2 transformed. There are 4 groups and each group has 3 biological replicates. For each sample, the median value of the three technical replicates was used for statistical analysis. Two-way ANOVA was used to test the main effects of the different treatments and their interactions. A p value < 0.05 was considered statistically significant. Red indicates higher intensity while green which indicates lower intensity.
Table 3.
Proteins whose levels are significantly altered by combination treatment with metformin and BMS-754807 compared with single agent treatment
| Proteins | BMS-754807 + Metformin vs. BMS-754807 | BMS- 754807 + Metformin vs. Metformin | ||
|---|---|---|---|---|
| p value | Fold Change | p value | Fold Change | |
| p27/KIP1 | <0.0001 | 2.60 | <0.0001 | 13.81 |
| p-p27(T187) | <0.0001 | 2.27 | <0.0001 | 5.11 |
| KLF4 | 0.0010 | 1.71 | <0.0001 | 2.32 |
| c-Jun | 0.0145 | 1.60 | <0.0001 | 2.65 |
| p-c-Jun(S63) | 0.0001 | 1.53 | <0.0001 | 2.73 |
| Cleaved caspase-7 | <0.0001 | 1.45 | <0.0001 | 1.52 |
| RRM2 | <0.0001 | −3.94 | <0.0001 | −6.05 |
| Ki-67 | <0.0001 | −3.31 | <0.0001 | −8.22 |
| Slug | <0.0001 | −2.43 | <0.0001 | −3.51 |
| Aurora A | <0.0001 | −2.13 | <0.0001 | −4.56 |
| p-Rb(S807/811) | <0.0001 | −1.91 | <0.0001 | −4.73 |
| CHAF1A | 0.0122 | −1.88 | <0.0001 | −9.76 |
| p-Rb(S780) | <0.0001 | −1.87 | <0.0001 | −4.16 |
| HexokinaseII | <0.0001 | −1.82 | <0.0001 | −1.84 |
| BRCA1 | <0.0001 | −1.70 | <0.0001 | −3.28 |
| Caveolin-1 | 0.0353 | −1.65 | <0.0001 | −2.76 |
| HIF-1a | 0.0087 | −1.49 | 0.0246 | −1.49 |
| LRP6 | 0.0101 | −1.46 | 0.0002 | −1.80 |
| PIAS1 | 0.0211 | −1.44 | <0.0001 | −2.81 |
| p-Src(Y527) | 0.0054 | −1.43 | <0.0001 | −3.33 |
| ATM | 0.0002 | −1.42 | <0.0001 | −2.06 |
| p-EGFR(Y1068) | 0.0117 | −1.38 | <0.0001 | −1.98 |
| p-SHC(Y317) | 0.0032 | −1.36 | <0.0001 | −1.70 |
| Ezh2 | 0.0351 | −1.32 | <0.0001 | −2.07 |
| p-ALK(Y1604) | 0.0370 | −1.30 | <0.0001 | −2.06 |
| p-FAK(Y576/577) | <0.0001 | −1.29 | 0.0006 | −1.24 |
| STAT3 | 0.0008 | −1.28 | 0.0012 | −1.26 |
| Notch1 | 0.0151 | −1.26 | <0.0001 | −1.63 |
| p-Beta-Catenin(S33/37/T41) | 0.0069 | −1.25 | <0.0001 | −1.49 |
| ILK1 | 0.0015 | −1.23 | <0.0001 | −1.42 |
| STAT1 | 0.0042 | −1.21 | <0.0001 | −1.43 |
| c-Myc | 0.0352 | −1.21 | <0.0001 | −1.63 |
| p-c-Myc(T58) | 0.0396 | −1.21 | <0.0001 | −2.21 |
| Rb | 0.0287 | −1.17 | <0.0001 | −3.51 |
| SCD1 | 0.0410 | −1.17 | 0.0041 | −1.26 |
The normalized data were log2 transformed for hypothesis testing. For each sample, the median value of three technical replicates was used for statistical analysis. Two-way ANOVA was used to test main effects of the different treatments and their interactions. p value < 0.05 was considered statistically significant.
Metformin and BMS-754807 combination treatment enhances p27 protein levels and reduces Skp2 protein levels in HCC1806
Among the proteins that are upregulated by the combination treatment, the important cell cycle inhibitor p27KIP1 and its phosphorylated form stood out (Fig 3 and Table 3). We performed Western blotting to verify the expression changes of p-p27(T187) in HCC1806 in the presence of single drugs and the combination. As shown in Fig 4, metformin alone did not appear to have a significant impact on p27 and p-p27(T187) expression levels, BMS-754807 alone led to an increase in both total and phosphorylated levels of p27, and the drug combination caused a further increase of the total and phosphorylated levels of p27, consistent with the findings from RPPA (Fig 3b). p27, a well-recognized inhibitor of the cyclin/cyclin-dependent kinase (CDK) complex, is phosphorylated at Thr187, ubiquitinated and degraded in the late G1 phase of the cell cycle [41,42]. The responsible E3 ligase is SCFSkp2, which targets p-p27(T187) for proteasome-mediated degradation [43–45]. To test whether this abnormal accumulation of p-p27(T187) as well as total p27 was due to depleted levels of Skp2, we quantified Skp2 levels in these cells. We found that BMS-754807 treatment led to a reduced level of Skp2, and that the combination treatment caused a further reduction of Skp2 (Fig 4b). Together, these data suggest that combination treatment with BMS-754807 and metformin leads to diminished levels of Skp2, therefore allowing p27 to accumulate and suppress cell cycle progression in TNBC cells.
Fig. 4. Combined treatment with metformin and BMS-754807 synergistically enhances p27 and p-p27(T187) but reduces Skp2.
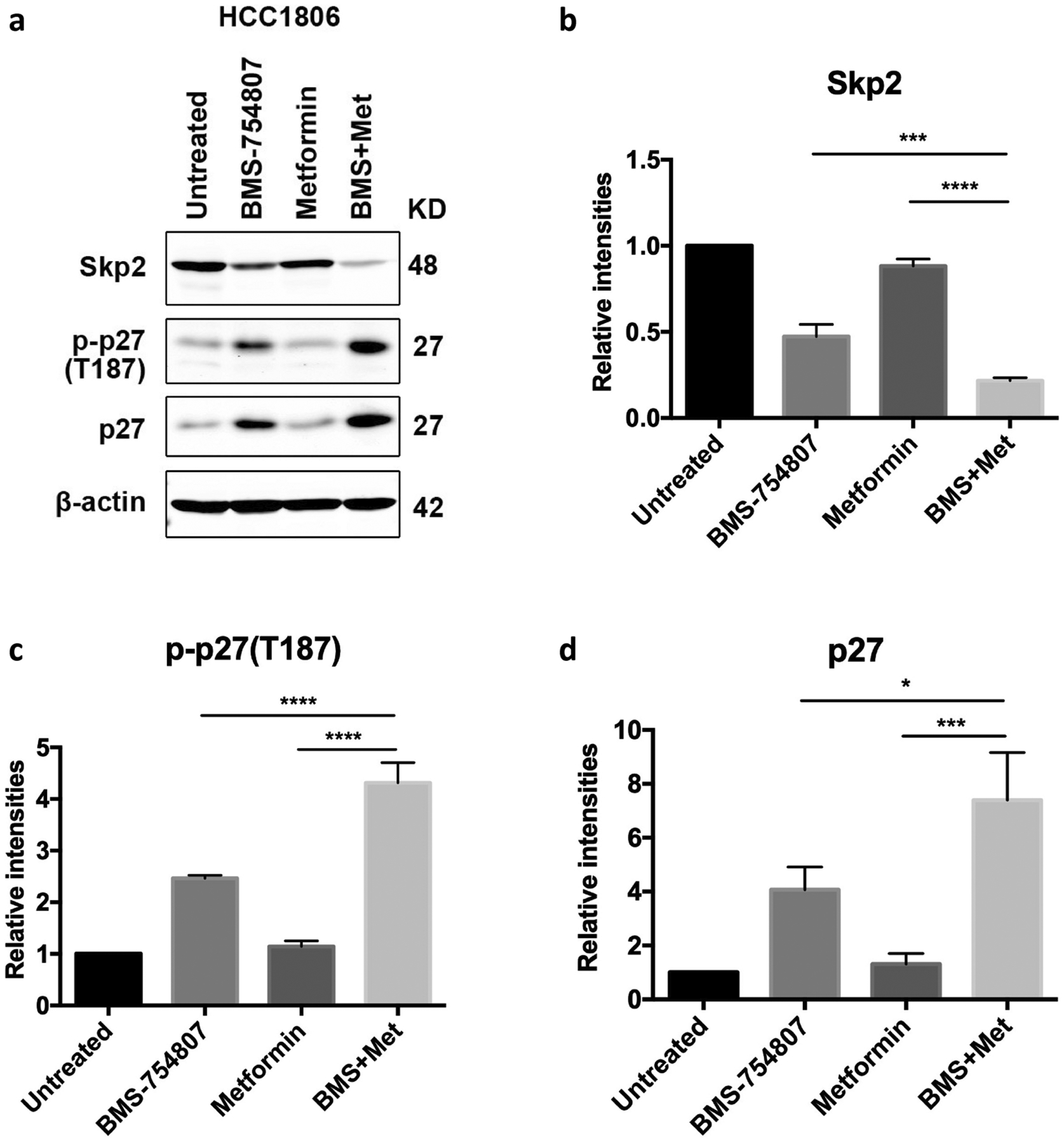
Immunoblots (a) and quantification (b-d) of the indicated proteins in the HCC1806 cell line. Cells were treated for 48 hours with metformin (5mM), BMS-754807 (15μM), or the drug combination.
Discussion
We demonstrate that treatment with the combination of metformin and BMS-754807 is synergistic in inhibiting cell proliferation in 11 out of 13 (85%) TNBC cell lines tested, representing 6 of the 7 known subtypes of TNBC [40]. We found that treatment with either drug alone led to changes of many proteins, but that the combination treatment led to changes of additional proteins and to increased intensities of changes. Notably, the expression levels of both total p27 and p-p27(T187) were increased more when the two drugs were combined. p27 plays a crucial role in suppressing cell cycle progression by blocking the activity of Cyclin/CDK complexes [46,47]. Although rarely mutated, p27 protein levels are commonly decreased in breast cancer and other types of cancer, and low p27 levels are often correlated with poor prognosis [48].
In progressing cancer, the p27 protein level is kept low primarily by CDK-mediated phosphorylation at Thr187 which then can be recognized and ubiquitinated by the Skp2 E3 ligase [49]. Combination treatment with metformin and BMS-754807 led to increased levels of both total and p-p27(T187), excluding inhibition of phosphorylation as the underlying reason for increased levels of total p27. Rather, Skp2 levels were found to be decreased. Downregulation of Skp2, with associated accumulation of p27, has been reported to drive cells into quiescence [50]. On the other hand, overexpression of Skp2 is frequently observed in human cancer and metastases [51]. Therefore, it is likely that the drug-induced suppression of Skp2 protein levels allowed p27 and p-p27 to accumulate in the treated cells. However, it is yet to be determined how Skp2 is reduced by BMS-754807 and further diminished by combination treatment with metformin and BMS-754807.
It also remains to be determined whether increased levels of phosphorylated p27 are entirely due to blockage of the degradation process mediated by Skp2. Enhanced kinase activities may also play a role. CDKs are the primary kinase that phosphorylate p27. AKT, a key downstream component of IGF signaling, has also been reported to phosphorylate p27 (at Thr157/Thr157) and exclude it from the nucleus [52–54]. Src has been reported to phosphorylate p27 (at Y78/88) and target it for degradation [55,56]. But our RPPA analysis did not show an increase in any of these kinases, although we found a decrease of p-Src with an inhibitory phosphorylation at Y527 in cells treated with the drug combination (Table 3). These data suggest that increased levels of p27 phosphorylation are primarily the outcome of blockaded degradation.
Increased levels of p27 may explain why the proportion of unphosphorylated Rb is increased in HCC1806 treated with these two drugs (Table 3). p27 binds to the complex of Cyclin D/CDK4, prevents CDK4 from phosphorylating pRb protein, and increases the fraction of unphosphorylated Rb. Unphosphorylated Rb is the form that is able to bind to and sequester the transcription factor E2F, thereby blocking cell cycle progression [57]. Therefore, this p27-Rb signaling axis may be another mechanism underlying the therapeutic effect of the drug combination.
There are likely other mechanisms by which the combination treatment slows cell growth. In particular, the Wnt signaling pathway appears to be downregulated by the drug combination: the Wnt co-receptor LRP6, p-β-Catenin(S33/37/T41), a classic Wnt signaling target c-Myc and its phosphorylated form p-c-Myc(T58), were all downregulated and they were in the same sub-cluster of the heatmap (Fig 3b), suggesting that they are coordinately controlled. Wnt signaling is known to play a critical role in TNBC formation, progression and therapeutic resistance [58]. Besides the Wnt signaling pathway, other notable proteins that may contribute to the growth inhibitory effects of the drug combination include p-EGFR(Y1068), Aurora, and RRM2 (Ribonucleoside-diphosphate reductase subunit M2). EGFR is amplified and overexpressed in TNBC and is a known core player of TNBC growth and progression [59]. Aurora A dysregulation occurs frequently in many human cancers [60], and inhibition of this kinase has been reported to inhibit TNBC growth [61]. RRM2 high expression predicts worse prognosis in TNBC [62].
In conclusion, we discovered that the combination of metformin and an insulin/IGF-1 inhibitor are synergistic in blocking the proliferation of the majority of the TNBC cell lines tested. One probable major mechanism of this synergy is the significant impact of the drug combination on the Skp2-p27 signaling axis and other proteins regulating the cell cycle and cell proliferation, leading to blocked cell growth. Although chemotherapy remains the mainstay of treatment for TNBC, research into targeted therapies has recently led to the addition of novel agents such as PARP inhibitors and immune checkpoint inhibitors as treatment options for a subset of TNBC patients [63–65]. Our in vitro data call for robust in vivo preclinical evaluation of the combination of metformin and an insulin/IGF-1 inhibitor in treating TNBC, which will provide critical data for a possible clinical trial to test this drug combination in TNBC patients.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported in part by the following grants: Susan G. Komen Foundation CCR13263802 (S.J.), National Institutes of Health R01CA204926 (Y.L) and P50CA186784 Project 3 (Y.L.), Cancer Prevention & Research Institute of Texas Proteomics & Metabolomics Core Facility Support Award (RP170005) (S.H.), and NCI Cancer Center Support Grant to Antibody-based Proteomics Core/Shared Resource (P30CA125123) (S.H.). L.X. was supported in part by a scholarship from the Chinese Scholarship Council (CSC) (Grant number:201406860002). We thank Drs. Kimal Rajapakshe and Cristian Coarfa for RPPA data processing and normalization, and Ms. Fuli Jia and Dr. Danli Wu from the Antibody-based Proteomics Core/Shared Resource for their excellent technical assistant in performing RPPA experiments.
Abbreviations
- TNBC
Triple-negative breast cancer
- IGF-1R
Insulin-like growth factor 1 receptor
- IGF
Insulin-like growth factor
- IR
Insulin receptor
- ER
Estrogen receptor
- PR
Progesterone receptor
- HER2
Human epidermal growth factor receptor 2
- RPPA
Reverse phase protein array
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest: The authors declare that they have no conflict of interest.
Ethical approval: This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, Jemal A, Kramer JL, Siegel RL (2019) Cancer treatment and survivorship statistics, 2019. CA: a cancer journal for clinicians 69 (5):363–385. doi: 10.3322/caac.21565 [DOI] [PubMed] [Google Scholar]
- 2.Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. The New England journal of medicine 363 (20):1938–1948. doi: 10.1056/NEJMra1001389 [DOI] [PubMed] [Google Scholar]
- 3.Yee D (2018) Anti-insulin-like growth factor therapy in breast cancer. Journal of molecular endocrinology 61 (1):T61–T68. doi: 10.1530/JME-17-0261 [DOI] [PubMed] [Google Scholar]
- 4.Lee AV, Yee D (2011) Targeting IGF-1R: at a crossroad. Oncology (Williston Park) 25 (6):535–536; discussion 551 [PMC free article] [PubMed] [Google Scholar]
- 5.Belfiore A, Frasca F (2008) IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia 13 (4):381–406. doi: 10.1007/s10911-008-9099-z [DOI] [PubMed] [Google Scholar]
- 6.Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, Hartwick W, Hoffman B, Hood N (2002) Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 20 (1):42–51. doi: 10.1200/JCO.2002.20.1.42 [DOI] [PubMed] [Google Scholar]
- 7.Papa V, Gliozzo B, Clark GM, McGuire WL, Moore D, Fujita-Yamaguchi Y, Vigneri R, Goldfine ID, Pezzino V (1993) Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res 53 (16):3736–3740 [PubMed] [Google Scholar]
- 8.Osborne CK, Bolan G, Monaco ME, Lippman ME (1976) Hormone responsive human breast cancer in long-term tissue culture: effect of insulin. Proceedings of the National Academy of Sciences of the United States of America 73 (12):4536–4540. doi: 10.1073/pnas.73.12.4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D (2011) RAS oncogenes: weaving a tumorigenic web. Nature reviews Cancer 11 (11):761–774. doi: 10.1038/nrc3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nature reviews Cancer 2 (7):489–501. doi: 10.1038/nrc839 [DOI] [PubMed] [Google Scholar]
- 11.Wittman MD, Carboni JM, Yang Z, Lee FY, Antman M, Attar R, Balimane P, Chang C, Chen C, Discenza L, Frennesson D, Gottardis MM, Greer A, Hurlburt W, Johnson W, Langley DR, Li A, Li J, Liu P, Mastalerz H, Mathur A, Menard K, Patel K, Sack J, Sang X, Saulnier M, Smith D, Stefanski K, Trainor G, Velaparthi U, Zhang G, Zimmermann K, Vyas DM (2009) Discovery of a 2,4-disubstituted pyrrolo[1,2-f][1,2,4]triazine inhibitor (BMS-754807) of insulin-like growth factor receptor (IGF-1R) kinase in clinical development. Journal of medicinal chemistry 52 (23):7360–7363. doi: 10.1021/jm900786r [DOI] [PubMed] [Google Scholar]
- 12.Carboni JM, Wittman M, Yang Z, Lee F, Greer A, Hurlburt W, Hillerman S, Cao C, Cantor GH, Dell-John J, Chen C, Discenza L, Menard K, Li A, Trainor G, Vyas D, Kramer R, Attar RM, Gottardis MM (2009) BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Molecular cancer therapeutics 8 (12):3341–3349. doi: 10.1158/1535-7163.MCT-09-0499 [DOI] [PubMed] [Google Scholar]
- 13.Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE (2012) BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Molecular cancer therapeutics 11 (12):2644–2653. doi: 10.1158/1535-7163.MCT-12-0447 [DOI] [PubMed] [Google Scholar]
- 14.Dayyani F, Parikh NU, Varkaris AS, Song JH, Moorthy S, Chatterji T, Maity SN, Wolfe AR, Carboni JM, Gottardis MM, Logothetis CJ, Gallick GE (2012) Combined Inhibition of IGF-1R/IR and Src family kinases enhances antitumor effects in prostate cancer by decreasing activated survival pathways. PLoS One 7 (12):e51189. doi: 10.1371/journal.pone.0051189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franks SE, Jones RA, Briah R, Murray P, Moorehead RA (2016) BMS-754807 is cytotoxic to non-small cell lung cancer cells and enhances the effects of platinum chemotherapeutics in the human lung cancer cell line A549. BMC research notes 9:134. doi: 10.1186/s13104-016-1919-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang F, Hurlburt W, Greer A, Reeves KA, Hillerman S, Chang H, Fargnoli J, Graf Finckenstein F, Gottardis MM, Carboni JM (2010) Differential mechanisms of acquired resistance to insulin-like growth factor-i receptor antibody therapy or to a small-molecule inhibitor, BMS-754807, in a human rhabdomyosarcoma model. Cancer Res 70 (18):7221–7231. doi: 10.1158/0008-5472.CAN-10-0391 [DOI] [PubMed] [Google Scholar]
- 17.Litzenburger BC, Creighton CJ, Tsimelzon A, Chan BT, Hilsenbeck SG, Wang T, Carboni JM, Gottardis MM, Huang F, Chang JC, Lewis MT, Rimawi MF, Lee AV (2011) High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin Cancer Res 17 (8):2314–2327. doi: 10.1158/1078-0432.CCR-10-1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desai J, Solomon BJ, Davis ID, Lipton LR, Hicks R, Scott AM, Park J, Clemens PL, Gestone TA, Finckenstein FG (2010) Phase I dose-escalation study of daily BMS-754807, an oral, dual IGF-1R/insulin receptor (IR) inhibitor in subjects with solid tumors. Journal of Clinical Oncology 28 (15_suppl):3104–3104. doi: 10.1200/jco.2010.28.15_suppl.310420516430 [DOI] [Google Scholar]
- 19.Kolb EA, Gorlick R, Lock R, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Billups C, Smith MA, Houghton PJ (2011) Initial testing (stage 1) of the IGF-1 receptor inhibitor BMS-754807 by the pediatric preclinical testing program. Pediatric blood & cancer 56 (4):595–603. doi: 10.1002/pbc.22741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunt TW, Mariani GL (2013) Novel approaches for molecular targeted therapy of breast cancer: interfering with PI3K/AKT/mTOR signaling. Current cancer drug targets 13 (2):188–204. doi: 10.2174/1568009611313020008 [DOI] [PubMed] [Google Scholar]
- 21.Zardavas D, Baselga J, Piccart M (2013) Emerging targeted agents in metastatic breast cancer. Nature reviews Clinical oncology 10 (4):191–210. doi: 10.1038/nrclinonc.2013.29 [DOI] [PubMed] [Google Scholar]
- 22.Bailey CJ, Turner RC (1996) Metformin. The New England journal of medicine 334 (9):574–579. doi: 10.1056/NEJM199602293340906 [DOI] [PubMed] [Google Scholar]
- 23.Salpeter SR, Buckley NS, Kahn JA, Salpeter EE (2008) Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. The American journal of medicine 121 (2):149–157 e142. doi: 10.1016/j.amjmed.2007.09.016 [DOI] [PubMed] [Google Scholar]
- 24.Pernicova I, Korbonits M (2014) Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol 10 (3):143–156. doi: 10.1038/nrendo.2013.256 [DOI] [PubMed] [Google Scholar]
- 25.Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell metabolism 1 (1):15–25. doi: 10.1016/j.cmet.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 26.Rattan R, Giri S, Singh AK, Singh I (2005) 5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem 280 (47):39582–39593. doi: 10.1074/jbc.M507443200 [DOI] [PubMed] [Google Scholar]
- 27.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD (2005) Metformin and reduced risk of cancer in diabetic patients. Bmj 330 (7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D (2010) Diabetes and cancer: a consensus report. Diabetes care 33 (7):1674–1685. doi: 10.2337/dc10-0666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, Gennari A, Trabacca MS, Galimberti V, Veronesi P, Johansson H, Aristarco V, Bassi F, Luini A, Lazzeroni M, Varricchio C, Viale G, Bruzzi P, Decensi A (2012) Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 30 (21):2593–2600. doi: 10.1200/JCO.2011.39.3769 [DOI] [PubMed] [Google Scholar]
- 30.Bayraktar S, Hernadez-Aya LF, Lei X, Meric-Bernstam F, Litton JK, Hsu L, Hortobagyi GN, Gonzalez-Angulo AM (2012) Effect of metformin on survival outcomes in diabetic patients with triple receptor-negative breast cancer. Cancer 118 (5):1202–1211. doi: 10.1002/cncr.26439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD (2009) Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle 8 (13):2031–2040. doi: 10.4161/cc.8.13.8814 [DOI] [PubMed] [Google Scholar]
- 32.Lee JO, Kang MJ, Byun WS, Kim SA, Seo IH, Han JA, Moon JW, Kim JH, Kim SJ, Lee EJ, In Park S, Park SH, Kim HS (2019) Metformin overcomes resistance to cisplatin in triple-negative breast cancer (TNBC) cells by targeting RAD51. Breast cancer research : BCR 21 (1):115. doi: 10.1186/s13058-019-1204-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi P, Liu W, Tala, Wang H, Li F, Zhang H, Wu Y, Kong Y, Zhou Z, Wang C, Chen W, Liu R, Chen C (2017) Metformin suppresses triple-negative breast cancer stem cells by targeting KLF5 for degradation. Cell discovery 3:17010. doi: 10.1038/celldisc.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strekalova E, Malin D, Rajanala H, Cryns VL (2017) Metformin sensitizes triple-negative breast cancer to proapoptotic TRAIL receptor agonists by suppressing XIAP expression. Breast cancer research and treatment 163 (3):435–447. doi: 10.1007/s10549-017-4201-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N (2007) Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 67 (22):10804–10812. doi: 10.1158/0008-5472.CAN-07-2310 [DOI] [PubMed] [Google Scholar]
- 36.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M (2006) Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66 (21):10269–10273. doi: 10.1158/0008-5472.CAN-06-1500 [DOI] [PubMed] [Google Scholar]
- 37.Creighton CJ, Huang S (2015) Reverse phase protein arrays in signaling pathways: a data integration perspective. Drug design, development and therapy 9:3519–3527. doi: 10.2147/DDDT.S38375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chou TC (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70 (2):440–446. doi: 10.1158/0008-5472.CAN-09-1947 [DOI] [PubMed] [Google Scholar]
- 39.Bu W, Liu Z, Jiang W, Nagi C, Huang S, Edwards DP, Jo E, Mo Q, Creighton CJ, Hilsenbeck SG, Leavitt AD, Lewis MT, Wong STC, Li Y (2019) Mammary Precancerous Stem and Non-Stem Cells Evolve into Cancers of Distinct Subtypes. Cancer Res 79 (1):61–71. doi: 10.1158/0008-5472.CAN-18-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. The Journal of clinical investigation 121 (7):2750–2767. doi: 10.1172/JCI45014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Podust VN, Brownell JE, Gladysheva TB, Luo RS, Wang C, Coggins MB, Pierce JW, Lightcap ES, Chau V (2000) A Nedd8 conjugation pathway is essential for proteolytic targeting of p27Kip1 by ubiquitination. Proceedings of the National Academy of Sciences of the United States of America 97 (9):4579–4584. doi: 10.1073/pnas.090465597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vlach J, Hennecke S, Amati B (1997) Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 16 (17):5334–5344. doi: 10.1093/emboj/16.17.5334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carrano AC, Eytan E, Hershko A, Pagano M (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature cell biology 1 (4):193–199 [DOI] [PubMed] [Google Scholar]
- 44.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H (1999) p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Current biology : CB 9 (12):661–664. doi: 10.1016/s0960-9822(99)80290-5 [DOI] [PubMed] [Google Scholar]
- 45.Nakayama KI, Nakayama K (2006) Ubiquitin ligases: cell-cycle control and cancer. Nature reviews Cancer 6 (5):369–381. doi: 10.1038/nrc1881 [DOI] [PubMed] [Google Scholar]
- 46.Chu IM, Hengst L, Slingerland JM (2008) The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nature reviews Cancer 8 (4):253–267. doi: 10.1038/nrc2347 [DOI] [PubMed] [Google Scholar]
- 47.Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ (1998) The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 396 (6707):177–180. doi: 10.1038/24179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blain SW, Scher HI, Cordon-Cardo C, Koff A (2003) p27 as a target for cancer therapeutics. Cancer Cell 3 (2):111–115. doi: 10.1016/s1535-6108(03)00026-6 [DOI] [PubMed] [Google Scholar]
- 49.Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, Malek NP (2004) Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes & development 18 (21):2602–2607. doi: 10.1101/gad.321004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M (2004) Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428 (6979):190–193. doi: 10.1038/nature02330 [DOI] [PubMed] [Google Scholar]
- 51.Pagano M, Benmaamar R (2003) When protein destruction runs amok, malignancy is on the loose. Cancer Cell 4 (4):251–256. doi: 10.1016/s1535-6108(03)00243-5 [DOI] [PubMed] [Google Scholar]
- 52.Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E, Slingerland JM (2002) PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nature medicine 8 (10):1153–1160 [DOI] [PubMed] [Google Scholar]
- 53.Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, Arteaga CL (2002) PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nature medicine 8 (10):1145–1152 [DOI] [PubMed] [Google Scholar]
- 54.Viglietto G, Motti ML, Bruni P, Melillo RM, D’Alessio A, Califano D, Vinci F, Chiappetta G, Tsichlis P, Bellacosa A, Fusco A, Santoro M (2002) Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nature medicine 8 (10):1136–1144 [DOI] [PubMed] [Google Scholar]
- 55.Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, Jakel H, Kullmann M, Kriwacki RW, Hengst L (2007) Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 128 (2):269–280. doi: 10.1016/j.cell.2006.11.047 [DOI] [PubMed] [Google Scholar]
- 56.Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, Sun P, Tan CK, Hengst L, Slingerland J (2007) p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 128 (2):281–294. doi: 10.1016/j.cell.2006.11.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murphree AL, Benedict WF (1984) Retinoblastoma: clues to human oncogenesis. Science 223 (4640):1028–1033. doi: 10.1126/science.6320372 [DOI] [PubMed] [Google Scholar]
- 58.Lindvall C, Bu W, Williams BO, Li Y (2007) Wnt signaling, stem cells, and the cellular origin of breast cancer. Stem cell reviews 3 (2):157–168 [DOI] [PubMed] [Google Scholar]
- 59.Nakai K, Hung MC, Yamaguchi H (2016) A perspective on anti-EGFR therapies targeting triple-negative breast cancer. American journal of cancer research 6 (8):1609–1623 [PMC free article] [PubMed] [Google Scholar]
- 60.Crane R, Gadea B, Littlepage L, Wu H, Ruderman JV (2004) Aurora A, meiosis and mitosis. Biol Cell 96 (3):215–229. doi: 10.1016/j.biolcel.2003.09.008 [DOI] [PubMed] [Google Scholar]
- 61.Xu J, Wu X, Zhou WH, Liu AW, Wu JB, Deng JY, Yue CF, Yang SB, Wang J, Yuan ZY, Liu Q (2013) Aurora-A identifies early recurrence and poor prognosis and promises a potential therapeutic target in triple negative breast cancer. PLoS One 8 (2):e56919. doi: 10.1371/journal.pone.0056919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang H, Liu X, Warden CD, Huang Y, Loera S, Xue L, Zhang S, Chu P, Zheng S, Yen Y (2014) Prognostic and therapeutic significance of ribonucleotide reductase small subunit M2 in estrogen-negative breast cancers. BMC cancer 14:664. doi: 10.1186/1471-2407-14-664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, Wu W, Goessl C, Runswick S, Conte P (2017) Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. The New England journal of medicine 377 (6):523–533. doi: 10.1056/NEJMoa1706450 [DOI] [PubMed] [Google Scholar]
- 64.Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, Fehrenbacher L, Yerushalmi R, Mina LA, Martin M, Roche H, Im YH, Quek RGW, Markova D, Tudor IC, Hannah AL, Eiermann W, Blum JL (2018) Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. The New England journal of medicine 379 (8):753–763. doi: 10.1056/NEJMoa1802905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA (2018) Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. The New England journal of medicine 379 (22):2108–2121. doi: 10.1056/NEJMoa1809615 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.