

Abstract
Mechanical deformations of DNA such as bending are ubiquitous and implicated in diverse cellular functions1. However, the lack of high-throughput tools to directly measure the mechanical properties of DNA limits our understanding of whether and how DNA sequences modulate DNA mechanics and associated chromatin transactions genome-wide. We developed an assay called loop-seq to measure the intrinsic cyclizability of DNA – a proxy for DNA bendability – in high throughput. We measured the intrinsic cyclizabilities of 270,806 50 bp DNA fragments that span the entire length of S. cerevisiae chromosome V and other genomic regions, and also include random sequences. We discovered sequence-encoded regions of unusually low bendability upstream of Transcription Start Sites (TSSs). These regions disfavor the sharp DNA bending required for nucleosome formation and are co-centric with known Nucleosome Depleted Regions (NDRs). We show biochemically that low bendability of linker DNA located about 40 bp away from a nucleosome edge inhibits nucleosome sliding into the linker by the chromatin remodeler INO80. The observation explains how INO80 can create promoter-proximal nucleosomal arrays in the absence of any other factors2 by reading the DNA mechanical landscape. We show that chromosome wide, nucleosomes are characterized by high DNA bendability near dyads and low bendability near the linkers. This contrast increases for nucleosomes deeper into gene bodies, suggesting that DNA mechanics plays a previously unappreciated role in organizing nucleosomes far from the TSS, where nucleosome remodelers predominate. Importantly, random substitution of synonymous codons does not preserve this contrast, suggesting that the evolution of codon choice has been impacted by selective pressure to preserve sequence-encoded mechanical modulations along genes. We also provide evidence that transcription through the TSS-proximal nucleosomes is impacted by local DNA mechanics. Overall, this first genome-scale map of DNA mechanics hints at a ‘mechanical code’ with broad functional implications.
DNA mechanics in high throughput
DNA looping (or cyclization) assays have long been used to measure DNA bendability3,4. Recently, a single molecule Fluorescence Resonance Energy Transfer5 (smFRET)-based DNA looping assay was developed6, whereby looping of a ~100 basepair (bp) DNA duplex flanked by complementary 10 nucleotide (nt) single-stranded overhangs is detected via an increase in FRET between fluorophores located at each end of DNA (Fig. 1a). The looping rate thus obtained has been interpreted as a measure of DNA bendability. In this assay, chemically synthesized single strands of DNA had to be annealed directly without PCR amplification to generate a duplex region flanked by long 10 nt overhangs. We simplified the process by developing a nicking-based method that allows the in situ conversion of a 120 bp duplex DNA (which can be produced via PCR amplification) into a 100 bp duplex flanked by 10 nt single-stranded overhangs (Fig. 1b). Using FRET6, we measured the looping times of ten DNA fragments with different sequences (Supplementary Note 1). The looping times varied by more than an order of magnitude (Fig. 1c), confirming the previously reported result that DNA sequence can have a profound effect on DNA looping at the ~100 bp length scale6,7. However, looping assays and all previous methods to directly measure DNA bendability have limited throughput, which greatly limits our knowledge of how DNA mechanics is modulated by sequence, varies along genomes, and influences chromosome transactions.
Figure 1: A high-throughout method to measure DNA mechanics.
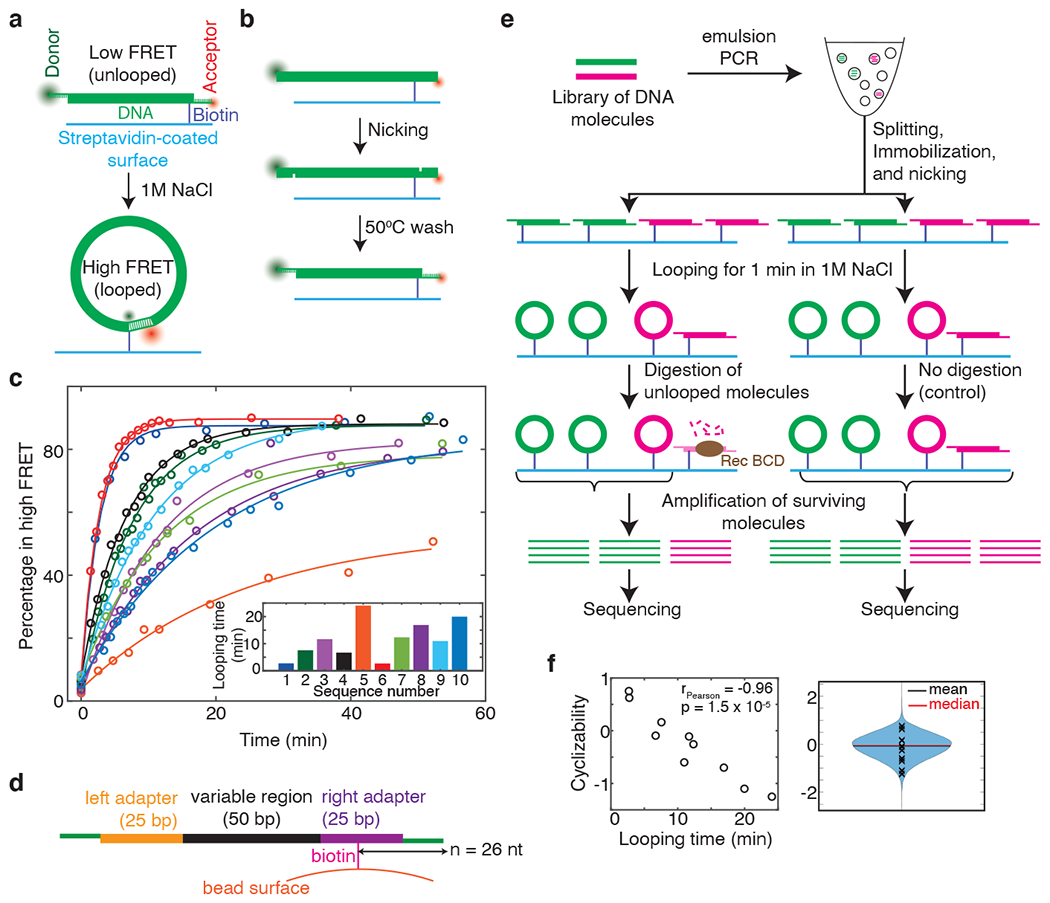
a, Schematic of the single-molecule looping assay. b, In situ nicking of 120 bp duplex DNA 10 nt from either end, following by washing with buffer at 50 °C, results in 100 bp duplex molecules flanked by 10 nt single-stranded overhangs (Supplementary Note 1). c, Percentage of DNA molecules in the high FRET (looped) state as a function of time after adding high salt, for 10 DNA sequences (Supplementary Note 1). Looping times (time constants of exponential decay fits (solid lines)) are displayed in the bar plot. (d) Schematic of a typical DNA fragment in a library just prior to looping. ‘n’ is the distance in nucleotides of the biotin tether from the end of the molecule (e) Schematic of loop-seq performed on a hypothetical library comprising only two sequences – green and pink. The library is amplified, immobilized on beads via biotin-streptavidin interactions and nicked in situ to generate loopable molecules (Fig. 1d, Methods). After looping for 1 minute in high salt followed by the digestion of unlooped molecules and the amplification of surviving molecules, the relative populations of green and pink in the digested fraction are 2/3 and 1/3 respectively. The corresponding values in the control fraction are 1/2 and 1/2. The cyclizability of green and pink are thus and respectively. (f) Left panel: Cyclizabilities of 10 sequences (listed in Supplementary Note 1) which were part of the Cerevisiae Nucleosomal Library (Supplementary Note 4) vs their looping times obtained via smFRET (Fig. 1c). Pearson’s r = −0.96. 95% CI = [−0.99, −0.81]. p = 1.5E-5 (t-test, two-sided). Right panel: Violin plot of the cyclizabilities of all 19,907 sequences in the Cerevisiae Nucleosomal Library. Cyclizabilities of sequences whose looping times were measured via smFRET are indicated by ‘x’.
Systematic Enrichment of Ligands by Exponential Enrichment (SELEX) has been used to enrich DNA sequences that are more bent8 or more loopable9 through many rounds of selection of rapidly looping DNA from a vast random pool of loopable molecules and PCR amplification of selected molecules. These assays revealed, for example, certain periodic dinucleotide distributions found in the variable regions of highly loopable DNA. However, direct bendability measurements of specified sequences of interest, such as those than span genomic regions, have never been reported in high throughput.
In order to extend direct DNA bendability measurements to a much larger sequence space, we established a sequencing-based approach termed loop-seq, which builds on previous low-throughput single-molecule looping6 and SELEX selection methods9. Using the nicking approach, we generated a library of up to ~90,000 different specified template sequences immobilized on streptavidin-coated beads. Library members had a central 50 bp duplex region of variable sequence flanked by 25 bp left and right duplex adapters and 10 nt single-stranded complementary overhangs (Fig. 1d). Looping was initiated in high salt for 1 minute, after which unlooped DNA molecules were digested with an exonuclease9 (RecBCD) that requires free DNA ends, thus preserving the looped molecules. The enriched library was sequenced, and the cyclizability of each sequence was defined as the natural logarithm of the ratio of the relative population of that sequence in the enriched library to that in an identically treated control in which only the digestion step was omitted (Fig. 1e, Supplementary Note 2).
The looping times of the 10 sequences determined via smFRET (Fig. 1c) were strongly anti-correlated with their cyclizability values obtained by performing loop-seq on a large library containing those 10 (along with 19,897 other) sequences (Fig. 1f). This confirmed that cyclizability is a good measure of looping rate. Additionally, varying the time for which looping is permitted before RecBCD digestion allowed for the measurement of the full looping kinetic curves of all sequences in the library (‘Timecourse loop-seq’, Supplementary Note 3 and Extended Data Fig. 1). The looped population could comprise closed structures with alternate shapes and basepairing geometries10,11 (Extended Data Fig. 2). However, irrespective of looped geometry, control experiments indicate that most looped molecules are protected from RecBCD digestion, and also serve to validate several other aspects of the assay (Extended Data Fig. 3).
We found that the distance of the biotin tether from the end of each molecule (n, Fig. 1d) imposed an oscillatory modulation on cyclizability, possibly owing to a sequence-dependent preference for the rotational orientation of the biotin tether (Supplementary Note 7). By varying n and performing loop-seq multiple times, we measured the mean, amplitude, and phase associated with this oscillation for every library sequence. We called the mean term the “intrinsic cyclizability” and showed that it is independent of the tethering geometry and rotational phasing (Supplementary Notes 7–8, Extended Data Fig. 4). Both dynamic flexibility and static bending may contribute to intrinsic cyclizability. Regardless of interpretation, intrinsic cyclizability is a measurable mechanical property that can be compared to functional properties of chromosomal DNA.
DNA at yeast NDRs is rigid
We used loop-seq to query the role of DNA mechanical properties in establishing characteristic features of genes that regulate expression, such as Nucleosome Depleted Regions (NDRs) upstream of the Transcription Start Sites (TSSs) and well-ordered arrays of downstream nucleosomes positioned at characteristic distances from the TSSs12. Although several lines of evidence had suggested that DNA mechanics, in addition to transcription factors and chromatin remodelers2,13,14, plays a role in this regard by modulating nucleosome organization2,9,15,16, the mechanical properties of DNA along promoters and genes have never been directly measured. We measured the intrinsic cyclizabilities of DNA fragments (‘Tiling Library’, Supplementary Note 9) that tile the region from 600 bp upstream to 400 bp downstream of the +1 nucleosome dyads of 576 genes in S. cerevisiae at 7 bp resolution (Fig. 2a). We discovered a sharply defined region of rigid DNA (i.e. with unusually low intrinsic cyclizability) located in the NDR17 (Fig. 2b). Further, there are many genes where our measurements are sensitive enough to detect this region of high rigidity without the need to average across multiple genes (Fig. 2c, Extended Data Fig. 5). As nucleosome assembly requires extensive DNA bending, the low intrinsic cyclizability of DNA around the NDR is likely to favor nucleosome depletion.
Figure 2: DNA mechanics contributes to nucleosome depletion at the NDR and modulates remodeler activities.
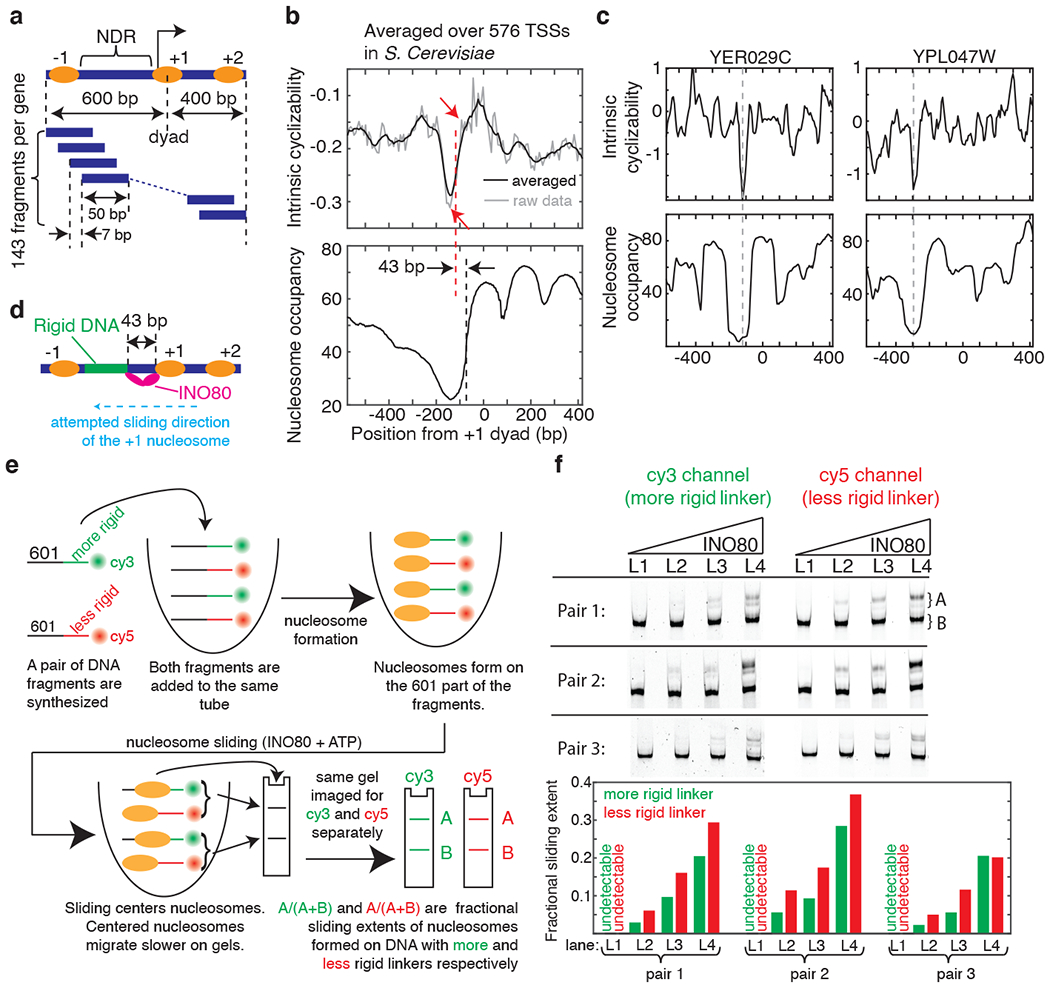
(a) Schematic of the Tiling Library. The regions around the TSSs of 576 genes were tiled at 7 bp resolution (Supplementary Note 9). DNA: blue bars, nucleosomes: ovals. (b) Mean intrinsic cyclizability (with (black) and without (grey) any smoothening) and nucleosome occupancy17 vs position from the canonical location32 of the dyad of the +1 nucleosome, averaged over all 576 genes in the Tiling Library. Blue dashed line (at −73 bp): edge of the +1 nucleosome, red dashed line: start of the rigid DNA region (approximated as the midpoint between the two red arrows). See supplementary note 10. (c) Intrinsic cyclizability and nucleosome occupancy vs position from the dyads of the +1 nucleosomes of two individual genes (see Extended Data Fig. 5 for more examples). (d) INO80 attempting to slide a +1 nucleosome upstream of its canonical location would be poised to contact the rigid DNA region via its Arp821,23. (e) Schematic of the experiment comparing the extent of nucleosome sliding by INO80 on a pair of constructs comprising a nucleosome attached to a rigid or flexible linker region, and distinguished by different fluorophores. Sliding results in centered nucleosomes, which migrate more slowly23. See Supplementary Note 11. (f) Three nucleosome sliding experiments were performed involving three pairs of nucleosome constructs as described in figure 2e and supplementary note 11. For each pair, four [INO80] were used (lanes L1 – L4): 2, 6, 9, and 13 nM. Post sliding, nucleosomes were run along a 6% TBE gel, which was imaged separately for Cy3 and Cy5 fluorescence. For each lane ([INO80]), the nucleosome sliding extents in the two constructs in the pair were quantified (bar plots). See figure 2e and supplementary note 11.
Chromatin remodelers sense DNA mechanics
Chromatin remodelers have been proposed to be critical in establishing the well-ordered array of nucleosomes downstream of TSSs by stacking nucleosomes against a barrier just upstream of the TSS14. What could constitute such a barrier has been a matter of debate, and transcription factors13 and paused polymerases18 have been suggested to contribute. Notably, in vitro chromatin reconstitution experiments2 showed that the remodeler INO80 can both position the +1 (and −1) nucleosomes and establish the NDRs in S. cerevisiae even in the absence of any such factors. We therefore asked whether the sequence-encoded rigid DNA region in the NDR can contribute to nucleosome positioning near promoters by serving as a barrier to the sliding activities of INO80.
To effect sliding, INO80 requires at least 40 – 50 bp of free extranucleosomal DNA ahead of the nucleosome19,20. The region around 40 – 50 bp ahead of the sliding nucleosome’s edge is enganged by the Arp8 module of INO8021–23, and disrupting the module’s DNA binding via mutation abolishes sliding and reduces +1 positioning genome-wide23. Intriguingly, we found that the region of rigid DNA also starts ~43 bp upstream of the edge of the +1 nucleosome (Fig. 2b). This would place the Arp8 module in contact with highly rigid DNA if INO80 were to slide the +1 nucleosome upstream from its canonical position (Fig. 2d). If highly rigid DNA interferes with Arp8 module binding, further upstream sliding of the +1 nucleosome would be hindered, helping position the +1 nucleosome and define the NDR.
To directly test the role of upstream DNA rigidity in +1 nucleosome positioning by INO80, we biochemically measured the effect of rigid DNA located ~40 bp ahead of a nucleosome on sliding by INO80. Using gel shift, we assayed sliding of nucleosomes formed on the 147 bp 601 sequence into adjacent 80 bp linkers. We chose three pairs of such constructs, each containing one construct with a linker that was uniformly flexible and another with a linker that had a significantly more rigid region near the middle (Supplementary Note 11, Extended Data Fig. 6a). In all three pairs, the extent of sliding (Supplementary Note 11) was lower for the nucleosome formed on the construct with the rigid linker (Fig. 2f, Extended Data Figs. 6b–c, 7). Various factors could cause this reduced sliding (Supplementary Note 11). Regardless, the observation is consistent with a model where the rigid DNA region starting ~43 bp upstream of the canonical +1 nucleosome’s edge (Fig. 2b) serves as a barrier that hinders further upstream sliding of the +1 nucleosome by INO80, possibly aided by other barriers set up by factors such as RSC, gene regulatory factors, and transcription factors2,13. Structural details behind rigidity sensing by the Arp8 module must await future studies.
DNA mechanics arranges nucleosomes
As nucleosomes involve extensive DNA bending, we asked if modulations in intrinsic cyclizability may directly contribute to nucleosome organization, in addition to the stacking action of remodelers14. Indeed, DNA at the canonical dyad locations of the +/−1 nucleosomes, and to a lesser extent the +2, +3 and +4 nucleosomes, have significantly higher intrinsic cyclizability than surrounding DNA (Fig. 2b). Consistent with this observation, promoters classified as having a fragile −1 nucleosome24 have more rigid DNA at the location of the −1 nucleosome (Fig. 3a).
Figure 3. DNA mechanics impacts chromosome-wide nucleosome organization.
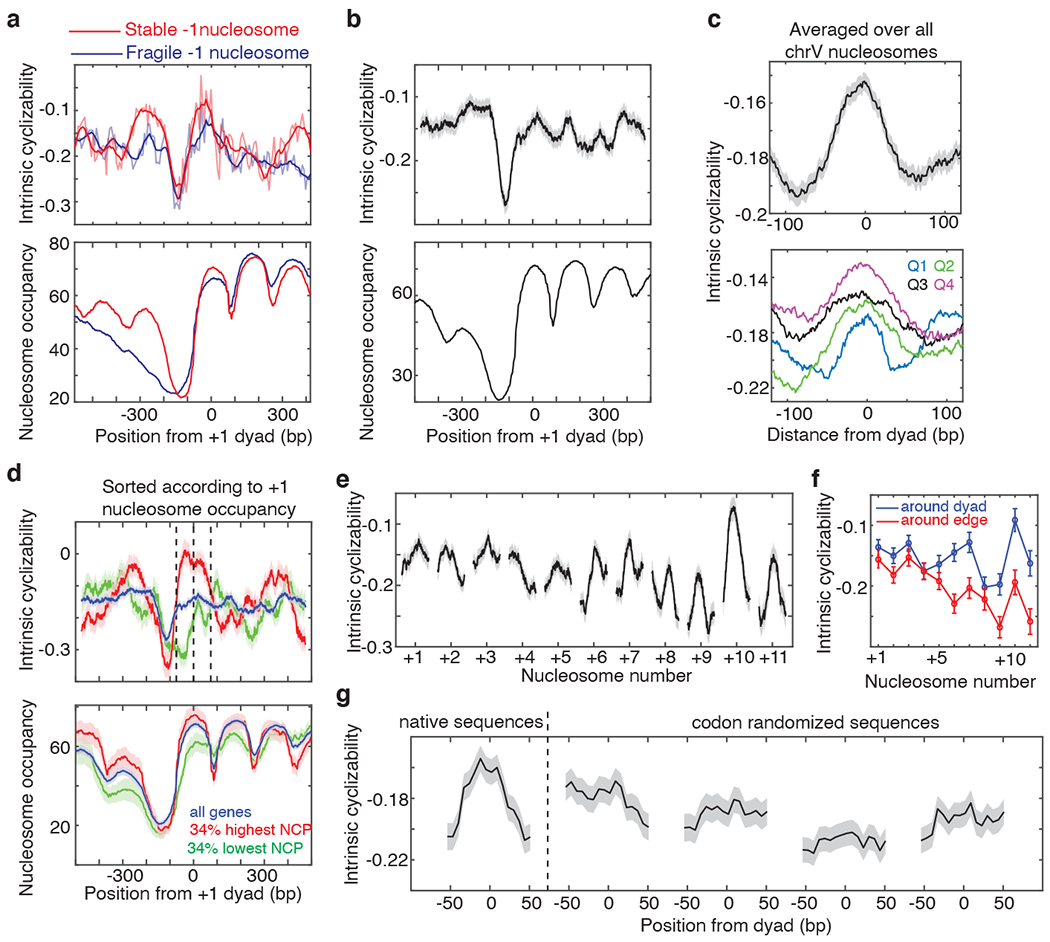
Mean intrinsic cyclizability and nucleosome occupancy vs position from the dyad of the +1 nucleosome, (a) averaged over 185 and 345 genes in the Tiling Library which possess stable and fragile −1 nucleosomes respectively24 (plotted as in Fig 2b), and (b) averaged over all 227 identified genes along S. cerevisiae chromosome V (Supplementary Note 13). Grey background: s.e.m. (c) Intrinsic cyclizability vs position from the dyad, averaged over all 3,192 nucleosomes along chromosome V (top), or over nucleosomes sorted into quartiles based on reported NCP scores32 (bottom). See also Extended Data Fig. 8a–b. Solid lines: mean, grey background: s.e.m. (d) Intrinsic cyclizability and nucleosome occupancy (solid lines) vs position along all chromosome V genes (blue), and among 34% of genes with the highest (red) and lowest (green) +1 nucleosome NCP scores32. Plots were obtained as in panel b. Dashed lines: edges and dyad of the +1 nucleosome. Shaded backgrounds: s.e.m. (e) Intrinsic cyclizability around nucleosomal dyads that lie within the transcribed region of all identified 227 genes along chromosome V in S. cerevisiae. Solid lines: mean, grey background: s.e.m. See supplementary Note 13 for N values. (f) Mean and s.e.m. of intrinsic cyclizability in a 50 bp window around the dyads (blue) and edges (red, from position −73 till −56 and from +56 till +73) of gene-body nucleosomes. N values are the same as in panel e. (g) Intrinsic cyclizability of the native sequences around the dyads of the 500 +7 nucleosomes represented in Library L (supplementary note 14), and along four sets of codon-altered sequences generated by randomly selecting synonymous codons while considering (the first two) or not considering (the next two) the natural codon-usage frequency. Solid line: mean, smoothened over a 7-fragment rolling window, grey background: s.e.m.
Several earlier studies have shed light on the role of DNA mechanics in nucleosome formation25. The fact that bendable DNA forms good substrates for nucleosomes and vice versa, has been demonstrated for various selected sequences4,26–30. Further, DNA selected for high loopability from a large random pool possess periodic distribution in dinucleotide contents9, which is also a feature found in ~3% of native yeast nucleosomal sequences31. However, the mechanical properties of known nucleosomal DNA sequences have never been directly measured in high throughput. To achieve this for nucleosomes along an entire yeast chromosome, we measured intrinsic cyclizability along S. cerevisiae chromosome V at 7 bp resolution (‘ChrV Library’, Supplementary Note 12, Extended Data Fig. 8). We first confirmed that intrinsic cyclizability shows the characteristic pronounced dip around the NDR when averaged over the 227 genes along chromosome V that have both ends mapped with high confidence25 (Fig. 3b). We found that chromosome-wide, DNA at nucleosomal dyad locations tends to have significantly higher intrinsic cyclizability than the surrounding linker DNA (Fig. 3c), suggesting that sequence-dependent modulations in DNA mechanics contribute to global nucleosome organization. We also found that nucleosomes are better positioned in vivo on more intrinsically cyclizable DNA (Fig. 3c, Extended Data Fig. 9a–c). Among TSS proximal nucleosomes, the correlation is strongest for +1 nucleosomes (Fig. 3d, Extended Data Fig. 9d–e).
Among nucleosomes that lie along transcribed regions, TSS-distal nucleosomes have a higher intrinsic cyclizability contrast between the dyad and the edges than TSS-proximal nucleosomes (Figs. 3e–f). This was contrary to expectation because TSS-proximal nucleosomes are known to be better positioned than TSS-distal nucleosomes14,32. TSS-proximal nucleosomes are likely primarily organized by chromatin remodelers into ordered arrays via stacking against the NDR barrier2,14. However, beyond the +4 nucleosome, the stacking effect has been shown to dissipate14, whereas our data shows that modulations in intrinsic cyclizability become more prominent (Fig. 3e–f). Thus nucleosomes that lie deeper in gene-bodies may rely more on sequence-encoded intrinsic cyclizability modulations for positioning.
DNA mechanics impacts codon selection
We next asked whether the strong modulation in intrinsic cyclizability for nucleosomes deep in the gene body would be preserved if the sequences were altered by using alternate codons that code for the same amino acids. We selected 500 +7 nucleosomes in S. cerevisiae and generated four sets of codon-altered sequences spanning the region around these nucleosomes, while preserving the amino acid sequences encoded. The natural codon usage frequency was considered when choosing synonymous codons in the first two sets, and was ignored in the next two (supplementary note 14). By performing loop-seq (‘Library L’, supplementary note 14), we measured intrinsic cyclizability at 7 bp resolution in the 200 bp region flanking the 500 +7 nucleosome dyads and their codon-altered sequences. Native sequences have a characteristic intrinsic cyclizability pattern – high near the dyads and low near the edges – which is absent in the four codon-altered sets (Fig. 3g). Thus naturally occurring codons are optimized to establish sequence-dependent intrinsic cyclizability modulations along genes that are favorable to the organization of gene body nucleosomes, suggesting that the evolution of codon choice in S. cerevisiae has been impacted by a selective pressure to preserve such modulations. The observation also points to a hitherto unappreciated significance of positioning nucleosomes that lie deeper in the gene body.
TSS-proximal nucleosomes are asymmetric
Several critical processes such as transcription and DNA replication require nucleosome unraveling. DNA could potentially peel off from either end, in a manner modulated by bendability. Indeed asymmetry in DNA bendability across the 601 nucleosome leads to asymmetric unraveling under tension33. Biochemical analysis has shown that yeast RNA polymerase II transcribing a 601 nucleosome produces four times more full-length transcripts when it enters the nucleosome through the ‘TA-rich’ side that contains the phased TA repeats34. Using loop-seq, we also found that the TA-rich side has significantly higher intrinsic cyclizability (Fig. 4a, Supplementary Note 18). This observation is consistent with an idea that RNA polymerase might better negotiate with a nucleosomal barrier when it first interacts with the side of the nucleosome containing DNA with higher intrinsic cyclizability. We constructed a library containing the 50 bp DNA fragments immediately to the left and right of the dyads of ~10,000 well-positioned S. cerevisiae nucleosomes (‘Cerevisiae Nucleosomal Library’, Supplementary Note 4, Fig. 4b). We found that DNA at well-occupied +1 and +2 nucleosomes indeed has, on average, higher intrinsic cyclizability on the promoter-proximal face than the distal face (Fig. 4c), thereby suggesting that this asymmetry may favor polymerase translocation. Consistently, this asymmetry is accentuated among the highly expressed genes and absent among poorly expressed genes (Fig. 4d).
Figure 4: TSS-proximal nucleosomes are asymmetric.

(a) Black: Intrinsic cyclizability as a function of position along 601 DNA (supplementary note 18) Blue: abscissa value of the solid horizontal line (−0.196) is the mean intrinsic cyclizability along the 500 native +7 nucleosomal sequences represented in Library L (supplementary note 14). The height of the light blue background (0.011) is twice the s.e.m. (b) Schematic representing the design of the Cerevisiae Nucleosomal Library (supplementary note 4). The library contains DNA fragments taken from the 50 bp immediately to the left and right of the dyads of the ~10,000 nucleosomes in S. cerevisiae that have the highest NCP scores. (c) Mean intrinsic cyclizabilities of the 50 bp DNA fragments that lie immediately adjacent to the TSS proximal (red) or distal (blue) side of the dyads of various categories of nucleosomes (−9 through +9) (see supplementary note 17). Error bars are s.e.m. See supplementary Note 17 for the number of nucleosomes in each category over which data was averaged. (d) A subset of the data as in panel c, where the means were calculated considering only genes among the 10% most (left panel) or least (middle panel) expressed in S. cerevisiae. The right panel is identical to panel b, except for an altered y-axis scale. Error bars are s.e.m. See supplementary note 17.
Conclusions
Intrinsic cyclizability is, thus far, the only mechanical property of DNA to be directly measured in high throughput, and will likely aid our understanding of how DNA mechanics influences chromatin transactions involving diverse factors such as topoisomerases, transcription factors, polymerases, structure maintenance of chromatin proteins and so on. The large dataset enabled by loop-seq should make it possible to develop comprehensive models to predict intrinsic cyclizability and other physical properties from DNA sequence. Preliminary analysis showed that simple sequence features such as GC content, polyA tracts, and dinucleotide parameters are generally poor or insufficient predictors of intrinsic cyclizability (Supplementary Note 16, Extended Data Fig. 10).
Our measurements suggest that intrinsic cyclizability is functionally important and must have applied selective pressure throughout the evolution of genomes. It remains to be investigated how genetic information content and the mechanical properties of DNA are linked, and how the sequence-dependent mechanical response of DNA to molecular-scale forces in its immediate environment may have influenced both the slow divergence of organisms and rapid mutations in contexts such as cancer.
METHODS:
smFRET based single-molecule DNA looping assay
Templates were purchased (IDT DNA) and converted into loopable molecules with 10 bp complementary overhangs on either side, Cy3 and Cy5 fluorophores at the ends, and a biotin molecule (Supplementary Note 1) via PCR amplification with KAPA Hi Fi Polymerase (Roche) and nicking near the ends by the site-specific nicking enzyme Nt.BspQ1 (NEB). Molecules were immobilized on a PEG-coated quartz surface (JHU slide production core for microscopy) functionalized with a small amount of biotin-PEG, via a streptavidin sandwich, as described previously6. Immobilized molecules were incubated with T2.5 (2.5 mM NaCl, 10 mM Tris-HCl pH 8) for 1.5 hours. Low salt imaging buffer (20 mM Tris-HCl pH 8, 3 mM Trolx, 0.8% dextrose, 0.1 mg/ml glucose oxidase, 0.02 mg/ml catalase) was flowed into the channel and the molecules were imaged on a TIRF microscope to determine the initial histogram of FRET values. High salt imaging buffer (1 M NaCl, and all components of the low salt imaging buffer) was then introduced into the channel at time 0, and FRET histograms were measured at various time points as done previously6. The plot of the percentage of molecules with both donor-acceptor pairs in high FRET as a function of time was fit to an exponential. Its time constant was defined as the looping time. The inverse of this was defined to be the looping rate.
Loop-seq
Instead of individual templates, entire libraries representing as many as ~90,000 individual DNA sequences, with the central 50 bp variable and flanked by identifcal 25 bp adapters, were obtained (Genscript), and amplified using KAPA Hi Fi polymerase (Roche) in 20 cycles of emulsion PCR35 (ePCR) using the Micellula DNA emulsion and purification kit (CHIMERx). The manufacturer’s guidelines were followed during ePCR. ePCR prevents improper annealing among different template molecules via the common adapter sequences. Amplification converted the library into 120 bp duplex molecules with a biotin near one end, and the recognition sequence for the nicking enzyme Nt.BspQ1 (NEB) near both ends (Supplementary Note 1). 20 μl of streptavidin-coated magnetic beads (Dynabeads MyOne Streptavidin T1, Thermo Fisher Scientific) were washed 2x with 400 μl T50 BSA (1 mg/ml BSA (Invitrogen) in T50 (50 mM NaCl, 10 mM Tris-HCl pH 8.0)) and resuspended in 20 μl T50 BSA. 2 μl of ~4 ng/ul amplified DNA was mixed with 5 μl of water, and 20 μl of the washed magnetic beads were added. After incubation for 10 minutes, the DNA bound beads were washed 2x with 200 μl T50BSA and 1x with 200 μl T10BSA (1 mg/ml BSA (Invitrogen) in T10 (10 mM Tris-HCl pH 8.0, 10 mM NaCl)). Digestion mix (84 μl water, 10 μl 10x NEB Buffer 3.1, 6 μl Nt.BspQ1 (NEB)) was prepared and heated to 50 °C for 5 minutes. Digestion resulted in an immobilized library, where every DNA molecule has a central 50 bp duplex variable region, flanked by 25 bp left and right adapters and 10 nt complementary single-stranded overhangs (Fig. 1d, Supplementary Note 1). The beads were pulled down and incubated with the heated digestion mix for 25 mins at 37 °C. The beads were then washed 2x with 100 μl of T10BSA preheated to 50 °C, followed by 200 μl of T2.5BSA (1 mg/ml BSA (Invitrogen) in T2.5 (10 mM Tris-HCl pH 8.0, 2.5 mM NaCl)). The beads were incubated in 200 μl T2.5BSA for 1.5 hours on a rotor at room temperature. The bead sample was then split into two 95 μl fractions denoted ‘sample’ and ‘control’. The beads in the sample fraction were pulled down and resuspended in 200 μl looping buffer (1M NaCl, 1 mg/ml BSA, 10 mM Tris-HCl pH 8) for 40 seconds. High salt (1M NaCl) initiates looping, which allows the complementary single-stranded overhangs at the ends to stably hybridize6. Apparent DNA bendability has been shown to be independent of the salt concentration used6.The tube containing the sample was then placed on magnets for an additional 35 seconds. The looping buffer was replaced with 200 μl of digestion buffer (6.66 μl of RecBCD (NEB), 20 μl NEB 10X Buffer 4, 20 μl of 10x ATP (NEB), 154 ul water) for 20 minutes. This was defined as looping for 1 minute. In general, looping for n minutes implies incubation in looping buffer for up to 20 seconds prior to the completion of n minutes, followed by 35 seconds over magnets before the solution was replaced with digestion buffer. After 20 minutes, digestion buffer was removed by pulling down the beads and replaced with 200 μl of looping buffer. The control was treated in exactly the same way, except the digestion buffer had 6.66 μl of water instead of RecBCD. Beads in the sample and control fractions were then pulled down and the looping buffer was replaced with 50 μl of PCR mix (25 μl 2x HiFi KAPA Hot Start ready mix (Roche), 1 ul each of 100 μM primers (supplementary note 1), 23 μl water) and PCR amplified (16 cycles). If the library contained less than 20,000 sequences, the products were sequenced on an Illumina MiSeq machine. For more complex libraries a HiSeq machine was used. Library preparation for sequencing was done using the Nextera XT primer kit and followed a protocol similar to the Illumina protocol for 16S metagenomic sequencing library preparation.
Sequencing results were mapped to the known sequences in the library using Bowtie 136. The number of times each sequence was represented in the sample and control was obtained and 1 was added to all counts. The relative population of each sequence in the digested and control pools was calculated. Cyclizability of a sequence was defined as the natural logarithm of the ratio of the relative population of a sequence in the sample pool to that in the control. In addition to Bowtie 136, SAMtools37, smCamera, and MATLAB (Matworks) versions 9.0, 9.2, 9.4, 9.6 were used to analyze the data.
Purification of INO80:
INO80 was purified according to a protocol published earlier38. Briefly, S. cerevisiae cells were grown in 12 liters of YPD medium to an O.D. of 1.5. Frozen yeast cells were lysed in a SPEX freezer mill (15 cycles: precool 2 min, run time 1 min, cool time 1 min, rate 15 cps). Ino80–3Flag was affinity-purified from whole lysate using anti-Flag M2 agarose beads and eluted with Flag peptide (0.5 mg/ml). The complex was further purified by sedimentation over a 20–50% glycerol gradient. Peak INO80 fractions were pooled and concentrated using Centricon filters (50 kDa cut off), and buffer changed to 25 mM HEPES–KOH (pH 7.6), 1 mM EDTA, 2 mM MgCl2, 10% glycerol, 0.01% NP-40, 0.1 M KCl. Aliquots of purified INO80 were flash-frozen and stored at −80°C. Recombinant INO80 was also purified as per earlier protocols21.
Nucleosome sliding by INO80:
Nucleosome preparation and sliding by INO80 was performed under conditions as reported earlier19. Sliding for in the presence of various [INO80] as reported in Fig. 2f (for 1 minute) and Extended Data Fig. 7 (for 1 minute and 2.5 minutes) was performed in 10 μl reaction volumes containing 8 nM nucleosomes (nucleosomes formed on both constructs in the pair were present in equimolar proportion), 2 mM ATP, 24 mM tris-HCl pH 7.5, 43 mM KCl, 2.86 mM MgCl2, 0.55% glycerol and indicated concentration of INO80. The mixture was incubated without ATP at 30 °C for 7 minutes. After addition of ATP, the reaction was allowed to proceed for 1 minute at 30 °C, and was then quenched by the addition of lambda DNA and ADP to final concentrations of 66.7 μg/ml and 20 mM respectively. For all sliding experiments reported in Extended Data Fig. 6b (timecourse of INO80 sliding), conditions were the same except incubations prior to ATP addition and the subsequent sliding reaction were carried out at room temperature. The reaction was continued for the indicated amounts of time in presence of saturating INO80 prior to quenching. Quenched reactions were loaded on to 6% TBE gels (Invitrogen) in presence of 10% glycerol and run at 150 V for 1.5 hrs. The gel was imaged separately for Cy3 and Cy5 fluorescence.
Statistics and Reproducibility
All presented loop-seq data in figures (unless explicitly comparing between multiple loop-seq runs on the same library, as in Extended Data Fig. 3c) were compiled from a single run of loop-seq on the library in question. However, some sequences in every library were included as part of at least one other library. Pearson’s coefficient of correlation for the intrinsic cyclizability values of these common sequences (and the 95% confidence interval and p values), as obtained via the two independent loop-seq runs on the two libraries, was measured to confirm reproducibility (Extended Data Fig. 3a–c, Supplementary Notes 4, 5, 6, 9, 12, 14). All such pearson’s correlation coefficients were greater than or similar to the correlation coefficient of cyclizability values of the two sets of reverse complement sequences in the “Mixed Reverse Complement of the Random Library and the Random Library” (Extended Data Fig. 3f). Further, measurements of intrinsic cyclizabilities of common sequences in different libraries constitute completely independent measurements starting from independently purchased libraries from the manufacturer.
All Pearson’s r have been calculated using the corrcoef function in MATLAB (Matworks). For two random variables A and B, Pearson’s r is the covariance of A and B, divided by the product of their standard deviations. p values have always been calculated using the MATLAB (matworks) function corrcoef, which calculates the p value by transforming the correlation to create a t-statistic having n-2 degrees of freedom, where n is the number of measurements. The test was always two-sided. Further, extremely small p values have always been indicated as p < 0.00001.
Reproducibility of the result that nucleosome sliding by INO80 is favored by flexible linker DNA was verified by repeating one condition for each of the three pairs shown in Fig. 2f 5 independent times (Extended Data Figs. 7a, c). The same preparations of nucleosome constructs and INO80 enzyme were used, although all subsequent steps were performed independently. However, swapping the Cy5 and Cy3 fluorophores between the two constructs in pair 1 (Extended Data Fig. 7b) required re-preparing of nucleosome constructs.
Direct reproducibility of smFRET experiments to measure looping kinetics (Fig. 1c) was limited to repeating the measurement once in the case of one of the sequences (Extended Data Fig. 3i). Our goal was to establish correlation between a large number of smFRET measurements on different sequences and their corresponding cyclizability values derived by loop-seq. Thus every time we performed a new smFRET experiment, we used a different sequence, rather than repeat an earlier measurement. High correlation between looping times measured via smFRET and cyclizability measured via the independent loop-seq method (Fig. 1f) establishes cyclizability as an accurate measure of looping time.
Extended Data
Extended Data Fig. 1: Timecourse loop-seq.
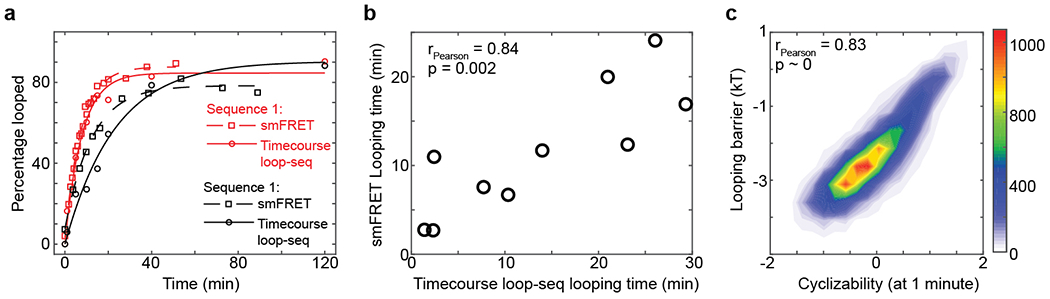
a, Looping kinetic curves of two individual sequences that were part of the Cerevisiae Nucleosomal Library (Supplementary Note 4), obtained by performing two individual smFRET experiments (Fig. 1a–c) as well as timecourse loop-seq (Supplementary Note 3) on the library. b, Looping times of 10 sequences that were part of the Cerevisiae Nucleosomal Library obtained from 10 individual smFRET experiments (Fig. 1c) vs looping times obtained by performing timecourse loop-seq on the library. Pearson’s r = 0.84. 95% CI = [0.44, 0.96]. p = 0.002 (t-test, two-sided). c, Looping barriers (natural logarithm of the looping times, see Supplementary Note 2) of all 19,907 sequences in the Cerevisiae Nucleosomal Library obtained by performing timecourse loop-seq vs the corresponding cyclizability values obtained by performing regular loop-seq involving 1 minute of DNA looping prior to RecBCD digestion. Pearson’s r = 0.833. 95% CI = [0.829, 0.837]. p < 0.00001 (t-test, two sided).
Extended Data Fig. 2: Pre-looped and looped geometries.
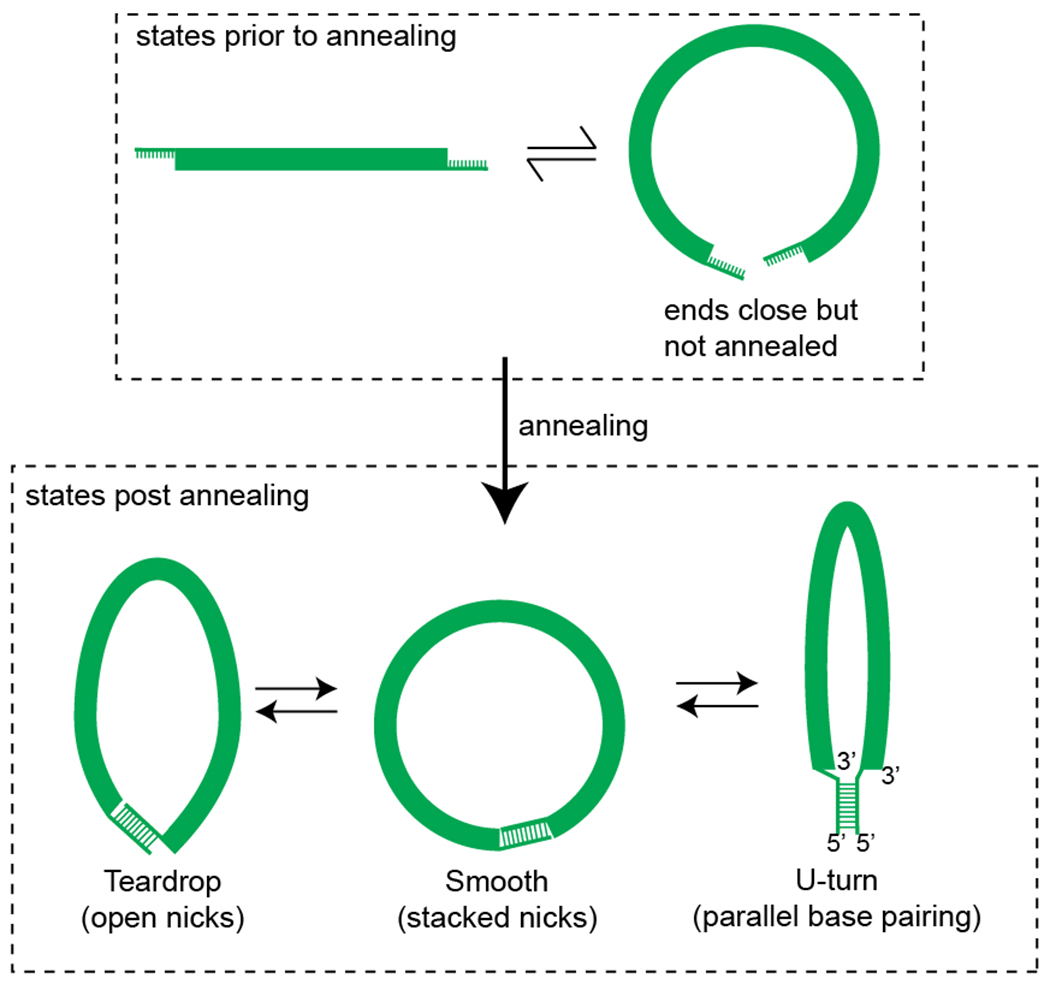
Prior to annealing of the ends, the DNA rapidly samples various configurations where the ends are far apart or closer together, described here for simplicity as a rapid equilibrium between two representative states. As described earlier6, annealing captures the state where the ends are close together. Thus the rate of looping as measured in the FRET based assay reports on the equilibrium population fraction of the state where the ends are close but not annealed, irrespective of the exact shape and geometry of the subsequent annealed state. It thus addresses the biological question of how quickly regions of DNA can approach, which can then be stabilized by protein binding6. However, nucleoprotein complex formation may require not just the ends to approach, but the intervening DNA to also adopt a certain shape, and the readout of looping rate does not distinguish between these possible shapes. Various shapes have been proposed for the subsequent annealed state, such as a teardrop configuration where the nicks are open and basepair stacking across nicks is disrupted, and a smooth state where basepair stacking is preserved across the nicks11. Other non-canonical geometries may also be possible, such as a U-turn geometry, where the sticky overhangs interact via reverse Watson-Crick basepairing in a parallel stranded configuration. Conventional Watson-Crick base pairing between the overhangs, but in a geometry similar to the U-turn configuration, has been achieved in the past (hairpin loop10). Presence of mismatches or transient defects in the duplex region may influence whether such a geometry is preferred over the teardrop or smooth configuration10. Because all members of various libraries in our study used the same overhang sequences, relative differences between them in their looping kinetics are unlikely to be affected by a potential conformational heterogeneity of the looped state.
Extended Data Fig. 3: Controls pertaining to the loop-seq assay.
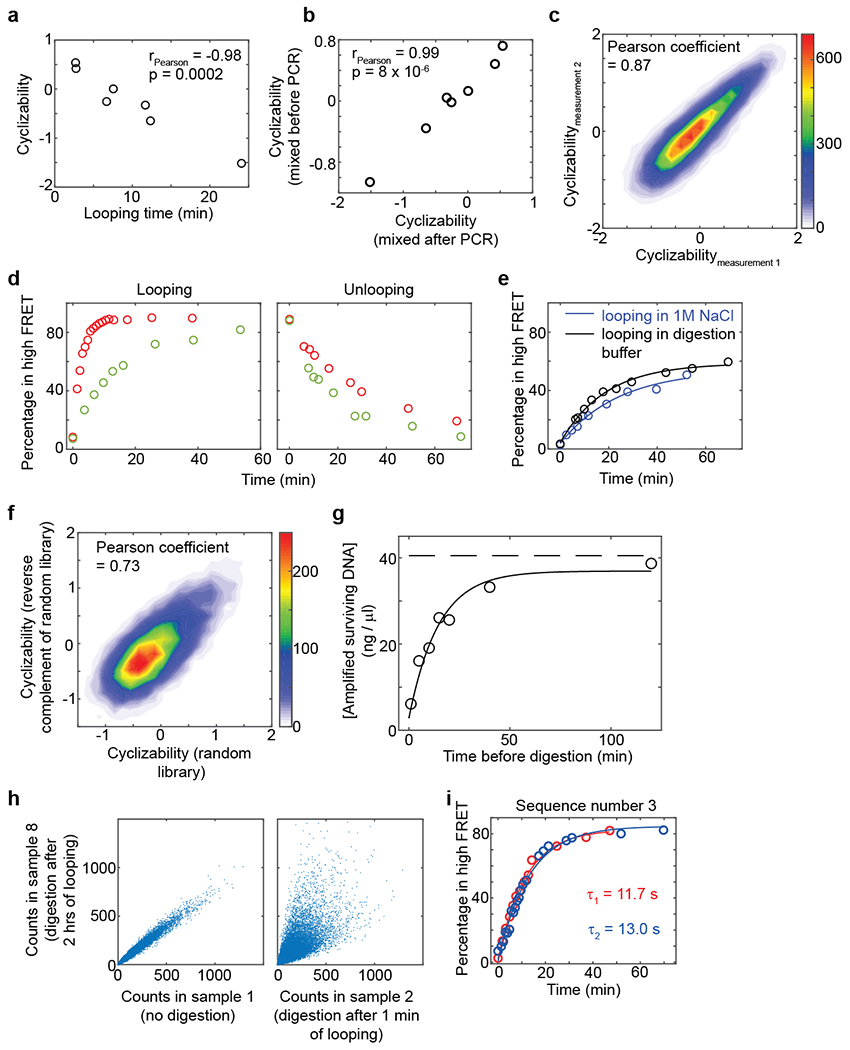
a, Seven of the ten sequences whose looping times were measured using smFRET (Sequences 1 – 7 in Fig. 1c, and listed in Supplementary Note 1) were combined to form a small library. Plotted are the cyclizability values obtained by performing loop-seq on this library vs the looping times obtained from the individual smFRET experiments. Pearson’s r = −0.977. 95% CI = [−0.848 −0.997]. p = 0.0002 (t-test, two-sided). This plot is similar to that in Fig. 1f, except that loop-seq was performed on a much smaller library comprising only these seven sequences. This control serves to revalidate the anti-correlative relation between looping time and cyclizability and confirm reproducibility of loop-seq measurements. b, Regular PCR of the entire library containing multiple templates can generate incorrectly annealed products that are annealed via the 25 bp identical adapters at the ends but have 50 bp bubbles in the middle. Such constructs would likely be extremely flexible and would be protected from digestion due to rapid cyclization. Emulsion PCR separates the templates in individual droplets, thereby preventing incorrect annealing between different templates. We performed a control experiment to verify that emulsion PCR of the library does not affect the measured value of cyclizability. In one case, seven template sequences (Sequences 1 – 7 as listed in supplementary note 1 and Fig. 1c) were mixed and then a single round of ePCR was performed to form a small library. In another case, seven separate regular PCR amplifications were carried out for the seven template molecules. The amplified products were then mixed in equimolar proportions to form the library. Loop-seq was performed on these two 7 member libraries and two sets of cyclizabilities of the seven sequences were measured. As indicated by the plot here, these values are highly correlated. Pearson’s r = 0.992. 95% CI = [0.951, 0.999]. p = 8E-6 (t-test, two-sided). c, Technical replicates of loop-seq performed on the Cerevisiae Nucleosomal Library (Supplementary Note 4). Pearson’s r = 0.869 95% CI = [0.865, 0.872]. p < 0.00001 (calculated using two-sided t-test). p value was obtained from a two-sided t-test. The same original library as provided by the manufacturer (Genscript) was used, but all subsequent steps were performed independently. d, This control was performed to verify the expectation that DNA sequences which are more bendable and thus loop quickly under high salt conditions are slow to unloop under low salt conditions. In red is the looping kinetics of Sequence 6, and in green is that of Sequence 7 (Fig. 1c) measured using smFRET. For the unlooping measurements, the slide containing nicked DNA was incubated for 2 hours with high salt imaging buffer containing 1M NaCl. After that, low salt imaging buffer containing no added NaCl was flowed in and the percentage of molecules in high FRET as a function of time was measured. e, This control was performed to verify that while performing loop-seq, molecules do not significantly unloop during the 20 minutes of digestion with RecBCD (see methods). If they do unloop, they would be immediately digested, which would affect the measurement of cyclizability. Sequence 5 (Supplementary Note 1) was used in this experiment. We find that digestion buffer (without the RecBCD enzyme) is in itself capable of looping molecules, and that too at a slightly faster rate than in the presence of looping buffer (which has 1M NaCl, see methods). This is owing to the presence of Mg2+ ions in the digestion buffer, which we know to also effect looping by allowing stable hybridization of the ends6. Thus, molecules looped in the presence of 1M NaCl in 1 minute are expected to stay looped during the subsequent 20 minutes of digestion with RecBCD. f, This control was performed to understand the effect of the orientation of the central 50 bp sequence on the measured value of cyclizability. First the Random Library was constructed (where the sequence of DNA in the 50 bp central variable region are randomly selected – see Supplementary Note 5). Then a library called “Mixed Reverse Complement of the Random Library and Random Library” was constructed by mixing the Random Library with another library, where every sequences in the Random Library was present, but had its central 50 bp variable region flipped (Supplementary Note 6). Loop-seq was performed on this new library. We found that the cyclizability of a sequence represented in the Random Library half of this new library was correlated with that of the corresponding sequence in the other half, where the central 50 bp region was flipped. Pearson’s r = 0.73. 95% CI = [0.72, 0.74]. p < 0.00001, calculated using t-test (two-sided). g, This plot serves to confirm the expectations that RecBCD does not digest looped molecules, and that over sufficient time, most molecules, even rigid ones, will loop. During timecourse loop-seq (Supplementary Note 3), the original sample was split into 8 identical fractions. Looping for various amounts of time and subsequent digestion was carried out for 7 of the 8 fractions, while one fraction was not subject to any digestion. All fractions were then PCR amplified (16 cycles) under identical conditions (see methods). Plotted are the concentrations of DNA obtained after PCR vs the corresponding times the samples were subject to the looping condition. These data points were fit to an exponential curve (solid line). The [DNA] obtained when no digestion was performed is represented as the dashed horizontal line. The fact that the fitted exponential approaches the dashed line indicates that for very long looping times, almost all molecules, even very rigid ones, have had sufficient time to loop and hence are protected from digestion. Thus, in this case, the concentration of DNA obtained after PCR of all surviving molecules approaches that of the fraction where no digestion was performed at all. Whether RecBCD would digest molecules sealed via non-canonical parallel basepairing or other geometries (Extended Data Fig. 1) is not known. However, this control suggests that either it does not, or such unconventional basepairings are rare. h, These plots serve to further demonstrate that if the library is permitted to loop for a very long time, most molecules, even very rigid ones, will loop, and that looped molecules are protected from subsequent digestion with RecBCD. In this case, the relative populations of various sequences measured after digestion should be similar to the case where no digestion was performed at all. We find this to indeed be the case: there is good correlation between the relative population of a sequence in sample 1 of timecourse loop-seq (no digestion) and sample 8 (2 hours of looping followed by digestion). However, correlation between the relative population of a sequence in sample 1 and that of a sequence in sample 2 (1 minute of looping prior to digestion) is much weaker. This is because in sample 2, only those molecules whose sequences render them bendable enough to loop under 1 minute are protected from subsequent RecBCD digestion. i, Technical replicates of the looping kinetic curve of sequence number 3 (Supplementary Note 1) measured using smFRET.
Extended Data Fig. 4: Dependence of cyclizability on tether geometry and rotational phasing.
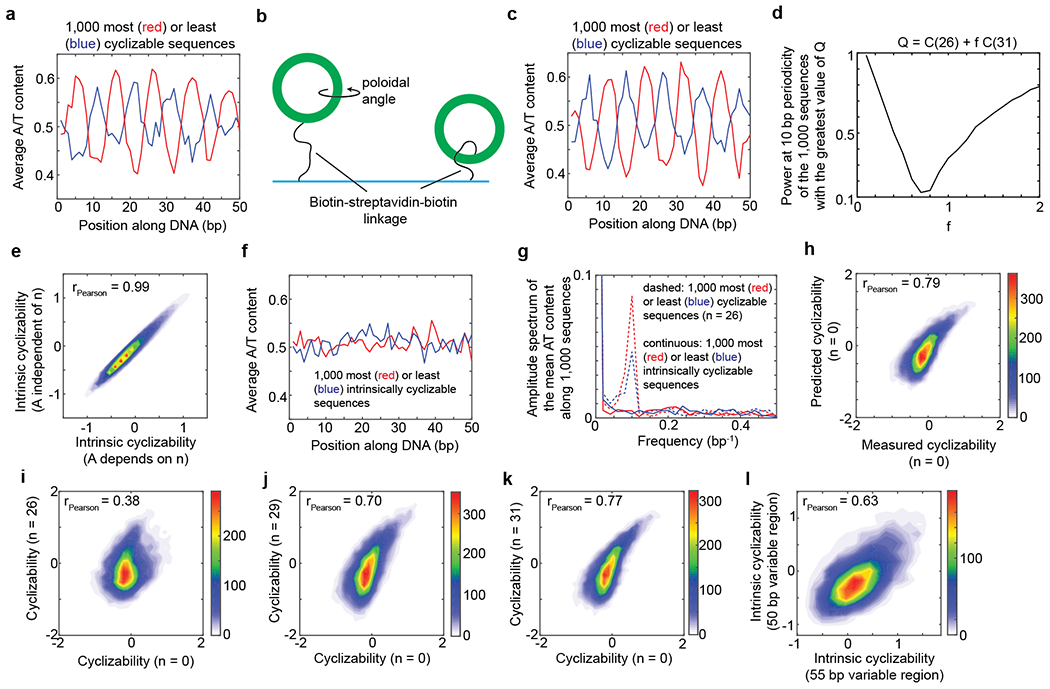
a, Loop-seq was performed on the Random Library (Supplementary Note 5). Plotted is the mean A/T content as a function of position along the central variable 50 bp region, where the mean is calculated by averaging over the 1,000 most cyclizable (red) or least cyclizable (blue) sequences. The value of n (n = 26) is the distance in nucleotides of the biotin tether from the end of each molecule in the library (Fig. 1d). b, In an untethered geometry, sequence features such as the phase of the oscillations in A/T content may result in the looped configuration having a preference for a certain poloidal angle (rotation along the long axis of DNA). Preference for a certain poloidal angle translates to a preference for a certain orientation of the biotin-streptavidin tether. Shown above are two extreme cases – in one case, the poloidal angle preference of the sequence results in a preferential orientation of the tether on the outside, while in the other case, the tether points to the inside at the point of contact with the DNA. As the biotin-streptavidin-complex is quite large, the outside orientation may be more favored for looping owing to steric considerations. The outside orientation can be converted to the inside orientation by moving the biotin tether point to a base that is half the DNA helical repeat away. This may explain why the phase of the oscillation of A/T content among the most or least cyclizable sequences shifts by half the helical repeat of DNA when the tether point is also shifted by half the helical repeat of DNA (about 5 bases) (compare panels a and c). c, The random library was re-prepared, placing the biotin 31 nt away from the ends (n = 31) and loop-seq was performed. Plotted are the same quantities as in panel a, except the 1,000 most and least cyclizable sequences of the library were identified based on the newly obtained cyclizability values under the n = 31 condition. d – e, See context in which these panels are referred to in Supplementary Note 7. In panel e, Pearson’s r = 0.987, 95% CI = [0.986, 0.987]. p < 0.00001 (t-test, two-sided). f, Mean A/T content as a function of position along the variable region of the Random Library, where the averaging is done over the 1,000 sequences that have the highest (red) or lowest (blue) values of intrinsic cyclizability. The scale of the axes is the same as in panels a and c. g, Amplitude spectra obtained from the fast Fourier transforms of the plots in panel f (solid lines) and panel a (dashed lines). h, 2D histogram of the scatter plot of measured cyclizability of sequences in the Random Library prepared with the biotin at the very end of the molecule (n = 0 condition) vs its predicted value based on the oscillatory model (equation 1 in Supplementary Note 7). Pearson’s r = 0.787. 95% CI = [0.78, 0.793]. p < 0.00001 (t-test, two-sided). i – k, 2D histogram of scatter plot of measured cyclizabilities of sequences in the Random library prepared at n = 0 vs prepared at n = 26, 29, 31 nt. For panel i, Pearson’s r = 0.38. 95% CI = [0.37, 0.40]. p < 0.00001 (t-test, two-sided). For panel j, Pearson’s r = 0.70. 95% CI = [0.69, 0.71]. p < 0.00001 (t-test, two-sided). For panel k, Pearson’s r = 0.77. 95% CI = [0.76, 0.78]. p < 0.00001 (t-test, two-sided). l, The use of long 10 nt overhangs has been shown to eliminate the need for ligase and to reduce the dependence of looping on rotational phasing between the ends6. Shown here is a 2D histogram of the scatter plot of intrinsic cyclizability of a sequence in the random library (which had 50 bp of DNA along the central variable region) vs the corresponding sequence in library L (Supplementary Note 8) where 5 bases were added to the variable region. A correlation coefficient only slightly poorer than the correlation between cyclizability values of the Random Library and the Reverse Complement of the Random Library (Extended Data Fig. 3f) suggests that rotational phasing of the ends does not significantly influence intrinsic cyclizability. Pearson’s r = 0.63. 95% CI = [0.61, 0.65]. p < 0.00001 (t-test, two-sided). See Supplementary Note 8.
Extended Data Fig. 5: Intrinsic cyclizability and nucleosome occupancy vs position from the dyads of the +1 nucleosomes of various individual genes in S. cerevisiae.
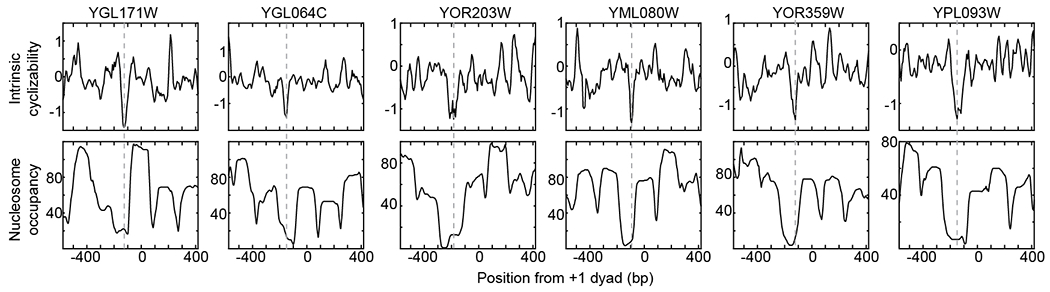
Plots are as shown for the two individual genes in Fig. 2c. The dashed line marks the ordinate value where intrinsic cyclizability is lowest.
Extended Data Fig. 6: DNA pair selection and timecourse of INO80 remodeling.
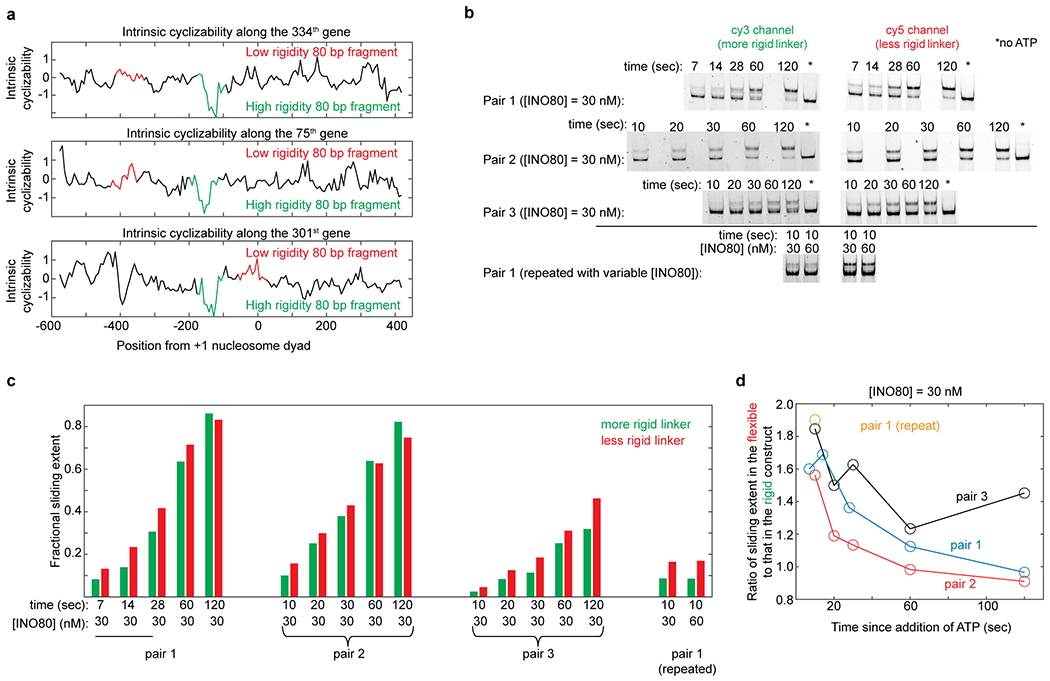
a, Intrinsic cyclizability as a function of position from the +1 nucleosomal dyad, along the 334th, 75th, and 301st genes in the list of 576 genes along which intrinsic cyclizability was measured (Supplementary Note 9). The 80 bp linker regions of both constructs (with rigid and flexible linkers extending from the 601 sequence) in pairs 1, 2, and 3 along which INO80 sliding extent was measured, were selected from the 334th, 75th, and 301st genes respectively. Genes are oriented in the upstream to downstream direction. Red and green denote the selected 80 bp less rigid and more rigid linker regions respectively. See Supplementary Note 11. b, The remodeling reactions shown in Fig. 2f were all performed for 1 minute of remodeling, under various enzyme concentrations. Here we show, instead, remodeling reaction timecourses at saturating [INO80] for all three pairs. Conditions are identical to that used in Fig. 2f, except that saturating (30 nM) INO80 is used and remodeling is permitted to progress for various amounts of time after addition of ATP (see methods). In all cases, the two constructs in a pair are present simultaneously, and distinguished by imaging the gel once for Cy3 and once for Cy5 fluorescence. Thus, although the sliding extent can be very sensitive to sliding time (especially for short sliding times), robust comparisons of sliding extents can be made between the two constructs in a pair. Quantification is done as explained in supplementary note 11. Sliding on the constructs in pair 1 was repeated in a separate experiment in presence of 30 nM enzyme, and also side by side in presence of 60 nM enzyme. Near identical extents of sliding indicate saturation has been reached at 30 nM INO80. c, Ratio of the fractional sliding extent in the construct formed on the more flexible linker to that formed on the more rigid linker, at various timepoints since addition of ATP, and in presence of 30 nM INO80. The dashed line indicates a ratio of 1. The ratio is computed from the data in panel b. The extent of sliding under saturating enzyme conditions is consistently higher for the construct involving more flexible linker (except, as expected, when the sliding extent approaches 100%). Solid lines connect observations that were made from the same initial reaction volume by sampling its fractions at various timepoints.
Extended Data Fig. 7: Control experiments pertaining to the INO80-mediated sliding of nucleosomes as reported in Fig. 2f.
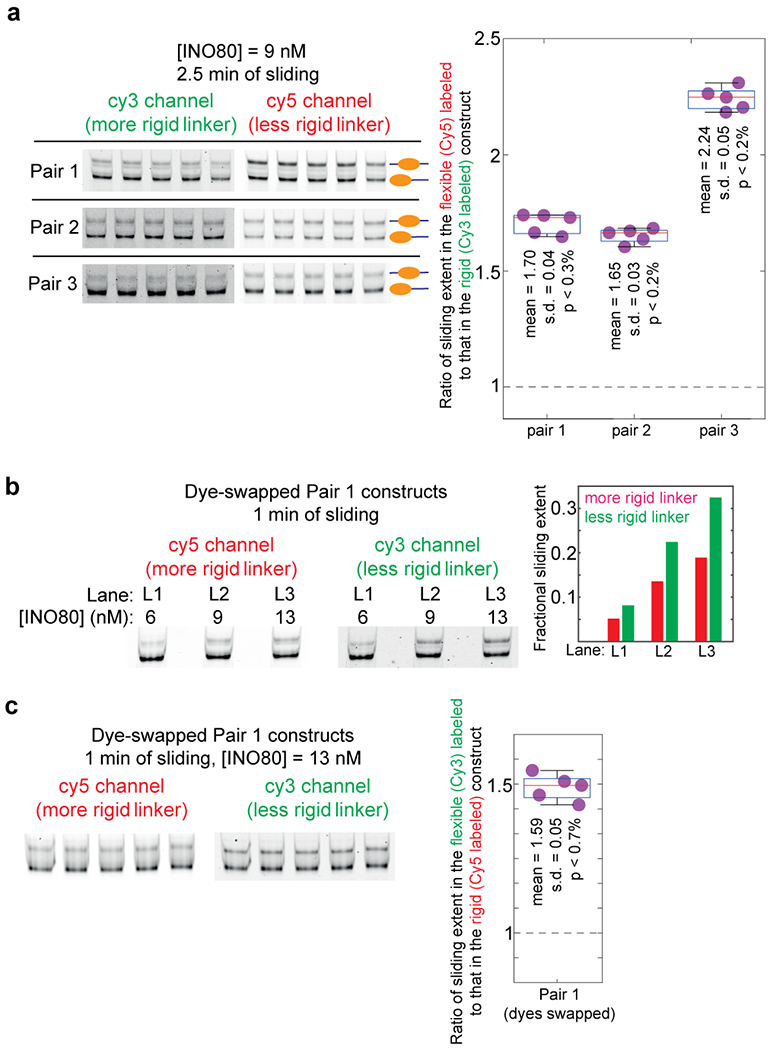
a, In order to assess our confidence in the result that INO80 mediated sliding is greater in the construct with the less rigid linker, we performed nucleosome sliding experiments similar to those reported in Fig. 2f five times for each pair in the presence of 9 nM INO80 and for 2.5 mins of sliding. These constitute technical replicates. The products of sliding were analyzed on a 6% TBE gel as done in Fig. 2f. Each gel was imaged separately for Cy3 and for Cy5 fluorescence and quantified to calculate the fold-difference in sliding extent between the flexible and the rigid construct in each pair. The measurements of fold-differences for each pair are displayed in the box plots, along with the actual data points. The central mark in each box (red) represents the median and the bottom and edges represent the 25th and 75th percentile respectively. The whiskers extent to the most extreme datapoints. Also indicated are the mean, standard deviation (s.d.), and the upper limit of the p value (defined here as the probability of obtaining a fold-difference of 1 if the distribution of fold differences has the same mean and s.d. as that of these 5 measurements) as obtained by the application of Chebyshev’s inequality. Dashed line represents a fold-difference of 1. b, In the experiment described in Fig. 2f, the more rigid construct in all pairs was labeled with Cy3, while the less rigid construct was labeled with Cy5. This control verifies that the result that sliding extent in greater in the less rigid construct is not influenced by different dye properties. We swapped the dyes between the two constructs in pair 1. We then performed nucleosome sliding experiments on this modified pair 1 constructs for the three INO80 concentrations that yielded detectible sliding in Fig. 2f (6, 9, 13 nM), and for 1 minute of sliding as done in Fig. 2f. The products of sliding were analyzed on a 6% TBE gel, and the sliding extents quantified as done in Fig. 2f. We indeed find that even when the dyes are swapped, sliding extent is greater for nucleosomes formed on the less rigid construct. c, To obtain better statistics of sliding along the dye-swapped pair 1 constructs, we repeated one of the conditions in panel b (13 nM INO80, 1 min of sliding) five times. These constitute technical replicates. The measured fold-difference values are displayed in the box plot. The central mark in each box (red) represents the median and the bottom and edges represent the 25th and 75th percentile respectively. The whiskers extent to the most extreme datapoints. Also indicated are the mean, standard deviation (s.d.), and the upper limit of the p value (defined as in panel a) as obtained by the application of Chebyshev’s inequality. Dashed line represents a fold-difference of 1.
Extended Data Fig. 8: Intrinsic cyclizability along S. cerevisiae chromosome V at 7 bp resolution.
Data was obtained by performing loop-seq on the ChrV Library (Supplementary note 12).
Extended Data Fig. 9: Intrinsic cyclizability along nucleosomes.
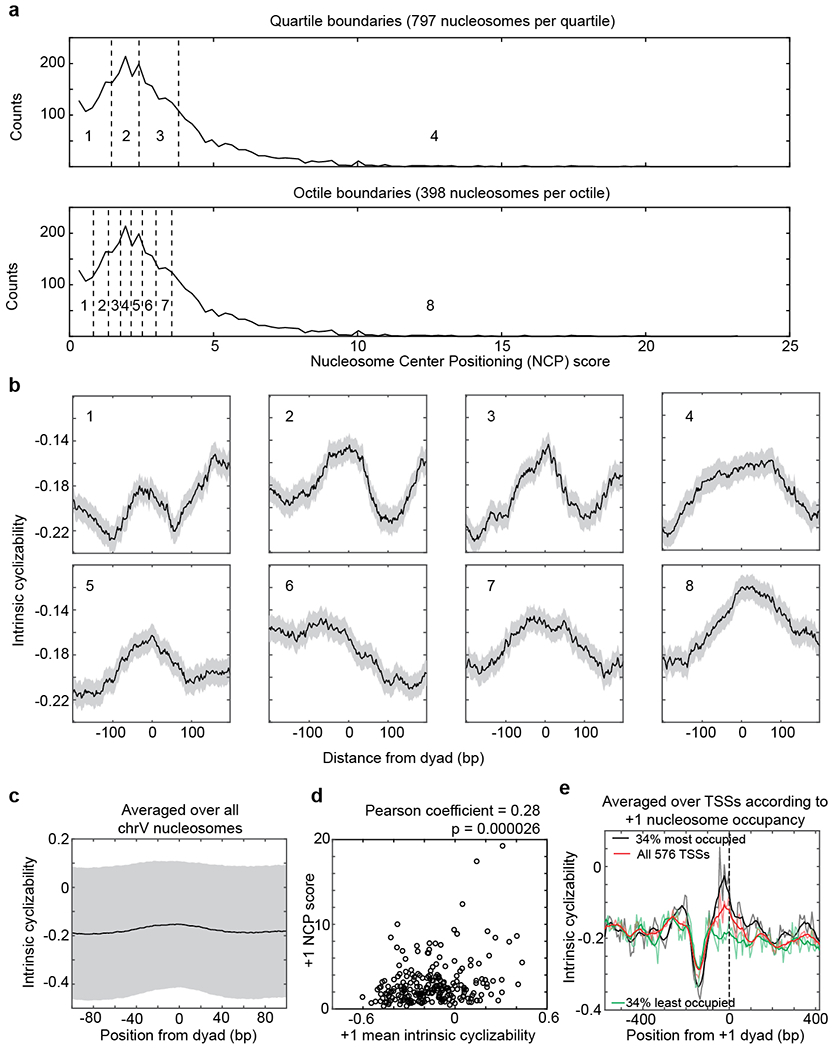
a, Distribution of Nucleosome Center Positioning (NCP) scores32 of all 3,192 S. cerevisiae chromosome V nucleosomes. Quartile and octile boundaries of the distribution are shown in dashed lines and numbered (1 through 4 for quartiles and 1 through 8 for octiles). b, Mean intrinsic cyclizability of DNA as a function of position from the dyads of nucleosomes along yeast chromosome V, averaged over nucleosomes in each octile indicated in panel a. Error extents (shaded background) are s.e.m. c, Mean intrinsic cyclizability as a function of position, averaged over all 3,192 S. cerevisiae chromosome V nucleosomes (solid line). Height of the shaded region is the standard deviation of measurements. d, Scatter plot of the NCP scores of the 227 +1 nucleosomes of the 227 genes identified along chromosome V vs the mean intrinsic cyclizabilities of DNA along the 147 bp that span these nucleosomes. Intrinsic cyclizability values were obtained by performing loop-seq on the ChrV Library. Pearson’s r = 0.28. 95% CI = [0.15, 0.39]. p = 2.6E-5 (t-test, two-sided). e, Plot of intrinsic cyclizability as a function of position along all the 576 genes in the Tiling Library (red), and among 34% of these genes that had the highest (black) or lowest (green) NCP score value of the gene’s +1 nucleosome. Plots were obtained in a manner identical to that in Fig. 2b. Intrinsic cyclizability on either side of the dyad of +1 nucleosomes of genes that have high +1 nucleosome NCP score (black) is asymmetric, being higher on the TSS proximal (i.e. “left”) side of the dyad.
Extended Data Fig. 10: Loop-seq measurements compared to expectations based on earlier measurements and models (see Supplementary Note 16).
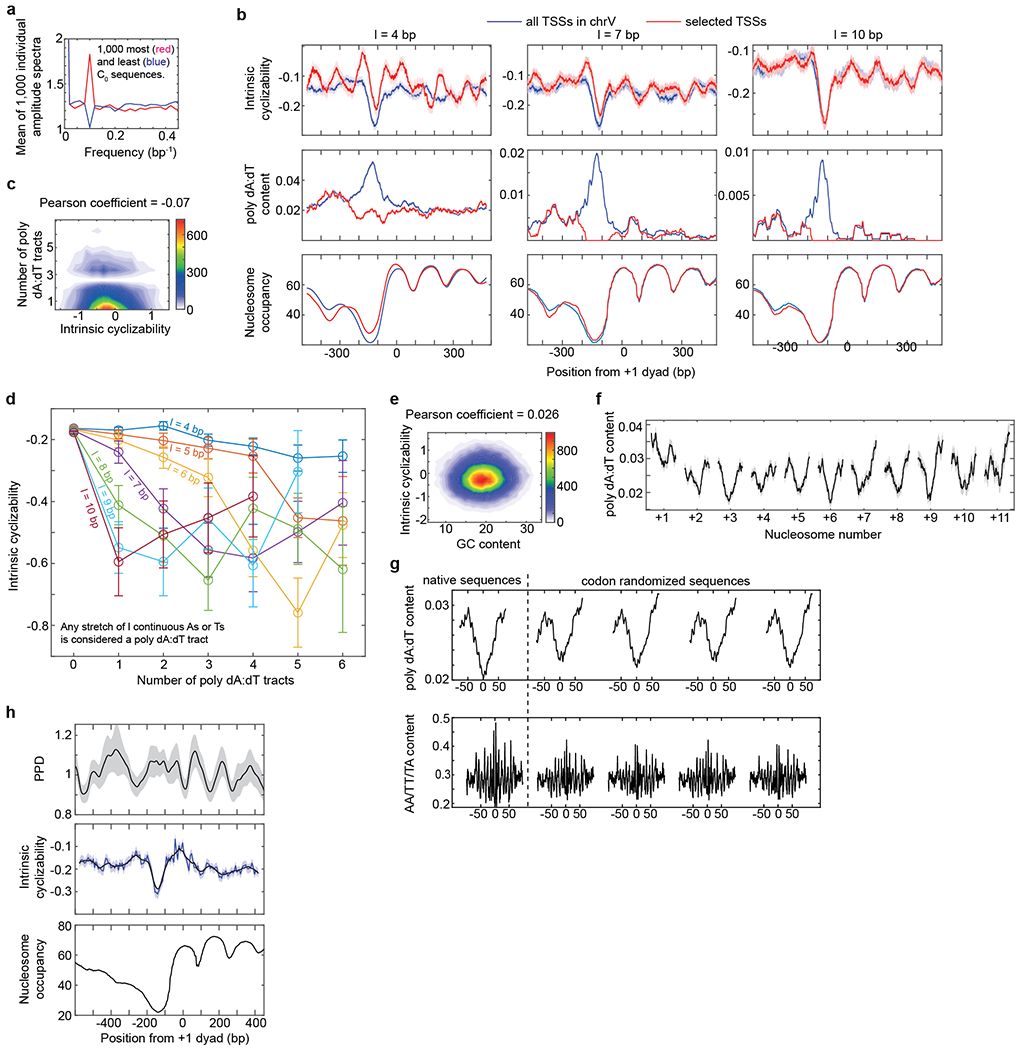
a, Two sets of 1,000 plots each of A/T content as a function of position along the central 50 bp variable region of 1,000 sequences in the Random Library with the highest and lowest values of intrinsic cyclizability were generated. Fast Fourier transforms of these two sets of 1,000 plots were taken individually and used to calculate a total of 2,000 amplitude spectra. Plotted is the mean of the 1,000 amplitude spectra for the 1,000 sequences that have the highest (red) or lowest (blue) intrinsic cyclizability values. The plot indicates that sequences that have very high or low intrinsic cyclizabilities also tend to be characterized by enhanced or suppressed periodic modulations in AT content respectively at the DNA helical repeat. b, We consider a poly dA:dT stretch which has at least l consecutive A or T nucleotides to be a poly dA:dT tract. For various values of l, plotted are intrinsic cyclizability (top panel), poly dA:dT tract content (middle panel), and nucleosome occupancy (bottom panel) vs position from the dyad of the +1 nucleosome, averaged over all 227 identified genes in S. cerevisiae chromosome V (blue) or over a selected subset of genes that show no peak in poly dA:dT content at the NDR (red; 30%, 62%, and 86% of genes for l = 4 bp, 7 bp, 10 bp respectively). See supplementary note 15 for plotting details, including how poly dA:dT content is defined. c, 2D histogram of the scatter plot between the number of poly dA:dT tracts in the 50 bp variable region and intrinsic cyclizability of sequences in the ChrV library. Any stretch of l or more consecutive As or Ts (here l = 4) is considered a poly dA:dT tract. Thus a sequence with one stretch of 5 As, and no other As or Ts, in the 50 bp variable region has 2 poly dA:dT tracts if l is considered to be 4. The scatter plot indicates that the overall correlation between intrinsic cyclizability and poly dA:dT content is very poor. Only non-overlapping sequences in the chrV library were considered. Pearson’s r = −0.07. 95% CI = [−0.09, −0.05]. p < 0.00001 (t-test, two-sided). d, Binned histogram of the data in panel c (which represents the l = 4 bp case), as well as for more restrictive definitions of poly dA:dT stretches (l = 5 to 10 bp). The y-axis values are the mean intrinsic cyclizabilities of those sequences in the ChrV library which contain the number of poly dA:dT tracts in the central 50 bp variable region as specified along the x-axis. Error bars are s.e.m. For l = 4, there were N = 5081, 2801, 1705, 948, 521, 268, 170 non-overlapping sequences in the ChrV library which had a poly dA:dT content of 0, 1, 2, 3, 4, 5, 6 respectively. For l = 5, the corresponding N values were 8771, 1594, 695, 323, 150, 89, 36. For l = 6, the corresponding N values were 10,523, 655, 295, 117, 62, 25, 32. For l = 7, the corresponding N values were 11,203, 290, 109, 56, 27, 30, 16. For l = 8, the corresponding N values were 11,497, 109, 58, 23, 30, 17, 11. For l = 9, the corresponding N values were 11,608, 57, 23, 29, 19, 13, 7. Because the count of poly dA:dT = 6 is less than 10, this point was left out from the plot. For l = 10, the corresponding N values were 11,665, 23, 30, 21, 11, 6, 1. Again, the intrinsic cyclizability for poly dA:dT content = 5, 6 were left out from the plot because the n value for these were less than 10. e, 2D histogram of the scatter plot of mean GC content along the central 50 bp variable region of sequences in the ChrV library vs their intrinsic cyclizabilities. Pearson’s r = 0.026. 95% CI = [0.019, 0.033]. p < 0.00001 (t-test, two-sided). f, A plot of mean poly dA:dT content (l = 4) as a function of position around the dyads of gene body nucleosomes along chromosome V in S. cerevisiae. The points along the horizontal axis where the nucleosome categories (+1, +2, etc) are marked represent the dyads of the nucleosomes. Light shaded region represents s.e.m. Poly dA:dT content was calculated as described in Supplementary Note 15. g, Mean poly dA:dT content (l = 4) and AA/TT/TA vs position along the native and codon-randomized nucleosomal DNA sequences of the same 500 +7 nucleosomes along which intrinsic cyclizability profiles are reported in figure 3g. h, Predicted Plectoneme Density (PPD, see Supplementary Note 16) (top panel), intrinsic cyclizability (middle panel) and nucleosome occupancy (bottom panel) vs position from the +1 nucleosomal dyad, averaged over all 576 genes in the Tiling Library (supplementary note 9). PPD along each gene was first smoothened using a rolling window of 51 bp. The smoothened PPDs were then averaged at each position across all 576 genes, normalized by the mean, and plotted as the solid line in the top panel. The shaded background represents s.e.m. Intrinsic cyclizability and nucleosome occupancy were plotted exactly as in figure 2b. For the plot of intrinsic cyclizability, the solid blue and black lines represent data without and with a 7-fragment smoothening respectively (as in Fig. 2b). The shaded blue background represents s.e.m of intrinsic cyclizability values in the unsmoothed data.
Supplementary Material
Acknowledgements
A.B. and T.H. would like to thank Carl Wu, Xinyu Feng, and Matthew F. Poyton for insights and help related to INO80 biochemistry, Quicen Zhang for help with initial assay development efforts, and Aditi Biswas for providing passivated glass and quartz slides for smFRET experiments. K.P.H and S.E. would like to thank Manuela Moldt for purification of recombinant INO80. This work was supported by the National Science Foundation Grants PHY-1430124 and EFMA 1933303 (to T.H.), the National Institutes of Health Grants GM122569 (to T.H.), the National Institutes of Health grant NIH R01CA163336 (to J.S.S.), the European Research Council (Advanced Grant INO3D to K.P.H), and by the Deutsche Forschungsgemeinschaft (CRC1064 and Gottfried Wilhelm Leibniz-Prize to K.P.H). A.B. was a Simons Foundation Fellow of the Life Sciences Research Foundation. T.H. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
Author Information Statement: The authors declare no financial or non-financial competing interests. All correspondence and requests for materials should be directed to Taekjip Ha (tjha@jhu.edu).
Data Availability: All sequencing data obtained as part of this study are deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession number PRJNA667271. Nucleosome positions and NCP scores along the genome of S. cerevisiae as reported earlier have been accessed from NCBI Gene Expression Omnibus (GEO) under accession number GSE36063. Nucleosome occupancy data in S. cerevisiae as reported earlier have been accessed from NCBI GEO under accession number GSE97290. Figs. 1 – 4, as well as Extended Data Figs. 2–5, 8–10 have associated raw data. There are no restrictions on data availability.
Code availability: No sequencing analysis in this study depends on the use of specialized code. Simple custom scripts were written in MATLAB (Matworks) versions 9.0, 9.2, 9.4, 9.6 for analysis of sequencing data, and will be made available upon request. smFRET data acquisition was carried out by custom scripts that can be obtained from http://ha.med.jhmi.edu/resources/ or upon request.
REFERENCES:
- 1.Garcia HG et al. Biological consequences of tightly bent DNA: The other life of a macromolecular celebrity. Biopolymers 85, 115–130 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krietenstein N et al. Genomic Nucleosome Organization Reconstituted with Pure Proteins. Cell 167, 709–721.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shore D, Langowski J & Baldwin RL DNA flexibility studied by covalent closure of short fragments into circles. PNAS 78, 4833–4837 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cloutier TE & Widom J Spontaneous sharp bending of double-stranded DNA. Mol. Cell 14, 355–362 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Ha T et al. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. U.S.A 93, 6264–6268 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vafabakhsh R & Ha T Extreme Bendability of DNA Less than 100 Base Pairs Long Revealed by Single-Molecule Cyclization. Science 337, 1097–1101 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geggier S & Vologodskii A Sequence dependence of DNA bending rigidity. PNAS 107, 15421–15426 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beutel BA & Gold L In vitro evolution of intrinsically bent DNA. Journal of Molecular Biology 228, 803–812 (1992). [DOI] [PubMed] [Google Scholar]
- 9.Rosanio G, Widom J & Uhlenbeck OC In vitro selection of DNAs with an increased propensity to form small circles. Biopolymers 103, 303–320 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Jeong J & Kim HD Base-Pair Mismatch Can Destabilize Small DNA Loops through Cooperative Kinking. Phys. Rev. Lett 122, 218101 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jeong J & Kim HD Determinants of cyclization–decyclization kinetics of short DNA with sticky ends. Nucleic Acids Res 48, 5147–5156 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee W et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet 39, 1235–1244 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Bai L, Ondracka A & Cross FR Multiple sequence-specific factors generate the nucleosome depleted region on CLN2 promoter. Mol Cell 42, 465–476 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z et al. A Packing Mechanism for Nucleosome Organization Reconstituted Across a Eukaryotic Genome. Science 332, 977–980 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segal E & Widom J Poly(dA:dT) tracts: major determinants of nucleosome organization. Current Opinion in Structural Biology 19, 65–71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Segal E et al. A genomic code for nucleosome positioning. Nature 442, 772–778 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chereji RV, Ramachandran S, Bryson TD & Henikoff S Precise genome-wide mapping of single nucleosomes and linkers in vivo. Genome Biology 19, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilchrist DA et al. Pausing of RNA Polymerase II Disrupts DNA-Specified Nucleosome Organization to Enable Precise Gene Regulation. Cell 143, 540–551 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou CY et al. The Yeast INO80 Complex Operates as a Tunable DNA Length-Sensitive Switch to Regulate Nucleosome Sliding. Mol. Cell 69, 677–688.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brahma S et al. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nature Communications 8, 15616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eustermann S et al. Structural basis for ATP-dependent chromatin remodelling by the INO80 complex. Nature 556, 386–390 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brahma S, Ngubo M, Paul S, Udugama M & Bartholomew B The Arp8 and Arp4 module acts as a DNA sensor controlling INO80 chromatin remodeling. Nature Communications 9, 3309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knoll KR et al. The nuclear actin-containing Arp8 module is a linker DNA sensor driving INO80 chromatin remodeling. Nat. Struct. Mol. Biol 25, 823–832 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Kubik S et al. Nucleosome Stability Distinguishes Two Different Promoter Types at All Protein-Coding Genes in Yeast. Mol. Cell 60, 422–434 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Widom J Role of DNA sequence in nucleosome stability and dynamics. Quarterly Reviews of Biophysics 34, 269–324 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Drew HR & Travers AA DNA bending and its relation to nucleosome positioning. Journal of Molecular Biology 186, 773–790 (1985). [DOI] [PubMed] [Google Scholar]
- 27.Hayes JJ, Tullius TD & Wolffe AP The structure of DNA in a nucleosome. PNAS 87, 7405–7409 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Widlund HR et al. Nucleosome structural features and intrinsic properties of the TATAAACGCC repeat sequence. J. Biol. Chem 274, 31847–31852 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Shrader TE & Crothers DM Artificial nucleosome positioning sequences. PNAS 86, 7418–7422 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lowary PT & Widom J New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276, 19–42 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Jin H, Rube HT & Song JS Categorical spectral analysis of periodicity in nucleosomal DNA. Nucleic Acids Res 44, 2047–2057 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brogaard K, Xi L, Wang J-P & Widom J A map of nucleosome positions in yeast at base-pair resolution. Nature 486, 496–501 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ngo TTM, Zhang Q, Zhou R, Yodh JG & Ha T Asymmetric Unwrapping of Nucleosomes under Tension Directed by DNA Local Flexibility. Cell 160, 1135–1144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bondarenko VA et al. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol. Cell 24, 469–479 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Nakano M et al. Single-molecule PCR using water-in-oil emulsion. Journal of Biotechnology 102, 117–124 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The Sequence Alignment/Map format and SAMtools. - PubMed - NCBI. https://www.ncbi.nlm.nih.gov/pubmed/19505943.
- 38.Mizuguchi G, Wu W-H, Alami S & Luk E Chapter Twelve - Biochemical Assay for Histone H2A.Z Replacement by the Yeast SWR1 Chromatin Remodeling Complex in Methods in Enzymology (eds. Wu C & Allis CD) vol. 512 275–291 (Academic Press, 2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.