

Abstract
We report a means to achieve the addition of two disparate nucleophiles to the amide carbonyl carbon in a single operational step. Our method takes advantage of non-precious metal catalysis and allows for the facile conversion of amides to chiral alcohols via a one-pot Suzuki–Miyaura cross-coupling / transfer hydrogenation process. This study is anticipated to promote the development of new transformations that allow for the conversion of carboxylic acid derivatives to functional groups bearing stereogenic centers via cascade processes.
Keywords: base metal catalysis, amides, Suzuki–Miyaura coupling, transfer hydrogenation, cascade reaction
Graphical Abstract
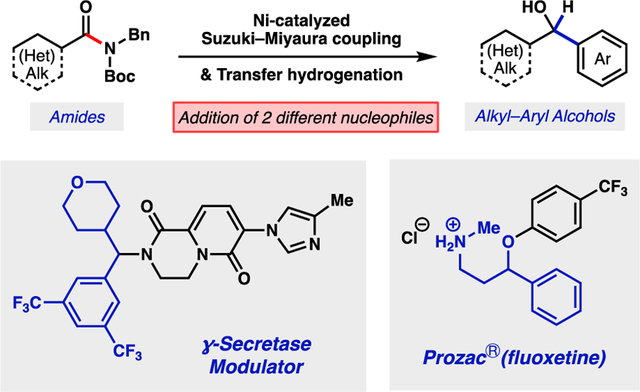
The first catalytic intermolecular addition of two disparate nucleophiles to the amide carbonyl carbon in a single operational step is reported. The method relies on nickel catalysis and enables the conversion of amides to chiral alcohols via a Suzuki–Miyaura cross-coupling / transfer hydrogenation cascade process.
The synthetic manipulation of carboxylic acid derivatives has become central to organic chemistry after more than a century of methodological development.[1] Though the field has a rich history, strategies for nucleophilic addition to carboxylic acid derivatives may be largely characterized by two primary mechanisms (Figure 1a). The first involves an addition-elimination sequence to produce carbonyl derivatives via a tetrahedral intermediate.[2] Notably, this traditional strategy has limitations in the context of organometallic nucleophiles, as the ketone products resulting from initial acyl substitution are susceptible to further nucleophilic attack to give achiral alcohol products. Specialized acyl derivatives such as N-methyl-N-methoxy amides, or “Weinreb amides,” are often required to avoid such undesired reactivity and necessitate two-step protocols.[2,3] A complementary approach employs transition metal catalysis,[4] where mild substrate activation affords an acyl-metal intermediate and allows for cross-coupling with a variety of nucleophiles.[4a,5] This alternative pathway differentiates the reactivity of the substrate from the product carbonyl to overcome the selectivity challenges mentioned above. An exciting opportunity offered by the latter strategy is the addition of a second, different nucleophile to the intermediate resulting from the initial cross-coupling reaction to generate chiral products. Cascade reactions of this type would provide efficient access to important chiral products in racemic or enantioenriched form from achiral starting materials.[6]
Figure 1.
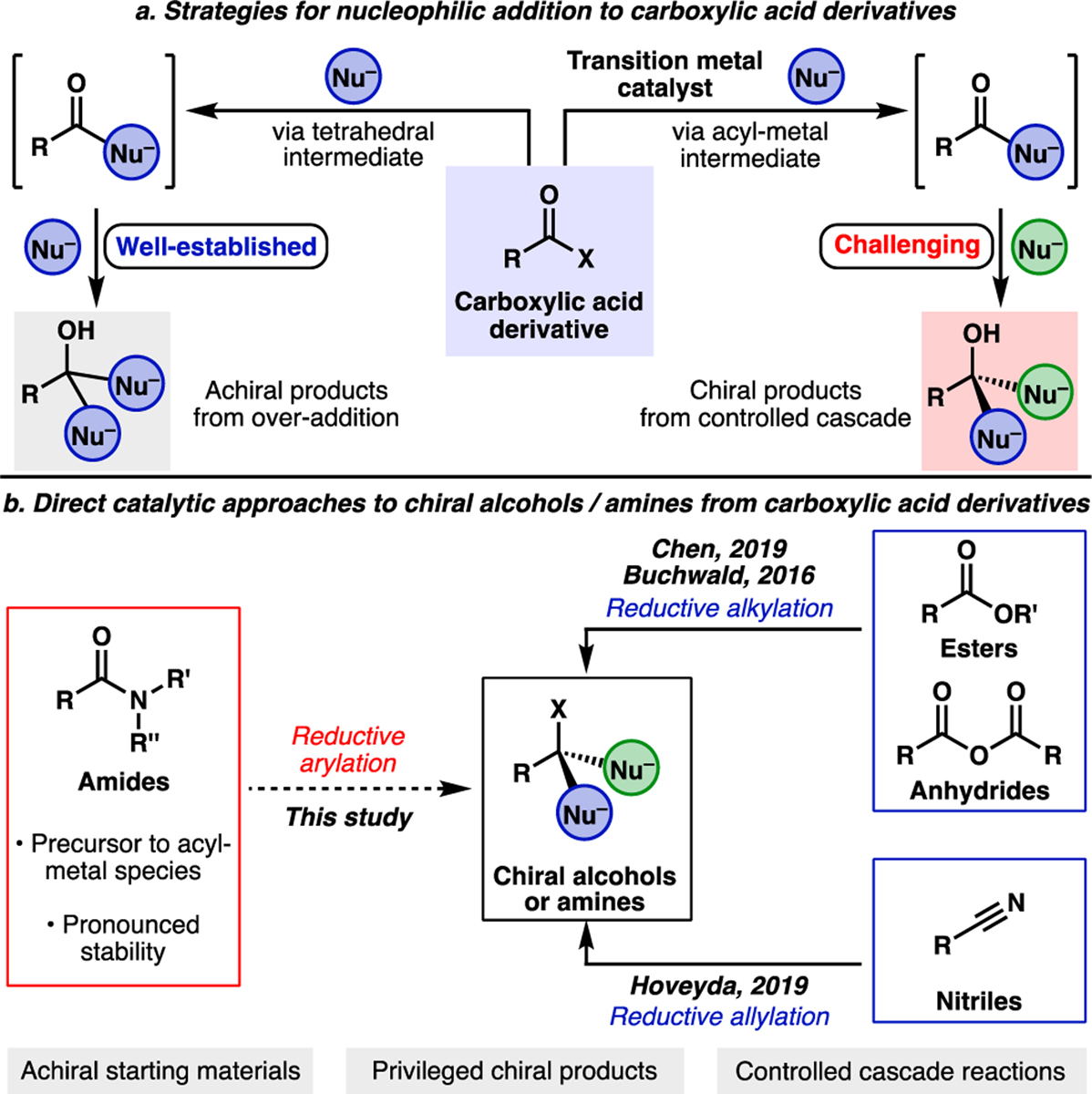
(a) Common reaction pathways for nucleophilic additions to carboxylic acid derivatives. (b) Direct catalytic approaches to chiral amines or alcohols from carboxylic acid derivatives.
Despite the widely recognized importance of cross-couplings, methods to leverage this platform for the addition of disparate nucleophiles to carboxylic acid derivatives remain underexplored.[7] We envisioned that amides could provide a viable entry to address this challenge, given their recent popularization as cross-coupling handles.[4j–o,8,9] Amides have been shown to undergo a variety of couplings through the intermediacy of acyl-metal species using either non-precious or precious metal catalysis (e.g., Ni or Pd). Additionally, we viewed them as ideal substrates for one-pot cascade reactions, as their stability under non-metal catalyzed conditions could allow for the orchestration of orthogonal bond-forming events.[10] Dixon has reported an elegant intramolecular reductive cyclization of a tertiary lactam substrate mediated by Vaska’s Ir complex,[11] however, no examples exist for the intermolecular addition of two distinct nucleophiles to amides using catalysis in a single operation.[12,13] Indeed, a reductive alkylation of aryl pyridyl esters reported by Chen and coworkers in 2019 represents the only known example of a carboxylic acid derivative undergoing direct catalytic addition of two nucleophiles through a cross-coupling approach (Figure 1b).[14] Though mechanistically distinct and not involving acyl metal species, two additional relevant methodologies should be highlighted. Buchwald and coworkers have reported a copper-catalyzed reductive alkylation of symmetric anhydrides to afford enantioenriched secondary alcohols,[15] and more recently the Hoveyda group reported a copper-catalyzed asymmetric reductive allylation of nitriles to access enantioenriched homoallylic amines (Figure 1b).[16] Together, these examples illustrate some of the potential advantages of cascade reactions that add disparate nucleophiles to a single reactive center of an achiral substrate and uncover synergistic reactivity beyond the capabilities of one reaction manifold.[17]
In this manuscript, we describe a synthetic method for achieving the addition of two different nucleophiles to a carboxylic acid derivative using nickel catalysis.[18] The overall transformation relies on a Suzuki–Miyaura cross-coupling / transfer hydrogenation cascade reaction of amide starting materials to form a C–C and C–H bond,[19,20] consecutively, and ultimately furnish alcohol products (Figure 2).[21] The results presented herein not only reinforce the notion that amides are versatile building blocks for transition-metal catalyzed reactions, but also validate their utility as synthons for directly generating sp3 carbon centers from the amide carbonyl.
Figure 2.
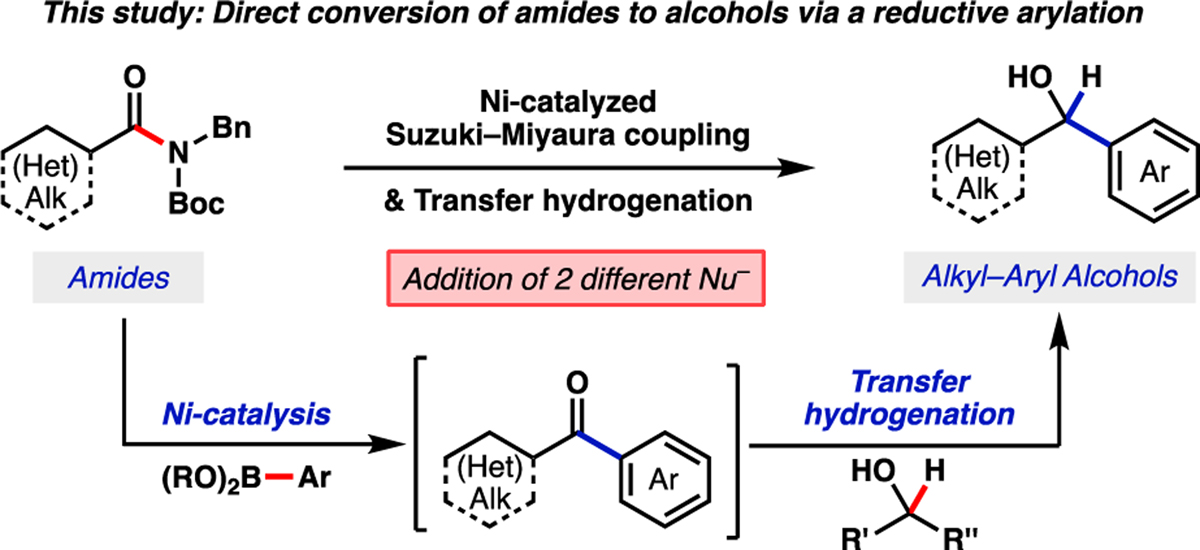
Overview of current study involving the conversion of aliphatic amides to alkyl–aryl alcohols via a Suzuki–Miyaura coupling / transfer hydrogenation cascade.
We initiated our study by examining the Ni-catalyzed Suzuki–Miyaura coupling and in situ reduction of dihydrocinnamic acid-derived amide 1 as shown in Figure 3.[19b] In the absence of a reducing agent, the Suzuki–Miyaura coupling with boronate 5 delivered ketone 3 in nearly quantitative yield (entry 1).[22,23,24] With the aim of developing the reductive variant, we questioned whether the use of a secondary alcohol reductant could effect the in situ transfer hydrogenation of ketone 3 to deliver alcohol 4. In this regard, we attempted the use of i-PrOH as solvent, reminiscent of common Meerwein–Ponndorf–Verley (MPV) reduction conditions.[25,26] Unfortunately, this change resulted in low yields of 4 (entries 2 and 3).[27] By shifting to the use of i-PrOH as an additive while using toluene as solvent, we obtained the desired product 4 in a slightly improved yield of 32%, with 18% of ketone 3 remaining (entry 4). Given our lab’s recent success in using 1–4-(dimethylamino)phenyl)-1-ethanol (DMPE, 7) in base-catalyzed MPV reductions,[28] we also tested this benzylic alcohol in our system.[29] By simply replacing i-PrOH with 7, alcohol 4 was obtained in 51% yield (entry 5). Finally, switching the solvent to 1,4-dioxane (entry 6) and using boronate 6 in place of boronate 5 (entry 7) led to further improvements, delivering alcohol 4 in 82% yield.[30,31,32]
Figure 3.
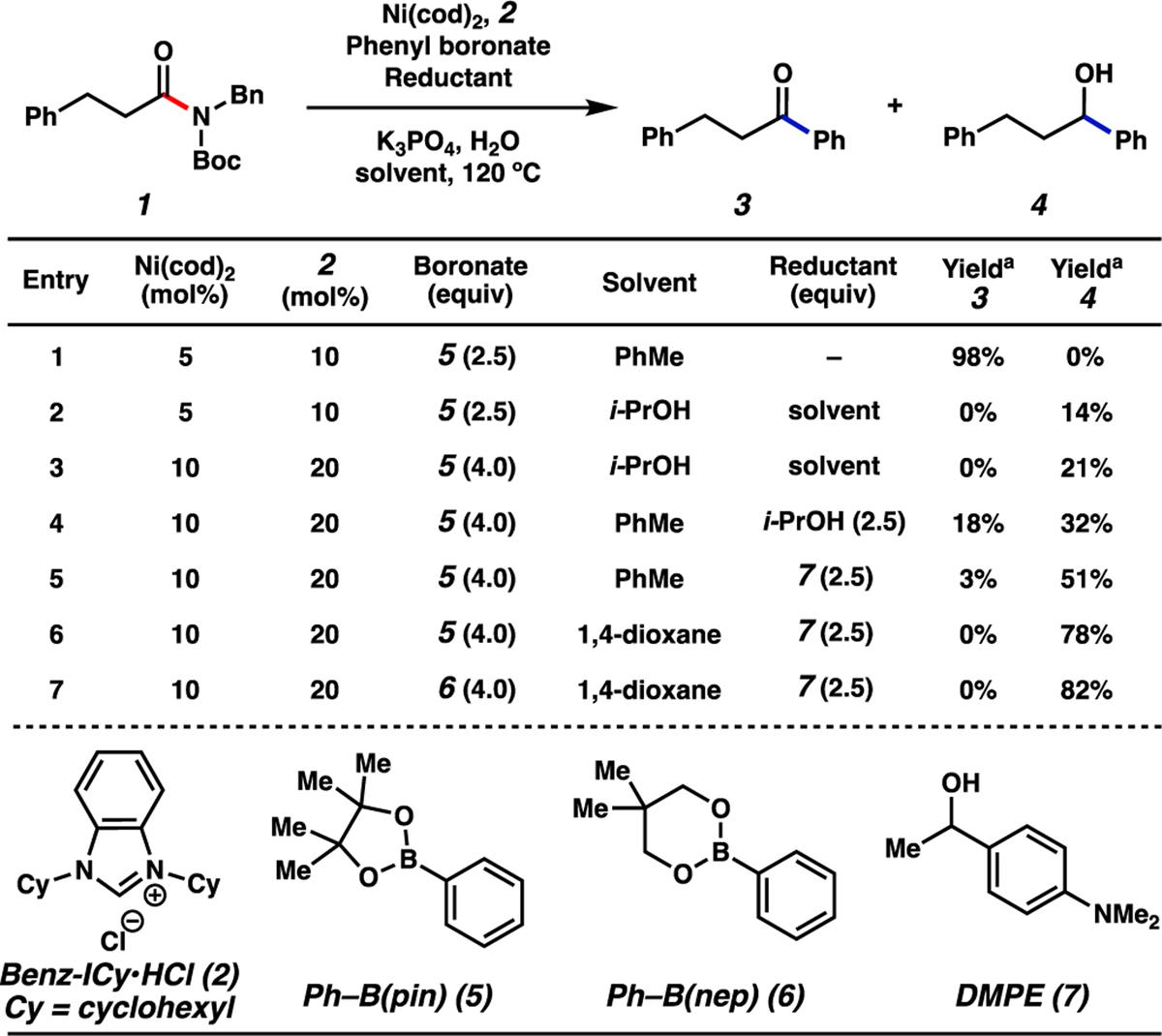
Evaluation of reaction conditions for the nickel-catalyzed Suzuki–Miyaura coupling / transfer hydrogenation cascade of amide 1 with phenyl boronates and reductants. Standard conditions unless otherwise noted: amide substrate (0.20 mmol, 1.0 equiv); phenyl boronate (0.50–0.80 mmol, 2.5–4.0 equiv); reductant (0.50 equiv, 2.5 equiv); K3PO4 (0.80 mmol, 4.0 equiv); H2O (0.40 mmol, 2.0 equiv); Ni(cod)2 (0.010–0.020 mmol, 5–10 mol%); 2 (0.020–0.040 mmol, 10–20 mol%); solvent (1.0 M); 120 °C; 16 h in a sealed vial. [a] Yield determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an external standard.
It is worth noting that these optimized conditions satisfy a challenging balance of reactivity required for the success of the amide to alcohol conversion. Specifically, reducing agent 7 does not significantly impede the nickel-catalyzed cross-coupling step, yet is reactive enough to efficiently reduce ketone 3. Furthermore, as will be shown, other carbonyl functional groups are tolerated by the methodology’s mild reducing conditions.
With optimized conditions in hand, we evaluated the scope of the reaction with respect to the aliphatic amide[33] coupling partner using phenyl boronate 6, which afforded a range of alkyl–aryl alcohol products (Figure 4). Beginning with the parent dihydrocinnamic acid-derived amide substrate used in optimization studies (i.e., 1), the reductive arylation furnished alcohol 4 in 76% isolated yield. Additionally, the use of an unbranched amide derived from decanoic acid provided alcohol 8 in 82% yield. Carrying out the reaction at 130 °C allowed for the reductive arylation of sterically encumbered substrates, as demonstrated by the formation of alcohol 9 in 61% yield. The compatibility of carbocyclic amides with boronate 6 was explored as well and gave alcohols 10–14 in good yields. We also evaluated an amide substrate bearing an epimerizable stereocenter α to the amide carbonyl. As shown by the formation of alcohol 15 from the corresponding trans amide substrate, minimal erosion of stereochemistry was observed.[34] Of note, the ester moiety was not disturbed, demonstrating both the preferential cleavage of the amide C–N bond over the ester C–O bond and the mildness of the reducing conditions.35 The tolerance of the methodology toward heterocycles was also determined. Notably, tetrahydropyrans, pyrrolidines, and piperidines, all of which are valuable in medicinal chemistry,36 could be employed as evidenced by the synthesis of alcohols 16–20, respectively.
Figure 4.
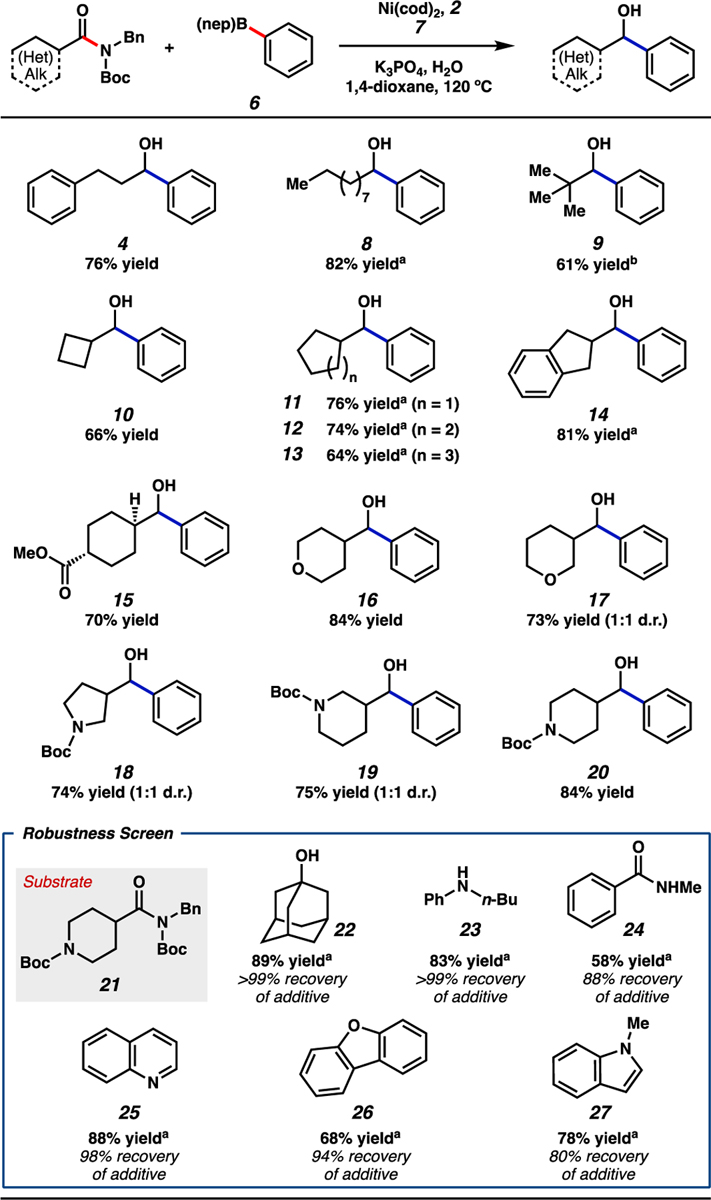
Scope of the reductive arylation of aliphatic amides and boronate 6. Standard conditions unless otherwise noted: amide substrate (0.20 mmol, 1.0 equiv); phenyl boronate 6 (0.80 mmol, 4.0 equiv); 7 (0.50 mmol, 2.5 equiv); K3PO4 (0.80 mmol, 4.0 equiv); H2O (0.40 mmol, 2.0 equiv); Ni(cod)2 (0.020 mmol, 10 mol%); 2 (0.040 mmol, 20 mol%); solvent (1.0 M); 120 °C; 16 h. Unless otherwise noted, yields reflect the average of two isolation experiments. [a] Yield determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an external standard. [b] Reaction ran at 130 °C.
With the aim of further improving the synthetic utility of the reductive arylation, we performed a robustness screen to assess the compatibility of the reaction with various functional groups and heterocycles (Figure 4).[37] Results indicated the tolerance of functional groups including tertiary alcohols, secondary anilines, and secondary amides, as demonstrated by moderate to good yields of alcohol 20 and appreciable recoveries of additives 22–24, respectively. Additionally, heterocycles such as quinoline (25), dibenzofuran (26), and N-methyl indole (27) were found to be stable under our standard reductive arylation conditions with minimal to no inhibition of reactivity.[38] These results complement those presented in the scope of the reaction and further demonstrate the methodology’s robustness toward several heteroatom-containing functional groups.
The scope of the aryl boronate component was also examined by coupling pinacol boronates with various amides (Figure 5).[31,39] Methyl substitution at the ortho, meta, or para positions of the aryl boronate was tolerated, as demonstrated by the formation of alcohols 28–30 in synthetically useful yields. We also evaluated aryl boronic ester nucleophiles bearing either a trimethylsilyl or trifluoromethyl group, which furnished alcohols 31 and 32, respectively, in good yields. Additionally, a naphthyl boronate ester underwent the reductive arylation to afford alcohol 33 in 58% yield. We also tested several boronates that possess functional groups that have been demonstrated to be reactive to nickel catalysis. To our delight, an aryl ester,[35] an ether,[40] and a dimethyl amine were tolerated,[41] thus giving rise to alcohols 34–36, respectively. Furthermore, a boronic ester containing a morpholinopyridine motif was employed to furnish alcohol 37, showing the reaction’s tolerance of this heteroatom-rich unit.[42]
Figure 5.
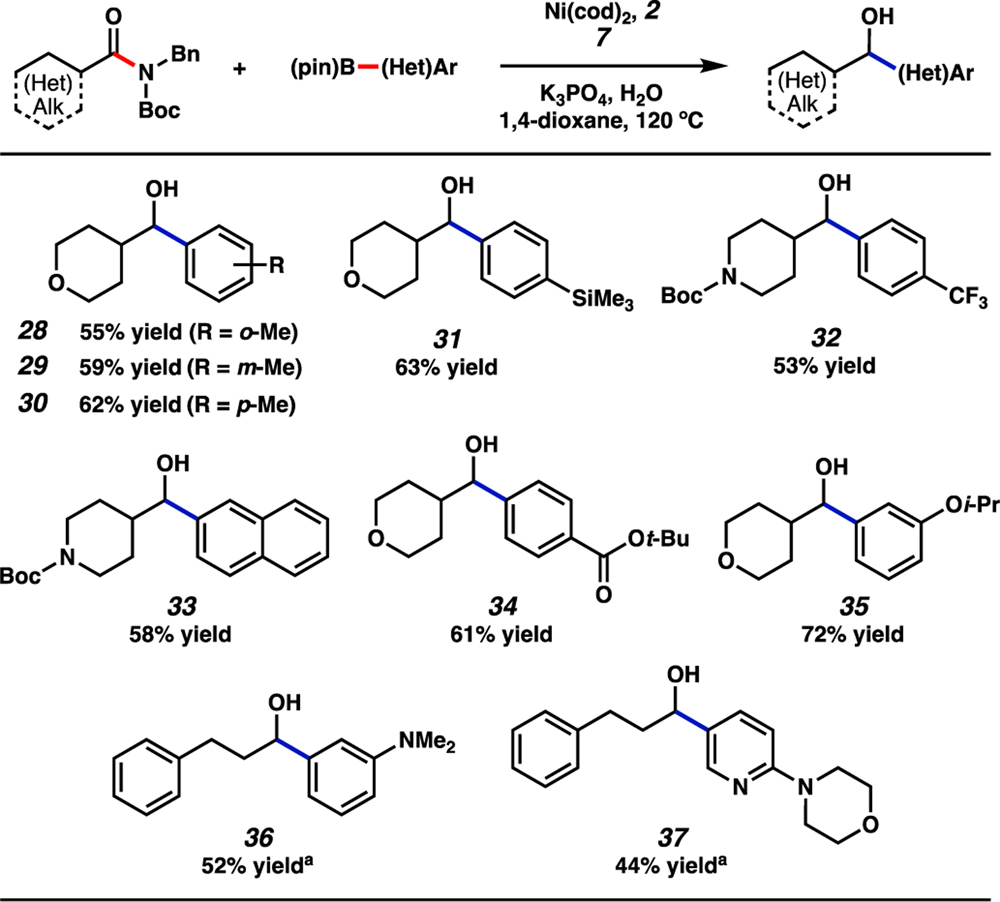
Scope of the reductive arylation of aliphatic amides and aryl boronates. Standard conditions unless otherwise noted: amide substrate (0.20 mmol, 1.0 equiv); aryl boronate (0.80–1.2 mmol, 4.0–6.0 equiv); 7 (0.50 mmol, 2.5 equiv); K3PO4 (0.80 mmol, 4.0 equiv); H2O (0.40 mmol, 2.0 equiv); Ni(cod)2 (0.020–0.040 mmol, 10–20 mol%); 2 (0.040–0.080 mmol, 20–40 mol%); solvent (1.0 M); 120 °C; 16–24 h. Unless otherwise noted, yields reflect the average of two isolation experiments. [a] Yield determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an external standard.
The utility of this methodology was evaluated in the synthesis of known intermediates toward two bioactive compounds (Figure 6). In the first example (Figure 6a), amide 38 underwent reductive arylation with boronate 39, despite the notable electron deficiency of this nucleophile. This delivered alcohol 40, a precursor to a known γ-secretase modulator.[43] We also targeted the interception of a known route to fluoxetine,[44] the active ingredient in the blockbuster drug Prozac®. Toward this end, amide 42, derived from the corresponding commercially available carboxylic acid, was coupled with boronate 6. This transformation furnished alcohol 43 in 69% yield, providing facile access to a known intermediate in the synthesis of 44 from commercially available materials.[44] These results not only further demonstrate the viability of leveraging a cross-coupling approach to add two disparate nucleophiles into an amide carbonyl carbon, but also showcase the practical utility of this reductive arylation protocol in the synthesis of complex chiral molecules.
Figure 6.
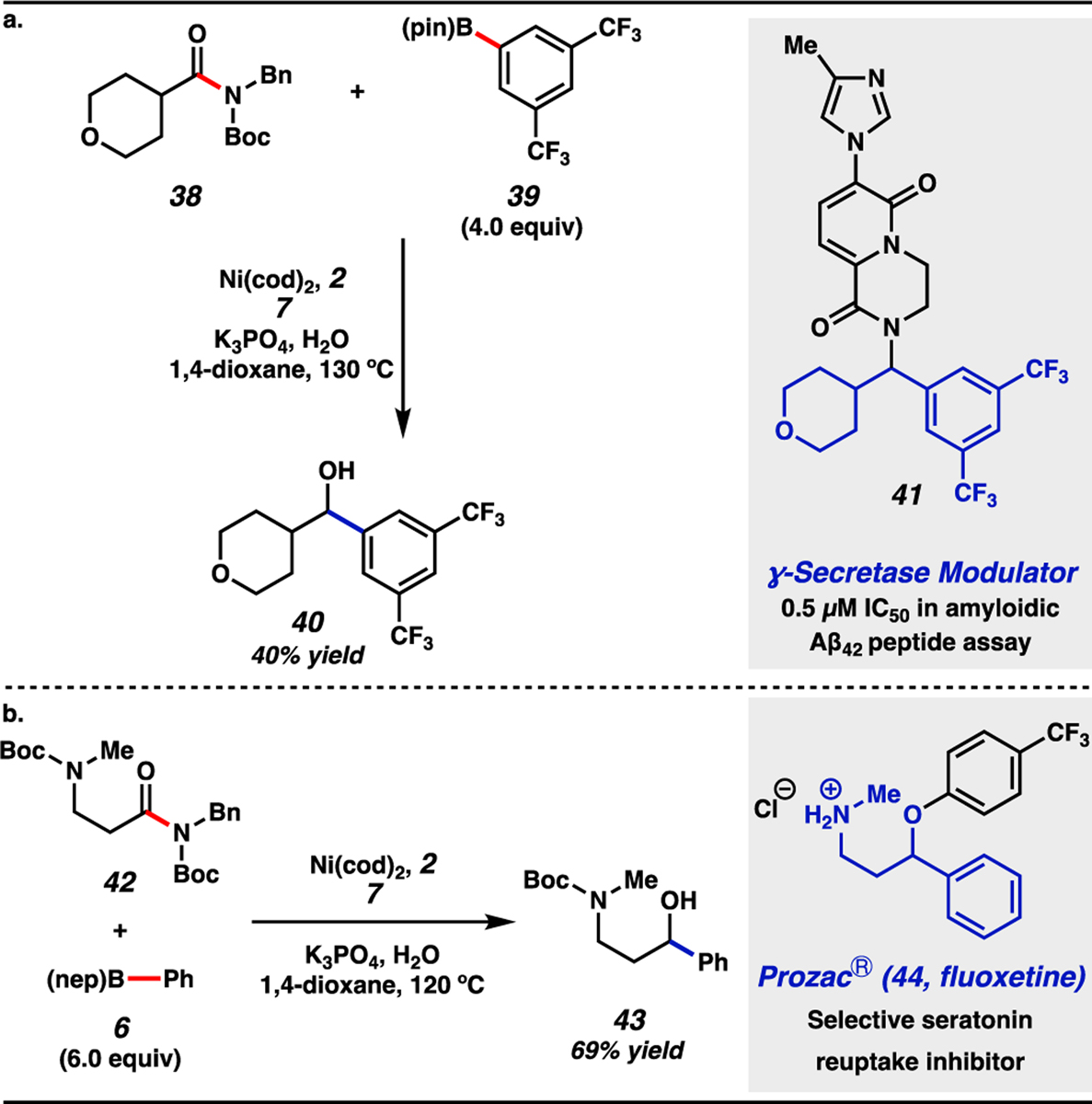
(a) Synthesis of alcohol 40, an intermediate in the synthesis of γ-secretase modulator 41. (b) Synthesis of alcohol 43, intercepting a known synthetic route toward Prozac® (44, fluoxetine). See Supporting Information for details.
In summary, we have developed the first catalytic method for the direct intermolecular addition of two distinct nucleophiles to the amide carbonyl carbon. This transformation takes advantage of non-precious metal catalysis and allows for the facile conversion of amides to chiral alcohols via a cascade reaction involving Suzuki–Miyaura cross-coupling and subsequent transfer hydrogenation. The methodology has a broad scope with respect to both the amide and boronate cross-coupling partners. Additionally, it shows tolerance toward epimerizable stereocenters, select functional groups (i.e., alcohols, amines, esters, ethers, and secondary amides,) and a range of heterocycles. Moreover, the methodology can be used to access scaffolds of value to medicinal chemistry, as shown by the syntheses of 40 and 43. This study validates the use of a cross-coupling approach to construct sp3 carbon centers from the amide carbonyl carbon in a single operational step. We hope this study will prompt the development of additional processes that allow for the direct conversion of carboxylic acid derivatives to functional groups bearing stereogenic centers[45] via catalytic cascade processes.
Supplementary Material
Acknowledgements
The authors thank the NIH-NIGMS (R01-GM117016 to N.K.G.), the California Tobacco-Related Disease Research Program (28DT-0006 to T.B.B.), the National Science Foundation GRFP (DGE-1650604 to M.M.M. and DGE-1144087 for E.L.B.), the Trueblood Family (N.K.G.), the Graduate Division of UCLA (J. K.), and the University of California, Los Angeles, for financial support. These studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the National Center for Research Resources (S10RR025631).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Larock RC Comprehensive Organic Transformations: A Guide to Functional Group Preparation; 2nd Ed. John Wiley & Sons: New York, 1997. [Google Scholar]
- [2].For the classic Weinreb amide synthesis of ketones from amides via a tetrahedral intermediate, see: Nahm S, Weinreb SM, Tetrahedron Lett 1981, 22, 3815–3818. [Google Scholar]
- [3].For a related synthetic method that allows for the addition of two nucleophiles to substituted ureas, thus affording ketone products, see: Heller ST, Newton JN, Fu T, Sarpong R, Angew. Chem., Int. Ed 2015, 54, 9839–9843. [DOI] [PubMed] [Google Scholar]
- [4].(a) For representative reviews and publications on the non-decarbonylative transition metal-catalyzed cross-coupling of carboxylic acid derivatives, see: Liebeskind LS, Srogl J, J. Am. Chem. Soc 2000, 122, 45, 11260–11261; [Google Scholar]; (b) Cheng H-G, Chen H, Liu Y, Qianghui Z, Asian J. Org. Chem 2008, 7, 490–508; [Google Scholar]; (c) Johnson JB, Rovis T, Acc. Chem. Res 2008, 41, 327–338; [DOI] [PubMed] [Google Scholar]; (d) Gooßen LJ, Rodriguez N, Gooßen K, Angew. Chem. Int. Ed 2008, 47, 3100–3120; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 3144–3164; [Google Scholar]; (e) Albano G, Aronica LA, Catalysts 2020, 10, 25–61; [Google Scholar]; (f) Blangetti M, Rosso H, Prandi C, Deagostino A, Venturello P, Molecules 2013, 18, 1188–1213; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hirschbeck V, Gehrtz PH, Fleischer I, Chem. Eur. J 2018, 24, 7092–7107; [DOI] [PubMed] [Google Scholar]; (h) Ogiwara Y, Sakai N, Angew. Chem. Int. Ed 2020, 59, 574–594; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2020, 132, 584–605; [Google Scholar]; (i) Gooßen LJ, Gooßen K, Rodriguez N, Blanchot M, Linder C, Zimmermann B, Pure Appl. Chem 2008, 80, 1725–1733; [Google Scholar]; (j) Meng G, Shi S, Szostak M, Synlett 2016, 27, 2530–2540; [Google Scholar]; (k) Liu C, Szostak M, Chem. Eur. J 2017, 23, 7157–7173; [DOI] [PubMed] [Google Scholar]; (l) Dander JE, Garg NK, ACS Catal 2017, 7, 1413–1423; [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Takise R, Muto K, Yamaguchi J, Chem. Soc. Rev 2017, 46, 5864–5888; [DOI] [PubMed] [Google Scholar]; (n) Meng G, Szostak M, Eur. J. Org. Chem 2018, 2352–2365; [Google Scholar]; (o) Buchspies J, Szostak M, Catalysts 2019, 9, 53–75. [Google Scholar]
- [5].Hu J, Zhao Y, Liu J, Zhang Y, Shi Z, Angew. Chem. Int. Ed 2016, 55, 8718–8722; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 8860–8864. [Google Scholar]
- [6].(a) These cascade reactions could involve either one or two catalytic steps. For discussion on the definitions of cascade and tandem processes, see: Fogg DE, dos Santos EN, Coord. Chem. Rev 2004, 248, 2365–2379; [Google Scholar]; (b) Hayashi Y, Chem. Sci 2016, 7, 866–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].(a) For representative examples of one-pot, sequential protocols for the reductive functionalization of amides, see: Meyers A, Comins D, Tetrahedron Lett 1978, 19, 5179–5182; [Google Scholar]; (b) Oda Y, Sato T, Chida N, Org. Lett 2012, 14, 950–953; [DOI] [PubMed] [Google Scholar]; (c) Zheng X, Liu J, Ye C-X, Wang A, Wang A-E, Huang P-Q, J. Org. Chem 2015, 80, 1034–1041; [DOI] [PubMed] [Google Scholar]; (d) Dander JE, Giroud M, Racine S, Darzi ER, Alvizo O, Entwistle D, Garg NK, Commun. Chem 2019, 2, 82; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ong DY, Fan D, Dixon DJ, Chiba S, Angew. Chem. Int. Ed 2020, 59, 11903–11907; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2020, 132, 12001–12005. [Google Scholar]
- [8].Kaiser D, Bauer A, Lemmerer M, Maulide N, Chem. Soc. Rev 2018, 47, 7899–7925. [DOI] [PubMed] [Google Scholar]
- [9].Hie L, Fine Nathel NF, Shah T, Baker EL, Hong X, Yang Y-F, Liu P, Houk KN, Garg NK, Nature 2015, 524, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pauling L, Corey RB, Proc. Natl. Acad. Sci. USA 1951, 37, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gabriel P, Gregory AW, Dixon DJ, Org. Lett 2019, 21, 6658–6662. [DOI] [PubMed] [Google Scholar]
- [12].(a) For examples of one-pot, sequential reductive functionalizations of tertiary amides, see: Gregory AW, Chambers A, Hawkins A, Jakubec P, Dixon DJ, Chem. Eur. J 2015, 21, 111–114; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tan PW, Seayad J, Dixon DJ, Angew. Chem. Int. Ed 2016, 55, 13436–13440; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 13634–13638; [Google Scholar]; (c) Xie L-G, Dixon DJ, Chem. Sci 2017, 8, 7492–7497; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fuentes de Arriba ÁL, Lenci E, Sonawane M, Formery O, Dixon DJ, Angew. Chem. Int. Ed 2017, 56, 3655–3659; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 3709–3713; [Google Scholar]; (e) Xie L-G, Dixon DJ, Nat. Commun 2018, 9, 2841; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Gabriel P, Xie L-G and Dixon DJ, Org. Synth 2019, 96, 511–527; [Google Scholar]; (g) Rogova T, Gabriel P, Zavitsanou S, Leitch JA, Duarte F, Dixon DJ, ChemRxiv preprint 2020, DOI: 10.26434/chemrxiv.12657410.v1; [DOI] [Google Scholar]; (h) For a review, see: Matheau-Raven D, Gabriel P, Leitch JA, Almehmadi YA, Yamazaki K, Dixon DJ, ACS Catal 2020, 10, 8880–8897. [Google Scholar]
- [13]. Although definitions for step count vary widely in the literature, here we employ the term “operational step” to indicate the number of reagent additions. Cascade reactions involving a single operational step enjoy certain practical advantages, such as operational simplicity, over complementary cascade reactions requiring multiple reagent additions. Moreover, cascade reactions involving one operational step present additional challenges to the organic chemist including the design of reaction conditions suitable for both transformations.
- [14].Wu X, Li X, Huang W, Wang Y, Xu H, Cai L, Qu J, Chen Y, Org. Lett 2019, 21, 2453–2458. [DOI] [PubMed] [Google Scholar]
- [15].Bandar JS, Ascic E, Buchwald SL, J. Am. Chem. Soc 2016, 138, 5821–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhang S, del Pozo J, Romiti F, Mu Y, Torker S, Hoveyda A, Science 2019, 364, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].(a) Further advantages of cascade reactions in regard to efficiency include the avoidance of intermediate purification, improved atom economy, and reduced waste generation. For general reviews on cascade reactions, see: Ref. 6;; (b) Tietze LF, Brasche G Gericke K, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006; [Google Scholar]; (c) Tietze LF, Beifuss U, Angew. Chem. Int. Ed 1993, 32, 131–163; [Google Scholar]; Angew. Chem 1993, 105, 137–170; [Google Scholar]; (d) Tietze LF, Chem. Rev 1996, 96, 115–136; [DOI] [PubMed] [Google Scholar]; (e) Ho T-L, Tandem Organic Reactions, Wiley, New York, 1992; [Google Scholar]; (f) Bunce RA, Tetrahedron 1995, 51, 13103–13159; [Google Scholar]; (g) Trost BM, Science 1991, 254, 1471–1477; [DOI] [PubMed] [Google Scholar]; (h) Trost BM Angew. Chem. Int. Ed 1995, 34, 259–281; [Google Scholar]; Angew. Chem 1995, 107, 285–307; [Google Scholar]; (i) Nicolaou KC, Edmonds DJ, Bulger PG, Angew. Chem. Int. Ed 2006, 45, 7134–7186; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2006, 118, 7292–7344; [Google Scholar]; (j) Romiti F, del Pozo J, Paioti PHS, Gonsales SA, Li X, Hartrampf FWW, Hoveyda A, J. Am. Chem. Soc 2019, 141, 17952–17961. [DOI] [PubMed] [Google Scholar]; (k) Jones A, Stoltz BM, May JA, Sarpong RS, Angew. Chem. Int. Ed 2014, 535, 2546–2591; [Google Scholar]; Angew. Chem 2014, 126, 2590–2628. [Google Scholar]
- [18].(a) For reviews on nickel catalysis see: Tasker SZ, Standley EA, Jamison TF, Nature 2014, 509, 299–309; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita A-M, Garg NK, Percec V, Chem. Rev 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].(a) For our laboratory’s nickel-catalyzed Suzuki–Miyaura couplings of amides, see: Weires NA, Baker EL, Garg NK, Nat. Chem 2016, 8, 75–79; [DOI] [PubMed] [Google Scholar]; (b) Boit TB, Weires NA, Kim J, Garg NK, ACS Catal 2018, 8, 1003–1008; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mehta MM, Boit TB, Dander JE, Garg NK, Org. Lett 2020, 22, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Johnstone RAW, Wilby AH, Entwistle ID, Chem. Rev 1985, 85, 129–170. [Google Scholar]
- [21]. This catalytic reductive arylation protocol complements the known catalytic reductive alkylation and reductive allylation methods shown in Figure 1b.
- [22].(a) Despite its air-sensitivity, Ni(cod)2 has been used in more than 800 synthetic methodology studies (see ref 23a). Moreover, it has been used in several process research studies, suggesting its value in manufacturing. For select studies, see: Dawson DD, Jarvo ER, Org. Process Res. Dev 2015, 19, 1356–1359; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu J, Gao S, Chen M, Org. Process Res. Dev 2019, 23, 1659–1662. [Google Scholar]
- [23].a) For the use of Ni(cod)2 on the benchtop, paraffin wax encapsulation has proven to be an effective strategy, including for the Suzuki–Miyaura coupling of aliphatic amides; see: Dander JE, Weires NA, Garg NK, Org. Lett 2016, 18, 3934–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mehta MM, Boit TB, Dander JE, Org. Lett 2020, 22, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. In our prior studies, we have shown that the Suzuki–Miyaura coupling of aliphatic amides requires high temperatures, presumably to facilitate the transmetalation step.
- [25].(a) For seminal publications, see: Meerwein H, Schmidt R, Liebigs Ann 1925, 444, 221–238; [Google Scholar]; (b) Verley A, Bull. Soc. Chim. Fr 1925, 37, 871–874; [Google Scholar]; (c) Ponndorf WZ, Angew. Chem 1926, 39, 138–143. [Google Scholar]
- [26].(a) For reviews, see: Cha JS, Org. Process Res. Dev 2006, 10, 1032–1053; [Google Scholar]; (b) de Graauw CF, Peters JA, van Bekkum H, Huskens J, Synthesis 1994, 1007–1017; [Google Scholar]; (c) Inch TD, Synthesis 1970, 466–473. [Google Scholar]; (d) Nishide K, Node M, Chirality 2002, 14, 759–767; [DOI] [PubMed] [Google Scholar]; (e) Ooi T, Miura T, Itagaki Y, Ichikawa H, Maruoka K, Synthesis 2002, 279–291; [Google Scholar]; (f) Wilds AL, Org. React 2011, 2, 178–223; [Google Scholar]; (g) Djerassi, C. Org. React 2011, 6, 207–272. [Google Scholar]
- [27]. When using i-PrOH as the solvent, up to 39% yield of the corresponding ester product was observed by 1H NMR.
- [28].Boit TB, Mehta MM, Garg NK, Org. Lett 2019, 21, 6447–6451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. We anticipated that the stability of the doubly vinylogous amide byproduct resulting from oxidation of DMPE (7) would drive the transfer hydrogenation reaction forward.
- [30].(a) Verheyen T, van Turnhout L, Vandavasi JK, Isbrandt ES, De Borggraeve WM, Newman SG, J. Am. Chem. Soc 2019, 141, 6869–6874; [DOI] [PubMed] [Google Scholar]; (b) Bera S, Bera A, Banerjee D, Chem. Commun 2020, 56, 6850–6853; [DOI] [PubMed] [Google Scholar]; (c) Berini C, Brayton DF, Mocka C, Navarro O, Org. Lett 2009, 11, 4244–4247. [DOI] [PubMed] [Google Scholar]
- [31]. It should be noted that for subsequent evaluation of the scope of the methodology, boronates derived from both pinacol and neopentyl glycol were used. We generally recommend the use of boronates derived from neopentyl glycol. However, the superior commercial availability of boronates derived from pinacol can sometimes be advantageous.
- [32]. Notably, the reductive arylation of amide 1 could be performed on the benchtop by employing either (a) a paraffin wax capsule charged with the precatalyst/ligand combination, Ni(cod)2/Benz-ICy•HCl, or (b) the air-stable Ni(II) precatalyst [(TMEDA)Ni(o-tolyl)Cl]. See Supporting Information for details.
- [33]. When amides derived from benzoic acids were employed, using a Ni(cod)2 / SIPr catalyst/ligand system and 3-pentanol as the alcohol reductant in toluene at 50 °C for 16 h, the corresponding ester was observed in 85% yield as determined by 1H NMR analysis using hexamethylbenzene as an external standard.
- [34]. Notably, under our standard Suzuki–Miyaura coupling conditions of the parent amide, epimerization of the stereocenter α to the amide carbonyl is observed affording a mixture of cis and trans diastereomers (see ref. 19b.)
- [35].(a) For transition metal-catalyzed cleavage of the acyl C–O bond of esters, see: Muto K, Yamaguchi J, Musaev DG, Itami K, Nat. Commun 2015, 6, 7508; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) LaBerge NA, Love JA, Eur. J. Org. Chem 2015, 5546–5553; [Google Scholar]; (c) Halima TB, Vandavasi JK, Shkoor M, Newman SG, ACS Catal 2017, 7, 2176–2180; [Google Scholar]; (d) Kruckenberg A, Wadepohl H, Gade LH, Organometallics 2013, 32, 5153–5170; [Google Scholar]; (e) Yue H, Guo L, Liao H-H, Cai Y, Zhu C, Rueping M, Angew. Chem. Int. Ed 2017, 56, 4282–4285; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 4346–4349; [Google Scholar]; (f) Yu H, Guo L, Lee S-C, Liu X, Rueping M, Angew. Chem. Int. Ed 2017, 56, 3972–3976; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2017, 129, 4030–4034; [Google Scholar]; (g) Halima TB, Zhang W, Yalaoui I, Hong X, Yang Y, Houk KN, Newman SG, J. Am. Chem. Soc 2017, 139, 1311–1318; [DOI] [PubMed] [Google Scholar]; (h) Halima TB, Masson-Makdissi J, Newman SG, Angew. Chem. Int. Ed 2018, 57, 12925–12929; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 13107–3111; [Google Scholar]; (i) Zheng Y-L, Newman SG, ACS Catal 2019, 9, 4426–4433; [Google Scholar]; (j) Zheng Y-L, Newman SG, Angew. Chem. Int. Ed 2019, 58, 18159–18164; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 18327–18322. [Google Scholar]
- [36].Vitaku E, Smith DT, Njardarson JT, J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- [37].Collins KD, Glorius F, Nat. Chem 2013, 5, 597–601. [DOI] [PubMed] [Google Scholar]
- [38]. The robustness screen was carried out using amide 21 and boronate 6, with one equivalent of additive. Additives 22–27 were recovered in near quantitative yields and did not significantly hinder the reductive arylation reaction. See Supporting Information for details.
- [39]. The amide coupling partner was varied in order to simplify isolation of purified products.
- [40].(a) For nickel-catalyzed cleavage of the aryl C–O bond of aryl ethers, see: Wenkert E, Michelotti EL, Swindell CS, J. Am. Chem. Soc 1979, 101, 2246–2247; [Google Scholar]; (b) Wenkert E, Michelotti EL, Swingdell CS, Tingoli M, J. Org. Chem 1984, 49, 4894–4899; [Google Scholar]; (c) Dankwardt JW, Angew. Chem. Int. Ed 2004, 43, 2428–2432; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2004, 116, 2482–2486; [Google Scholar]; (d) Guan B-T, Xiang S-K, Wu T, Sun Z-P, Wang B-Q, Zhao K-Q, Shi Z-J, Chem. Commun 2008, 1437–1439; [DOI] [PubMed] [Google Scholar]; (e) Tobisu M, Shimasaki T, Chatani N, Angew. Chem. Int. Ed 2008, 47, 4866–4869; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 4944–4947; [Google Scholar]; (f) Sergeev AG, Hartwig JF, Science 2011, 322, 439–443. [DOI] [PubMed] [Google Scholar]
- [41].For the nickel-catalyzed cleavage of the aryl C–N bond of dimethylanilines, see: Cao Z-C, Xie S-J, Fang H, Shi Z-J, J. Am. Chem. Soc 2018, 140, 13575–13579. [DOI] [PubMed] [Google Scholar]
- [42]. When boronates bearing para electron-donating groups were employed, a significant amount of the ketone intermediate was observed. For example, the coupling of amide 1 with p-NMe2Ph–B(pin) under our standard reaction conditions gave the corresponding ketone and alcohol in 17% and 42% yields, respectively.
- [43].Rombouts FJR, Trabanco-Suárez AA, Gijsen HJM, Macdonald GJ, Bischoff FP, Alonso-de Diego S-A, Velter AI, Van Roosbroeck YEM (Janssen Pharmaceuticals, Inc.), WO2013171712A1, 2013. [Google Scholar]
- [44].Zhao S, Guo Y, Han E-J, Luo J, Liu H-M, Liu C, Xie W, Zhang W, Wang M, Org. Chem. Front 2018, 5, 1143–1147. [Google Scholar]
- [45]. An exciting opportunity lies in catalytic asymmetric variants of our strategy. In an encouraging preliminary result, we have found that the use of a chiral NHC ligand allows for the synthesis of 4 in 36% yield and 20% ee. See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.