

Abstract
Traumatic brain injury (TBI) is a leading cause of death and disability. Patients with isolated TBI lose a limited amount of blood to primary injury, but they often develop secondary coagulopathy, resulting in delayed or recurrent intracranial and intracerebral hematoma. TBI-induced coagulopathy is closely associated with poor outcomes for these patients, including death. This secondary coagulopathy is consumptive in nature, involving not only brain-derived molecules, coagulation factors, and platelets, but also endothelial cells in a complex process now called endotheliopathy. A key question is how a localized injury to the brain is rapidly disseminated to affect hemostasis systemically, as this is not directly affected the way it is in trauma to the body and limbs, especially with hemorrhagic shock. Increasing evidence suggests that the adhesive ligand von Willebrand factor (VWF), which is synthesized in and released from endothelial cells, plays a paradoxical role in both facilitating local hemostasis at the site of injury and also propagating TBI-induced endotheliopathy and coagulopathy systemically. This review discusses recent progress in understanding these diverse activities of VWF and the knowledge gaps in defining their roles in TBI and associated coagulopathy.
Keywords: Traumatic brain injury, coagulopathy, endotheliopathy, VWF, ADAMTS-13, extracellular vesicles
Trauma is the leading cause of death among people under 50 years of age, more than cancer and cardiovascular diseases combined, and contributes substantially to healthcare costs and lost productivity. Uncontrolled hemorrhage accounts for 30–50% of all trauma fatalities [1, 2] and is caused by direct injury to the vasculature and secondary coagulopathy. This secondary coagulopathy is increasingly recognized as an integral part of hemorrhage-induced dysregulation of microvascular endothelial cells (endotheliopathy), which can progress into hemorrhagic blood failure [3, 4], leading to multiple organ failure, and late death in survivors of hemorrhage [5, 6]. The new term “hemorrhagic blood failure” defines the complex interplay among blood components (cells and soluble factors), the endothelium, and oxygen deficits (shock) during acute trauma [4]. This term combines endotheliopathy, which defines primary and secondary endothelial injuries after trauma, with coagulopathy and tissue ischemia due to severe blood loss. Although trauma prevention is primarily a public safety issue, preventing and treating secondary coagulopathy is a major goal of trauma resuscitation. Secondary coagulopathy after trauma and hemorrhagic shock is caused by significant blood loss due to vascular injury (hemorrhagic shock), hemodilution and hypothermia due to fluid resuscitation, metabolic acidosis due to tissue ischemia, and dysfunctional platelets, coagulation, and fibrinolysis [7–11].
TRAUMATIC BRAIN INJURY-INDUCED COAGULOPATHY
Secondary coagulopathy is equally common in patients with TBI [12–14], even though these patients do not suffer significant blood loss; are restricted in fluid resuscitation to prevent high intracranial pressure, cerebral edema, and tissue ischemia; rarely develop hypothermia (but often develop fever); and are unlikely to develop systemic metabolic acidosis. These differences suggest that TBI-induced coagulopathy (TBI-IC) differs mechanistically from coagulopathy arising after extracranial trauma and hemorrhagic shock. TBI-IC has been extensively reported in the literature for its clinical presentations, confounding factors, and association with poor outcomes [15]. While traditional laboratory hemostatic assays (e.g., INR and platelet counts), which focus on individual components of hemostatic system, have been widely used to diagnose TBI-IC, there is increasing use of viscoelastic assays (e.g., thromboelastogram) to globally evaluate hemostasis in patients with acute TBI. In a study of 572 patients with either TBI, extracranial injures or combined, TBI-IC is independently associated with activated clotting time (<128 seconds), angle (<65 degrees), and functional fibrinogen levels (<356) but not with maximum amplitude, hyperfibrinolysis, fibrinolysis shutdown, or partial thromboplastin time [16]. However, the underlying mechanism of TBI-IC remains poorly understood. This lack of understanding has significantly hindered the development of more effective and targeted treatments. A critical question is how a localized brain injury is rapidly disseminated to alter hemostasis systemically, which is not directly affected as in patients with extracranial trauma and hemorrhagic shock. While critical for local hemostasis at the site of injury, increasing evidence suggests that the adhesive ligand VWF is also a key causal and disseminating factor in secondary endotheliopathy and coagulopathy. These paradoxical activities may be attributed to the unique structures and adhesive activities of VWF in resting state and during the acute phase reaction of TBI.
STRUCTURE AND FUNCTION OF VWF
VWF is a large multimeric glycoprotein, encoded by the VWF gene on the short arm of chromosome 12 [17, 18]. It is synthesized as a single-chain propolypeptide of 2,813 amino acids and contains multiple and repeated domains [19, 20] . The A1 domain contains the binding site for glycoprotein (GP) Ibα, which is the ligand binding subunit of the VWF receptor GP Ib-IX-V complex on the surface of platelets. The A3 domain binds collagen, which is a major component of the subendothelial matrix. Through these two domains, VWF captures platelets to the endothelial matrix exposed at the site of vascular injury to initiate the process of forming a hemostatic plug. The A2 domain contains the Y1605-M1606 peptide bond, which is cleaved by the metalloprotease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, number 13) to reduce the size and adhesive activity of VWF [21, 22]. The C domain contains an RGD sequence that binds integrins.
VWF is synthesized primarily in endothelial cells and megakaryocytes. In the endoplasmic reticulum of these cells, newly synthesized pro-VWF monomers first dimerize through disulfide bonds in the CK domain [23]. A variable number of dimers then multimerize through N-terminal disulfide bonds in the Golgi and post-Golgi compartments, where posttranslational modifications also occur [24, 25]. This multi-domain multimeric structure allows VWF to bind multiple receptors on a platelet to induce activation (i.e., receptor crosslinking) [26, 27] and on different platelets and other cells (i.e., cell-coupling) to aggregate them, for example under high fluid shear stress [28]. Upon synthesis, VWF is either secreted constitutively as smaller multimers or stored in Weibel-Palade bodies of endothelial cells and α-granules of platelets, where multimerization continues. As a result, the VWF in these storage granules is enriched in large and ultra-large forms of multimers (ULVWF) [24, 28].
When activated or injured, human endothelial cells release stored ULVWF multimers [28–30] and anchor them to their surfaces with CD62p [31] and/or integrin αvβ3 [32] in vitro. ULVWF multimers are also anchored to the endothelial surface in vivo in a mouse model, but the anchorage appears to require other molecules [33]. It has recently been reported in a mouse model of ischemic stroke that the cytoskeletal protein vimentin is translocated to the surface of activated endothelial cells and anchors ULVWF multimers [34]. The anchored ULVWF multimers are stretched by the hydrodynamic forces of blood flow (unfolding), and then rapidly but partially cleaved by ADAMTS13 before release into circulation [35]. Hydrodynamic forces generated by blood flow are therefore a key factor in regulating the rate and location of VWF cleavage [35–37]. Because of cleavage, circulating VWF multimers are smaller in molecular mass and, more importantly, bind platelets poorly in solution unless they are immobilized onto the subendothelial collagen at the site of vascular injury or activated by high fluid shear stress [38–41]. By contrast, ULVWF multimers freshly released from activated endothelial cells and platelets are intrinsically active and hyper-adhesive, capable of spontaneously binding platelets in solution and endothelial cells [42, 43]. This hyperadhesive activity can be defined as forming multivalent and thus stronger bonds with the receptor [44]. This difference in adhesive activity also suggests that the receptor-binding A1 domain is hidden in the globular structure of less active plasma VWF multimers but exposed on the surface of hyper-adhesive ULVWF multimers [45, 46]. The fact that both VWF and constitutively active ADAMTS13 circulate in blood during homeostasis but VWF is not completely cleaved to minimal fragments, suggests that ADAMTS13 primarily cleaves ULVWF anchored to the endothelial surface and has less or no activity toward the circulating plasma type of VWF multimers. The limited and selective cleavage of ULVWF multimers by ADAMTS-13 therefore serves as a safeguard against spontaneous intravascular platelet aggregation induced by hyperadhesive ULVWF multimers and resultant thromboembolism, as is seen in patients with thrombotic thrombocytopenic purpura [22, 47, 48], while maintaining the basic hemostatic activity of VWF. This safeguard is disrupted during acute TBI.
VWF IN TRAUMATIC BRAIN INJURY-INDUCED COAGULOPATHY
As an acute phase reactant, VWF is a key part of the hemostatic defense system for preventing blood loss upon vascular injury. Individuals with VWF deficiency (von Willebrand disease [49]) not only develop spontaneous mucocutaneous bleeding (in rare cases, intracranial hemorrhage) but also suffer much severe hemorrhage upon injury [50–52]. The level of plasma VWF is approximately 10 μg/ml at baseline [53], but it can increase more than 100% during acute TBI, and it is thus widely used as a marker for endothelial injury [54–56]. A high level of circulating VWF is closely associated with severity of injury and poor prognosis [54–57]. However, there is increasing evidence that VWF is not merely a marker but may also promote TBI-IC by activating platelets to express procoagulant activity and produce procoagulant extracellular vesicles to propagate consumptive coagulopathy. VWF can also mediate the adhesion of platelets, leukocytes, and extracellular vesicles to endothelial cells to facilitate inflammatory endotheliopathy at the site of injury and elsewhere [35, 58–65]. These non-hemostatic activities may be associated with hyperadhesive VWF multimers found during the acute phase reaction of TBI.
We recently demonstrated in mouse models of lateral fluid percussion injury that TBI induces a rapid but transient increase in plasma VWF, reaching its peak 3 hours post-injury [59]. VWF found during acute TBI is also hyper-adhesive, as defined by its enhanced ability to bind collagen and by the elevated levels of circulating VWF-bound extracellular vesicles from platelets [59, 66]. This hyper-adhesive activity continues to be detected after adjustment for high VWF antigen, suggesting that VWF is intrinsically active, as compared to the plasma VWF found in healthy subjects. Consistent with these findings, VWF-containing microvascular thrombosis has been detected in the peri-contusion cortex and the hippocampal CA3 region of rats subjected to severe TBI, potentially contributing to TBI-induced cerebral ischemia [67]. These findings suggest that the measurement of plasma antigen alone is not sufficient to define the pathological activity of VWF, and it raises the question why VWF becomes hyperadhesive during acute TBI. While no direct evidence available in the literature, circumstantial evidence suggests that VWF may become hyperadhesive (or activated) through several distinct but also closely related pathways.
Slowing the kinetics of VWF cleavage by ADAMTS-13
The molar ratio of ADAMTS-13 and VWF is approximately 1:7 in health subjects during homeostasis, sufficient for maintaining basal hemostasis without causing either bleeding or thrombosis. However, patients with acute TBI have a lower ratio of ADAMTS-13 activity to VWF antigen in their plasma [56]. This clinical finding is supported by the enhanced VWF adhesive activity in mice subjected to severe TBI [59, 66]. One likely explanation for this TBI-induced imbalance between VWF and ADAMTS-13 is the substantial release of VWF from injured or activated endothelial cells. The VWF release is more significant during TBI because the vascular network of the brain is far denser than that of other organs in order to accommodate 20% of total cardiac output, even though the human brain makes up only 2% of total body weight [68]. Cerebral endothelial cells also appear to express a higher level of VWF than other vascular beds [69, 70]. In contract, plasma ADAMTS-13 is not increased but rather moderately reduced during acute TBI [56] This moderate reduction in circulating ADAMTS-13 is associated with coagulopathy, endothelial damage, and mortality [56]. There is no an intracellular storage pool of ADAMTS-13, as it is for VWF and, while not examined specifically in TBI, an analysis of the ADAMTS13 gene promotor has indicated that the ADAMTS13 transcription is not changed after being exposed to proinflammatory stimuli such as endotoxin, TNF-α, IL-6, and IL-1β, which are known to trigger the acute phase reaction [71]. Endothelial cells and platelets also synthesize or contain ADAMTS-13 and may release it upon activation [72, 73], but their contribution to plasma ADAMTS-13 is presently not known. In addition, TBI patients may have reduced ADAMTS-13 synthesis because of preexisting or trauma-induced pathologies of the liver [74], the primary site of ADAMTS-13 synthesis [75, 76]. Finally, TBI and resultant cerebral edema increase intracranial pressure, which reduces cerebral blood flow and thus the fluid shear stress critical for cleaving ULVWF multimers. As a result, the amount of ULVWF multimers released from injured endothelial cells may significantly overwhelm the capacity of ADAMTS-13 to cleave them, slowing the kinetics of VWF cleavage.
Consistent with this TBI-induced VWF and ADAMTS-13 imbalance, we have shown that exogenous recombinant ADAMTS-13 given 30 min after TBI reduces injury-induced vascular leakage, consumptive coagulopathy, and neurological deficits in wild-type VWF sufficient C57Bl/6J mice, without impairing the baseline hemostasis [59]. We also found that mice with ADAMTS-13 deficiency on the background of high plasma VWF [77] develop worse coagulopathy, have severe neurological deficiencies, and suffer more death during acute TBI [59]. The size of VWF multimers has not been examined in patients with acute TBI and hemorrhagic shock, but the proportion of large VWF multimers (>5000 kDa) is increased in patients with trauma compared to small multimers (≤5000 kDa). These trauma patients also develop moderate ADAMTS-13 deficiency [78], suggesting that VWF multimers are less cleaved.
Modifying VWF adhesive activity by inflammatory and oxidative mediators
TBI is known to induce extensive oxidative stress [79, 80], which can chemically modify VWF multimers and ADAMTS-13, making the former hyperadhesive and resistant to cleavage and the latter less active in cleaving VWF in vitro [81–83]. The oxidation can occur on both cysteine and methionine residues at multiple locations. Free cysteine thiols in the C domains can be oxidized to form intermultimer disulfide bonds and laterally associated fibrils [84, 85]. The methionine residue 1606 at the cleavage site can be oxidized, rendering VWF resistant to ADAMTS-13 [82]. Furthermore, ADAMTS-13 can also be oxidized to become less active in its enzymatic activity [83] and potentially lose its disulfide bond-reducing activity [86], aggravating the deficit of VWF cleavage caused by increased VWF release. Though not specifically studied in a TBI setting, these in vitro observations provide extensive but circumstantial evidence to suggest that TBI-induced oxidative stress may be a factor in the development of TBI-induced imbalance between VWF and ADAMTS-13. Our recent study provides a source of oxidative stress in blood during acute TBI. We detected morphologically intact or partially damaged extracellular mitochondria in the peripheral blood of mice subjected to acute TBI [87]. These cell-free mitochondria expose the anionic phospholipid cardiolipin to promote coagulation [87], and are also metabolically competent to produce reactive oxygen species, which activate platelets to express anionic phospholipids and undergo microvesiculation to release procoagulant extracellular vesicles [88].
In addition, ADAMTS-13 activity may also be inhibited by inflammatory cytokines such as IL-6 [89], which is significantly elevated in the peripheral blood of TBI patients [90, 91]. These individual reports call for more comprehensive studies to mechanistically define the overall influence of oxidative stress and inflammation on the pathogenesis of TBI-IC, beyond clinical association studies.
Altering VWF structure
One structural difference between intrinsically active ULVWF and less active plasma VWF multimers is that the receptor-binding A1 domain is hidden in the globular structure of plasma VWF but exposed on the surface of ULVWF multimers [45, 46, 81, 92]. The A1 domain is hidden in plasma VWF because it forms a complex with the adjacent A2 domain [93–95]. High fluid shear stress appears to disrupt this complex because it facilitates VWF binding to its receptor GP Ibα on platelets [96, 97] and its cleavage by ADAMTS-13 [36, 37]; however, the differential requirements for the two processes are not known. By contrast, the A1 domain is exposed on ULVWF multimers, as it is readily detected in VWF from TTP patients using a nanobody made against the A1 domain [92]. Another nanobody that blocks VWF and GP Ibα interaction [98] has been developed into a humanized single-variable domain immunoglobulin (caplacizumab) and has been approved for treating patients with TTP [99, 100]. We have recently shown that the recombinant VWF A2 protein binds to the exposed A1 domain to form complex with active VWF multimers in plasma of TBI mice. The binding blocks the hyper-adhesive activity of VWF to reduce vascular permeability and cerebral edema, increase cerebral blood flow, prevent TBI-induced consumptive coagulopathy, and improve the outcome of TBI [66]. Two recent studies have provided a structural basis for the two states of the A1-A2 interaction. They show that an A1-A2 complex is formed when the two vicinal cysteine residues (C1669-C1670) in the A2 domain are reduced (free thiols), but that it disassociates when the two cysteines are oxidized [101, 102]. This finding offers a novel pathway for activating plasma VWF in an oxidative stress environment, which is well-documented in acute and severe TBI and means to selectively block the activated VWF.
Although they have been discussed individually, these potential causes of VWF hyperadhesive activity are not mutually exclusive, but indeed are closely associated and in some cases synergic. However, we are still at an early stage in distinguishing the roles of VWF multimers in ensuring hemostasis and contributing to the pathogenesis of TBI-IC. For example, thromboelastography detects multiple variations in its hemostatic profile and is increasingly used to evaluate hemostasis during acute TBI [16, 103] , but the impact of VWF on these thromboelastographic parameters is not known. Changes in platelet reactivity identified by thromboelastography in patients with TBI could be further examined for the impact of VWF.
VWF ON VASCULAR PERMEABILITY
A hallmark of trauma-induced endotheliopathy is dysfunction of the endothelial barrier. We have recently shown that extracellular vesicles released from injured brains are not only procoagulant and responsible for the development of consumptive coagulation, but also disrupt the integrity of the endothelial barrier, resulting in vascular leakage and interstitial bleeding in the brain and lungs [62]. This extracellular vesicle-induced vascular permeability is mediated by hyperadhesive VWF bound to the vesicles [66] because it is blocked by a VWF polyclonal antibody [59] and by the recombinant A2 protein, which forms a complex with the A1 domain exposed on hyperadhesive VWF found in TBI mice [66]. Furthermore, VWF on the surface of extracellular vesicles from platelets, endothelial cells, glial cells, and neurons may mediate these vesicles to interact with other target cells and deliver biologically active cargo molecules carried by these vesicles [66]. In addition to mediating the adhesion of extracellular vesicles to endothelial cells, VWF can also induce endothelial permeability by recruiting platelets, which have been shown to induce vascular permeability in conditions of inflammation [60, 61]. We have shown that extracellular vesicle-induced endothelial permeability is enhanced by live but not lyophilized platelets [62], suggesting that VWF activates platelets to release biologically active molecules such as vascular endothelial growth factor, that are known to disrupt the endothelial barrier [104, 105].
In contrast to the detrimental effects of hyperadhesive VWF, several reports have demonstrated the importance of VWF for maintaining endothelial homeostasis. VWF-deficient mice have an elevated expression of the tight junction protein claudin-5 and improved integrity of the blood-brain barrier, but paradoxically are sensitized for hypoxia-induced endothelial injury and permeability [106]. VWF-deficient mice and VWF-efficient mice treated with a VWF antibody both developed an alleviated permeability of the blood-brain barrier after being subjected to acute ischemic and hemorrhagic stroke [107, 108]. One potential explanation for the phenotype is that VWF deficiency results in the defective biogenesis of Weibel-Palade bodies, where CD62p also resides, leading to reduced CD62p expression and recruitment of leukocytes to the endothelium [109]. Studying VWF or ADAMTS-13-deficient mice may provide vital information about the likely outcomes for patients with von Willebrand disease or TTP, but the findings may not be directly applicable to TBI patients with significantly elevated plasma VWF and moderately reduced ADAMTS-13 activity [55, 56]. For example, VWF deficiency inevitably results in more severe bleeding upon traumatic injury, which is a confounding variable that is difficult to stratify for the purpose of evaluating endothelial barrier function and secondary coagulopathy after injury. In VWF-sufficient mice, both exogenous ADAMTS13 and the blocking of active VWF protect the integrity of the blood-brain barrier after TBI [59, 66] and acute ischemic stroke [107, 110].
Given the significant variation in VWF expression among different regions of the brain [69, 70], the impact of VWF may also depend on the type and location of injury. For example, ischemic stroke induces primarily cortical tissue ischemia, whereas hemorrhagic stroke often occurs in the basal ganglia. Traumatic injury induces an initial physical impact to the cortical tissue followed by disseminated ischemic and inflammatory secondary injuries to the cortical and grey matter tissues.
SUMMARY
Secondary coagulopathy is a life-threatening complication of TBI that remains difficult to manage clinically. Current laboratory tests can accurately diagnose coagulopathy after it has occurred but cannot accurately predict when it will occur or how severe it will become. As a result, there are no effective and targeted prophylactic measures for preventing TBI-IC or reducing its severity. Recent studies have begun to distinguish consumptive coagulopathy induced by TBI from that induced by extracranial injuries and hemorrhagic shock. They have also begun recognizing the diverse activities of VWF for facilitating trauma hemostasis and promoting TBI-IC, as summarized in Figure 1. However, key knowledge gaps remain. For example, can hemostatic and proinflammatory activities be attributed to different types of VWF multimers? What laboratory tests are appropriate for accurately differentiating these diverse activities of VWF? Studies of VWF structure and function correlation are mostly focused on the homologous A domains, and the question remains whether structural changes in other domains influence VWF interaction with other partners, such as FVIII and integrins, in an acute injury? Can the hyperadhesive activity of VWF be selectively blocked with minimal influence on its hemostatic activity? These questions can be answered by examining not only VWF antigen, but also changes in structure and activity in combination with the evaluation of ADAMTS-13 synthesis, release, and activity in patients. Findings from clinical studies could help to develop a panel of complementary laboratory tests that globally evaluate VWF functions. These clinical evaluations can provide new markers for predict the role of VWF in causing TBI-IC and also identify new agents that can be evaluated in vitro and in animal models for reducing the hyperadhesive activity VWF while maintaining its basic hemostasis activity.
Figure 1: The role of VWF in TBI-induced coagulopathy.
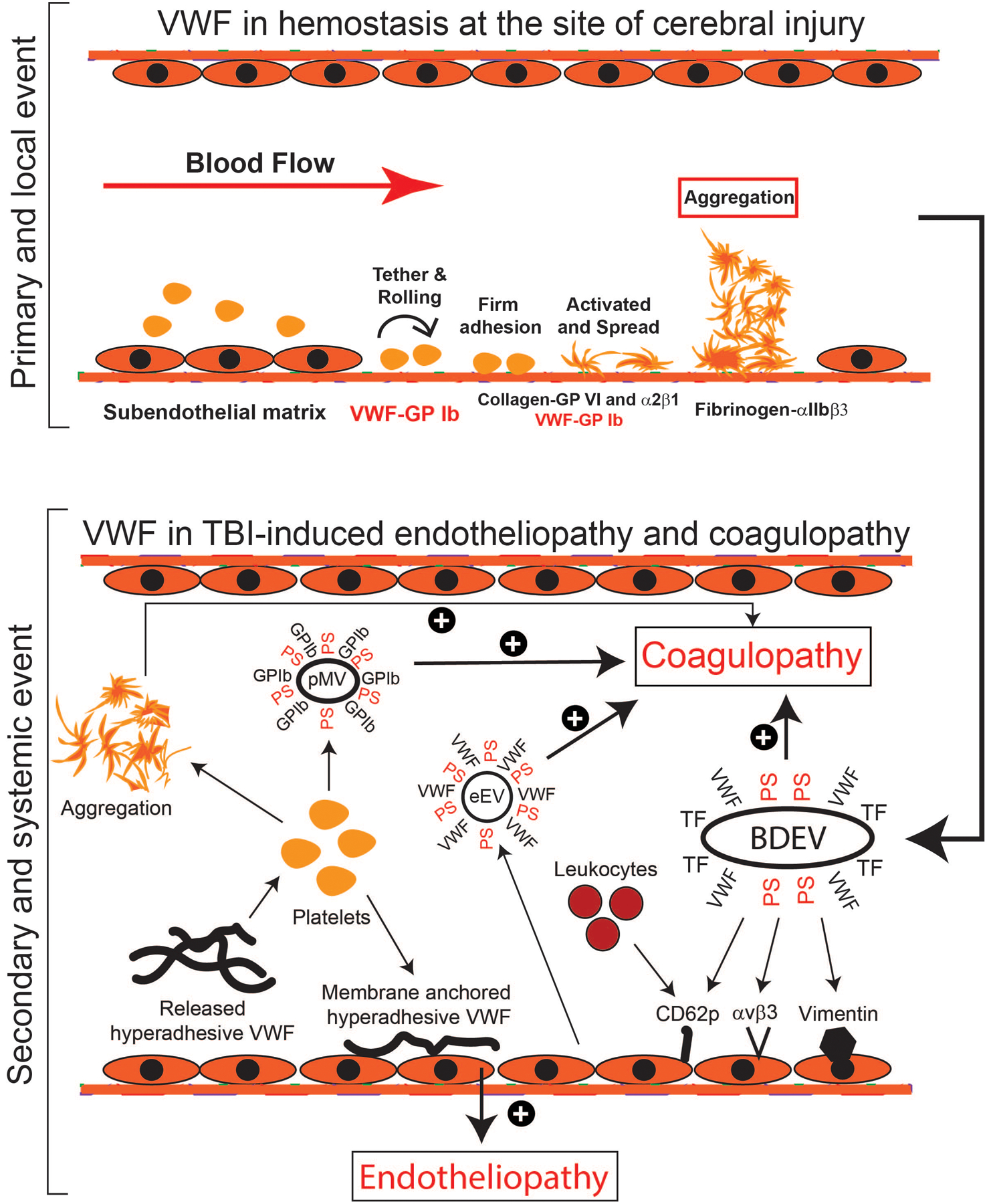
TBI induces two closely related, but distinct processes: local hemostasis to seal the vascular injury (top panel) and systemic endotheliopathy and coagulopathy that affect remote cerebral tissue and remote organs (bottom panel). VWF is critical for both processes. For the former, VWF immobilized to the subendothelium captures platelets to the site of injury and contributes to the subsequent platelet activation and aggregation to seal the vascular injury. For the latter, trauma and resultant cerebral ischemia induce the injured brain cells to release brain-derived extracellular vesicles (BDEVs) that express the procoagulant anionic phosphatidylserine (PS), tissue factor (TF), and potentially other biologically active molecules, activate endothelial cells to release uncleaved and hyperadhesive (UL)VWF, and disrupts the blood-brain barrier. The hyperadhesive VWF binds BDEVs to become VWF-bound BDEVs that are released into circulation. These VWF-bound BDEVs bind and activate endothelial cells in the vascular beds remote from the injury site to release procoagulant extracellular vesicles (eEVs) and more intrinsically active VWF multimers. These active VWF multimers can be either anchored to endothelial surface through anchor molecules (e.g., CD62p, integrin αvβ3, and vimentin) to capture platelets and leukocytes, inducing secondary endotheliopathy or released into circulation, where they bind and activate platelets to express procoagulant activity and to release procoagulant extracellular vesicles (pEVs). Extracellular vesicles from multiple types of cells express abundant procoagulant anionic phospholipids and some also express tissue factor, causing a systemic hypercoagulable state that rapidly turns into consumptive coagulopathy.
Acknowledgments
The work is supported by NIH grant HL152200 (JFD), GM107482 (RAK), and National Natural Science Foundation of China grants 81930031 and 81720108015 (JNZ)
Footnotes
CONFLICT OF INTEREST STATEMENT
All authors claim no relevant conflict of interest
References
- 1.Niles SE, McLaughlin DF, Perkins JG, Wade CE, Li Y, Spinella PC, Holcomb JB. Increased mortality associated with the early coagulopathy of trauma in combat casualties. J Trauma. 2008; 64: 1459–63; discussion 63–5. 10.1097/TA.0b013e318174e8bc. [DOI] [PubMed] [Google Scholar]
- 2.Frith D, Brohi K. The acute coagulopathy of trauma shock: clinical relevance. The surgeon : journal of the Royal Colleges of Surgeons of Edinburgh and Ireland. 2010; 8: 159–63. 10.1016/j.surge.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 3.Bjerkvig CK, Strandenes G, Eliassen HS, Spinella PC, Fosse TK, Cap AP, Ward KR. “Blood failure” time to view blood as an organ: how oxygen debt contributes to blood failure and its implications for remote damage control resuscitation. Transfusion. 2016; 56 Suppl 2: S182–9. 10.1111/trf.13500. [DOI] [PubMed] [Google Scholar]
- 4.White NJ, Ward KR, Pati S, Strandenes G, Cap AP. Hemorrhagic blood failure: Oxygen debt, coagulopathy, and endothelial damage. The journal of trauma and acute care surgery. 2017; 82: S41–S9. 10.1097/TA.0000000000001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carroll RC, Craft RM, Langdon RJ, Clanton CR, Snider CC, Wellons DD, Dakin PA, Lawson CM, Enderson BL, Kurek SJ. Early evaluation of acute traumatic coagulopathy by thrombelastography. Translational research : the journal of laboratory and clinical medicine. 2009; 154: 34–9. 10.1016/j.trsl.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Floccard B, Rugeri L, Faure A, Saint Denis M, Boyle EM, Peguet O, Levrat A, Guillaume C, Marcotte G, Vulliez A, Hautin E, David JS, Negrier C, Allaouchiche B. Early coagulopathy in trauma patients: an on-scene and hospital admission study. Injury. 2012; 43: 26–32. 10.1016/j.injury.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Wafaisade A, Wutzler S, Lefering R, Tjardes T, Banerjee M, Paffrath T, Bouillon B, Maegele M, Trauma Registry of DGU. Drivers of acute coagulopathy after severe trauma: a multivariate analysis of 1987 patients. Emergency medicine journal : EMJ. 2010; 27: 934–9. 10.1136/emj.2009.088484. [DOI] [PubMed] [Google Scholar]
- 8.Maani CV, DeSocio PA, Holcomb JB. Coagulopathy in trauma patients: what are the main influence factors? Current opinion in anaesthesiology. 2009; 22: 255–60. 10.1097/ACO.0b013e32832922be. [DOI] [PubMed] [Google Scholar]
- 9.Shaz BH, Winkler AM, James AB, Hillyer CD, MacLeod JB. Pathophysiology of early trauma-induced coagulopathy: emerging evidence for hemodilution and coagulation factor depletion. J Trauma. 2011; 70: 1401–7. 10.1097/TA.0b013e31821266e0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport RA, Brohi K. Coagulopathy in trauma patients: importance of thrombocyte function? Current opinion in anaesthesiology. 2009; 22: 261–6. 10.1097/ACO.0b013e328325a6d9. [DOI] [PubMed] [Google Scholar]
- 11.Mitra B, Cameron PA, Mori A, Fitzgerald M. Acute coagulopathy and early deaths post major trauma. Injury. 2012; 43: 22–5. 10.1016/j.injury.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 12.Hulka F, Mullins RJ, Frank EH. Blunt brain injury activates the coagulation process. ArchSurg. 1996; 131: 923–7. [DOI] [PubMed] [Google Scholar]
- 13.Hoots WK. Experience with antithrombin concentrates in neurotrauma patients. Seminars in thrombosis and hemostasis. 1997; 23 Suppl 1: 3–16. [PubMed] [Google Scholar]
- 14.Harhangi BS, Kompanje EJ, Leebeek FW, Maas AI. Coagulation disorders after traumatic brain injury. Acta neurochirurgica. 2008; 150: 165–75; discussion 75. 10.1007/s00701-007-1475-8. [DOI] [PubMed] [Google Scholar]
- 15.Yuan Q, Sun YR, Wu X, Yu J, Li ZQ, Du ZY, Wu XH, Zhou LF, Hu J. Coagulopathy in Traumatic Brain Injury and Its Correlation with Progressive Hemorrhagic Injury: A Systematic Review and Meta-Analysis. Journal of neurotrauma. 2016; 33: 1279–91. 10.1089/neu.2015.4205. [DOI] [PubMed] [Google Scholar]
- 16.Samuels JM, Moore EE, Silliman CC, Banerjee A, Cohen MJ, Ghasabyan A, Chandler J, Coleman JR, Sauaia A. Severe traumatic brain injury is associated with a unique coagulopathy phenotype. The journal of trauma and acute care surgery. 2019; 86: 686–93. 10.1097/TA.0000000000002173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadler JE, Shelton-Inloes BB, Sorace JM, Titani K. Cloning of cDNA and genomic DNA for human von Willebrand factor. Cold Spring HarbSympQuantBiol. 1986; 51 Pt 1:515.-.: 515–23. [DOI] [PubMed] [Google Scholar]
- 18.Collins CJ, Underdahl JP, Levene RB, Ravera CP, Morin MJ, Dombalagian MJ, Ricca G, Livingston DM, Lynch DC. Molecular cloning of the human gene for von Willebrand factor and identification of the transcription initiation site. ProcNatlAcadSciUSA. 1987; 84: 4393–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Titani K, Kumar S, Takio K, Ericsson LH, Wade RD, Ashida K, Walsh KA, Chopek MW, Sadler JE, Fujikawa K. Amino acid sequence of human von Willebrand factor. Biochemistry. 1986; 25: 3171–84. [DOI] [PubMed] [Google Scholar]
- 20.Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood. 2012; 120: 449–58. 10.1182/blood-2012-01-405134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furlan M, Robles R, Lammle B. Partial purification and characterization of a protease from human plasma cleaving von Willebrand factor to fragments produced by in vivo proteolysis. Blood. 1996; 87: 4223–34. [PubMed] [Google Scholar]
- 22.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD Jr., Ginsburg D, Tsai HM. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature. 2001; 413: 488–94. [DOI] [PubMed] [Google Scholar]
- 23.Wagner DD, Lawrence SO, Ohlsson-Wilhelm BM, Fay PJ, Marder VJ. Topology and order of formation of interchain disulfide bonds in von Willebrand factor. Blood. 1987; 69: 27–32. [PubMed] [Google Scholar]
- 24.Vischer UM, Wagner DD. von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994; 83: 3536–44. [PubMed] [Google Scholar]
- 25.Mayadas TN, Wagner DD. In vitro multimerization of von Willebrand factor is triggered by low pH. Importance of the propolypeptide and free sulfhydryls. JBiolChem. 1989; 264: 13497–503. [PubMed] [Google Scholar]
- 26.Wu Y, Asazuma N, Satoh K, Yatomi Y, Takafuta T, Berndt MC, Ozaki Y. Interaction between von Willebrand factor and glycoprotein Ib activates Src kinase in human platelets: role of phosphoinositide 3-kinase. Blood. 2003; 101: 3469–76. 10.1182/blood-2002-03-0806. [DOI] [PubMed] [Google Scholar]
- 27.Satoh K, Asazuma N, Yatomi Y, Fujimura Y, Miura S, Titani K, Ozaki Y. Activation of protein-tyrosine kinase pathways in human platelets stimulated with the A1 domain of von Willebrand factor. Platelets. 2000; 11: 171–6. 10.1080/095371000403116. [DOI] [PubMed] [Google Scholar]
- 28.Moake JL, Turner NA, Stathopoulos NA, Nolasco LH, Hellums JD. Involvement of large plasma von Willebrand factor (vWF) multimers and unusually large vWF forms derived from endothelial cells in shear stress-induced platelet aggregation. JClinInvest. 1986; 78: 1456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mayadas T, Wagner DD, Simpson PJ. von Willebrand factor biosynthesis and partitioning between constitutive and regulated pathways of secretion after thrombin stimulation. Blood. 1989; 73: 706–11. [PubMed] [Google Scholar]
- 30.Sporn LA, Marder VJ, Wagner DD. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell. 1986; 46: 185–90. [DOI] [PubMed] [Google Scholar]
- 31.Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, Nolasco L, Turner N, Berndt MC, Anvari B, Lopez JA, Dong JF. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood. 2004; 103: 2150–6. [DOI] [PubMed] [Google Scholar]
- 32.Huang J, Roth R, Heuser JE, Sadler JE. Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood. 2009; 113: 1589–97. 10.1182/blood-2008-05-158584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chauhan AK, Goerge T, Schneider SW, Wagner DD. Formation of platelet strings and microthrombi in the presence of ADAMTS-13 inhibitor does not require P-selectin or beta3 integrin. JThrombHaemost. 2007; 5: 583–9. [DOI] [PubMed] [Google Scholar]
- 34.Fasipe TA, Hong SH, Da Q, Valladolid C, Lahey MT, Richards LM, Dunn AK, Cruz MA, Marrelli SP. Extracellular Vimentin/VWF (von Willebrand Factor) Interaction Contributes to VWF String Formation and Stroke Pathology. Stroke. 2018; 49: 2536–40. 10.1161/STROKEAHA.118.022888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002; 100: 4033–9. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009; 324: 1330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu T, Lin J, Cruz MA, Dong JF, Zhu C. Force-induced cleavage of single VWFA1A2A3 tridomains by ADAMTS-13. Blood. 2010; 115: 370–8. 10.1182/blood-2009-03-210369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadler JE. Biochemistry and genetics of von Willebrand factor. AnnuRevBiochem. 1998; 67:395.-.: 395–424. [DOI] [PubMed] [Google Scholar]
- 39.Fowler WE, Fretto LJ, Hamilton KK, Erickson HP, McKee PA. Substructure of human von Willebrand factor. JClinInvest. 1985; 76: 1491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moake JL, Chow TW. Increased von Willebrand factor (vWf) binding to platelets associated with impaired vWf breakdown in thrombotic thrombocytopenic purpura. JClinApheresis. 1998; 13: 126–32. [DOI] [PubMed] [Google Scholar]
- 41.Counts RB, Paskell SL, Elgee SK. Disulfide bonds and the quaternary structure of factor VIII/von Willebrand factor. JClinInvest. 1978; 62: 702–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furlan M Von Willebrand factor: molecular size and functional activity. AnnHematol. 1996; 72: 341–8. [DOI] [PubMed] [Google Scholar]
- 43.Sporn LA, Marder VJ, Wagner DD. von Willebrand factor released from Weibel-Palade bodies binds more avidly to extracellular matrix than that secreted constitutively. Blood. 1987; 69: 1531–4. [PubMed] [Google Scholar]
- 44.Arya M, Anvari B, Romo GM, Cruz MA, Dong JF, McIntire LV, Moake JL, Lopez JA. Ultralarge multimers of von Willebrand factor form spontaneous high-strength bonds with the platelet glycoprotein Ib-IX complex: studies using optical tweezers. Blood. 2002; 99: 3971–7. [DOI] [PubMed] [Google Scholar]
- 45.Siedlecki CA, Lestini BJ, Kottke-Marchant KK, Eppell SJ, Wilson DL, Marchant RE. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. 1996; 88: 2939–50. [PubMed] [Google Scholar]
- 46.Singh I, Shankaran H, Beauharnois ME, Xiao Z, Alexandridis P, Neelamegham S. Solution structure of human von Willebrand factor studied using small angle neutron scattering. J Biol Chem. 2006; 281: 38266–75. 10.1074/jbc.M607123200. [DOI] [PubMed] [Google Scholar]
- 47.Moake JL, Rudy CK, Troll JH, Weinstein MJ, Colannino NM, Azocar J, Seder RH, Hong SL, Deykin D. Unusually large plasma factor VIII:von Willebrand factor multimers in chronic relapsing thrombotic thrombocytopenic purpura. NEnglJMed. 1982; 307: 1432–5. [DOI] [PubMed] [Google Scholar]
- 48.Joly BS, Coppo P, Veyradier A. Thrombotic thrombocytopenic purpura. Blood. 2017; 129: 2836–46. 10.1182/blood-2016-10-709857. [DOI] [PubMed] [Google Scholar]
- 49.Lillicrap D von Willebrand disease: advances in pathogenetic understanding, diagnosis, and therapy. Blood. 2013; 122: 3735–40. 10.1182/blood-2013-06-498303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leebeek FW, Eikenboom JC. Von Willebrand’s Disease. The New England journal of medicine. 2016; 375: 2067–80. 10.1056/NEJMra1601561. [DOI] [PubMed] [Google Scholar]
- 51.Labarque V, Stain AM, Blanchette V, Kahr WH, Carcao MD. Intracranial haemorrhage in von Willebrand disease: a report on six cases. Haemophilia : the official journal of the World Federation of Hemophilia. 2013; 19: 602–6. 10.1111/hae.12142. [DOI] [PubMed] [Google Scholar]
- 52.Tosetto A, Badiee Z, Baghaipour MR, Baronciani L, Battle J, Berntorp E, Bodo I, Budde U, Castaman G, Eikenboom JCJ, Eshghi P, Ettorre C, Goodeve A, Goudemand J, Charles Richard Morris H, Hoorfar H, Karimi M, Keikhaei B, Lassila R, Leebeek FWG, Lopez Fernandez MF, Mannucci PM, Mazzucconi MG, Morfini M, Oldenburg J, Peake I, Parra Lopez R, Peyvandi F, Schneppenheim R, Tiede A, Toogeh G, Trossaert M, Zekavat O, Zetterberg EMK, Federici AB. Bleeding symptoms in patients diagnosed as type 3 Von Willebrand Disease: results from 3WINTERS-IPS, an international and collaborative cross-sectional study. Journal of thrombosis and haemostasis : JTH. 2020. 10.1111/jth.14886. [DOI] [PubMed] [Google Scholar]
- 53.Horbett TA, Counts RB. von Willebrand Factor/Factor VIII adsorption to surfaces from human plasma. Thrombosis research. 1984; 36: 599–608. [DOI] [PubMed] [Google Scholar]
- 54.Yokota H, Naoe Y, Nakabayashi M, Unemoto K, Kushimoto S, Kurokawa A, Node Y, Yamamoto Y. Cerebral endothelial injury in severe head injury: the significance of measurements of serum thrombomodulin and the von Willebrand factor. Journal of neurotrauma. 2002; 19: 1007–15. 10.1089/089771502760341929. [DOI] [PubMed] [Google Scholar]
- 55.De Oliveira CO, Reimer AG, Da Rocha AB, Grivicich I, Schneider RF, Roisenberg I, Regner A, Simon D. Plasma von Willebrand factor levels correlate with clinical outcome of severe traumatic brain injury. Journal of neurotrauma. 2007; 24: 1331–8. 10.1089/neu.2006.0159. [DOI] [PubMed] [Google Scholar]
- 56.Kumar MA, Cao W, Pham HP, Raju D, Nawalinski K, Maloney-Wilensky E, Schuster J, Zheng XL. Relative Deficiency of Plasma A Disintegrin and Metalloprotease with Thrombospondin Type 1 Repeats 13 Activity and Elevation of Human Neutrophil Peptides in Patients with Traumatic Brain Injury. Journal of neurotrauma. 2019; 36: 222–9. 10.1089/neu.2018.5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kutcher ME, Ferguson AR, Cohen MJ. A principal component analysis of coagulation after trauma. The journal of trauma and acute care surgery. 2013; 74: 1223–9; discussion 9–30. 10.1097/TA.0b013e31828b7fa1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.A B, Ball C, Nolasco L, Choi H, Moake JL, Dong JF. Platelets adhered to endothelial cell-bound ultra-large von willebrand factor strings support leukocyte tethering and rolling under high shear stress. JThrombHemost. 2005; 3: 562–70. [DOI] [PubMed] [Google Scholar]
- 59.Wu Y, Liu W, Zhou Y, Hilton T, Zhao Z, Liu W, Wang M, Yeon J, Houck K, Thiagarajan P, Zhang F, Shi FD, Wu X, Li M, Dong JF, Zhang J. von Willebrand factor enhances microvesicle-induced vascular leakage and coagulopathy in mice with traumatic brain injury. Blood. 2018; 132: 1075–84. 10.1182/blood-2018-03-841932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gros A, Ollivier V, Ho-Tin-Noe B. Platelets in inflammation: regulation of leukocyte activities and vascular repair. Frontiers in immunology. 2014; 5: 678 10.3389/fimmu.2014.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circulation research. 2018; 122: 337–51. 10.1161/CIRCRESAHA.117.310795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, Wu X, Zhang Y, Konkle BA, Thiagarajan P, Li M, Zhang J, Dong JF. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. 2015; 125: 2151–9. 10.1182/blood-2014-09-598805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Denorme F, Vanhoorelbeke K, De Meyer SF. von Willebrand Factor and Platelet Glycoprotein Ib: A Thromboinflammatory Axis in Stroke. Frontiers in immunology. 2019; 10: 2884 10.3389/fimmu.2019.02884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Denorme F, Martinod K, Vandenbulcke A, Denis CV, Lenting PJ, Deckmyn H, Vanhoorelbeke K, De Meyer SF. The von Willebrand Factor A1 domain mediates thromboinflammation, aggravating ischemic stroke outcome in mice. Haematologica. 2020. 10.3324/haematol.2019.241042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andre P, Denis CV, Ware J, Saffaripour S, Hynes RO, Ruggeri ZM, Wagner DD. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood. 2000; 96: 3322–8. [PubMed] [Google Scholar]
- 66.Xu X, Wang C, Wu Y, Houck KL, Hilton T, Zhou A, Wu X, Han C, Yang M, Yang W, Shi FD, Stolla M, Cruz MA, Li M, Zhang J, Dong JF. Conformation-Dependent Blockage of Activated VWF Improved Outcomes of Traumatic Brain Injury in Mice. Blood. 2020. 10.1182/blood.2020007364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu D, Mahmood A, Goussev A, Qu C, Zhang ZG, Chopp M. Delayed thrombosis after traumatic brain injury in rats. Journal of neurotrauma. 2004; 21: 1756–66. 10.1089/neu.2004.21.1756. [DOI] [PubMed] [Google Scholar]
- 68.Sokoloff DDCL. Circulation and Energy Metabolism of the brain. Philadelphia: Lippincott-Raven, 1999. [Google Scholar]
- 69.Yamamoto K, de Waard V, Fearns C, Loskutoff DJ. Tissue distribution and regulation of murine von Willebrand factor gene expression in vivo. Blood. 1998; 92: 2791–801. [PubMed] [Google Scholar]
- 70.Mbagwu SI, Filgueira L. Differential Expression of CD31 and Von Willebrand Factor on Endothelial Cells in Different Regions of the Human Brain: Potential Implications for Cerebral Malaria Pathogenesis. Brain sciences. 2020; 10 10.3390/brainsci10010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Claus RA, Bockmeyer CL, Kentouche K, Sieber MW, Oberle V, Kaufmann R, Deigner HP, Losche W. Transcriptional regulation of ADAMTS13. Thromb Haemost. 2005; 94: 41–5. 10.1160/TH04-08-0498. [DOI] [PubMed] [Google Scholar]
- 72.Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS-13. Journal of thrombosis and haemostasis : JTH. 2006; 4: 1396–404. 10.1111/j.1538-7836.2006.01959.x. [DOI] [PubMed] [Google Scholar]
- 73.Liu L, Choi H, Bernardo A, Bergeron AL, Nolasco L, Ruan C, Moake JL, Dong JF. Platelet-derived VWF-cleaving metalloprotease ADAMTS-13. Journal of thrombosis and haemostasis : JTH. 2005; 3: 2536–44. 10.1111/j.1538-7836.2005.01561.x. [DOI] [PubMed] [Google Scholar]
- 74.McDonald SJ, Sharkey JM, Sun M, Kaukas LM, Shultz SR, Turner RJ, Leonard AV, Brady RD, Corrigan F. Beyond the Brain: Peripheral Interactions after Traumatic Brain Injury. Journal of neurotrauma. 2020; 37: 770–81. 10.1089/neu.2019.6885. [DOI] [PubMed] [Google Scholar]
- 75.Zheng X, Chung D, Takayama TK, Majerus EM, Sadler JE, Fujikawa K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. JBiolChem. 2001; 276: 41059–63. [DOI] [PubMed] [Google Scholar]
- 76.Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Matsuyama T, Ishikawa M, Iwamoto TA, Mori T, Wanaka A, Fukui H, Fujimura Y. Localization of ADAMTS13 to the stellate cells of human liver. Blood. 2005; 106: 922–4. [DOI] [PubMed] [Google Scholar]
- 77.Motto DG, Chauhan AK, Zhu G, Homeister J, Lamb CB, Desch KC, Zhang W, Tsai HM, Wagner DD, Ginsburg D. Shigatoxin triggers thrombotic thrombocytopenic purpura in genetically susceptible ADAMTS13-deficient mice. The Journal of clinical investigation. 2005; 115: 2752–61. 10.1172/JCI26007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dyer MR, Plautz WE, Ragni MV, Alexander W, Haldeman S, Sperry JL, Guyette FX, Zuckerbraun BS, Rollins-Raval MA, Raval JS, Neal MD, Publication AT. Traumatic injury results in prolonged circulation of ultralarge von Willebrand factor and a reduction in ADAMTS13 activity. Transfusion. 2020; 60: 1308–18. 10.1111/trf.15856. [DOI] [PubMed] [Google Scholar]
- 79.Rodriguez-Rodriguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A. Oxidative stress in traumatic brain injury. Current medicinal chemistry. 2014; 21: 1201–11. 10.2174/0929867321666131217153310. [DOI] [PubMed] [Google Scholar]
- 80.Khatri N, Thakur M, Pareek V, Kumar S, Sharma S, Datusalia AK. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS & neurological disorders drug targets. 2018; 17: 689–95. 10.2174/1871527317666180627120501. [DOI] [PubMed] [Google Scholar]
- 81.Fu X, Chen J, Gallagher R, Zheng Y, Chung DW, Lopez JA. Shear stress-induced unfolding of VWF accelerates oxidation of key methionine residues in the A1A2A3 region. Blood. 2011; 118: 5283–91. 10.1182/blood-2011-01-331074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen J, Fu X, Wang Y, Ling M, McMullen B, Kulman J, Chung DW, Lopez JA. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2010; 115: 706–12. 10.1182/blood-2009-03-213967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Chen J, Ling M, Lopez JA, Chung DW, Fu X. Hypochlorous acid generated by neutrophils inactivates ADAMTS13: an oxidative mechanism for regulating ADAMTS13 proteolytic activity during inflammation. J Biol Chem. 2015; 290: 1422–31. 10.1074/jbc.M114.599084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi H, Aboulfatova K, Pownall HJ, Cook R, Dong JF. Shear-induced disulfide bond formation regulates adhesion activity of von willebrand factor. JBiolChem. 2007; 282: 35604–11. [DOI] [PubMed] [Google Scholar]
- 85.Li Y, Choi H, Zhou Z, Nolasco L, Pownall HJ, Voorberg J, Moake JL, Dong JF. Covalent regulation of ULVWF string formation and elongation on endothelial cells under flow conditions. JThrombHaemost. 2008; 6: 1135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yeh HC, Zhou Z, Choi H, Tekeoglu S, May W III, Wang C, Turner N, Scheiflinger F, Moake JL, Dong JF. Disulfide bond reduction of von Willebrand factor by ADAMTS-13. JThrombHaemost. 2010; 8: 2778–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhao Z, Wang M, Tian Y, Hilton T, Salsbery B, Zhou EZ, Wu X, Thiagarajan P, Boilard E, Li M, Zhang J, Dong JF. Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood. 2016; 127: 2763–72. 10.1182/blood-2015-12-688838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao Z, Zhou Y, Hilton T, Li F, Han C, Liu L, Yuan H, Li Y, Xu X, Wu X, Zhang F, Thiagarajan P, Cap A, Shi FD, Zhang J, Dong JF. Extracellular mitochondria released from traumatized brains induced platelet procoagulant activity. Haematologica. 2020; 105: 209–17. 10.3324/haematol.2018.214932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bernardo A, Ball C, Nolasco L, Moake JF, Dong JF. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow. Blood. 2004; 104: 100–6. [DOI] [PubMed] [Google Scholar]
- 90.Aisiku IP, Yamal JM, Doshi P, Benoit JS, Gopinath S, Goodman JC, Robertson CS. Plasma cytokines IL-6, IL-8, and IL-10 are associated with the development of acute respiratory distress syndrome in patients with severe traumatic brain injury. Critical care. 2016; 20: 288 10.1186/s13054-016-1470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rodney T, Osier N, Gill J. Pro- and anti-inflammatory biomarkers and traumatic brain injury outcomes: A review. Cytokine. 2018; 110: 248–56. 10.1016/j.cyto.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 92.Hulstein JJ, de Groot PG, Silence K, Veyradier A, Fijnheer R, Lenting PJ. A novel nanobody that detects the gain-of-function phenotype of von Willebrand factor in ADAMTS13 deficiency and von Willebrand disease type 2B. Blood. 2005; 106: 3035–42. [DOI] [PubMed] [Google Scholar]
- 93.Martin C, Morales LD, Cruz MA. Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibalpha. JThrombHaemost. 2007; 5: 1363–70. [DOI] [PubMed] [Google Scholar]
- 94.Karoulia Z, Papadopoulos G, Nomikos M, Thanassoulas A, Papadopoulou TC, Nounesis G, Kontou M, Stathopoulos C, Leonidas DD. Studies on the essential intramolecular interaction between the A1 and A2 domains of von Willebrand factor. Protein and peptide letters. 2013; 20: 231–40. 10.2174/092986613804725226. [DOI] [PubMed] [Google Scholar]
- 95.Aponte-Santamaria C, Huck V, Posch S, Bronowska AK, Grassle S, Brehm MA, Obser T, Schneppenheim R, Hinterdorfer P, Schneider SW, Baldauf C, Grater F. Force-sensitive autoinhibition of the von Willebrand factor is mediated by interdomain interactions. Biophys J. 2015; 108: 2312–21. 10.1016/j.bpj.2015.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lof A, Muller JP, Brehm MA. A biophysical view on von Willebrand factor activation. Journal of cellular physiology. 2018; 233: 799–810. 10.1002/jcp.25887. [DOI] [PubMed] [Google Scholar]
- 97.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. 1996; 88: 1525–41. [PubMed] [Google Scholar]
- 98.Callewaert F, Roodt J, Ulrichts H, Stohr T, van Rensburg WJ, Lamprecht S, Rossenu S, Priem S, Willems W, Holz JB. Evaluation of efficacy and safety of the anti-VWF Nanobody ALX-0681 in a preclinical baboon model of acquired thrombotic thrombocytopenic purpura. Blood. 2012; 120: 3603–10. 10.1182/blood-2012-04-420943. [DOI] [PubMed] [Google Scholar]
- 99.Scully M, Cataland SR, Peyvandi F, Coppo P, Knobl P, Kremer Hovinga JA, Metjian A, de la Rubia J, Pavenski K, Callewaert F, Biswas D, De Winter H, Zeldin RK, Investigators H. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. The New England journal of medicine. 2019; 380: 335–46. 10.1056/NEJMoa1806311. [DOI] [PubMed] [Google Scholar]
- 100.Peyvandi F, Scully M, Kremer Hovinga JA, Cataland S, Knobl P, Wu H, Artoni A, Westwood JP, Mansouri Taleghani M, Jilma B, Callewaert F, Ulrichts H, Duby C, Tersago D, Investigators T. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. The New England journal of medicine. 2016; 374: 511–22. 10.1056/NEJMoa1505533. [DOI] [PubMed] [Google Scholar]
- 101.Butera D, Passam F, Ju L, Cook KM, Woon H, Aponte-Santamaria C, Gardiner E, Davis AK, Murphy DA, Bronowska A, Luken BM, Baldauf C, Jackson S, Andrews R, Grater F, Hogg PJ. Autoregulation of von Willebrand factor function by a disulfide bond switch. Science advances. 2018; 4: eaaq1477 10.1126/sciadv.aaq1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Interlandi G Destabilization of the von Willebrand factor A2 domain under oxidizing conditions investigated by molecular dynamics simulations. PloS one. 2018; 13: e0203675 10.1371/journal.pone.0203675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Albert V, Subramanian A, Pati HP, Agrawal D, Bhoi SK. Efficacy of Thromboelastography (TEG) in Predicting Acute Trauma-Induced Coagulopathy (ATIC) in Isolated Severe Traumatic Brain Injury (iSTBI). Indian journal of hematology & blood transfusion : an official journal of Indian Society of Hematology and Blood Transfusion. 2019; 35: 325–31. 10.1007/s12288-018-1003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wartiovaara U, Salven P, Mikkola H, Lassila R, Kaukonen J, Joukov V, Orpana A, Ristimaki A, Heikinheimo M, Joensuu H, Alitalo K, Palotie A. Peripheral blood platelets express VEGF-C and VEGF which are released during platelet activation. Thromb Haemost. 1998; 80: 171–5. [PubMed] [Google Scholar]
- 105.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005; 437: 497–504. 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 106.Suidan GL, Brill A, De Meyer SF, Voorhees JR, Cifuni SM, Cabral JE, Wagner DD. Endothelial Von Willebrand factor promotes blood-brain barrier flexibility and provides protection from hypoxia and seizures in mice. Arteriosclerosis, thrombosis, and vascular biology. 2013; 33: 2112–20. 10.1161/ATVBAHA.113.301362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu H, Cao Y, Yang X, Cai P, Kang L, Zhu X, Luo H, Lu L, Wei L, Bai X, Zhu Y, Zhao BQ, Fan W. ADAMTS13 controls vascular remodeling by modifying VWF reactivity during stroke recovery. Blood. 2017; 130: 11–22. 10.1182/blood-2016-10-747089. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 108.Zhu X, Cao Y, Wei L, Cai P, Xu H, Luo H, Bai X, Lu L, Liu JR, Fan W, Zhao BQ. von Willebrand factor contributes to poor outcome in a mouse model of intracerebral haemorrhage. Scientific reports. 2016; 6: 35901 10.1038/srep35901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Denis CV, Andre P, Saffaripour S, Wagner DD. Defect in regulated secretion of P-selectin affects leukocyte recruitment in von Willebrand factor-deficient mice. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98: 4072–7. 10.1073/pnas.061307098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhao BQ, Chauhan AK, Canault M, Patten IS, Yang JJ, Dockal M, Scheiflinger F, Wagner DD. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood. 2009; 114: 3329–34. 10.1182/blood-2009-03-213264. [DOI] [PMC free article] [PubMed] [Google Scholar]