

Abstract
Mitochondrial dysfunction – including increased apoptosis, calcium and protein dyshomeostasis within the organelle, and dysfunctional bioenergetics and oxidative status – is a common and early feature in all the major neurodegenerative diseases, including Alzheimer’s Disease (AD) and Parkinson’s Disease (PD). However, the exact molecular mechanisms that drive the organelle to dysfunction and ultimately to failure in these conditions are still not well described. Different authors have shown that inorganic polyphosphate (polyP), an ancient and well-conserved molecule, plays a key role in the regulation of mitochondrial physiology under basal conditions. PolyP, which is present in all studied organisms, is composed of chains of orthophosphates linked together by highly energetic phosphoanhydride bonds, similar to those found in ATP. This polymer shows a ubiquitous distribution, even if a high co-localization with mitochondria has been reported. It has been proposed that polyP might be an alternative to ATP for cellular energy storage in different organisms, as well as the implication of polyP in the regulation of many of the mitochondrial processes affected in AD and PD, including protein and calcium homeostasis. Here, we conduct a comprehensive review and discussion of the bibliography available regarding the role of polyP in the mitochondrial dysfunctional present in AD and PD. Taking into account the data presented in this review, we postulate that polyP could be a valid, innovative and plausible pharmacological target against mitochondrial dysfunction in AD and PD. However, further research should be conducted to better understand the exact role of polyP in neurodegeneration, as well as the metabolism of the polymer, and the effect of different lengths of polyP in cellular and mitochondrial physiology.
Keywords: Mitochondria, inorganic polyphosphate, polyP, neurodegeneration, Alzheimer’s disease, Parkinson’s disease
1. Introduction
Mitochondrial dysfunction is an early and common feature in all of the main neurodegenerative disorders [1]. While the complex mechanisms that induce the dysfunction of the organelle are still far from being entirely understood, different authors have proven a role for inorganic polyphosphate (polyP) – a polymer which is well-conserved throughout evolution and present in all studied organisms [2] – in the regulation of several processes involved in maintaining mitochondrial physiology and affected in neurodegeneration. These processes include apoptosis, energy metabolism and oxidative stress status, inflammation, calcium homeostasis, and proteostasis [3–10]. In this review, we aim to summarize and discuss the finding of these works. We will focus our discussion on the main processes that are related to mitochondria and on the two main neurodegenerative disorders in terms of incidence and prevalence, that is, Alzheimer’s Disease (AD) and Parkinson’s Disease (PD).
AD is the most incident and prevalent neurodegenerative disorder. In fact, accordingly with the 2020 Alzheimer’s Disease Facts and Figures, published by the Alzheimer’s Association, 1 in 3 seniors die with either AD or other types of dementia in the US. Moreover, AD is one of the costliest medical conditions [11] and it is projected that up to 14 million people in the US alone will be diagnosed with AD by 2050, with a subsequent cost increase for healthcare systems and patients. While the vast majority of the cases of AD are sporadic, familial forms of the disease associated with mutations in specific genes have also been reported [12].
The main pathophysiological characteristics of all types of AD are the presence of extracellular aggregations of amyloid plaques (especially amyloid-β, Aβ), and of intracellular neurofibrillary tangles (NFTs, mainly composed by the aggregation of the protein Tau hyperphosphorylated) in specific areas of the brain (such as the cortex and the hippocampus). While the role of NFTs as a causal factor in the cytotoxicity observed in AD is still very controversial, the clear relationship between increased amyloids aggregation, such as Aβ, and increased cytotoxicity and cell death, is well-known. In fact, through mostly unknown mechanisms, the accumulation of amyloid plaques in the extracellular space around neurons can disrupt the transmission of certain chemical signals, which leads to increased cell death. In the case of Aβ, the toxicity is exerted by the oligomeric aggregation of small chains of peptides derived from the breakdown of the amyloid precursor protein (APP), resulting in the formation of plaques. While different lengths of the peptide are present simultaneously in the brain of AD patients, Aβ1–42 is one of the most aggressive forms, in regards to causing mitochondrial dysfunction [13, 14].
PD is the second most common neurodegenerative disease and the most common movement disorder [15]. Symptoms of PD include tremor, bradykinesia, and rigidity due to the progressive apoptotic loss of dopaminergic neurons in specific brain areas associated with movement, such as the substantia nigra and the striatum. PD patients also present with non-motor symptoms, including cognitive disorders and hallucinations [16]. While approximately 20% of PD cases have a genetic etiology, induced by relatively well-known mutations in specific genes [17], the majority of cases are sporadic, and the causes remain mostly unknown. The main pathophysiological feature of PD is the presence of Lewy bodies, which are intracellular aggregations of proteins, mainly of α-synuclein (α-syn) amyloids [18–20]. Other proteins, such as ubiquitins, are also commonly found in these structures [21]. Aggregated Tau can also be present in Lewy bodies and in these cases, the structures may be surrounded by NFTs [22, 23]. Interestingly, the deleterious effect of aggregated α-syn and Tau upstream of mitochondrial dysfunction has already been proposed [24, 25]. In the case of α-syn, this detrimental effect could be explained by the direct action of α-syn on bioenergetics [26].
As previously mentioned, both AD and PD share some pathophysiological features, including mitochondrial dysfunction, which is the immediate cause of increased apoptosis, the main cause of neuronal death in AD and PD [27–29]. Interestingly, the literature is rich in examples of mitochondrial processes in which polyP could play a key regulatory role. Many of these processes are affected in AD and PD.
2. What is polyP?
PolyP is a strongly negatively charged and multifunctional polymer, composed of chains of orthophosphates bound together by high-energy phosphoanhydride bonds, similar to those found in ATP [10]. In fact, the quick inter-conversion between ATP and polyP in bacteria is well-known [30]. Moreover, it has recently been proven that the mammalian mitochondrial F0F1-ATP synthase plays a crucial role in the metabolism of polyP [31], which for the most part is still unknown, especially in eukaryotic organisms. Pavlov et al., in a different manuscript, also demonstrated that the levels of this polymer are dependent on the metabolic state of the mitochondria [32].
2.1. Physiological roles
In 1999, the late Nobel Laureate Arthur Kornberg and his colleagues published a complete review compelling the physiological roles of polyP, including the protective effect of the polymer against different insults in bacteria [33]. For example, Rao et al., described that bacteria with decreased levels of polyP are hypersensitive to a wide variety of stressors, including heat shock and chemical insults [34, 35]. Today, more than 20 years later, we know that polyP is involved in the regulation of multiple biochemical processes related to the stress response in a wide-range of organisms. For example, in prokaryotes, specifically in bacteria, different researchers have described significantly increased levels of polyP in response to common insults that induce stress, such as osmotic variations [36], nutrient deprivation [36, 37], decreased pH [38], increased temperature [3], and increased oxidative stress [39]. Moreover, the increased presence of polyP could also be involved in the modulation of the prokaryotic response to these stressors, through the regulation of biofilm production, motility, protein homeostasis and chaperoning, quorum-sensing, and virulence [3, 40–44].
In eukaryotic organisms, polyP is also involved in a wide-range of processes, ranging from blood coagulation to chelation of calcium ions during bone mineralization [9, 45, 46]. Interestingly, this polymer showed effects in different systems and cellular processes that are clearly affected in AD and PD and related to mitochondrial dysfunction. These includes the activation and regulation of apoptosis (mostly by its effect on the mitochondrial permeability transition pore (mPTP)), the stimulation of the mammalian target to rapamycin (mTOR, a protein kinase essential involved in mitochondrial physiology and affected in neurodegeneration); the regulation of the levels of ATP and growth; the homeostasis of calcium; and the chaperoning ability of the cells, especially with amyloids [4, 5, 47–55].
2.2. PolyP metabolism
As previously mentioned, the exact metabolism of polyP in many organisms, including mammalian cells, is still unknown. In Saccharomyces cerevisiae, polyP, which has been proposed to play a dual role in energy and phosphate storage in these organisms, is synthesized by the vacuolar transporter chaperone 4 (VTC4), in a process that consumes ATP. After being synthesized, polyP is transported into the vacuole, where it is stored [56, 57]. In bacteria, polyP can be directly generated from ATP by the enzyme polyphosphate kinase 1 (PPK1), which is present in several pathogenic species. In this reaction, PPK1 catalyzes the removal of the terminal γ-phosphate, which is the third phosphate group farthest from the ribose sugar of ATP, and its reversible transfer to polyP [58]. The γ-phosphate has the highest energy of hydrolysis, compared to the ɑ and β-phosphates. Other authors, also using bacteria, described that the enzyme polyphosphate kinase 2 (PPK2), which belongs to the same enzymatic family as PPK1, is involved in the metabolism of polyP. PPK2, however, catalyzes the removal of the terminal phosphate from polyP and its addition to GDP to form the final product GTP [59–61]. Interestingly, most bacteria that encode PPK1 also show exopolyphosphatase (PPX) activity as well [62]. In yeast and bacteria, PPX is the enzyme in charge of degrading polyP by its free-end into single orthophosphates [62, 63].
2.3. Cellular location of polyP
PolyP is extremely ubiquitous and can be found throughout eukaryotic cells, including the nucleus, cytoplasm, cell membrane and organelles [2]. In mammalian cells, there is a high co-localization of polyP and mitochondria [5, 50], which could be related to the important role proposed for polyP in maintenance of mitochondrial physiology. PolyP is also present in the extracellular space [64]. In fact, it has been proven that astrocytes are able to secrete polyP which can be later imported by neurons [6, 65]. This variety of subcellular and cytoplasmic locations seems to suggest that PolyP could play different roles in cellular metabolism depending on its location.
2.4. PolyP length and quantification methods
Despite the drastic variations in the concentrations and chain lengths of polyP in various cellular locations, the concentrations of polyP are usually within the micromolar range and the size length does not exceed a few hundred orthophosphate units in mammalian cells [2]. For example, the concentration of polyP in the brains of mammalian animal models can range up to 100 μM [2, 66]. Interestingly, some studies have shown that variations in the intracellular concentration of polyP could correlate with age and disease, which highlights the possible impact of this polymer in aging-related changes in mammalian cellular physiology [67]. Moreover, some cells, such as mast cells and platelets, show higher concentrations of polyP. In these systems, medium-sized polyP is present in dense granules, at concentrations up to 130 mM. Different authors have hypothesized that these high concentrations are necessary for the activation of the inflammatory and coagulation cascades [10, 68, 69].
The in vivo quantification of polyP remains a challenge because of 1) the technical difficulty of finding specific methods to study this polymer, 2) the lack of knowledge regarding its metabolism, and 3) its pleiotropic roles of the polymer in cellular physiology, ranging from bone formation to cell signaling. In fact, every known method used to quantify the presence of polyP in biological samples presents disadvantages. For example, the detection of Phosphorus-31 using NMR detects molecules that contain phosphate units linked by phosphoanhydride bonds. While this method is very sensitive, it does not solely recognize polyP, but also nucleotides or any other molecule containing the same bonds as polyP [70]. DAPI staining, which is usually used for nucleic acids labeling, can also be used to detect and quantify polyP, through the use of specific emission and excitation wavelengths that differ from the wavelengths used to measure nucleic acids [50, 71, 72]. In this case, fluorescence intensity is directly proportional to the concentration of polyP present in a sample. This method, which is less sensitive than NMR, also presents many pitfalls, such as the possible interference of various nucleic acids, nucleotides and amorphous calcium phosphates. Another method used to assay the levels of polyP is targeting the polyP Binding Domain (PPBD) of the PPX enzyme through immunocytochemistry [73]. This technique takes advantage of the affinity of the PPBD in the C-terminal of the PPX from Escherichia coli (E. coli) by polyP. While this assay shows great sensitivity and specificity, it can be exclusively conducted in fixed tissue and thus, does not provide any information about the dynamics of polyP. Moreover, PPX is only able to bind polyP by its free ends. Therefore, if polyP is bound to some cellular element, PPBD will not mark that molecule of the polymer. This can seriously affect the visualization of polyP, since it can attract monovalent, divalent or trivalent cations, as well as other cellular structures, depending on the conformation that it adopts [74].
3. PolyP, Amyloid Aggregation, and Chaperone Function
Amyloids are aggregates of specific proteins that show beta-sheet secondary structures. These aggregates, that are capable of accumulating and forming deposits in organs and tissues, are clearly implicated in the development of PD and AD, amongst other diseases. Current evidence suggests that amyloid-induced toxicity is due to oligomeric intermediates formed prior to mature aggregates [75–77], which may alter mitochondrial functions, target the cytoskeleton, or increase cell membrane permeability among other effects [78]. In fact, the deleterious effects of amyloids in mitochondrial physiology through at incompletely understood mechanism has already been broadly described [79–83]. To further prove the importance of mitochondria in amyloid-induced damage, it has been shown that most cytoplasmic aggregated proteins are imported into the organelle to be eliminated through a mechanism that, although still poorly understood, could involve the activation of the mitochondrial specific autophagy (mitophagy) and the mitochondrial unfolded protein response (UPRmt) [84, 85]. Moreover, in addition to the increased presence of amyloids in AD and PD, the dysfunction of the main cellular systems involved in proteostasis, including the ubiquitin-proteasomal and the endosomal-lysosomal pathways, have also been described [86–88].
The specific effects of polyP in amyloidogenesis have been explored in different organisms and systems. For example, Cremers et al., described that polyP accelerates the folding of the CsgA protein (Curli-specific gene A) from the easily degradable, unfolded conformation that the protein has when synthesized to a beta-sheet secondary structure, thereby enhancing its ability to aggregate and to form amyloid structures [89]. CsgA is present in the uropathogenic strains of E. coli, where it is required for robust biofilm formation, a common stress-response in these organisms. The same authors proved similar results regarding the role of polyP in accelerating amyloid formation in other model systems. For example, they supplemented the food source of mutant Caenorhabditis elegans (C. elegans GMC101) with fluorescent-labeled polyP. These worms expressed Aβ1–42 and accumulated fibrils in their muscular cells, causing muscular paralysis in early adulthood [90]. The worms treated with polyP demonstrated a better response to amyloid stress than the control group. Specifically, 45% of control worms displayed significant movement defects by adulthood due to amyloid accumulation, whereas polyP treated worms were paralyzed in fewer than 30% of cases [89]. Moreover, the authors also explored the interaction between polyP, α-syn, Aβ1–42, and Tau40 – a member of the Tau family which is implicated in the development of AD [91]. To accomplish this, they used different in vitro systems. In the case of the α-syn fibril formation – an intricate process, often requiring shaking, a beaded matrix, and protein concentrations as high as 20-folds more than those found at physiological levels – the authors proved that the presence of physiological concentrations of polyP, accelerated the process [89, 92]. A similar effect was observed when they tested Aβ1–42 and Tau40. Lastly, using different mammalian cell lines, including differentiated SH-SY5Y, PC12 and HeLa, the authors corroborated the protective effect of polyP against α-syn-induced toxicity. This data suggests that polyP might play an important role in the protection exerted against the proteotoxicity induced by AD and PD, by enhancing and accelerating protein fibrillation through deleterious oligomer intermediates.
Supporting the role of polyP in amyloidegenesis, Khong et al., using human SaOS-2 cell lines, showed that this polymer is crucial for the proper folding of collagen, in a process mediated by cyclophilin B (CypB) [93]. Collagen, which relationship with mitochondrial physiology has already been demonstrated [94], is a structural protein widely present in all the connective tissue in mammalians. Interestingly, it is also found in the central nervous system [95–97]. Moreover, some specific types of collagen, such as type XXV collagen which is also known as collagen-like amyloidogenic component, are a component of the amyloids plaques, affecting the amyloid fibril elongation [98, 99]. Khong et al., focused their studies in the Endoplasmic Reticulum (ER), where they showed the direct and specific bind of polyP and CypB. This binding inhibits the peptidyl-prolyl cis-trans isomerase of CypB, an enzyme which activity is critical for the correct collagen folding. Interestingly, treatment of the cells with spermine (a pharmacological inhibitor of polyP [100]) and the expression of the ER-addressed PPX enzyme reverted the effects induced by polyP on collagen misfolding [93].
Interestingly, polyP might play a dual role in cellular and more specifically, mitochondrial proteostasis. While we have just discussed the effects of polyP in the amylogenic process, polyP might also have a regulatory action in the molecular mechanisms that protect against proteotoxicity, which are still mostly unknown, especially in mammalian cells. In fact, different authors have shown the protective effects of polyP on protein homeostasis against oxidative, heat, and chemical stress, using different non-mammalian models [39, 101–104]. Moreover, it has also been demonstrated that polyP stabilizes different proteins in bacteria [3]. Specifically, Yoo et al., explored the effects of the polymer on preventing thermal unfolding. Using the lactate dehydrogenase protein (LDH) extracted from rabbit muscle – a well-known model for protein folding studies [105] – they showed that polyP stabilizes LDH protein. This effect was more prominent when polyP forms complexes with LDH unfolding intermediates. Additionally, they showed that the longer chains of polyP were the most effective, as well as that LDH-polyP complexes remain soluble at physiologically viable conditions and allow for the refolding capability.
One of the main protein families involved in the cellular and mitochondrial responses to increased levels of aggregated amyloids are chaperones. Gray et al., have shown that polyP could be an ancient, well-conserved, highly efficient, and inorganic chaperone in different organisms [106]. The authors of this study claimed that the chaperoning effect of polyP could be one of the main factors that contributes to the wide-variety of pleiotropic phenotypes associated with deficiencies of the polymer. Specifically, using E. Coli models, they identified the key role played by polyP in counteracting oxidative stress conditions caused by the proteotoxic effects of the fast-acting, oxidant, hypochlorous acid (HOCl). HOCl-treated E. coli cells stained with DAPI showed significantly higher levels of DAPI bound to polyP, compared to non-treated E. coli. However, lower levels of polyP were observed in E. coli treated with the non-proteotoxic oxidant hydrogen peroxide (H2O2). Interestingly, once the stress insult is over, the same authors have demonstrated that polyP was found to refold, in an ATP-dependent manner [106].
The main source of protein dyshomeostasis in AD and PD is the accumulation of amyloids and the failure of the protection systems against proteotoxicity. As previously mentioned, the relationship between mitochondrial dysfunction and protein dyshomeostasis has been broadly described. Interestingly, the studies presented in this section show a clear role for polyP on stabilizing different amyloids found in AD and PD, as well as a chaperoning action of the polymer. Thus, it is feasible to propose that by modulating the levels of polyP, we could prevent the increased presence of toxic amyloids and maintain protein homeostasis. This will contribute to the preservation of mitochondrial physiology in these conditions and hopefully prevent the increased apoptotic death observed in AD and PD. However, further studies should be conducted in this field.
4. PolyP, and Calcium Signaling and Homeostasis
Calcium buffering and signaling, as well as calcium-dependent cell signaling, are crucial for the proper functioning of glial and neuronal systems [107]. In fact, the dysregulation of these processes, through mechanisms not yet properly described, has been broadly reported in AD and PD as one of the main mechanisms that induces increased apoptosis [108, 109]. Mitochondria perform a crucial role in calcium buffering and signaling [110], and polyP is clearly involved in the regulation of the concentrations of this cation within the organelle [4, 5]. In this case, by targeting polyP to prevent calcium dyshomeostasis in AD and PD, mitochondrial physiology could be protected and cell death could be reduced.
The importance of polyP in calcium-mediated signaling has been demonstrated in the mammalian central nervous system, through the study of gliotransmitters, which are neuroactive chemicals released from any glial cell and are key to the preservation of neuronal homeostasis. Their release may be calcium-dependent and thus dysfunctional calcium homeostasis may affect this system. Interestingly, dysfunctional release of these molecules has been described in both AD and PD [111, 112]. The action of ATP as a gliotransmitter has already been shown [113] and some authors have reported that PolyP can also act as a calcium-dependent gliotransmitter in mammalian astrocytes [65]. The same authors described that the migration of polyP from astrocytes to neurons is calcium-dependent and mediated by a P2Y G-protein coupled receptor [65]. In fact, the presence of the receptor phospholipase-C (PLC), a selective P2Y antagonist, inhibits the migration of polyP from astrocytes to neurons [65]. Interestingly, PLC is a membrane-associated enzyme that cleaves phospholipids from phosphate groups, and regulates the mobilization of intracellular calcium by modulating the release of stored calcium from organelles. Moreover, the authors of the study show that the migration of polyP from astrocytes to neurons might be dependent on a transport mechanism, as the neuronal membrane seems to be impermeable to polyP, even if the uptake of the polymer from the extracellular space by neurons has been probed.
The exact buffering system in mitochondria remains poorly understood. While other cellular compartments, such as the endoplasmic reticulum, have specific proteins in charge of calcium regulation, the presence of similar proteins in mitochondria has not yet been described. Moreover, while the total intra-mitochondrial calcium concentration can increase by several orders of magnitude under certain stress stimuli, the concentration of free calcium within the organelle has to be strictly maintained within the micromolar range, to prevent the activation of the apoptotic cell death [78, 114]. In this scenario, the potent buffering effect of polyP in mitochondria has been demonstrated [4, 5]. The authors of these studies combined experimental and mathematical models to show that the union between polyP and calcium is reversible, thus allowing for a buffering action. Interestingly, the bond between orthophosphate and calcium is irreversible, forming crystals [115]. As a cellular model for their studies, the authors used cells lacking mitochondrial polyP (MitoPPX). These cells were created by the stable transfection of different mammalian cell lines with a plasmid containing the sequence for expressing the PPX enzyme in mammalian cells. The plasmid also contained a mitochondrial-targeting sequence and the sequence for expressing the fluorescent protein GFP. The authors demonstrated the lack of polyP in cells expressing the PPX enzyme, as a consequence of the activity of the PPX to degrade polyP.
Mitochondria are one of the main cellular locations of both polyP and calcium. Moreover, these organelles play a key role in regulating the cellular levels of the cation. Additionally, intra-mitochondrial calcium buffering is crucial for cell survival and is clearly affected in AD and PD. Thus, by regulating the levels of polyP, mitochondrial calcium buffering could be improved and calcium-induced cell death could be potentially reduced in neurodegenerative diseases. Regulating the levels of this polymer could also contribute to improve some specific types of signaling within the central nervous system, which are affected by the mitochondrial levels of ATP, polyP and calcium.
5. PolyP and Energy Metabolism
Dysfunctional energy metabolism has been broadly described in many diseases. Interestingly, inappropriate energy production is especially deleterious within the central nervous system, due to the enormous amount of energy that this system needs to function. In fact, the human brain consumes approximately 20% of total organismal energy. For example, it has been demonstrated that one single cortical neuron utilizes approximately 4.7 billion ATPs/second, in a resting human brain [116]. Thus, it is not surprising that dysfunctional energy production has been broadly reported in all the major aging-related diseases and neurodegenerative pathologies, including both AD and PD [117, 118].
While extramitochondrial pathways, such as glycolysis, account for some of the total energy produced in mammalian cells, the vast majority of the energy is produced in the mitochondrial electron transfer chain. Thus, mitochondrial dysfunction and insufficient energy supply are closely related in these organisms. The main activated carrier of energy in mammals is ATP. However, other compounds might also play an important role in energy carry and storage. Interestingly, the molecular structure of polyP – which bonds are isoenergetic to the bounds found in ATP – and its preferred location within mitochondria makes polyP a perfect candidate to contribute to the regulation of energy metabolism in different scenarios, including AD and PD.
In fact, the role of polyP as an energy storage molecule has already been demonstrated by different authors [119–121]. For example, in metazoa, polyP plays a crucial role as metabolic fuel (reviewed in [122]). Specifically, in this model, the evidence seems to point towards the involvement of polyP in an extracellular system in charge of energy transport and delivery. Through the use of human cell lines (SaOS-2), Muller and his collaborators demonstrated that the enzymatic hydrolysis of polyP contributes to the synthesis of ADP and ATP, thus increasing the levels of these molecules in the extracellular matrix of mammalian cell cultures [120]. In this study, the authors, also showed the effects of Na-polyP in inducing the formation of matrix vesicles on the cell surface, which drives the accumulation of ATP and ADP. These vesicles contain alkaline phosphatase (ALP) and adenylate kinase (AK1) which translocate to the cell membrane in response to polyP. In the membrane, ALP hydrolyzes polyP and synthesizes ADP by a catabolic reaction. This process is then followed by an anabolic reaction that catalyzes the formation of ATP by AK1-mediated hydrolysis of polyP, wherein AK1 acts as metabolic fuel in the extracellular space. The action of AK1 in the extracellular space could help to control the proliferation capacity of mammalian cells, by regulating the rates of energy metabolism. In fact, the effects of the extracellular energy state in modulating cell proliferation, the opposite process of cell death, has already been suggested in cancer [123].
The action of polyP in the regulation of the intracellular levels of energy has also been broadly demonstrated. For example, using yeast from the YKO collection, Freimoser et al., showed a clear relationship between polyP and the energetic metabolism of these cells. In this study, the levels of the polymer were quantified in different yeast strains expressing individual Knock-Out (KO) of 235 genes, encoding various vacuolar proteins.
Interestingly, the researchers found that the specific strains that contain lower levels of polyP as a consequence of the KO of some specific genes, including the VPH1, VTCA, and VTC1, also exhibited reduced acid phosphatase, ATP and glycogen levels [121]. This effect was proven to be caused by the action of the ATP-dependent kinase activity of polyP. Moreover, upon genetic screening of the KO yeast strains, 4% of their genome was found to be involved in maintaining polyP levels, which supports the hypothesis implying the importance of polyP in the overall organism energy homeostasis. The authors also propose that the maintenance of orthophosphate/polyP and polyP/ATP equilibria is key for the survival of the organism. A similar equilibrium between cellular levels of ATP and polyP has been demonstrated in different mammalian tissues and cells, including fibroblasts, kidney and adrenal cells [2].
Interestingly, Muller et al., using different mammalian cellular models, including neurons, have shown that polyP is able to reverse the compromised energy state of the cells, induced by the treatment with Aβ25–35 [119]. Moreover, recently it was reported that polyP can be produced and hydrolyzed by the F0F1-ATP synthase in mammalian mitochondria [31]. The same authors showed that polyP can stimulate the activity of the F0F1-ATP synthase, as well as activate mitochondrial respiration, which, interestingly, was shown not to be coupled with ATP production in their model. Pavlov et al., also using mammalian cellular models, have demonstrated that the intracellular levels of polyP are not static and seem to be closely related to the presence of ATP and to the metabolic state of the cell. In fact, when mammalian cells were treated with substrates of the mitochondrial respiration, increased levels of polyP have been reported. The opposite effect was observed when the cells were treated with inhibitors of the mitochondrial respiration, such as rotenone (which interestingly, has been broadly used as a cellular model of PD [124]) or with uncouplers of the electron transfer chain (such as FCCP). Moreover, the use of oligomycin, a well-known inhibitor of the mitochondrial F0F1-ATP synthase, completely blocked the production of polyP [32].
A common consequence of dysfunctional bioenergetics observed in many pathologies, including AD and PD, is the increased presence of Reactive Oxygen Species (ROS), which causes oxidative damage in neuronal populations and affects the structures of a wide-variety of cellular components, ranging from nucleic acids to proteins [125–127]. Moreover, increased ROS is also a well-established key activator of the mPTP and of apoptotic cell death in different pathologies that affect the central nervous system, including neurodegenerative diseases [128–132].
The laboratory of Dr. Arthur Kornberg described the deleterious effects of the disruption of the gene coding for the PPX in E. coli in the cellular protection against different stressors, including increased oxidative stress induced by H2O2 [101]. This increased sensitivity to ROS due to the lack of PPX expression has been probed by diverse groups using different microorganisms [101, 103, 104, 106].
In mammals, ROS are mostly produced in mitochondria, during the process of energy generation. Thus, mitochondria are exposed to higher concentrations of ROS than other subcellular compartments [133]. To further explore the correlation between cellular polyP levels and ROS, Seidlmayer et al., used a model of ischemia/reperfusion (I/R), (known to increase oxidative stress conditions [128]) in left ventricular myocytes from rabbit. The authors of this study reported that during the reperfusion phase, massive endogenous polyP production was observed. This effect was simultaneous with a decline in superoxide generation and with the acceleration of H2O2 production, which in turn, contributed to the restoration of the mitochondrial pH, decreased during the ischemia phase of the I/R, and to the increased cell death observed in the myocytes during the reperfusion phase. This data suggests a clear correlation between the cellular levels of ROS and the presence of polyP. Interestingly, during the ischemia phase of the injury, the authors compared the production of ROS and the levels of cell death in Wild-type (Wt) and cardiomyocytes overexpressing mitochondrial-targeted PPX. They reported increased levels of ROS and cell death in the mutant samples, compared with the Wt cells, which supports the role of polyP in the regulation of ROS production. However, increased superoxide production was not prevented by the treatment of the cells with Cyclosporine A, a well-known inhibitor of the opening of the mPTP [134], in any of the conditions. This data seems to point that polyP’s ability to regulate superoxide production is not linked to its role in the regulation of calcium homeostasis. Lastly, the authors showed that the use of MnTBAP [135], a potent antioxidant, exerted a similar protective effect in Wt and mutated samples [136].
All of these studies show that polyP might play a multifaceted mechanism in the control of cell bioenergetics and oxidative status, both clearly related to mitochondria and affected in AD and PD. However, some of the data regarding the effects of polyP in these parameters may look contradictory. Thus, further studies using specific lengths and cellular locations of polyP should be conducted to explicate the exact mechanism of action of the polymer on energy metabolism and consequently, on the regulation of the oxidative status of the cell.
6. PolyP and Inflammation
While acute inflammation is a fairly well-defined protective response against tissue injury, orchestrated by a cascade of pro and anti-inflammatory mediators [137], chronic inflammation, a much less known process involving a progressive change in the cell types at the inflammation site, can cause detrimental and irreversible damage to the surrounding healthy tissues [138]. Neuroinflammation, a specific type of chronic inflammation, is localized in tissues of the central nervous system.
Both α-syn and Aβ aggregates have been reported as pro-inflammatory molecules in mammalian brains, exacerbating the progression of the neurodegeneration [139–141]. Moreover, the presence of neuroinflammation has been broadly identified in the brain of AD and PD patients [142–145]. For example, the activation of the microglia (non-neuronal cells, acting as phagocytes in the central nervous system) has been associated with AD and PD, among other disorders [146]. While the activation of microglia is protective against neurodegeneration in the early stages, it might play a deleterious effect in disease progression, as well as in the late stages of the disorders [147]. Interestingly, the effect of polyP on the physiology of astrocytes – a type of glia cells which are also well-known regulators of neuroinflammation [148] – has been demonstrated [6], and previously discussed in this review.
The relationship between mitochondria and inflammation has been increasingly recognized in the last years. For example, mice lacking Parkin and Pink, two of the main proteins involved in mitophagy and whose mutations are related with early onset PD [149–151], showed a strong inflammatory phenotype, after exercise. The same effects were observed in a mutated Parkin mouse which accumulates mutations in the mitochondrial DNA [152]. In both cases, inflammation was reverted by the concurrent loss of STING, which is a key molecule involved in the regulation of the interferon response to cytosolic DNA [153, 154].
Several studies have described the direct role of polyP in the modulation of inflammation [10, 100, 155–158]. Many of these studies use platelets as cellular models. Platelets not only are they key cells in the coagulation system, but they serve roles in the inflammatory response [159]. In fact, inflammation and hemostasis are closely linked through the kallikrein kinin pathway, in which platelets are involved in [158, 160–163].
Platelets contain high concentrations of polyP, which can be released into circulation [69]. Muller et al., demonstrated that activated platelets release polyP, which bind and activate the plasma protease factor XII [10]. The activation of the factor XII triggers the release of pro-coagulant and pro-inflammatory mediators, such as bradykinin [10]. In fact, the treatment with exogenous polyP at concentrations higher than1 μg/mL stimulated a high release of bradykinin (>750 ng/ml) from platelet-dense granules into the plasma. The same authors showed that polyP increases vascular permeability and initiates the contact system-mediated capillary leakage. To conduct their studies, the authors used mice deficient in factor XII or in bradykinin.
Dinarvand et al., showed the effect of polyP on amplifying the proinflammatory response in primary human umbilical vein endothelial cells [155]. This effect was mediated by the direct binding of polyP to the histone H4 (H4) and the High Mobility Group Box 1 (HMGB1), two potent nuclear pro-inflammatory mediators which are released during the activation of inflammation [164–166]. PolyP and these proteins show a high affinity, potentiating the pro-inflammatory activity of both H4 and HMGB1, in different models. The authors propose that the mechanism of this interaction is the direct bind of polyP, the Receptor for Advanced Glycation End products (RAGE) and the P2Y1 receptors. Interestingly, this interaction also induced an intracellular calcium release. To further dig into the tight relationship between inflammation and coagulation, the authors showed an anticoagulant-induced inhibition of the inflammatory response activated by polyP [155]. Hassanian et al., using vascular endothelial cells, showed that the pro-inflammatory action of polyP was also mediated by the activation of mTOR complexes 1 and 2 [156]. In this case, the data show that polyP induced the phosphorylation of p70S6K, an mTOR1 substrate. Using an elegant approach, the authors also showed the link between the pro-inflammatory and the mTOR signaling functions of polyP. Lastly, using different lengths of polyP (45, 60, and 70 phosphate monomers) and human umbilical vein endothelial cells and mice models, Bae et al., showed that polyP activated the NF-κB factor (a key component of the mammalian immune system [167]) inducing the activation of a potent anti-inflammatory response [157]. They hypothesize that this effect could contribute to the proinflammatory action of polyP in platelets, that was previously reported [155].
In line with all these studies, it has bene proposed that the inhibition of polyP could be a valid strategy to prevent inflammation and thrombosis [100]. The authors, using in vitro and mouse models, identified several polyP inhibitors, including cationic proteins, small molecules and polymers. Generation 1.0 dendrimer and polymyxin B, two of these inhibitors, were administrated retro-orbitally to the mice. After 40 min, saline, polyP or bradykinin were injected intradermically into the same animals, for 30 min. The animals were then euthanized. While generation 1.0 dendrimers were able to reduce inflammation by significantly reducing vascular leakage in mice treated with polyP, this inhibitor did not show any effects on the animals treated with bradykinin [100]. In the same study, the authors identified the polyamines spermine, spermidine, and putrescine as potent inhibitors of polyP.
The data presented here shows that manipulating the metabolism of polyP could be a promising strategy to prevent or to counteract the neuroinflammation observed in AD and PD, perhaps through the improvement of mitochondrial physiology. While the effects of polyP on inflammation have been studied in different systems, further studies addressing the effects of polyP in neuroinflammation should be conducted. Moreover, the study of the specific effects of different lengths of polyP will also here help us to understand the exact role of polyP in this process.
7. PolyP and Apoptosis
The adequate and tight equilibrium between cell growth and death is key to maintaining homeostasis at the cellular level. Apoptosis, the most common type of programmed cell death found in the central nervous systems, is essential to various physiological processes, ranging from early embryonic development to cellular turnover. However, increased levels of this type of cell death are in the bases of the vast majority of the neuronal and glial loss in neurodegenerative disorders, including AD and PD.
Apoptosis can proceed through two pathways, either intrinsic or extrinsic. The intrinsic pathway, also known as the mitochondrial pathway, accounts for the majority of apoptotic events present in mammalian cells. This pathway includes the opening of the mPTP (a point of no return in apoptosis) in response to specific intracellular stimuli generated under physiological conditions. The opening of the pore induces massive influx of calcium in the mitochondria, disruption of the mitochondrial membrane potential and eventual swelling of the organelle [168]. Thus, the relationship between mitochondrial dysfunction and increased apoptotic cell death in AD and PD is, once again, clear. In fact, different efforts to prevent these processes have been conducted in models of AD and PD [13, 14, 169].
Various authors have proposed that polyP could play a dual role in the regulation of apoptosis. On one hand, proteotoxicity, calcium dyshomeostasis, increased inflammation, and defective bioenergetics (all processes in which regulation polyP is involved in) are well-known triggers of increased apoptosis. On the other hand, this polymer could be a structural component of the mPTP [170, 171]. In fact, Elustondo et al., using mitochondria isolated from rats, demonstrated that the induction of the pore is closely linked to the formation of a complex, which components are polyhydroxybutyrate, polyP and the C-subunit from the ATP synthase, [172]. Interestingly, in a recent manuscript, Amodeo et al., have demonstrated that this subunit is an amyloidogenic channel-forming peptide [173].
The pleiotropic effect of polyP on apoptosis can likely explain the different results regarding the role of polyP in the activation and regulation of apoptosis that several authors have reported. Also, in this case the length of the polymer seems to be crucial to understand the effects exerted by polyP. In fact, Angelova et al., showed that caspase 3 – one of the main proteins involved in apoptosis – was only activated when human cortical neurons and astrocytes were treated with long chain polyP (polyP120), but not with short (polyP15) or medium (polyP60) chain lengths [174].
Interestingly, the intracellular levels of polyP show clear differences between cancer cells – where apoptosis is clearly decreased – and non-cancer cells. Specifically, Jimenex-Nunez et al., showed that myeloma cells (U266) present increased levels of polyP, compared to non-cancer cells and other B-lymphocytes [175]. The authors of this manuscript also showed a four to five-fold increase in the presence of polyP in the nucleus of U266 cells, compared with the cytoplasm. Using confocal microscopy, they demonstrated the accumulation of polyP in the nucleoli within the nuclear compartment, as well as a strong co-localization of polyP with nucleolar markers (fibrillin and RNA pol I). This suggests that polyP may act as a transcription modulator in the mammalian cell. The authors also showed that increased presence of polyP can induce apoptosis per se. In fact, when U266 cells were incubated in the presence or absence of 3 mM exogenous polyP75 for 6 hours, flow cytometry showed that caspase 3 levels were four times higher compared with control conditions. Consequently, U266 cells treated with polyP75 developed a higher percentage of cells with apoptotic bodies.
More recently, the co-localization of polyP in the fibrillar center of the nucleolus in HeLa cells has also been demonstrated [176]. The authors of this study probed that the accumulation of polyP in the nucleolar structures is a consequence of the cellular response to the cytotoxicity induced by the chemotherapy drug cisplatin, a well-known inducer of apoptosis and a common treatment for different types of cancer in humans. The authors corroborated their results in OVCAR3 cells, proving that the observed effect is not cell specific. Moreover, they correlated their results with the chain length of polyP. Specifically, longer chains of polyP were shown to have a greater effect on proliferation and apoptosis rates in cisplatin-treated cells, compared to shorter chains of the polymer [176]. Lastly, Sakatani et al., demonstrated that polyP derived from Lactobacillus brevis, a well-known probiotic, inhibits the progression of colon cancer by inducing apoptosis. This was observed in both in vivo patient derived tumor xenografts and in vitro using human colon cancer cells (SW620). However, polyP did not affect the viability of intestinal cells under physiological conditions. Apoptosis in the SW620 cells were found to be induced via the phosphorylated extracellular signal-regulated kinase (ERK) pathway [177].
As previously mentioned, polyP might play a dual role in apoptosis, as this polymer could also be a structural component of the mPTP [54, 55, 136]. Interestingly, the opening of this pore is dependent on the accumulation of mitochondrial calcium. Seidlmayer et al., showed that decreased polyP levels can have a protective effect in cardiomyocytes by preventing the opening of mPTP [54]. In fact, other authors showed that when polyP was depleted in the mitochondria, mPTP opening was inhibited [55, 178]. They also showed decreased mitochondrial levels of polyP in myocytes isolated from murine models, after I/R, proposing that the reduction of mitochondrial polyP may serve an adaptive and cardioprotective feature to increase myocyte resistance towards mechanical stress and cell death in cardiac conditions.
One plausible explanation for the observed differences regarding the role of polyP in mitochondria may be due to the cellular model used by the different authors. While stable cell lines are cancer cells with the apoptosis mechanisms already compromised, primary cultures should have this mechanism intact. In any case, it seems clear that polyP plays a key role in the activation and regulation of apoptosis, even if the exact mechanism of this relationship has not yet been elucidated.
8. Conclusions
AD and PD are the two main neurodegenerative disorders in our societies, affecting an increasing number of patients year after year. Currently, the pharmacological strategies available to delay, stop or revert these disorders are very limited, mostly due to the lack of knowledge about the etiopathological mechanisms that lead to the increased cell death observed in these diseases.
The presence of mitochondrial dysfunction is an early and well-known event, shared by all the major neurodegenerative disorders. In this review, we discussed the effects of polyP in the modulation of different aspects related to the dysfunction of the organelle, which could potentially constitute a valid and extremely promising strategy against the deleterious effects of AD and PD in mitochondrial physiology and thus, in the advance of these diseases. However, while all these studies seem to point towards an effect for polyP in mitochondrial dysfunction in AD and PD, more research needs to be conducted to propose polyP as a target in these conditions, as well as to clarify the studies showing data apparently contradictory. For example, while increased levels of polyP have shown to be protective against protein and calcium dyshomeostasis, the action of these increased levels is deleterious when inflammation and apoptosis are assayed. These differences could be due to the specific lengths of polyP used in the experiments; the plausible association of the polymer with other subcellular and mitochondrial components, such as proteins or positively charged molecules; or the exact location of polyP. The only path to elucidate the exact role of polyP in the physiology of the organelle will be to increase our knowledge of the metabolism of the polymer, by developing new analytical tools. This way, we will be able to conduct further studies, specifically addressing mitochondrial polyP, that could pave the way for future pharmacological strategies against the dysfunction and failure of the organelle in AD and PD, as well as in regular aging.
Figure 1. Role of polyP in different cellular processes in which mitochondrial physiology is affected and are dysfunctional in AD an PD.
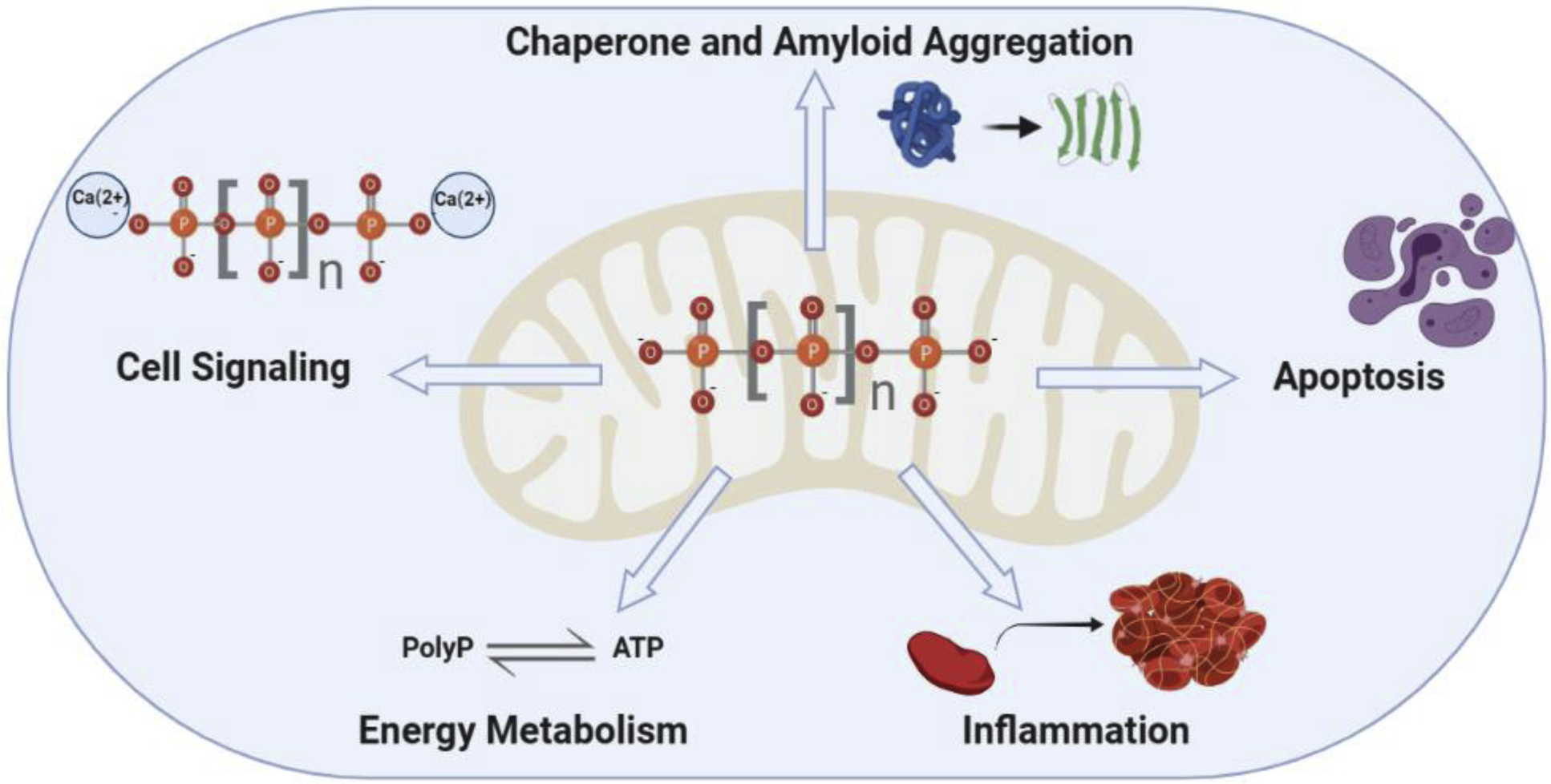
Various authors have shown the important role played by polyP in cell signaling (acting as a gliotransmitter and regulating mitochondrial calcium buffering and thus calcium signaling; energy metabolism) as an alternative energy storage form; inflammation (mainly located in the extracellular space and connecting inflammation and coagulation; and in the chaperoning and amyloid aggregation regulation) contributes to proteostasis within the organelle and the cell. Moreover, polyP has also been proposed to play a role in the direct regulation of apoptosis, which is the main cause of cell death within the central nervous system in all neurodegenerative diseases. All these processes share some common features: they are affected in AD and PD, mitochondria play a crucial role in them, and they are tightly inter-connected and inter-regulated. While many authors have demonstrated the effect of polyP on these processes, some of the data regarding the protective or deleterious action of the polymer seem to be contradictory. The size and the exact intra-cellular location of polyP could explain these differences. However, more research should be conducted to determine the exact metabolism of the polymer, as well as the optimal conditions in which polyP protects cellular populations against the mitochondrial dysfunction and the subsequent increased apoptotic cell death that is observed in AD and PD.
Figure 2. Role of polyP in protein biology.
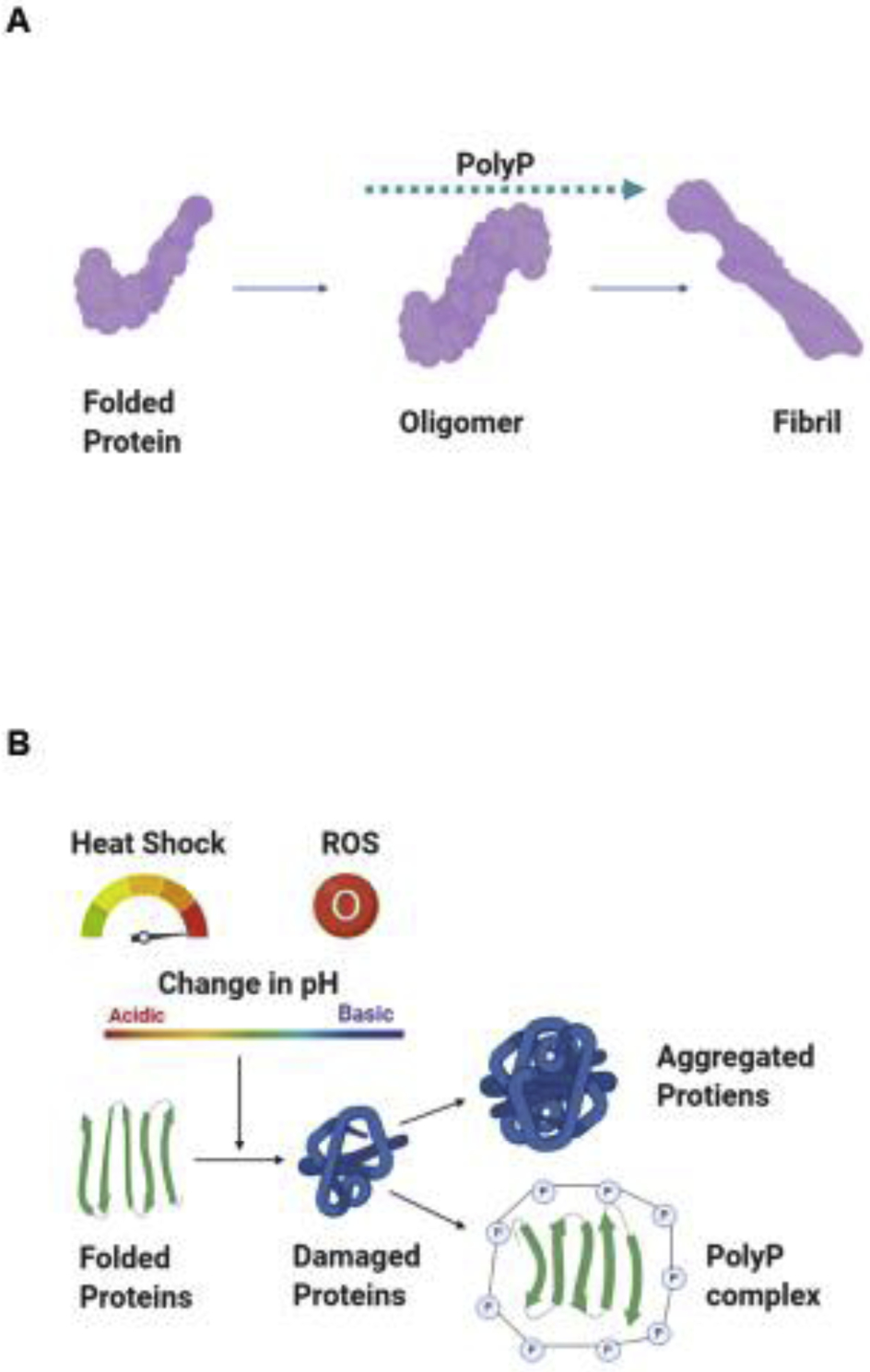
Various authors, using in vitro and in vivo models, have shown the protective effect of polyP against increased proteotoxicity in both A. amyloidogenic and, B. non-amyloidogenic proteins. While in the first case, polyP promotes the quick formation of insoluble fibrils, avoiding the presence of the toxic, soluble, oligomeric intermediates, in the non-amyloidogenic proteins, polyP promotes their refolding, into their functional conformation. Thereafter, polyP can be recycled, conserving its chaperoning activity. The most common insults known to increase protein dyshomeostasis are heat shock, variations in the cellular and/or mitochondrial pH, and increased presence of ROS. In the case of AD and PD, increased ROS – which is tightly related to mitochondrial dysfunction – have been broadly reported in different cellular and animal models of the disease.
Figure 3: Role of polyP in mitochondrial calcium buffering.
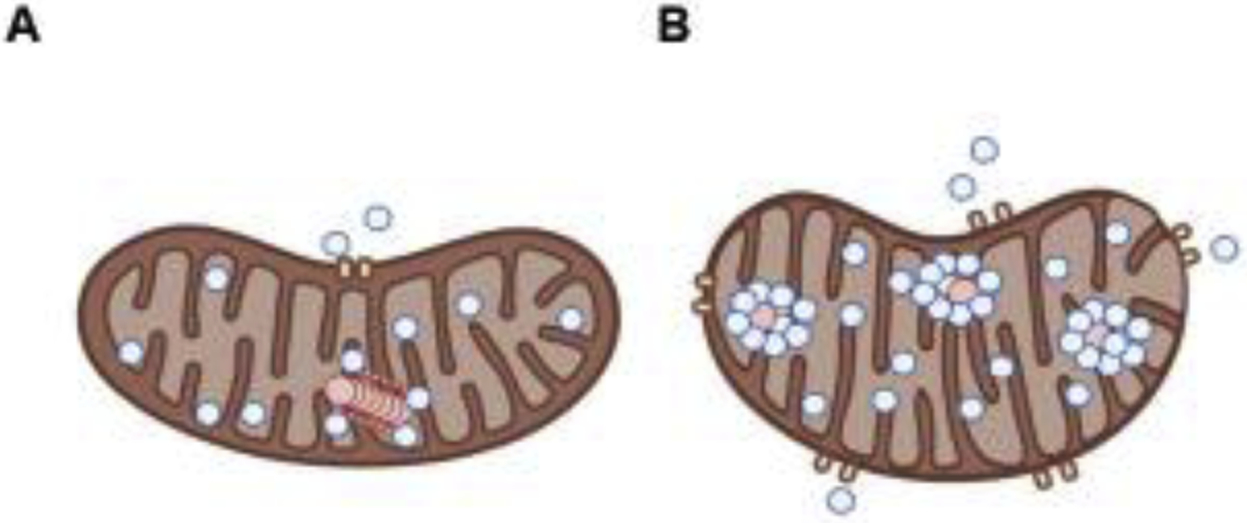
Mitochondria is a key organelle in the calcium buffering system, which is closely related to the signaling mediated by the cation. Interestingly, both calcium buffering and signaling are affected and dysfunctional in various neurodegenerative disorders, including AD and PD. A. While polyP and calcium are capable of forming reversible bonds, which can dissociate and contribute to buffering the levels of the cation, under the proper physiological conditions, B. the bound of orthophosphate and calcium is irreversible and it forms a crystal, which precipitates. Dysregulated calcium homeostasis within mitochondria will induce the opening of the mPTP and the consequent apoptotic cell death. Please, note that blue dots simbolize calcium anions, while red dots represent polyP.
ACKNOWLEDGMENTS
We kindly thank Mr. Mitch Maleki, Esq., for editing the manuscript. The writing of this review was supported by the National Institutes of Health (4R00AG055701-03 to MES) and by Rutgers University (Start Up funds to MES). Figures were created with BioRender.com.
ABBREVIATIONS:
- α-syn
α-synuclein
- Aβ
amyloid-β
- AD
Alzheimer’s Disease
- PPX
exopolyphosphatase enzyme
- polyP
inorganic polyphosphate
- I/R
ischemia/reperfusion
- mPTP
mitochondrial permeability transition pore
- MitoPPX
mitochondrial polyP(−) cells
- UPRmt
mitochondrial unfolded protein response
- PD
Parkinson’s Disease
- PPK
polyphosphate kinase
- ROS
reactive oxygen species
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare no conflict of interest
BIBLIOGRAPHY
- 1.Lin MT and Beal MF, Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 2006. 443(7113): p. 787–95. [DOI] [PubMed] [Google Scholar]
- 2.Kumble KD and Kornberg A, Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem, 1995. 270(11): p. 5818–22. [DOI] [PubMed] [Google Scholar]
- 3.Yoo NG, et al. , Polyphosphate Stabilizes Protein Unfolding Intermediates as Soluble Amyloid-like Oligomers. J Mol Biol, 2018. 430(21): p. 4195–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solesio ME, et al. , Inorganic polyphosphate is required for sustained free mitochondrial calcium elevation, following calcium uptake. Cell Calcium, 2020. 86: p. 102127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solesio ME, et al. , Contribution of inorganic polyphosphate towards regulation of mitochondrial free calcium. Biochim Biophys Acta, 2016. 1860(6): p. 1317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angelova PR, et al. , Signal transduction in astrocytes: Localization and release of inorganic polyphosphate. Glia, 2018. 66(10): p. 2126–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maiolino M, et al. , Inorganic Polyphosphate Regulates AMPA and NMDA Receptors and Protects Against Glutamate Excitotoxicity via Activation of P2Y Receptors. J Neurosci, 2019. 39(31): p. 6038–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie A, et al. , Shared mechanisms of neurodegeneration in Alzheimer’s disease and Parkinson’s disease. Biomed Res Int, 2014. 2014: p. 648740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morrissey JH, Choi SH, and Smith SA, Polyphosphate: an ancient molecule that links platelets, coagulation, and inflammation. Blood, 2012. 119(25): p. 5972–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller F, et al. , Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell, 2009. 139(6): p. 1143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurd MD, et al. , Monetary costs of dementia in the United States. N Engl J Med, 2013. 368(14): p. 1326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanzi RE, et al. , The gene defects responsible for familial Alzheimer’s disease. Neurobiol Dis, 1996. 3(3): p. 159–68. [DOI] [PubMed] [Google Scholar]
- 13.Solesio ME, et al. , Carbonic anhydrase inhibition selectively prevents amyloid beta neurovascular mitochondrial toxicity. Aging Cell, 2018. 17(4): p. e12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fossati S, et al. , The carbonic anhydrase inhibitor methazolamide prevents amyloid beta-induced mitochondrial dysfunction and caspase activation protecting neuronal and glial cells in vitro and in the mouse brain. Neurobiol Dis, 2016. 86: p. 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Lau LM and Breteler MM, Epidemiology of Parkinson’s disease. Lancet Neurol, 2006. 5(6): p. 525–35. [DOI] [PubMed] [Google Scholar]
- 16.Guo Y, et al. , Predictors of cognitive impairment in Parkinson’s disease: a systematic review and meta-analysis of prospective cohort studies. J Neurol, 2020. [DOI] [PubMed] [Google Scholar]
- 17.Klein C and Westenberger A, Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med, 2012. 2(1): p. a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spillantini MG, et al. , alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A, 1998. 95(11): p. 6469–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spillantini MG, et al. , Alpha-synuclein in Lewy bodies. Nature, 1997. 388(6645): p. 839–40. [DOI] [PubMed] [Google Scholar]
- 20.Baba M, et al. , Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol, 1998. 152(4): p. 879–84. [PMC free article] [PubMed] [Google Scholar]
- 21.Kuzuhara S, et al. , Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol, 1988. 75(4): p. 345–53. [DOI] [PubMed] [Google Scholar]
- 22.Ishizawa T, et al. , Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol, 2003. 62(4): p. 389–97. [DOI] [PubMed] [Google Scholar]
- 23.Braak H, et al. , Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna), 2003. 110(5): p. 517–36. [DOI] [PubMed] [Google Scholar]
- 24.Mullin S and Schapira A, alpha-Synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol Neurobiol, 2013. 47(2): p. 587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng Y and Bai F, The Association of Tau With Mitochondrial Dysfunction in Alzheimer’s Disease. Front Neurosci, 2018. 12: p. 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambon F, et al. , Cellular alpha-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum Mol Genet, 2019. 28(12): p. 2001–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreira PI, et al. , Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta, 2010. 1802(1): p. 2–10. [DOI] [PubMed] [Google Scholar]
- 28.Wang X, et al. , Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta, 2014. 1842(8): p. 1240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abou-Sleiman PM, Muqit MM, and Wood NW, Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci, 2006. 7(3): p. 207–19. [DOI] [PubMed] [Google Scholar]
- 30.Kornberg SR, Adenosine triphosphate synthesis from polyphosphate by an enzyme from Escherichia coli. Biochim Biophys Acta, 1957. 26(2): p. 294–300. [DOI] [PubMed] [Google Scholar]
- 31.Bayev AY, Angelova PR, and Abramov AY, Inorganic polyphosphate is produced and hydrolysed in F0F1-ATP synthase of mammalian mitochondria. Biochem J, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pavlov E, et al. , Inorganic polyphosphate and energy metabolism in mammalian cells. J Biol Chem, 2010. 285(13): p. 9420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kornberg A, Rao NN, and Ault-Riche D, Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem, 1999. 68: p. 89–125. [DOI] [PubMed] [Google Scholar]
- 34.Rao NN, Gomez-Garcia MR, and Kornberg A, Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem, 2009. 78: p. 605–47. [DOI] [PubMed] [Google Scholar]
- 35.Rao NN and Kornberg A, Inorganic polyphosphate regulates responses of Escherichia coli to nutritional stringencies, environmental stresses and survival in the stationary phase. Prog Mol Subcell Biol, 1999. 23: p. 183–95. [DOI] [PubMed] [Google Scholar]
- 36.Ault-Riche D, et al. , Novel assay reveals multiple pathways regulating stress-induced accumulations of inorganic polyphosphate in Escherichia coli. J Bacteriol, 1998. 180(7): p. 1841–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shiba T, et al. , Inorganic polyphosphate and the induction of rpoS expression. Proc Natl Acad Sci U S A, 1997. 94(21): p. 11210–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mullan A, Quinn JP, and McGrath JW, Enhanced phosphate uptake and polyphosphate accumulation in Burkholderia cepacia grown under low pH conditions. Microb Ecol, 2002. 44(1): p. 69–77. [DOI] [PubMed] [Google Scholar]
- 39.Gray MJ and Jakob U, Oxidative stress protection by polyphosphate--new roles for an old player. Curr Opin Microbiol, 2015. 24: p. 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao NN, Liu S, and Kornberg A, Inorganic polyphosphate in Escherichia coli: the phosphate regulon and the stringent response. J Bacteriol, 1998. 180(8): p. 2186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao NN and Kornberg A, Inorganic polyphosphate supports resistance and survival of stationary-phase Escherichia coli. J Bacteriol, 1996. 178(5): p. 1394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rashid MH, Rao NN, and Kornberg A, Inorganic polyphosphate is required for motility of bacterial pathogens. J Bacteriol, 2000. 182(1): p. 225–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi X, Rao NN, and Kornberg A, Inorganic polyphosphate in Bacillus cereus: motility, biofilm formation, and sporulation. Proc Natl Acad Sci U S A, 2004. 101(49): p. 17061–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim KS, et al. , Inorganic polyphosphate is essential for long-term survival and virulence factors in Shigella and Salmonella spp. Proc Natl Acad Sci U S A, 2002. 99(11): p. 7675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Travers RJ, Smith SA, and Morrissey JH, Polyphosphate, platelets, and coagulation. Int J Lab Hematol, 2015. 37 Suppl 1: p. 31–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Omelon S, et al. , Control of vertebrate skeletal mineralization by polyphosphates. PLoS One, 2009. 4(5): p. e5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Azevedo C, et al. , Screening a Protein Array with Synthetic Biotinylated Inorganic Polyphosphate To Define the Human PolyP-ome. ACS Chem Biol, 2018. 13(8): p. 1958–1963. [DOI] [PubMed] [Google Scholar]
- 48.Azevedo C, et al. , Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 Gag release. Proc Natl Acad Sci U S A, 2009. 106(50): p. 21161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, et al. , Inorganic polyphosphate stimulates mammalian TOR, a kinase involved in the proliferation of mammary cancer cells. Proc Natl Acad Sci U S A, 2003. 100(20): p. 11249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abramov AY, et al. , Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci U S A, 2007. 104(46): p. 18091–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie L and Jakob U, Inorganic polyphosphate, a multifunctional polyanionic protein scaffold. J Biol Chem, 2019. 294(6): p. 2180–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hernandez-Ruiz L, et al. , Inorganic polyphosphate and specific induction of apoptosis in human plasma cells. Haematologica, 2006. 91(9): p. 1180–6. [PubMed] [Google Scholar]
- 53.Dedkova EN, Inorganic polyphosphate in cardiac myocytes: from bioenergetics to the permeability transition pore and cell survival. Biochem Soc Trans, 2016. 44(1): p. 25–34. [DOI] [PubMed] [Google Scholar]
- 54.Seidlmayer LK, et al. , Dual role of inorganic polyphosphate in cardiac myocytes: The importance of polyP chain length for energy metabolism and mPTP activation. Arch Biochem Biophys, 2019. 662: p. 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seidlmayer LK, et al. , Inorganic polyphosphate is a potent activator of the mitochondrial permeability transition pore in cardiac myocytes. J Gen Physiol, 2012. 139(5): p. 321–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ogawa N, DeRisi J, and Brown PO, New components of a system for phosphate accumulation and polyphosphate metabolism in Saccharomyces cerevisiae revealed by genomic expression analysis. Mol Biol Cell, 2000. 11(12): p. 4309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hothorn M, et al. , Catalytic core of a membrane-associated eukaryotic polyphosphate polymerase. Science, 2009. 324(5926): p. 513–6. [DOI] [PubMed] [Google Scholar]
- 58.Ahn K and Kornberg A, Polyphosphate kinase from Escherichia coli. Purification and demonstration of a phosphoenzyme intermediate. J Biol Chem, 1990. 265(20): p. 11734–9. [PubMed] [Google Scholar]
- 59.Ishige K, Zhang H, and Kornberg A, Polyphosphate kinase (PPK2), a potent, polyphosphate-driven generator of GTP. Proc Natl Acad Sci U S A, 2002. 99(26): p. 16684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang H, Ishige K, and Kornberg A, A polyphosphate kinase (PPK2) widely conserved in bacteria. Proc Natl Acad Sci U S A, 2002. 99(26): p. 16678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nocek B, et al. , Polyphosphate-dependent synthesis of ATP and ADP by the family-2 polyphosphate kinases in bacteria. Proc Natl Acad Sci U S A, 2008. 105(46): p. 17730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akiyama M, Crooke E, and Kornberg A, An exopolyphosphatase of Escherichia coli. The enzyme and its ppx gene in a polyphosphate operon. J Biol Chem, 1993. 268(1): p. 633–9. [PubMed] [Google Scholar]
- 63.Lichko LP, Kulakovskaya TV, and Kulaev IS, Partial purification and characterization of nuclear exopolyphosphatase from Saccharomyces cerevisiae strain with inactivated PPX1 gene encoding a major yeast exopolyphosphatase. Biochemistry (Mosc), 2004. 69(3): p. 270–4. [DOI] [PubMed] [Google Scholar]
- 64.Suess PM, et al. , Extracellular polyphosphate signals through Ras and Akt to prime Dictyostelium discoideum cells for development. J Cell Sci, 2017. 130(14): p. 2394–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Holmstrom KM, et al. , Signalling properties of inorganic polyphosphate in the mammalian brain. Nat Commun, 2013. 4: p. 1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gabel NW and Thomas V, Evidence for the occurrence and distribution of inorganic polyphosphates in vertebrate tissues. J Neurochem, 1971. 18(7): p. 1229–42. [DOI] [PubMed] [Google Scholar]
- 67.Angelova PR, et al. , In situ investigation of mammalian inorganic polyphosphate localization using novel selective fluorescent probes JC-D7 and JC-D8. ACS Chem Biol, 2014. 9(9): p. 2101–10. [DOI] [PubMed] [Google Scholar]
- 68.Moreno-Sanchez D, et al. , Polyphosphate is a novel pro-inflammatory regulator of mast cells and is located in acidocalcisomes. J Biol Chem, 2012. 287(34): p. 28435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruiz FA, et al. , Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem, 2004. 279(43): p. 44250–7. [DOI] [PubMed] [Google Scholar]
- 70.Glonek T, et al. , Studies of biological polyphosphate through the use of phosphorus-31 nuclear magnetic resonance. Arch Biochem Biophys, 1971. 142(2): p. 508–13. [DOI] [PubMed] [Google Scholar]
- 71.Tijssen JP, Beekes HW, and Van Steveninck J, Localization of polyphosphates in Saccharomyces fragilis, as revealed by 4’,6-diamidino-2-phenylindole fluorescence. Biochim Biophys Acta, 1982. 721(4): p. 394–8. [DOI] [PubMed] [Google Scholar]
- 72.Aschar-Sobbi R, et al. , High sensitivity, quantitative measurements of polyphosphate using a new DAPI-based approach. J Fluoresc, 2008. 18(5): p. 859–66. [DOI] [PubMed] [Google Scholar]
- 73.Saito T, et al. , Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci, 2014. 17(5): p. 661–3. [DOI] [PubMed] [Google Scholar]
- 74.Muller WEG, Schroder HC, and Wang X, Inorganic Polyphosphates As Storage for and Generator of Metabolic Energy in the Extracellular Matrix. Chem Rev, 2019. 119(24): p. 12337–12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walsh DM and Selkoe DJ, Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett, 2004. 11(3): p. 213–28. [DOI] [PubMed] [Google Scholar]
- 76.Haass C and Selkoe DJ, Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol, 2007. 8(2): p. 101–12. [DOI] [PubMed] [Google Scholar]
- 77.Roberts HL and Brown DR, Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules, 2015. 5(2): p. 282–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Canevari L, Abramov AY, and Duchen MR, Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem Res, 2004. 29(3): p. 637–50. [DOI] [PubMed] [Google Scholar]
- 79.Hashimoto M, et al. , Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med, 2003. 4(1–2): p. 21–36. [DOI] [PubMed] [Google Scholar]
- 80.Lu B and Guo S, Mechanisms Linking Mitochondrial Dysfunction and Proteostasis Failure. Trends Cell Biol, 2020. 30(4): p. 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X, et al. , Insights into amyloid-beta-induced mitochondrial dysfunction in Alzheimer disease. Free Radic Biol Med, 2007. 43(12): p. 1569–73. [DOI] [PubMed] [Google Scholar]
- 82.Smith MA, et al. , Amyloid-beta, tau alterations and mitochondrial dysfunction in Alzheimer disease: the chickens or the eggs? Neurochem Int, 2002. 40(6): p. 527–31. [DOI] [PubMed] [Google Scholar]
- 83.Pinho CM, Teixeira PF, and Glaser E, Mitochondrial import and degradation of amyloid-beta peptide. Biochim Biophys Acta, 2014. 1837(7): p. 1069–74. [DOI] [PubMed] [Google Scholar]
- 84.Ruan L, et al. , Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature, 2017. 543(7645): p. 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu W, et al. , Mitochondrial protein import regulates cytosolic protein homeostasis and neuronal integrity. Autophagy, 2018. 14(8): p. 1293–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ciechanover A and Brundin P, The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron, 2003. 40(2): p. 427–46. [DOI] [PubMed] [Google Scholar]
- 87.Nixon RA, Mathews PM, and Cataldo AM, The neuronal endosomal-lysosomal system in Alzheimer’s disease. J Alzheimers Dis, 2001. 3(1): p. 97–107. [DOI] [PubMed] [Google Scholar]
- 88.Pan T, et al. , The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain, 2008. 131(Pt 8): p. 1969–78. [DOI] [PubMed] [Google Scholar]
- 89.Cremers CM, et al. , Polyphosphate: A Conserved Modifier of Amyloidogenic Processes. Mol Cell, 2016. 63(5): p. 768–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McColl G, et al. , Utility of an improved model of amyloid-beta (Abeta(1)(−)(4)(2)) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol Neurodegener, 2012. 7: p. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goedert M, et al. , Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature, 1996. 383(6600): p. 550–3. [DOI] [PubMed] [Google Scholar]
- 92.Lempart J, et al. , Mechanistic insights into the protective roles of polyphosphate against amyloid cytotoxicity. Life Sci Alliance, 2019. 2(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Khong ML, et al. , Inorganic polyphosphate controls cyclophilin B-mediated collagen folding in osteoblast-like cells. FEBS J, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rizzuto R, The collagen-mitochondria connection. Nat Genet, 2003. 35(4): p. 300–1. [DOI] [PubMed] [Google Scholar]
- 95.Neo SH and Tang BL, Collagen 1 signaling at the central nervous system injury site and astrogliosis. Neural Regen Res, 2017. 12(10): p. 1600–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nagy N, et al. , Collagen 18 and agrin are secreted by neural crest cells to remodel their microenvironment and regulate their migration during enteric nervous system development. Development, 2018. 145(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gregorio I, et al. , Collagen VI in healthy and diseased nervous system. Dis Model Mech, 2018. 11(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Forsell C, et al. , Genetic association to the amyloid plaque associated protein gene COL25A1 in Alzheimer’s disease. Neurobiol Aging, 2010. 31(3): p. 409–15. [DOI] [PubMed] [Google Scholar]
- 99.Tong Y, et al. , COL25A1 triggers and promotes Alzheimer’s disease-like pathology in vivo. Neurogenetics, 2010. 11(1): p. 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Smith SA, et al. , Inhibition of polyphosphate as a novel strategy for preventing thrombosis and inflammation. Blood, 2012. 120(26): p. 5103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Akiyama M, Crooke E, and Kornberg A, The polyphosphate kinase gene of Escherichia coli. Isolation and sequence of the ppk gene and membrane location of the protein. J Biol Chem, 1992. 267(31): p. 22556–61. [PubMed] [Google Scholar]
- 102.Alcantara C, et al. , Accumulation of polyphosphate in Lactobacillus spp. and its involvement in stress resistance. Appl Environ Microbiol, 2014. 80(5): p. 1650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jahid IK, Silva AJ, and Benitez JA, Polyphosphate stores enhance the ability of Vibrio cholerae to overcome environmental stresses in a low-phosphate environment. Appl Environ Microbiol, 2006. 72(11): p. 7043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nikel PI, et al. , Accumulation of inorganic polyphosphate enables stress endurance and catalytic vigour in Pseudomonas putida KT2440. Microb Cell Fact, 2013. 12: p. 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qiu L, Gulotta M, and Callender R, Lactate dehydrogenase undergoes a substantial structural change to bind its substrate. Biophys J, 2007. 93(5): p. 1677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gray MJ, et al. , Polyphosphate is a primordial chaperone. Mol Cell, 2014. 53(5): p. 689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brini M, et al. , Neuronal calcium signaling: function and dysfunction. Cell Mol Life Sci, 2014. 71(15): p. 2787–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lautenschlager J, et al. , C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat Commun, 2018. 9(1): p. 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang Y, Shi Y, and Wei H, Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J Alzheimers Dis Parkinsonism, 2017. 7(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rizzuto R, et al. , Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol, 2012. 13(9): p. 566–78. [DOI] [PubMed] [Google Scholar]
- 111.Joe EH, et al. , Astrocytes, Microglia, and Parkinson’s Disease. Exp Neurobiol, 2018. 27(2): p. 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harada K, Kamiya T, and Tsuboi T, Gliotransmitter Release from Astrocytes: Functional, Developmental, and Pathological Implications in the Brain. Front Neurosci, 2015. 9: p. 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Butt AM, ATP: a ubiquitous gliotransmitter integrating neuron-glial networks. Semin Cell Dev Biol, 2011. 22(2): p. 205–13. [DOI] [PubMed] [Google Scholar]
- 114.Ludtmann MHR and Abramov AY, Mitochondrial calcium imbalance in Parkinson’s disease. Neurosci Lett, 2018. 663: p. 86–90. [DOI] [PubMed] [Google Scholar]
- 115.Dorozhkin SV, Calcium orthophosphates: occurrence, properties, biomineralization, pathological calcification and biomimetic applications. Biomatter, 2011. 1(2): p. 121–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhu XH, et al. , Quantitative imaging of energy expenditure in human brain. Neuroimage, 2012. 60(4): p. 2107–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mattson MP, et al. , Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer’s and Parkinson’s diseases. Ann N Y Acad Sci, 1999. 893: p. 154–75. [DOI] [PubMed] [Google Scholar]
- 118.Quansah E, et al. , Targeting energy metabolism via the mitochondrial pyruvate carrier as a novel approach to attenuate neurodegeneration. Mol Neurodegener, 2018. 13(1): p. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Muller WEG, et al. , Rebalancing beta-Amyloid-Induced Decrease of ATP Level by Amorphous Nano/Micro Polyphosphate: Suppression of the Neurotoxic Effect of Amyloid beta-Protein Fragment 25–35. Int J Mol Sci, 2017. 18(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Muller WEG, et al. , Polyphosphate as a donor of high-energy phosphate for the synthesis of ADP and ATP. J Cell Sci, 2017. 130(16): p. 2747–2756. [DOI] [PubMed] [Google Scholar]
- 121.Freimoser FM, et al. , Systematic screening of polyphosphate (poly P) levels in yeast mutant cells reveals strong interdependence with primary metabolism. Genome Biol, 2006. 7(11): p. R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang X, Schroder HC, and Muller WE, Polyphosphate as a metabolic fuel in Metazoa: A foundational breakthrough invention for biomedical applications. Biotechnol J, 2016. 11(1): p. 11–30. [DOI] [PubMed] [Google Scholar]
- 123.Loo JM, et al. , Extracellular metabolic energetics can promote cancer progression. Cell, 2015. 160(3): p. 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tanner CM, et al. , Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect, 2011. 119(6): p. 866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Burton GJ and Jauniaux E, Oxidative stress. Best Pract Res Clin Obstet Gynaecol, 2011. 25(3): p. 287–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Markesbery WR, Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med, 1997. 23(1): p. 134–47. [DOI] [PubMed] [Google Scholar]
- 127.Jenner P and Olanow CW, Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology, 1996. 47(6 Suppl 3): p. S161–70. [DOI] [PubMed] [Google Scholar]
- 128.Halestrap AP, Mitochondria and reperfusion injury of the heart--a holey death but not beyond salvation. J Bioenerg Biomembr, 2009. 41(2): p. 113–21. [DOI] [PubMed] [Google Scholar]
- 129.Angelova PR and Abramov AY, Alpha-synuclein and beta-amyloid - different targets, same players: calcium, free radicals and mitochondria in the mechanism of neurodegeneration. Biochem Biophys Res Commun, 2017. 483(4): p. 1110–1115. [DOI] [PubMed] [Google Scholar]
- 130.Friberg H and Wieloch T, Mitochondrial permeability transition in acute neurodegeneration. Biochimie, 2002. 84(2–3): p. 241–50. [DOI] [PubMed] [Google Scholar]
- 131.Adam-Vizi V and Starkov AA, Calcium and mitochondrial reactive oxygen species generation: how to read the facts. J Alzheimers Dis, 2010. 20 Suppl 2: p. S413–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Muller M, et al. , Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front Neurosci, 2018. 12: p. 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zorov DB, Juhaszova M, and Sollott SJ, Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev, 2014. 94(3): p. 909–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bernardi P, Broekemeier KM, and Pfeiffer DR, Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr, 1994. 26(5): p. 509–17. [DOI] [PubMed] [Google Scholar]
- 135.Faulkner KM, Liochev SI, and Fridovich I, Stable Mn(III) porphyrins mimic superoxide dismutase in vitro and substitute for it in vivo. J Biol Chem, 1994. 269(38): p. 23471–6. [PubMed] [Google Scholar]
- 136.Seidlmayer LK, et al. , Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc Res, 2015. 106(2): p. 237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ward PA and Lentsch AB, The acute inflammatory response and its regulation. Arch Surg, 1999. 134(6): p. 666–9. [DOI] [PubMed] [Google Scholar]
- 138.Lawrence T and Gilroy DW, Chronic inflammation: a failure of resolution? Int J Exp Pathol, 2007. 88(2): p. 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee HJ, et al. , Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem, 2010. 285(12): p. 9262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kitazawa M, Yamasaki TR, and LaFerla FM, Microglia as a potential bridge between the amyloid beta-peptide and tau. Ann N Y Acad Sci, 2004. 1035: p. 85–103. [DOI] [PubMed] [Google Scholar]
- 141.Couch Y, et al. , The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation, 2011. 8: p. 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sokolova A, et al. , Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s disease. Brain Pathol, 2009. 19(3): p. 392–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walter S, et al. , Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem, 2007. 20(6): p. 947–56. [DOI] [PubMed] [Google Scholar]
- 144.Monahan AJ, Warren M, and Carvey PM, Neuroinflammation and peripheral immune infiltration in Parkinson’s disease: an autoimmune hypothesis. Cell Transplant, 2008. 17(4): p. 363–72. [PubMed] [Google Scholar]
- 145.Tansey MG and Goldberg MS, Neuroinflammation in Parkinson’s disease: its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis, 2010. 37(3): p. 510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Perry VH, Nicoll JA, and Holmes C, Microglia in neurodegenerative disease. Nat Rev Neurol, 2010. 6(4): p. 193–201. [DOI] [PubMed] [Google Scholar]
- 147.Lull ME and Block ML, Microglial activation and chronic neurodegeneration. Neurotherapeutics, 2010. 7(4): p. 354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Colombo E and Farina C, Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol, 2016. 37(9): p. 608–620. [DOI] [PubMed] [Google Scholar]
- 149.Kitada T, et al. , Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature, 1998. 392(6676): p. 605–8. [DOI] [PubMed] [Google Scholar]
- 150.Valente EM, et al. , PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol, 2004. 56(3): p. 336–41. [DOI] [PubMed] [Google Scholar]
- 151.Pickrell AM and Youle RJ, The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron, 2015. 85(2): p. 257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sliter DA, et al. , Parkin and PINK1 mitigate STING-induced inflammation. Nature, 2018. 561(7722): p. 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 153.Chen Q, Sun L, and Chen ZJ, Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol, 2016. 17(10): p. 1142–9. [DOI] [PubMed] [Google Scholar]
- 154.Ishikawa H and Barber GN, STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature, 2008. 455(7213): p. 674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dinarvand P, et al. , Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y1 purinergic receptor. Blood, 2014. 123(6): p. 935–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hassanian SM, et al. , Inorganic polyphosphate elicits pro-inflammatory responses through activation of the mammalian target of rapamycin complexes 1 and 2 in vascular endothelial cells. J Thromb Haemost, 2015. 13(5): p. 860–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bae JS, Lee W, and Rezaie AR, Polyphosphate elicits pro-inflammatory responses that are counteracted by activated protein C in both cellular and animal models. J Thromb Haemost, 2012. 10(6): p. 1145–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith SA and Morrissey JH, Polyphosphate: a new player in the field of hemostasis. Curr Opin Hematol, 2014. 21(5): p. 388–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Foley JH and Conway EM, Cross Talk Pathways Between Coagulation and Inflammation. Circ Res, 2016. 118(9): p. 1392–408. [DOI] [PubMed] [Google Scholar]
- 160.Bjorkqvist J, Jamsa A, and Renne T, Plasma kallikrein: the bradykinin-producing enzyme. Thromb Haemost, 2013. 110(3): p. 399–407. [DOI] [PubMed] [Google Scholar]
- 161.DiScipio RG, The activation of the alternative pathway C3 convertase by human plasma kallikrein. Immunology, 1982. 45(3): p. 587–95. [PMC free article] [PubMed] [Google Scholar]
- 162.Wiggins RC, Giclas PC, and Henson PM, Chemotactic activity generated from the fifth component of complement by plasma kallikrein of the rabbit. J Exp Med, 1981. 153(6): p. 1391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ghebrehiwet B, et al. , Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J Clin Invest, 1983. 71(5): p. 1450–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Xu J, et al. , Extracellular histones are major mediators of death in sepsis. Nat Med, 2009. 15(11): p. 1318–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang H, et al. , HMG-1 as a late mediator of endotoxin lethality in mice. Science, 1999. 285(5425): p. 248–51. [DOI] [PubMed] [Google Scholar]
- 166.Sama AE, et al. , Bench to bedside: HMGB1-a novel proinflammatory cytokine and potential therapeutic target for septic patients in the emergency department. Acad Emerg Med, 2004. 11(8): p. 867–73. [DOI] [PubMed] [Google Scholar]
- 167.Hayden MS and Ghosh S, NF-kappaB in immunobiology. Cell Res, 2011. 21(2): p. 223–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Halestrap AP and Davidson AM, Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J, 1990. 268(1): p. 153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Solesio ME, et al. , The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochim Biophys Acta, 2013. 1832(1): p. 174–82. [DOI] [PubMed] [Google Scholar]
- 170.Amodeo GF, Solesio ME, and Pavlov EV, From ATP synthase dimers to C-ring conformational changes: unified model of the mitochondrial permeability transition pore. Cell Death Dis, 2017. 8(12): p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Solesio ME, et al. , Inorganic polyphosphate (polyP) as an activator and structural component of the mitochondrial permeability transition pore. Biochem Soc Trans, 2016. 44(1): p. 7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Elustondo PA, et al. , Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov, 2016. 2: p. 16070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Amodeo GF, et al. , C subunit of the ATP synthase is an amyloidogenic channel-forming peptide: possible implications in mitochondrial pathogenesis. bioRxiv, 2020: p. 2020.01.16.908335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Angelova PR, et al. , Role of inorganic polyphosphate in mammalian cells: from signal transduction and mitochondrial metabolism to cell death. Biochem Soc Trans, 2016. 44(1): p. 40–5. [DOI] [PubMed] [Google Scholar]
- 175.Jimenez-Nunez MD, et al. , Myeloma cells contain high levels of inorganic polyphosphate which is associated with nucleolar transcription. Haematologica, 2012. 97(8): p. 1264–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xie L, et al. , Accumulation of Nucleolar Inorganic Polyphosphate Is a Cellular Response to Cisplatin-Induced Apoptosis. Front Oncol, 2019. 9: p. 1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sakatani A, et al. , Polyphosphate Derived from Lactobacillus brevis Inhibits Colon Cancer Progression Through Induction of Cell Apoptosis. Anticancer Res, 2016. 36(2): p. 591–8. [PubMed] [Google Scholar]
- 178.Seidlmayer LK, et al. , Inorganic polyphosphate--an unusual suspect of the mitochondrial permeability transition mystery. Channels (Austin), 2012. 6(6): p. 463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]