

Abstract
Atopic dermatitis is a common, chronic, inflammatory skin condition characterized by recurrent and pruritic skin eruptions. Multiple factors contribute to the pathogenesis of atopic dermatitis including skin barrier dysfunction, microbial dysbiosis and immune dysregulation. Interactions between these factors form a complex, multidirectional network that can reinforce atopic skin disease, but can also be ameliorated by targeted therapies. This review summarizes the complex interactions between contributing factors in atopic dermatitis and the implications on disease development and therapeutic interventions.
INTRODUCTION
Atopic dermatitis (AD) is the most common inflammatory skin disorder, with an estimated worldwide prevalence of 3–5% in adults and 15–20% in children (Asher et al. 2006; Weidinger et al. 2018). AD is characterized by eczematous, pruritic skin patches and plaques that can severely affect quality of life and result in high socioeconomic costs (Chung and Simpson 2019). AD has also been proposed to initiate the “atopic march”, a sequential development of allergic disorders including food allergies, allergic rhinitis, and asthma, though this concept has been challenged as a clustering of diseases rather than a sequential march (Dharmage et al. 2014; Gustafsson et al. 2000; Paller et al. 2019)._Therefore, a deeper understanding and targeted treatments against the pathogenic mechanisms in AD could have implications in the prevention of other allergic diseases.
Several models have been proposed to explain the pathogenesis of AD. Initial studies focused on the role of altered immune responses, especially Th2 and IgE responses, to be major drivers of the disease (van der Heijden et al. 1991; Vestergaard et al. 1999). The so-called “inside-out” hypothesis postulated that immune dysregulation leads to compromised skin barrier function, thus permitting allergens and pathogens to penetrate the skin (Leung 1999). However, the discovery of inherited defects in factors that contribute to the skin barrier posited the alternative “outside-in” hypothesis, where an underlying barrier dysfunction allows antigen penetration to induce altered immune responses (Proksch et al. 2006). Recently, models have incorporated aspects of both, including an “outside-inside-outside” model where the skin microbiome and environmental factors penetrate the body from the outside through epidermal barrier defects and trigger immune dysregulation, which further exacerbates skin barrier defects (Elias 2008).
The complexity of current AD models indicates significant potential for interaction and crosstalk between epidermal barrier defects, the skin microbiome and immune dysregulation. This review will summarize the current state of research into the complex interactions of these factors in the immunopathology of AD and the implications of this complexity for the development of therapeutics.
CONTRIBUTING FACTORS: INSIDE AND OUT
Barrier dysfunction
The skin provides a key barrier between the body and the outside world, preventing transepidermal water loss (TEWL) and excluding pathogens and environmental antigens (De Benedetto et al. 2012). A major component of this barrier function is the multifunctional epidermal protein filaggrin. Filaggrin is produced as the large, insoluble profilaggrin in the keratohyalin granules of keratinocytes (Brown and Irwin McLean 2012). These granules form liquid-liquid phase separation droplets that contribute to barrier formation as they reach the skin surface (Quiroz et al. 2020). Additionally, profilaggrin undergoes processing and crosslinking to ultimately contribute to a tightly interwoven lipid/protein matrix that acts as the “mortar” that holds keratinocyte bricks together in a strong, impermeable “brick and mortar” structure (Nemes and Steinert 1999). In addition to this structural role, filaggrin is degraded by proteases to release its component amino acids as one component of the so called natural moisturizing factors (NMF) that help to keep the epidermis hydrated (Rawlings and Harding 2004).
Lipids play an important role in barrier formation by maintaining lubrication and preventing dehydration, as well as having antimicrobial activity (Bhattacharya et al. 2019). Epidermal lipids are mainly composed of ceramides, cholesterol and free fatty acids, though the composition and amount vary across different body surfaces (Greene et al. 1970; Pappas 2009). These lipids make up a significant proportion of the extracellular matrix surrounding the crosslinked filaggrin, helping to make up the “mortar” of the brick and mortar structure (Elias and Friend 1975).
Disruption of barrier function can have profound implications for the development of AD. Filaggrin mutations are the strongest genetic risk factor in the development of AD (Palmer et al. 2006), and correlate with increased disease severity, allergic sensitization, and overall higher healthcare costs (Heede et al. 2017; Soares et al. 2018). Filaggrin mutations have also served as the basis for AD mouse models, including the flaky tail model (ft/ft mice) and the monogenic filaggrin-knockout mice (Flgft/ft mice) (Nakajima et al. 2019). Despite this strong association, filaggrin mutations are only present in a minority of the population and are unevenly distributed across racial and ethnic groups. For example, filaggrin mutations are present in approximately 30–50% of European Americans with AD, 27% of Asians with AD and 3–6% of African Americans with AD (Margolis et al. 2012; Czarnowicki et al. 2019), and the R501X and 2282del4 mutations account for 80% of filaggrin mutations in Ireland but only 1% in Singaporean Chinese (Chen et al. 2011).
In addition to filaggrin, other components of the skin barrier have been implicated in AD, including tight junction proteins (Zaniboni et al. 2016). For example, decreased claudin-1 expression in the affected skin of AD patients correlated with disease severity (De Benedetto et al. 2011). The skin also produces host defense peptides (HDPs) to regulate and control skin microbes and trigger host immune responses including inflammatory cytokine production (Schauber and Gallo 2008). Levels of these skin barrier proteins are decreased or dysregulated in atopic skin, further disrupting the barrier (Kuo et al. 2013). Furthermore, alterations in skin lipid composition are associated with barrier dysfunction in AD, including decreased ceramide chain length (Ishikawa et al. 2010; Janssens et al. 2012), increased cholesterol-3-sulfate levels (Li et al. 2017), and increased short chain and mono-unsaturated free fatty acids (Macheleidt et al. 2002; van Smeden et al. 2014)._A delicate balance between proteases, both intrinsic and environmental, and host-derived protease inhibitors regulate barrier formation through cleavage of structural proteins to induce desquamation (Veer et al. 2014). For example, excessive protease activity in the skin of patients with Netherton syndrome (that is caused by mutations in the protease inhibitor SPINK5) contributes to atopic skin disease and other systemic allergic conditions (Chavanas et al. 2000).
AD is an intensely pruritic condition driven by hyperinnervation, hypersensitivity and the release of pruritogens including IL-31, histamine, TSLP, bradykinin, and substance P (Furue et al. 2017; Hosogi et al. 2006; Kantor and Silverberg 2017; Kido-Nakahara et al. 2017; Mollanazar et al. 2016). As a result, skin injury from scratching significantly contributes towards impairment of skin barrier function via mechanical damage and upregulation of host proteases (Hachem et al. 2006). Additionally, microbial and/or host proteases produced or induced by common environmental skin exposures to house dust mites, cockroaches, fungi, pollen and bacteria can degrade intracellular junctions, disrupt the epithelial barrier and decrease lamellar body secretion critical for recovery of the epidermis (Takai and Ikeda 2011) (Jeong et al. 2008). Weather conditions, especially cold temperatures and low humidity, can also increase the permeability of the skin through a variety of mechanisms including decreased skin hydration, decreased extensibility and increased sensation of itch (Engebretsen et al. 2016).
Dysbiosis
As part of the interface with the environment, the skin is normally colonized by diverse commensal microorganisms while also preventing penetration by pathogenic microorganisms (Byrd et al. 2018; Paller et al. 2019). The balance of microorganisms that compose the skin microbiome is dynamic and there are differences in composition, depending on the anatomic skin site, age, sex and disease state (Findley et al. 2013; Grice et al. 2009). AD is associated with drastic shifts of the skin microbiome, notably the loss of commensal diversity and the dominant colonization with pathogenic Staphylococcus aureus and commensal S. epidermidis (Byrd et al. 2017). AD skin lesions have an estimated 90% S. aureus colonization rate that correlates with increased disease burden (Higaki et al. 1999). Clonotypic analysis of S. aureus clinical isolates shows that highly pathogenic, antimicrobial resistant, toxigenic strains predominate in AD skin that correlate with disease severity (Guzik et al. 2005; Pascolini et al. 2011). Conversely, recovery of commensal diversity can precede and indicate resolution of disease (Kong et al. 2012; Shi et al. 2018).
Immune dysregulation
AD is an inflammatory skin disease in which immune dysregulation results in subsequent systemic immune complications (Gavrilova 2018). Atopic dermatitis is strongly associated with type 2 immunity_(Brandt and Sivaprasad 2011). Robust Th2 polarization is seen as early as in the cord blood of infants that develop AD and continues to be present through adulthood (Herberth et al. 2010). This polarization towards Th2 immunity promotes the development of IgE producing B cell and plasma cells, thus contributing to other allergic diseases like food allergies, allergic rhinitis and asthma (Dharmage et al. 2014).
Recent studies have expanded on this Th2 paradigm to include roles for other immune subsets (Berker et al. 2017), such as Th17 (Esaki et al. 2016; Sugaya 2020), Th22 (Gittler et al. 2012), T regulatory cells (Tregs) (Fyhrquist et al. 2012) and Th9 cells (Ciprandi et al. 2013; Ma et al. 2014) in AD pathogenesis. Even Th1 immunity, while downregulated in acute AD, plays a crucial role in the maintenance of chronic AD (Su et al. 2017). Components of the innate immune system contribute significantly to the development of AD skin inflammation, including IL-5 and IL-13 from group 2 innate lymphoid cells (ILC2s), IL-4 from basophils, and IL-25, IL-33, and TSLP from epithelial cells (Divekar and Kita 2015; Hussain et al. 2018; Mashiko et al. 2017; Salimi et al. 2013). This immune milieu is dependent on a patient’s age, sex, and ethnic background, thereby adding to the complexity of this disease (Brunner et al. 2019; Brunner and Guttman-Yassky 2019; Kanda et al. 2019).
As an inflammatory skin condition, associations between human leukocyte antigen (HLA) loci and AD were thought to be likely. Multiple large studies have identified HLA loci including HLA-DRB1, HLA-DQA1, and HLA-DQB1, that demonstrate linkage to AD (Margolis et al. 2015; Paternoster et al. 2015; Saeki et al. 1995). Interestingly, all co-associated HLA loci between psoriasis and AD display opposing effects, in accordance with lower than expected concomitance of these two disparate diseases in patients (Baurecht et al. 2015; Nanda 1995).
MULTIDIRECTIONAL INTERACTIONS BETWEEN CONTRIBUTING FACTORS IN ATOPIC DERMATITIS PATHOGENESIS
Barrier dysfunction and dysbiosis
Barrier dysfunction alters the skin microbiome
One of the crucial roles of the skin barrier is to exclude pathogenic microorganisms and maintain a healthy layer of commensal bacteria (Gallo 2017). Disruptions to barrier function can result in deleterious changes to the microbiome known as dysbiosis (Figure 1a) (Zeeuwen et al. 2012). Patients with loss-of-function filaggrin mutations have profound shifts in their microbiome, including increased colonization with S. aureus as well as decreased microbial diversity (Clausen et al. 2017; Zeeuwen et al. 2017). Loss-of-function mutations in filaggrin results in increased surface accessibility of fibronectin and fibrinogen, which S. aureus uses for surface adherence (Cho et al. 2001). Additionally, filaggrin deficiency decreases skin hydrating natural moisturizing factors (NMFs), further disrupting the skin barrier and increasing accessibility of S. aureus binding proteins (Kezic et al. 2011). S. aureus clonotypes with increased avidity to adherence proteins are shown to be enriched in the skin of patients with filaggrin mutations (Clausen et al. 2017; Fleury et al. 2017). In addition to structural roles, filaggrin also plays a role in the expression and release of enzymes crucial to antimicrobial defense. Loss-of-function mutations in filaggrin results in decreased sphingomyelinase, an enzyme that can decrease levels of sphingomyelin. Sphingomyelin is employed by S. aureus in the binding of its pore forming α-toxin and the loss of sphingomyelinase results in increased lysis of keratinocytes and dermonecrosis (Brauweiler et al. 2013). In addition to the important role of filaggrin in impacting the skin microbiome, disruptions in the lipid envelope have also been implicated in promoting dysbiosis, including increases in S. aureus burden (Baurecht et al. 2018).
Figure 1. Complex interactions in atopic dermatitis.
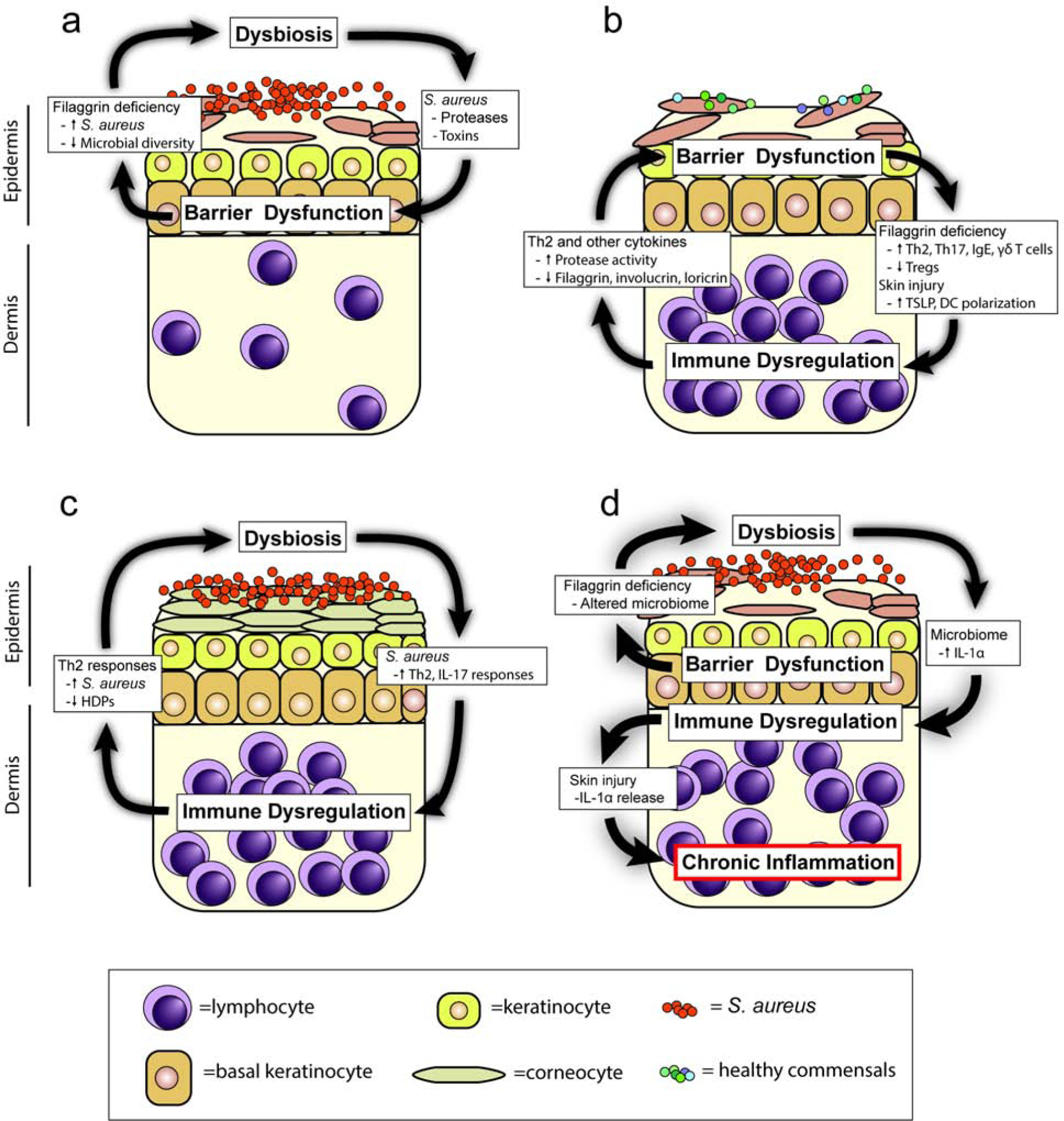
Atopic dermatitis pathogenesis involves directional interactions between (a) dysbiosis and barrier dysfunction, (b) barrier dysfunction and immune dysregulation, (c) dysbiosis and immune dysregulation, or (d) barrier dysfunction, dysbiosis, and immune dysregulation. For example, barrier dysfunction mediated by filaggrin deficiency promotes dysbiosis, resulting in dysregulated IL-1α production that is released upon skin injury to drive chronic skin inflammation (Archer et al 2019). NMF=natural moisturizing factors; HDP=host defense peptides.
Microbiome composition affects skin barrier function
Dysbiosis can also have consequences for skin barrier function. Clinical studies have shown that TEWL is increased with S. aureus colonization and positively correlates with bacterial burden (Jinnestål et al. 2014), though the directionality of this interaction is unknown. S. aureus utilizes a variety of secreted factors to disrupt the barrier, including proteases that degrade intercellular connections and pore-forming toxins to induce dermonecrosis (Williams et al. 2019). S. aureus also induces the activity of host proteases, which exacerbate barrier dysfunction by degrading structural proteins such as filaggrin and desmoglein-1 (Williams et al. 2017).
Barrier dysfunction and immune dysregulation
Barrier dysfunction induces immune dysregulation
Defective barrier function also plays a role in the immune dysregulation seen in AD (Figure 1b). Classical AD immune responses are elevated in patients with filaggrin loss-of-function mutations, with increased Th2 polarization, IgE levels and enhanced responses to epicutaneous allergen challenge (Brough et al. 2014). In mice, filaggrin deficiency induced similar polarization (Fallon et al. 2009; Scharschmidt et al. 2009). For example, increased levels of thymic stromal lymphopoietin (TSLP) are seen in skin equivalent cultures with filaggrin deficiency, which resulted in increased Th2 polarization (Wallmeyer et al. 2017). Filaggrin inhibits phospholipase A2 (PLA2) which participates in the generation of neolipid antigens that bind CD1a, resulting in a decrease in T cell proliferation and ultimately a decrease in Th2 immune responses (Jarrett et al. 2016). Conversely, patients with mutations in filaggrin lack inhibition of PLA2 dependent neolipid generation, resulting in significant increases in both total CD1a reactive T cells and percentage of those cells expressing IL-13 compared with patients without filaggrin mutations (Jarrett et al 2016). Excessive protease activity in filaggrin-deficient (ft/ft) mice may also play a role in TSLP production, with inhibition of the protease activating receptor 2 (PAR2) inhibiting this response (Moniaga et al. 2013). The excessive protease activity in Netherton syndrome (described above) also resulted in elevated skin TSLP and Th2 responses (Briot et al. 2009). In addition to filaggrin, other barrier deficiencies can induce immune dysregulation. Claudin-1, a tight junction protein, has been shown to be inversely correlated with Th2 polarization, though directionality is unclear (De Benedetto 2011). Tape-stripping of mouse skin is an experimental approach to artificially mimic skin barrier dysfunction from scratching behavior, which provides a model for the itch-scratch cycle in AD and results in induction of TSLP and polarizes dendritic cells to elicit Th2 responses in draining lymph nodes (Angelova-Fischer et al. 2010; Kondo et al. 1998; Oyoshi and Geha 2008).
Beyond the traditional Th2 paradigm, barrier dysfunction also induces other T cell subsets seen in AD. Children with loss-of-function filaggrin mutations have been shown to have increased peripheral Th17 responses, while filaggrin-deficient mice have been shown to have increased Th17 cells and IL-17-producing γδ T cells (Bonefeld et al. 2016; Jee et al. 2018; Oyoshi et al. 2009). Adults with loss-of-function filaggrin mutations also have decreased numbers of Tregs with impaired function and decreased IL-10 production (Moosbrugger-Martinz et al. 2019).
Immune dysregulation alters skin barrier function
Immune dysregulation can also have profound effects on skin barrier function. Treatment of keratinocytes with IL-4 and IL-13 decreases expression of filaggrin and other epidermal structural barrier proteins, including involucrin and loricrin (Howell et al. 2009; Kim et al. 2008). Filaggrin processing is also decreased by the downregulation of caspase-14, which processes filaggrin to profilaggrin (Hvid et al. 2011). Treatment of keratinocytes with IL-4 upregulates the production of proteases that contribute to desquamation, resulting in decreased barrier function as measured by TEWL (Hatano et al. 2013). Additionally, Th2-polarized skin has decreased levels of sphingomyelinase, an enzyme that depletes the sphingomyelin used by S. aureus α-toxin to cause keratinocyte lysis and skin barrier impairment (Brauweiler et al. 2013). The effect of inflammation on barrier function can also synergize with genetic defects. In filaggrin-deficient skin equivalent cultures there is upregulation of involucrin, loricrin and β-defeinsin-2, all of which are abrogated in the presence of Th2 cytokines (Hönzke et al. 2016). In addition to Th2 cytokines, other cytokines implicated in the pathogenesis of AD have been shown to downregulate filaggrin and other factors involved in skin barrier function, including IL-17 (Gutowska-Owsiak et al. 2012), IL-31(Cornelissen et al. 2012) and IL-22 (Gutowska-Owsiak et al. 2011).
Dysbiosis and immune dysregulation
Skin microbiome alters immune responses in AD
The pathogenesis of AD involves key interactions between immune responses and microbial communities (Figure 1c). The skin microbiome of AD patients has considerable variability, with various microorganisms associated with exacerbating or alleviating the skin inflammation (Byrd et al. 2017). Colonization with S. aureus is associated with both Th2 polarization and IgE levels in AD patients (Simpson et al. 2018). A variety of S. aureus-derived components have been implicated in Th2 polarization, including S. aureus superantigen B (SEB), protein A, δ-toxin and diacylated lipopeptides (Forbes-Blom et al. 2012; Nakamura et al. 2013; Terada et al. 2006). Penetration of the epidermis by S. aureus-derived proteases is crucial for this induction of Th2 immunity (Nakatsuji et al. 2016). In addition to S. aureus, Corynebacterium bovis induces Th2 polarization in a mouse model of AD (Kobayashi et al. 2015). Conversely, the secretome of S. epidermidis blocks CD4 proliferation and induces Tregs (Laborel-Préneron et al. 2015). Commensal skin microbiota also produce tryptophan metabolites that block Th2 induction through the aryl hydrocarbon receptor (Yu et al. 2019).
AD patients have been identified with antigen-specific IgE directed against S. aureus components, which correlates with increased disease severity (Sonesson et al. 2013). Specifically, IgE directed against S. aureus exotoxins can activate mast cells, basophils and other cells bearing FcεR1 to exacerbate the immune response (Leung et al. 1993).
In addition to inducing the canonical Th2/IgE responses, dysbiosis can promote other cell subsets implicated in AD pathogenesis. S. aureus exotoxins can induce IL-22 production from human PBMC-derived T cells, with increased production observed from AD patients when compared to healthy controls or psoriasis patients (Niebuhr et al. 2014; Niebuhr et al. 2010). Epicutaneous application of S. aureus induces AD-like skin inflammation in an IL-17 dependent manner (Liu et al. 2017; Nakamura et al. 2013). In addition to bacteria, commensal yeast (Malassezia spp.) can induce Th17 dependent skin inflammation on tape-stripped skin (Sparber et al. 2019)
Immune dysregulation alters the skin microbiome
Immune alterations can also influence the skin microbiome in AD. S. aureus binds more avidly to the Th2-polarized skin of AD due to increased accessibility of surface proteins fibronectin and fibrinogen as compared to skin in psoriasis or in healthy controls (Cho et al. 2001). Finally, expression of HDPs with anti-staphylococcal activity by cultured human keratinocytes, including human β-defensin 2 (HBD2), HBD3 and cathelicidin (LL-37), are inhibited by Th2 cytokines (Albanesi et al. 2007; Howell et al. 2008). Similar decreases in HDP expression were seen with AD skin explants and could be reversed with anti-IL-4, anti-IL-10 and anti-IL-13 neutralizing antibodies (Howell et al. 2009; Howell et al. 2006). The skin of patients with the Th2 predominant AD showed decreased levels of HDPs when compared to healthy controls and patients with psoriasis (Nomura et al. 2003; Ong et al. 2002).
Barrier dysfunction, dysbiosis, and immune dysregulation
Despite the interactions amongst barrier dysfunction, dysbiosis and immune dysregulation, relatively few studies have made the connection between all the contributing factors in a single model. We recently found in a mouse model of AD-like skin inflammation mimicking tissue injury from scratching that: (1) filaggrin-deficient (ft/ft) mice exhibited homeostatic dysbiosis and IL-1α dysregulation in skin compared to wildtype mice, and (2) injury-induced skin inflammation was driven by dysbiosis-mediated release of keratinocyte-derived IL-1α (Archer et al. 2019). Therefore, we discovered a mechanism wherein the barrier dysfunction caused by filaggrin deficiency resulted in dysbiosis that along with skin injury led to IL-1α-mediated immune dysregulation and chronic AD-like skin inflammation (Figure 1d). The interconnectedness of these contributing factors demonstrates the importance of understanding the complex interactions in the pathogenesis of AD.
THERAPEUTICS: TAKING COMPLEX INTERACTIONS INTO ACCOUNT
The complex interactions of barrier dysfunction, microbial dysbiosis, and immune dysregulation are important considerations in the development and use of therapeutics for AD (Figure 2). Targeting barrier dysfunction has long been a major aspect of clinical therapy, with topical emollients being first line initial treatment for AD (Eichenfield et al. 2017). Topical emollients treatment can improve the dysbiosis present in AD, decreasing the prevalence of S. aureus and restoring the normally diverse skin microbiome that is also observed in unaffected skin of AD patients (Seite et al. 2014). Barrier directed topical therapies are often combined with topical corticosteroids or calcineurin inhibitors, helping to address the immune dysfunction in AD. However, these immunomodulatory compounds can compromise skin barrier function, an effect that must be balanced with their ability to suppress inflammation (Kao et al. 2003; Kim et al. 2010)
Figure 2. The role of therapeutics in regulating interactions in atopic dermatitis pathogenesis.
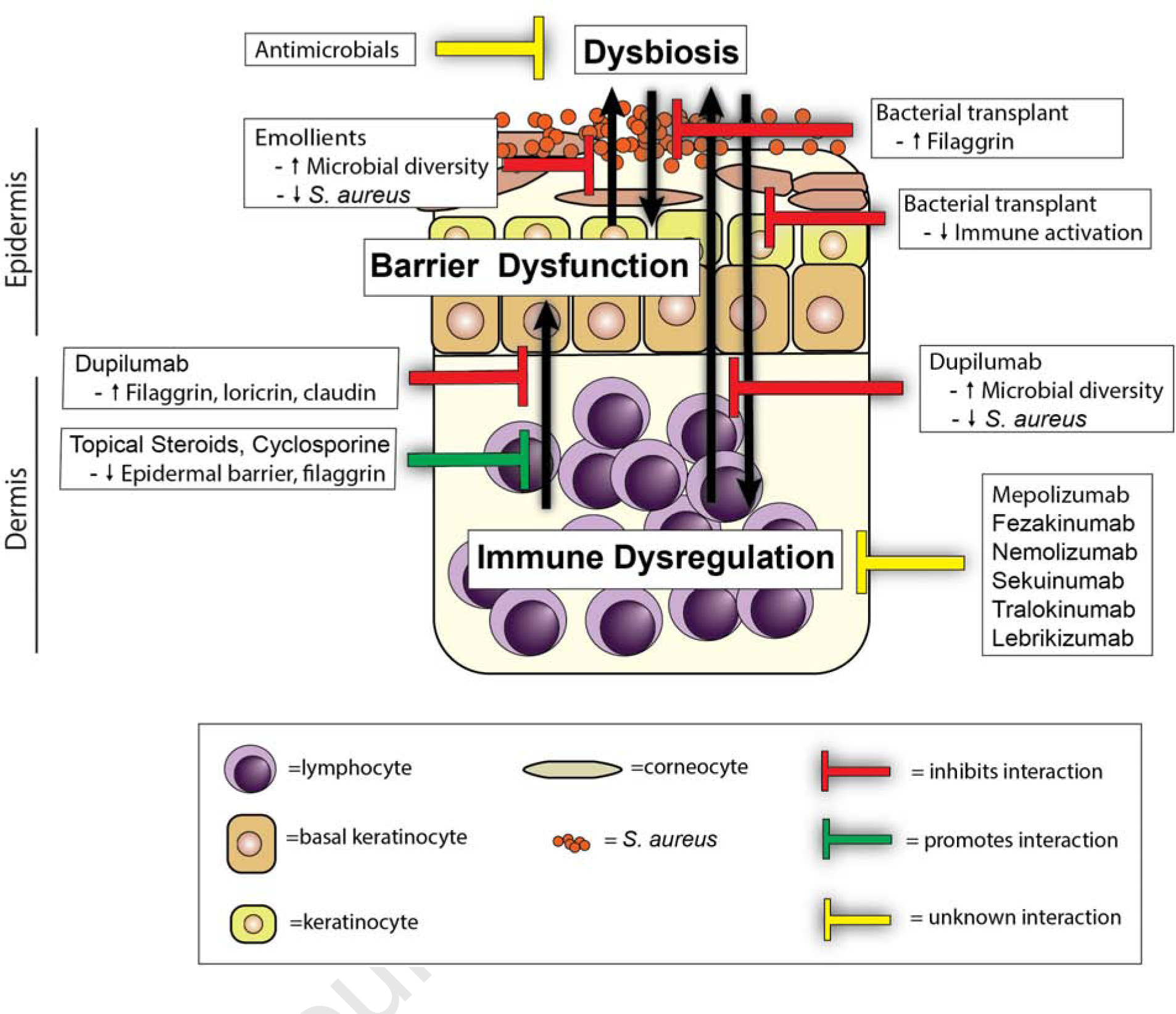
Therapeutic strategies can disrupt (e.g., dupilumab, bacterial transplant, and emollients) or strengthen (e.g., steroids and cyclosporine) the interactions between dysbiosis, barrier dysfunction, and immune dysregulation in atopic dermatitis pathogenesis, whereas the consequences of other therapeutics in these interaction are unknown (e.g., antimicrobials, mepolizumab, etc.).
Numerous therapies attempting to target the microbial dysfunction have been attempted, though none have yet been included in clinical guidelines in the absence of clinical findings consistent with cutaneous bacterial superinfection (Eichenfield et al. 2017). Trials of topical antibiotics in AD have been performed but results have been mixed and the use of antibiotics raises concern for the development of antibiotic resistance (Błażewicz et al. 2017; Breuer et al. 2002; Broberg and Faergemann 1995; Hung et al. 2007). Dilute bleach baths have been suggested by several studies, but in comparative studies were found to not be more effective than water baths alone (Hon et al. 2016; Huang et al. 2009). In addition to reducing pathogenic bacteria, treatments addressing the role of commensals have been evaluated. Commensal bacteria can produce antimicrobial peptides, decreasing the colonization of S. aureus, improving barrier function and decreasing immune activation (Myles et al. 2016; Nakatsuji et al. 2017). Coagulase-negative staphylococci produce autoinducing peptides that inhibit S. aureus agr-regulated toxin and virulence factor production (Williams et al. 2019). Transplant with Roseamonas mucosa, a bacteria derived from the skin of healthy volunteers, showed a benefit in reducing disease severity and S. aureus burden (Myles et al. 2018).
Therapies targeting the immune dysregulation often involve broad, systemic immunosuppression, including the use of methotrexate, cyclosporine, mycophenolate mofetil and azathioprine, though these drugs are usually reserved for severe, refractory cases due to their adverse side effect profile (Megna et al. 2016). Therapies directed towards specific dysregulated immune pathways have also become important in the treatment of severe AD. Dupilumab is a fully human monoclonal antibody targeting the shared receptor subunit for IL-4 and IL-13. Dupilumab is approved in the U.S. for the treatment of adults and children (above the age of 6 years-old) with poorly controlled AD (Gooderham et al. 2018; Licari et al. 2020). In phase III trials, AD patients treated with dupilumab achieved 36–38% improvement in the primary endpoint of the investigator global assessment (IGA) (compared with 8–10% of the placebo group) and nearly 50% of AD patients treated with dupilumab achieved at least 75% on the Eczema Area and Severity Index (EASI-75) (compared with ~15% of the placebo group) as a secondary endpoint (Simpson et al. 2016). Treatment has been shown to improve bacterial dysbiosis, reducing the prevalence of S. aureus and increasing microbial diversity (Callewaert et al. 2020). Dupilumab treatment also resulted in improved barrier function measures, including increased expression of filaggrin, loricrin, and claudins (Guttman-Yassky et al. 2019). However, the sizable population of partial or nonresponsive dupilumab-treated patients may be partially explained by the heterogenous nature of this disease and suggests a need for a personalized therapeutic approach (Hendricks et al. 2019).
Antibodies targeting additional cytokines have been attempted with varying degrees of success. For example, mepolizumab (anti-IL-5 mAb) is currently in phase 3 clinical trials after demonstrating improvements in pruritus whereas nemolizumab (anti-IL-31R mAb) also showed a significant benefit in pruritus in AD patients but little or no efficacy on the skin inflammation (Hajdarbegovic and Balak 2017; Oldhoff et al. 2005). Tralokinumab and Lebrikizumab, both anti-IL-13 mAbs though targeting different epitopes, have also shown efficacy in moderate to severe AD in phase 2 studies and are both currently undergoing phase 3 trials (Guttman-Yassky et al. 2020; Wollenberg et al. 2019). Fezakinumab, an anti-IL-22 mAb showed modest but significant efficacy in reducing skin inflammation in moderate to severe AD patients; however, the improvement persisted beyond the final treatment dose (Guttman-Yassky et al. 2018) Secukinumab (anti-IL-17A mAb) and MOR106 (anti-IL-17C mAb) both lacked efficacy against the skin inflammation in AD in phase II clinical trials (Galapagos NV 2020; Ungar et al. 2020). Furthermore, anti-IgE responses have been targeted in atopic dermatitis, but they either lacked efficacy or only had a modest effect in reducing skin inflammation in AD (Gomez et al. 2007; Krathen and Hsu 2005; Thaiwat and Sangasapaviliya 2011; Wang et al. 2016). Conversely, several clinical trials have shown potential for exacerbation of AD, including recombinant IL-4 therapy that increased Th2 polarization in psoriasis and the use of anti-TNF therapies that induced sporadic and worsening AD-like lesions (Flendrie et al. 2005; Nakamura et al. 2017; Rahier et al. 2010). However, additional studies are needed to understand how treatment efficacy is influenced by the variations in AD disease between patients.
CONCLUSIONS
The interactions between barrier dysfunction, dysbiosis, and immune dysregulation are crucial factors in the pathogenesis of AD. Further research is necessary into the roles and directionality of these interactions to better understand and treat this inflammatory skin disease. An improved understanding will help to usher in an era of directed, multifactorial treatments to this complex and heterogeneous disease.
AKNOWLEDGEMENTS
This work was funded in part by grants R01AR073665 (L.S.M. and N.K.A.) and R01AR069502 (L.S.M. and N.K.A.) from the U.S. National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
L.S.M. is a full-time employee at Janssen Research and Development and may own Johnson & Johnson stock and stock options. L.S.M. has received grant support from AstraZeneca, MedImmune (a subsidiary of AstraZeneca), Pfizer, Boerhinger Ingelheim, Regeneron Pharmaceuticals, and Moderna Therapeutics, is a shareholder of Noveome Biotherapeutics, was a paid consultant for Armirall and Janssen Research and Development and was on the scientific advisory board of Integrated Biotherapeutics, which are all developing therapeutics against infections (including S. aureus and other pathogens) and/or inflammatory conditions. N.K.A. has received grant support from Pfizer.
REFERENCES
- Albanesi C, Fairchild HR, Madonna S, Scarponi C, Pità OD, Leung DYM, et al. IL-4 and IL-13 Negatively Regulate TNF-α- and IFN-γ-Induced β-Defensin Expression through STAT-6, Suppressor of Cytokine Signaling (SOCS)-1, and SOCS-3. J. Immunol 2007;179(2):984–92 [DOI] [PubMed] [Google Scholar]
- Angelova-Fischer I, Fernandez IM, Donnadieu M-H, Bulfone-Paus S, Zillikens D, Fischer TW, et al. Injury to the stratum corneum induces in vivo expression of human thymic stromal lymphopoietin in the epidermis. J. Invest. Dermatol 2010;130(10):2505–7 [DOI] [PubMed] [Google Scholar]
- Archer NK, Jo J-H, Lee SK, Kim D, Smith B, Ortines RV, et al. Injury, dysbiosis, and filaggrin deficiency drive skin inflammation through keratinocyte IL-1α release. J. Allergy Clin. Immunol 2019;143(4):1426–1443.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher MI, Montefort S, Björkstén B, Lai CKW, Strachan DP, Weiland SK, et al. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 2006;368(9537):733–43 [DOI] [PubMed] [Google Scholar]
- Baurecht H, Hotze M, Brand S, Büning C, Cormican P, Corvin A, et al. Genome-wide Comparative Analysis of Atopic Dermatitis and Psoriasis Gives Insight into Opposing Genetic Mechanisms. Am. J. Hum. Genet 2015;96(1):104–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baurecht H, Rühlemann MC, Rodríguez E, Thielking F, Harder I, Erkens A-S, et al. Epidermal lipid composition, barrier integrity, and eczematous inflammation are associated with skin microbiome configuration. J. Allergy Clin. Immunol 2018;141(5):1668–1676.e16 [DOI] [PubMed] [Google Scholar]
- Berker M, Frank LJ, Geßner AL, Grassl N, Holtermann AV, Höppner S, et al. Allergies - A T cells perspective in the era beyond the TH1/TH2 paradigm. Clin. Immunol 2017;174:73–83 [DOI] [PubMed] [Google Scholar]
- Błażewicz I, Jaśkiewicz M, Bauer M, Piechowicz L, Nowicki RJ, Kamysz W, et al. Decolonization of Staphylococcus aureus in patients with atopic dermatitis: a reason for increasing resistance to antibiotics? Adv. Dermatol. Allergol 2017;34(6):553–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonefeld CM, Petersen TH, Bandier J, Agerbeck C, Linneberg A, Ross-Hansen K, et al. Epidermal filaggrin deficiency mediates increased systemic T-helper 17 immune response. Br. J. Dermatol 2016;175(4):706–12 [DOI] [PubMed] [Google Scholar]
- Brandt EB, Sivaprasad U. Th2 Cytokines and Atopic Dermatitis. J. Clin. Cell. Immunol 2011;2(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauweiler AM, Bin L, Kim BE, Oyoshi MK, Geha RS, Goleva E, et al. Filaggrin-dependent secretion of sphingomyelinase protects against staphylococcal α-toxin–induced keratinocyte death. J. Allergy Clin. Immunol 2013;131(2):421–427.e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer K, HÄussler S, Kapp A, Werfel T. Staphylococcus aureus: colonizing features and influence of an antibacterial treatment in adults with atopic dermatitis. Br. J. Dermatol 2002;147(1):55–61 [DOI] [PubMed] [Google Scholar]
- Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med 2009;206(5):1135–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broberg A, Faergemann J. Topical antimycotic treatment of atopic dermatitis in the head/neck area. A double-blind randomised study. Acta Derm. Venereol 1995;75(1):46–9 [DOI] [PubMed] [Google Scholar]
- Brough HA, Simpson A, Makinson K, Hankinson J, Brown S, Douiri A, et al. Peanut allergy: Effect of environmental peanut exposure in children with filaggrin loss-of-function mutations. J. Allergy Clin. Immunol 2014;134(4):867–875.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SJ, Irwin McLean WH. One Remarkable Molecule: Filaggrin. J. Invest. Dermatol 2012;132(3, Part 2):751–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner PM, Guttman-Yassky E. Racial differences in atopic dermatitis. Ann. Allergy Asthma Immunol 2019;122(5):449–55 [DOI] [PubMed] [Google Scholar]
- Brunner PM, He H, Pavel AB, Czarnowicki T, Lefferdink R, Erickson T, et al. The blood proteomic signature of early-onset pediatric atopic dermatitis shows systemic inflammation and is distinct from adult long-standing disease. J. Am. Acad. Dermatol 2019;81(2):510–9 [DOI] [PubMed] [Google Scholar]
- Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat. Rev. Microbiol 2018;16(3):143–55 [DOI] [PubMed] [Google Scholar]
- Byrd AL, Deming C, Cassidy SKB, Harrison OJ, Ng W-I, Conlan S, et al. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci. Transl. Med 2017;9(397) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert C, Nakatsuji T, Knight R, Kosciolek T, Vrbanac A, Kotol P, et al. IL-4Rα Blockade by Dupilumab Decreases Staphylococcus aureus Colonization and Increases Microbial Diversity in Atopic Dermatitis. J. Invest. Dermatol 2020;140(1):191–202.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet 2000;25(2):141–2 [DOI] [PubMed] [Google Scholar]
- Cho S-H, Strickland I, Tomkinson A, Fehringer AP, Gelfand EW, Leung DYM. Preferential Binding of Staphylococcus aureus to Skin Sites of Th2-Mediated Inflammation in a Murine Model. J. Invest. Dermatol 2001;116(5):658–63 [DOI] [PubMed] [Google Scholar]
- Chung J, Simpson EL. The socioeconomics of atopic dermatitis. Ann. Allergy. Asthma. Immunol 2019;122(4):360–6 [DOI] [PubMed] [Google Scholar]
- Ciprandi G, De Amici M, Giunta V, Marseglia A, Marseglia G. Serum interleukin-9 levels are associated with clinical severity in children with atopic dermatitis. Pediatr. Dermatol 2013;30(2):222–5 [DOI] [PubMed] [Google Scholar]
- Clausen M-L, Edslev SM, Andersen PS, Clemmensen K, Krogfelt KA, Agner T. Staphylococcus aureus colonization in atopic eczema and its association with filaggrin gene mutations. Br. J. Dermatol 2017;177(5):1394–400 [DOI] [PubMed] [Google Scholar]
- Cornelissen C, Marquardt Y, Czaja K, Wenzel J, Frank J, Lüscher-Firzlaff J, et al. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J. Allergy Clin. Immunol 2012;129(2):426–433.e8 [DOI] [PubMed] [Google Scholar]
- Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J. Allergy Clin. Immunol 2019;143(1):1–11 [DOI] [PubMed] [Google Scholar]
- De Benedetto A, Kubo A, Beck LA. Skin Barrier Disruption: A Requirement for Allergen Sensitization? J. Invest. Dermatol 2012;132(3, Part 2):949–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J. Allergy Clin. Immunol 2011;127(3):773–786.e1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmage SC, Lowe AJ, Matheson MC, Burgess JA, Allen KJ, Abramson MJ. Atopic dermatitis and the atopic march revisited. Allergy. 2014;69(1):17–27 [DOI] [PubMed] [Google Scholar]
- Divekar R, Kita H. Recent advances in epithelium-derived cytokines (IL-33, IL-25 and TSLP) and allergic inflammation. Curr. Opin. Allergy Clin. Immunol 2015;15(1):98–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichenfield LF, Ahluwalia J, Waldman A, Borok J, Udkoff J, Boguniewicz M. Current guidelines for the evaluation and management of atopic dermatitis: A comparison of the Joint Task Force Practice Parameter and American Academy of Dermatology guidelines. J. Allergy Clin. Immunol 2017;139(4S):S49–57 [DOI] [PubMed] [Google Scholar]
- Elias PM. Skin Barrier Function. Curr. Allergy Asthma Rep 2008;8(4):299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engebretsen KA, Johansen JD, Kezic S, Linneberg A, Thyssen JP. The effect of environmental humidity and temperature on skin barrier function and dermatitis. J. Eur. Acad. Dermatol. Venereol. JEADV 2016;30(2):223–49 [DOI] [PubMed] [Google Scholar]
- Esaki H, Brunner PM, Renert-Yuval Y, Czarnowicki T, Huynh T, Tran G, et al. Early-onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J. Allergy Clin. Immunol 2016;138(6):1639–51 [DOI] [PubMed] [Google Scholar]
- Fallon PG, Sasaki T, Sandilands A, Campbell LE, Saunders SP, Mangan NE, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat. Genet 2009;41(5):602–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498(7454):367–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury OM, McAleer MA, Feuillie C, Formosa-Dague C, Sansevere E, Bennett DE, et al. Clumping Factor B Promotes Adherence of Staphylococcus aureus to Corneocytes in Atopic Dermatitis. Infect. Immun 2017;85(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes-Blom E, Camberis M, Prout M, Tang S-C, Gros GL. Staphylococcal-derived superantigen enhances peanut induced Th2 responses in the skin. Clin. Exp. Allergy 2012;42(2):305–14 [DOI] [PubMed] [Google Scholar]
- Fyhrquist N, Lehtimäki S, Lahl K, Savinko T, Lappeteläinen A-M, Sparwasser T, et al. Foxp3+ Cells Control Th2 Responses in a Murine Model of Atopic Dermatitis. J. Invest. Dermatol 2012;132(6):1672–80 [DOI] [PubMed] [Google Scholar]
- Galapagos NV. A Phase II, Randomized, Double-blind, Placebo-controlled Repeated-dose Study to Evaluate the Efficacy, Safety, Tolerability, and PK/PD of Intravenously Administered MOR106 in Adult Subjects With Moderate to Severe Atopic Dermatitis. clinicaltrials.gov; 2020. March Report No.: NCT03568071 Available from: https://clinicaltrials.gov/ct2/show/NCT03568071
- Gallo RL. Human Skin Is the Largest Epithelial Surface for Interaction with Microbes. J. Invest. Dermatol 2017;137(6):1213–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova T Immune Dysregulation in the Pathogenesis of Atopic Dermatitis. Dermat. Contact Atopic Occup. Drug 2018;29(2):57–62 [DOI] [PubMed] [Google Scholar]
- Gittler JK, Shemer A, Suárez-Fariñas M, Fuentes-Duculan J, Gulewicz KJ, Wang CQF, et al. Progressive activation of TH2/TH22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol 2012;130(6):1344–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez G, Jogie-Brahim S, Shima M, Schwartz LB. Omalizumab reverses the phenotypic and functional effects of IgE-enhanced Fc epsilonRI on human skin mast cells. J. Immunol. 2007;179(2):1353–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooderham MJ, Hong HC-H, Eshtiaghi P, Papp KA. Dupilumab: A review of its use in the treatment of atopic dermatitis. J. Am. Acad. Dermatol 2018;78(3 Suppl 1):S28–36 [DOI] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and Temporal Diversity of the Human Skin Microbiome. Science. 2009;324(5931):1190–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson D, Sjöberg O, Foucard T. Development of allergies and asthma in infants and young children with atopic dermatitis – a prospective follow-up to 7 years of age. Allergy. 2000;55(3):240–5 [DOI] [PubMed] [Google Scholar]
- Gutowska-Owsiak D, Schaupp AL, Salimi M, Selvakumar TA, McPherson T, Taylor S, et al. IL-17 downregulates filaggrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp. Dermatol 2012;21(2):104–10 [DOI] [PubMed] [Google Scholar]
- Gutowska-Owsiak D, Schaupp AL, Salimi M, Taylor S, Ogg GS. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol 2011;165(3):492–8 [DOI] [PubMed] [Google Scholar]
- Guttman-Yassky E, Bissonnette R, Ungar B, Suárez-Fariñas M, Ardeleanu M, Esaki H, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol 2019;143(1):155–72 [DOI] [PubMed] [Google Scholar]
- Guttman-Yassky E, Blauvelt A, Eichenfield LF, Paller AS, Armstrong AW, Drew J, et al. Efficacy and Safety of Lebrikizumab, a High-Affinity Interleukin 13 Inhibitor, in Adults With Moderate to Severe Atopic Dermatitis: A Phase 2b Randomized Clinical Trial. JAMA Dermatol. 2020;156(4):411–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman-Yassky E, Brunner PM, Neumann AU, Khattri S, Pavel AB, Malik K, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: A randomized, double-blind, phase 2a trial. J. Am. Acad. Dermatol 2018;78(5):872–881.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, Bzowska M, Kasprowicz A, Czerniawska-Mysik G, Wójcik K, Szmyd D, et al. Persistent skin colonization with Staphylococcus aureus in atopic dermatitis: relationship to clinical and immunological parameters. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol 2005;35(4):448–55 [DOI] [PubMed] [Google Scholar]
- Hajdarbegovic E, Balak DMW. Anti-Interleukin-31 Receptor A Antibody for Atopic Dermatitis. N. Engl. J. Med 2017;376(21):2092. [DOI] [PubMed] [Google Scholar]
- Hatano Y, Adachi Y, Elias PM, Crumrine D, Sakai T, Kurahashi R, et al. The Th2 cytokine, interleukin-4, abrogates the cohesion of normal stratum corneum in mice: implications for pathogenesis of atopic dermatitis. Exp. Dermatol 2013;22(1):30–5 [DOI] [PubMed] [Google Scholar]
- Heede NG, Thyssen JP, Thuesen BH, Linneberg A, Szecsi PB, Stender S, et al. Health-related quality of life in adult dermatitis patients stratified by filaggrin genotype. Contact Dermatitis. 2017;76(3):167–77 [DOI] [PubMed] [Google Scholar]
- van der Heijden FL, Wierenga EA, Bos JD, Kapsenberg ML. High Frequency of IL-4–Producing CD4+ Allergen-Specific T Lymphocytes in Atopic Dermatitis Lesional Skin. J. Invest. Dermatol 1991;97(3):389–94 [DOI] [PubMed] [Google Scholar]
- Hendricks AJ, Lio PA, Shi VY. Management Recommendations for Dupilumab Partial and Non-durable Responders in Atopic Dermatitis. Am. J. Clin. Dermatol 2019;20(4):565–9 [DOI] [PubMed] [Google Scholar]
- Herberth G, Heinrich J, Röder S, Figl A, Weiss M, Diez U, et al. Reduced IFN-γ- and enhanced IL-4-producing CD4+ cord blood T cells are associated with a higher risk for atopic dermatitis during the first 2 yr of life. Pediatr. Allergy Immunol 2010;21(1-Part-I):5–13 [DOI] [PubMed] [Google Scholar]
- Higaki S, Morohashi M, Yamagishi T, Hasegawa Y. Comparative study of staphylococci from the skin of atopic dermatitis patients and from healthy subjects. Int. J. Dermatol 1999;38(4):265–9 [DOI] [PubMed] [Google Scholar]
- Hon KL, Tsang YCK, Lee VWY, Pong NH, Ha G, Lee ST, et al. Efficacy of sodium hypochlorite (bleach) baths to reduce Staphylococcus aureus colonization in childhood onset moderate-to-severe eczema: A randomized, placebo-controlled cross-over trial. J. Dermatol. Treat 2016;27(2):156–62 [DOI] [PubMed] [Google Scholar]
- Hönzke S, Wallmeyer L, Ostrowski A, Radbruch M, Mundhenk L, Schäfer-Korting M, et al. Influence of Th2 Cytokines on the Cornified Envelope, Tight Junction Proteins, and β-Defensins in Filaggrin-Deficient Skin Equivalents. J. Invest. Dermatol 2016;136(3):631–9 [DOI] [PubMed] [Google Scholar]
- Howell MD, Boguniewicz M, Pastore S, Novak N, Bieber T, Girolomoni G, et al. Mechanism of HBD-3 deficiency in atopic dermatitis. Clin. Immunol. Orlando Fla 2006;121(3):332–8 [DOI] [PubMed] [Google Scholar]
- Howell MD, Fairchild HR, Kim BE, Bin L, Boguniewicz M, Redzic JS, et al. Th2 Cytokines Act on S100/A11 to Downregulate Keratinocyte Differentiation. J. Invest. Dermatol 2008;128(9):2248–58 [DOI] [PubMed] [Google Scholar]
- Howell MD, Kim BE, Gao P, Grant AV, Boguniewicz M, DeBenedetto A, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol 2009;124(3, Supplement 2):R7–12 [DOI] [PubMed] [Google Scholar]
- Huang JT, Abrams M, Tlougan B, Rademaker A, Paller AS. Treatment of Staphylococcus aureus Colonization in Atopic Dermatitis Decreases Disease Severity. Pediatrics. 2009;123(5):e808–14 [DOI] [PubMed] [Google Scholar]
- Hung S-H, Lin Y-T, Chu C-Y, Lee C-C, Liang T-C, Yang Y-H, et al. Staphylococcus colonization in atopic dermatitis treated with fluticasone or tacrolimus with or without antibiotics. Ann. Allergy. Asthma. Immunol 2007;98(1):51–6 [DOI] [PubMed] [Google Scholar]
- Hussain M, Borcard L, Walsh KP, Rodriguez MP, Mueller C, Kim BS, et al. Basophil-derived IL-4 promotes epicutaneous antigen sensitization concomitant with the development of food allergy. J. Allergy Clin. Immunol 2018;141(1):223–234.e5 [DOI] [PubMed] [Google Scholar]
- Hvid M, Johansen C, Deleuran B, Kemp K, Deleuran M, Vestergaard C. Regulation of caspase 14 expression in keratinocytes by inflammatory cytokines--a possible link between reduced skin barrier function and inflammation? Exp. Dermatol 2011;20(8):633–6 [DOI] [PubMed] [Google Scholar]
- Jarrett R, Salio M, Lloyd-Lavery A, Subramaniam S, Bourgeois E, Archer C, et al. Filaggrin inhibits generation of CD1a neolipid antigens by house dust mite–derived phospholipase. Sci. Transl. Med 2016;8(325):325ra18–325ra18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jee MH, Johansen JD, Buus TB, Petersen TH, Gadsbøll A-SØ, Woetmann A, et al. Increased Production of IL-17A-Producing γδ T Cells in the Thymus of Filaggrin-Deficient Mice. Front. Immunol. Frontiers; 2018;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinnestål CL, Belfrage E, Bäck O, Schmidtchen A, Sonesson A. Skin barrier impairment correlates with cutaneous Staphylococcus aureus colonization and sensitization to skin-associated microbial antigens in adult patients with atopic dermatitis. Int. J. Dermatol 2014;53(1):27–33 [DOI] [PubMed] [Google Scholar]
- Kanda N, Hoashi T, Saeki H. The Roles of Sex Hormones in the Course of Atopic Dermatitis. Int. J. Mol. Sci 2019;20(19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao JS, Fluhr JW, Man M-Q, Fowler AJ, Hachem J-P, Crumrine D, et al. Short-term glucocorticoid treatment compromises both permeability barrier homeostasis and stratum corneum integrity: inhibition of epidermal lipid synthesis accounts for functional abnormalities. J. Invest. Dermatol 2003;120(3):456–64 [DOI] [PubMed] [Google Scholar]
- Kezic S, O’Regan GM, Yau N, Sandilands A, Chen H, Campbell LE, et al. Levels of filaggrin degradation products are influenced by both filaggrin genotype and atopic dermatitis severity. Allergy. 2011;66(7):934–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Jung M, Hong S-P, Jeon H, Kim M-J, Cho M-Y, et al. Topical calcineurin inhibitors compromise stratum corneum integrity, epidermal permeability and antimicrobial barrier function. Exp. Dermatol 2010;19(6):501–10 [DOI] [PubMed] [Google Scholar]
- Kim BE, Leung DYM, Boguniewicz M, Howell MD. Loricrin and involucrin expression is downregulated by Th2 cytokines through STAT-6. Clin. Immunol 2008;126(3):332–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, et al. Dysbiosis and Staphylococcus aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity. 2015;42(4):756–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo H, Ichikawa Y, Imokawa G. Percutaneous sensitization with allergens through barrier-disrupted skin elicits a Th2-dominant cytokine response. Eur. J. Immunol 1998;28(3):769–79 [DOI] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22(5):850–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krathen RA, Hsu S. Failure of omalizumab for treatment of severe adult atopic dermatitis. J. Am. Acad. Dermatol 2005;53(2):338–40 [DOI] [PubMed] [Google Scholar]
- Kuo I-H, Yoshida T, Benedetto AD, Beck LA. The cutaneous innate immune response in patients with atopic dermatitis. J. Allergy Clin. Immunol 2013;131(2):266–78 [DOI] [PubMed] [Google Scholar]
- Laborel-Préneron E, Bianchi P, Boralevi F, Lehours P, Fraysse F, Morice-Picard F, et al. Effects of the Staphylococcus aureus and Staphylococcus epidermidis Secretomes Isolated from the Skin Microbiota of Atopic Children on CD4+ T Cell Activation. 2015;10(10):e0141067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DYM. Pathogenesis of atopic dermatitis. J. Allergy Clin. Immunol 1999;104(3):S99–108 [DOI] [PubMed] [Google Scholar]
- Leung DY, Harbeck R, Bina P, Reiser RF, Yang E, Norris DA, et al. Presence of IgE antibodies to staphylococcal exotoxins on the skin of patients with atopic dermatitis. Evidence for a new group of allergens. J. Clin. Invest 1993;92(3):1374–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licari A, Castagnoli R, Marseglia A, Olivero F, Votto M, Ciprandi G, et al. Dupilumab to Treat Type 2 Inflammatory Diseases in Children and Adolescents. Pediatr. Drugs 2020; Available from: 10.1007/s40272-020-00387-2 [DOI] [PubMed]
- Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, et al. Staphylococcus aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe. 2017;22(5):653–666.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Xue H-B, Guan X-H, Shu C-M, Zhang J-H, Yu J. Possible pathogenic role of T helper type 9 cells and interleukin (IL)-9 in atopic dermatitis. Clin. Exp. Immunol 2014;175(1):25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Mitra N, Kim B, Gupta J, Hoffstad OJ, Papadopoulos M, et al. Association of HLA-DRB1 genetic variants with the persistence of atopic dermatitis. Hum. Immunol 2015;76(8):571–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashiko S, Mehta H, Bissonnette R, Sarfati M. Increased frequencies of basophils, type 2 innate lymphoid cells and Th2 cells in skin of patients with atopic dermatitis but not psoriasis. J. Dermatol. Sci 2017;88(2):167–74 [DOI] [PubMed] [Google Scholar]
- Megna M, Napolitano M, Patruno C, Villani A, Balato A, Monfrecola G, et al. Systemic Treatment of Adult Atopic Dermatitis: A Review. Dermatol. Ther 2016;7(1):1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniaga CS, Jeong SK, Egawa G, Nakajima S, Hara-Chikuma M, Jeon JE, et al. Protease Activity Enhances Production of Thymic Stromal Lymphopoietin and Basophil Accumulation in Flaky Tail Mice. Am. J. Pathol 2013;182(3):841–51 [DOI] [PubMed] [Google Scholar]
- Moosbrugger-Martinz V, Gruber R, Ladstätter K, Bellutti M, Blunder S, Schmuth M, et al. Filaggrin null mutations are associated with altered circulating Tregs in atopic dermatitis. J. Cell. Mol. Med 2019;23(2):1288–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles IA, Earland NJ, Anderson ED, Moore IN, Kieh MD, Williams KW, et al. First-in-human topical microbiome transplantation with Roseomonas mucosa for atopic dermatitis. JCI Insight. 2018;3(9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles IA, Williams KW, Reckhow JD, Jammeh ML, Pincus NB, Sastalla I, et al. Transplantation of human skin microbiota in models of atopic dermatitis. JCI Insight. 2016;1(10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, Jenisch S, et al. Sequence and Haplotype Analysis Supports HLA-C as the Psoriasis Susceptibility 1 Gene. Am. J. Hum. Genet 2006;78(5):827–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S, Nomura T, Common J, Kabashima K. Insights into atopic dermatitis gained from genetically defined mouse models. J. Allergy Clin. Immunol 2019;143(1):13–25 [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Muñoz-Planillo R, Hasegawa M, et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature. 2013;503(7476):397–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med 2017;9(378) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Two AM, Chun KA, Narala S, Geha RS, et al. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J. Invest. Dermatol 2016;136(11):2192–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda A Concomitance of psoriasis and atopic dermatitis. Dermatol. 1995;191(1):72. [DOI] [PubMed] [Google Scholar]
- Nemes Z, Steinert PM. Bricks and mortar of the epidermal barrier. Exp. Mol. Med 1999;31(1):5–19 [DOI] [PubMed] [Google Scholar]
- Niebuhr M, Mainardy J, Heratizadeh A, Satzger I, Werfel T. Staphylococcal exotoxins induce interleukin 22 in human th22 cells. Int. Arch. Allergy Immunol 2014;165(1):35–9 [DOI] [PubMed] [Google Scholar]
- Niebuhr M, Scharonow H, Gathmann M, Mamerow D, Werfel T. Staphylococcal exotoxins are strong inducers of IL-22: A potential role in atopic dermatitis. J. Allergy Clin. Immunol 2010;126(6):1176–1183.e4 [DOI] [PubMed] [Google Scholar]
- Nomura I, Goleva E, Howell MD, Hamid QA, Ong PY, Hall CF, et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J. Immunol 2003;171(6):3262–9 [DOI] [PubMed] [Google Scholar]
- Oldhoff JM, Darsow U, Werfel T, Katzer K, Wulf A, Laifaoui J, et al. Anti-IL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy. 2005;60(5):693–6 [DOI] [PubMed] [Google Scholar]
- Ong PY, Ohtake T, Brandt C, Strickland I, Boguniewicz M, Ganz T, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med 2002;347(15):1151–60 [DOI] [PubMed] [Google Scholar]
- Oyoshi MK, Geha RS. Mechanical injury by tape stripping causes Th2 responses in mice. FASEB J. Federation of American Societies for Experimental Biology; 2008;22(1_supplement):670.4–670.4 [Google Scholar]
- Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J. Allergy Clin. Immunol 2009;124(3):485–493.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paller AS, Kong HH, Seed P, Naik S, Scharschmidt TC, Gallo RL, et al. The microbiome in patients with atopic dermatitis. J. Allergy Clin. Immunol 2019;143(1):26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CNA, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet 2006;38(4):441–6 [DOI] [PubMed] [Google Scholar]
- Pascolini C, Sinagra J, Pecetta S, Bordignon V, De Santis A, Cilli L, et al. Molecular and Immunological Characterization of Staphylococcus aureus in Pediatric Atopic Dermatitis: Implications for Prophylaxis and Clinical Management. Clin. Dev. Immunol 2011;2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, et al. Multi-ethnic genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet 2015;47(12):1449–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proksch E, Fölster-Holst R, Jensen J-M. Skin barrier function, epidermal proliferation and differentiation in eczema. J. Dermatol. Sci 2006;43(3):159–69 [DOI] [PubMed] [Google Scholar]
- Quiroz FG, Fiore VF, Levorse J, Polak L, Wong E, Pasolli HA, et al. Liquid-liquid phase separation drives skin barrier formation. Science. 2020;367(6483) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings AV, Harding CR. Moisturization and skin barrier function. Dermatol. Ther 2004;17 Suppl 1:43–8 [DOI] [PubMed] [Google Scholar]
- Saeki H, Kuwata S, Nakagawa H, Etoh T, Yanagisawa M, Miyamoto M, et al. Analysis of disease-associated amino acid epitopes on HLA class II molecules in atopic dermatitis. J. Allergy Clin. Immunol 1995;96(6, Supplement):1061–8 [DOI] [PubMed] [Google Scholar]
- Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med 2013;210(13):2939–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharschmidt TC, Man M-Q, Hatano Y, Crumrine D, Gunathilake R, Sundberg JP, et al. Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J. Allergy Clin. Immunol 2009;124(3):496–506.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauber J, Gallo RL. Antimicrobial peptides and the skin immune defense system. J. Allergy Clin. Immunol 2008;122(2):261–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seite S, Flores GE, Henley JB, Martin R, Zelenkova H, Aguilar L, et al. Microbiome of affected and unaffected skin of patients with atopic dermatitis before and after emollient treatment. J. Drugs Dermatol. JDD 2014;13(11):1365–72 [PubMed] [Google Scholar]
- Shi B, Leung DYM, Taylor PA, Li H. Methicillin-Resistant Staphylococcus aureus Colonization Is Associated with Decreased Skin Commensal Bacteria in Atopic Dermatitis. J. Invest. Dermatol 2018;138(7):1668–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two Phase 3 Trials of Dupilumab versus Placebo in Atopic Dermatitis. N. Engl. J. Med 2016;375(24):2335–48 [DOI] [PubMed] [Google Scholar]
- Simpson EL, Villarreal M, Jepson B, Rafaels N, David G, Hanifin J, et al. Patients with Atopic Dermatitis Colonized with Staphylococcus aureus Have a Distinct Phenotype and Endotype. J. Invest. Dermatol 2018;138(10):2224–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P, Fidler K, Felton J, Tavendale R, Hövels A, Bremner SA, et al. Individuals with filaggrin-related eczema and asthma have increased long-term medication and hospital admission costs. Br. J. Dermatol 2018;179(3):717–23 [DOI] [PubMed] [Google Scholar]
- Sonesson A, Bartosik J, Christiansen J, Roscher I, Nilsson F, Schmidtchen A, et al. Sensitization to skin-associated microorganisms in adult patients with atopic dermatitis is of importance for disease severity. Acta Derm. Venereol 2013;93(3):340–5 [DOI] [PubMed] [Google Scholar]
- Sparber F, De Gregorio C, Steckholzer S, Ferreira FM, Dolowschiak T, Ruchti F, et al. The Skin Commensal Yeast Malassezia Triggers a Type 17 Response that Coordinates Anti-fungal Immunity and Exacerbates Skin Inflammation. Cell Host Microbe. 2019;25(3):389–403.e6 [DOI] [PubMed] [Google Scholar]
- Su C, Yang T, Wu Z, Zhong J, Huang Y, Huang T, et al. Differentiation of T-helper cells in distinct phases of atopic dermatitis involves Th1/Th2 and Th17/Treg. Eur. J. Inflamm 2017;15(1):46–52 [Google Scholar]
- Sugaya M The Role of Th17-Related Cytokines in Atopic Dermatitis. Int. J. Mol. Sci 2020;21(4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada M, Tsutsui H, Imai Y, Yasuda K, Mizutani H, Yamanishi K, et al. Contribution of IL-18 to atopic-dermatitis-like skin inflammation induced by Staphylococcus aureus product in mice. Proc. Natl. Acad. Sci 2006;103(23):8816–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiwat S, Sangasapaviliya A. Omalizumab treatment in severe adult atopic dermatitis. Asian Pac. J. Allergy Immunol 2011;29(4):357–60 [PubMed] [Google Scholar]
- Ungar B, Pavel AB, Li R, Kimmel G, Nia J, Hashim P, et al. Phase 2 Randomized, Double-blind Study of IL-17-Targeting with Secukinumab in Atopic Dermatitis. J. Allergy Clin. Immunol 2020; Available from: http://www.sciencedirect.com/science/article/pii/S0091674920306849 [DOI] [PubMed] [Google Scholar]
- Veer SJ de, Furio L, Harris JM, Hovnanian A. Proteases: common culprits in human skin disorders. Trends Mol. Med 2014;20(3):166–78 [DOI] [PubMed] [Google Scholar]
- Vestergaard C, Yoneyama H, Murai M, Nakamura K, Tamaki K, Terashima Y, et al. Overproduction of Th2-specific chemokines in NC/Nga mice exhibiting atopic dermatitis–like lesions. J. Clin. Invest 1999;104(8):1097–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallmeyer L, Dietert K, Sochorová M, Gruber AD, Kleuser B, Vávrová K, et al. TSLP is a direct trigger for T cell migration in filaggrin-deficient skin equivalents. Sci. Rep 2017;7(1):1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H-H, Li Y-C, Huang Y-C. Efficacy of omalizumab in patients with atopic dermatitis: A systematic review and meta-analysis. J. Allergy Clin. Immunol 2016;138(6):1719–1722.e1 [DOI] [PubMed] [Google Scholar]
- Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat. Rev. Dis. Primer 2018;4(1):1. [DOI] [PubMed] [Google Scholar]
- Williams MR, Costa SK, Zaramela LS, Khalil S, Todd DA, Winter HL, et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci. Transl. Med. American Association for the Advancement of Science; 2019;11(490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Nakatsuji T, Sanford JA, Vrbanac AF, Gallo RL. Staphylococcus aureus Induces Increased Serine Protease Activity in Keratinocytes. J. Invest. Dermatol 2017;137(2):377–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J. Allergy Clin. Immunol 2019;143(1):135–41 [DOI] [PubMed] [Google Scholar]
- Yu J, Luo Y, Zhu Z, Zhou Y, Sun L, Gao J, et al. A tryptophan metabolite of the skin microbiota attenuates inflammation in patients with atopic dermatitis through the aryl hydrocarbon receptor. J. Allergy Clin. Immunol 2019;143(6):2108–2119.e12 [DOI] [PubMed] [Google Scholar]
- Zaniboni MC, Samorano LP, Orfali RL, Aoki V, Zaniboni MC, Samorano LP, et al. Skin barrier in atopic dermatitis: beyond filaggrin. An. Bras. Dermatol 2016;91(4):472–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeuwen PL, Boekhorst J, van den Bogaard EH, de Koning HD, van de Kerkhof PM, Saulnier DM, et al. Microbiome dynamics of human epidermis following skin barrier disruption. Genome Biol. 2012;13(11):R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeuwen PLJM, Ederveen THA, van der Krieken DA, Niehues H, Boekhorst J, Kezic S, et al. Gram-positive anaerobe cocci are underrepresented in the microbiome of filaggrin-deficient human skin. J. Allergy Clin. Immunol 2017;139(4):1368–71 [DOI] [PubMed] [Google Scholar]