

Abstract
In cutaneous leishmaniasis (CL) the immune response is not only protective, but also mediates immunopathology. We previously found that cytolytic CD8 T cells promote inflammatory responses that are difficult to treat with conventional therapies that target the parasite. Therefore, we hypothesized that inhibiting CD8 T cell cytotoxicity would reduce disease severity in patients. IL-15 is a potential target for such treatment, as it is highly expressed in human CL patients’ lesions and promotes granzyme B (GzmB) dependent CD8 T cell cytotoxicity. Here we tested whether tofacitinib, which inhibits IL-15 signaling by blocking janus kinase (jak) 3, might decrease CD8 dependent pathology. We found that tofacitinib reduced expression of GzmB by CD8 T cells in vitro, and in vivo systemic and topical treatment with tofacitinib protected mice from developing severe CL lesions. Importantly, tofacitinib treatment did not alter Th1 responses or parasite control. Collectively, our results suggest that host-directed therapies do not need to be limited to autoimmune disorders and that topical tofacitinib application should be considered strategy for the treatment of CL disease in combination with anti-parasitic drugs.
Introduction
CD8 T cells promote protection by killing infected or transformed cells, but in certain situations this can have an undesired outcome by perpetuating inflammatory responses. For example, CD8 T cell cytotoxicity has been linked with enhanced disease in autoimmune diseases, such as type 1 diabetes (Young et al., 1989; Kägi et al., 1997; Trivedi et al., 2016) and alopecia universalis (Xing et al., 2014), in infectious diseases, such as cerebral malaria (Junqueira et al., 2018; Claser et al., 2019; Kaminski et al., 2019; Riggle et al., 2019), myocarditis caused by coxsackievirus (Gebhard et al., 1998) and cutaneous leishmaniasis (CL) (Brodskyn et al., 1997; Faria et al., 2009; Novais et al., 2013; Santos et al., 2013; Cardoso et al., 2015; Crosby et al., 2015; Novais et al., 2015; Novais et al., 2017; Amorim et al., 2019; Campos et al., 2019; Covre et al., 2020). Therefore, understanding how to dampen cytotoxicity is of major clinical importance in several human disorders.
Cutaneous leishmaniasis (CL) is caused by protozoans of the genus Leishmania with 1.5 million people living at risk of contracting the disease. Standard treatment for CL is pentavalent antimony, a toxic drug with high failure rates and growing antimony resistance (Croft et al., 2006; Arevalo et al., 2007; Unger et al., 2009; Ponte-Sucre et al., 2017). One of the major problems of this treatment is that it only targets the parasite, but fails to limit the immunopathologic responses that have a major role in disease severity (Scott and Novais, 2016). Using murine models of CL, we identified CD8 T cells as prime drivers of pathology due to cytolysis and release of damage associated molecular patterns that activate the NLRP3 inflammasome, which processes IL-1β ultimately promoting severe inflammation (Novais et al., 2013; Novais et al., 2017) (Novais et al., 2015). Recently, we validated the importance of this pathway in humans by showing that the levels of components of the CD8 T cell cytolytic granules in lesions predict treatment failure with pentavalent antimony (Amorim et al., 2019). Therefore, pharmacological strategies that block the cytotoxic response of CD8 T cells have the potential to dampen inflammation while preventing the development of severe cases of mucosal leishmaniasis, which develops when parasites metastasize to the nasal-pharyngeal mucosa.
Cytokines of the common gamma (γc) chain (or CD132) family, including IL-2, IL-15 and IL-21, induce granzyme B (GzmB), a serine protease responsible for the activation of the cell death pathway mediated by cytotoxic granules (Golstein and Griffiths, 2018)(Gadina et al., 2018). (Ye et al., 1996; Alves et al., 2003; Janas et al., 2005; Özdemir et al., 2005; Rausch et al., 2006; Tamang et al., 2006; Ebert, 2009; Sutherland et al., 2013; Younes et al., 2016). Therefore, we hypothesized that blocking these cytokines might lessen the development of severe CL lesions. Since elevated levels of IL15 were found in lesions we tested if the jak 1/3 inhibitor tofacitinib, which blocks IL-15 signaling, might lessen CD8 T cell mediated pathology. Systemic and topical treatment with tofacitinib prevented immunopathology in two different CL models of CD8 T cell-mediated disease, but did not abrogate IFN-γ production or parasite control. Collectively, our results suggest that blocking jak1/3 using topical tofacitinib should be considered as a strategy for the treatment of severe cases of CL in combination with pentavalent antimony.
Results
IL-15 expression is induced in CL lesions.
To investigate if cytokines involved in the expression of GzmB were expressed in CL lesions, we took advantage of our previously published transcriptional analysis performed in 10 normal skin biopsies and 25 L. braziliensis-infected lesions from patients in which we found that cytotoxicity is a major signature of CL (Novais et al., 2015). Using this published dataset, we performed a new analysis to look for the expression of the γc-chain cytokines and found that only IL-15 and IL-15Ra mRNA expression was increased in lesions compared to normal skin (Fig. 1a). Similarly, gene expression analysis of lesions from mice infected with Leishmania at the peak of disease (5 weeks) showed that expression of Il15 and Il15ra is higher in lesions compared to normal skin (Fig. 1b). Thus, our analysis suggest that IL-15 mRNA expression is increased in CL lesions from mice and humans and we hypothesize that the IL-15 rich environment of CL lesions promotes GzmB expression in CD8 T cells.
Figure 1:
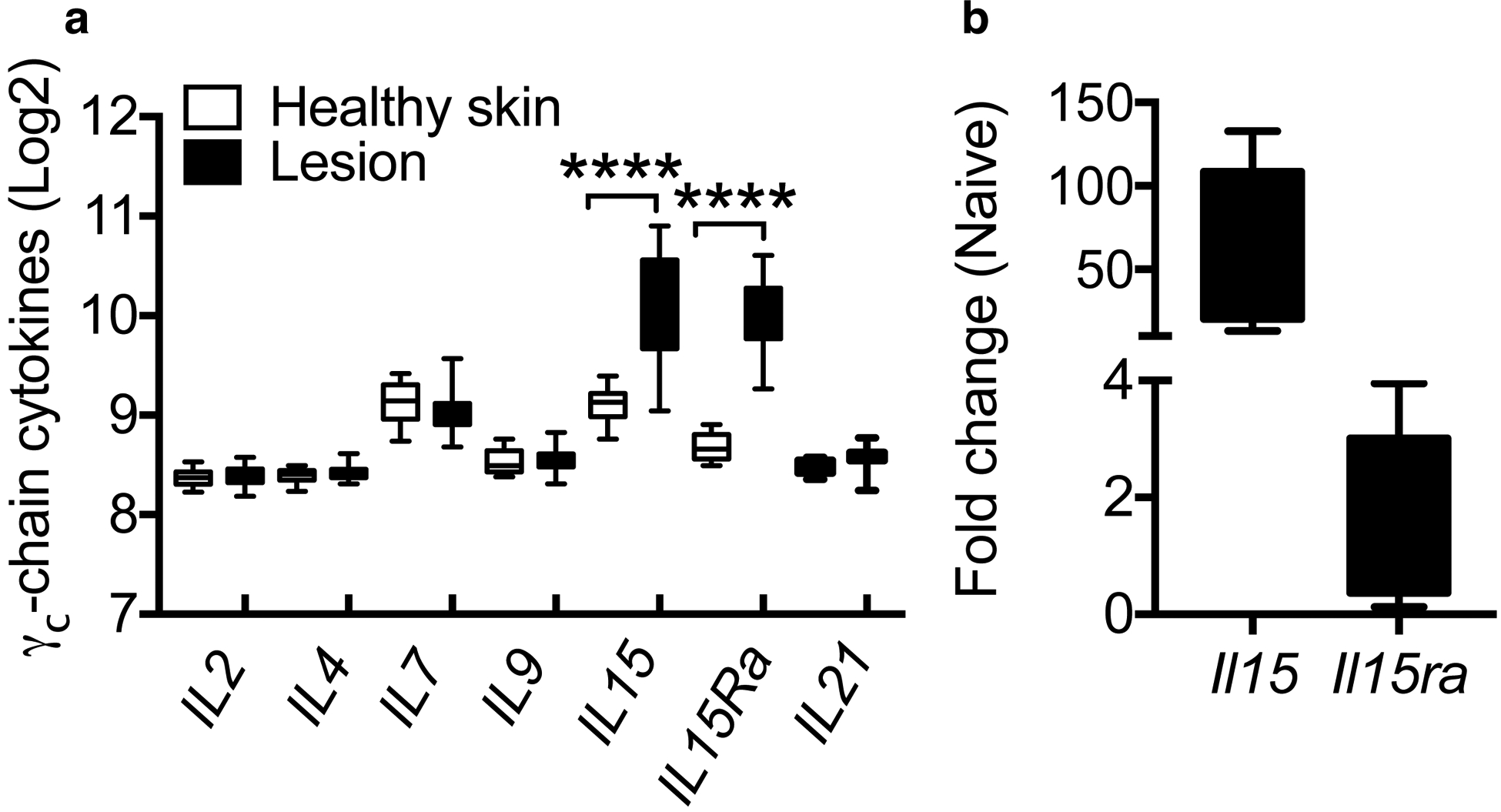
IL-15 mRNA is induced in human and mouse lesions compared to healthy skin. (a) Log2 expression of γc-chain cytokines in human healthy skin or L. braziliensis-infected lesions. (b) Il15 and Il15ra expression of L. major-infected lesions at 5 weeks post-infection. mRNA data is represented as a fold change over expression in naive mice. ***p ≤ 0.001 and ****p ≤ 0.0001
Tofacitinib prevents GzmB expression induced by IL-15.
To determine if IL-15 was sufficient to induce the expression of GzmB by CD8 T cells, we cultured splenocytes from naïve mice in the presence of IL-15 for 30 and 72 hours (Fig. 2a and b). CD8 T cells exposed to IL-15 expressed GzmB as early as 30 hours and at 72 hours all CD8 T cells in IL-15 cultures were GzmB+. Since IL-15 signals through jaks 1 and 3, we next asked if we could block GzmB expression by adding the jak1/3 inhibitor tofacitinib to cultures. Our results showed that GzmB expression induced by IL-15 was completely blocked by the addition of tofacitinib. Since tofacitinib might also impact jak 2 required for IL-12 signaling and therefore induction of the differentiation of CD4 T cells into IFN-γ-producing Th1 cells (Vignali and Kuchroo, 2012), we cultured splenocytes under Th1 polarizing conditions in the presence of IL-12 and IL-18. In CD8 T cells, we observed that tofacitinib treatment significantly decreased IFN-γ production by 20% cells at 30h (Fig. 2a and c). Tofacitinib treatment had a more discrete impact in the ability of CD4 T cells to produce IFN-γ (10% decrease) detectable at 30h though not significant (Fig. 2a and d). Therefore, our results suggest that while tofacitinib was effective at blocking GzmB production by CD8 T cells in response to the γc-chain cytokine IL-15, it had a smaller impact on the production of IFN-γ, especially in CD4 T cells. To further investigate the influence of tofacitinib on GzmB and IFN-γ production, we harvested splenocytes from L. major infected mice and stimulated them with leishmanial antigen in the presence or absence of tofacitinib. Supernatants were collected and IFN-γ levels assessed by ELISA. We found a complete block in GzmB production (Fig. 2e), and approximately a 50% reduction in IFN-γ responses from cells treated in vitro with tofacitinib (Fig. 2f).
Figure 2:
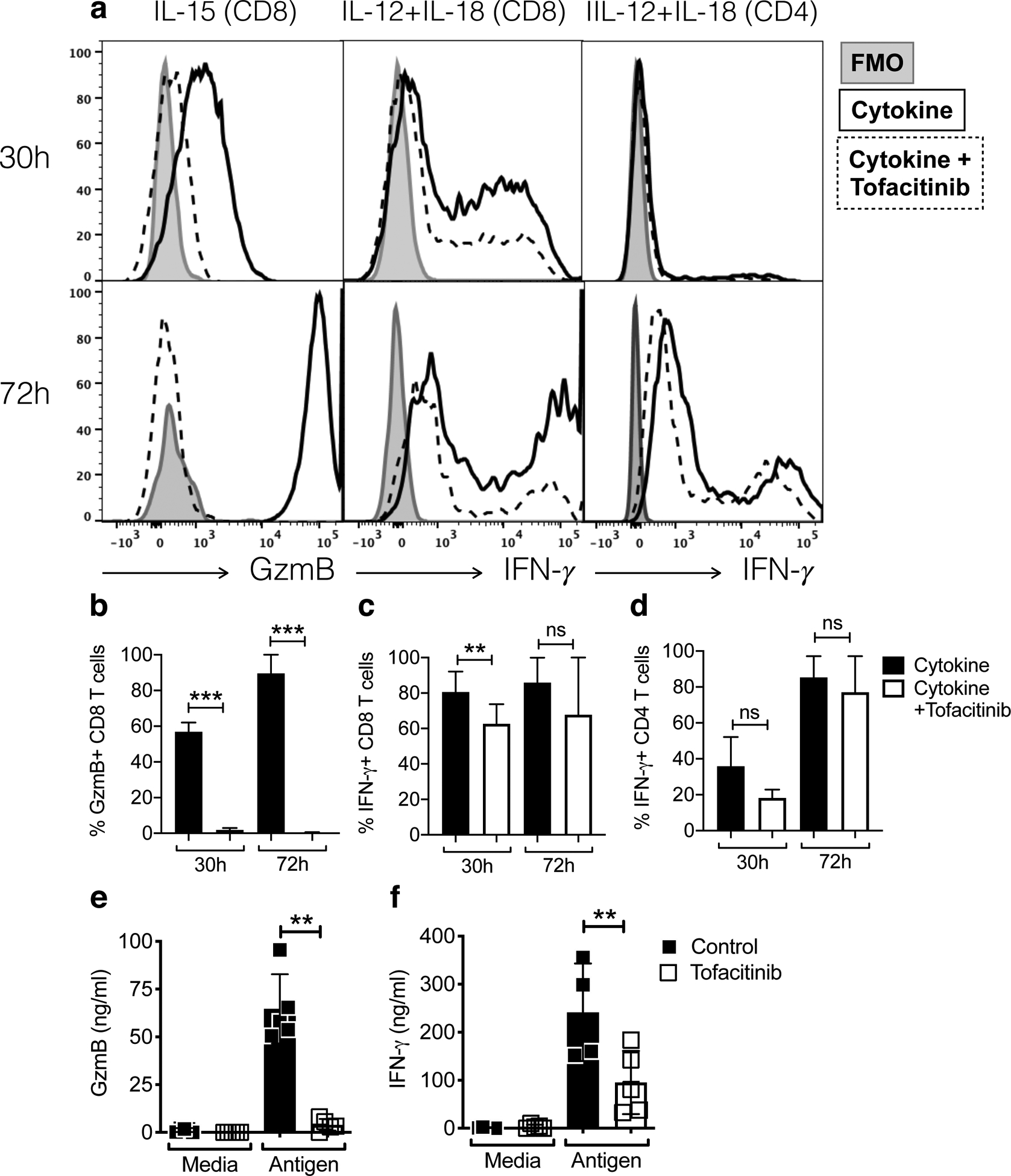
Jak1/3 inhibition with tofacitinib suppresses IL-15-induced granzyme B production by CD8 T cells. (a-d) Splenocytes from naive mice were stimulated for 30 or 72 hours with tofacitinib or vehicle in the presence of various cytokines. Cells were stimulated with PMA and ionomycin in the presence of brefeldin A for the last 4 hours and analyzed by flow cytometry for the expression of intracellular granzyme B and IFN-γ. Data are representative of at least 3 individual experiments performed with duplicates. Gating strategy: live, singlets, CD3, CD44high, CD8β or CD4. (e and f) Splenocytes from L. major-infected mice were stimulated with tofacitinib or vehicle in the presence or leishmania antigen. Supernatants were collected at 24h post-antigen stimulation and used to perform ELISA for (e) GzmB and (f) IFN-γ. Bar graphs are representative of 2 individual experiments with 5 mice per group with similar results.
Tofacitinib does not alter Th1 responses in vivo during Leishmania infection.
Our in vitro studies indicated that tofacitinib would block GzmB but might also decrease IFN-γ responses. Therefore, to test if tofacitinib might influence protective responses in vivo, we first tested if tofacitinib treatment inhibited a classical delayed type hypersensitivity (DTH) response. We infected mice with L. major in one ear and after the lesion resolved (>10 weeks post-infection), we challenged mice with L. major in the contralateral ear and assessed the DTH response (Fig. 3a). As expected, previously infected mice exhibited a DTH response, while naïve mice did not (Fig. 3b). DTH was accompanied by IFN-γ expression in CD4 T cells, while IFN-γ was not detected in the naïve mice (Fig. 3c and d). Importantly, naïve or immune mice treated with tofacitinib at days −1, 0, 1 and 2 post-challenged had similar responses to their control groups (Fig. 3b), and immune mice expressed similar levels of IFN-γ whether they were treated or not with tofacitinib (Fig. 3c and d). These data indicate that tofacitinib does not alter classical DTH responses or affect the ability of CD4 T cells to express IFN-γ in leishmaniasis.
Figure 3:
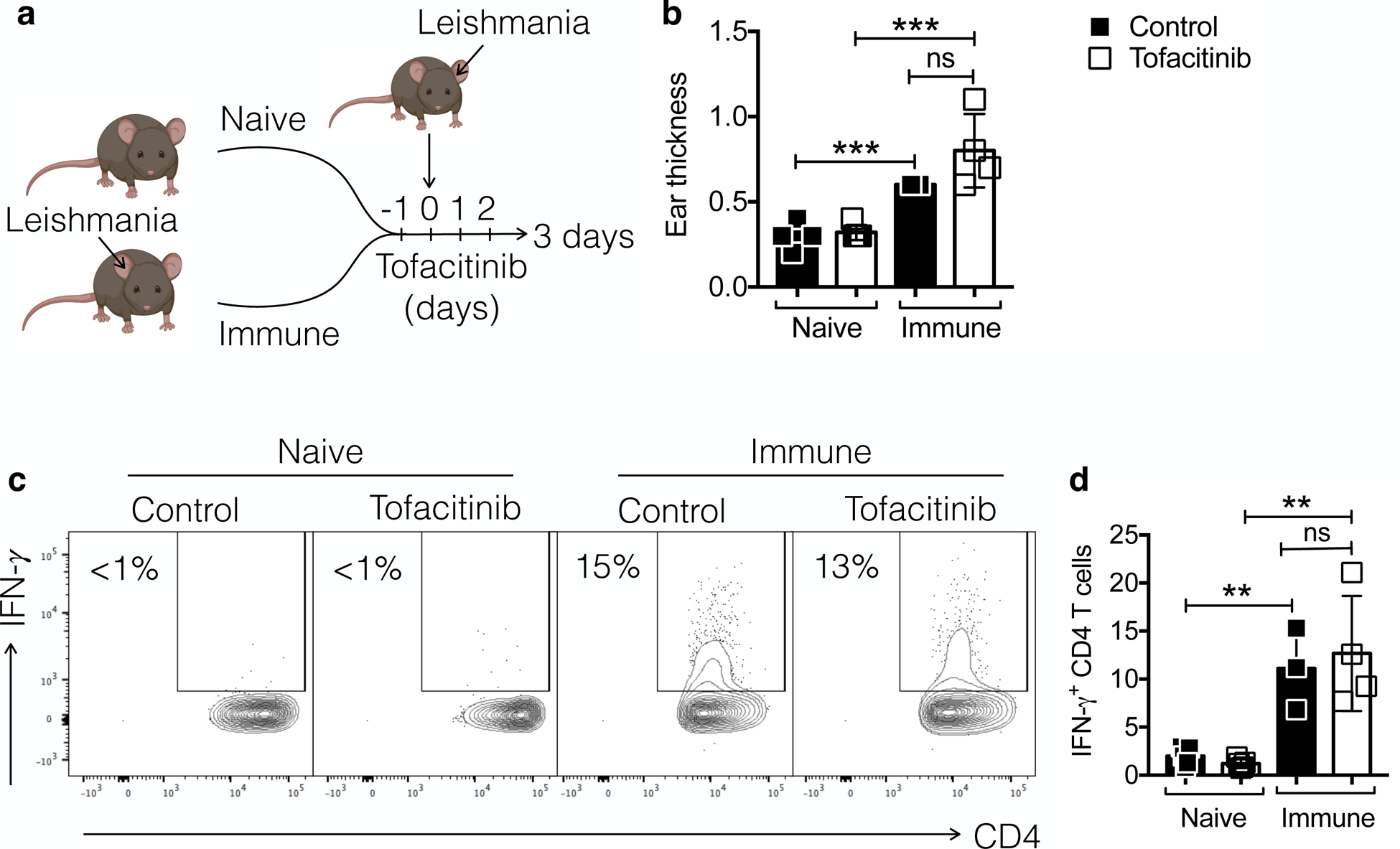
DTH responses to Leishmania challenge are intact after treatment with tofacitinib. (a) Naive or immune mice (infected for >10 weeks) were challenged with Leishmania at the contralateral ear and mice were treated with tofacitinib or vehicle. (b) DTH reaction was measured at 72 hours post-challenge. (c and d) Cells were stimulated with PMA and ionomycin in the presence of brefeldin A for 4 hours and analyzed by flow cytometry for the expression of intracellular IFN-γ. (c) Representative contour plots and (d) bar graphs are representative of 2 individual experiments with 4–5 mice per group with similar results. Gating strategy: live, singlets, CD3, CD4. **p ≤ 0.01 and ***p ≤ 0.001
To test if tofacitinib treatment would impact Th1 responses during infection, we infected C57BL/6 mice with L. major and, when lesions and immune responses were established, at 2.5 weeks post-infection, we began systemic (i.p.) daily injections with tofacitinib until mice were euthanized at 5 weeks post-infection. We chose this timepoint since lesions are large and visible and would simulate when patients would look for treatment. Treatment with tofacitinib had no impact on lesion development (Fig. 4a), or parasite numbers (Fig. 4b). Also, the frequency of IFN-γ producing Th1 cells in draining LN (dLN) (Fig. 4c and d) or lesions (Fig. 4c and e) was unchanged by treatment with tofacitinib. We also analyzed the expression of IFN-γ in other CD45+ cells that were negative for CD3 and we were unable to find differences in the expression of IFN-γ in this population (data not shown). Hence, treatment with tofacitinib did not impact the ability of already differentiated Th1 cells to produce IFN-γ and control Leishmania parasites in CL, which suggests that tofacitinib treatment would be safe for treating CL.
Figure 4:
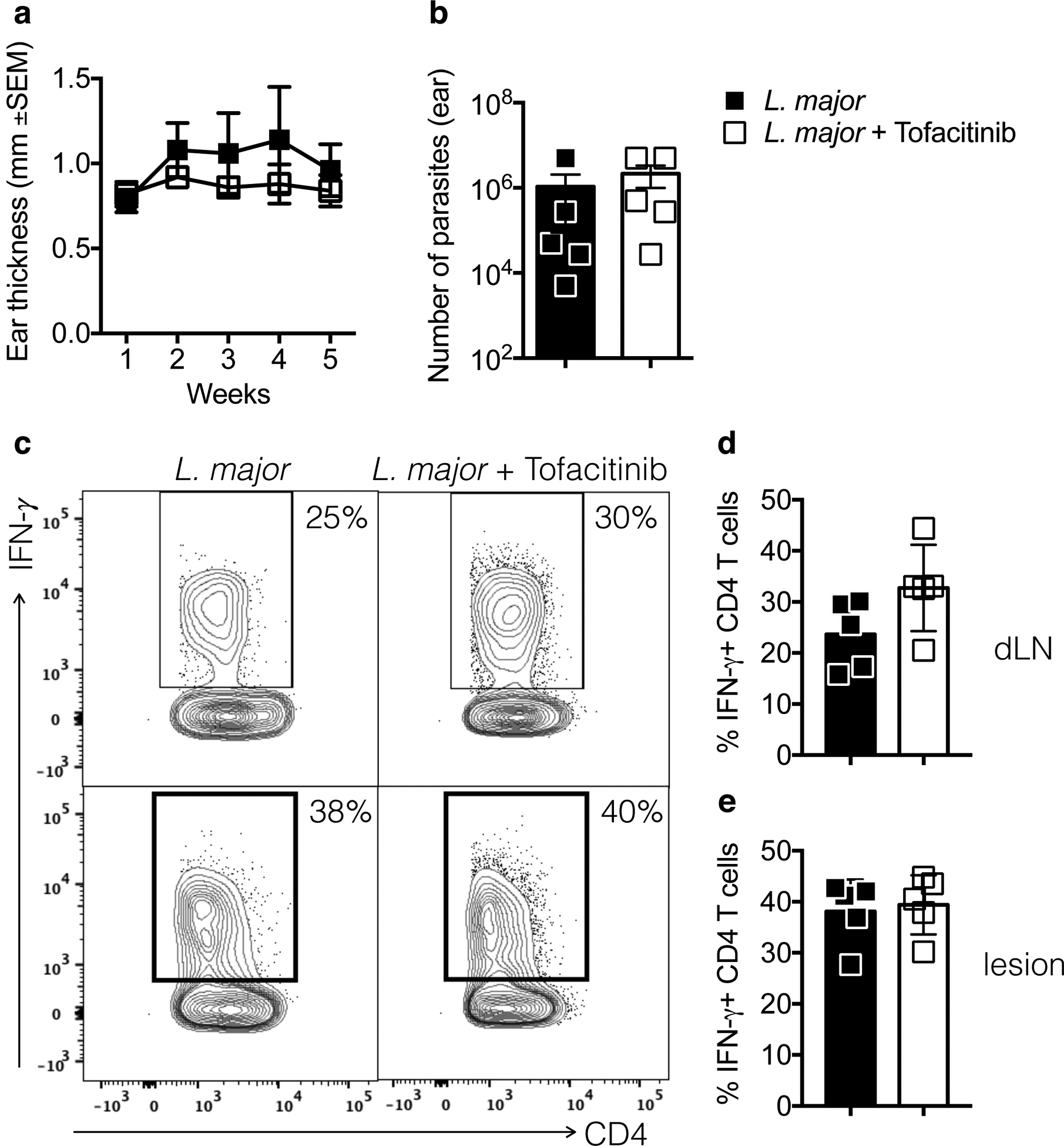
IFN-γ production by CD4 T cells is not altered by treatment with tofacitinib. Mice were infected with Leishmania and 2.5 weeks later daily i.p. treatment with tofacitinib or vehicle started. (a) Ear thickness was measured weekly and at 5 weeks (b) parasite numbers (c) and IFN-γ production by CD4 T cells was assessed by flow cytometry. Cells from (d) infected ears or (e) draining LN (dLN) were stimulated with PMA and ionomycin in the presence of brefeldin A for 4 hours and analyzed by flow cytometry for the expression of intracellular IFN-γ. (c) Representative contour plots and (d and e) bar graphs are representative of 2 individual experiments with 5 mice per group. Gating strategy: live, singlets, CD3, CD4, CD44high.
Tofacitinib inhibits CD8 T cell-mediated disease.
To determine if tofacitinib might lessen disease severity, we infected RAG deficient mice with L. braziliensis and reconstituted the mice with CD8 T cells (RAG+CD8). As previously described (Novais et al., 2013), RAG+CD8 develop severe pathology characterized by GzmB expressing CD8 T cells in lesions and a persistent influx of neutrophils. When RAG+CD8 mice were treated with tofacitinib starting at 2 weeks post-infection, the treated mice developed significantly smaller lesions compared to vehicle-treated controls (Fig. 5a), while parasite numbers in the infected skin were similar between groups (Fig. 5b). The decrease in lesion size was accompanied by a significant reduction in the number of neutrophils recruited to the lesions (Fig. 5c and d) and the frequency of CD8 T cells expressing GzmB (Fig. 5e and f). These results demonstrate that tofacitinib prevents immunopathology by CD8 T cells without affecting the number of parasites in the skin.
Figure 5:
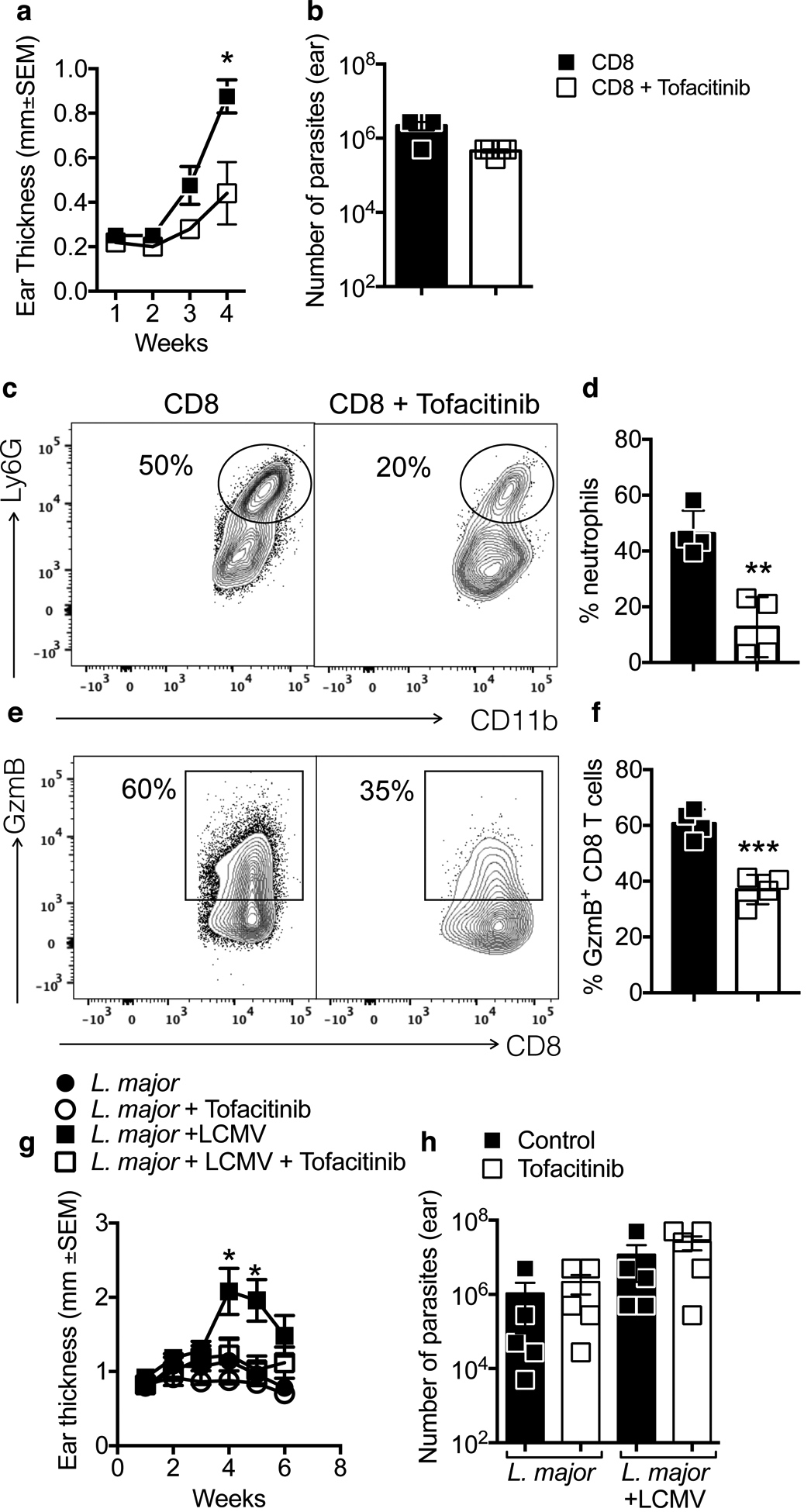
Treatment with tofacitinib prevents CD8 T cells from inducing pathology in Leishmania infection. RAG−/− mice were infected with Leishmania and reconstituted with CD8 T cells. Two weeks post-infection, mice were treated with tofacitinib or vehicle for 2 weeks. (a) Ear thickness was measured weekly and at 4 weeks (b) parasite numbers was assessed. (c and d) neutrophil recruitment and (e and f) granzyme B expression by CD8 T cells was assessed directly ex vivo by flow cytometry at 4 weeks post-infection. (c and e) Representative contour plots and (d and f) bar graphs are representative of 2 individual experiments with 5 mice per group. Gating strategy: live, singlets, CD11b, Ly6G or CD3, CD8β. (g and h) Mice were infected with Leishmania in the skin and 2 weeks post-infection a subset of mice were co-infected with LCMV. 10 days post-LCMV infection mice were treated with tofacitinib or vehicle. (g) Ear thickness was measured weekly and at 6 weeks (h) parasite numbers was assessed. *p ≤ 0.05 and **p ≤ 0.01
CD8 T cell-mediated disease can be caused by both Leishmania-specific (Novais et al., 2013) as well as CD8 T cells activated by a co-infection (Crosby et al., 2015), here called bystander CD8 T cells. Thus, Leishmania-infected mice that are co-infected with an acute strain of LCMV expand a population of cytolytic CD8 T cells that are non-specifically recruited to leishmanial lesions, leading to the development of more severe lesions compared with controls due to NKG2D lysis of NKG2D ligand expressing cells in the lesion (Crosby et al., 2014; Crosby et al., 2015). This NKG2D-dependent killing mechanism of CD8 T cell killing is IL-15-dependent in celiac disease (Meresse et al., 2004), and we hypothesized that tofacitinib would prevent the pathology caused by bystander CD8 T cells. C57BL/6 mice with L. major were infected with LCMV 2 weeks post-infection, and systemic treatment with tofacitinib or vehicle commenced 10 days post LCMV infection, by which time the virus was cleared (Crosby et al., 2015). Treatment with tofacitinib blocked the pathology induced by bystander CD8 T cells (Fig. 5g), while the number of parasites in lesions was unaltered (Fig. 5h). Together, these results show that tofacitinib treatment blocks disease in two mouse models of CD8 T cell-mediated disease in CL and that tofacitinib treatment does not alter the parasite load in the skin.
Topical tofacitinib treatment prevents CD8 T cell-mediated disease in CL.
While systemic tofacitinib administration was effective, topical application is ideal since it can be administered with ease and would reduce the chances of any potential side effects. Tofacitinib was previously used topically in several skin conditions (Schwartz et al., 2017), hence we tested if local treatment with tofacitinib was sufficient to prevent CD8 T cell-mediated inflammation. RAG+CD8 mice were infected with L. braziliensis and 2 weeks post-infection topical application of tofacitinib began. Topical tofacitinib was able to significantly reduce lesion development (Fig. 6a) without affecting parasite numbers (Fig. 6b). Correspondingly, there was a significant decrease in the recruitment of neutrophils to the lesion (Fig. 6c and d) and suppressed expression of GzmB by CD8 T cells (Fig. 6e and f). Together, our results demonstrate that CD8 T cells promote disease in CL in a jak1/3-dependent manner, and that treatment with tofacitinib prevents pathogenic responses from developing while keeping protective responses intact.
Figure 6:
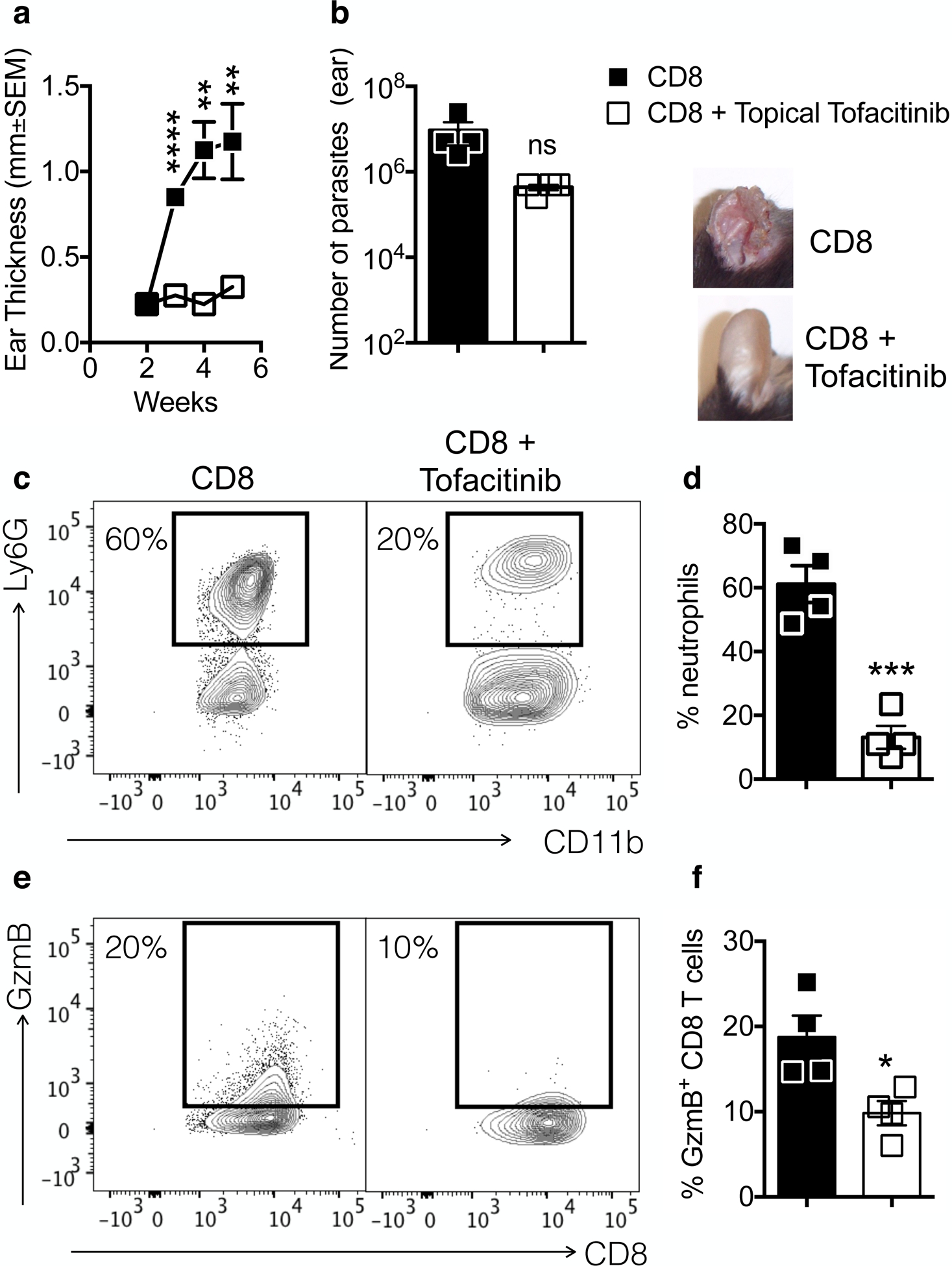
Topical treatment with tofacitinib is sufficient to inhibit pathology induced by CD8 T cells during Leishmania infection. RAG−/− mice were infected with Leishmania and reconstituted with CD8 T cells. Two weeks post-infection, mice were topically treated with tofacitinib or vehicle for 3 weeks. (a) Ear thickness was measured weekly and at 4 weeks (b) parasite numbers was assessed. Depicted are pictures of lesions at 5 weeks post-infection. (c and d) neutrophil recruitment and (e and f) granzyme B expression by CD8 T cells was assessed directly ex vivo by flow cytometry at 4 weeks post-infection. (c and e) Representative contour plots and (d and f) bar graphs are representative of 2 individual experiments with 5 mice per group. Gating strategy: live, singlets, CD11b, Ly6G or CD3, CD8β. *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001
Discussion
Pentavalent antimony is the first line of treatment for CL, but has numerous limitations such as cardiotoxicity, pancreatitis and hepato/nephrotoxicity (Ponte-Sucre et al., 2017), lengthy treatment, intravenous administration and an association with treatment failure (Machado et al., 2002; Croft et al., 2006; Unger et al., 2009; Ponte-Sucre et al., 2017). While new drugs are needed, since the severity of the disease is also mediated by immunopathologic responses, therapies that block such responses might augment disease resolution. Our work demonstrates that treatment of mice with tofacitinib ameliorates pathologic aspects of the disease, while leaving IFN-γ production by Th1 cells intact. Importantly, both systemic and topical delivery of tofacitinib was effective, and since topical application of jak inhibitors has been used in other dermatologic conditions with no evidence of systemic toxicity (Schwartz et al., 2017), topical application of tofacitinib should be considered as a strategy for the treatment of CL in conjunction with standard anti-parasitic drugs.
In some CL patients, the magnitude of the disease is due to an ineffective immune response, while in others, severe disease occurs concomitantly with a Th1 response and control of the parasites (Scott and Novais, 2016). In L. braziliensis patients and murine models, we found that cytolytic CD8 T cells were the driving force in the development of pathology (Novais et al., 2013; Crosby et al., 2014; Crosby et al., 2015), leading to NLRP3 activation, IL-1β release and an exaggerated inflammatory response (Novais et al., 2017). Here we show that blocking the initiation of the pathway, mediated by cytolytic CD8 T cells, limits disease. We found elevated levels of IL-15 mRNA in lesions, and therefore sought to block IL-15 signaling using tofacitinib, an inhibitor of the jak molecules downstream of the IL-15 receptor. Further supporting a focus on IL-15 is the observation that NKG2D/IL-15-dependent killing of target cells promote disease in celiac patients (Meresse et al., 2004; Tang et al., 2009; Tang et al., 2015). Our results show that blocking CD8 cytolysis with tofacitinib is effective in lessening the magnitude of disease, and that this therapeutic effect was evident in models where CD8 T cells were known to be killing Leishmania-infected cells (Novais et al., 2013), as well in our model where NKG2D expressing bystander CD8 T cells promoted disease (Crosby et al., 2015).
Tofacitinib, that inhibits primarily jaks 3 and 1, and to some extent jak 2, was the first jak inhibitor developed for treatment of autoimmune diseases (Schwartz et al., 2017). Clinical trials in rheumatoid arthritis (RA) showed high efficacy in the treatment of disease (Fleischmann et al., 2012; van Vollenhoven et al., 2012; Lee et al., 2014; Conaghan et al., 2016; Moodley et al., 2016). In addition, tofacitinib has been used for the treatment of ulcerative colitis with some success (Sandborn et al., 2014; Panés et al., 2017; Sandborn et al., 2017). In a mouse model of lupus nephritis, tofacitinib treatment impaired CD8 T cell-mediated disease and was able to improve kidney function (Zhou et al., 2020). The major concern with using jak inhibitors is the obvious risk of developing side effects since the cytokines it inhibits are essential for protecting the host from certain pathogens. For example, patients treated with jak inhibitors are at higher risk of developing tuberculosis and herpes zoster (Yamaoka, 2019; Zhang et al., 2019). Though this should be a major risk for patients that receive long-term treatment for inflammatory diseases such as RA (Kubo et al., 2017; Schwartz et al., 2017), in CL patients treatment would be limited, and any undesirable effects of tofacinitib would be significantly reduced by topical delivery. In the case of leishmaniasis, the concern would be blockade of IFN-γ production that might lead to loss of parasite control. While in vitro studies indicated an effect on IFN-γ, we found that in vivo treatment with tofacitinib did not block IFN-γ or protection. We do not know why there is a discrepancy in the in vitro and in vivo results, although it may reflect the limitations of recapitulating what is happening in vivo with in vitro studies. Considering all of our findings, we conclude that topical treatment with tofacitinib has the potential to reduce the deleterious effects of cytotoxicity in CL lesions without overtly increasing the risk of side-effects.
Tofacitinib has also been used for the treatment of numerous dermatologic conditions (Shreberk-Hassidim et al., 2017). For example, psoriasis is an autoimmune skin disorder in which cytokines such as TNF, IL-17 and IL-23 play a major role (Lowes et al., 2014), and in several clinical trials tofacitinib treated patients showed significant improvement over controls (Schwartz et al., 2017). In alopecia areata, CD8 T cells kill hair follicles in an NKG2D-dependent manner as a result of an environment rich in IFN-γ and IL-15 (Xing et al., 2014), and murine studies showed that tofacitinib leads to disease improvement (Xing et al., 2014). Similarly, in humans, the treatment of alopecia areata, totalis and universalis with tofacitinib was effective at reducing symptoms (Kennedy Crispin et al., 2016; Scheinberg and Ferreira, 2016; Craiglow et al., 2017; Liu and King, 2018; Hernandez-Montoya and Ruiz-Villaverde, 2019). In vitiligo, an autoimmune disorder also caused by cytotoxic CD8 T cells that kill melanocytes (van den Boorn et al., 2009; Zloza et al., 2011; Cheuk et al., 2017; Frisoli et al., 2020), treatment with tofacitinib was also shown to be efficacious (Craiglow and King, 2015; Kim et al., 2018). In addition, tofacitinib has been successfully used in atopic dermatitis and idiopathic erythema multiform (Bissonnette et al., 2016; Damsky and King, 2016). Thus, our results are consistent with the use of tofacitinib to prevent several inflammatory skin conditions, and is likely to be safe given the extensive experience with this drug.
Here we used mouse models to ask if tofacitinib is able to block pathology induced by cytolytic CD8 T cells while sparing protective Th1 responses. Because individual mouse models have limitations in their ability to address how tofacitinib impacts every aspect of the immune response in CL, we used transcriptional profiling, in vitro, ex vivo and 3 different in vivo models to analyze each individual question by different angles. Collectively, these series of experiments suggest tofacitinib is an inhibitor that should be considered for the treatment of CL. The use of host-directed therapies that block inflammatory responses might be considered problematic in infectious diseases, since it is possible that protective responses might simultaneously be dampened. Since Leishmania killing requires the development of a Th1 response, blocking either IL-12 or IFN-γ might lead to uncontrolled parasite growth (Ghoreschi et al., 2011; Almanzar et al., 2019). However, we found that tofacitinib had a mild impact on IFN-γ production by CD4 T cells stimulated with IL-12 and IL-18, and in vivo tofacitinib treatment had no impact on the production of IFN-γ during infection. Furthermore, in CL we have identified an immunopathologic response that can be blocked without affecting parasite control. Thus, we found that tofacitinib inhibited CD8 T cell mediated pathology, while Th1 responses remained intact. Thus, our results indicate that tofacitinib, used in combination with anti-parasitic drugs, would be a successful and to our knowledge a previously unreported strategy for the treatment of CL. In another host-directed therapy using a jak inhibitor, inhibition of type I IFN was shown to enhance protection in visceral leishmaniasis (Kumar et al., 2020). More broadly, these results suggest that host-directed therapies in infectious diseases should be considered as a strategy for lessening disease manifestations in situations where the therapy does not compromise control of the pathogen.
Materials & Methods
Ethics statement.
This study was carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee, University of Pennsylvania.
Patients and lesion biopsies.
Patient data in Fig. 1a is derived from published transcriptional profiling (Novais et al., 2015).
Mice.
C57BL/6 mice (6 weeks old) were purchased from Charles River, and RAG−/− (B6.12957-RAG1tm1Mom) were purchased from The Jackson Laboratory. All mice were maintained in a specific pathogen-free environment at the University of Pennsylvania Animal Care Facilities.
RNA isolation and quantitative real-time PCR.
Skin was homogenized and RNA was extracted using the RNeasy Mini kit (QIAGEN) according to the manufacturer’s instructions. RNA was reverse transcribed using high capacity cDNA Reverse Transcription (Applied Biosystems). Real-time PCR was performed on a ViiA 7 Real-Time PCR System (Applied Biosystems) using SYBR Green PCR Master Mix (Applied Biosystems). mRNA levels for each sample were normalized to ribosomal protein S11 genes and displayed as fold change over uninfected skin. Primers: Il15, forward, 50 - GCAGAGTTGGACGAAGAC - 30, and reverse 50 - AGCACGAGATGGATGTATT - 30; Il15ra, forward, 50 - TCTCCCCACAGTTCCAAA - 30, and reverse 50 - GGCACCCAGGCTCAGTAA - 30.
Parasites and virus.
L. braziliensis (MHOM/BR/01/BA788) and L. major (Friedlin) were grown in Schneider’s insect medium (GIBCO) supplemented with 20% heat-inactivated FBS and 2 mM glutamine. Metacyclic enriched promastigotes were used for infection. Mice were infected with either 105 L. braziliensis or 106 L. major intradermally in the ear, and the lesion progression was monitored weekly by measuring the diameter of ear. For lymphocytic choriomeningitis virus (LCMV) infections, mice were infected with 2×105 PFU of Armstrong strain by intraperitoneal injection.
Cell purification and adoptive transfer.
Splenocytes from C57BL/6 mice were collected, red blood cells lysed with ACK lysing buffer (LONZA) and CD8 T cells were purified using a magnetic bead separation kit (Miltenyi Biotec). Three million CD8 T cells were transferred into RAG−/− mice that were subsequently infected with L. braziliensis. Mice reconstituted with CD8 T cells received 4 injections of 250 μg of anti-CD4 within the first 2 weeks.
Ear preparation.
Infected ears were harvested, the dorsal and ventral layers of the ear separated, and the ears incubated in RPMI (Gibco) with 250 μg/mL of Liberase (Roche) for 90 mins at 37°C/5% CO2. Ears were dissociated using a cell strainer (40 μm, BD Pharmingen) and an aliquot of the cell suspension was used for parasite titration.
Parasite titration.
The parasite burden in the ears was quantified as described previously (Uzonna et al., 2004). Briefly, the homogenate was serially diluted and incubated at 26°C. The number of viable parasites was calculated from the highest dilution at which parasites were observed after 7 days.
Flow cytometric analysis.
Cell suspensions were incubated for 4–6h with PMA (50 ng/mL), ionomycin (500 ng/mL) and Brefeldin A (10 μg /mL) (all from SIGMA) for intracellular staining. Before surface and intracellular staining, cells were stained with live/dead fixable aqua dead cell stain kit (Molecular Probes), according to manufacturer instructions. Analysis was performed using the FlowJo Software and gates were drawn based on fluorescence minus one (FMO) controls. Antibodies: anti-CD45 APC-AlexaFluor 780, anti-CD11b eF450, anti-CD3 eFluor 450 and anti-IFN-γ PeCy7 (all from eBioscience). Anti-CD4 APC-Cy7 (BD Pharmingen), anti-CD8β PerCPCy5.5 and anti-Ly6G APC (Biolegend) and anti-granzyme B APC (Invitrogen).
In vitro and in vivo treatment with tofacitinib.
In vitro (Fig. 2 a–d): splenocytes from mice were collected, red blood cells lysed with ACK lysing buffer (LONZA), cells were counted and plated in a 96-well plate “u” bottom. Cells were stimulated with IL-2 (10 ng/mL), IL-15 (50 ng/mL) or IL-12 (10 ng/mL) + IL-18 (25 ng/mL) with 2 μM of tofacitinib citrate (Selleckchem) or vehicle for 30 or 72 hours at 37°C/5% CO2. The 2μM dose was determined by dose-response assay in which we measured GzmB expression by treating splenocytes with 0.5–2μM (data not shown). All doses were able to block GzmB expression by CD8 T cells in vitro. During the last 4 hours, cells were incubated with PMA, ionomycin and brefeldin A as described above. In vitro (Fig. 2e and f): splenocytes from 2 week-infected mice were stimulated for 24h with media or antigen (5:1) +/− 2μM of tofacitinib and supernatants were collected for ELISA. IFN-γ (eBioscience) and GzmB (R&D Systems) ELISA were performed according with the manufacturer’s instructions. In vivo systemic: 2 weeks post infection of RAG−/− mice with L. braziliensis or 10 days post LCMV infection in C57BL/6 mice infected with L. major, treatment with 30 mg/Kg of tofacitinib per day commenced. In vivo topical: 2 weeks post infection of RAG−/− mice with L. braziliensis treatment with 30 mg/Kg of tofacitinib per day commenced. Tofacitinib or vehicle was combined with moisturizing lotion (Cetaphil) and applied to the skin. For the in vivo studies, we use the dose suggested by the manufacturer.
Statistical analysis.
Data are presented as mean ± standard error. Statistical significance was determined using the two-tailed unpaired Student’s t-test. Pearson correlation coefficient was used to determine correlation between LOG2 expressions of genes from human skin transcripts. Statistical analysis was calculated using Prism software (GraphPad).
Acknowledgements:
The authors would like to thank Dr. John Wherry for providing LCMV. We also thank the co-authors of our original publication profiling leishmanial lesions: Drs. Lucas P. Carvalho, Sara Passos, David S. Roos, Edgar M. Carvalho and Daniel P. Beiting. Grants: R01-AI-150606
Abreviations:
- CL
Cutaneous leishmaniasis
- RA
Rheumatoid arthritis
- LCMV
Lymphocytic choriomeningitis virus
- jak
Janus kinase
- DTH
Delayed type hypersensitivity
Footnotes
Data availability: Datasets related to this article can be found at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE55664, hosted at the GEO database (Novais et al., 2015).
Conflict of Interest: The authors state no conflict of interest.
References
- Almanzar G, Kienle F, Schmalzing M, Maas A, Tony H-P, Prelog M. Tofacitinib modulates the VZV-specific CD4+ T cell immune response in vitro in lymphocytes of patients with rheumatoid arthritis. Rheumatology 2019;58:2051–60. [DOI] [PubMed] [Google Scholar]
- Alves NL, Hooibrink B, Arosa FA, van Lier RAW. IL-15 induces antigen-independent expansion and differentiation of human naive CD8+ T cells in vitro. Blood 2003;102:2541–6. [DOI] [PubMed] [Google Scholar]
- Amorim CF, Novais FO, Nguyen BT, Misic AM, Carvalho LP, Carvalho EM, et al. Variable gene expression and parasite load predict treatment outcome in cutaneous leishmaniasis. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo J, Ramirez L, Adaui V, Zimic M, Tulliano G, Miranda-Verástegui C, et al. Influence of Leishmania (Viannia) species on the response to antimonial treatment in patients with American tegumentary leishmaniasis. J Infect Dis 2007;195:1846–51. [DOI] [PubMed] [Google Scholar]
- Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol 2016;175:902–11. [DOI] [PubMed] [Google Scholar]
- van den Boorn JG, Konijnenberg D, Dellemijn TAM, van der Veen JPW, Bos JD, Melief CJM, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol 2009;129:2220–32. [DOI] [PubMed] [Google Scholar]
- Brodskyn CI, Barral A, Boaventura V, Carvalho E, Barral-Netto M. Parasite-driven in vitro human lymphocyte cytotoxicity against autologous infected macrophages from mucosal leishmaniasis. J Immunol 1997;159:4467–73. [PubMed] [Google Scholar]
- Campos TM, Novais FO, Saldanha M, Costa R, Lordelo M, Celestino D, et al. Granzyme B produced by natural killer cells enhances inflammatory response and contributes to the immunopathology of cutaneous leishmaniasis. J Infect Dis 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso TM, Machado Á, Costa DL, Carvalho LP, Queiroz A, Machado P, et al. Protective and pathological functions of CD8+ T cells in Leishmania braziliensis infection. Infect Immun 2015;83:898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, et al. CD49a Expression Defines Tissue-Resident CD8+ T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017;46:287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claser C, Nguee SYT, Balachander A, Wu Howland S, Becht E, Gunasegaran B, et al. Lung endothelial cell antigen cross-presentation to CD8+T cells drives malaria-associated lung injury. Nat Commun 2019;10:4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaghan PG, Østergaard M, Bowes MA, Wu C, Fuerst T, van der Heijde D, et al. Comparing the effects of tofacitinib, methotrexate and the combination, on bone marrow oedema, synovitis and bone erosion in methotrexate-naive, early active rheumatoid arthritis: results of an exploratory randomised MRI study incorporating semiquantitative and quantitative techniques. Ann Rheum Dis 2016;75:1024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covre LP, Devine O, Garcia de Moura R, Vukmanovic-Stejic M, Dietze R, Rodrigues RR, et al. Compartmentalized cytotoxic immune response leads to distinct pathogenic roles of natural killer and senescent CD8+ T cells in human cutaneous leishmaniasis. Immunology 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craiglow BG, King BA. Tofacitinib Citrate for the Treatment of Vitiligo: A Pathogenesis-Directed Therapy. JAMA Dermatol 2015;151:1110–2. [DOI] [PubMed] [Google Scholar]
- Craiglow BG, Liu LY, King BA. Tofacitinib for the treatment of alopecia areata and variants in adolescents. J Am Acad Dermatol 2017;76:29–32. [DOI] [PubMed] [Google Scholar]
- Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev 2006;19:111–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby EJ, Clark M, Novais FO, Wherry EJ, Scott P. Lymphocytic Choriomeningitis Virus Expands a Population of NKG2D+CD8+ T Cells That Exacerbates Disease in Mice Coinfected with Leishmania major. J Immunol 2015;195:3301–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby EJ, Goldschmidt MH, Wherry EJ, Scott P. Engagement of NKG2D on bystander memory CD8 T cells promotes increased immunopathology following Leishmania major infection. PLoS Pathog 2014;10:e1003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damsky W, King BA. Idiopathic erythema multiforme: Evidence of underlying Janus kinase-signal transducer and activator of transcription activation and successful treatment with tofacitinib. JAAD Case Reports 2016;2:502–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert EC. Interleukin 21 up-regulates perforin-mediated cytotoxic activity of human intra-epithelial lymphocytes. Immunology 2009;127:206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria DR, Souza PEA, Durães FV, Carvalho EM, Gollob KJ, Machado PR, et al. Recruitment of CD8(+) T cells expressing granzyme A is associated with lesion progression in human cutaneous leishmaniasis. Parasite Immunol 2009;31:432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med 2012;367:495–507. [DOI] [PubMed] [Google Scholar]
- Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol 2020; [DOI] [PubMed] [Google Scholar]
- Gadina M, Johnson C, Schwartz D, Bonelli M, Hasni S, Kanno Y, et al. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J Leukoc Biol 2018;104:499–514. [DOI] [PubMed] [Google Scholar]
- Gebhard JR, Perry CM, Harkins S, Lane T, Mena I, Asensio VC, et al. Coxsackievirus B3-induced myocarditis: perforin exacerbates disease, but plays no detectable role in virus clearance. Am J Pathol 1998;153:417–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol 2011;186:4234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golstein P, Griffiths GM. An early history of T cell-mediated cytotoxicity. Nat Rev Immunol 2018;18:527–35. [DOI] [PubMed] [Google Scholar]
- Hernandez-Montoya C, Ruiz-Villaverde R. The role of tofacitinib in the management of alopecia totalis. Sultan Qaboos Univ Med J 2019;19:e77–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janas ML, Groves P, Kienzle N, Kelso A. IL-2 regulates perforin and granzyme gene expression in CD8+ T cells independently of its effects on survival and proliferation. J Immunol 2005;175:8003–10. [DOI] [PubMed] [Google Scholar]
- Junqueira C, Barbosa CRR, Costa PAC, Teixeira-Carvalho A, Castro G, Sen Santara S, et al. Cytotoxic CD8+ T cells recognize and kill Plasmodium vivax-infected reticulocytes. Nat Med 2018;24:1330–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kägi D, Odermatt B, Seiler P, Zinkernagel RM, Mak TW, Hengartner H. Reduced incidence and delayed onset of diabetes in perforin-deficient nonobese diabetic mice. J Exp Med 1997;186:989–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski L-C, Riehn M, Abel A, Steeg C, Yar DD, Addai-Mensah O, et al. Cytotoxic T Cell-Derived Granzyme B Is Increased in Severe Plasmodium Falciparum Malaria. Front Immunol 2019;10:2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy Crispin M, Ko JM, Craiglow BG, Li S, Shankar G, Urban JR, et al. Safety and efficacy of the JAK inhibitor tofacitinib citrate in patients with alopecia areata. JCI Insight 2016;1:e89776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SR, Heaton H, Liu LY, King BA. Rapid Repigmentation of Vitiligo Using Tofacitinib Plus Low-Dose, Narrowband UV-B Phototherapy. JAMA Dermatol 2018;154:370–1. [DOI] [PubMed] [Google Scholar]
- Kubo S, Yamaoka K, Amano K, Nagano S, Tohma S, Suematsu E, et al. Discontinuation of tofacitinib after achieving low disease activity in patients with rheumatoid arthritis: a multicentre, observational study. Rheumatology 2017;56:1293–301. [DOI] [PubMed] [Google Scholar]
- Kumar R, Bunn PT, Singh SS, Ng SS, Montes de Oca M, De Labastida Rivera F, et al. Type I Interferons Suppress Anti-parasitic Immunity and Can Be Targeted to Improve Treatment of Visceral Leishmaniasis. Cell Rep 2020;30:2512–2525.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Fleischmann R, Hall S, Wilkinson B, Bradley JD, Gruben D, et al. Tofacitinib versus methotrexate in rheumatoid arthritis. N Engl J Med 2014;370:2377–86. [DOI] [PubMed] [Google Scholar]
- Liu LY, King BA. Tofacitinib for the treatment of severe alopecia areata in adults and adolescents. J Investig Dermatol Symp Proc 2018;19:S18–20. [DOI] [PubMed] [Google Scholar]
- Lowes MA, Suárez-Fariñas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol 2014;32:227–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado P, Araújo C, Da Silva AT, Almeida RP, D’Oliveira A Jr, Bittencourt A, et al. Failure of early treatment of cutaneous leishmaniasis in preventing the development of an ulcer. Clin Infect Dis 2002;34:E69–73. [DOI] [PubMed] [Google Scholar]
- Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity 2004;21:357–66. [DOI] [PubMed] [Google Scholar]
- Moodley D, Yoshida H, Mostafavi S, Asinovski N, Ortiz-Lopez A, Symanowicz P, et al. Network pharmacology of JAK inhibitors. Proc Natl Acad Sci USA 2016;113:9852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novais FO, Carvalho AM, Clark ML, Carvalho LP, Beiting DP, Brodsky IE, et al. CD8+ T cell cytotoxicity mediates pathology in the skin by inflammasome activation and IL-1β production. PLoS Pathog 2017;13:e1006196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novais FO, Carvalho LP, Graff JW, Beiting DP, Ruthel G, Roos DS, et al. Cytotoxic T cells mediate pathology and metastasis in cutaneous leishmaniasis. PLoS Pathog 2013;9:e1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novais FO, Carvalho LP, Passos S, Roos DS, Carvalho EM, Scott P, et al. Genomic profiling of human Leishmania braziliensis lesions identifies transcriptional modules associated with cutaneous immunopathology. J Invest Dermatol 2015;135:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özdemir Ö, Sorensen R, Ochoa A, Savasan S. Interleukin-15 induces superior cytotoxic lymphokine activated killer cells in vitro with higher perforin, granzyme B, and cytokine expressions. Journal of Allergy and Clinical Immunology 2005;115:S15. [Google Scholar]
- Panés J, Sandborn WJ, Schreiber S, Sands BE, Vermeire S, D’Haens G, et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: results of two phase IIb randomised placebo-controlled trials. Gut 2017;66:1049–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte-Sucre A, Gamarro F, Dujardin J-C, Barrett MP, López-Vélez R, García-Hernández R, et al. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl Trop Dis 2017;11:e0006052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch A, Hessmann M, Hölscher A, Schreiber T, Bulfone-Paus S, Ehlers S, et al. Interleukin-15 mediates protection against experimental tuberculosis: a role for NKG2D-dependent effector mechanisms of CD8+ T cells. Eur J Immunol 2006;36:1156–67. [DOI] [PubMed] [Google Scholar]
- Riggle BA, Manglani M, Maric D, Johnson KR, Lee M-H, Lopes Abath Neto O, et al. CD8+ T cells target cerebrovasculature in children with cerebral malaria. J Clin Invest 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandborn WJ, Ghosh S, Panes J, Vranic I, Wang W, Niezychowski W, et al. A phase 2 study of tofacitinib, an oral Janus kinase inhibitor, in patients with Crohn’s disease. Clin Gastroenterol Hepatol 2014;12:1485–93.e2. [DOI] [PubMed] [Google Scholar]
- Sandborn WJ, Su C, Sands BE, D’Haens GR, Vermeire S, Schreiber S, et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2017;376:1723–36. [DOI] [PubMed] [Google Scholar]
- Santos C da S, Boaventura V, Ribeiro Cardoso C, Tavares N, Lordelo MJ, Noronha A, et al. CD8(+) granzyme B(+)-mediated tissue injury vs. CD4(+)IFNγ(+)-mediated parasite killing in human cutaneous leishmaniasis. J Invest Dermatol 2013;133:1533–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinberg M, Ferreira SB. Reversal of alopecia universalis by tofacitinib: A case report. Ann Intern Med 2016;165:750–1. [DOI] [PubMed] [Google Scholar]
- Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 2017;16:843–62. [DOI] [PubMed] [Google Scholar]
- Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Rev Immunol 2016;16:581–92. [DOI] [PubMed] [Google Scholar]
- Shreberk-Hassidim R, Ramot Y, Zlotogorski A. Janus kinase inhibitors in dermatology: A systematic review. J Am Acad Dermatol 2017;76:745–753.e19. [DOI] [PubMed] [Google Scholar]
- Sutherland APR, Joller N, Michaud M, Liu SM, Kuchroo VK, Grusby MJ. IL-21 promotes CD8+ CTL activity via the transcription factor T-bet. J Immunol 2013;190:3977–84. [DOI] [PubMed] [Google Scholar]
- Tamang DL, Redelman D, Alves BN, Vollger L, Bethley C, Hudig D. Induction of granzyme B and T cell cytotoxic capacity by IL-2 or IL-15 without antigens: multiclonal responses that are extremely lytic if triggered and short-lived after cytokine withdrawal. Cytokine 2006;36:148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Chen Z, Ciszewski C, Setty M, Solus J, Tretiakova M, et al. Cytosolic PLA2 is required for CTL-mediated immunopathology of celiac disease via NKG2D and IL-15. J Exp Med 2009;206:707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Sally B, Lesko K, Discepolo V, Abadie V, Ciszewski C, et al. Cysteinyl leukotrienes mediate lymphokine killer activity induced by NKG2D and IL-15 in cytotoxic T cells during celiac disease. J Exp Med 2015;212:1487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi P, Graham KL, Krishnamurthy B, Fynch S, Slattery RM, Kay TWH, et al. Perforin facilitates beta cell killing and regulates autoreactive CD8+ T-cell responses to antigen in mouse models of type 1 diabetes. Immunol Cell Biol 2016;94:334–41. [DOI] [PubMed] [Google Scholar]
- Unger A, O’Neal S, Machado PRL, Guimarães LH, Morgan DJ, Schriefer A, et al. Association of treatment of American cutaneous leishmaniasis prior to ulcer development with high rate of failure in northeastern Brazil. Am J Trop Med Hyg 2009;80:574–9. [PMC free article] [PubMed] [Google Scholar]
- Uzonna JE, Joyce KL, Scott P. Low dose Leishmania major promotes a transient T helper cell type 2 response that is down-regulated by interferon gamma-producing CD8+ T cells. J Exp Med 2004;199:1559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali DAA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol 2012;13:722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med 2012;367:508–19. [DOI] [PubMed] [Google Scholar]
- Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med 2014;20:1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka K Tofacitinib for the treatment of rheumatoid arthritis: an update. Expert Rev Clin Immunol 2019;15:577–88. [DOI] [PubMed] [Google Scholar]
- Ye W, Young JD, Liu CC. Interleukin-15 induces the expression of mRNAs of cytolytic mediators and augments cytotoxic activities in primary murine lymphocytes. Cell Immunol 1996;174:54–62. [DOI] [PubMed] [Google Scholar]
- Younes S-A, Freeman ML, Mudd JC, Shive CL, Reynaldi A, Panigrahi S, et al. IL-15 promotes activation and expansion of CD8+ T cells in HIV-1 infection. J Clin Invest 2016;126:2745–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young LH, Peterson LB, Wicker LS, Persechini PM, Young JD. In vivo expression of perforin by CD8+ lymphocytes in autoimmune disease. Studies on spontaneous and adoptively transferred diabetes in nonobese diabetic mice. J Immunol 1989;143:3994–9. [PubMed] [Google Scholar]
- Zhang Z, Deng W, Wu Q, Sun L. Tuberculosis, hepatitis B and herpes zoster in tofacitinib-treated patients with rheumatoid arthritis. Immunotherapy 2019;11:321–33. [DOI] [PubMed] [Google Scholar]
- Zhou M, Guo C, Li X, Huang Y, Li M, Zhang T, et al. JAK/STAT signaling controls the fate of CD8+CD103+ tissue-resident memory T cell in lupus nephritis. J Autoimmun 2020;102424. [DOI] [PubMed] [Google Scholar]
- Zloza A, Lyons GE, Chlewicki LK, Kohlhapp FJ, O’Sullivan JA, Lacek AT, et al. Engagement of NK receptor NKG2D, but not 2B4, results in self-reactive CD8+ T cells and autoimmune vitiligo. Autoimmunity 2011;44:599–606. [DOI] [PubMed] [Google Scholar]