

Summary
Macrophages continuously survey their environment in search of pathogens or apoptotic corpses or debris. Targets intended for clearance expose ligands that initiate their phagocytosis (“eat me” signals), while others avoid phagocytosis by displaying inhibitory ligands (“don’t eat me” signals). We report that such ligands can be obscured by the glycosaminoglycans and glycoproteins that coat pathogenic as well as malignant phagocytic targets. In addition, a reciprocal barrier of self-synthesized or acquired glycocalyx components on the macrophage surface shrouds phagocytic receptors, curtailing their ability to engage particles. The coating layers of macrophages and their targets hinder phagocytosis by both steric and electrostatic means. Their removal by enzymatic means is shown to markedly enhance phagocytic efficiency. In particular, we show that removal of mucins, which are overexpressed in cancer cells, facilitates their clearance. These results shed light on the physical barriers that modulate phagocytosis, which have been heretofore underappreciated.
Graphical Abstract

eTOC
Imbert et al. report that ligands that stimulate phagocytosis can be obscured by the glycocalyx of pathogenic and malignant targets. A reciprocal barrier conceals phagocytic receptors. Removal of these barriers enhances phagocytic efficiency and facilitates target clearance. The results shed light on physical barriers that modulate phagocytosis.
Introduction
Phagocytosis, the ingestion of particulate matter, plays an essential role in the maintenance of tissue homeostasis. It serves as a first line of defense in the elimination of invading pathogens [1]. Phagocytosis also prevents secondary necrosis and unwanted inflammation by efficiently recognizing and disposing of apoptotic bodies and debris [2]. In addition, phagocytic clearance of malignant cells is fundamental in the innate immune surveillance for cancerous growth; indeed, suppression of phagocytosis facilitates tumor-mediated immune evasion [3–5]. To fulfil these essential functions phagocytes reside in virtually all tissues of the body, where they constantly survey their surroundings for prey [6].
In the course of surveillance, phagocytes must rapidly distinguish harmful from healthy components by detecting features exposed on the surface of their putative targets. Features that trigger phagocytosis can be intrinsic to the target –like the phosphatidylserine exposed on the surface of apoptotic cells [7] or microbial-associated molecular patterns, like β-glucans [8]. Phagocytosis is often also facilitated by the deposition of soluble opsonins on the target, notably complement components or antibodies. In all cases, intrinsic or deposited “eat me” signals function as ligands for phagocytic receptors that trigger extensive remodeling of the plasma membrane (PM) and of the actin cytoskeleton, culminating in the extension of pseudopods that surround and engulf the target [9].
In addition to scanning for “eat me” signals, phagocytes also recognize surface molecules that serve as “don’t eat me” signals. These include CD47, PD-L1, and CD24 that engage their cognate receptors SIRPα, PD-1, and Siglec-10, respectively, to exert an inhibitory effect on phagocytosis [4, 5, 10]. When engaged, these inhibitory receptors arrest signaling pathways by recruiting otherwise cytosolic phosphatases that suppress phagocytic signaling [11]. In certain anti-inflammatory contexts, cytokines mediate the transcriptional upregulation of these inhibitory receptors [12]. Pro-inflammatory mediators, conversely, can prime or stimulate phagocytic receptors [9]. Clearly, phagocytosis is a tunable process dependent on the features of the target and the microenvironment of the phagocyte that modulate receptor expression and activation.
An overlooked aspect of innate immune surveillance is that the receptors that sense both “eat me” and “don’t eat me” ligands are generally short (≈5 nm), and therefore require intimate contact with the target for their engagement. Such contacts are expected to be influenced by the glycocalyx, a gel-like layer consisting of glycolipids, glycoproteins, and surface-associated glycosaminoglycans that surrounds the plasma membrane of all mammalian cells. In addition to its function in preserving membrane integrity, the glycocalyx serves as a diffusional barrier, restricting (filtering) the encounters made between molecules in solution and the membrane [13–15]. The thickness and composition of the glycocalyx can vary widely between cell types, shows remarkable polarization (e.g. in epithelial and endothelial tissues), and can be modulated by enzymes to alter its barrier function, especially in inflammatory settings [16, 17].
Given its under-appreciated role in regulating phagocyte biology, we systematically tested the electrical and mechanical properties of the glycocalyx and the impact of these properties on phagocytosis. Such considerations are particularly apparent in biological contexts where phagocytes acquire additional glycocalyx components, as is the case for macrophages exposed to hyaluronan in the synovium. Coating of the cell surface by gigantic hyaluronan molecules is anticipated to obstruct access to phagocytic receptors [18], particularly those with short extracellular domains such as Dectin-1 and Fc receptors. In this study, we devised approaches to build or break down the glycocalyx of host cells, pathogenic particles, or inert particles in these biological settings. We report that the size and charge of the glycocalyx on macrophages and of phagocytic targets limits the engagement of phagocytic receptors and thereby serves as a “don’t eat” or “don’t eat me” signal, respectively. Importantly, by removing the glycocalyx of tumor cells, we established a means for sensitizing their efferocytosis, findings that should inform strategies that employ IgG-based biologics [19].
Results
High molecular weight forms of hyaluronan in synovial fluid obstruct particle binding without curtailing membrane ruffling
In addition to endogenously synthesized components of the glycocalyx, cells can acquire glycosaminoglycans and/or proteoglycans from their surroundings to elaborate their pericellular coating. An extreme example occurs in articulations where chondrocytes and fibroblasts synthesize and secrete high molecular weight forms of hyaluronan (>4 million Da [20]) along with proteoglycans into the synovial fluid that fills and lubricates the joint cavity. On the synovial membrane that delimits the joint cavity reside macrophages that are exposed to the synovial fluid and its soluble hyaluronan (HA). HA –a linear polymer of disaccharides composed of D-glucuronic acid and D-N-acetylglucosamine– is in fact a major constituent of synovial fluid, present at 3–4 mg/mL in healthy joints [21, 22]. The HA of synovial fluid contributes to tissue hydrodynamics and impacts cell movement and proliferation and, most germane to this study, can form barriers to receptor access [23].
Given the unique and physiologically important setting of the synovium, we devised an experimental system to test if the macrophage glycocalyx –partially acquired from the synovial fluid– affects the ability for the cells to probe, survey, and ultimately bind particulates in their surroundings. To this end, we cultured primary murine bone marrow-derived macrophages (BMDM) that were differentiated in macrophage-colony stimulating factor (M-CSF) and bathed them in synovial fluid. We found that macrophages bathed in synovial fluid largely failed to engage IgG-opsonized particles that were normally bound by cells in regular tissue culture medium (Figure 1A–B). The inability to engage phagocytic targets did not appear to be caused by a defect in the ability of the cells to ruffle, as actin-rich extensions of the membrane were observed in macrophages even in the presence of synovial fluid (Figure 1A). We nevertheless formally tested if membrane ruffling was impacted by synovial fluid by assessing its effect on macropinocytosis, a process that requires membrane extension and remodelling. Macropinocytosis was measured as the uptake of tetramethylrhodamine-labeled 70 kDa dextran, a fluid-phase marker, determined by serial confocal imaging followed by 3-dimensional reconstruction and quantitation of the dextran trapped by the cells. As illustrated in Figure 1C and quantified in Figure 1D, the number and size of macropinosomes formed by cells incubated with the dextran in synovial fluid were indistinguishable from those formed in culture medium. Taken together, these results imply that while macrophages in synovial fluid showed normal sampling and probing behaviour, they nevertheless failed to engage IgG-opsonized phagocytic targets.
Figure 1. High molecular weight forms of hyaluronan (HA) in synovial fluid curtail particle binding without impairing membrane ruffling.
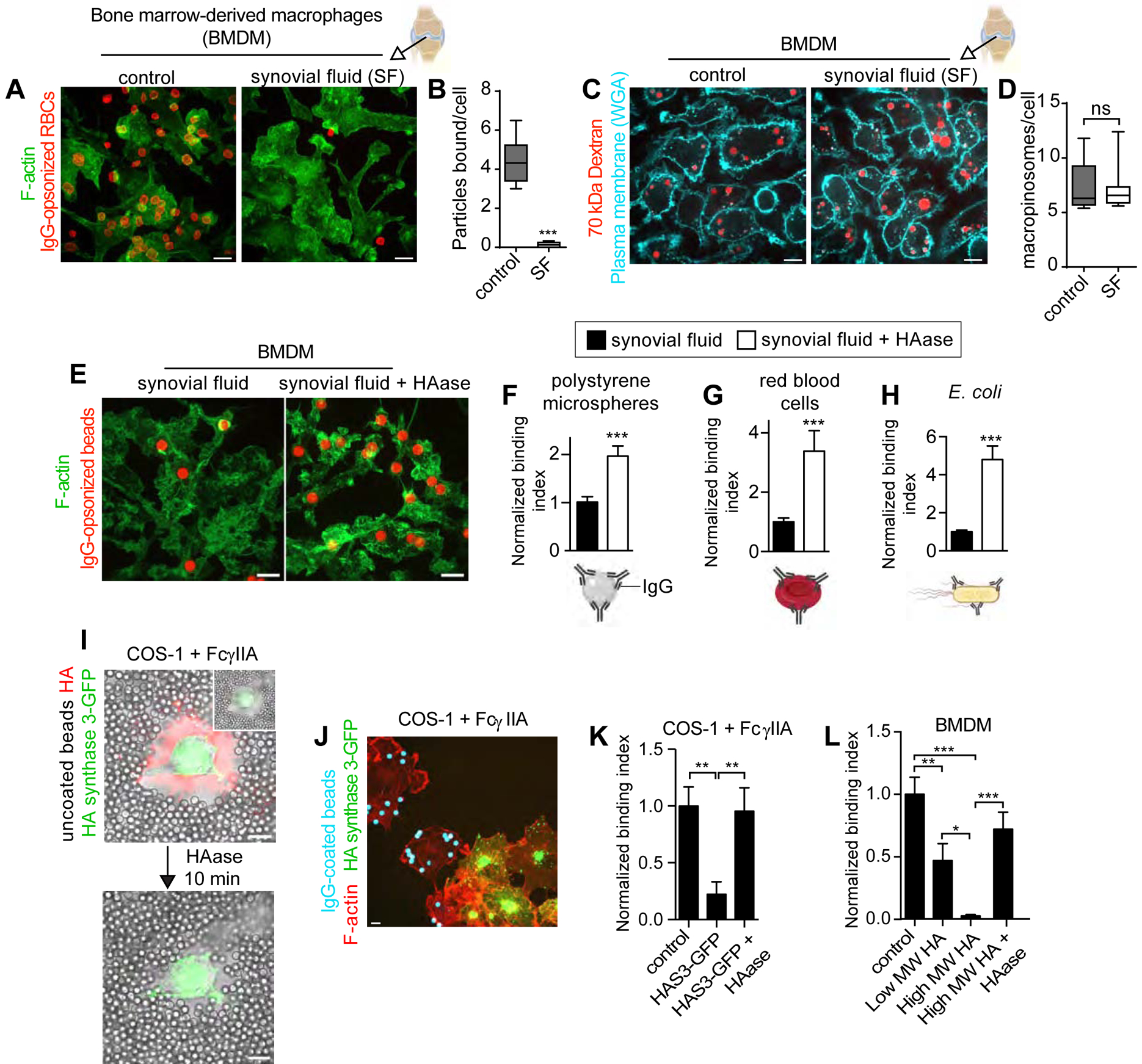
A-B) Primary, bone marrow-derived macrophages (BMDM) bathed in medium or synovial fluid were incubated with IgG-opsonized red blood cells (RBCs) for 20 min before fixation. B) Mean number of RBCs bound per cell determined for 5 fields of 3–10 cells, n=3. C-D) Cells were incubated with medium or synovial fluid containing tetramethylrhodamine-labelled 70 kDa dextran for 15 min, subsequently labeled with Alexa 647-wheat germ agglutinin for 2 min and imaged live. See also Figure S1 for phagocytic efficiency determinations and additional controls. D) Mean number of macropinosomes per cell determined for 5 fields of 5–10 cells, n=3. E-H) BMDM were incubated with synovial fluid or synovial fluid, then treated with hyaluronidase (HAase) where indicated, and finally challenged with the indicated IgG-opsonized phagocytic targets, fixed and stained for F-actin and target IgG. A representative image is shown in E. Normalized binding indices in F-H are from >5 fields of 15–30 cells, n=3. Bars represent means ± SEM. I-K) COS1 cells stably expressing FcγRIIA transiently expressing GFP-tagged HA synthase 3 (HAS3-GFP). I) Uncoated polystyrene microspheres added to cells (DIC). HA was imaged using an HA-binding complex covalently labelled with biotin followed by Alexa 555-streptavidin, before (left panel) and after (right panel) HAase treatment. J-K) IgG-opsonized beads added to cells for 15 min before fixation and staining with rhodamine phalloidin. Normalized binding index in K is from >5 fields of 5–10 cells, n=3. Bars represent means ± SEM. L) BMDM, preincubated with indicated forms of HA, then treated with or without HAase and challenged with IgG-opsonized beads. Normalized binding index is from >5 fields of >10 cells each, n=3. Bars represent means ± SEM. All scale bars, 10 μm. Here and elsewhere, Mann-Whitney tests were used; *p<0.05, **p<0.01, ***p<0.001.
Given the high expression of CD44 –the pre-eminent HA receptor– on the surface of macrophages [23], we hypothesized that binding of the glycosaminoglycan to the cell surface was responsible for the inability of the cells to bind phagocytic targets. However, in addition to HA, synovial fluid (an ultrafiltrate of plasma) contains blood plasma proteins and proteoglycans, as well as enzymes produced locally in the joint. This complex mixture renders synovial fluid viscous. Such viscosity, rather than HA binding, could have altered the ability of the cells to bind phagocytic targets. To parse the specific contribution of HA from that of other components/properties of the synovial fluid, we incubated macrophages with synovial fluid and removed unbound components before assessing the binding of phagocytic targets. The inhibition of binding persisted despite removal of the synovial fluid, implying that components of the fluid bound to the membrane, as opposed to increased viscosity, were responsible for the inhibitory effect. That HA was the bound component of synovial fluid that impaired the binding was tested using hyaluronidase. This approach was revealing: degradation of HA following incubation with synovial fluid was sufficient to increase the binding of IgG-opsonized targets by 2 to 5-fold (Figures 1E–H, S1A–B).
Further evidence that HA association with the surface of phagocytes suffices to depress particle binding was obtained by stimulating the de novo biosynthesis of the glycosaminoglycan. We found that overexpression of the HA synthase (HAS3-GFP) in phagocytic cells created a large (micron-sized) HA coat (stained red in Figure 1I) that obstructed the approach of particles, preventing the formation of close contacts with the plasma membrane. As a result, HAS3-expressing cells did not bind IgG-opsonized targets, an encumbrance that was reversed by treating the cells with hyaluronidase (Figure 1I–K). Finally, we added purified preparations of low (<50 kDa) and high (>1000 kDa) molecular weight HA to macrophages and found a decrease in particle binding that was proportional to the size of the glycosaminoglycan and that, as before, was reverted by degrading the HA with hyaluronidase (Figure 1L). In all cases, obstruction to phagocytosis by the added synovial fluid and HA occurred at the very initial, particle-binding stage; the engulfment of particles that bound successfully was ostensibly unimpeded by an HA-glycocalyx (Figure S1C–F).
Taken together, acquired components of the macrophage glycocalyx, such as HA, can limit the ability of the cells to bind phagocytic targets.
Acquired and endogenous glycocalyx components contribute to the negative zeta potential of macrophages that impairs binding of like-charged particles
HA is negatively charged; 50% of its constituent sugars have acidic groups. Because phagocytic targets are often negatively charged (including the polystyrene microspheres used above; see Figure S2A), electrostatic repulsion may contribute to the barrier function of HA and possibly also of other glycocalyx components [24]. The charge of the glycocalyx at the interface with the surrounding fluid, where the Stern counter-ion layer is generated, is the most relevant for target particle engagement. We therefore measured the zeta potential, i.e. the potential difference between the dispersion medium and the Stern layer of both cells and their targets using electrophoretic light scattering [25]. By lifting macrophages into suspension, we were able to reliably measure their zeta potential. As expected, the zeta potential of the BMDM was dependent on the ionic strength of the solution (data not shown); in PBS it was determined to be between −6.5 and −7.5 mV (Figure 2A,C), decreasing gradually over time after the cells were lifted. Preincubation of the macrophages with HA caused a robust increase in the negativity of the potential to −9 mV, which returned to control levels when the cells were treated with hyaluronidase (Figure 2A). Clearly, HA binding to the cell surface was sufficient to augment the negative surface charge of the cells.
Figure 2. Acquired and endogenous glycocalyx components contribute to the negative zeta potential of macrophages.

A) Zeta potential of suspended BMDMs coated or not with high molecular weight HA, followed by HAase treatment where noted. Bars represent means ± SEM. n=3. B) BMDM left untreated or treated with intact or denatured α2–3,6,8 neuraminidase for 30 min. Cells stained with Cy5-conjugated Sambucus nigra agglutinin (SNA) or Alexa 488-conjugated peanut agglutinin (PNA), lectins that recognize terminal sialic acid or galactose, respectively. Scale bar, 10 μm. C) Cells treated as in B, but lifted into suspension and measured for zeta potential as in A. D) Zeta potential determinations for indicated IgG-opsonized particles. Bars represent means ± SEM. n=3. E) BMDM, treated as in B and challenged with phagocytic targets. Normalized binding index of IgG-opsonized beads is from >5 fields of >10 cells each, n=3. Bars represent means ± SEM. See also Figure S2 for additional zeta potential measurements, Siglec expression, and the functional effect of SHP1/2.
In the absence of synovial fluid, and especially when lifted into suspension, macrophages have only marginally detectable self-synthesized hyaluronan (data not shown). Their negative surface potential must therefore be conferred by other anionic charges, presumably terminal sugars (e.g. sialic acid) and sulfates present in surface proteoglycans. Sialic acid is found at the end of branched N- and O-linked glycans and can be readily removed from cells with neuraminidase. We treated BMDM with the enzyme and validated its efficiency by staining macrophages with fluorescent Sambucus nigra agglutinin, a lectin that binds to terminal sialic acid. While control cells labeled brightly with the agglutinin, staining was virtually eliminated by treatment with neuraminidase. The removal of sialic acid was accompanied by exposure of galactose moieties that were visualized using peanut agglutinin, a different lectin that detects terminal, but not underlying (internal) galactose moieties (Figures 2B, S2B). Denatured (boiled) neuraminidase was without effect (Figure 2B). We next measured the effects of neuraminidase on zeta potential by electrophoretic light scattering. Sialic acid was found to account for a substantial fraction of the zeta potential (Figure 2C).
For a negative surface charge to contribute an electrostatic barrier to the engagement of phagocytic targets, such targets would need to also be negatively charged. We therefore compared the zeta potential of a panel of phagocytic targets –polystyrene beads, red cells, and E. coli– by electrophoretic light scattering. Albeit to different degrees, the zeta potential of all of these particles was found to be negative, regardless of whether they were opsonized with IgG or not (Figures 2D, S2A). To test the hypothesis that a negative outer layer comprised of glycoproteins could impose an electrostatic barrier to the engagement of Fc receptors, we challenged untreated macrophages and those with sialic acid removed from their surface with IgG-opsonized targets. As shown in Figure 2E, treatment of the cells with neuraminidase increased the binding of phagocytic targets 2-fold, supporting the idea that a negative surface potential impedes receptor access to like-charged targets. Of note, a denatured preparation of the enzyme was without effect (Figure 2E), indicating that the observed changes were not caused by contaminating lipopolysaccharide, which is heat-resistant. The treatment of macrophages with neuraminidase may have additional effects on the cytoskeleton that could impact particle binding. However, we found that the augmented binding observed when sialic acid was removed from the surface of phagocytes persisted at 4°C, conditions that prevent cytoskeletal remodelling and particle internalization (Figure S2G).
Ectopic surface expression of CD43 or syndecan-1 reduces particle binding
Based on the preceding observations, we predicted that increasing the density of endogenous sialic acid on the cell surface would have the converse effect, i.e. would reduce the ability of the macrophages to engage negatively charged targets. A molecular approach, namely ectopic expression of the transmembrane sialomucin, CD43, was used to test this premise. CD43, which is poorly expressed in macrophages (data not shown), bears ≈80 glycans (Figure 3A) and is therefore a rich source of surface sialic acid. When a GFP-tagged form of CD43 was expressed in macrophages it was properly targeted to the plasma membrane and was apparent in ruffles, sites where particles are first captured [26, 27]. We found that CD43-GFP itself did not impact membrane ruffling, macropinocytosis, or surface expression of phagocytic receptors (Figure S3A,C–E) but imposed a significant hindrance to the engagement of IgG-coated particles (Figure 3B–C), which was reversed when the cells were treated with neuraminidase (Figure 3B–C). This suggested that sialic acid was the major contributor to the barrier.
Figure 3. Ectopic surface expression of CD43 or syndecan-1 reduces particle binding.
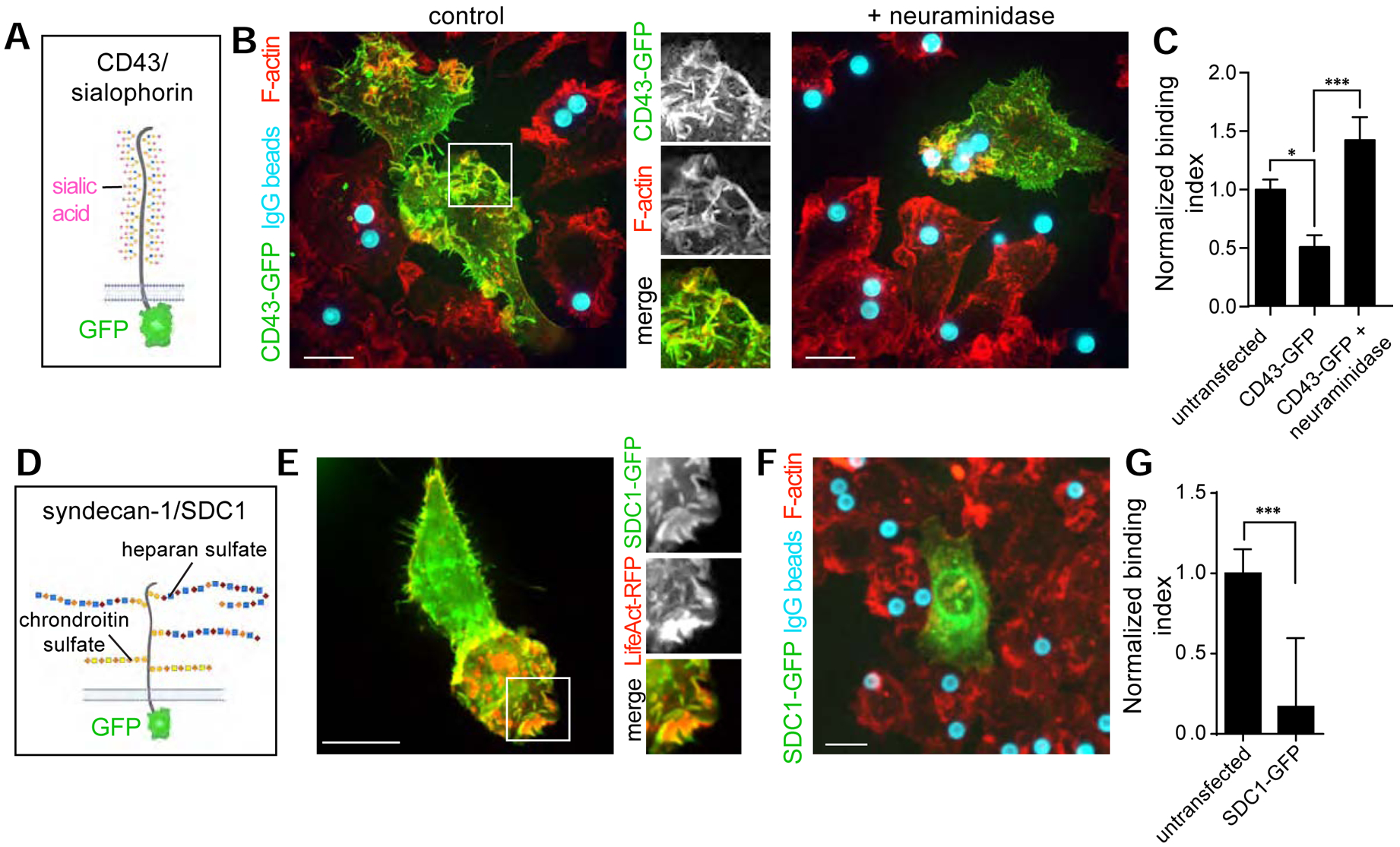
A) Diagrammatic representation of CD43, a large transmembrane mucin-type glycoprotein bearing ≈80 sialylated O-glycan sites. The predicted molecular weight of the polypeptide backbone is 44 kDa, while the observed molecular weight determined by SDS-PAGE is 115–130 kDa, suggesting a large contribution to the mass by glycans. B) RAW cells expressing CD43-GFP, treated or not with α2–3,6,8 neuraminidase, challenged with IgG-opsonized particles for 15 min then fixed and stained for F-actin and target IgG. C) Quantification from B. >20 cells, n=3. Bars represent means ± SEM. D) Diagrammatic representation of syndecan-1, a proteoglycan containing two extracellular attachment sites for chondroitin sulfate and three for heparan sulfate. E) RAW 264.7 cells expressing syndecan-1(SDC1)-GFP and LifeAct-RFP. F) RAW 264.7 cells expressing syndecan-1(SDC1)-GFP challenged with IgG-coated particles for 15 min and stained for F-actin and target IgG. G) Quantification from experiments like that in F; >15 cells, n=4. All bars represent means ± SEM. All scale bars, 10 μm. See also Figure S3 demonstrating that surface receptor expression is not impacted by CD43 or SDC1 overexpression.
As discussed in more detail in the Discussion, sialic acid can alter phagocyte function by means other than its electrostatic contribution, e.g. by interacting with inhibitory receptors. In an effort to modify the surface charge of the cells without changing their sialic acid content, we sought to express other charged molecules. Syndecans, proteoglycans that are variably expressed in macrophages [28], were an attractive candidate as their charge stems primarily from their sulfation. Syndecans generally have multiple chains of heparan sulfate and of chrondroitin sulfate (Figure 3D) [29]. We attempted the overexpression of GFP-tagged forms of syndecan-1,−3, and −4, and found that syndecan-1 showed the best targeting to the plasma membrane (Figure 3E and data not shown). Like CD43, syndecan-1 was found in actin-rich membrane ruffles (Figure 3E) which were unaltered relative to untransfected cells, implying that the ability for cells to probe their surroundings was not impacted (Figure S3B). The ectopic expression of syndecan-1 did, however, reduce binding of IgG-opsonized phagocytic targets 4- fold (Figure 3F–G), without altering surface expression of Fc receptors (Figure S3C–E) indicating that proteoglycans can also generate electrostatic barriers to the engagement of Fc receptors.
Charge selectivity in phagocytic target binding
If the negative surface potential presented by the glycocalyx forms an electrostatic barrier for the engagement of phagocytic receptors, such an effect would be expected to be proportional to the magnitude and sign of the charge on the targets. To validate this notion, we functionalized polystyrene microspheres with cationic or anionic adducts. Avidin-coated beads were first opsonized with anti-avidin antibodies to provide a comparable array of ligands for macrophage Fc receptors, and then decorated the particles with either biotinylated-poly(L)lysine (PLL) or with biotinylated-heparin. The PLL used was ≈25 kDa and contained ≈100 lysine units; the heparin was of a comparable size (≈15 kDa). Thus, our experimental system was devised to confer a high density of positive or negative charges to the particles without imposing major steric barriers to the Fc region of the anti-avidin antibody (Figure 4A). Measurements of the zeta potential of the particles at physiological ionic strength (PBS) indicated that, as expected, the PLL-coated particles became positive while the heparin-coated ones became more negative (Figure 4B). We next challenged macrophages with these beads in serum-free medium to avoid interference by adherent soluble proteins. The positively-charged particles bound 2.5-fold more effectively than did beads coated with IgG only, while negatively-charged beads bound 2-fold less. Interestingly, the preferential binding of positively charged targets to the macrophages persisted even without IgG opsonization, provided the particles were incubated for sufficient periods of time (Figure S4B). The electrostatic selectivity was also confirmed by exposing the macrophages to 1:1 ratios of differently charged beads. In these experiments, macrophages preferentially bound the PLL-coated targets over the heparin targets (Figure 4D). Lastly, decreasing the ionic strength of the medium while maintaining its osmolarity with mannitol accentuated the selectivity for positively-charged target particles, as expected for an electrostatic interaction (Figure S4C,D).
Figure 4. Charge selectivity in phagocytic target binding by macrophages.
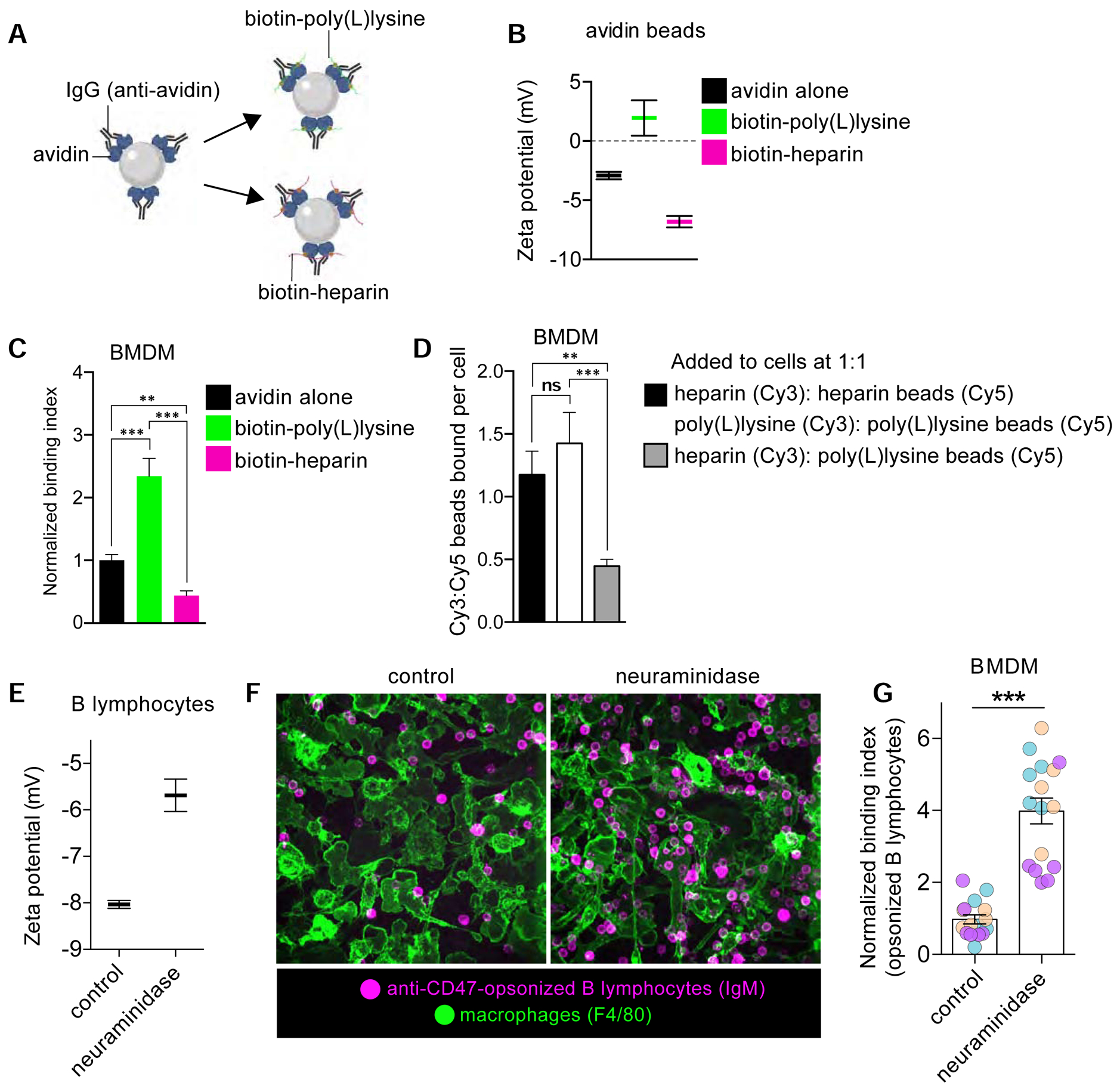
A-B) Diagrammatic representation of experimental setup (A), where polystyrene microspheres were coated with avidin followed by anti-avidin antibodies and biotinylated polymers. B) Zeta potential measurements of avidin beads and those containing biotinylated poly(L)lysine or heparin. Bars represent means ± SEM. C) Normalized binding index of indicated particles by BMDM. D) Ratio of beads bound per cell given to BMDM at an equal (1:1) ratio. >5 fields of 15–30 cells, n=3. Bars represent means ± SEM. E) Zeta potential determination of B lymphocytes either untreated or treated with α2–3,6,8-neuraminidase. Bars represent means ± SEM. F-G) Anti-CD47 opsonized B cells with or without sialic acid were added to BMDM and subsequently fixed and stained for F-actin and IgM-BCR (B cells). Representative image shown in F. Normalized binding indices in G are from >5 fields of 15–30 cells, n=3. Bars represent means ± SEM. See also Figure S4 demonstrating that force overcomes electrical barriers to particle engagement.
Programmed cell death is coincident with a marked decrease in the abundance of surface sialic acid residues, an effect that contributes to the engulfment of corpses by phagocytes [30]. The IgG-based opsonization of live targets, that retain and even enhance their surface sialic acid content, has emerged as a therapeutic strategy to facilitate phagocytosis of tumor cells and lipid-laden foam cells. We sought an experimental system to determine if the enzymatic removal of sialic acid from live, IgG-opsonized cells could improve the efficacy of their capture by macrophages. To this end we isolated primary murine B lymphocytes and incubated them with an IgG antibody targeted against CD47, a “don’t eat me” ligand that can otherwise depress the signaling pathways that trigger phagocytosis. The CD47-blocking antibody chosen has been shown to facilitate phagocytosis in vitro and in vivo [31]. Note that while the variable region of the anti-CD47 IgG serves to block the inhibitory ligand, its Fc region in fact enhances engulfment, as it will be recognized by phagocytic Fcγ receptors [3]. To remove sialic acid from their surface, we treated the suspended opsonized B cells with neuraminidase, which was sufficient to reduce their negative surface charge (Figure 4E). We then challenged primary murine macrophages with such opsonized B cells. Remarkably, we found that the removal of sialic acid was sufficient to increase binding of the lymphocytes 4-fold (Figure 4F–G). Taken together with the previous data, these results demonstrated that macrophages selectively engage their targets in accordance with their surface charge, contributed to a large extent by sialic acid in live cell targets.
Steric hindrance contributes to the barrier opposing ligand recognition and engagement by phagocytes.
In addition to their net negative charge, components of the glycocalyx present a physical barrier for ligands to access the short phagocytic receptors on the cell surface. Indeed, the binding of HA to the macrophage surface and the heterologous expression of CD43 or syndecan-1 must have increased the density and height of the glycocalyx, not only its charge. We therefore designed a series of experiments in an effort to distinguish the role of steric hindrance without a confounding electrostatic contribution. We opsonized avidin-coated microspheres with anti-avidin IgG and functionalized them with rigid polyethylene glycol (PEG) of varied sizes containing a single terminal biotin (Figure 5A). Long forms of PEG, which is uncharged, were sufficient to obstruct particle binding (Figure 5B).
Figure 5. The glycocalyx of phagocytic targets constitutes a mechanical barrier to ligand recognition and engagement by phagocytic receptors.
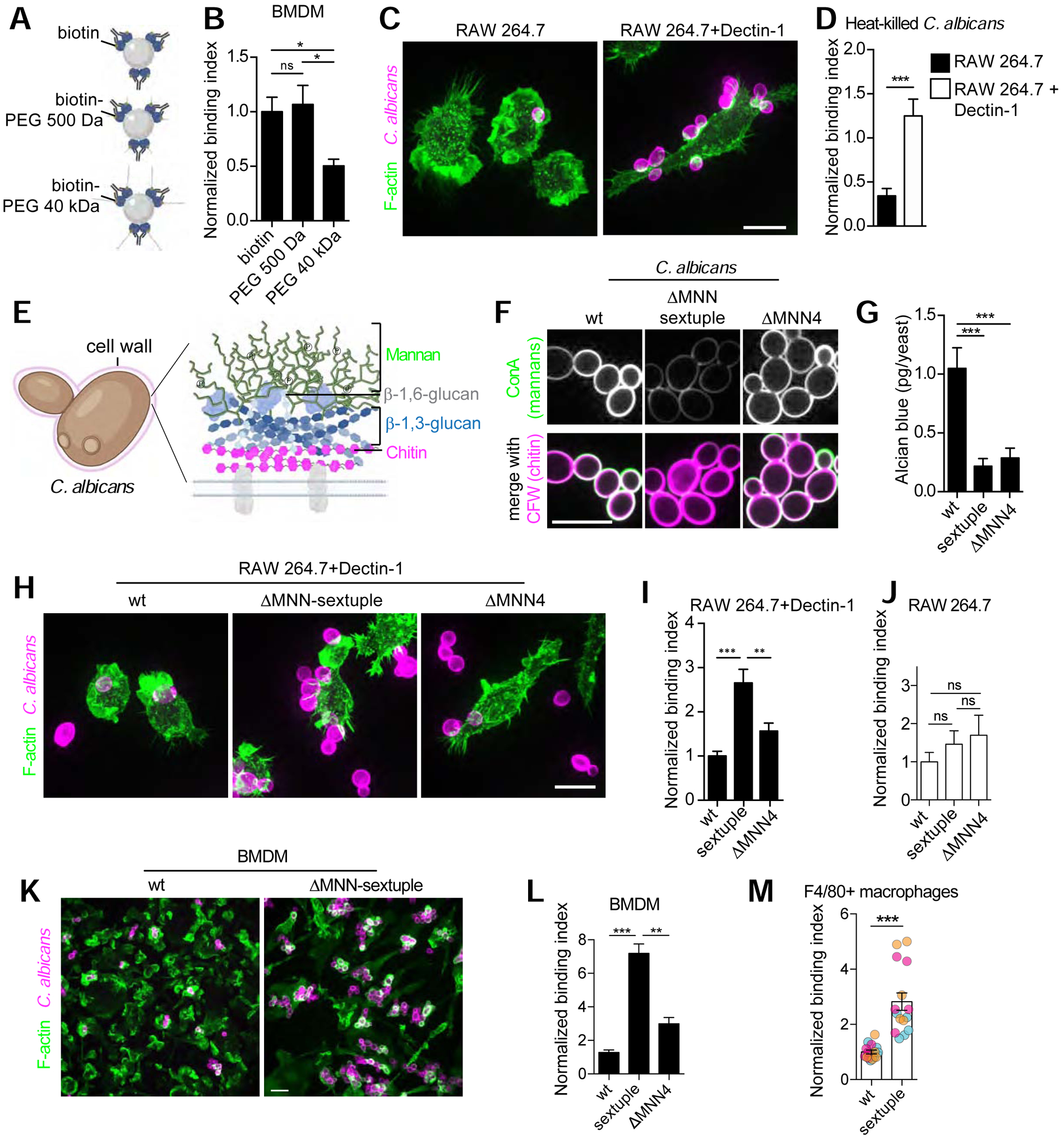
A) Diagrammatic representation of experimental model. B) Avidin beads opsonized with anti-avidin antibodies and containing biotin-PEG of indicated sizes were incubated with BMDM for 15 min. Normalized binding index is from >5 fields of 5–10 cells, n=3. Bars represent means ± SEM. C) RAW 264.7 cells and those stably expressing Dectin-1 challenged with heat-killed Candida albicans and stained with calcofluor-white (CFW) and Alexa488-phalloidin (F-actin) after fixation and permeabilization. D) The mean normalized binding index determined for >5 fields of >10 cells each, n=3. E) Diagrammatic representation of C. albicans cell wall. F) Wildtype (wt) and indicated MNN deletion strains of C. albicans live-stained with CFW to detect chitin and concanavalin A (ConA) to detect mannans. G) Quantitation of Alcian blue staining for C. albicans strains shown in F. H) Representative images of RAW 264.7 cells stably expressing Dectin-1 challenged with indicated C. albicans strains stained with calcofluor-white (C. albicans) and Alexa488-phalloidin (F-actin) after fixation and permeabilization. I-J) Quantification of RAW 264.7 cells stably expressing Dectin-1 in I, or parental RAW 264.7 cells in J, and challenged with indicated C. albicans strains. K-L) BMDM challenged with wt and ΔMNN-sextuple C. albicans strains. Representative images are shown in K. Normalized binding index determined for >5 fields of >5 cells, n=3. Bars represent means ± SEM. M) Macrophages elicited to the peritoneal cavity by periodate were challenged with C. albicans strains in vivo for 1 h followed by fixation and staining for F4/80. Normalized binding index determined for >5 fields of >5 cells each, n=3. Bars represent means ± SEM. All scale bars, 10 μm.
Pathogens and microbes of our normal microbiome synthesize capsules [32, 33] or cell walls rich in polysaccharides and mannosylated proteins [34], functionally analogous to the glycocalyx of mammalian cell. This coating is immunologically relevant, as it shields the microbe from digestive enzymes and neutralizing antibodies. It is also likely to interfere with phagocytosis. Detailed analysis of the role of wall thickness and charge required a system where its synthesis could be modulated, and where a defined phagocytic receptor is responsible for engulfment. We opted to use Candida albicans, a commensal fungus that can become an opportunistic pathogen, which is engaged by the phagocytic pattern-recognition receptor Dectin-1 that binds to fungal β−1,3-glucans [35]. Importantly, the β−1,3-glucans of C. albicans are normally found deep within the yeast’s wall, where the outermost layer is comprised of proteins attached to a large mannan structure (of up to 200 mannose residues) (Figure 5E). Under normal circumstances, such glucans are revealed only upon heat-induced denaturation, which enables binding of the killed yeast by Dectin-1-expressing macrophages (Figure 5C–D).
The mannan structures –which comprise 40% of the dry weight of the C. albicans cell wall– are synthesized by the MNN2 gene family of glycosyl transferases, which is comprised of six members [36]. A phosphate is attached to the first mannose on some of the branches, conferring negative charge to the yeast surface that can be detected with polyvalent cationic dyes like Alcian blue (Figure 5G). To ascertain how the various components of the wall affect the ability for Dectin-1 to engage β−1,3-glucans, we utilized mutant strains of C. albicans lacking all MNN2 genes (ΔMNN sextuple) or a gene that regulates the phosphate transfer (ΔMNN4). We initially confirmed the described phenotypes of the mutants [36]: the ΔMNN sextuple mutant strain had fewer mannans, as judged by decreased concanavalin A staining and increased access to the deep-seated chitin by calcofluor white, and less negative charge, as determined by Alcian blue binding (Figure 5F–G). The ΔMNN4 strain, on the other hand, only showed deficiency in its charge (Figure 5F–G).
The wildtype, ΔMNN sextuple and ΔMNN4 mutant strains of C. albicans provided an auspicious system where the contribution of size and charge to particle engagement could be assessed separately. When Dectin-1-expressing macrophages were challenged with these strains, we found that removal of the mannan layer (including the phosphorylated mannans that confer charge to the wall) markedly increased particle binding (2.5-fold; Figure 5H–I). Selective removal of the phosphates using the ΔMNN4 mutant also increased particle binding, albeit to a lesser extent (Figure 5H–I). These observations are in good agreement with those reported by [36]. We also confirmed that, as anticipated, phagocytosis of the fungi was dependent on the expression of Dectin-1 (Figure 5J). Primary macrophages, which express the Dectin-1 receptor endogenously [37], captured the C. albicans mutant strains in vitro following a similar pattern (Figure 5K–L). This prompted us to test if these findings would apply to a more complex in vivo setting where primary macrophages are elicited to the peritoneal cavity. As shown in Figure 5M, we found that yeast devoid of a mannan layer we more readily bound and engulfed by the elicited macrophages than were wildtype yeast. Taken together, these data imply that a combination of electrical and steric factors protect the microorganism from being bound and internalized by macrophages.
Transmembrane mucins override antibody blockade of CD47-SIRPα “don’t eat me” signals
Like the phagocytes themselves, animal cells that are targets of phagocytosis are also endowed with a glycocalyx that could in principle obstruct access to phagocytic determinants. A hallmark in malignancy is immune evasion, but a role for the tumor glycocalyx in this strategy has not been formally tested. Multiple tumor types upregulate bulky transmembrane mucins (e.g. Muc-1, Muc-16, podocalyxin) that drive growth and invasion of the cancer. In light of our preceding findings, we therefore asked if the tumor glycocalyx participates in the immune evasion strategy by protecting malignant cells from detection and phagocytosis by neighbouring macrophages. In that event, both physical and electrical obstruction would need to be considered. To this end, we used a metastatic breast tumor line, MCF7, reported to highly express transmembrane mucins [38], as well as an ovarian adenocarcinoma line, SK-OV-3, that is also rich in transmembrane mucins (data not shown). To first determine the surface potential of the cells, we generated karyocytes as previously described (Figure S5) [39]. Karyocytes (cell fragments devoid of a large fraction of their cytoplasm) are smaller small (≈7–8 μm) than the intact cells, and therefore better suited to electrophoretic estimation of the zeta potential. As expected, the zeta potentials of MCF7 and SK-OV-3 cells were negative (Figure 6A,D). Sialic acid residues provided a sizable fraction of the negative charge, which decreased markedly upon treatment with neuraminidase (Figure 6A). Because of their enhanced expression and since they extend tens and even hundreds of nanometers from the plasmalemmal bilayer, mucins are likely to contribute heavily to the zeta potential, which assesses the potential at the slippage plane. This prediction was tested by cleaving mucins using a recently described recombinant mucinase [40]. Treatment with mucinase reduced the zeta potential to a level comparable to that attained with neuraminidase, suggesting that mucins contain the bulk of the sialic acids that contribute to the negative surface charge (Figure 6A,D).
Figure 6. Transmembrane mucins override antibody blockade of CD47-SIRPα “don’t eat me” signals.
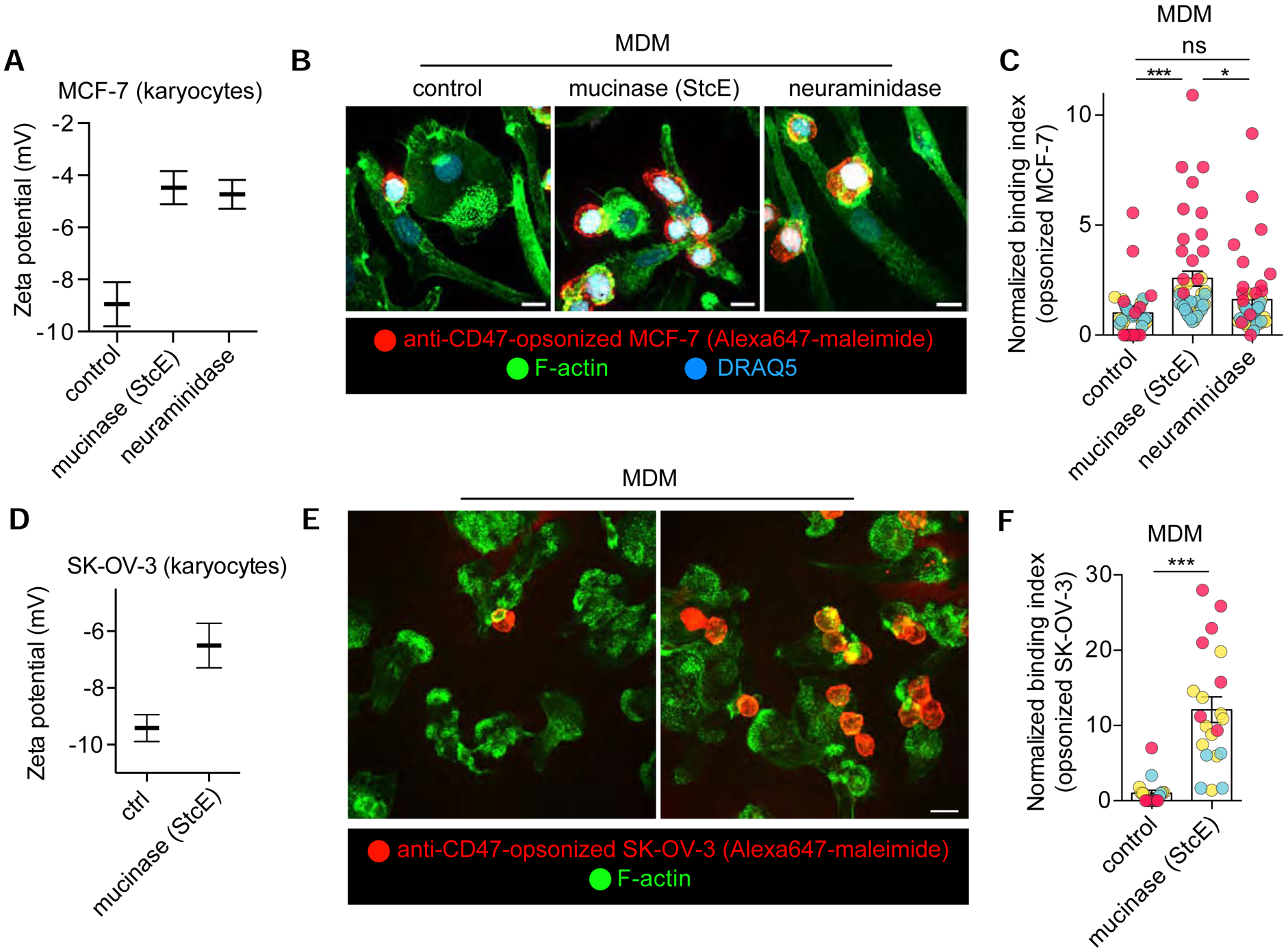
A) Zeta potential measurements of suspended karyoplasts derived from human breast adenocarcinoma (MCF-7) cells treated with a recombinant mucinase (StcE), α2–3,6,8 neuraminidase, or left untreated. Bars represent means ± SEM. n=3. B) Human monocyte-derived macrophages (hMDM) challenged with anti-CD47-opsonized MCF-7 cells treated with an Alexa Fluor 647 maleimide derivative. Cells are stained with Alexa 488 phalloidin (F-actin) and DRAQ5 (nuclei). C) Normalized binding index from >5 fields of >10 cells, n=3. Bars represent means ± SEM. D-F) Performed as in A-C but with SK-OV-3 cells. See also Figure S5 for target particle size determinations.
In addition to targeting major T-cell inhibitory pathways, IgG antibodies are used in the treatment of cancer to block “don’t eat me” signals that prevent ingestion of malignant cells by phagocytes. Antibodies currently in use are targeted against CD47, as described above, and PD-L1, ligands expressed on tumor cells that depress phagocytosis. To test if the tumor cell glycocalyx functions as a barrier to phagocytosis, we opsonized MCF7 and SK-OV-3 cells with the CD47-blocking antibody as in Figure 4. We found that primary human macrophages did not bind or internalize mucinous tumor cells in the absence of the opsonin (data not shown), but showed moderate phagocytic efficiency when the cancer cells were opsonized with the anti- CD47 antibody (Figure 6B–C,E–F). In contrast, when the mucins were cleaved from the tumor cells, the macrophages often bound and internalized several targets at once, an occurrence not observed under control conditions. Some of the increased binding and phagocytic ability was attributable to the removal of the charges contributed by mucin-associated sialic acid, as treatment of the tumor cells with neuraminidase also increased their binding by macrophages, albeit to a lesser extent (Figure 6B–C). Taken together, these findings suggest that the mucins enriching the cancer cell glycocalyx impede the engagement of trans-acting phagocytic receptors; this obstruction is in part electrostatic, although steric blockade seems to play a bigger part.
Discussion
The first point of contact between phagocytic targets –including invading pathogens and malignant/apoptotic cells– and the host immune system are their outermost layers: the cell walls/glycocalyces of the targets and the glycocalyx of phagocytes. By systematically breaking down or building up the coating layers of macrophages or their targets, we demonstrated that heparan sulfate proteoglycans, mannosylated proteins, transmembrane mucins, and the sialic acids can constitute a barrier to the physical contacts required to initiated phagocytosis. This likely constitutes the earliest event in a hierarchical sequence of “don’t eat” and/or “don’t eat me” signals that curtail the effectiveness of the phagocytic process, conferring selectivity to the process. Supporting this idea, we found that even when we blocked the canonical “don’t eat me” molecule, CD47, with antibodies that simultaneously provide ligands for Fc receptors, phagocytosis was still remarkably sensitized by the removal of the tumor cell glycocalyx, at least in mucinous tumour lines. Clearly, by obstructing phagocytosis, tumors can limit antigen presentation and thus evade immune attack.
There are two components to the outermost barrier: one that is electrostatic, the other steric. We found that sialic acid residues contribute a large fraction of the negative charge that reduces phagocytic effectiveness, presumably by electrostatically repulsing anionic targets. In principle, terminal sialic acid residues may exert their effects via Siglecs, a family of receptors that in some instances harbour cytosolic inhibitory immunoreceptor tyrosine motifs (ITIMs) that could antagonize phagocytic signaling. For example, Siglec G/10 which is expressed by tumor-associated macrophages, is engaged in trans by CD24, a sialoglycoprotein that is upregulated in breast and ovarian cancers [10]. Similarly, blocking the CD22 Siglec that is upregulated in ageing phagocytes of the brain can restore their phagocytosis of myelin debris, amyloid-β oligomers and α-synuclein fibrils [41]. However, we found that the ITIM containing Siglecs G, F, E, CD22, and CD33 were expressed at rather low levels in BMDM (Figure S2H). Moreover, the inhibition of Shp1/2 phosphatases –reported to mediate the inhibitory effect of ITIMs when Siglecs are engaged by sialic acids– did not enhance particle binding (Figure S2I–J). These observations, together with the dependence of phagocytosis on the charge of the targets regardless of their sialic acid content, led us to conclude that the primary effects of terminal sialic acid are electrostatic in nature.
We initiated our studies with the notion that articular macrophages can acquire components of their glycocalyx from synovial fluid, in particular, from HA. Large polymers of HA had previously been described to inhibit phagocytosis of latex microspheres [18] and IgG-opsonized targets [23]. This effect was reported even for solutions of relatively low concentrations of HA (0.25 mg/mL), much below that found in healthy synovial fluid (3–4 mg/mL). Importantly, the large glycosaminoglycan chains that function to maintain tissue integrity and provide lubrication [17] can become fragmented in inflammatory conditions. Fragmented HA can have pro-inflammatory effects on macrophages, leading to their production and release of inflammatory mediators including TNF-α, IL-1β, and nitric oxide [42]. How a ubiquitous molecule –that has high biocompatibility and is produced in abundance in every tissue– can elicit such effects has been mysterious [43, 44]. A role for hyaluronan in forming barriers to phagocytosis may help to shed light on this apparent paradox. Rather than simply increasing the viscosity of the medium surface [18], HA bound to the macrophage surface may maintain physical separation between particulates and the phagocyte. Additionally, polymers affixed to the surface of macrophages by its receptor, CD44, can limit receptor diffusion and activation and may thereby dampen inflammatory signaling [23, 45, 46]. HA fragmentation could promote closer contacts between macrophages and their cellular surroundings, while simultaneously fostering reorganization of surface receptors that promote inflammation.
Pathogens have experienced evolutionary pressure to protect themselves from immune attack. Formation of an elaborate cell wall contributes to the evasive response. Accordingly, the exofacial mannosylated proteins of fungal cell walls and the hyaluronic acid in the group A streptococcal capsule play roles in virulence [33, 36]. Phagocytes have coevolved to recognize microbial-associated molecular patterns, but these are often buried within the microbial protective wall. In a similar fashion, malignant cells manage to avoid detection and ingestion by amplifying the expression of bulky, anionic mucins.
Considering their prevalence, how are the physical and electrostatic barriers to phagocytosis overcome? Two mechanisms can be readily envisaged. Momentum provided to target particles by hydraulic flow, such as that of lymph pulsing over resident phagocytes of the lymph nodes or lymphatic vessels [47], could outstrip the repulsive effect of the glycocalyx. In this regard, we find that forcing the apposition of target particles onto adherent macrophages by centrifugation (Figure S4G), or gravitationally by increasing the size of the particles (Figure 4E,F,H) overcomes a barrier formed by the glycocalyx. Because target centrifugation has been often used to synchronize phagocytosis and to increase its efficiency, physiological barriers to phagocytosis may be underappreciated. Secondly, the active surveillance mechanisms of macrophages may also facilitate particle engagement, by generating outward force via cytoskeletal rearrangements. It is also likely that the structure of the glycocalyx may be different at the tips of the pseudopodial extensions used by phagocytes to probe their environment. Lastly, it must be borne in mind that barriers to phagocytosis are dynamic. Upon inflammatory insults, cells undergo extensive remodelling that involves the breakdown, cross-linking, and general restructuring of the glycocalyx [16]. We would predict these events to coincide with changes in phagocytic ability. Moreover, once innate immune cells complete surveillance and target ingestion and become antigen-presenting cells, they switch from a phagocytic program to a migratory one. Remodelling of the phagocyte glycocalyx may serve as a mechanism to influence these behaviours.
STAR ★ METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Spencer Freeman (spencer.freeman@sickkids.ca).
Materials Availability
The plasmids generated in this study are available on request. Other reagents can also be made available on request.
Data and Code Availability
This study did not generate unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mouse protocols were approved by the Animal Care Committee at the Hospital for Sick Children. 6–12 week-old C57BL/6 mice were bred in-house. Males and females were used. For each n, mice were age and sex matched or experiments were done from macrophages derived from the same mouse.
Primary cells and cell lines
Primary bone marrow-derived macrophages (BMDM) were derived from the femoral bones from 6–12-week-old C57BL/6 wild-type mice. Cells were washed before culturing in DMEM with L-glutamine containing 10% heat-inactivated FBS (Wisent) and 100 U/mL penicillin and 100 mg/mL streptomycin. BMDM were seeded and differentiated on Petri dishes using murine M-CSF (PeproTech) at 0.1 μg/mL for 5–7 days at 37°C, 5% CO2.
Primary human monocytes were isolated from heparinized blood of healthy donors aged 20–35 who provided informed consent, following a protocol approved by the Research Ethics Board of The Hospital for Sick Children. Peripheral blood mononuclear cells were first isolated using Lympholyte-H (Cedarlane), resuspended in RPMI, and seeded onto tissue culture-treated plastic dishes for 30 min to select adherent cells. Non-adherent cells were removed by washing with RPMI and adherent cells were then incubated in RPMI with L-glutamine containing 10% heat-inactivated serum, 100 U/mL penicillin, 100 mg/mL streptomycin, and 10 ng/mL hM-CSF (PeproTech) for 5–7 days.
Primary B lymphocytes were isolated from the spleens of 6–12-week-old C57BL/6 wild-type mice by negative selection with an EasySep Mouse B Cell Isolation Kit (StemCell, cat. # 19854A) and used immediately.
COS-1 fibroblasts stably expressing human FcγRIIA were generated and cultured as previously described (52). RAW 264.7 cells obtained from American Type Culture Collection were grown in DMEM with 10% heat-inactivated FBS. RAW 264.7 stably expressing HA-Dectin-1 were described previously (49).
MCF-7 and SK-OV-3 cell lines were grown in DMEM with 10% heat-inactivated FBS.
Microbe strains
C. albicans strains (WT, ΔMNN4, and ΔMNN sextuple) were generously provided by Drs. Leah E. Cowen and Neil A. R. Gow and are described previously (36). The yeast were grown in YPD Broth or agar media at 30°C, supplemented with 100 ug/mL of uridine. The E.coli DH5α strain used in this study was cultured in LB Broth or agar media at 37°C.
METHOD DETAILS
Particle binding and phagocytosis
Macrophages, adhered to uncoated 18 mm coverglass for 24–48 h, were incubated with 500 μL healthy bovine synovial fluid (LAMPIRE Biological Laboratories, cat. # 8600853) on ice for 20 min. Where indicated, the synovial fluid was pretreated with hyaluronidase isolated from bovine testes (Sigma, cat. # H3506–100MG) used at 20 μg/mL (10 units/mL) for 30 min at 37°C before addition to the macrophages. In experiments using exogenous hyaluronan, cells were incubated with 5 mg/mL of either high (1–1.5 million Da) or low (<50 kDa; Lotioncrafter) molecular weight forms of the glycosaminoglycan on ice for 20 min. Where indicated, neuraminidase (α2–3,6,8) from New England Biolabs (cat. # P0720L) was used at 500 units/mL for 30 min at 37°C in DMEM.
Bacterial-derived mucinase (StcE) was expressed and His-tag purified as previously described (40). To remove endotoxin, purified protein was first treated using High Capacity Endotoxin Removal Spin Columns (Pierce, cat. # 88274) following manufacturer’s instructions. A second round of endotoxin removal was performed using EndoTrap® HD endotoxin removal columns (Hyglos, cat. # 800063) following manufacturer instructions. Column eluate was filter-sterilized using a 0.2 μm Acrodisc syringe filter (VWR cat. # 28143–310), then diluted to 2 mg/mL in sterile PBS. Endotoxin levels were confirmed to be < 5 EU/mL using the HEK-Blue™ LPS Detection Kit (InVivoGen, cat. # rep-lps2). Tumor cells were treated with 5 μg/mL of the enzyme for 1.5 h in DMEM or PBS.
In all cases, particle binding and phagocytosis of targets was done as previously described (23). Briefly, IgG-opsonized polystyrene microspheres (3.8 μm, Bangs Laboratories Inc., cat. # PS05003) were opsonized with human IgG from human serum (Sigma, cat. # I4506) at 2 mg/mL. Sheep erythrocytes (MP Biomedicals, cat. # 55876) were opsonized with rabbit anti-sheep red blood cell antibodies (MP Biomedicals, cat. # 55806) at 0.5 mg/mL. E. coli (DH5α) were grown overnight, centrifuged and opsonized with anti-E. coli antibodies (EastCoast Bio, cat. # J124). Experiments where macrophages were challenged with C. albicans were done using a culture of either WT, ΔMNN4 or ΔMNN sextuple strains of yeast grown at 30°C to an OD600 of 1 in YPD broth supplemented with 100 μg/mL of uridine to prevent the formation of hyphae. MCF-7 and SK-OV-3 cells were lifted and fixed with 4 μg/mL Alexa 546-conjugated maleimide (Molecular Probes, cat. # A-10258) and 5 mM N-ethylmaleimide for 20 min at room temperature. Cells were subsequently opsonized with anti-CD47 antibodies (BioXCell, cat. # BE0283) at 20 μg/mL for 45 min. All particles were added to the macrophages for indicated times (≈10–20 min) at 37°C before fixation with 4% paraformaldehyde except in the case of MCF-7 cells which were added for ≈5 min.
Zeta potential determinations
BMDM were lifted into suspension with 5 mM EDTA in PBS. Suspended cells were incubated with 5 mg/mL of either high or low molecular weight hyaluronan for 20 min on ice that had been pretreated or not with 20 μg/mL (10 units/mL) of hyaluronidase. Cells were then washed and treated with 1 mM of N-ethylmaleimide at RT for 20 min. Karyocytes were generated from intact MCF-7 or SK-OV-3 cells as described previously (39). Briefly, cells were lifted into suspension with 5 mM EDTA in PBS and gentle scraping. Cells were centrifuged and resuspended in warmed 12% Ficoll 70 solution diluted in PBS containing 1 μM of Latrunculin A (LatA) (ThermoFisher scientific, cat. # L12370). The cell suspension was layered on a pre-warmed discontinuous gradient of 20% to 80% Ficoll 70 also mixed in PBS and 1 μM LatA before centrifugation at 70,000 G for 30 min at 37°C. The karyocyte fraction, found at the bottom of the Ficoll layer, was subsequently resuspended, allowed to recover in warm DMEM, and then fixed with 1 mM of N-ethylmaleimide for 20 min. All other particle suspensions were used directly. In all cases, these suspensions were transferred into a Capillary Zeta Cell (Malvern), and subsequently measured using a Zetasizer Nano ZS (Malvern). For cells, karyoplasts, and bacteria, 15 to 30 charge pulses were used to determine the mean Zeta potential. For beads, ≈100 pulses were used.
Macropinocytosis
RAW 264.7 cells were incubated with 3–5 μL (1:500) of neutral 70 kDa TMR-Dextran and 10 ng/mL of LPS, in culture medium. Cells were incubated at 37°C for 20 min and washed with HBSS before imaging. The summed fluorescence intensity per cell was determined using Volocity software (Perkin-Elmer).
Peritoneal infection with C. albicans
Macrophages were elicited to the peritoneal cavity of C57BL/6 with a single injection of 5 mM periodate in a saline solution > 24 h before infection with C. albicans. The yeast strains were grown overnight, washed and resuspended in PBS at a concentration of 1×106 yeast per mL, and then 500 μL was injected for 1 h. The peritoneal cavity was lavaged and cell solutions were washed and fixed. Macrophages were stained with APC-F4/80 (Biolegend, cat # 123116) and C. albicans were stained with calcofluor white (Sigma, cat # 18909).
Additional Reagents
For Western blotting analysis, adherent macrophages that had been serum-starved overnight were incubated for 3 h with 50 μM of the NSC-87877 SHP1/2 inhibitor (Millipore Sigma, cat. # 565851), neuraminidase with or without polymyxin B (Sigma, cat. # P1004) for 30 min, or aggregated IgG (Sigma, cat. # I4506) for 20 min before lysis with RIPA lysis buffer. 4G10 anti-phosphotyrosine was from Millipore Sigma and used at 1:2000. Anti-α-tubulin was from Millipore Sigma and used at 1:1000. Antibodies were incubated with membranes overnight at 4°C in blocking buffer (3% BSA in TBST). Rabbit anti-mouse conjugated with HRP (Abcam, cat. # ab6728) was used at 1:5000 for 30 min at room temperature.
For staining cells, Alexa 488- or 647-labeled wheat germ agglutinin (Invitrogen) or Cy5-labeled Sambucus nigra agglutinin (Vector Laboratories, Inc, cat. # CL-1305) or Alexa 488-Peanut agglutinin (Molecular Probes, cat. # L21409), were added to the cells at 1:200. Rhodamine or Alexa 488-phalloidin (Invitrogen) were used at 1:500. DRAQ5 (BioStatus, cat. # DR50050) for nuclear staining was used at 1:1000. Calcofluor white was from Millipore Sigma and used at 1:500. Concanavalin A was from Millipore Sigma and used at 1:500. Mouse anti-human CD43 was from STEMCELL was used at 1:1000. Conjugated secondary antibodies including Cy3-, or Alexa 647-conjugated donkey anti-mouse, anti-rabbit, anti-goat or anti-human (Jackson ImmunoResearch) were each used at 1:500. Peritoneal macrophages were stained with APC-F4/80 at 1:300.
Streptavidin beads were made by incubating 3.8 μm polystyrene microspheres (Bangs Laboratories, Inc., cat. # PS06005) with 120 μg/mL streptavidin (ThermoFisher, cat. # S32351) for 45 min at 37°C with shaking. To then functionalize the streptavidin coated beads, they were first washed and resuspended with rabbit anti-avidin (Rockland Immunochemicals, Inc., cat. # 100–4197) at 1.6 mg/mL in PBS for 45 min at 37°C with vigorous shaking. Beads were then washed and resuspended in PBS, divided and incubated with 250 μg/mL biotinylated-poly-L-lysine (NANOCS, cat. # PL1-BN-1) or 400 μg/mL biotinylated-heparin (EMD Millipore, cat. # 375054). Alternatively, beads were incubated with 12.5 mg/mL biotin, biotinylated 550 Da PEG, or biotinylated 40 kDa PEG (Creative PEGWorks, cat. # PLS-2057, PSB-2050).
Particle size determination
The size of B lymphocytes, whole human cancer lines or karyocytes, and yeast strains, was determined using a Coulter counter (Beckman Coulter) as previously described [48].
Transfection
RAW 264.7 cells were transfected with FuGeneHD (Promega); COS-1 cells with FuGene6 (Promega) as per the manufacturer’s protocol. Generally, cells were imaged 16 h after transfection.
Plasmid DNA
CD44-GFP and HAS3-GFP were previously described (23, 51). The CD43-GFP plasmid was generated by PCR amplification of human CD43 using the forward primer 5′ gatctcgagctcaagcttcgatggccacgcttctccttc and reverse primer 5′ gtaccgtcgactgcagaattggaggggcagccccgtctcc, and insertion into EcoRI-digested pEGFPN1 using Gibson Assembly (E2611L, New England Biolabs). The plasmid was sequence-verified prior to use. Syndecan-1-GFP was purchased from Sinobiological (cat. # HG11429-ACG). LifeAct-RFP was from Michael Davidson (Addgene plasmid #54586).
mRNA expression
Quantitative reverse-transcription PCR was used to determine relative mRNA levels in macrophages. Where stated, BMDM were stimulated with LPS at 1 μg/mL overnight. RNA extraction was done using a GeneJET RNA Purification Kit (ThermoFisher Scientific). cDNA was made using SuperScript™ VILO™ (ThermoFisher Scientific) reverse transcriptase. Taq-man probes used for CD22, CD33, Siglec-E, Siglec-F, Siglec-G, and Abt1 were from Thermo Scientific.
Immunofluorescence
Cells were fixed with 4% PFA (Electron Microscopy Sciences) for 10–20 min at RT. Where specified, cells were then permeabilized with 0.5% Triton X-100 in PBS for 5–15 min, blocked with 2% BSA in PBS, and stained with the indicated antibodies or dyes in blocking solution. After staining, coverslips were mounted onto glass slides using ProLong Diamond mounting medium (ThermoFisher scientific). For surface staining of CD16/CD32, transfected RAW 264.7 cells were stained without permeabilization and the mean fluorescence intensity of the receptors per cell, with the background subtracted, was determined using Volocity software (Perkin-Elmer).
Microscopy and image analysis
Imaging was performed using a Quorum spinning disc mounted on a Zeiss Axiovert 200M microscope, using 63x or 25x objectives, and a back-thinned EM-CCD camera (C9100–13, Hamamatsu), or performed using an EVOS Cell Imaging System (ThermoFisher) with a 20x objective. Acquisitions were controlled by the Volocity software (Perkin-Elmer), exported, analysed and quantified using Volocity or Fiji (NIH) software.
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of experiments and cells quantified are indicated in individual Figure Legends. Briefly, each experiment was performed with macrophages derived from different animals or human donors (each being one n). For macrophage cell lines used, each n represents their use on different passage numbers. Phagocytic targets were also made on different days for tumor cell lines, from different clones for bacteria and yeast, and from different mice in the case of B cells. Quantification for binding and phagocytic indices was done by acquiring >5 fields of cells using a 25x lens (≈5–20 cells per field on the Zeiss Axiovert 200M which contains a x2 custom lens and ≈10–40 cells per field on the EVOS system). To express normalized binding indices, control samples were first determined and each field from the experimental conditions were compared and expressed as a fold difference from the mean of the control. A Mann-Whitney test was used to determine p values between populations of single cell measurements. Otherwise, one-way ANOVA test was used to compare multi-population sets.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-sheep red blood cell | MP Biomedicals | cat. # 55806 |
| IgG from human serum | Sigma | cat. # I4506 RRID:AB_1163606 |
| Goat anti-E. coli antibodies | EastCoast Bio | cat. # J124 |
| Mouse anti-CD47 | BioXCell | cat. # BE0283 RRID:AB_2687806 |
| Mouse anti-phosphotyrosine, clone 4G10 | Millipore Sigma | cat. # 05–321 RRID:AB_309678 |
| Mouse anti-α-tubulin | Millipore Sigma | cat. # T6199 RRID:AB_477583 |
| Rabbit anti-mouse conjugated with HRP | Abcam | cat. # ab6728 RRID:AB_955440 |
| Mouse anti-human CD43, clone CD43–10G7 | STEMCELL | cat. # 60085 |
| Rat anti-mouse CD16/CD32, clone 2.4G2 | BD Biosciences | cat. # 553141 RRID:AB_394656 |
| Donkey anti-mouse Cy3 | Jackson ImmunoResearch | Code: 715-165-150 RRID: AB_2340813 |
| Donkey anti-mouse Alexa 647 | Jackson ImmunoResearch | Code: 715-605-150 RRID: AB_2340862 |
| Donkey anti-rabbit Cy3 | Jackson ImmunoResearch | Code: 711-165-152 RRID: AB_2307443 |
| Donkey anti-rabbit Alexa 647 | Jackson ImmunoResearch | Code: 711-605-152 RRID: AB_2492288 |
| Donkey anti-goat Cy3 | Jackson ImmunoResearch | Code: 705-165-003 RRID: AB_2340411 |
| Donkey anti-goat Alexa 647 | Jackson ImmunoResearch | Code: 705-605-003 RRID: AB_2340436 |
| Donkey anti-human Cy3 | Jackson ImmunoResearch | Code: 709-165-149 RRID: AB_2340535 |
| Donkey anti-human Alexa 647 | Jackson ImmunoResearch | Code: 709-605-149 RRID: AB_2340578 |
| Rabbit anti-avidin | Rockland Immunochemicals, Inc. | cat. # 100–4197 RRID:AB_219937 |
| Bacterial and Virus Strains | ||
| E.coli DH5α | ThermoFisher Scientific | cat. # 18265017 |
| Biological Samples | ||
| Bovine synovial fluid | LAMPIRE Biological Laboratories | cat. # 8600853 |
| Sheep erythrocytes | MP Biomedicals | cat. # 55876 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NSC-87877 SHP1/2 inhibitor | Millipore Sigma | cat. # 565851 |
| α2–3,6,8 Neuraminidase | New England BioLabs | cat. # P0720L |
| Polymyxin B | Sigma | cat. # P1004 |
| Alexa 488-labeled wheat germ agglutinin | Invitrogen | cat. # W11261 |
| Alexa 647-labeled wheat germ agglutinin | Invitrogen | cat. # W32466 |
| Cy5-labeled Sambucus Nigra agglutinin | Vector Laboratories, Inc, | cat. # CL-1305 |
| Alexa 488-Peanut agglutinin | Molecular Probes | cat. # L21409 RRID:AB_2315178 |
| Rhodamine-phalloidin | Invitrogen | cat. # R415 RRID:CVCL_R415 |
| Alexa 488-phalloidin | Invitrogen | cat. # A12379 |
| DRAQ5 | BioStatus | cat. # DR50050 RRID:AB_2314341 |
| Calcofluor white stain | Millipore Sigma | cat. # 18909 |
| Concanavalin A FITC conjugate | Millipore Sigma | cat. # C7642 |
| Streptavidin | ThermoFisher | cat. # S32351 |
| Biotinylated-poly-L-lysine | NANOCS | cat. # PL1-BN-1 |
| Biotinylated-heparin | EMD Millipore | cat. # 375054 |
| Biotinylated 550 Da PEG | Creative PEGWorks | cat. # PLS-2057 |
| Biotinylated 40 kDa PEG | Creative PEGWorks | cat. # PSB-2050 |
| Hyaluronidase | Sigma | cat. # H3506 |
| Purified hyaluronan Low MW (<50 kDa) | Lotioncrafter | cat. # 9067-32-7 |
| Purified hyaluronan High MW (1–1.5 million Da) | Lotioncrafter | cat. # 9067-32-7 |
| Bacterial-derived mucinase (StcE) | Source for this enzyme [40] | N/A |
| Alexa 546-conjugated maleimide | Molecular Probes | cat. # A-10258 |
| Latrunculin A | ThermoFisher scientific | cat. # L12370 |
| Dextran, Tetramethylrhodamine (70 kDa) | ThermoFisher scientific | cat. # D1818 |
| APC- F4/80 | Biolegend | cat # 123116 RRID:AB_893481 |
| ProLong™ Diamond Antifade Mountant | ThermoFisher scientific | cat # P36970 |
| Critical Commercial Assays | ||
| EasySep Mouse B Cell Isolation Kit | StemCell | cat. # 19854A |
| High Capacity Endotoxin Removal Spin Columns | Pierce | cat. # 88274 |
| EndoTrap® HD endotoxin removal columns | Hyglos | cat. # 800063 |
| HEK-Blue™ LPS Detection Kit | InVivoGen | cat. # rep-lps2 |
| FuGeneHD | Promega | cat. # E2311 |
| FuGene6 | Promega | cat. # E2691 |
| GeneJET RNA Purification Kit | ThermoFisher Scientific | cat. # K0731 |
| SuperScript™ VILO™ cDNA Synthesis Kit | ThermoFisher Scientific | cat. # 11754050 |
| Experimental Models: Cell Lines | ||
| Mouse : RAW 264.7 | American Type Culture Collection | ATCC® TIB-71™ RRID:CVCL_0493 |
| Mouse : RAW 264.7, stably expressing HA-Dectin-1 | Source for these cells [49] | N/A |
| Primate : COS-1, stably expressing human FcγRIIA | Source for these cells [50] | N/A |
| Human : MCF-7 | American Type Culture Collection | ATCC® CRL-3435™ RRID:CVCL_0031 |
| Human : SK-OV-3 | American Type Culture Collection | ATCC® HTB-77™ RRID:CVCL_0532 |
| Experimental Models: Organisms/Strains | ||
| C. albicans, WT | Kind gift from Drs. Leah E. Cowen and Neil A. R. Gow. Source for this strain [36] | N/A |
| C. albicans, ΔMNN4 | Kind gift from Drs. Leah E. Cowen and Neil A. R. Gow. Source for this strain [36] | N/A |
| C. albicans, ΔMNN sextuple | Kind gift from Drs. Leah E. Cowen and Neil A. R. Gow. Source for this strain [36] | N/A |
| Mice, C57BL/6 | Jackson | RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| PCR amplification of human CD43, forward primer 5’ gatctcgagctcaagcttcgatggccacgcttctccttc | This paper | N/A |
| PCR amplification of human CD43, reverse primer 5’ gtaccgtcgactgcagaattggaggggcagccccgtctcc | This paper | N/A |
| Taq-man mouse probe CD22 | ThermoFisher scientific | cat. # 4331182 |
| Taq-man mouse probe CD33 | ThermoFisher scientific | cat. # 4331182 |
| Taq-man mouse probe Siglec-E | ThermoFisher scientific | cat. # 4331182 |
| Taq-man mouse probe Siglec-F | ThermoFisher scientific | cat. # 4331182 |
| Taq-man mouse probe Siglec-G | ThermoFisher scientific | cat. # 4331182 |
| Taq-man mouse probe Abt1 | ThermoFisher scientific | cat. # 4331182 |
| Recombinant DNA | ||
| CD44-GFP | Source for this plasmid [23] | N/A |
| HAS3-GFP | Source for this plasmid [51] | N/A |
| CD43-GFP | This study | N/A |
| Syndecan-1-GFP | Sinobiological | cat. # HG11429-ACG |
| LifeAct-RFP | Kindly provide from Michael Davidson. | Addgene plasmid #54586 RRID:Addgene_54586 |
| Software and Algorithms | ||
| Volocity software | Perkin-Elmer | https://www.quorumtechnologies.com/volocity |
| FIJI | NIH software | https://imagej.net/Fiji/Downloads |
| Other | ||
| Polystyrene microspheres 3.8–4 μm | Bangs Laboratories, Inc. | cat. # PS06005 |
| Acrodisc syringe filter | VWR | cat. # 28143–310 |
| Capillary Zeta Cell | Malvern | cat. # DTS1070 |
Highlights.
Large hyaluronans in synovial fluid obstruct particle binding by macrophages
The glycocalyx contributes to negative surface potential of macrophages
Macrophages selectively bind targets according to their charge
Bulky transmembrane mucins of tumors override antibody blockade of CD47-SIRPα
ACKNOWLEDGMENTS
P.I. was supported by an EMBO Postdoctoral Fellowship (ALTF 36-2018). Supported by Canadian Institutes of Health Research grants FDN-143202 (to S.G.) and PJT-169180 (to S.F.) and U.S. National Cancer Institute Grant R01CA227942 (to C.R.B.). K.P. was supported by a U.S. National Science Foundation Graduate Research Fellowship, a Stanford Graduate Fellowship, and the Stanford ChEM-H Chemistry/Biology Interface Predoctoral Training Program. We thank Dr. Warren Chan for reagents and use of his dynamic light scattering system and Dr. Calvin Roskelley for providing SK-OV-3 cells and for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
C.R.B is a co-founder and Scientific Advisory Board member of Lycia Therapeutics, Palleon Pharmaceuticals, Enable Bioscience, Redwood Biosciences (a subsidiary of Catalent) and InterVenn Biosciences, and a member of the Board of Directors of Eli Lilly & Company.
References
- 1.Flannagan RS, Jaumouille V, and Grinstein S (2012). The cell biology of phagocytosis. Annu Rev Pathol 7, 61–98. [DOI] [PubMed] [Google Scholar]
- 2.Arandjelovic S, and Ravichandran KS (2015). Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, and Weissman IL (2019). Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 19, 568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. (2017). PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, et al. (2013). Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A 110, 11103–11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies LC, Jenkins SJ, Allen JE, and Taylor PR (2013). Tissue-resident macrophages. Nat Immunol 14, 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravichandran KS (2010). Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 207, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown GD, Willment JA, and Whitehead L (2018). C-type lectins in immunity and homeostasis. Nat Rev Immunol 18, 374–389. [DOI] [PubMed] [Google Scholar]
- 9.Freeman SA, and Grinstein S (2014). Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev 262, 193–215. [DOI] [PubMed] [Google Scholar]
- 10.Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, Krishnan V, Hatakeyama J, Dorigo O, Barkal LJ, et al. (2019). CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrissey MA, Kern N, and Vale RD (2020). CD47 Ligation Repositions the Inhibitory Receptor SIRPA to Suppress Integrin Activation and Phagocytosis. Immunity 53, 290–302 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nimmerjahn F, and Ravetch JV (2008). Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8, 34–47. [DOI] [PubMed] [Google Scholar]
- 13.Alphonsus CS, and Rodseth RN (2014). The endothelial glycocalyx: a review of the vascular barrier. Anaesthesia 69, 777–784. [DOI] [PubMed] [Google Scholar]
- 14.Henry CB, and Duling BR (1999). Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am J Physiol 277, H508–514. [DOI] [PubMed] [Google Scholar]
- 15.Carlsson SR, Roth J, Piller F, and Fukuda M (1988). Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Major sialoglycoproteins carrying polylactosaminoglycan. J Biol Chem 263, 18911–18919. [PubMed] [Google Scholar]
- 16.Day AJ, and de la Motte CA (2005). Hyaluronan cross-linking: a protective mechanism in inflammation? Trends Immunol 26, 637–643. [DOI] [PubMed] [Google Scholar]
- 17.Petrey AC, and de la Motte CA (2014). Hyaluronan, a crucial regulator of inflammation. Front Immunol 5, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forrester JV, and Balazs EA (1980). Inhibition of phagocytosis by high molecular weight hyaluronate. Immunology 40, 435–446. [PMC free article] [PubMed] [Google Scholar]
- 19.Bakalar MH, Joffe AM, Schmid EM, Son S, Podolski M, and Fletcher DA (2018). Size-Dependent Segregation Controls Macrophage Phagocytosis of Antibody-Opsonized Targets. Cell 174, 131–142 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee HG, and Cowman MK (1994). An agarose gel electrophoretic method for analysis of hyaluronan molecular weight distribution. Anal Biochem 219, 278–287. [DOI] [PubMed] [Google Scholar]
- 21.Decker B, Mc GW, Mc KB, and Slocumb CH (1959). Concentration of hyaluronic acid in synovial fluid. Clin Chem 5, 465–469. [PubMed] [Google Scholar]
- 22.Cowman MK, Lee HG, Schwertfeger KL, McCarthy JB, and Turley EA (2015). The Content and Size of Hyaluronan in Biological Fluids and Tissues. Front Immunol 6, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freeman SA, Vega A, Riedl M, Collins RF, Ostrowski PP, Woods EC, Bertozzi CR, Tammi MI, Lidke DS, Johnson P, et al. (2018). Transmembrane Pickets Connect Cyto- and Pericellular Skeletons Forming Barriers to Receptor Engagement. Cell 172, 305–317 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arias-Alpizar G, Kong L, Vlieg RC, Rabe A, Papadopoulou P, Meijer MS, Bonnet S, Vogel S, van Noort J, Kros A, et al. (2020). Light-triggered switching of liposome surface charge directs delivery of membrane impermeable payloads in vivo. Nat Commun 11, 3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharjee S (2016). DLS and zeta potential - What they are and what they are not? J Control Release 235, 337–351. [DOI] [PubMed] [Google Scholar]
- 26.Kress H, Stelzer EH, Holzer D, Buss F, Griffiths G, and Rohrbach A (2007). Filopodia act as phagocytic tentacles and pull with discrete steps and a load-dependent velocity. Proc Natl Acad Sci U S A 104, 11633–11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flannagan RS, Harrison RE, Yip CM, Jaqaman K, and Grinstein S (2010). Dynamic macrophage “probing” is required for the efficient capture of phagocytic targets. J Cell Biol 191, 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Angsana J, Chen J, Smith S, Xiao J, Wen J, Liu L, Haller CA, and Chaikof EL (2015). Syndecan-1 modulates the motility and resolution responses of macrophages. Arterioscler Thromb Vasc Biol 35, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sarrazin S, Lamanna WC, and Esko JD (2011). Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meesmann HM, Fehr EM, Kierschke S, Herrmann M, Bilyy R, Heyder P, Blank N, Krienke S, Lorenz HM, and Schiller M (2010). Decrease of sialic acid residues as an eat-me signal on the surface of apoptotic lymphocytes. J Cell Sci 123, 3347–3356. [DOI] [PubMed] [Google Scholar]
- 31.Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, Direnzo D, Nanda V, Ye J, Connolly AJ, et al. (2016). CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kass EH, and Seastone CV (1944). The Role of the Mucoid Polysaccharide (Hyaluronic Acid) in the Virulence of Group a Hemolytic Streptococci. J Exp Med 79, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wessels MR, Moses AE, Goldberg JB, and DiCesare TJ (1991). Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proc Natl Acad Sci U S A 88, 8317–8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erwig LP, and Gow NA (2016). Interactions of fungal pathogens with phagocytes. Nat Rev Microbiol 14, 163–176. [DOI] [PubMed] [Google Scholar]
- 35.Brown GD (2006). Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol 6, 33–43. [DOI] [PubMed] [Google Scholar]
- 36.Hall RA, Bates S, Lenardon MD, Maccallum DM, Wagener J, Lowman DW, Kruppa MD, Williams DL, Odds FC, Brown AJ, et al. (2013). The Mnn2 mannosyltransferase family modulates mannoprotein fibril length, immune recognition and virulence of Candida albicans. PLoS Pathog 9, e1003276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SY, and Gordon S (2002). Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med 196, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, et al. (2014). The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511, 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grinstein S, Elder B, and Furuya W (1985). Phorbol ester-induced changes of cytoplasmic pH in neutrophils: role of exocytosis in Na+-H+ exchange. Am J Physiol 248, C379–386. [DOI] [PubMed] [Google Scholar]
- 40.Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, Yu J, Woods EC, Kramer JR, Westerlind U, et al. (2019). The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc Natl Acad Sci U S A 116, 7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pluvinage JV, Haney MS, Smith BAH, Sun J, Iram T, Bonanno L, Li L, Lee DP, Morgens DW, Yang AC, et al. (2019). CD22 blockade restores homeostatic microglial phagocytosis in ageing brains. Nature 568, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, and Noble PW (1996). Hyaluronan (HA) fragments induce chemokine gene expression in alveolar macrophages. The role of HA size and CD44. J Clin Invest 98, 2403–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong Y, Arif A, Olsson M, Cali V, Hardman B, Dosanjh M, Lauer M, Midura RJ, Hascall VC, Brown KL, et al. (2016). Endotoxin free hyaluronan and hyaluronan fragments do not stimulate TNF-alpha, interleukin-12 or upregulate co-stimulatory molecules in dendritic cells or macrophages. Sci Rep 6, 36928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee-Sayer SS, Dong Y, Arif AA, Olsson M, Brown KL, and Johnson P (2015). The where, when, how, and why of hyaluronan binding by immune cells. Front Immunol 6, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mylvaganam SM, Grinstein S, and Freeman SA (2018). Picket-fences in the plasma membrane: functions in immune cells and phagocytosis. Semin Immunopathol 40, 605–615. [DOI] [PubMed] [Google Scholar]
- 46.Mylvaganam S, Riedl M, Vega A, Collins RF, Jaqaman K, Grinstein S, and Freeman SA (2020). Stabilization of Endothelial Receptor Arrays by a Polarized Spectrin Cytoskeleton Facilitates Rolling and Adhesion of Leukocytes. Cell Rep 31, 107798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Randolph GJ, Ivanov S, Zinselmeyer BH, and Scallan JP (2017). The Lymphatic System: Integral Roles in Immunity. Annu Rev Immunol 35, 31–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Freeman SA, Uderhardt S, Saric A, Collins RF, Buckley CM, Mylvaganam S, Boroumand P, Plumb J, Germain RN, Ren D, et al. (2020). Lipid-gated monovalent ion fluxes regulate endocytic traffic and support immune surveillance. Science 367, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Esteban A, Popp MW, Vyas VK, Strijbis K, Ploegh HL, and Fink GR (2011). Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc Natl Acad Sci U S A 108, 14270–14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Downey GP, Botelho RJ, Butler JR, Moltyaner Y, Chien P, Schreiber AD, and Grinstein S (1999). Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcgammaRIIA receptors. J Biol Chem 274, 28436–28444. [DOI] [PubMed] [Google Scholar]
- 51.Rilla K, Siiskonen H, Spicer AP, Hyttinen JM, Tammi MI, and Tammi RH (2005). Plasma membrane residence of hyaluronan synthase is coupled to its enzymatic activity. J Biol Chem 280, 31890–31897. [DOI] [PubMed] [Google Scholar]
- 52.Downey GP, Botelho RJ, Butler JR, Moltyaner Y, Chien P, Schreiber AD, and Grinstein S (1999). Phagosomal maturation, acidification, and inhibition of bacterial growth in nonphagocytic cells transfected with FcgammaRIIA receptors. J Biol Chem 274, 28436–28444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate unique datasets or code.