

Abstract
Background:
Traditional methods for disease risk prediction and assessment, such as diagnostic tests using serum, urine, blood, saliva or imaging biomarkers, have been important for identifying high-risk individuals for many diseases, leading to early detection and improved survival. For pancreatic cancer, traditional methods for screening have been largely unsuccessful in identifying high-risk individuals in advance of disease progression leading to high mortality and poor survival. Electronic health records (EHR) linked to genetic profiles provide an opportunity to integrate multiple sources of patient information for risk prediction and stratification. We leverage a constellation of temporally associated diagnoses available in the EHR to construct a summary risk score, called a phenotype risk score (PheRS), for identifying individuals at high-risk for having pancreatic cancer. The proposed PheRS approach incorporates the time with respect to disease onset into the prediction framework. We combine and contrast the PheRS with more well-known measures of inherited susceptibility, namely, the polygenic risk scores (PRS) for prediction of pancreatic cancer.
Methodology:
We first calculated pairwise, unadjusted associations between pancreatic cancer diagnosis and all possible other diagnoses across the medical phenome. We call these pairwise associations co-occurrences. After accounting for cross-phenotype correlations, the multivariable association estimates from a subset of relatively independent diagnoses were used to create a weighted sum PheRS. We constructed time-restricted risk scores using data from 38,359 participants in the Michigan Genomics Initiative (MGI) based on the diagnoses contained in the EHR at 0, 1, 2, and 5 years prior to the target pancreatic cancer diagnosis. The PheRS was assessed for predictability in the UK Biobank (UKB). We tested the relative contribution of PheRS when added to a model containing a summary measure of inherited genetic susceptibility (PRS) plus other covariates like age, sex, smoking status, drinking status, and body mass index (BMI).
Results:
Our exploration of co-occurrence patterns identified expected associations while also revealing unexpected relationships that may warrant closer attention. Solely using the pancreatic cancer PheRS at 5 years before the target diagnoses yielded an AUC of 0.60 (95% CI = [0.58, 0.62]) in UKB. A larger predictive model including PheRS, PRS, and the covariates at the 5-year threshold achieved an AUC of 0.74 (95% CI = [0.72, 0.76]) in UKB. We note that PheRS does contribute independently in the joint model. Finally, scores at the top percentiles of the PheRS distribution demonstrated promise in terms of risk stratification. Scores in the top 2% were 10.20 (95% CI = [9.34, 12.99]) times more likely to identify cases than those in the bottom 98% in UKB at the 5-year threshold prior to pancreatic cancer diagnosis.
Conclusions:
We developed a framework for creating a time-restricted PheRS from EHR data for pancreatic cancer using the rich information content of a medical phenome. In addition to identifying hypothesis-generating associations for future research, this PheRS demonstrates a potentially important contribution in identifying high-risk individuals, even after adjusting for PRS for pancreatic cancer and other traditional epidemiologic covariates. The methods are generalizable to other phenotypic traits.
Keywords: Electronic health records, EHR, Michigan Genomics Initiative, pancreatic cancer, risk prediction, UK Biobank
Graphical Abstract
1. Introduction
Identifying individuals or sub-groups who are at increased risk of developing an adverse health condition is an overarching goal of public health. [1–4] Many successful screening tests and diagnostic procedures exist, particularly for cancers such as breast, colorectal, and cervical [5–7] for early detection and targeted prevention. For example, it is estimated that Pap smear screening reduced cervical cancer incidence roughly in half from 1976 to 2009 in the US (from 9.8 to 4.9 and from 5.3 to 3.7 per 100,000 for early-and late-stage diagnoses, respectively). [8] Identified high-risk individuals can initiate targeted primary prevention strategies, including chemoprevention, lifestyle changes, and sometimes prophylactic surgical interventions. In addition, such individuals can be screened more frequently to detect disease prior to manifestation of symptoms in hopes of altering the clinical course of disease. Increasingly, laboratory and genetic testing are essential tools for identifying high-risk individuals so that interventions can be initiated at earlier stages of the natural history of disease.
The identification of pathogenic variants and high penetrance genes like BRCA1, BRCA2, and MMR and the development of tumor profiling assays like Oncotype DX® have pioneered the use of genetic information for risk and treatment assessment. [9–11] As costs for genotyping have decreased, [12] large patient sample sets have been collected (e.g., biobanks), [13] and computational and methodological tools have been developed, [14–19] tremendous opportunities have arisen for identifying genetic variant-disease associations. The establishment of genome-wide association studies (GWAS) has enabled methodologies to assess genetic associations for virtually any heritable, common complex trait. Leveraging these data, condition-specific polygenic risk scores (PRS) have emerged as a promising avenue for risk prediction and stratification. PRS summarize an individual’s genetic risk based on identified associations and have demonstrated predictive abilities for common complex diseases. [20] However, some rare conditions such as pancreatic cancer have not yet benefitted from this PRS-based approach to risk stratification.
Pancreatic cancer is a particularly devastating diagnosis. It is increasing in incidence in the United States (from 10.9 per 100,000 people in 1999 to 12.9 per 100,000 people in 2016), [21] often diagnosed at late stage, and has poor survival at a time when improvements for most cancers are evident. [22,23] Due to a lack of curative treatments and suitable screening mechanism, primary prevention focuses on modifiable risk factors for pancreatic cancer including smoking, diet, physical activity, and other lifestyle choices [24–26]. Once diagnosed, the 5-year relative survival ranges from 37% for localized to 3% for distant disease, [27] reflecting the relative effectiveness of the standard surgery followed by chemotherapy and radiation treatment regimen at earlier stages. However, overall 5-year survival is only 9.3% because half of all pancreatic cancer diagnoses occur after the cancer has metastasized. [27] For pancreatic cancer, PRS for risk prediction has not been successful because currently identified GWAS loci only explain 4.1% of the phenotypic variation. [28] Novel methods for risk prediction in pancreatic cancer will improve our ability to identify at-risk individuals and improve outcomes.
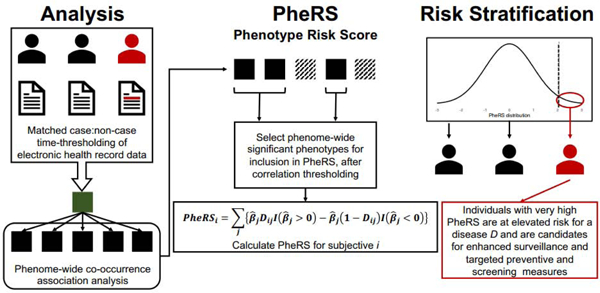
The explosive rise of electronic health record (EHR)-linked biobanks represents an emerging opportunity for the exploration and development of new risk prediction and stratification methods, which can be compared to traditional PRS-based approaches. We propose a new metric called a phenotype risk score (or PheRS), which utilizes time-stamped diagnosis codes recorded in the EHR to generate a summary score and identify individuals with high disease risk. If validated, PheRS could be incorporated into the EHR to help clinicians identify population subgroups that could most benefit from screening and early detection efforts. Our framework opens up a new avenue for “precision prevention” and looks for unexpected clues towards targeted risk prediction and screening.
In this study, we propose an analytic framework for developing a PheRS using EHR-linked biobank data in order to identify individuals at high-risk of pancreatic cancer. Using data from the Michigan Genomics Initiative (MGI) and the UK Biobank (UKB), we evaluate various PheRS constructs and assess their discriminatory ability, calibration, and accuracy. We also explore the relative contribution of PheRS in models including PRS, age, sex, smoking status, alcohol drinking status, and BMI after adjusting for covariates like length of EHR follow-up, genotyping batch, and the first four principal components of the genotype data. We hypothesize that our proposed PheRS framework can be used to identify potentially novel cross-phenotype associations and provide a meaningful contribution in identifying high-risk individuals, even in the presence of PRS, traditional covariates, and risk factors. Our development focuses on the particular setting of pancreatic cancer, but the methods developed in this paper can be applied for risk stratification across a broad spectrum of diseases.
2. Materials and Methods
2.1. Data
This study uses data from the Michigan Genomics Initiative (MGI) [29,30] and the UK Biobank Study (UKB). [31,32] The MGI cohort, for which recruitment is ongoing, consists of over 65,000 adults who have consented to allow research on both their biospecimens and EHR data, as well as linking their EHR data to national data sources such as medical and pharmaceutical claims data. Participants are recruited while awaiting a diagnostic or interventional procedure either at a preoperative appointment or on the day of their operative procedure at the University of Michigan Health System (Michigan Medicine). The MGI subset used in this analysis consists of 38,359 unrelated patients of inferred, recent European ancestry with available EHR data. [33] Analyses are restricted to patients of European ancestry to assess transportability of PRS across comparable biobank populations. Of particular interest to the current work are the age-stamped (operationalized as days since birth) International Classification of Disease (ICD)9-CM and ICD10-CM codes, which allow for the sequential and temporal analysis of diagnoses for each patient. We do not have dates of diagnoses, which means we cannot identify birth cohorts within our sample but only have the temporal ordering of diagnoses. The granular ICD9-CM and ICD10-CM codes were aggregated into clinically relevant groupings to form phenotype codes, called PheWAS codes or phecodes. [34] Collectively, we refer to all phecodes as the phenome. In our MGI phenome, we defined a total of 1,859 phecodes.
The UKB is a population-based cohort from 22 sites across the United Kingdom and includes over 500,000 individuals aged 40 to 69 years at enrollment for recruitment from 2006–2010. UKB data is accessible to researchers across the globe and include genotype data, ICD9 and ICD10 codes, sex, inferred White British ancestry, kinship estimates down to the third degree, birthyear, genotype array, and precomputed principal components of the genotypes. [35] The UKB cohort used in our analysis represents a subset of 393,092 unrelated individuals of inferred, recent European ancestry. We did not have access to time-stamped ICD code data for UKB, so we were not able to ascertain the time ordering of diagnoses. Instead, we have data on the presence or absence of ICD9 and ICD10 codes in each participant’s entire EHR. In our UKB defined phenome, using a similar ICD-to-phecode mapping as in MGI, we defined 1,686 phecodes.
Available phecodes might differ slightly between UKB and MGI because the underlying ICD code system used by UKB may not be specific enough to define all phecodes (e.g., basal cell carcinoma, a skin cancer subtype, is not available in ICD10 but is present in ICD10-CM). The ICD10-CM are clinical modifications (CM) made to the ICD10 coding system by the Centers for Medicare and Medicaid Services and the National Center for Health Statistics resulting in a more granular classification system. This means many of the US modified codes allow for finer mapping of more phecodes. The UK adopted ICD10 prior to the US adoption of ICD10-CM, which also has implications on clinician coding and subsequent phenotyping. Only the set of 1,683 phecodes present in both MGI and UKB are considered in these analyses.
For all traits, an individual is considered as having the trait if the phecode occurs at least once in their EHR. For analyses considering time-stamped data, the sequence of diagnoses is based on the first appearance of each phecode in the patient’s EHR. The pancreatic cancer phecode is constructed using ICD9 codes 157, 157.1, 157.2, 157.3, 157.4, 157.8, 157.9 and ICD10 codes C25, C25.0, C25.1, C25.2, C25.3, C25.4, C25.7, C25.8, C25.9. An interactive map of all ICD9(-CM) and ICD10(-CM) codes to their corresponding phecode used in this paper is based on a curated version of the phecodes available online from https://www.phewascatalog.org/phecodes and visualized here: https://shiny.sph.umich.edu/ICD_Coding/.
Descriptive statistics on patient characteristics in the MGI and UKB cohorts can be found in Table 1.
TABLE 1.
Description of MGI and UKB cohorts
| MGI (Discovery) | UKB (Validation) | |||
|---|---|---|---|---|
| Analytic cohort size | 38,359 | 393,092 | ||
| Phecodes | 1,859 | 1,686 | ||
| Pancreatic Cancer Cases | Controls | Pancreatic Cancer Cases | Controls | |
| n (%) | 429 (1.12) | 37,930 (98.88) | 659 (0.17) | 392,640 (99.83) |
| Age* | ||||
| Min | 32 | 18 | 52 | 49 |
| Mean | 66.36 | 56.70 | 72.67 | 67.70 |
| Median | 68 | 59 | 74 | 69 |
| Max | 92 | 103 | 82 | 85 |
| Sex | ||||
| Female | 181 (42.19) | 19,960 (52.62) | 307 (46.59) | 212,809 (54.23) |
| Male | 248 (57.81) | 17,970 (47.38) | 352 (53.41) | 179,624 (45.77) |
| Length of follow-up (in years)** | ||||
| Min | 0.05 | 0 | - | |
| Mean | 6.86 | 7.74 | - | |
| Median | 4.44 | 5.42 | 7† | |
| Max | 20.86 | 40.19 | - | |
| Number of unique phecodes | ||||
| Min | 11 | 1 | - | - |
| Mean | 99.59 | 66.74 | - | - |
| Median | 79 | 48 | - | - |
| Max | 495 | 536 | - | - |
Age represents age at last electronic health record encounter.
Follow-up data on UK Biobank participants comes from 413,591 individuals with time-stamped ICD code data. 86,717 individuals have only a single encounter. These data are not stratified by pancreatic cancer case status. This data was requested for the purposes of this comparison and was unavailable to us for our analysis.
Jani BD, Hanlon P, Nicholl BI, et al. Relationship between multimorbidity, demographic factors and mortality: findings from the UK Biobank cohort. BMC Med. 2019;17(1):74. doi:10.1186/s12916-019-1305-x
2.2. Co-occurrence analysis
Prior to constructing PheRS, we assessed the overall covariate-adjusted association between pairs of EHR-based diagnoses (e.g., acute pain and pancreatic cancer), which we will call co-occurrence analysis. This describes the likelihood of a patient to have a given diagnosis in their EHR based on whether they were diagnosed with another disease. For example, a co-occurrence analysis may show that patients with an acute pain diagnosis on their EHR are 1.7 times more likely to have a pancreatic cancer diagnosis than those without an acute pain diagnosis. It does not say whether the acute pain diagnosis preceded or first appeared after the pancreatic cancer diagnosis. The result is a numerical representation of their tendency to co-occur (i.e., both appear) in an individual’s EHR. This analysis was repeated between a target phenotype (in this case pancreatic cancer) and each of the remaining 1,682 phecodes in the phenome (i.e., 1,682 separate regression models). We evaluated the association between pairs of phenotypes using Firth-corrected logistic regression adjusting for sex, age, length of follow-up, genotyping array batch, and the first four principal components of the genotype data, where the log-odds ratio (beta) estimates and saddlepoint approximation p-values (ScoreTest_SPA function from the R package ‘SPAtest’) [36] describe the strength and significance of each pair’s co-occurrence relationship.
The key limitation to this overall broad-brush co-occurrence analysis is that, because it does not take into account the timing of diagnoses, it may capture associations that are driven by post-pancreatic cancer diagnoses. To overcome this limitation, we implemented a matching and time-thresholding strategy, which we call a time-restricted co-occurrence analysis. Cases were identified by the presence of a pancreatic cancer diagnosis in their EHR. Cases were then matched to two non-cases using Mahalanobis distance matching with age at first EHR diagnosis (nearest neighbor), sex (exact), and length of follow-up (nearest neighbor) as matching variables. Length of follow-up, the difference between the days since birth at last recorded EHR encounter and the earliest recorded EHR encounter, was included as a matching variable to achieve a fairer comparison between cases and non-cases. Individuals who have longer follow-up tend to have more phenotypes of any type in their EHR, which could spuriously impact the diagnostics for the predictive ability of the PheRS. Moreover, simply because a diagnosis does not appear in an individual’s EHR does not mean that they do not have a given phenotype. It could mean that the phenotype was not observed, diagnosed, or recorded in the EHR. Matching on length of follow-up gives the individuals in a matched group the same window of time for any diagnosis to appear on their EHR and potentially make the misclassification probabilities comparable.
To evaluate the association of conditions that occur before a pancreatic cancer diagnosis, we applied time thresholds to the matched, time-stamped phecode data. Take one matched group as an example: one case and two non-cases. If the case is first diagnosed with pancreatic cancer at the age of 55, we remove all diagnoses that occurred afterward. We also remove all diagnoses that occur after the age of 55 in the two matched non-cases. In this example, we are looking at diagnoses that occur before the day of the first pancreatic cancer diagnosis. However, it is more informative to determine an individual’s risk for pancreatic cancer more than a day in advance. To achieve this, we also removed diagnoses within a time window prior to the case’s pancreatic cancer diagnosis as well: one year, two years, and five years prior to the day of the first pancreatic cancer diagnosis in the matched case. We aim to identify diagnoses that are associated with future case status so that a well-discriminating risk score can be obtained years before a pancreatic cancer diagnosis and overt symptom manifestation. In the case of pancreatic cancer, a well-performing risk score even one year before a diagnosis would represent a meaningful improvement given the common late stage at diagnosis and poor prognosis. If any of these thresholds eliminate all EHR data for a case or both matched non-cases due to short follow-ups, the entire matched group is removed to maintain balance.
Using the matched time threshold EHR data, we created a phecode indicator matrix that indicates whether each patient has at least a single occurrence of each phecode in their time-restricted EHR data. With this matrix, we obtained the log-odds ratio (beta) estimates using Firth-corrected logistic regression and saddlepoint approximation p-values. These betas and p-values are adjusted for sex, age at time threshold, length of follow-up, genotyping array batch, and the first four principal components of the genotype data. Figure 1 provides a visual assessment of the resulting −log10(p-values), which are color-coded by disease category in a Manhattan plot. A Bonferroni-corrected phenome-wide p-value threshold is included for reference. Interactive versions of the plots in Figure 1 and their corresponding data can be viewed and downloaded online at: https://umich-biostatistics.shinyapps.io/pancan_cooccur/
Figure 1.
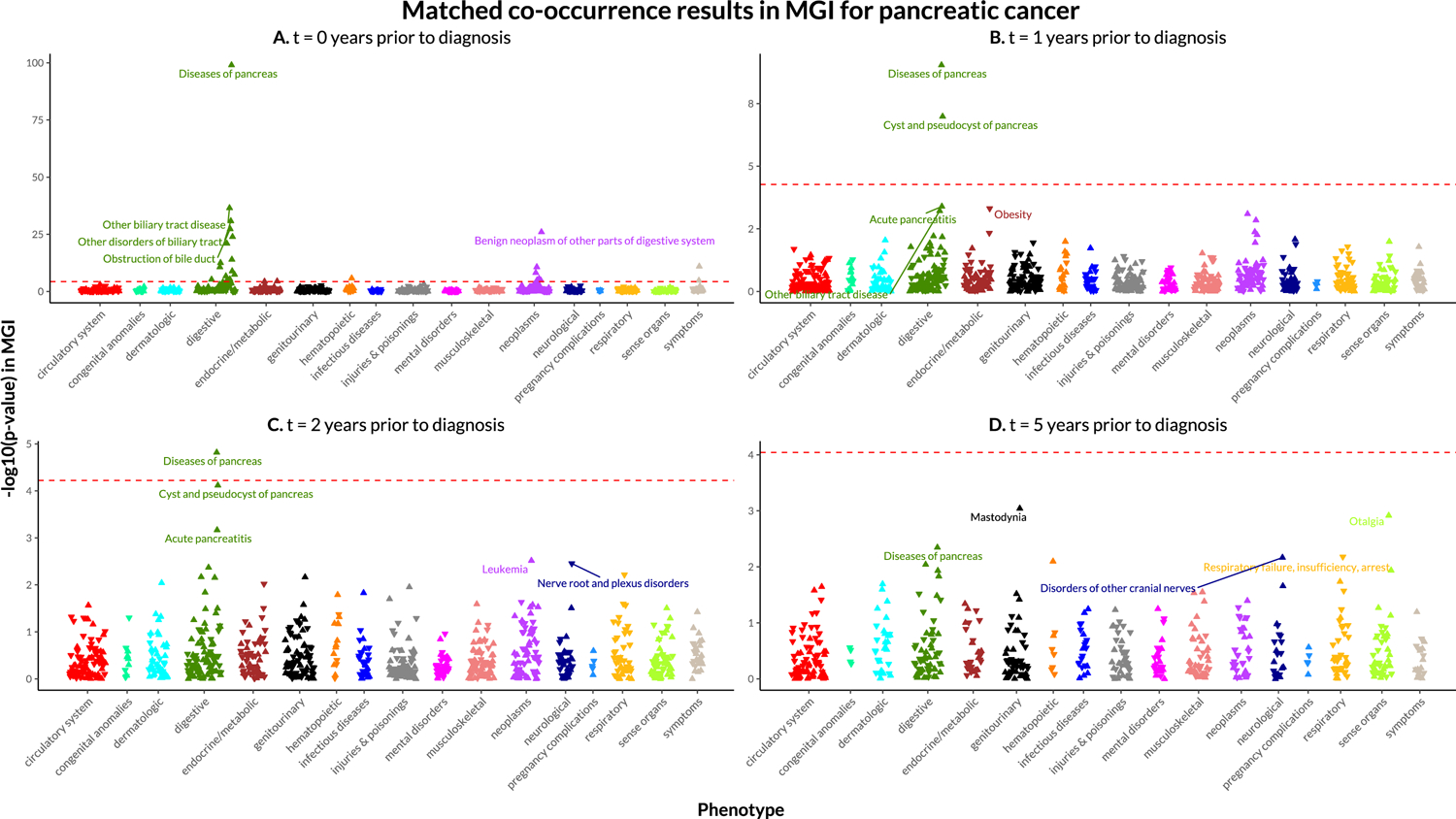
Manhattan plots for matched, time-based co-occurrence analyses in MGI.
Manhattan plots for time-unrestricted matched and unmatched co-occurrence analyses in MGI and for the time-unrestricted unmatched co-occurrence analysis in UKB can be found in Supplementary Materials Section 1.
2.3. Construction of Phenotype risk score
After obtaining beta estimates and p-values from the co-occurrence analysis, we identified an initial subset of phecodes to include in the PheRS. For each PheRS, our initial subset represents the phecodes with the 50 smallest p-values from the co-occurrence analysis. We selected these “top hits” as opposed to a Bonferroni-correction multiple testing p-value threshold because the small sample size in matched, time-restricted data are unlikely to find phenome-wide significant hits. We recognized that the proportion of false positive associations among the 50 top hits might increase with decreasing sample size.
Correlations between phecodes present a challenge for risk score construction. Take, for example, diseases of pancreas and acute pancreatitis. In MGI, the correlation coefficient between them is very high (0.72), and diseases of pancreas and acute pancreatitis are more moderately correlated with pancreatic cancer (0.37 and 0.18, respectively). To characterize the overall correlation in MGI, we present the average correlation within and across each of the 17 phenotype categories (previously defined in the R package “PheWAS”) [37] in Figure 2, which indicates it is necessary to consider phenome-wide correlations. We were reluctant to include several correlated phecodes in the PheRS, which could overwhelm the impact of more moderate but independently associated phenotypes on the overall risk score. To prevent this, we performed correlation thresholding (akin to the idea of clumping in genetics [38]). The pairwise Pearson correlation coefficients between the selected phecodes were calculated and ordered in terms of absolute magnitude. We sequentially examined the coefficients, keeping the phecode with the smaller p-value until all included phecodes had pairwise correlations of 0.25 or less. The resulting set of phecodes and their corresponding betas obtained from the co-occurrence analysis were used to calculate a covariate-adjusted PheRS. Despite covariate adjustment, we will refer to this as a “univariable” PheRS since the beta estimates did not directly account for other phenotypes. We further performed forward selection (forward function in the R package “logistf”; p < 0.05 entry threshold) using Firth-corrected logistic regression to identify an independent set of phecodes and multivariable phenotype-adjusted beta estimates. The phecodes and their corresponding point estimates from the joint model were used to calculate a “multivariable” PheRS, which serves as our primary analysis.
Figure 2. Phenome-wide correlation structure in MGI.
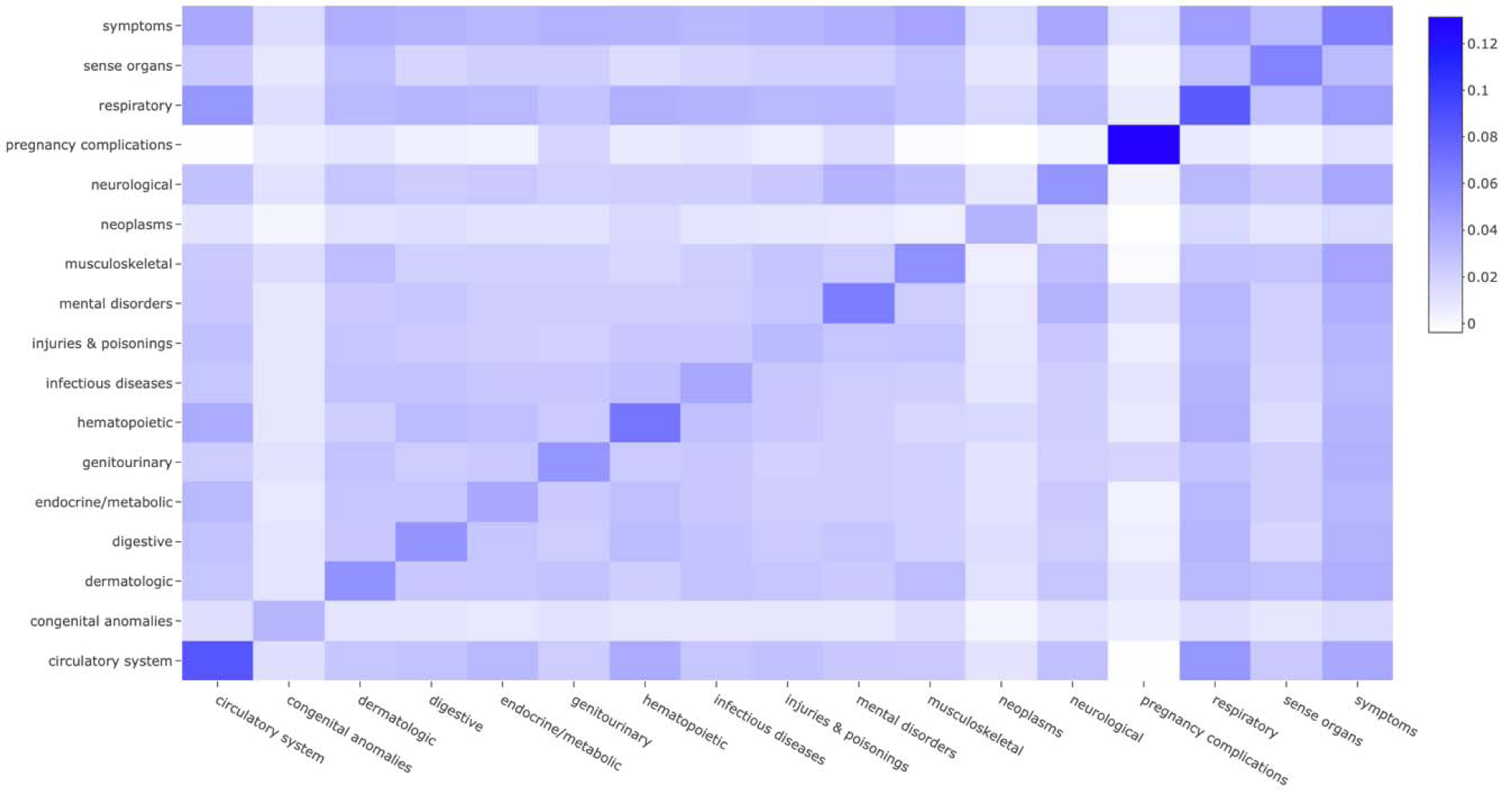
Each box represents the average pairwise Pearson correlation coefficients among every pair within the 17 broad phenotype categories.
The PheRS itself was defined as a summation of the multivariable log-odds estimates for the selected phecodes. Mathematically, the equation is
where is the log-odds estimate corresponding to phenotype j, and are indicator functions that equals 1 when the inequality is true and 0 when false, and Dij is an indicator variable equal to 1 when individual i has been diagnosed with phenotype j. This construction reverse-codes phenotypes with negative beta estimates to ensure that all PheRS are greater than or equal to 0, since it is more intuitive to discuss non-negative risk scores.
The multivariable betas obtained from the co-occurrence analysis in the matched MGI cohort are applied to the phecode indicator matrices in MGI and UKB to calculate PheRS. Because we have time-stamped data for MGI, we use time-restricted phecode indicator matrices when constructing PheRS in MGI. Since we do not have time-stamped data in UKB, the time-restricted PheRS defined using MGI betas are evaluated using each patient’s full, time-unrestricted EHR in UKB, which includes phenotypes that may have first occurred after a pancreatic cancer diagnosis.
To evaluate the association between the PheRS and pancreatic cancer diagnosis in MGI and UKB, we calculated covariate-adjusted odds ratios and 95% confidence intervals using a logistic generalized linear model (GLM) adjusted for age (or birthyear), sex, genotyping array, and the first four principal components of the genotype data. While the analysis is restricted to those of recent European ancestry, the first four principal components of the genotype data were used to account for any remaining ancestry differences. Recent ancestry was inferred by projecting genotyped samples into the space of the Human Genome Diversity Project reference panel as described elsewhere. [39] We also evaluated each PheRS’s (a) area under the receiver-operator characteristics (ROC) curve (AUC) to assess its discriminatory ability (using the R package “pROC”), (b) calibration using the Hosmer-Lemeshow χ2 Goodness of Fit test statistic and p-value (from the R package “ResourceSelection”) and (c) accuracy via the Brier score (from the R package “DescTools”). These diagnostics were not adjusted for covariates. In addition, we explored the odds ratios of the 1st, 2nd, 5th, 10th, and 25th PheRS percentiles compared to the rest. For example, an indicator variable for the top percentile compared to the bottom 99% of all PheRS was constructed. These percentile-based indicator variables are used to assess the ability of high PheRS scores to identity high-risk individuals. [40] In the case where the beta estimates are applied to the same data source from which they are derived, no cross validation is performed, implying that the results are overfit and projections are overoptimistic in MGI. Deriving beta estimates in MGI and applying it to UKB provides a more honest exploration though the latter lacks temporal information. All analyses were conducted using R 3.6.3. [41] These estimates and diagnostics are presented in Table 3.
TABLE 3a.
UKB PheRS estimate and diagnostics using beta estimates from MGI
| Time threshold (in years prior to diagnosis) |
||||
|---|---|---|---|---|
| 0 years | 1 year | 2 years | 5 years | |
| Odds Ratio | 1.66 | 1.51 | 1.57 | 1.56 |
| Odds Ratio 95% CI | (1.62, 1.70) | (1.48, 1.55) | (1.51, 1.63) | (1.50, 1.62) |
| P-value | 0.00E+00 | 1.61E-245 | 1.48E-111 | 1.31E-108 |
| AUC | 0.699 | 0.657 | 0.606 | 0.603 |
| AUC 95% CI | 0.679, 0.719 | 0.635, 0.679 | 0.583, 0.629 | 0.583, 0.623 |
| HL Stat, P-value | 45.44, 3.04e-07 | 89.75, 5.55e-16 | 100.72, 0.00e+00 | 38.71, 5.56e-06 |
| Brier score | 0.00168 | 0.00168 | 0.00167 | 0.00167 |
| Top %-ile OR (95% CI) | ||||
| 1st percentile | 15.77 (14.51, 20.05) | 21.24 (19.47, 27.44) | 14.07 (12.70, 17.93) | 1.75 (1.19, 2.58) |
| 2nd percentile | 20.14 (18.84, 25.67) | 12.20 (11.40, 15.93) | 9.60 (8.78, 12.23) | 10.20 (9.34, 12.99) |
| 5th percentile | 1.28 (0.90, 1.88) | 0.71 (0.55, 0.89) | 0.77 (0.63, 0.90) | 0.62 (0.47, 0.77) |
| 10th percentile | 1.28 (0.90, 1.88) | 0.71 (0.55, 0.89) | 0.77 (0.63, 0.90) | 0.62 (0.47, 0.77) |
| 25th percentile | 1.28 (0.90, 1.88) | 0.71 (0.55, 0.89) | 0.77 (0.63, 0.90) | 0.62 (0.47, 0.77) |
Abbreviations: CI, confidence interval; OR, odds ratio; HL, Hosmer-Lemeshow Goodness of Fit test; %-ile, percentile; PheRS, phenotype risk score
Notes:
- The estimates and diagnostic values in this table correspond to pancreatic cancer PheRS constructed in UKB using association estimates obtained from time-restricted co-occurrence analysis in the matched MGI discovery cohort.
- The odds ratio, corresponding 95% confidence interval estimate, and p-value come from a logistic GLM model for the PheRS on pancreatic cancer, adjusted for, sex, birthyear, genotyping array, and the first four principal components of the genotype data.
- The AUC and corresponding 95% confidence interval are from an unadjusted ROC model for pancreatic cancer case/control status.
- The Hosmer-Lemeshow test statistic and p-value and the Brier score are from a matched, unadjusted logistic GLM model for pancreatic cancer case/control status.
- The percentile-based odds ratios and confidence intervals come from a Firth-corrected logistic regression model on pancreatic cancer case/control status adjusted for sex, birthyear, genotyping array, and the first four principal components of the genotype data.
2.4. Polygenic risk score
We explore PheRS and their relative contribution in the presence of an increasingly popular risk score – the polygenic risk score (PRS). Pancreatic cancer PRS were calculated using linkage disequilibrium (LD) clumping and p-value thresholding in both MGI and UKB using 18 independent previously reported genetic risk variants reported in the GWAS Catalog (see Supplementary Materials Section 2). [42–47] The advantage of PRS is that they are assigned at birth and are unlikely to change over time. More information regarding the construction of these PRS can be found in Fritsche et al. (2018). [33]
2.5. Prediction models
For each PheRS, we created eight logistic GLM regression models to explore the performance of risk scores and covariates independently and alongside one another. We first fit a covariate-only model consisting of age (or birth year), sex, genotyping array, and the first four principal components of genotype data. We then fit a model including traditional covariates and three known risk factors for pancreatic cancer: BMI (continuous), alcohol (ever vs. never), and smoking (ever vs. never). We fit models adjusting for PheRS and/or PRS alone, and we fit models adjusting for PheRS and/or PRS and the set of covariates and three risk factors. Odds ratios and confidence intervals for all predictors are reported. Additionally, the AUC, Hosmer-Lemeshow χ2 Goodness of Fit statistic, and Brier score are evaluated on each model’s fitted values. The results from these models are in Table 4 and AUCs are plotted in Figure 3.
TABLE 4a.
Model estimates, predictive diagnostics, and relative contribution of PheRS in the presence of covariates in UKB.
| t | Model |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Covariates only | Covariates + risk factors | PRS only | PRS + covariates + risk factors | PheRS only | PheRS + covariates + risk factors | PheRS + PRS only | PheRS, PRS, covariates + risk factors | |||
| 0 | PheRS | Odds Ratio | - | - | - | - | 1.69 | 1.67 | 1.69 | 1.67 |
| 95% CI | - | - | - | - | 1.65, 1.73 | 1.63, 1.71 | 1.65, 1.72 | 1.63, 1.71 | ||
| PRS | Odds Ratio | - | - | 2.15 | 2.17 | - | - | 2.11 | 2.16 | |
| 95% CI | - | - | 1.76, 2.62 | 1.77, 2.65 | - | - | 1.72, 2.59 | 1.76, 2.65 | ||
| Diagnostics | AUC | 0.692 | 0.694 | 0.581 | 0.711 | 0.701 | 0.805 | 0.741 | 0.812 | |
| AUC 95% CI | 0.673, 0.711 | 0.676, 0.713 | 0.560, 0.603 | 0.693, 0.729 | 0.680, 0.721 | 0.786, 0.824 | 0.718, 0.765 | 0.794, 0.830 | ||
| HL Stat (P-value) | 37.50 (9.22E-06) | 43.10 (8.44E-07) | 10.90 (2.05E-01) | 69.8 (5.39E-12) | 1010 (0.00E+00) | 821 (0.00E+00) | 607 (0.00E+00) | 781 (0.00E+00) | ||
| Brier Score | 0.00166 | 0.00166 | 0.00166 | 0.00166 | 0.00166 | 0.00167 | 0.00167 | 0.00168 | ||
| 1 | PheRS | Odds Ratio | - | - | - | - | 1.55 | 1.52 | 1.55 | 1.52 |
| 95% CI | - | - | - | - | 1.51, 1.59 | 1.48, 1.56 | 1.51, 1.58 | 1.48, 1.56 | ||
| PRS | Odds Ratio | - | - | 2.15 | 2.17 | - | - | 2.12 | 2.15 | |
| 95% CI | - | - | 1.76, 2.62 | 1.77, 2.65 | - | - | 1.73, 2.59 | 1.76, 2.64 | ||
| Diagnostics | AUC | 0.692 | 0.694 | 0.581 | 0.711 | 0.660 | 0.772 | 0.695 | 0.779 | |
| AUC 95% CI | 0.673, 0.711 | 0.676, 0.713 | 0.560, 0.603 | 0.693, 0.729 | 0.638, 0.682 | 0.751, 0.793 | 0.669, 0.720 | 0.759, 0.800 | ||
| HL Stat (P-value) | 37.50 (9.22E-06) | 43.10 (8.44E-07) | 10.90 (2.05E-01) | 69.8 (5.39E-12) | 1570 (0.00E+00) | 966 (0.00E+00) | 653 (0.00E+00) | 889 (0.00E+00) | ||
| Brier Score | 0.00166 | 0.00166 | 0.00166 | 0.00166 | 0.00166 | 0.00167 | 0.00166 | 0.00167 | ||
| 2 | PheRS | Odds Ratio | - | - | - | - | 1.64 | 1.59 | 1.65 | 1.59 |
| 95% CI | - | - | - | - | 1.58, 1.71 | 1.52, 1.65 | 1.58, 1.71 | 1.53, 1.65 | ||
| PRS | Odds Ratio | - | - | 2.15 | 2.17 | - | - | 2.16 | 2.18 | |
| 95% CI | - | - | 1.76, 2.62 | 1.77, 2.65 | - | - | 1.77, 2.64 | 1.78, 2.66 | ||
| Diagnostics | AUC | 0.692 | 0.694 | 0.581 | 0.711 | 0.612 | 0.737 | 0.647 | 0.748 | |
| AUC 95% CI | 0.673, 0.711 | 0.676, 0.713 | 0.560, 0.603 | 0.693, 0.729 | 0.589, 0.635 | 0.715, 0.758 | 0.621, 0.674 | 0.727, 0.769 | ||
| HL Stat (P-value) | 37.50 (9.22E-06) | 43.10 (8.44E-07) | 10.90 (2.05E-01) | 69.8 (5.39E-12) | 1260 (0.00E+00) | 599 (0.00E+00) | 483 (0.00E+00) | 617 (0.00E+00) | ||
| Brier Score | 0.00166 | 0.00166 | 0.00166 | 0.00166 | 0.00167 | 0.00167 | 0.00167 | 0.00167 | ||
| 5 | PheRS | Odds Ratio | - | - | - | - | 1.62 | 1.57 | 1.62 | 1.57 |
| 95% CI | - | - | - | - | 1.56, 1.68 | 1.50, 1.63 | 1.56, 1.68 | 1.51, 1.63 | ||
| PRS | Odds Ratio | - | - | 2.15 | 2.17 | - | - | 2.16 | 2.18 | |
| 95% CI | - | - | 1.76, 2.62 | 1.77, 2.65 | - | - | 1.76, 2.63 | 1.78, 2.67 | ||
| Diagnostics | AUC | 0.692 | 0.694 | 0.581 | 0.711 | 0.605 | 0.732 | 0.651 | 0.742 | |
| AUC 95% CI | 0.673, 0.711 | 0.676, 0.713 | 0.560, 0.603 | 0.693, 0.729 | 0.585, 0.626 | 0.710, 0.754 | 0.626, 0.676 | 0.720, 0.763 | ||
| HL Stat (P-value) | 37.50 (9.22E-06) | 43.10 (8.44E-07) | 10.90 (2.05E-01) | 69.8 (5.39E-12) | 613 (0.00E+00) | 509 (0.00E+00) | 301 (0.00E+00) | 437 (0.00E+00) | ||
| Brier Score | 0.00166 | 0.00166 | 0.00166 | 0.00166 | 0.00167 | 0.00167 | 0.00167 | 0.00167 | ||
Abbreviations: AUC, area under the receiver-operator characteristics curve; CI, confidence interval; HL, Hosmer-Lemeshow Chi-Square test; PheRS, phenotype risk score; PRS, polygenic risk score.
Notes: Variable estimates come from a logistic GLM model. PheRS constructed using multivariate betas obtained in MGI and applied to UKB data. Diagnostic results are obtained on using the fitted values from the corresponding model. The covariates used are sex, birthyear, genotyping batch, and the first four principal components from the genotype data. The risk factors are BMI (continuous), drinker (ever/never), and smoker (ever/never).
Figure 3.
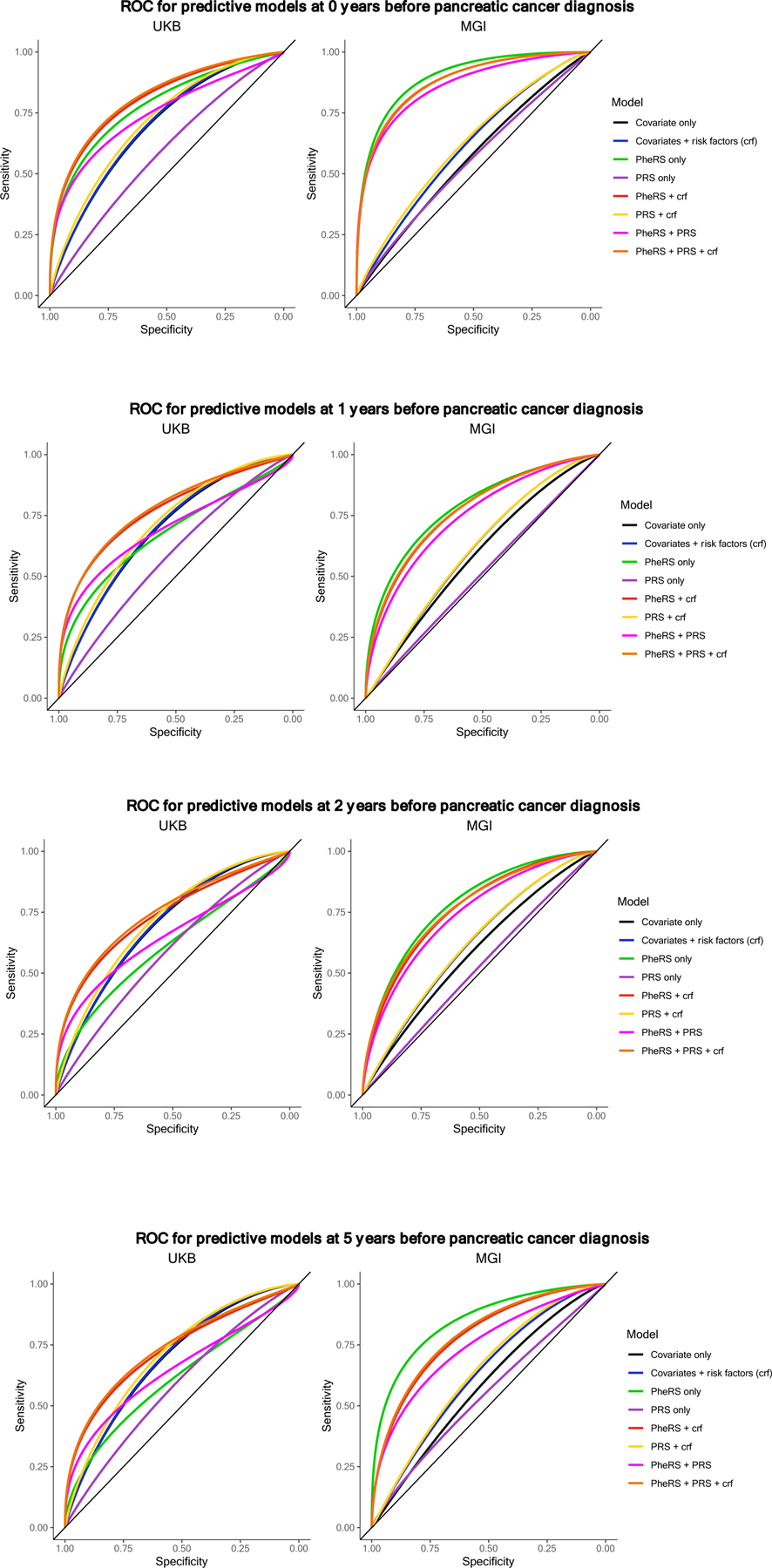
AUC plot comparing model cascade
a. AUC curves by model in MGI (left) and UKB (right) at 0 years before pancreatic cancer diagnosis threshold. Corresponding AUC and 95% CI values for UKB and MGI can be found in Table 4.
b. AUC curves by model in MGI (left) and UKB (right) at 1 year before pancreatic cancer diagnosis threshold. Corresponding AUC and 95% CI values for UKB and MGI can be found in Table 4.
c. AUC curves by model in MGI (left) and UKB (right) at 2 years before pancreatic cancer diagnosis threshold. Corresponding AUC and 95% CI values for UKB and MGI can be found in Table 4.
d. AUC curves by model in MGI (left) and UKB (right) at 5 years before pancreatic cancer diagnosis threshold. Corresponding AUC and 95% CI values for UKB and MGI can be found in Table 4.
2.6. Sensitivity analyses
In additional sensitivity analyses, we applied three alternative PheRS construction strategies. First, as further exploration of the impact of timing on PheRS performance, we constructed a PheRS using time-unrestricted weights obtained from UKB co-occurrence analyses and evaluated this PheRS in both UKB and in time-restricted MGI data. The PheRS construction methods considered so far are data-driven and do not directly build in prior knowledge. We also evaluate two additional PheRS construction strategies which are less agnostic, where we (1) incorporate established risk factors for pancreatic cancer [48] or (2) use PubMed publication results as a secondary source to inform trait selection. The description of how these were calculated along with their estimates and diagnostics for these PheRS are presented in Supplementary Materials Section 3.
3. Results
3.1. Cohort description
There were 429 pancreatic cancer cases in the MGI cohort and 659 in the UKB cohort, although the proportion of cases was substantially greater in MGI (1.12% vs 0.17%). Both cases and controls were younger in MGI than in UKB, consistent with their recruitment protocols and criteria (see Data section above). In both cohorts, there was a higher proportion of males among the cases and a higher proportion of females among the controls. The median length of follow-up is slightly longer in UKB compared to MGI. Detailed cohort characteristics are described in Table 1.
3.2. Co-occurrence, phenotype discovery, and identification
We plotted the −log10(p-values) from the matched co-occurrence analyses in MGI at each time threshold in a Manhattan plot to visualize the phenome-wide associations (Figure 1). The p-values are adjusted for sex, age, length of follow-up, genotyping array batch, and the first four principal components of the genotype data. At the 0-years prior to pancreatic cancer diagnosis threshold, there are a lot of digestive system phenotypes along with some neoplasms and symptoms. The strongest association found was with diseases of pancreas which include both acute and chronic pancreatitis. Other strongly associated digestive phenotypes were related to the biliary tract (e.g., other biliary tract disease), liver (e.g., jaundice), and pancreas (e.g., cyst or pseudocyst of pancreas). Benign neoplasm of the digestive system, abdominal pain, and secondary malignant neoplasm of digestive system were the strongest non-digestive phenotype associations. With increasing time before the cancer diagnosis, the strength of the associations decreases, with digestive system phenotypes being the most commonly associated with future pancreatic cancer diagnosis. After correcting for multiple testing, there were no phenome-wide significant associations found at 5 years prior to pancreatic cancer diagnosis. An interactive version of the plots in Figure 1 along with accompanying tables can be accessed online at https://umich-biostatistics.shinyapps.io/pancan_cooccur/.
The plots show −log10-transformed p-values obtained from a saddlepoint approximation test in MGI using the ScoreTest_SPA function from the “SPAtest” R package. These pairwise comparisons are adjusted for age, sex, genotyping batch, and the first four principal components of the genotype data.
The −log10(p-values) are plotted along the y-axis with the colors corresponding to the broad 17 phenotype categories indicated along the x-axis. The points are triangles with the upward-facing triangles indicating a positive relationship and the downward-facing triangles indicating a negative relationship. The red dashed line represents the Bonferroni-corrected phenome-wide p-value threshold for reference.
Interactive versions of the plots in Figure 1 and their corresponding data can be viewed and downloaded online at: https://umich-biostatistics.shinyapps.io/pancan_cooccur/
After selecting the top 50 candidate phecodes for each PheRS, performing pairwise Pearson correlation thresholding, and applying forward selection using Firth-corrected logistic regression, the final set of phecodes and their weights were identified at each time threshold and are summarized in Table 2. The PheRS constructed include between 8 and 12 phecodes. Thirty-three unique phecodes from 11 of the 17 phecode categories were identified across all time thresholds. Digestive (7) and neoplasm (6) phecodes were the most common, followed by genitourinary, circulatory system, and endocrine/metabolic (3 each) phenotypes. Five traits appeared in PheRS in at least two time thresholds (listed with corresponding phecode): diseases of pancreas (577), obesity (278.1), leukemia (204), nerve root and plexus disorders (353), and respiratory failure, insufficiency, arrest (509). Of those that appear in PheRS at multiple time thresholds, the weights are consistent in sign and magnitude. Diseases of pancreas was the only phecode to appear in the PheRS at all time thresholds.
TABLE 2.
Phenotypes included in multivariable PheRS construction with corresponding beta estimates
| Phenotype [PheWAS code] | Category | Time threshold (in years prior to diagnosis) |
|||
|---|---|---|---|---|---|
| 0 years | 1 year | 2 years | 5 years | ||
| Diseases of pancreas [577] | Digestive | 3.765 | 1.811 | 1.544 | 1.479 |
| Secondary malignant neoplasm of digestive systems [198.3] | Neoplasms | 2.462 | |||
| Cancer of other lymphoid, histiocytic tissue [202] | Neoplasms | 1.517 | |||
| Cholelithiasis with other cholecystitis [574.12] | Digestive | 3.503 | |||
| Genital prolapse [618] | Genitourinary | −2.114 | |||
| Colon cancer [153.2] | Neoplasms | 0.944 | |||
| Gingival and periodontal diseases [523] | Digestive | 1.798 | |||
| Cerebral artery occlusion, with cerebral infarction [433.21] | Circulatory system | −1.093 | |||
| Obesity [278.1] | Endocrine/Metabolic | −0.861 | −0.725 | ||
| Cancer of other male genital organs [187] | Neoplasms | 3.206 | |||
| Leukemia [204] | Neoplasms | 2.321 | 2.239 | ||
| Constipation [563] | Digestive | 0.682 | |||
| Nerve root and plexus disorders [353] | Neurological | −1.463 | −2.856 | ||
| Respiratory failure, insufficiency, arrest [509] | Respiratory | −0.985 | −1.405 | −2.699 | |
| Diseases of spleen [289.5] | Hematopoietic | 1.720 | |||
| Type 1 diabetes [250.1] | Endocrine/Metabolic | −1.138 | |||
| Ventral hernia [550.5] | Digestive | −1.081 | |||
| Other disorders of intestine [569] | Digestive | 0.828 | |||
| Fracture of tibia and fibula [800.3] | Injuries & poisonings | 1.397 | |||
| Other symptoms referable to back [724.8] | Musculoskeletal | 1.548 | |||
| Tachycardia NOS [427.7] | Circulatory system | 0.907 | |||
| Lymphadenitis [289.4] | Hematopoietic | −0.889 | |||
| Asthma [495] | Respiratory | −0.643 | |||
| Mastodynia [613.5] | Genitourinary | 2.325 | |||
| Otalgia [382] | Sense organs | 3.091 | |||
| Disorders of other cranial nerves [352] | Neurological | 2.008 | |||
| Coagulation defects [286] | Hematopoietic | 1.538 | |||
| Occlusion and stenosis of precerebral arteries [433.1] | Circulatory system | 1.406 | |||
| Dysphagia [532] | Digestive | −1.567 | |||
| Other disorders of male genital organs [608] | Genitourinary | 1.745 | |||
| Nontoxic nodular goiter [241] | Endocrine/Metabolic | 1.268 | |||
| Fracture of vertebral column without mention of spinal cord injury [805] | Injuries & poisonings | 1.343 | |||
| Lipoma [214] | Neoplasms | −1.329 | |||
Notes:
- Phenotype is bolded if it appears in multiple phenotype constructions.
- For a phenotype to be included at a given time threshold they first had to have one of the 50 smallest p-values from the co-occurrence analysis in MGI. Next, pairwise Pearson correlation coefficient threshold was performed, starting with the strongest absolute value of the coefficient and phenotypes with the smaller p-value were kept for all coefficients with an absolute value correlation coefficient greater than 0.25. Finally, the final set of phenotypes were identified after performing forward selection using Firth-corrected logistic regression with an entry p-value threshold of 0.05. The select phenotypes and their corresponding beta values from this joint model appear in the table.
3.3. Operating characteristics of PheRS and PRS
The PheRS alone demonstrates some discriminatory ability in MGI, achieving AUCs ranging from 0.69 (95% CI: 0.65, 0.76) at 5-years prior to pancreatic cancer diagnosis to 0.85 (95% CI: 0.83, 0.88) at the 0-years threshold, just prior to diagnosis. As expected, with weights transferred from MGI, the PheRS does not perform as well in UKB, with AUCs ranging from 0.60 (95% CI: 0.58, 0.62) at 5-years prior to pancreatic cancer diagnosis to 0.70 (95% CI: 0.68, 0.72) at the 0-years threshold. In both cohorts, AUCs increase closer to the time of diagnosis. These PheRS estimates across time thresholds in UKB and MGI are reported in Table 3a and 3b, respectively.
TABLE 3b.
MGI PheRS estimate and diagnostics using beta estimates from MGI
| Time threshold (in years prior to diagnosis) |
||||
|---|---|---|---|---|
| 0 years | 1 year | 2 years | 5 years | |
| Odds Ratio | 7.86 | 3.69 | 3.71 | 5.54 |
| Odds Ratio 95% CI | (6.22, 10.13) | (2.73, 5.22) | (2.69, 5.33) | (3.37, 10.46) |
| P-value | 4.91E-62 | 2.61E-15 | 5.33E-14 | 1.65E-09 |
| AUC | 0.853 | 0.711 | 0.702 | 0.694 |
| AUC 95% CI | 0.829, 0.877 | 0.673, 0.749 | 0.660, 0.743 | 0.652, 0.736 |
| HL Stat, P-value | 6.30, 6.14e-01 | 0.54, 1.00e+00 | 1.57, 9.91e-01 | 0.52, 1.00e+00 |
| Brier score | 0.11420 | 0.18076 | 0.18506 | 0.17231 |
| Top %-ile OR (95% CI) | ||||
| 1st percentile | 23.86 (20.22, 28.84) | 14.14 (7.02, 48.17) | 10.36 (5.64, 14.62) | 26.96 (10.44, 104.47) |
| 2nd percentile | 23.86 (20.22, 28.84) | 10.08 (6.19, 23.41) | 9.16 (5.12, 27.90) | 17.77 (8.06, 45.88) |
| 5th percentile | 33.53 (23.19, 49.45) | 6.50 (4.34, 12.91) | 7.17 (4.20, 13.22) | 11.28 (2.92, 101.50) |
| 10th percentile | 21.88 (5.87, 194.23) | 4.06 (2.81, 9.97) | 4.58 (2.74, 8.50) | 11.28 (2.92, 101.50) |
| 25th percentile | 21.88 (5.87, 194.23) | 4.06 (2.81, 9.97) | 4.58 (2.74, 8.50) | 11.28 (2.92, 101.50) |
Abbreviations: CI, confidence interval; OR, odds ratio; HL, Hosmer-Lemeshow Goodness of Fit test; %-ile, percentile; PheRS, phenotype risk score
Notes:
- The estimates and diagnostic values in this table correspond to pancreatic cancer PheRS constructed in MGI using association estimates obtained from time-restricted co-occurrence analysis in the matched MGI discovery cohort.
- The odds ratio, corresponding 95% confidence interval estimate, and p-value come from a logistic GLM model for the PheRS on pancreatic cancer, adjusted for, sex, birthyear, genotyping array, and the first four principal components of the genotype data.
- The AUC and corresponding 95% confidence interval are from an unadjusted ROC model.
- The Hosmer-Lemeshow test statistic and p-value and the Brier score are from a matched, unadjusted logistic GLM model for pancreatic cancer case/control status.
- The percentile-based odds ratios and confidence intervals come from a Firth-corrected logistic regression model on pancreatic cancer case/control status adjusted for sex, age at time threshold, genotyping array, and the first four principal components of the genotype data.
The PRS alone demonstrates generally lower discriminatory ability in MGI, achieving an AUC of 0.56 (95% CI: 0.52, 0.59) at the 0-years time threshold (95% CI contains 0.5 at all other time threshold in MGI). Due to the generally unchanging nature of an individual’s germline genetics profile, the PRS estimate is consistent across time thresholds in MGI with slight variation due to changing sample size). In UKB, the PRS alone performs slightly better, achieving an AUC of 0.58 (95% CI: 0.56, 0.60). The PRS performance estimates are consistent with the literature to date where reported PRS alone does not have good discriminatory abilities for pancreatic cancer diagnosis. These PRS estimates across time thresholds in UKB and MGI are reported in the PRS only column of Table 4a and 4b, respectively.
TABLE 4b.
Model estimates, predictive diagnostics, and relative contribution of PheRS in the presence of covariates in MGI.
| Model |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Covariates only | Covariates + risk factors | PRS only | PRS + covariates + risk factors | PheRS only | PheRS + covariates + risk factors | PheRS + PRS only | PheRS, PRS, covariates + risk factors | |||
| 0 | PheRS | Odds Ratio | - | - | - | - | 7.58 | 7.70 | 7.57 | 7.69 |
| 95% CI | - | - | - | - | 6.01, 9.75 | 6.08, 9.97 | 6.00, 9.76 | 6.06, 9.97 | ||
| PRS | Odds Ratio | - | - | 1.59 | 1.59 | - | - | 1.51 | 1.47 | |
| 95% CI | - | - | 1.14, 2.24 | 1.13, 2.25 | - | - | 0.94, 2.43 | 0.90, 2.39 | ||
| Diagnostics | AUC | 0.557 | 0.609 | 0.555 | 0.619 | 0.852 | 0.885 | 0.864 | 0.887 | |
| AUC 95% CI | 0.520, 0.594 | 0.573, 0.645 | 0.517, 0.593 | 0.583, 0.655 | 0.827, 0.876 | 0.862, 0.908 | 0.838, 0.891 | 0.865, 0.910 | ||
| HL Stat (P-value) | 3.59 (8.92E-01) | 8.28 (4.06E-01) | 12.59 (1.27E-01) | 4.09 (8.49E-01) | 0.78 (1.00E+00) | 12.63 (1.25E-01) | 9.97 (2.67E-01) | 16.05 (4.17E-02) | ||
| Brier Score | 0.22870 | 0.22330 | 0.22890 | 0.22150 | 0.11600 | 0.11290 | 0.11630 | 0.1130 | ||
| 1 | PheRS | Odds Ratio | - | - | - | - | 3.68 | 3.74 | 3.69 | 3.75 |
| 95% CI | - | - | - | - | 2.72, 5.21 | 2.74, 5.35 | 2.72, 5.22 | 2.75, 5.36 | ||
| PRS | Odds Ratio | - | - | 1.11 | 1.19 | - | - | 1.18 | 1.23 | |
| 95% CI | - | - | 0.70, 1.77 | 0.74, 1.92 | - | - | 0.71, 1.95 | 0.73, 2.09 | ||
| Diagnostics | AUC | 0.575 | 0.604 | 0.524 | 0.606 | 0.717 | 0.755 | 0.730 | 0.758 | |
| AUC 95% CI | 0.525, 0.624 | 0.556, 0.652 | 0.473, 0.575 | 0.557, 0.654 | 0.678, 0.755 | 0.713, 0.798 | 0.686, 0.774 | 0.716, 0.800 | ||
| HL Stat (P-value) | 9.25 (3.22E-01) | 11.34 (1.83E-01) | 18.77 (1.62E-02) | 14.04 (8.07E-02) | 2.16 (9.76E-01) | 6.95 (5.43E-01) | 10.07 (2.60E-01) | 5.48 (7.06E-01) | ||
| Brier Score | 0.22060 | 0.21820 | 0.22430 | 0.21790 | 0.18100 | 0.17590 | 0.18070 | 0.17560 | ||
| 2 | PheRS | Odds Ratio | - | - | - | - | 3.62 | 3.68 | 3.72 | 3.79 |
| 95% CI | - | - | - | - | 2.63, 5.17 | 2.66, 5.29 | 2.70, 5.35 | 2.73, 5.50 | ||
| PRS | Odds Ratio | - | - | 1.21 | 1.32 | - | - | 1.55 | 1.67 | |
| 95% CI | - | - | 0.74, 1.97 | 0.80, 2.20 | - | - | 0.91, 2.65 | 0.96, 2.91 | ||
| Diagnostics | AUC | 0.576 | 0.609 | 0.530 | 0.611 | 0.704 | 0.744 | 0.729 | 0.750 | |
| AUC 95% CI | 0.523, 0.628 | 0.558, 0.661 | 0.476, 0.584 | 0.560, 0.663 | 0.663, 0.746 | 0.699, 0.789 | 0.682, 0.775 | 0.705, 0.795 | ||
| HL Stat (P-value) | 13.21 (1.05E-01) | 9.96 (2.68E-01) | 14.98 (5.96E-01) | 11.59 (1.70E-01) | 4.31 (8.28E-01) | 17.37 (2.65E-02) | 12.29 (1.39E-01) | 8.32 (4.03E-01) | ||
| Brier Score | 0.21980 | 0.21660 | 0.22370 | 0.21610 | 0.18720 | 0.18140 | 0.18570 | 0.17940 | ||
| 5 | PheRS | Odds Ratio | - | - | - | - | 5.62 | 5.65 | 5.60 | 5.59 |
| 95% CI | - | - | - | - | 3.42, 10.60 | 3.43, 10.70 | 3.41, 10.60 | 3.39, 10.50 | ||
| PRS | Odds Ratio | - | - | 1.52 | 1.70 | - | - | 1.61 | 1.81 | |
| 95% CI | - | - | 0.87, 2.68 | 0.95, 3.09 | - | - | 0.85, 3.06 | 0.93, 3.57 | ||
| Diagnostics | AUC | 0.575 | 0.621 | 0.552 | 0.629 | 0.697 | 0.766 | 0.728 | 0.774 | |
| AUC 95% CI | 0.514, 0.636 | 0.562, 0.679 | 0.488, 0.615 | 0.571, 0.687 | 0.655, 0.739 | 0.715, 0.816 | 0.672, 0.784 | 0.725, 0.824 | ||
| HL Stat (P-value) | 8.30 (4.04E-01) | 7.45 (4.89E-01) | 6.79 (5.60E-01) | 9.40 (3.10E-01) | 1.64 (9.90E-01) | 8.00 (4.34E-01) | 12.84 (1.18E-01) | 13.81 (8.68E-01) | ||
| Brier Score | 0.22020 | 0.21500 | 0.22230 | 0.21350 | 0.17330 | 0.16790 | 0.17180 | 0.16670 | ||
Abbreviations: AUC, area under the receiver-operator characteristics curve; CI, confidence interval; HL, Hosmer-Lemeshow Chi-Square test; PheRS, phenotype risk score; PRS, polygenic risk score.
Notes: Variable estimates come from a logistic GLM model. PheRS constructed using multivariate betas obtained in MGI and applied to time-restricted, matched MGI data. Diagnostic results are obtained on using the fitted values from the corresponding model. The covariates used are sex, age at time threshold, genotyping batch, and the first four principal components from the genotype data. The risk factors are BMI (continuous), drinker (ever/never), and smoker (ever/never).
3.4. Combined models – PheRS + PRS + covariates
In both MGI and UKB, predictive models that do not contain PheRS performed poorer than those that do contain PheRS. In both cohorts, we see that a model’s discriminatory ability increases along with its complexity. The full model (PheRS, PRS, age at time threshold, genotyping batch, first four principal components from the genotype data, BMI, drinking status, and smoking status) in MGI has an AUC ranging from 0.77 (95% CI: 0.73, 0.82) at the 5-years threshold to 0.89 (95% CI: 0.87, 0.91) at the 0-years threshold. Similar patterns are observed in UKB, although at lower AUC levels. In UKB, the full model as an AUC ranging from 0.74 (95% CI: 0.72, 0.76) at the 5-years threshold to 0.81 (95% CI: 0.79, 0.83) at the 0-years threshold. Overall, the pancreatic cancer PRS (alone) used in our analyses did not perform better than covariates and risk factors (alone). PheRS, PRS, and covariates and risk factors independently and jointly contribute to models of risk prediction for pancreatic cancer. These estimates in UKB and MGI are shown in Table 4a and 4b and are visualized in Figure 3a and 3b, respectively.
3.5. PheRS stratification
Percentile-based odds ratios were calculated to assess the ability of PheRS scores at the top (1st, 2nd, 5th, 10th, and 25th percentiles) end of the distribution to identify individuals at high risk for future pancreatic cancer diagnosis (Table 3). In MGI, we observe increasing odds ratios for higher percentiles (i.e., going from 25th to 1st percentile) at all time thresholds. For example, if at the 5-year threshold, an individual’s PheRS is in the top 2% of the PheRS distribution, they are 18.70 (95% CI: 3.51, 8160.11) times more likely to be diagnosed with pancreatic cancer in the future. This trend is not apparent in UKB. In UKB, only PheRS in the top 1% or 2% of the PheRS are associated with future pancreatic cancer diagnosis. This can be explained by the relative rarity of pancreatic cancer in a very large cohort. In UKB, at the 5-year threshold, an individual with a PheRS in the top 2% is 10.20 (95% CI: 9.32, 12.99) times more likely to be diagnosed with pancreatic cancer in the future. These results show the promise of using very elevated PheRS scores in risk stratification and identifying high-risk individuals.
3.6. Sensitivity analysis
We carried out several sensitivity analyses to ensure our conclusions are robust to perturbation of assumptions or analytic choices we have made. In the following we summarize the findings from these ensemble of sensitivity analyses.
Co-occurrence analyses: time-unrestricted unmatched (Supplementary Figure S1) and matched (Supplementary Figure S2) co-occurrence analyses in MGI were consistent with each other and were similar to the matched co-occurrence analysis results at the time thresholding at the time of pancreatic cancer diagnosis (time=0). Moreover, they were similar to the time-unrestricted, unmatched co-occurrence analysis in UKB (Supplementary Figure S3), suggesting consistency across cohorts.
UKB-based PheRS: Applying effect estimates from the UKB co-occurrence analysis to the time-unrestricted UKB data performed very well as expected since the data is overfit (Supplementary Table S5a). The application of this PheRS in MGI shows weaker performance, achieving an AUC of 0.53 (95% CI: 0.50, 0.55) at the 5-year threshold as well as enrichment in the top percentile of the distribution (OR 3.65 [95% CI: 1.39, 13.33])
Established Risk factor PheRS: Using the established pancreatic cancer risk factors [48] to inform trait selection for inclusion in the PheRS and effect estimates from time-restricted MGI data, the PheRS performed reasonably well in the UKB (Supplementary Table S6a), particularly at the 5-year threshold (AUC: 0.65 [95% CI: 0.63, 0.67]); however, it lacked enrichment in the top percentiles of the PheRS distributions at the 0- and 1-year thresholds. Conversely, the PheRS performed fairly at the 0-, 1-, and 2-year thresholds in MGI, but not at the 5-year threshold (Supplementary Table S6b).
PubMed-informed PheRS: Of the alternative PheRS construction methods, this performed the most consistently across time thresholds in both cohorts (Supplementary Tables S7a and S7b). The 5-year AUC was 0.53 (95% CI: 0.51, 0.56) in UKB and 0.76 (95% CI: 0.71, 0.80) in MGI. Interestingly, this method appears to achieve poorer performance in UKB and slightly improved performance in MGI relative to the primary construction method (weights from MGI without PubMed thresholding).
Concordance: We found fair concordance across PheRS constructs (Supplementary Figure S4) and across time within the same construct (Supplementary Figure S5) in UKB. Our alternate PheRS constructs did not show meaningful improvements over the simpler, agnostic approach of the primary construct.
4. Discussion
Using EHR-linked biobank data from MGI and UKB, we constructed PheRS for pancreatic cancer using associations and weights obtained from a co-occurrence analysis in MGI. We discuss the results through two lenses: (1) PheRS as a general construct and (2) PheRS applied to pancreatic cancer.
4.1. Phenotype risk score as a construct
As diverse data streams become available for disease risk prediction, we are interested in exploring the potential for EHR data to identify individuals at-risk for diseases that have not yet benefitted from alternative risk stratification methods. Specifically, there has been significant growth in EHR-linked biobank-based research [13] aimed at repurposing clinical datasets for secondary research purposes. The goal of the PheRS is to create a single-number summary risk score to assess an individual’s relative risk for a future diagnosis using diagnoses from their EHR. We hypothesized that a PheRS can be used alongside other risk predictors like PRS, covariates, and risk factor status to improve our ability to improve overall risk stratification. In the short term, the clinical utility of PheRS would be to inform waiting, watching, and “flagging” individuals. In the long term, PheRS can be incorporated with additional data for absolute risk prediction where the focus is more on individual risk and less on effect estimates.
The PheRS is designed as a metric to guide precision prevention strategies in advance of the cancer diagnosis, thus having time restricted EHR with temporal ordering of disease codes is key here. Unfortunately, in UKB we did not have access to time-stamped data and as such diagnoses after cancer diagnoses also contributed to PheRS, likely leading to inflated predictive power. Our goal is not to discover individual phecode association with pancreatic cancer but to predict disease risk years in advance so that targeted screening can be intensified for high risk individuals.
The co-occurrence analysis identifies phenotypes and corresponding weights across the medical phenome in association with a diagnosis of interest. Phecodes may be correlated, though, because a doctor may assign multiple ICD codes for the same diagnosis or because there are clusters of diseases that occur together or in progression. We believe the proposed ranked pairwise Pearson correlation thresholding procedure and subsequent Firth-corrected logistic regression forward selection algorithm adequately account for correlation among phecodes.
A key feature of the framework developed in our paper is the implementation of case-control matching and time-thresholding. It is crucial that a risk score considers conditions that occur prior to a diagnosis of interest. Hanauer et al. (2013) explored assessing temporal relationships in EHR data between one condition and a diagnosis of interest while also exploring different time windows. [49,50] Additional related work includes exploring time-oriented medical records. [51–57] However, the challenge is maintaining comparability (notably, age and length of follow-up) with individuals who do not have the diagnosis of interest. By matching and thresholding EHR data and by varying time thresholds within each matched group, we can more directly observe these temporal relationships among people who are similar prior to pancreatic cancer diagnosis.
Because the co-occurrence analysis is a hypothesis-free exploration of the phenome, it provides a simple approach to identify many associations in a short amount of time. It may replicate known associations or identify potentially novel associations. Unfortunately, an agnostic approach can identify spurious associations. Hanauer et al. (2014) [58] attempted to automate the process of distinguishing known from unknown associations by linking to Medline citations using MetaMap [59], but their effort emphasized that this remains an open challenge. At this time, manual review of all individual pairwise associations is required to assess their validity and/or medical plausibility.
In summary: (1) the co-occurrence analysis represents a simple, agnostic approach towards exploring phecode-phecode relationships at a phenome-wide level, (2) the matching and time-thresholding mechanism overcomes key conceptual barriers necessary for constructing a score for future disease risk, (3) the simple PheRS construction framework allows one to explicitly identify the contributing traits and their contributions for future case status, and (4) the PheRS pipeline can be easily applied to any condition in the medical phenome (but may be particularly useful in clinical risk stratification for diseases without well-understood risk factors or well-performing PRS).
4.2. Pancreatic cancer phenotype risk score
Pancreatic cancer is a devastating disease with increasing incidence and mortality, late stage at diagnosis and poor survival, which is not yet adequately detected early in disease progression when treatments are more effective. For these reasons, it is an excellent candidate phenotype for PheRS application (however, the PheRS approach can be applied for any condition which there is a pressing need to develop a risk stratification tool with currently available data). Cancers like liver and ovarian cancer may also benefit from progress in risk stratification for similar reasons.
Looking at the series of models constructed in Table 4, we can see the PheRS contribution to risk prediction alongside PRS, covariates and risk factors. In both cohorts, PRS does not perform particularly well, although it makes a notable contribution to risk prediction. The overall heritability of pancreatic cancer is quoted around 5–10% [60] which is relatively low and indicates that a well-performing PRS for pancreatic cancer is difficult to achieve at this time.. Covariates and risk factors alone achieved fair AUCs (~0.69 in UKB and ~0.55–0.62 in MGI). In addition, PheRS in both MGI and UKB at all time thresholds markedly improve our ability to discriminate between cases and controls. AUCs of 0.70 (95% CI: 0.66, 0.74) and 0.61 (95% CI: 0.59, 0.63) were obtained in the PheRS-only model at the 5-year threshold in MGI and UKB, respectively. We believe that these results are promising given our current inability to accurately identify individuals at-risk in even a much shorter time window. Moreover, when PheRS is combined with PRS, covariates, and risk factors, we see marked increases in AUC (0.77 [95% CI: 0.73, 0.82] and 0.74 [95% CI: 0.72, 0.77] at the 5-year threshold in MGI and UKB, respectively). The highest AUC achieved was 0.89 (95% CI: 0.87, 0.91) in MGI at the 0-years threshold; however, these data are overfit and include diagnoses that tend to occur shortly before pancreatic cancer diagnosis, when it is already suspected. These results extend to the percentile-based odds ratios presented in Table 3, indicating that PheRS can be used to identify individuals at very high risk of pancreatic cancer diagnosis (e.g., top 5th percentile or higher). It should be noted that, while we are able to enrich the top percentiles, some cases (e.g., 18 of 128 cases (14.1%) in MGI at the 5-year threshold) were not identified to be at high risk and thus would not benefit from such a screening mechanism.
Phenotypes selected for inclusion in the PheRS using the described methods were intuitive, as summarized in Table 2. It is reasonable that phenotypes related to pancreatic and digestive conditions could be predictive of future pancreatic cancer (or, alternatively, potential misdiagnoses). Upon closer investigation, other identified phenotypes are also plausible. For example, there is some evidence that asthma (particularly long-standing asthma) is negatively associated with pancreatic cancer. [61,62] In some cases, like leukemia, despite there being a rather strong positive relationship across two PheRS constructions, we were only able to find limited literature suggesting an association with acute pancreatitis. [63,64] Other phenotypes identified are less convincing. For example, a 2012 literature review found that obesity tends to be positively associated with pancreatic cancer risk while our observed log-odds estimate included in two PheRS constructions is modestly negative. [65]
Our analyses complement recent work that has utilized the clinical and health data available in EHR for risk prediction in general and for pancreatic cancer specifically. Notably, Bastarache et al. (2018) constructed a phenotype risk score based on clinical features specially designed to identify associations between rare variants and phenotypes consistent with Mendelian diseases. [66] Boland et al. (2015) analyzed EHR data and found that birth month is significantly associated with 55 diseases, indicating season-dependent early development exposures may predispose individuals towards increased lifetime disease risk. [67] There are several studies that attempt to predict pancreatic cancer-related outcomes: diagnosis in general, [68–71] diagnosis in a new-onset diabetes cohort, [72] and malignant versus benign disease. [73] While some of these predictive models perform well, they either (a) do not restrict the analysis to data prior to pancreatic cancer diagnosis, (b) consider data immediately prior to pancreatic cancer diagnosis when it is already suspected, or (c) focus on trait-specific subsets of the population. However, together, these approaches demonstrate that leveraging different data sources (e.g., genetics, metabolomics, laboratory results) and tailoring to high-risk subpopulations (e.g., new-onset diabetes patients) alongside PheRS can improve our ability to identify high-risk individuals earlier.
Our pancreatic cancer PheRS performs well relative to existing pancreatic cancer risk prediction models while also overcoming some of the limitations present in those methods and demonstrates the importance of integrating heterogeneous data sources for making gains in risk prediction and stratification.
4.3. Limitations and challenges
We recognize that there are limitations to our analysis. Notably, the time-restricted association estimates (log-odds) obtained from the co-occurrence analysis in MGI are applied to two datasets in less than ideal circumstances. First, these association estimates obtained in MGI are applied to time-restricted data in MGI and thus the data are overfit. These results are presented to show an upper bound of the PheRS performance and offer a comparison to the PheRS constructed in UKB. Second, the rank-based selection of phecodes for inclusion crudely addresses the problem of correlation among phecodes, which may result in information loss. We believe the aggregation of related ICD codes into phecodes makes the proposed method somewhat defensible as we include ICD codes that could have been interchangeably used under the umbrella of one phecode. However, phecodes themselves are nested. Whether we should only include the “parent” phecode or “child” phecode in a PheRS is a topic that requires deeper exploration and needs to be addressed in more rigorous detail in future work. Third, the time-restricted association estimates from MGI are applied to time-unrestricted data in UKB. It is possible diagnoses that contribute to a case’s PheRS occur after the pancreatic cancer diagnosis. Fourth, the analysis was restricted to individuals of inferred recent European ancestry. This improves comparability across MGI and UKB and allows for a more direct comparison of genotype data and PRS construction, but it limits generalizability of results to other communities and does not provide insight to improve the health of all people. While sample sizes are often cited as statistically valid explanations for this restriction, the health and medicine communities need to prioritize investments in diversifying the collection and analysis of data on groups commonly excluded from research. [74,75]
We found that while the co-occurrence analysis identified sensible associations (like diseases of pancreas), it may also identify associations of questionable plausibility, such as the negative association found with obesity. It remains a challenge to distinguish between legitimate associations, valid yet previously uncovered associations, and pure noise [58]. While the phenotypes identified for inclusion in each PheRS were reviewed, we did not implement a mechanism for excluding potentially spurious phenotypes from being included (e.g., fracture of tibia and fibula appears to have a spurious association with pancreatic cancer, but is included in the PheRS at the 5 year threshold [see Table 2]), instead allowing the data to drive the PheRS construction.
Many challenges cited in previously published, EHR-based literature apply to our work. Rhodes et al. (2007) found that coded data from billing codes can be extremely inaccurate, [76] while Williams et al. (2007) found that clinicians may fail to report problems. [77] Beesley et al. (2019) hypothesized that length of follow-up may be associated with likelihood of receiving diagnoses but that serious conditions, like cancer, may be less subject to misclassification. [13] However, Wei et al. (2017) compared the use of manually curated phecodes (used in this paper) to ICD9 clinical modification (ICD-9-CM) codes and the Agency for Healthcare Research and Quality Clinical Classification Software (CCS) for ICD-9-CM codes and found that the phecodes best identified 100 selected phenotypes compared with the other phenotype classification coding systems. [78] Even still, there are differences among clinicians’ code assignments, [79–84] which are likely only exacerbated across biobanks and would impact generalizability of PheRS. At this time, manual review of results and follow-up explorations of all associations are required.
4.4. Conclusion
Ultimately, pancreatic cancer is a devastating diagnosis, and there are opportunities for improving our ability to identify high-risk individuals earlier, both through improved PheRS and through incorporating additional data. Future work can explore more statistically elegant, less p-value driven methods than the forward selection algorithm used here to overcome the challenge of variable selection in PheRS construction. Alternative methods that can account for multicollinearity like LASSO, elastic net, and principal components analysis could result in a more statistically robust PheRS. Incorporating interaction and non-linearity may be critical and as such modern machine learning based on nets, trees and ensemble methods could be used to construct risk scores. [85] In addition, other avenues of future exploration include adding domains of data, like behavioral and lifestyle survey data, and applying this framework to other diverse, multidimensional, time-stamped datasets, such as eMERGE. [86] The process of constructing PheRS is fairly simple. Modern machine learning tools may be used to capture cross-phecode interactions and enhance predictive power.
The framework proposed here for constructing a pancreatic cancer PheRS is straightforward, allowing for the identification of a constellation of phenotypes predictive of pancreatic cancer diagnosis at varying time horizons. Moreover, the PheRS alone has a non-negligible discriminatory ability and adds additional predictive ability in the presence of covariates, risk factors, and PRS. We believe our approach to handling time-stamped data by matching and time-thresholding represents an accessible approach in identifying temporal associations using EHR data. Additionally, it allows for time-based exploration of phenotype associations with a cautious potential for identifying new relationships.
To summarize our main takeaway messages, PheRS (1) on its own has fair discriminatory ability, (2) can be a useful risk stratification metric when comparing scores at the very top end of the distribution with the rest (e.g., top 1–2%), and (3) meaningfully adds to prediction in the presence of PRS and covariates.
Other important data sources can and should be incorporated into a pancreatic cancer prediction model. Metabolomics and laboratory data have proven useful for diagnostic and prediction purposes in studies about pancreatic cancer [68,69,72,73] and could be useful to include in risk prediction models in addition to PheRS, PRS, and covariates. Beyond pancreatic cancer, this PheRS framework can be applied to other phenotypes where there is relatively poor ability to identify high risk individuals but can also enhance stratification for traits that already have good prediction models. Further, this PheRS framework can be modified and applied to outcomes other than disease diagnoses, such as post-diagnosis complications and survival.
Large biobank datasets like those used in this paper provide a substantial opportunity to explore PheRS and integrate PheRS alongside many sources of data into a single model towards the goals of risk prediction and stratification. For example, biobanks are exploring ways of connecting to cancer and death registry data, local and national surgical databases, laboratory data, prescription claims data, and geo-coded data. [13] We join previous works [13,87–90] that call for the need to integrate many disparate data sources to allow for a more complete picture of the conditions that lead to disease- and health-related outcomes, particularly given the performance of metabolomics-based risk prediction.
Supplementary Material
Highlights.
Introduces an EHR diagnosis-based phenotype risk score (PheRS) framework
Applies phenome-wide association study (PheWAS)-derived unique to each diagnosis
Distills a patient’s entire EHR history into a time-dependent, diagnosis-specific risk score
Explores pancreatic cancer PheRS in two large biobanks: MGI and UKB
Evaluates the contribution of PheRS alongside polygenic risk score
Acknowledgements
This study was supported by the Center for Precision Health Data Science at the University of Michigan School of Public Health, The University of Michigan Rogel Cancer Center, the Michigan Institute of Data Science, CA 046592 and NSF DMS 1712933.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data statement
Data cannot be shared publicly due to patient confidentiality. The data underlying the results presented in the study are available from University of Michigan Medical School Central Biorepository at https://research.medicine.umich.edu/our-units/central-biorepository/get-access and from the UK Biobank at http://www.ukbiobank.ac.uk/register-apply/ for researchers who meet the criteria for access to confidential data.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Nelson HD, Tyne K, Naik A, et al. Screening for Breast Cancer: Systematic Evidence Review Update for the US Preventive Services Task Force. Rockville, MD; 2009. https://www.ncbi.nlm.nih.gov/books/NBK36392/?report=classic. [PubMed] [Google Scholar]
- 2.Lin JS, Piper MA, Perdue LA, et al. Screening for Colorectal Cancer: A Systemative Review for the U.S. Preventive Services Task Force. Rockville, MD; 2016. https://www.ncbi.nlm.nih.gov/books/NBK373584/. [Google Scholar]
- 3.Humphrey L, Deffebach M, Pappas M, et al. Screening for Lung Cancer: Systematic Review to Update the U.S. Preventive Services Task Force Recommendation; Rockville, MD; 2013. https://www.ncbi.nlm.nih.gov/sites/books/NBK154610/%0A. [PubMed] [Google Scholar]
- 4.Wilson PWF, D’Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of Coronary Heart Disease Using Risk Factor Categories. Circulation. 1998;97(18):1837–1847. doi: 10.1161/01.CIR.97.18.1837 [DOI] [PubMed] [Google Scholar]
- 5.Nelson HD, Fu R, Cantor A, Pappas M, Daeges M, Humphrey L. Effectiveness of Breast Cancer Screening: Systematic Review and Meta-analysis to Update the 2009 U.S. Preventive Services Task Force Recommendation. Ann Intern Med. 2016;164(4):244. doi: 10.7326/M15-0969 [DOI] [PubMed] [Google Scholar]
- 6.Bray C, Bell LN, Liang H, Collins D, Yale SH. Colorectal Cancer Screening. WMJ. 2017;116(1):27–33. http://www.ncbi.nlm.nih.gov/pubmed/29099566. [PubMed] [Google Scholar]
- 7.Melnikow J, Henderson JT, Burda BU, Senger CA, Durbin S, Weyrich MS. Screening for Cervical Cancer With High-Risk Human Papillomavirus Testing. JAMA. 2018;320(7):687. doi: 10.1001/jama.2018.10400 [DOI] [PubMed] [Google Scholar]
- 8.Yang DX, Soulos PR, Davis B, Gross CP, Yu JB. Impact of Widespread Cervical Cancer Screening: Number of Cancers Prevented and Changes in Race-specific Incidence. Am J Clin Oncol. 2018;41(3):289–294. doi: 10.1097/COC.0000000000000264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genomic Health Inc. oncotypeIQ. https://www.oncotypeiq.com/en-US.
- 10.National Comprehensive Cancer Network. NCCN Guidelines for Detection, Prevention, & Risk Reduction. https://www.nccn.org/professionals/physician_gls/default.aspx#detection.
- 11.US Preventive Services Task Force. Final Recommendation Statement: BRCA-Related Cancer: Risk Assessment, Genetic Counseling, and Genetic Testing. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/brca-related-cancer-risk-assessment-genetic-counseling-and-genetic-testing1.
- 12.National Human Genome Research Institute. DNA Sequencing Costs: Data. https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data. Published 2019. Accessed January 30, 2020.
- 13.Beesley LJ, Salvatore M, Fritsche LG, et al. The emerging landscape of health research based on biobanks linked to electronic health records: Existing resources, statistical challenges, and potential opportunities. Stat Med. December 2019:sim.8445. doi: 10.1002/sim.8445 [DOI] [PMC free article] [PubMed]
- 14.Ozaki K, Ohnishi Y, Iida A, et al. Functional SNPs in the lymphotoxin-α gene that are associated with susceptibility to myocardial infarction. Nat Genet. 2002;32(4):650–654. doi: 10.1038/ng1047 [DOI] [PubMed] [Google Scholar]
- 15.Ohnishi Y, Tanaka T, Ozaki K, Yamada R, Suzuki H, Nakamura Y. A high-throughput SNP typing system for genome-wide association studies. J Hum Genet. 2001;46(8):471–477. doi: 10.1007/s100380170047 [DOI] [PubMed] [Google Scholar]
- 16.Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24. doi: 10.1016/j.ajhg.2011.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Visscher PM, Wray NR, Zhang Q, et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017;101(1):5–22. doi: 10.1016/j.ajhg.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17(10):1520–1528. doi: 10.1101/gr.6665407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Privé F, Aschard H, Blum MGB. Efficient Implementation of Penalized Regression for Genetic Risk Prediction. Genetics. 2019;212(1):65–74. doi: 10.1534/genetics.119.302019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert SA, Abraham G, Inouye M. Towards clinical utility of polygenic risk scores. Hum Mol Genet. 2019;00(00):1–10. doi: 10.1093/hmg/ddz187 [DOI] [PubMed] [Google Scholar]
- 21.U.S. Cancer Statistics Working Group. U.S. Cancer Statistics Data Visualization Tool, based on November 2018 submission data (1999–2016). www.cdc.gov/cancer/dataviz Published 2019.
- 22.Ward EM, Sherman RL, Henley SJ, et al. Annual Report to the Nation on the Status of Cancer, Featuring Cancer in Men and Women Age 20–49 Years. JNCI J Natl Cancer Inst. 2019;111:1–19. doi: 10.1093/jnci/djz106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.National Cancer Institute. Cancer Stat Facts: Pancreatic Cancer.
- 24.Hart AR. Classic diseases revisited: Pancreatic cancer: any prospects for prevention? Postgrad Med J. 1999;75(887):521–526. doi: 10.1136/pgmj.75.887.521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hart AR, Kennedy H, Harvey I. Pancreatic Cancer: A Review of the Evidence on Causation. Clin Gastroenterol Hepatol. 2008;6(3):275–282. doi: 10.1016/j.cgh.2007.12.041 [DOI] [PubMed] [Google Scholar]
- 26.Kuroczycki-Saniutycz S, Grzeszczuk A, Zwierz ZW, et al. Prevention of pancreatic cancer. Współczesna Onkol. 2017;1(1):30–34. doi: 10.5114/wo.2016.63043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Surveillance Research Program. SEER*Explorer: An interactive website for SEER cancer statistics [Internet]. https://seer.cancer.gov/explorer. Accessed October 17, 2019.
- 28.Chen F, Childs EJ, Mocci E, et al. Analysis of Heritability and Genetic Architecture of Pancreatic Cancer: A PanC4 Study. Cancer Epidemiol Biomarkers Prev. 2019;28(7):1238–1245. doi: 10.1158/1055-9965.EPI-18-1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michigan Genomics Initiative Website. https://www.michigangenomics.org.
- 30.Fritsche LG, Gruber SB, Wu Z, et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. Am J Hum Genet. 2018;102(6):1048–1061. doi: 10.1016/j.ajhg.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.UK Biobank Website. http://www.ukbiobank.ac.uk.
- 32.Allen N, Sudlow C, Downey P, et al. UK Biobank: Current status and what it means for epidemiology. Heal Policy Technol. 2012;1(3):123–126. doi: 10.1016/j.hlpt.2012.07.003 [DOI] [Google Scholar]
- 33.Fritsche LG, Gruber SB, Wu Z, et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. Am J Hum Genet. 2018;102(6):1048–1061. doi: 10.1016/j.ajhg.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denny JC, Bastarache L, Ritchie MD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31(12):1102–1111. doi: 10.1038/nbt.2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sudlow C, Gallacher J, Allen N, et al. UK Biobank: An Open Access Resource for Identifying the Causes of a Wide Range of Complex Diseases of Middle and Old Age. PLOS Med. 2015;12(3):e1001779. doi: 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dey R, Schmidt EM, Abecasis GR, Lee S. A Fast and Accurate Algorithm to Test for Binary Phenotypes and Its Application to PheWAS. Am J Hum Genet. 2017;101(1):37–49. doi: 10.1016/j.ajhg.2017.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30(16):2375–2376. doi: 10.1093/bioinformatics/btu197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Privé F, Vilhjálmsson BJ, Aschard H, Blum MGB. Making the Most of Clumping and Thresholding for Polygenic Scores. Am J Hum Genet. 2019;105(6):1213–1221. doi: 10.1016/j.ajhg.2019.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fritsche LG, Patil S, Beesley LJ, et al. Cancer PRSweb: An Online Repository with Polygenic Risk Scores for Major Cancer Traits and Their Evaluation in Two Independent Biobanks. Am J Hum Genet. September 2020. doi: 10.1016/j.ajhg.2020.08.025 [DOI] [PMC free article] [PubMed]
- 40.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R Core Team. R: A language and environment for statistical computing. 2019. https://www.r-project.org/.
- 42.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41(9):986–990. doi: 10.1038/ng.429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu C, Kraft P, Stolzenberg-Solomon R, et al. Genome-wide association study of survival in patients with pancreatic adenocarcinoma. Gut. 2014;63(1):152–160. doi: 10.1136/gutjnl-2012-303477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolpin BM, Rizzato C, Kraft P, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46(9):994–1000. doi: 10.1038/ng.3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang M, Wang Z, Obazee O, et al. Three new pancreatic cancer susceptibility signals identified on chromosomes 1q32.1, 5p15.33 and 8q24.21. Oncotarget. 2016;7(41):66328–66343. doi: 10.18632/oncotarget.11041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klein AP, Wolpin BM, Risch HA, et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nat Commun. 2018;9(1):556. doi: 10.1038/s41467-018-02942-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buniello A, MacArthur JAL, Cerezo M, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–D1012. doi: 10.1093/nar/gky1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porta M, Fabregat X, Malats N, et al. Exocrine pancreatic cancer: Symptoms at presentation and their relation to tumour site and stage. Clin Transl Oncol. 2005;7(5):189–197. doi: 10.1007/BF02712816 [DOI] [PubMed] [Google Scholar]
- 49.Hanauer DA, Ramakrishnan N. Modeling temporal relationships in large scale clinical associations. J Am Med Informatics Assoc. 2013;20(2):332–341. doi: 10.1136/amiajnl-2012-001117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hanauer DA, Rhodes DR, Chinnaiyan AM. Exploring Clinical Associations Using ‘-Omics’ Based Enrichment Analyses Bajic VB, ed. PLoS One. 2009;4(4):e5203. doi: 10.1371/journal.pone.0005203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Klimov D, Shahar Y. A framework for intelligent visualization of multiple time-oriented medical records. AMIA . Annu Symp proceedings AMIA Symp. 2005:405–409. http://www.ncbi.nlm.nih.gov/pubmed/16779071%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC1560450. [PMC free article] [PubMed]
- 52.Moskovitch R, Shahar Y. Medical temporal-knowledge discovery via temporal abstraction. AMIA . Annu Symp proceedings AMIA Symp. 2009;2009:452–456. http://www.ncbi.nlm.nih.gov/pubmed/20351898%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2815492. [PMC free article] [PubMed] [Google Scholar]
- 53.Patnaik D, Butler P, Ramakrishnan N, Parida L, Keller BJ, Hanauer DA. Experiences with mining temporal event sequences from electronic medical records In: Proceedings of the 17th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining - KDD ‘11. New York, New York, USA: ACM Press; 2011:360. doi: 10.1145/2020408.2020468 [DOI] [Google Scholar]
- 54.Hripcsak G, Albers DJ, Perotte A. Exploiting time in electronic health record correlations. J Am Med Informatics Assoc. 2011;18(Supplement_1):i109–i115. doi: 10.1136/amiajnl-2011-000463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Batal I, Sacchi L, Bellazzi R, Hauskrecht M. A temporal abstraction framework for classifying clinical temporal data. AMIA . Annu Symp proceedings AMIA Symp. 2009;2009:29–33. http://www.ncbi.nlm.nih.gov/pubmed/20351817%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC2815443. [PMC free article] [PubMed] [Google Scholar]
- 56.Klimov D, Shahar Y. Intelligent querying and exploration of multiple time-oriented medical records. Stud Heal Technol Inf. 2007;129:1314–1318. [PubMed] [Google Scholar]
- 57.Klimov D, Shahar Y. Intelligent interactive visual exploration of temporal associations among multiple time-oriented patient records. Methods Inf Med. 2009;48:254–262. [DOI] [PubMed] [Google Scholar]
- 58.Hanauer DA, Saeed M, Zheng K, et al. Applying MetaMap to Medline for identifying novel associations in a large clinical dataset: a feasibility analysis. J Am Med Informatics Assoc. 2014;21(5):925–937. doi: 10.1136/amiajnl-2014-002767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aronson AR, Lang F-M. An overview of MetaMap: historical perspective and recent advances. J Am Med Informatics Assoc. 2010;17(3):229–236. doi: 10.1136/jamia.2009.002733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klein AP. Genetic susceptibility to pancreatic cancer. Mol Carcinog. 2012;51(1):14–24. doi: 10.1002/mc.20855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santibañez M, O’Rorke M, O’Leary E, De Camargo Cancela M, Murray L, Sharp L. Allergies, asthma and the risk of pancreatic cancer: A population-based case-control study in Ireland. Eur Respir J. 2015;46:PA3389. doi: 10.1183/13993003.congress-2015.PA3389 [DOI] [Google Scholar]
- 62.Gomez-Rubio P, Zock J-P, Sharp L, et al. Asthma and nasal allergies associate with reduced pancreatic cancer risk. Pancreatology. 2015;15(3):S124–S125. doi: 10.1016/j.pan.2015.05.439 [DOI] [Google Scholar]
- 63.de Castro JA, Vencer L, Espinosa W. Acute Myelogenous Leukemia Presenting as Acute Pancreatitis: A Case of Primary Pancreatic Extramedullary Acute Myeloid Leukemia. Clin Gastroenterol Hepatol. 2017;15(1):e30–e31. doi: 10.1016/j.cgh.2016.09.077 [DOI] [Google Scholar]
- 64.Stefanović M, Jazbec J, Lindgren F, Bulajić M, Löhr M. Acute pancreatitis as a complication of childhood cancer treatment. Cancer Med. 2016;5(5):827–836. doi: 10.1002/cam4.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bracci PM. Obesity and pancreatic cancer: Overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog. 2012;51(1):53–63. doi: 10.1002/mc.20778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bastarache L, Hughey JJ, Hebbring S, et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science (80- ). 2018;359(6381):1233–1239. doi: 10.1126/science.aal4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boland MR, Shahn Z, Madigan D, Hripcsak G, Tatonetti NP. Birth month affects lifetime disease risk: a phenome-wide method. J Am Med Informatics Assoc. 2015;22(5):1042–1053. doi: 10.1093/jamia/ocv046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao D, Weng C. Combining PubMed knowledge and EHR data to develop a weighted bayesian network for pancreatic cancer prediction. J Biomed Inform. 2011;44(5):859–868. doi: 10.1016/j.jbi.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kobayashi T, Nishiumi S, Ikeda A, et al. A Novel Serum Metabolomics-Based Diagnostic Approach to Pancreatic Cancer. Cancer Epidemiol Biomarkers Prev. 2013;22(4):571–579. doi: 10.1158/1055-9965.EPI-12-1033 [DOI] [PubMed] [Google Scholar]
- 70.Yu A, Woo SM, Joo J, et al. Development and Validation of a Prediction Model to Estimate Individual Risk of Pancreatic Cancer Obukhov AG, ed. PLoS One. 2016;11(1):e0146473. doi: 10.1371/journal.pone.0146473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakatochi M, Lin Y, Ito H, et al. Prediction model for pancreatic cancer risk in the general Japanese population Toland AE, ed. PLoS One. 2018;13(9):e0203386. doi: 10.1371/journal.pone.0203386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boursi B, Finkelman B, Giantonio BJ, et al. A Clinical Prediction Model to Assess Risk for Pancreatic Cancer Among Patients With New-Onset Diabetes. Gastroenterology. 2017;152(4):840–850.e3. doi: 10.1053/j.gastro.2016.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bathe OF, Shaykhutdinov R, Kopciuk K, et al. Feasibility of Identifying Pancreatic Cancer Based on Serum Metabolomics. Cancer Epidemiol Biomarkers Prev. 2011;20(1):140–147. doi: 10.1158/1055-9965.EPI-10-0712 [DOI] [PubMed] [Google Scholar]
- 74.Sirugo G, Williams SM, Tishkoff SA. The Missing Diversity in Human Genetic Studies. Cell. 2019;177(1):26–31. doi: 10.1016/j.cell.2019.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Popejoy AB. Diversity In Precision Medicine And Pharmacogenetics: Methodological And Conceptual Considerations For Broadening Participation. Pharmgenomics Pers Med. 2019;12:257–271. doi: 10.2147/PGPM.S179742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rhodes ET, Laffel LMB, Gonzalez TV, Ludwig DS. Accuracy of Administrative Coding for Type 2 Diabetes in Children, Adolescents, and Young Adults. Diabetes Care. 2007;30(1):141–143. doi: 10.2337/dc06-1142 [DOI] [PubMed] [Google Scholar]
- 77.Williams C, Mosley-Williams A, C M. Accuracy of provider generated computerized problem lists in the Veterans Administration. AMIA Annu Symp Proc. 2007:1155. [PubMed]
- 78.Wei W-Q, Bastarache LA, Carroll RJ, et al. Evaluating phecodes, clinical classification software, and ICD-9-CM codes for phenome-wide association studies in the electronic health record Rzhetsky A, ed. PLoS One. 2017;12(7):e0175508. doi: 10.1371/journal.pone.0175508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lorence D, Ibrahim I. Disparity in coding concordance: do physicians and coders agree? J Heal Care Financ. 2003;29:43–53. [PubMed] [Google Scholar]
- 80.Lorence D, Ibrahim I. Benchmarking variation in coding accuracy across the United States. J Heal Care Financ. 2003;29:29–42. [PubMed] [Google Scholar]
- 81.O’Malley KJ, Cook KF, Price MD, Wildes KR, Hurdle JF, Ashton CM. Measuring Diagnoses: ICD Code Accuracy. Health Serv Res. 2005;40(5p2):1620–1639. doi: 10.1111/j.1475-6773.2005.00444.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.SURJAN G Questions on validity of International Classification of Diseases-coded diagnoses. Int J Med Inform. 1999;54(2):77–95. doi: 10.1016/S1386-5056(98)00171-3 [DOI] [PubMed] [Google Scholar]
- 83.Shi X, Pashova H, Heagerty PJ. Comparing healthcare utilization patterns via global differences in the endorsement of current procedural terminology codes. Ann Appl Stat. 2017;11(3):1349–1374. doi: 10.1214/17-AOAS1028 [DOI] [Google Scholar]
- 84.Shi X, Li X, Cai T. Spherical Regression under Mismatch Corruption with Application to Automated Knowledge Translation. October 2018:1–60. http://arxiv.org/abs/1810.05679.
- 85.Park SK, Zhao Z, Mukherjee B. Construction of environmental risk score beyond standard linear models using machine learning methods: application to metal mixtures, oxidative stress and cardiovascular disease in NHANES. Environ Heal. 2017;16(1):102. doi: 10.1186/s12940-017-0310-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McCarty CA, Chisholm RL, Chute CG, et al. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4(1):13. doi: 10.1186/1755-8794-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goh K-I, Cusick ME, Valle D, Childs B, Vidal M, Barabasi A-L. The human disease network. Proc Natl Acad Sci. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schneeweiss S, Seeger JD, Smith SR. Methods for developing and analyzing clinically rich data for patient-centered outcomes research: an overview. Pharmacoepidemiol Drug Saf. 2012;21:1–5. doi: 10.1002/pds.3270 [DOI] [PubMed] [Google Scholar]
- 89.Schneeweiss S, Gagne JJ, Glynn RJ, Ruhl M, Rassen JA. Assessing the Comparative Effectiveness of Newly Marketed Medications: Methodological Challenges and Implications for Drug Development. Clin Pharmacol Ther. 2011;90(6):777–790. doi: 10.1038/clpt.2011.235 [DOI] [PubMed] [Google Scholar]
- 90.Tung JY, Do CB, Hinds DA, et al. Efficient Replication of over 180 Genetic Associations with Self-Reported Medical Data Reitsma PH, ed. PLoS One. 2011;6(8):e23473. doi: 10.1371/journal.pone.0023473 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.