

Abstract
The systematic evolution of ligands by exponential enrichment (SELEX) process enables the isolation of aptamers from random oligonucleotide libraries. However, it is generally difficult to identify the best aptamer from the resulting sequences, and the selected aptamers often exhibit suboptimal affinity and specificity. Post-SELEX aptamer engineering can improve aptamer performance, but current methods exhibit inherent bias and variable rates of success or require specialized instruments. Here, we describe a generalizable method that utilizes exonuclease III and exonuclease I to interrogate the binding properties of small-molecule-binding aptamers in a rapid, label-free assay. By analyzing an ochratoxin-binding DNA aptamer and six of its mutants, we determined that ligand binding alters the exonuclease digestion kinetics to an extent that closely correlates with the aptamer’s ligand affinity. We then utilized this assay to enhance the binding characteristics of a DNA aptamer which binds indiscriminately to ATP, ADP, AMP, and adenosine. We screened 13 mutants derived from this aptamer against all these analogues and identified two new high-affinity aptamers that solely bind to adenosine. We incorporated these two aptamers directly into an electrochemical aptamer-based sensor, which achieved a detection limit of 1 μM adenosine in 50% serum. We also confirmed the generality of our method to characterize target-binding affinities of protein-binding aptamers. We believe our approach is generalizable for DNA aptamers regardless of sequence, structure, and length and could be readily adapted into an automated format for high-throughput engineering of small-molecule-binding aptamers to acquire those with improved binding properties suitable for various applications.
Graphical Abstract
INTRODUCTION
Systematic evolution of ligands by exponential enrichment (SELEX) is used to isolate nucleic-acid-based bioaffinity elements known as aptamers from random oligonucleotide libraries.1,2 This multistep process involves iterative rounds of partitioning target-binding oligonucleotides from nonbinding sequences and amplification of the binders via polymerase chain reaction (PCR).3 Aptamers offer a number of advantages as molecular sensing reagents relative to antibodies, including greater thermostability, lower cost, excellent reproducibility of manufacturing, and the ease with which they can be engineered or chemically modified.4,5 However, SELEX sometimes fails to yield aptamers with suitable affinity and specificity for an intended application. This can occur for a variety of reasons. First, since starting oligonucleotide libraries can typically encompass anywhere from 1018 to 1040 possible unique sequences (for libraries containing 30–70 random nucleotides), it is unrealistic to screen every possible sequence in a SELEX experiment, and this will inevitably exclude high-quality aptamer candidates.6,7 Second, due to the low copy number of each sequence in the starting library, oligonucleotides with desirable binding properties can easily be lost during the early rounds of SELEX.8,9 Finally, these aptamers can be eliminated if they have low PCR amplification efficiency due to their sequence and/or structure.10–12
Several strategies can be employed to derive higher-performance aptamers upon the completion of SELEX. In doped-SELEX, selection is performed on a partially randomized library based on a parent aptamer sequence obtained from a prior SELEX effort.13,14 Doped-SELEX allows for the exploration of the sequence space close to the original aptamer, encompassing sequences that may have been entirely excluded or lost during earlier rounds.15 However, this method is greatly biased toward the original parent sequence. Based on calculations by Knight and Yarus, the copy number of individual unique sequences decreases exponentially with increasing numbers of mutations from the original sequence.16 As a result, sequences containing multiple mutations have a high likelihood of being excluded from the doped-SELEX pool. Although doped-SELEX has proven successful in a few instances, the extent of improvement is generally limited, and the success rate varies depending on the target and aptamer.15 An alternative approach is to design and synthesize a panel of aptamer mutants, and then individually test their binding properties to find the best sequence. Nucleotides may be altered in either the target-binding domain17,18 or surrounding scaffold regions,19–21 with guidance from genetic algorithms,22 computationally predicted secondary structures,19,20 or three-dimensional structures based on nuclear magnetic resonance (NMR) data.21,23 This method overcomes the competition problem associated with doped-SELEX because mutants are screened individually using instrument-based approaches or microarrays. Techniques such as surface plasmon resonance (SPR) spectroscopy,24 isothermal titration calorimetry (ITC),25 and microscale thermophoresis26 can provide accurate assessments of the binding parameters of the mutants. However, these experiments are costly and labor-intensive and also have low throughput as only a single aptamer-target pair can be tested at a time. Novel nanocalorimeters have been reported that allow for simultaneous high-throughput binding affinity determination.27,28 However, these instruments are not yet commercially available, and it is difficult to fabricate these devices without sufficient resources and expertise. Microarray-based techniques enable high-throughput characterization of large pools of aptamer mutants29 but require targets that are either inherently fluorescent or labeled with a fluorophore.29–31 This is feasible for protein targets29,30 but is generally impractical for the screening of small-molecule-binding aptamers because such labeling could alter binding affinity.
We demonstrate here an efficient and cost-effective exonuclease-based fluorescence assay for accurate, rapid, and label-free screening of the binding affinity and specificity of a panel of DNA aptamers. This assay is based on our previous finding that a mixture of exonuclease III (Exo III), a 3′-to-5′ double-strand DNA exonuclease, and exonuclease I (Exo I), a 3′-to-5′ single-strand DNA exonuclease, digests unbound aptamers into mononucleotides, but such digestion is inhibited for target-bound aptamers.32,33 We first digested an ochratoxin-binding DNA aptamer and its six mutants21 with a mixture of Exo III and Exo I and demonstrated that the kinetics of aptamer digestion are correlated with the aptamer’s relative target-binding strength. We then exploited this finding to sensitively screen the affinity and specificity of engineered aptamer mutants to identify and confirm high-performance mutants. To demonstrate the generality of this assay, we then used this method to improve the specificity of a previously described DNA aptamer that binds indiscriminately to ATP and its analogues.32 Specifically, we designed 13 aptamer mutants from this aptamer and performed our exonuclease-based assay with 59 aptamer-ligand pairs. We identified mutations at various positions surrounding the binding domain that greatly affect affinity and specificity and obtained two new structure-switching aptamers that retain the parent aptamer’s affinity for adenosine (ADE) but not for its phosphorylated analogues. We used these two aptamers to fabricate an electrochemical aptamer-based (E-AB) sensor that could sensitively detect ADE with a measurable limit of detection of 1 μM and minimal response to adenosine analogues in 50% serum. We finally demonstrated the generality of our method to sensitively profile the binding affinities of protein-binding aptamers. Our exonuclease-based screening strategy is a facile single-step fluorescence assay and therefore could be easily adapted into a high-throughput format using liquid-handling systems. Such a screening platform could greatly simplify and accelerate the identification of small-molecule-binding aptamers with desirable binding properties for various applications.
MATERIALS AND METHODS
Reagents.
Exonuclease III (Escherichia coli) (Exo III) (100 U/μL) and exonuclease I (E. coli) (Exo I) (20 U/μL) were purchased from New England Biolabs. Human α-thrombin and human factor X were purchased from Haematologic Technologies. Human plasma immunoglobulin G and human myeloma plasma λ immunoglobulin E were purchased from Athens Research and Technology. Deionized (DI) water with resistivity of 18 MΩ·cm was obtained from a Millipore water dispensing system. SYBR Gold was purchased from Invitrogen. Formamide, 0.5 M EDTA solution, glycerol, and sodium dodecyl sulfate (SDS) were purchased from Fisher Scientific. Ochratoxin A and B were purchased from Cayman Chemicals and dissolved in 100% DMSO to a final concentration of 4 mM. Adenosine-5′-triphosphate (ATP) disodium salt trihydrate was purchased from MP Biomedical. Adenosine-5′-diphosphate (ADP) sodium salt, adenosine-5′-monophosphate (AMP) sodium salt, and adenosine (ADE) were purchased from Sigma-Aldrich. ATP, ADP, AMP, and ADE stock solutions were prepared by dissolving in DI water to a final concentration of 2.5 mM followed by the addition of Tris base and NaCl to reach a molar equivalent of 2Na+·2Tris·1ATP/analogue. HCl was added as needed to neutralize the pH of the solution. A blank solution was created by dissolving Tris base and NaCl in DI water to a final concentration of 5 mM and neutralizing the solution with HCl. All other chemicals were purchased from Sigma-Aldrich. Unmodified oligonucleotides were purchased from Integrated DNA Technologies with standard desalting purification; they were dissolved in PCR-quality water, and their concentrations were measured using a NanoDrop 2000 (Thermo Fisher Scientific) spectrophotometer. Thiolated methylene blue (MB)-modified aptamers were purchased from LGC Biosearch Technologies with dual-HPLC purification and dissolved in TE Buffer (10 mM Tris-HCl + 1 mM EDTA, pH 8.0). All DNA sequences are listed in the Supporting Information, Table S1. Nunc 384-well black plates were purchased from Thermo Fisher Scientific.
Aptamer Digestion Experiments.
Unless otherwise specified, all digestion experiments were performed using the following procedure at 25 °C in 50 μL reaction volumes. Enzyme reaction buffer consisted of 8.1 mM Na2HPO4, 1.9 mM KH2PO4, 10 mM MgCl2, 0.1 mg/mL bovine serum albumin (BSA), 2.5% DMSO, pH 7.4, for ochratoxin-binding aptamers, 10 mM Tris-HCl, 20 mM NaCl, 1.5 mM MgCl2, 0.1 mg/mL BSA, pH 7.4, for ATP-analogue-binding aptamers, or 10 mM Tris-HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 0.1 mg/mL BSA, pH 7.4, for thrombin- and immunoglobulin E-binding aptamers. Aptamers were dissolved in their respective buffer (final concentration 1 μM for ochratoxin- and ATP-analogue-binding aptamers and 0.5 μM for thrombin- and immunoglobulin E-binding aptamers), heated to 95 °C for 10 min, and then immediately cooled on ice, followed by the addition of salts and BSA. Next, various concentrations of ligand or blank solution were added to the reaction mixture, and the mixture was incubated in a thermal cycler (C1000 Touch, Bio-Rad) at 25 °C for 60 min, after which 5 μL of enzymes (final concentrations: 0.025 U/μL Exo III and 0.05 U/μL Exo I) were added to each reaction mixture. An amount of 5 μL of sample was collected at various time-points and loaded directly into the wells of a Nunc 384-well black plate containing 10, 15, and 20 μL of quench solution (final concentration: 10 mM Tris-HCl, 12.5% formamide, 10 mM EDTA, 1× SYBR Gold) used for ochratoxin-, thrombin- and immunoglobulin E-, and ATP-analogue-binding aptamers, respectively. Fluorescence intensity at 545 nm was recorded using a Tecan Infinite M1000 PRO microplate reader with excitation at 495 nm. All error bars represent the standard error of fitting with the first-order rate equation.
Analysis of Aptamer Digestion Rates.
Data analysis was performed using the Origin 2019 software. The time-dependent fluorescence plots obtained from the aptamer digestion experiments were fit using first-order reaction kinetics described by eq 1:
| (1) |
where t is the time of digestion in minutes, Ft is the fluorescence intensity at time t, F0 is the maximum fluorescence intensity of the inhibition product, C is a constant to correct for background fluorescence, and t1/2 is the half-life of the reaction in minutes. The first time-point was excluded from fitting unless otherwise specified. During fitting, bounds were placed on F0 and C; these values could vary between 75–100% and 0–10% of the fluorescence intensity of the undigested aptamer, respectively. Error bars represent the standard error of fitting.
The three parameters used to determine target-binding-induced inhibition of enzyme digestion are the t1/2 ratio, first-order rate reduction, and resistance value. The t1/2 ratio was obtained by dividing the t1/2 obtained in the presence of ligand by the t1/2 obtained from the blank sample. On the basis of the fitted parameter of t1/2, the first-order reaction rates (k) can also be obtained by dividing -ln 2 by t1/2. Rate reduction was calculated by the expression 1 − (kligand/kblank). Finally, resistance values were calculated as previously described.35 The area under the curve (AUC) of the fluorescence digestion plots was determined using Origin 2019 software, and the resistance value was calculated using the expression (AUCligand/AUCblank) − 1.
Reverse ITC Experiments.
Reverse ITC experiments were performed with a MicroCal ITC200 instrument (Malvern) at 23 °C. For ochratoxin-binding aptamer titrations, the following buffer was used: 8.1 mM Na2HPO4, 1.9 mM KH2PO4, 10 mM MgCl2, and 2.5% DMSO, pH 7.4. For thrombin-binding aptamer titrations, the following buffer was used: 10 mM Tris–HCl, 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2, pH 7.4. For each experiment, 60 μL of aptamer solution was heated at 95 °C for 10 min in its respective buffer and cooled on ice, after which salts were added and then DMSO was added as appropriate. For ochratoxin-binding aptamer titrations, the cell was loaded with 300 μL of 15 μM ochratoxin A or ochratoxin B in reaction buffer, and the syringe was loaded with 250 or 500 μM ochratoxin-binding aptamer, respectively. For thrombin-binding aptamer titrations, 5 or 7.5 μM thrombin in buffer was loaded in the cell and 50 or 75 μM thrombin-binding aptamer was loaded in the syringe, respectively. Concentrations of aptamer and ligands used are listed in Table S2. Each titration consisted of an initial purge injection of 0.4 μL and 19 successive injections of 2 μL aptamer, with a spacing of 180 s between injections. The raw data was first corrected for the dilution heat of the aptamer, and then analyzed with the MicroCal analysis kit integrated into the Origin 7 and fitted using a single-site model to obtain KD.
Isothermal Titration Calorimetry Experiments for ATP-Analogue-Binding Aptamers.
ITC experiments for the ATP-binding aptamer and its mutants were performed with a MicroCal ITC200 instrument at 23 °C in reaction buffer of 10 mM Tris-HCl, 20 mM NaCl, and 1.5 mM MgCl2, pH 7.4. For each experiment, 300 μL of a 20 μM ATP-analogue-binding aptamer solution was heated at 95 °C for 10 min in Tris buffer and cooled down on ice, after which salt was added. The syringe was then loaded with ADE, AMP, ADP, or ATP in reaction buffer. Concentrations of aptamer and ligands are listed in Table S3. Each titration consisted of an initial purge injection of 0.4 μL and either 38 successive injections of 1 μL of ligand or 19 successive injections of 2 μL of ligand, with a spacing of 120–180 s between injections. For titrations of AMP, ADP, or ATP to A23T-29 and G10T-A23G-29, a total of three successive titrations were performed to saturate the aptamer with ligand. The raw data was first corrected for the dilution heat of the ligand, and then analyzed with the MicroCal analysis kit integrated into the Origin 7 software and fitted with a two-site sequential binding model to yield KD1 and KD2.
Electrochemical Aptamer-Based Adenosine Sensor Fabrication.
Gold disk electrodes (CH Instruments) (2 mm in diameter) were polished with 1 μm diamond slurry (Buehler) followed by 0.05 μm alumina suspension. To remove bound particulates, electrodes were sonicated in 70% ethanol solution for 5 min, and then in DI for another 5 min. Electrochemical cleaning was performed using a previously published protocol.36 The charge consumed during reduction of surface gold oxide in 0.05 M H2SO4 was used to calculate the surface area of each electrode, using the previously reported value of 390 ± 10 μC/cm−2.37 The roughness factor of the electrodes was calculated based on the ratio between the electro-chemically measured area and the geometric surface area and ranged from 1.0 to 1.05. Sensor fabrication involved a multistep process. First, 2 μL of 200 μM thiolated MB-modified aptamer (A23T-29-MB or G10T-A23G-29-MB) was mixed with 8 μL of 100 mM tris(2-carboxyethyl)phosphine (TCEP) at room temperature for 2 h to reduce disulfide bonds. The aptamer solution was then diluted with 1× PBS buffer (10 mM phosphate buffer, 1 M NaCl, 1 mM MgCl2, pH 7.2) to a final DNA concentration of 50 nM for A23T-29-MB or G10T-A23G-29-MB alone or 25 nM of each aptamer for the dual-aptamer electrode. Freshly cleaned gold electrodes were dried under a nitrogen stream, and then incubated in 250 μL of thiolated MB-modified aptamer solution overnight at room temperature. The electrodes were then backfilled with 1 mM 6-mercapto-1-hexanol for 2 h at room temperature. After sensor fabrication, the electrode surface coverage was determined as previously reported.38 The electrodes were stored in 10 mM Tris buffer (pH 7.4) for at least 1 h prior to use.
RESULTS AND DISCUSSION
Exonuclease Fluorescence Assay to Characterize Affinity and Specificity of Ochratoxin-Binding Aptamers.
We recently developed a sensitive and label-free aptamer-based assay for small-molecule detection using Exo III and Exo I.32 Specifically, we determined that a mixture of Exo III and Exo I digests aptamers into mononucleotides (Figure 1A, left), whereas the digestion of target-bound aptamers is stalled several bases prior to the binding domain (Figure 1A, right). We quantified the concentration of these partially digested aptamers at a single time-point using SYBR Gold (Figure 1A) and used this measurement to determine target concentration. On the basis of these findings, we hypothesized that these exonucleases could be used to rapidly profile aptamer-binding affinity and specificity and that the kinetics of aptamer digestion could potentially be used to accurately determine relative ligand-binding affinity.
Figure 1.
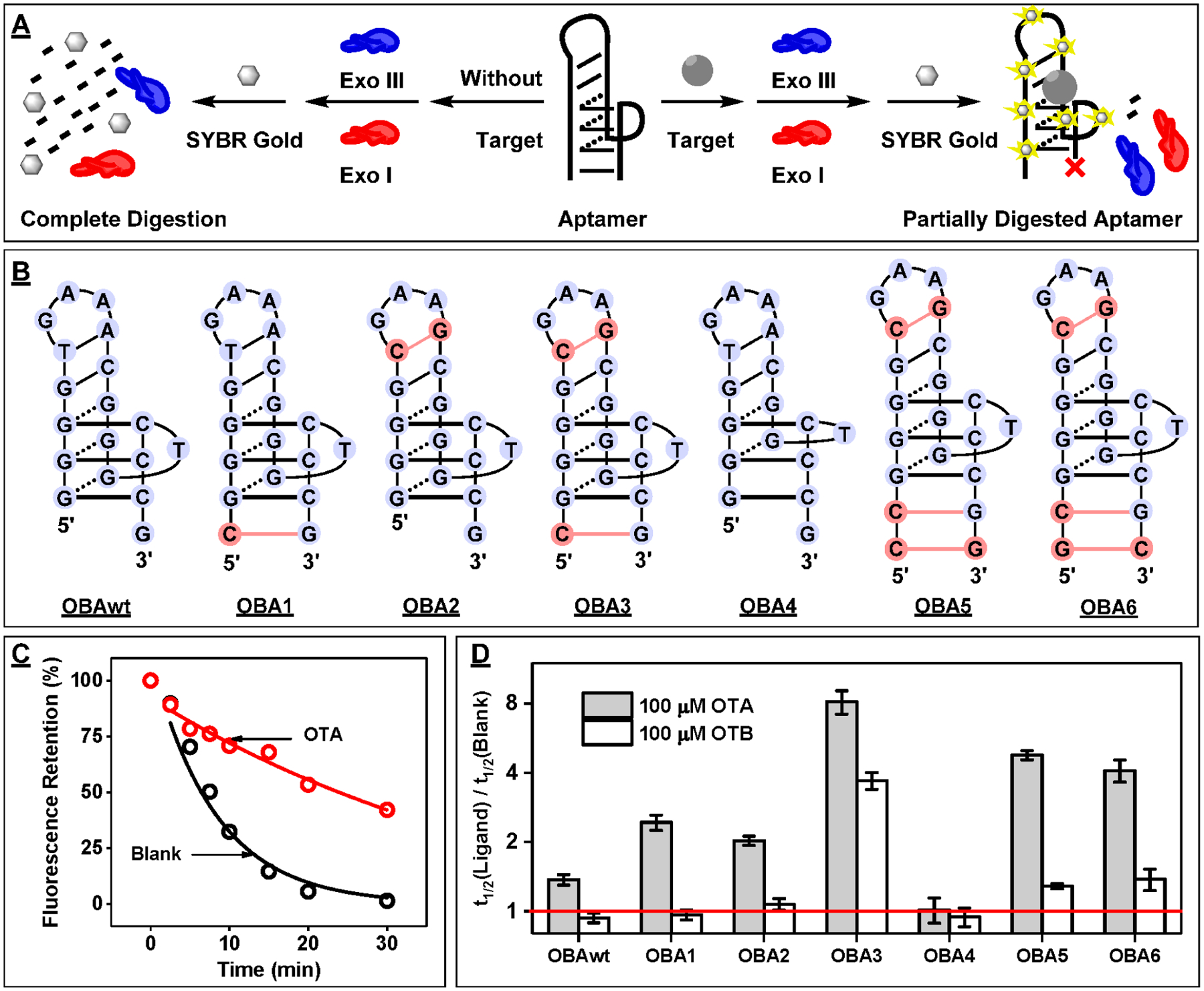
Design and characterization of binding properties of OBAwt and six mutant derivatives. (A) Schematic of the exonuclease digestion assay based on Exo III and Exo I. (B) Secondary structure of ligand-bound aptamers, with mutated nucleotides relative to the OBAwt parent sequence highlighted in red. (C) Time-course plot of OBA3 digestion by Exo III and Exo I in the absence and presence of 25 μM ochratoxin A (OTA). (D) The half-life (t1/2) ratio of the digestion reaction was used to determine the relative binding affinity of OBAwt and its six mutants to 100 μM OTA or OTB. The red line indicates a t1/2 ratio of 1, which means there was no inhibition of aptamer digestion.
To demonstrate this, we performed a series of exonuclease digestion experiments with a well-studied ochratoxin A (OTA) aptamer, OBAwt, and a variety of derivatives of this sequence (Figure 1B). Xu et al. reported that the binding domain of OBAwt is composed of a G-G-C DNA triplex with a short hairpin and a GAA loop.21 They subsequently engineered five mutants via base mutations and insertions that stabilize either the DNA triplex (OBA1), the short hairpin (OBA2), or both structures (OBA3, OBA5, and OBA6) and characterized their binding affinities. To demonstrate the importance of the DNA triplex, they also generated one additional mutant (OBA4) by deletion of a guanine within the triplex, resulting in complete loss of affinity. These sequences and DNA triplex structure have not been previously tested using the exonuclease mixture. We first digested the 19-nt OBA3 sequence, which tightly binds OTA with a dissociation constant (KD) of 1.4 μM.21,39 The digestion process was monitored by collecting aliquots of the reaction mixture at different time-points, followed by quenching with EDTA and fluorescence-based quantification of the remaining oligonucleotides with SYBR Gold. In the absence of the target, OBA3 was completely digested into mononucleotides and the fluorescence intensity rapidly decreased exponentially within 30 min (Figure 1C, blank). However, digestion of OBA3 was inhibited and the decrease of fluorescence intensity was greatly reduced in the presence of 25 μM OTA (Figure 1C, OTA), and a further increase in inhibition was observed when the concentration of OTA was raised to 100 μM (Figure S1A). This suggests that aptamer binding to OTA inhibits enzymatic digestion in a concentration-dependent manner.
To confirm that enzyme inhibition is a result of aptamer-target binding, we performed this same digestion with OBA4, which was reported to have no affinity for OTA.21 We observed no inhibition of digestion of OBA4 in the presence of OTA (Figure S1B), which demonstrates that OTA itself does not inhibit enzymatic activity in the case where the aptamer does not bind to the ligand. We observed that the enzymatic digestion of OBA3 occurred exponentially, possibly indicating first-order reaction kinetics. To confirm this, we digested various concentrations of OBA3 (0.25–2 μM) with or without 25 μM OTA. Both in the absence and presence of OTA, the natural logarithm of fluorescence plotted against time at each aptamer concentration followed a linear trend (Figure S2), which indicates that digestion obeys first-order reaction kinetics under the experimental conditions we employed.40 To determine the half-life (t1/2) of digestions, we fit each time-course plot using a first-order exponential decay equation (see the Material and Methods). Notably, the t1/2 of aptamer digestion in the presence of 25 μM OTA was approximately 4.8-fold higher relative to that in the absence of OTA, showing that OTA strongly binds to the aptamer and the binding inhibits aptamer digestion by the enzyme mixture (Figure 1C).
Having established a correlation between the kinetics of an aptamer’s digestion and its target-binding affinity, we utilized our exonuclease digestion assay to investigate the effects of mutations, insertions, and deletions on aptamer-ligand binding performance. We first digested OBAwt and its six mutants, OBA1–6, in the absence and presence of 100 μM OTA to determine their target-binding affinity. We observed a large variation in the t1/2 of the digestion reactions for each aptamer in the absence of OTA. For example, OBA2, OBA3, and OBA4 had t1/2 values between 4 and 8 min, whereas OBAwt, OBA1, OBA5, and OBA6 had t1/2 values between 15 and 33 min (Figure S3). These different digestion rates are most likely due to the aptamers having different tertiary structures as a result of their respective mutations, insertions, or deletions. We used the ratio of t1/2 in the presence versus the absence of target as a metric to determine the extent of binding-induced inhibition of enzyme digestion for other ligands. A larger t1/2 ratio indicates stronger enzymatic inhibition and presumably greater ligand-binding affinity, while a t1/2 ratio equal to 1 indicates no binding-related enzyme inhibition. Our experimental results demonstrated that OBA4 had no binding to OTA, with a t1/2 ratio of 1, while OBAwt, OBA1, OBA2, OBA3, OBA5, and OBA6 produced t1/2 ratios of 1.4, 2.4, 2.0, 8.3, 4.7, and 4.1, respectively (Figure 1D). These results indicate that OBA3 has the highest OTA affinity, while the other mutants have lower affinity (OBA5 > OBA6 > OBA1 > OBA2 > OBAwt > OBA4). These results closely correspond to the affinities measured by Xu et al. using a fluorescence polarization technique, and we also obtained similar results using ITC. For example, the KD values of OBA3, OBA5, and OBA1 for OTA were 1.8 ± 0.1, 3.4 ± 0.1, and 5.5 ± 0.2 μM, respectively (Figures S4–6). This confirms that our assay can accurately profile the relative target-binding affinity of aptamer mutants regardless of differences in their sequence, length, or structure.
We further tested the specificity of these six mutants and OBAwt against 100 μM ochratoxin B (OTB), which differs from OTA by a single chlorine atom on the coumarin ring. In the presence of OTB, OBAwt, OBA1, OBA2, and OBA4 had t1/2 ratios of nearly 1 (Figure 1D), indicating little or no affinity for OTB. However, OBA5 and OBA6 demonstrated moderate enzyme inhibition, with t1/2 ratios of 1.3 and 1.4, which indicated weak binding to OTB. Digestion of OBA3 was strongly inhibited by OTB, with a t1/2 ratio of 3.7, demonstrating tight binding (Figure 1D). These results again correlated well with our ITC results which determined the KD values for OTB of 25.5 ± 1.2, 59.9 ± 6.9, and 74.1 ± 9.5 μM for OBA3, OBA5, and OBA1, respectively (Figures S4–6). Overall, our digestion results show that, although OBA1 and OBA2 have low affinity for OTA, they are capable of distinguishing OTA from OTB with high specificity. And while OBA3 has greater affinity for OTA, its specificity is poorer relative to OBA1 and OBA2. Thus, t1/2 ratio can be used to discriminate high-affinity binding from weaker-binding ligands and to report the relative binding affinity of an aptamer to various ligands.
Characterizing a Cross-Reactive ATP-Binding Aptamer.
To demonstrate the generality of our exonuclease fluorescence assay, we next studied a well-characterized 33-nt ATP-binding DNA aptamer32,34 (ATPwt). This aptamer also binds to adensoine-5′-diphosphate (ADP), adenosine-5′-monophosphate (AMP), and ADE but not to uridine-5′-triphosphate (UTP), guanosine-5′-triphosphate (GTP), or cytosine-5′-triphosphate (CTP).34,41 In order to determine whether our assay could accurately profile the binding spectrum of ATPwt, we first confirmed that the digestion of this aptamer follows first-order kinetics by digesting various concentrations of ATPwt (0.25–2 μM) with or without 250 μM ATP (Figure S7). We then digested ATPwt in the absence and presence of 250 μM ATP or its analogues. The t1/2 ratio of digestion in the presence of UTP, GTP, and CTP was approximately 1, which corresponds to previous reports showing that ATPwt does not bind to these molecules.34,41 However, digestion of ATPwt was greatly reduced in the presence of ATP, ADP, AMP, and ADE (Figure 2A), with t1/2 ratios of 13.8, 14.0, 11.2, and 14.9, respectively (Figure 2B), confirming that binding to these ligands strongly inhibits exonuclease digestion. These t1/2 ratios were essentially indistinguishable, which is most likely due to the saturating target concentration (250 μM) used in this experiment. When the same experiment was performed with 100 μM ligand, we observed significant differences, with a t1/2 ratio of 4.0, 6.4, 2.4, and 13.8 for ATP, ADP, AMP, and ADE, respectively (Figure S8 and Figure 2B). This indicated the following binding preference: ADE > ADP > ATP > AMP. We confirmed this by ITC, obtaining a K1/2 (concentration of ligand required to reach half saturation) of 4.2 ± 0.1, 6.3 ± 0.1, 10.2 ± 0.2, and 14.1 ± 0.2 μM for ADE, ADP, ATP and AMP, respectively (Figure S9). Again, t1/2 ratio is clearly a reliable indicator of the relative binding affinities of an aptamer to various ligands, including those with similar affinities.
Figure 2.
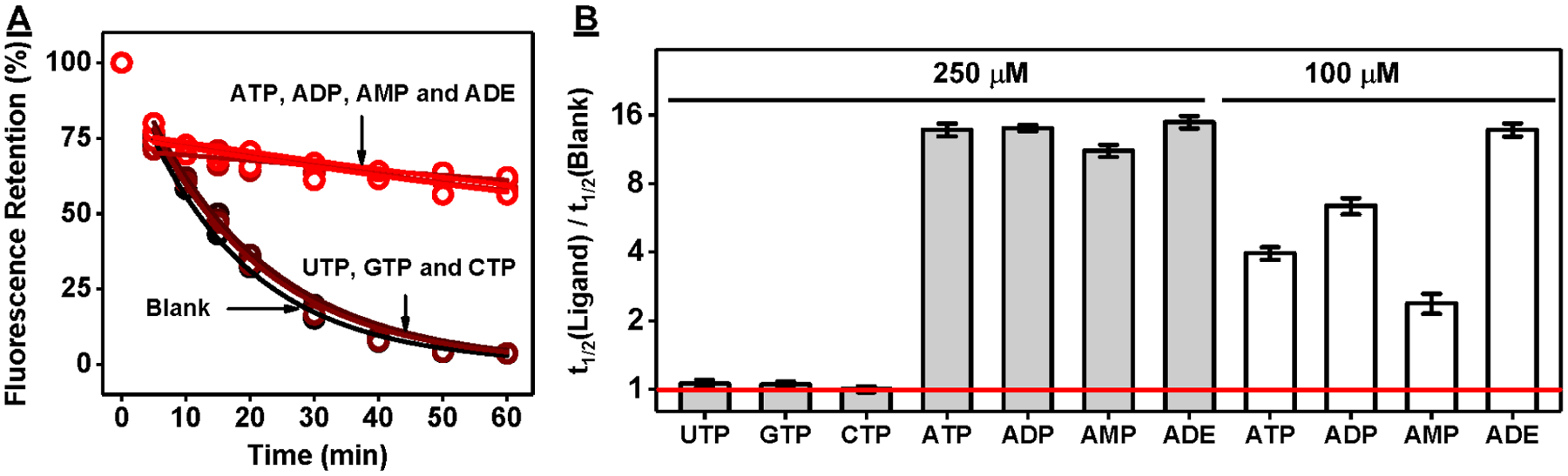
Exonuclease-based fluorescence profiling of ATPwt binding to various targets. (A) Time-course plot of ATPwt digestion by Exo III and Exo I in the absence and presence of various ribonucleotides at a concentration of 250 μM. (B) The t1/2 ratio was used to determine the relative binding affinity to each ligand at 100 and 250 μM. The red line indicates a t1/2 ratio of 1, which reflects no inhibition of aptamer digestion.
Having demonstrated accurate and sensitive profiling of aptamer-ligand binding, we next tested whether this method could be used to screen for new aptamer candidates with improved binding properties from a panel of mutants. ATPwt has limited analytical utility in that it binds to ADE, AMP, ADP, and ATP, which typically coexist at similar concentrations in biological media,42 and we therefore set out to identify a more specific aptamer that selectively binds only to one of these molecules. On the basis of its NMR structure,43 ATPwt possesses two binding domains within the minor groove of a DNA helix composed of G·G and A·G mismatches (Figure 3A). In the first binding site, G12 forms hydrogen bonds with the adenine base of the ligand. G11 and G21 base stack with G12 to stabilize this binding domain, and further base-stacking occurs between the adenine of the ligand and G22. In the second binding site, the adenine of the ligand hydrogen bonds with G25, which is supported by base-stacking with G8 and G24. Similar stacking is also observed between the ligand and G9. These G’s are clearly crucial for ligand binding, and there is a high likelihood that mutations at these positions would greatly reduce the affinity of the aptamer. However, mutating the nucleotides at the periphery of these binding sites could yield aptamer sequences with improved binding affinity and/or specificity. We therefore mutated nucleotides G10, A13, A23, and A26 adjacent to the binding domain of ATPwt, generating a set of 12 point mutants in which we substituted each nucleotide with the three alternate nucleobases (Figure 3A, and Table S1).
Figure 3.
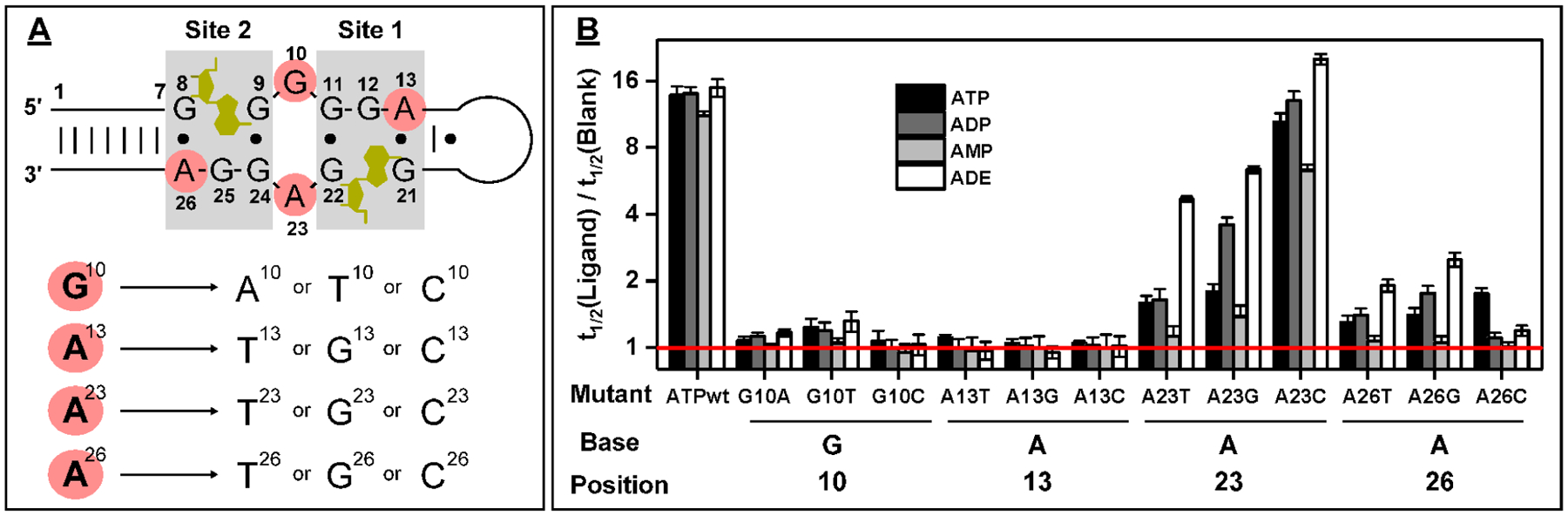
Design and characterization of ATPwt mutants. (A) Secondary structure of ligand-bound ATPwt, with mutated nucleotides highlighted in red. The ligand is highlighted in gold, and nucleotide positions are marked. Twelve different point mutants were generated by changing G10, A13, A23, A26 to either of the three alternative nucleobases. (B) The t1/2 ratio (with log2 scale) for each mutant in the presence of ATP, ADP, AMP, or ADE. The red line indicates a t1/2 ratio of 1, reflecting no binding-induced inhibition of enzyme digestion.
We then investigated the affinity and specificity of these mutants for ATP, ADP, AMP, and ADE using our exonuclease fluorescence assay. We digested each mutant as well as ATPwt in the absence of ligand and found that each had a different rate of digestion (Figures S10–S13). G10T, A13T, A13G, A13C, A23T, A23C, A26T, and A26G were digested slightly faster (t1/2 = 6–8 min) than ATPwt (t1/2 = 10 min). Two mutants (G10C and A23G) had the same t1/2 as ATPwt, while the remaining two (G10A and A26C) had much slower digestion rates (t1/2 = 25–35 min). We believe that these disparities can be attributed to differences in the tertiary structures of the aptamers. All of the mutants were then digested in the presence of 250 μM of each ligand, and we observed different digestion rates and levels of enzymatic inhibition (Figures S10–S13). Our results demonstrated that G10 and A13 mutants had no meaningful affinity for any of the analogues, with t1/2 ratios of ~1 (Figure 3B). A26 mutants demonstrated low levels of enzymatic inhibition for all tested ligands, with t1/2 ratios ranging from 1 to 2.4, indicating greatly reduced affinity relative to ATPwt, with t1/2 ratios of 11.2–14.9. Notably, we also observed that mutations at this site altered aptamer specificity: A26T and A26G showed a minor preference for ADE over the other analogues, while A26C had slightly better specificity for ATP (Figure 3B). A23 mutants displayed the highest levels of enzymatic inhibition among all mutants, with a notable preference for ADE. Notably, A23T had the highest ligand specificity, with a t1/2 ratio of 4.6 for ADE versus <1.7 for ATP, ADP, and AMP (Figure 3B). To more closely evaluate the binding spectra of the A23 mutants, we performed the same digestion experiment at a lower ligand concentration (100 μM). As expected, the level of exonuclease inhibition was lower, but we were better able to identify differences in mutant specificity. These results confirmed that A23T offered the greatest level of ligand discrimination, with a t1/2 ratio of 2.5 for ADE but nearly 1 for the other analogues (Figure S14). These results clearly show that this exonuclease fluorescence assay enables rapid post-SELEX identification of alternative aptamer sequences with altered binding properties. We further determined the relationship between the t1/2 ratios of the mutants and their ligand affinity. On the basis of the t1/2 ratios, we predicted that the affinity of the A23 and A26 mutants for ADE would be A23C > A23G ≈ A23T > A26G ≈ A26T. We confirmed an identical relative affinity profile for this set of mutants via ITC (Figure S15).
Identification of a New Highly ADE-Specific Aptamer.
On the basis of our point mutation experiments, G10 is essential for target binding, while T23 greatly enhances specificity toward ADE. According to the reported three-dimensional structure of ATPwt, nucleotides G10 and T23 base-stack with binding sites 1 and 2, respectively.43 Previous work has also reported that interchanging the nucleotides at these two positions in ATPwt does not impair aptamer affinity.34 To confirm that this held true for A23T, we designed a double mutant (G10T-A23G) in which we swapped the G at position 10 with the T at position 23 (Figure 4A). Digestion of G10T-A23G in the absence and presence of 250 μM ATP or its analogues demonstrated greater inhibition than A23T in the presence of ADE (Figure 4B), with a t1/2 ratio of 9.5 (Figure 4C). This double mutant also exhibited excellent specificity against other ligands, with a t1/2 ratio of 1.6, 2.4, and 1.4 for ATP, ADP and AMP, respectively (Figure 4C). Using ITC, we determined that G10T-A23G tightly binds to ADE, with a K1/2 of 15.3 ± 0.2 μM (Figure S16).
Figure 4.
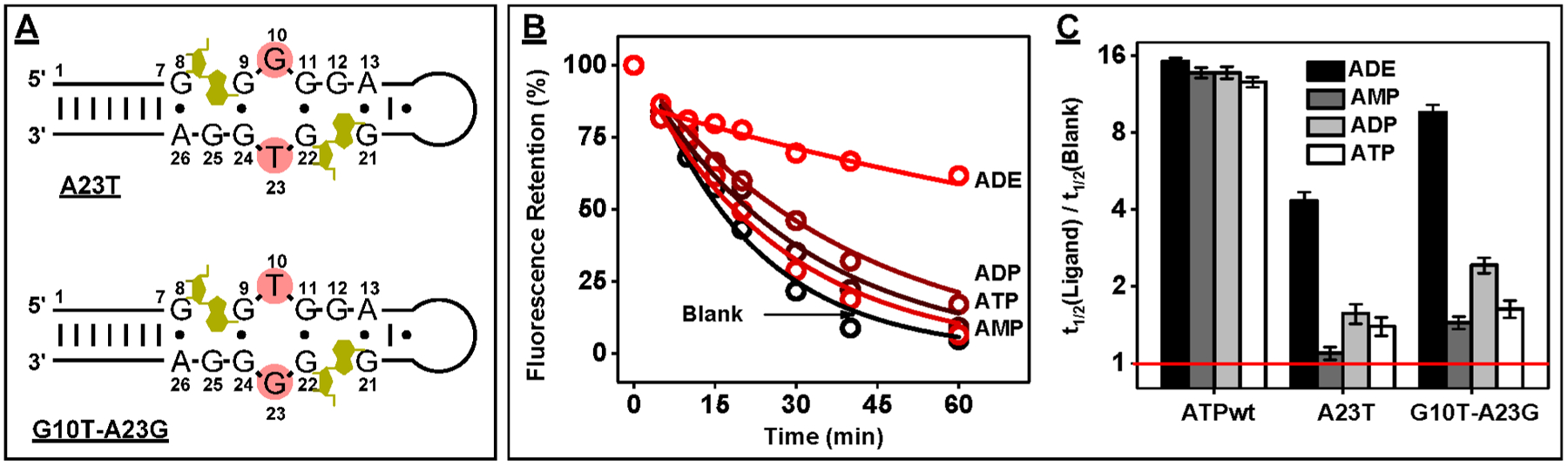
Design and characterization of a double-mutant aptamer. (A) Inversion of nucleotides G10 and T23 in construct A23T results in a double-mutant G10T-A23G. (B) Fluorescence time-course plot of G10T-A23G digestion by Exo III and Exo I in the absence and presence of 250 μM ADE, AMP, ADP, and ATP. (C) The t1/2 ratio (with log2 scale) for ATPwt, A23T, and G10T-A23G in the presence vs the absence of 250 μM of each ligand. The red line indicates a t1/2 ratio of 1, reflecting no inhibition of aptamer digestion. Error bars represent the standard error of fitting.
A23T and G10T-A23G possess similar target affinities but have different t1/2 ratios. To determine the reason for the greater ADE-induced enzymatic inhibition of G10T-A23G relative to A23T we identified the digestion products using polyacrylamide gel electrophoresis (PAGE). We observed that exonuclease digestion halted 3 or 4 nt from the 3′ end of both aptamers, resulting in two major products of 30 nt and 29 nt (Figure S17, parts A and B). We synthesized these digestion products of A23T (A23T-30 and A23T-29) and G10T-A23G (G10T-A23G-30 and G10T-A23G-29) (Figure S18) and determined their affinity for ADE using ITC (Figure S19). A23T-30 (K1/2 = 18.6 ± 1.7 μM), G10T-A23G-30 (K1/2 = 18.1 ± 0.4 μM), and G10T-A23G-29 (K1/2 = 18.5 ± 0.4 μM) had similar ADE affinities to their parent aptamers. However, A23T-29 had nearly 3-fold poorer affinity for this ligand (K1/2 = 37.4 ± 6.2 μM). Thus, the observed t1/2 ratios are not only linked to the binding affinity of the parent aptamer but the truncated products as well. This accounts for the lower enzymatic inhibition and smaller t1/2 ratio value displayed by A23T relative to G10T-A23G.
Fabrication of E-AB Sensors from ADE-Specific Aptamers.
ADE is a ubiquitous extracellular signaling molecule that has diagnostic value for cardiovascular diseases including cerebral ischemia, tissue ischemia, and cardiac ischemia.44–46 Basal ADE levels in the cerebrospinal fluid and circulatory system are in the nanomolar range but increase to 1–50 μM during ischemic episodes, with large variations.47–49 We anticipated that our engineered aptamers A23T and G10T-A23G could be useful for the clinical detection of ADE in serum due to their high specificity.
E-AB sensors represent an excellent platform for the sensitive and specific detection of small-molecule analytes in complex samples such as serum50 and whole blood.51 These consist of thiolated aptamers that are tagged with electroactive molecules (e.g., methylene blue, MB) and immobilized onto gold electrodes. Aptamer-ligand binding induces a conformational change that repositions the MB tag, resulting in a target-concentration-dependent change in current. We have previously determined that exonuclease-truncated aptamers have structure-switching functionality33,52 and thus determined that it should be feasible to directly incorporate the exonuclease-truncated ADE-specific aptamers into an E-AB sensor. We first assessed the structure-switching functionality of A23T-30, A23T-29, G10T-A23G-30, and G10T-A23G-29 based on circular dichroism (CD), a well-established method for studying conformational changes in aptamers.53 In the absence of target, we observed a negative peak at 245 nm and a broad positive peak ranging from 255 to 300 nm, with a maximum at 265 nm (Figure S20), indicating an unfolded single-stranded DNA structure.53 Upon addition of ADE, all aptamers produced similar spectra but with target-concentration-dependent increases in the intensity of all peaks (Figure S20). The observed increases in the intensities of the 245 and 265 nm peaks indicate a target-induced transition from a single-stranded structure to a folded structure with anti-anti stacking of guanine bases.54 This is consistent with the previously described NMR structure of the ligand-bound ATPwt aptamer.43 Since A23T-29 and G10T-A23G-29 displayed the largest target-induced conformational changes, we used these aptamers to fabricate the E-AB sensors. We further confirmed that these aptamers retain high specificity for ADE via ITC. A23T-29 exhibited binding affinities of 37.4 ± 6.2, >1000, 285.4 ± 5.7, and 406.3 ± 21.8 μM for ADE, AMP, ADP, and ATP, respectively (Figure S21). Similarly, G10T-A23G-29 exhibited binding affinities of 18.5 ± 0.4, 214.7 ± 7.2, 162.9 ± 2.8, and 171.9 ± 4.3 μM for ADE, AMP, ADP, and ATP, respectively (Figure S22).
We then synthesized 5′-thiolated/3′-MB-modified versions of G10T-A23G-29 and A23T-29 (G10T-A23G-29-MB and A23T-29-MB) and immobilized 50 nM of each aptamer onto individual gold disk electrodes to fabricate single-aptamer E-AB sensors. Using a previously reported method,38 we determined that both sensors had similar surface coverages of 4.6 ± 0.3 and 4.2 ± 0.3 pmol/cm2, respectively (Figure S23). We used both sensors to perform ADE detection (Figure S24A). The G10T-A23G-29-MB sensor produced a linear range from 1 to 25 μM with a limit of detection of 1 μM, whereas the A23T-29-MB sensor had a linear range from 25 to 500 μM with a limit of detection of 25 μM (Figure S24B). The lower sensitivity of the A23T-29-MB sensor can be attributed to the lower ADE affinity of this aptamer relative to G10T-A23G-29.
Clinically useful detection requires a sensor that responds to ADE across a large range of concentrations (1–50 μM),47–49 and we were unable to achieve this using our single-aptamer E-AB sensors. It has been reported that the linear range of an aptamer-based sensor can be expanded by using a mixture of different aptamers with varying target-binding affinities.55–57 We therefore fabricated a dual-aptamer E-AB sensor using a 1:1 ratio of A23T-29-MB and G10T-A23G-29-MB (Figure 5A). This dual-aptamer modified electrode had similar surface coverage (4.3 ± 0.3 pmol/cm2) to the single-aptamer E-AB sensors fabricated with each individual aptamer (Figure S23). Importantly, the dual-aptamer sensor produced a broader linear range from 1 to 100 μM ADE (Figure S24B), which was probably facilitated by the relatively low ADE affinity of A23T-29-MB. Importantly, this dual-aptamer sensor still retained a limit of detection of 1 μM, presumably due to the high affinity of G10T-A23G-29-MB (Figure S24B). Finally, we used our dual-aptamer E-AB sensor to perform detection in 50% fetal bovine serum and evaluated the sensor’s specificity against ADE as well as AMP, ADP, ATP, UTP, GTP, and CTP. The dual-aptamer sensor produced ~30% lower current in 50% serum (Figure S25), regardless of the absence or presence of ADE, presumably due to protein fouling of the electrode surface.58 However, the linear range and limit of detection remained the same in 50% serum as in buffer (Figure 5B and Figure S24B), demonstrating the sensor’s excellent performance in biological samples. We further determined that the dual-aptamer E-AB sensor had less than 10% cross-reactivity to the various nucleotide analogues in 50% serum relative to ADE at a concentration of 100 μM (Figure 5C and Figure S26), reflecting the high specificity of the aptamers we used.
Figure 5.
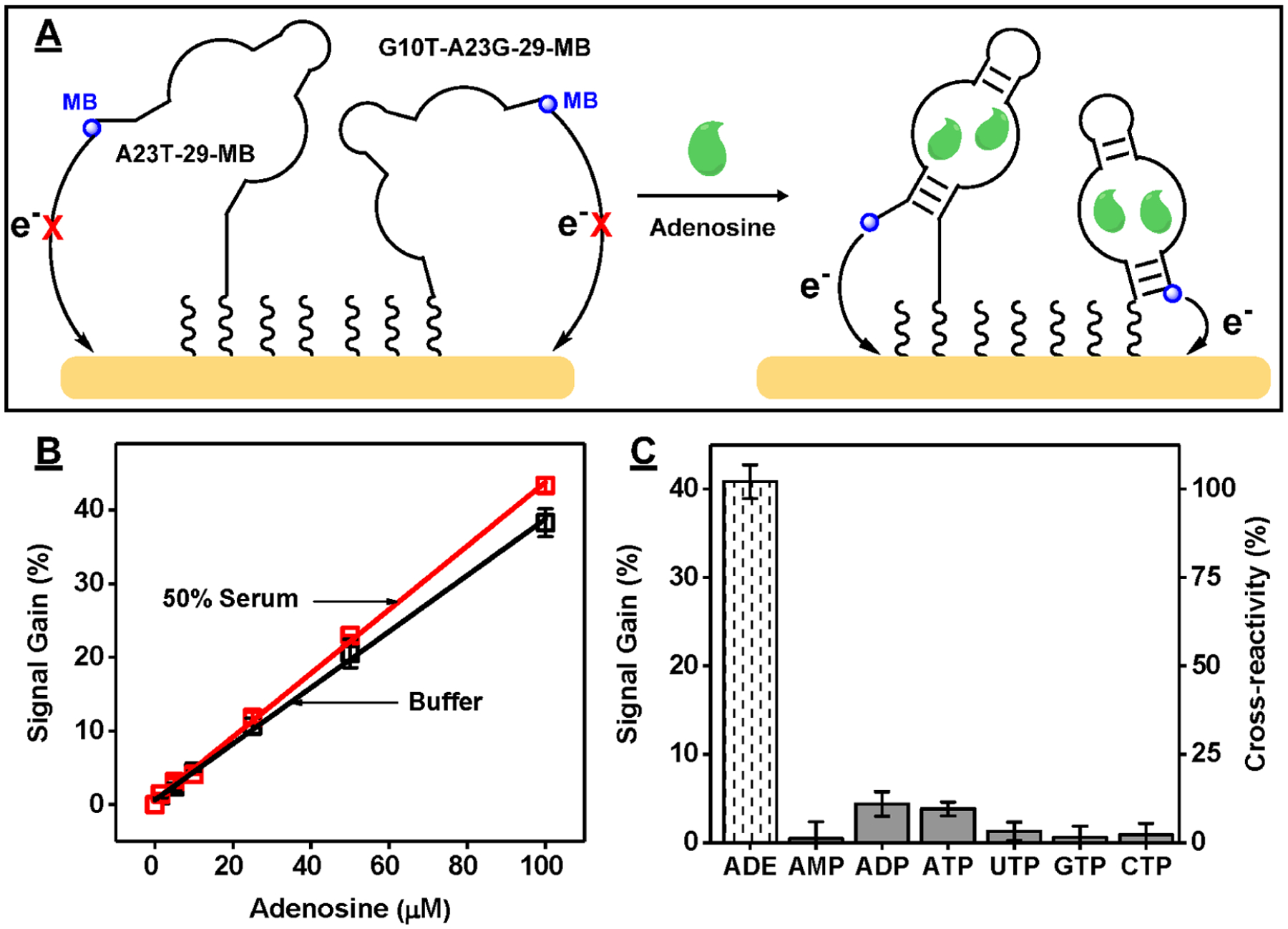
Specific detection of ADE in serum using the dual-aptamer E-AB sensor. (A) Schematic of the dual-aptamer-modified E-AB sensor. (B) Linear range of the sensor in buffer and 50% fetal bovine serum. (C) Signal gain and cross-reactivity (relative to ADE) of the sensor to 100 μM ADE, AMP, ADP, ATP, UTP, GTP, and CTP in 50% serum. Error bars represent the average standard deviation of three electrodes.
Generality of the Exonuclease Fluorescence Assay for Protein-Binding Aptamers.
We finally used our exonuclease fluorescence assay to determine the binding characteristics of three different G-quadruplex aptamers that bind to human α-thrombin: Tasset,59 Bock,60 and Bock-hang61 in Tris-buffered saline. We digested 500 nM aptamer in the absence and presence of 500 nM thrombin or human factor X, which was reported to have no binding affinity to these aptamers.62 For all three aptamers, relative fluorescence intensities decreased in the absence of target at an exponential rate down to 2–5% within 30 min, indicating complete digestion into mononucleotides (Figure 6A–C). The aptamers were likewise completely digested in the presence of factor X (t1/2 ratios: ~1), confirming that they did not have any affinity for this protein. In the presence of thrombin, the digestion of all three aptamers was greatly inhibited, with t1/2 ratios of 14.0, 10.4, and 3.6 for Tasset, Bock, and Bock-hang, respectively (Figure 6D). These results suggested that the thrombin affinity of these aptamers follows the order of Tasset > Bock > Bock- hang. We confirmed this by measuring the thrombin-binding affinity of these aptamers using ITC in Tris-buffered saline at 23 °C, observing KD’s of 13.6 ± 3.1, 23.1 ± 5.4, and 97 ± 19 nM for Tasset, Bock, and Bock-hang, respectively (Figure S27).
Figure 6.
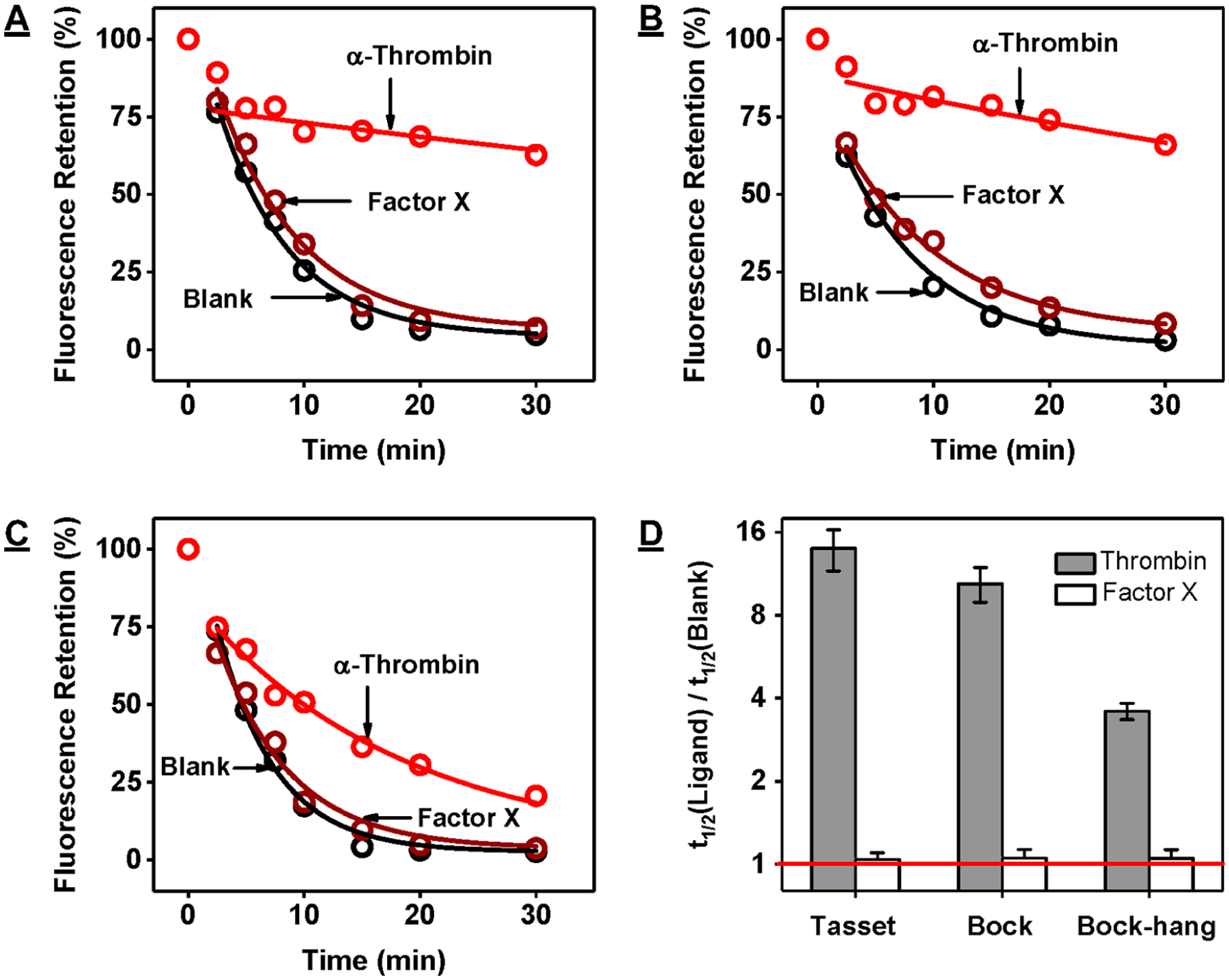
Exonuclease-based fluorescence profiling of thrombin-binding aptamers. Time-course plot of digestion of 500 nM (A) Tasset, (B) Bock, and (C) Bock-hang by Exo III and Exo I in the absence (black) and presence of 500 nM human α-thrombin (red) or human factor X (brown). (D) The half-life (t1/2) ratio of the digestion reaction was used to determine relative aptamer binding affinity to α-thrombin and factor X. The y-axis is log2-scaled. The red line indicates a t1/2 ratio of 1, which means there was no inhibition of aptamer digestion.
We then performed our exonuclease fluorescence assay to characterize a stem-loop-structured DNA aptamer that has nanomolar affinity for IgE but at least 1000-fold lower affinity for IgG.63 We digested the aptamer in the absence and presence of these proteins in Tris-buffered saline. This aptamer was completely digested in the absence of target and in the presence of 500 nM human IgG, confirming that the aptamer does not strongly bind this protein. However, digestion was greatly inhibited in the presence of 500 nM human IgE, with a t1/2 ratio of 5.4 (Figure S28), demonstrating that the aptamer binds IgE. Thus, our method is generalizable for protein-binding aptamers regardless of their binding affinity, secondary structure, or the size of the target protein.
CONCLUSION
Aptamers offer a variety of advantages relative to antibodies that make them desirable biorecognition elements for a variety of applications. The SELEX procedure efficiently partitions and amplifies target-binding sequences from large random oligonucleotide libraries but often yields aptamers with suboptimal binding properties due to the limited sequence capacity of the library employed for selection and the biases from PCR amplification. Post-SELEX mutagenesis can facilitate identification of new high-performance aptamers, but current characterization strategies are limited by low throughput and the need for specialized instrumentation (e.g., SPR or ITC) or require the use of aptamers or targets that are intrinsically fluorescent or fluorescently labeled (e.g., for microarray assays), which is unfeasible for small-molecule-binding aptamers.
We have developed a novel exonuclease-based fluorescence assay for characterizing the binding properties of small-molecule-binding aptamers in a high-throughput, label-free manner. We determined that there is a strong correlation between relative aptamer-ligand binding affinity and the kinetics of aptamer digestion by the exonucleases Exo III and Exo I. By profiling previously reported aptamers and mutants that bind to ochratoxin A, we determined that the ratio of exonuclease digestion half-lives (t1/2) in the presence versus the absence of target could be used to compare aptamer affinity in an unbiased fashion and the aptamer with a longer t1/2 ratio has a higher binding affinity compared to those that yielded shorter t1/2 ratios. After examining the affinity and specificity of 14 aptamer-ligand pairs, we were able to identify those with the highest affinity for OTA (OBA3) and the greatest capability to distinguish OTA from OTB (OBA1 and OBA2). Importantly, the results of our exonuclease assay closely matched those in a previous report as well as results from the gold-standard method, ITC.
We then characterized a DNA aptamer isolated by Huizenga and Szostak that binds to ATP, ADP, AMP, and ADE with similar binding affinities and improved its binding characteristics by designing and testing 13 mutants. We screened 59 aptamer-ligand pairs and identified two new aptamers with high specificity for ADE relative to its phosphorylated analogues while still retaining the high affinity of the parent aptamer sequence. Again, the results of our exonuclease-based fluorescence assay correlated well with ITC but offer a more high-throughput and cost-effective alternative. Notably, the Liu group64 recently engineered a new derivative of the ADE-binding DNA aptamer (A10-excised) that also specifically binds to ADE, but this has 20-fold lower ADE affinity compared to our aptamers under the same buffer conditions (Figure S29). Our assay can also generate structure-switching aptamers during the screening process, which can be directly incorporated into folding-based sensing platforms. As a demonstration, we employed two ADE-specific, structure-switching aptamers identified from our screen to construct a dual-aptamer E-AB sensor that achieved sensitive and specific detection of ADE with a measurable limit of detection of 1 μM and no cross-reactivity to ATP, ADP, or AMP in 50% serum.
Finally, we assessed the generality of our assay by determining the binding properties of protein-binding aptamers. We digested three G-quadruplex-structured DNA aptamers which bind to human α-thrombin and once again observed a clear correlation between aptamer-ligand binding affinity and t1/2 ratio. We also performed our assay with a stem-loop-structured IgE-binding aptamer and determined that the aptamer binds to IgE but not IgG, which is consistent with previously reported findings. These examples demonstrate that our assay is not only applicable for small-molecule-binding aptamers but also for protein-binding aptamers regardless of their binding affinity, secondary structure, or the size of the target protein.
On the basis of the systems we have studied, we provide a guide to interpreting t1/2 ratio values as well as using other methods of analyzing the kinetic data. The t1/2 ratio values are not necessarily directly proportional to the KD of the aptamer, but the t1/2 ratio can be used as a relative measure of affinity. Generally, lower t1/2 ratios imply weaker binding and vice versa. As described above, the t1/2 ratio is dependent on the affinity of both the parent and truncated products. Thus, for a single aptamer, t1/2 ratios can be used to compare the relative affinity of the aptamer to a set of ligands. Similarly, t1/2 ratios can also be used to evaluate the relative affinity of a set of aptamer mutants derived from a common parent aptamer for the same ligand. We have also identified two other parameters to assess exonuclease digestion assay data: first-order reaction rates of digestion and aptamer resistance to digestion. To demonstrate these different methods of analysis, we digested 1 μM OBA3 with Exo III and Exo I in the presence of 0–100 μM OTA (Figure S30A). We respectively plotted the reduction in first-order reaction rate of aptamer digestion (Figure S30B), aptamer resistance to digestion (calculated as resistance value)(Figure S30C), and t1/2 ratio (Figure S30D) at each ligand concentration. We found that the first-order reaction rate was the most sensitive parameter to ligand concentration, followed by aptamer resistance. However, the t1/2 ratio demonstrated a linear relationship across a broader range of target concentrations. We believe that all three parameters are interchangeable and can be used to accurately perform data analysis.
Together, these results indicate that our exonuclease fluorescence assay can be used to determine the binding profiles of aptamers regardless of their sequence, structure, binding affinity, or the properties of the ligand being tested. Although we studied aptamer mutants designed with the guidance from their known tertiary structures, we believe that our method can be generally applied to other small-molecule-binding aptamers without any need for prior knowledge of the target-binding site or overall structure. We recommend two methods to design mutants. First, for in silico maturation techniques, genetic algorithms create mutants via a combination of crossing over different aptamer sequences and point mutations, and these are then tested in vitro. The outcome of these experiments is used by the algorithm to generate a new batch of mutants, and the process is iterated several times until an aptamer with the desired binding characteristics is found.22 Second, for aptamers found through high-throughput sequencing, the sequencing data can be analyzed using clustering algorithms such as aptaMOTIF65 and MEME66 to identify consensus sequences and variable motifs or nucleotides. This information can be used to design mutants. Although it is difficult to estimate the likelihood of finding a better aptamer from a collection of mutants, this probability can be greatly increased by testing more mutants and higher-order mutants (i.e., double or triple mutants). Since our assay only requires a single mix-and-read step, it can readily be performed using multichannel pipettes and 384-microwell plates to screen hundreds of aptamer-ligand combinations simultaneously within <2 h or even adapted into a high-throughput screening format using an automated liquid handling system. As such, we believe our approach should allow for the greatly accelerated characterization of aptamers for a variety of applications.
Supplementary Material
Funding
This work was supported by the National Institutes of Health—National Institute on Drug Abuse (R15DA036821-01A1, R21DA045334–01A1) and the National Science Foundation (1905143).
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c09559.
Experimental details, oligonucleotide sequences used in this work, concentrations of aptamers and ligands used for ITC and determined KD, fluorescence time-course of the digestion of OTA-binding aptamers with Exo III and Exo I in the absence and presence of OTA or OTB, OBA3 or ATPwt digestion kinetics by Exo III and Exo I, OBA1, OBA3, and OBA5 affinity for OTA and OTB, digestion of ATPwt with Exo III and Exo I, affinity of ATPwt to ATP, ADP, AMP, and ADE, binding profile of various mutant aptamers and A23 mutants, affinity of single-site mutants and G10T-A23G for ADE, PAGE analysis of A23T and G10T-A23G products, sequence and secondary structure of A23T-30, A23T-29, G10T-A23G-30, and G10T-A23G-29, affinity of ADE-specific aptamer mutants, characterizing structure-switching functionality of A23T and G10T-A23G digestion products, A23T-29 and G10T-A23G-29 affinity, surface coverage, performance, and SWV curves of E-AB sensors fabricated using G10T-A23G-29-MB, A23T-29-MB, or both, dual-aptamer E-AB sensor response to ADE, AMP, ADP, or ATP or other nucleotide triphosphates, characterization of the thrombin-binding affinity of Tasset, Bock, and Bock-hang, exonuclease-based fluorescence profiling of an IgE-binding aptamer, characterization of ligand-binding affinity of a recently published ADE-binding aptamer to ADE, and characterization of OBA3 affinity for OTA (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c09559
REFERENCES
- (1).Tuerk C; Gold L Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- (2).Ellington AD; Szostak JW In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- (3).Yang K-A; Pei R; Stojanovic MN In Vitro Selection and Amplification Protocols for Isolation of Aptameric Sensors for Small Molecules. Methods 2016, 106, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dunn MR; Jimenez RM; Chaput JC Analysis of Aptamer Discovery and Technology. Nat. Rev. Chem 2017, 1, 0076. [Google Scholar]
- (5).Jayasena SD Aptamers: An Emerging Class of Molecules That Rival Antibodies in Diagnostics. Clin. Chem 1999, 45, 1628–1650. [PubMed] [Google Scholar]
- (6).McKeague M; McConnell EM; Cruz-Toledo J; Bernard ED; Pach A; Mastronardi E; Zhang X; Beking M; Francis T; Giamberardino A; Cabecinha A; Ruscito A; Aranda-Rodriguez R; Dumontier M; DeRosa MC Analysis of In Vitro Aptamer Selection Parameters. J. Mol. Evol 2015, 81, 150–161. [DOI] [PubMed] [Google Scholar]
- (7).Ruscito A; DeRosa MC Small-Molecule Binding Aptamers: Selection Strategies, Characterization, and Applications. Front. Chem 2016, 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Klug SJ; Famulok M All You Wanted to Know about SELEX. Mol. Biol. Rep 1994, 20, 97–107. [DOI] [PubMed] [Google Scholar]
- (9).Stoltenburg R; Reinemann C; Strehlitz B SELEX-a (R)evolutionary Method to Generate High-Affinity Nucleic Acid Ligands. Biomol. Eng 2007, 24, 381–403. [DOI] [PubMed] [Google Scholar]
- (10).Polz MF; Cavanaugh CM Bias in Template-to-Product Ratios in Multitemplate PCR. Appl. Environ. Microbiol 1998, 64, 3724–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Takahashi M; Wu X; Ho M; Chomchan P; Rossi JJ; Burnett JC; Zhou J High Throughput Sequencing Analysis of RNA Libraries Reveals the Influences of Initial Library and PCR Methods on SELEX Efficiency. Sci. Rep 2016, 6, 33697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Levay A; Brenneman R; Hoinka J; Sant D; Cardone M; Trinchieri G; Przytycka TM; Berezhnoy A Identifying High-Affinity Aptamer Ligands with Defined Cross-Reactivity Using High-Throughput Guided Systematic Evolution of Ligands by Exponential Enrichment. Nucleic Acids Res. 2015, 43, No. e82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bartel DP; Zapp ML; Green MR; Szostak JW HIV-1 Rev Regulation Involves Recognition of Non-Watson-Crick Base Pairs in Viral RNA. Cell 1991, 67, 529–536. [DOI] [PubMed] [Google Scholar]
- (14).Famulok M Molecular Recognition of Amino Acids by RNA-Aptamers: An L-Citrulline Binding RNA Motif and Its Evolution into an L-Arginine Binder. J. Am. Chem. Soc 1994, 116, 1698–1706. [Google Scholar]
- (15).Vorobyeva MA; Davydova AS; Vorobjev PE; Pyshnyi DV; Venyaminova AG Key Aspects of Nucleic Acid Library Design for in Vitro Selection. Int. J. Mol. Sci 2018, 19, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Knight R; Yarus M Analyzing Partially Randomized Nucleic Acid Pools: Straight Dope on Doping. Nucleic Acids Res. 2003, 31, No. e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang Z; Oni O; Liu J New Insights into a Classic Aptamer: Binding Sites, Cooperativity and More Sensitive Adenosine Detection. Nucleic Acids Res. 2017, 45, 7593–7601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zheng X; Hu B; Gao SX; Liu DJ; Sun MJ; Jiao BH; Wang LH A Saxitoxin-Binding Aptamer with Higher Affinity and Inhibitory Activity Optimized by Rational Site-Directed Mutagenesis and Truncation. Toxicon 2015, 101, 41–47. [DOI] [PubMed] [Google Scholar]
- (19).Neves MAD; Reinstein O; Saad M; Johnson PE Defining the Secondary Structural Requirements of a Cocaine-Binding Aptamer by a Thermodynamic and Mutation Study. Biophys. Chem 2010, 153, 9–16. [DOI] [PubMed] [Google Scholar]
- (20).Roncancio D; Yu H; Xu X; Wu S; Liu R; Debord J; Lou X; Xiao Y A Label-Free Aptamer-Fluorophore Assembly for Rapid and Specific Detection of Cocaine in Bio Fluids. Anal. Chem 2014, 86, 11100–11106. [DOI] [PubMed] [Google Scholar]
- (21).Xu G; Zhao J; Liu N; Yang M; Zhao Q; Li C; Liu M Structure-Guided Post-SELEX Optimization of an Ochratoxin A Aptamer. Nucleic Acids Res. 2019, 47, 5963–5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Nonaka Y; Yoshida W; Abe K; Ferri S; Schulze H; Bachmann TT; Ikebukuro K Affinity Improvement of a VEGF Aptamer by in Silico Maturation for a Sensitive VEGF-Detection System. Anal. Chem 2013, 85, 1132–1137. [DOI] [PubMed] [Google Scholar]
- (23).Biniuri Y; Albada B; Willner I Probing ATP/ATP-Aptamer or ATP-Aptamer Mutant Complexes by Microscale Thermophoresis and Molecular Dynamics Simulations: Discovery of an ATP-Aptamer Sequence of Superior Binding Properties. J. Phys. Chem. B 2018, 122, 9102–9109. [DOI] [PubMed] [Google Scholar]
- (24).Hendrix M; Priestley ES; Joyce GF; Wong CH Direct Observation of Aminoglycoside - RNA Interactions by Surface Plasmon Resonance. J. Am. Chem. Soc 1997, 119, 3641–3648. [DOI] [PubMed] [Google Scholar]
- (25).Cowan JA; Ohyama T; Wang D; Natarajan K Recognition of a Cognate RNA Aptamer by Neomycin B: Quantitative Evaluation of Hydrogen Bonding and Electrostatic Interactions. Nucleic Acids Res. 2000, 28, 2935–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Baaske P; Wienken CJ; Reineck P; Duhr S; Braun D Optical Thermophoresis for Quantifying the Buffer Dependence of Aptamer Binding. Angew. Chem., Int. Ed 2010, 49, 2238–2241. [DOI] [PubMed] [Google Scholar]
- (27).Torres FE; Kuhn P; De Bruyker D; Bell AG; Wolkin MV; Peeters E; Williamson JR; Anderson GB; Schmitz GP; Recht MI; Schweizer S; Scott LG; Ho JH; Elrod SA; Schultz PG; Lerner RA; Bruce RH Enthalpy Arrays. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 9517–9522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Torres FE; Recht MI; Coyle JE; Bruce RH; Williams G Higher Throughput Calorimetry: Opportunities, Approaches and Challenges. Curr. Opin. Struct. Biol 2010, 20, 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Katilius E; Flores C; Woodbury NW Exploring the Sequence Space of a DNA Aptamer Using Microarrays. Nucleic Acids Res. 2007, 35, 7626–7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Platt M; Rowe W; Knowles J; Day J; Kell DB Analysis of Aptamer Sequence Activity Relationships. Integr. Biol 2009, 1, 116–122. [DOI] [PubMed] [Google Scholar]
- (31).Ketterer S; Fuchs D; Weber W; Meier M Systematic Reconstruction of Binding and Stability Landscapes of the Fluorogenic Aptamer Spinach. Nucleic Acids Res. 2015, 43, 9564–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Canoura J; Wang Z; Yu H; Alkhamis O; Fu F; Xiao Y No Structure-Switching Required: A Generalizable Exonuclease-Mediated Aptamer-Based Assay for Small-Molecule Detection. J. Am. Chem. Soc 2018, 140, 9961–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Liu Y; Yu H; Alkhamis O; Moliver J; Xiao Y Tuning Biosensor Cross-Reactivity Using Aptamer Mixtures. Anal. Chem 2020, 92, 5041–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Huizenga DE; Szostak JW A DNA Aptamer That Binds Adenosine and ATP. Biochemistry 1995, 34, 656–665. [DOI] [PubMed] [Google Scholar]
- (35).Alkhamis O; Yang W; Farhana R; Yu H; Xiao Y Label-Free Profiling of DNA Aptamer-Small Molecule Binding Using T5 Exonuclease. Nucleic Acids Res. 2020, 48, No. e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xiao Y; Lai RY; Plaxco KW Preparation of Electrode-Immobilized, Redox-Modified Oligonucleotides for Electrochemical DNA and Aptamer-Based Sensing. Nat. Protoc 2007, 2, 2875–2880. [DOI] [PubMed] [Google Scholar]
- (37).Trasatti S; Petrii OA Real Surface Area Measurements in Electrochemistry. J. Electroanal. Chem 1992, 327, 353–376. [Google Scholar]
- (38).Herne TM; Tarlov MJ Characterization of DNA Probes Immobilized on Gold Surfaces. J. Am. Chem. Soc 1997, 119, 8916–8920. [Google Scholar]
- (39).Cruz-Aguado JA; Penner G Determination of Ochratoxin A with a DNA Aptamer. J. Agric. Food Chem 2008, 56, 10456–10461. [DOI] [PubMed] [Google Scholar]
- (40).Petrucci RH; Harwood WS; Herring GE; Madura J General Chemistry: Principles & Modern Applications; Pearson/Prentice Hall: Upper Saddle River, NJ, 2013. [Google Scholar]
- (41).Entzian C; Schubert T Studying Small Molecule-Aptamer Interactions Using MicroScale Thermophoresis (MST). Methods 2016, 97, 27–34. [DOI] [PubMed] [Google Scholar]
- (42).Traut TW Physiological Concentrations of Purines and Pyrimidines. Mol. Cell. Biochem 1994, 140, 1–22. [DOI] [PubMed] [Google Scholar]
- (43).Lin CH; Patel DJ Structural Basis of DNA Folding and Recognition in an AMP-DNA Aptamer Complex: Distinct Architectures but Common Recognition Motifs for DNA and RNA Aptamers Complexed to AMP. Chem. Biol 1997, 4, 817–832. [DOI] [PubMed] [Google Scholar]
- (44).Layland J; Carrick D; Lee M; Oldroyd K; Berry C Adenosine: Physiology, Pharmacology, and Clinical Applications. JACC: Cardiovasc. Interv 2014, 7, 581–591. [DOI] [PubMed] [Google Scholar]
- (45).Eltzschig HK; Warner DS; Warner MA Adenosine: An Old Drug Newly Discovered. Anesthesiology 2009, 111, 904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sommerschild HT; Kirkebøen KA Adenosine and Cardioprotection during Ischaemia and Reperfusion - An Overview. Acta Anaesthesiol. Scand 2000, 44, 1038–1055. [DOI] [PubMed] [Google Scholar]
- (47).Li Y; Wang W; Parker W; Clancy JP Adenosine Regulation of Cystic Fibrosis Transmembrane Conductance Regulator through Prostenoids in Airway Epithelia. Am. J. Respir. Cell Mol. Biol 2006, 34, 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fisher O; Benson RA; Imray CH The Clinical Application of Purine Nucleosides as Biomarkers of Tissue Ischemia and Hypoxia in Humans In Vivo. Biomarkers Med. 2019, 13, 953–964. [DOI] [PubMed] [Google Scholar]
- (49).Latini S; Pedata F Adenosine in the Central Nervous System: Release Mechanisms and Extracellular Concentrations. J. Neurochem 2001, 79, 463–484. [DOI] [PubMed] [Google Scholar]
- (50).Baker BR; Lai RY; Wood MS; Doctor EH; Heeger AJ; Plaxco KW An Electronic, Aptamer-Based Small-Molecule Sensor for the Rapid, Label-Free Detection of Cocaine in Adulterated Samples and Biological Fluids. J. Am. Chem. Soc 2006, 128, 3138–3139. [DOI] [PubMed] [Google Scholar]
- (51).Ferguson BS; Hoggarth DA; Maliniak D; Ploense K; White RJ; Woodward N; Hsieh K; Bonham AJ; Eisenstein M; Kippin TE; Plaxco KW; Soh HT Real-Time, Aptamer-Based Tracking of Circulating Therapeutic Agents in Living Animals. Sci. Transl. Med 2013, 5, 213ra165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Wang Z; Yu H; Canoura J; Liu Y; Alkhamis O; Fu F; Xiao Y Introducing Structure-Switching Functionality into Small-Molecule-Binding Aptamers via Nuclease-Directed Truncation. Nucleic Acids Res. 2018, 46, No. e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kypr J; Kejnovská I; Renčiuk D; Vorlíčková M Circular Dichroism and Conformational Polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Del Villar-Guerra R; Trent JO; Chaires JB G-Quadruplex Secondary Structure Obtained from Circular Dichroism Spectroscopy. Angew. Chem., Int. Ed 2018, 57, 7171–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Drabovich AP; Okhonin V; Berezovski M; Krylov SN Smart Aptamers Facilitate Multi-Probe Affinity Analysis of Proteins with Ultra-Wide Dynamic Range of Measured Concentrations. J. Am. Chem. Soc 2007, 129, 7260–7261. [DOI] [PubMed] [Google Scholar]
- (56).Porchetta A; Vallèe-Bèlisle A; Plaxco KW; Ricci F Using Distal-Site Mutations and Allosteric Inhibition to Tune, Extend, and Narrow the Useful Dynamic Range of Aptamer-Based Sensors. J. Am. Chem. Soc 2012, 134, 20601–20604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Schoukroun-Barnes LR; Glaser EP; White RJ Heterogeneous Electrochemical Aptamer-Based Sensor Surfaces for Controlled Sensor Response. Langmuir 2015, 31, 6563–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Hanssen BL; Siraj S; Wong DKY Recent Strategies to Minimise Fouling in Electrochemical Detection Systems. Rev. Anal. Chem 2016, 35, 1–28. [Google Scholar]
- (59).Tasset DM; Kubik MF; Steiner W Oligonucleotide Inhibitors of Human Thrombin That Bind Distinct Epitopes. J. Mol. Biol 1997, 272, 688–698. [DOI] [PubMed] [Google Scholar]
- (60).Bock LC; Griffin LC; Latham JA; Vermaas EH; Toole JJ Selection of Single-Stranded DNA Molecules That Bind and Inhibit Human Thrombin. Nature 1992, 355, 564–566. [DOI] [PubMed] [Google Scholar]
- (61).Xiao Y; Lubin AA; Heeger AJ; Plaxco KW Label-Free Electronic Detection of Thrombin in Blood Serum by Using an Aptamer-Based Sensor. Angew. Chem., Int. Ed 2005, 44, 5456–5459. [DOI] [PubMed] [Google Scholar]
- (62).Mani RJ; Dye RG; Snider TA; Wang S; Clinkenbeard KD Bi-Cell Surface Plasmon Resonance Detection of Aptamer Mediated Thrombin Capture in Serum. Biosens. Bioelectron 2011, 26, 4832–4836. [DOI] [PubMed] [Google Scholar]
- (63).Wiegand TW; Williams PB; Dreskin SC; Jouvin MH; Kinet JP; Tasset D High-Affinity Oligonucleotide Ligands to Human IgE Inhibit Binding to Fc Epsilon Receptor I. J. Immunol 1996, 157, 221–230. [PubMed] [Google Scholar]
- (64).Li Y; Liu B; Huang Z; Liu J Engineering Base-Excised Aptamers for Highly Specific Recognition of Adenosine. Chem. Sci 2020, 11, 2735–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Hoinka J; Zotenko E; Friedman A; Sauna ZE; Przytycka TM Identification of Sequence-Structure RNA Binding Motifs for SELEX-Derived Aptamers. Bioinformatics 2012, 28, i215–i223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Bailey TL; Elkan C Fitting a Mixture Model by Expectation Maximization to Discover Motifs in Biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol 1994, 2, 28–36. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.