

Abstract
There is strong evidence that inhibition of one or more Aldehyde Dehydrogenase 1A (ALDH1A) isoforms may be beneficial in chemotherapy-resistant ovarian cancer and other tumor types. While many previous efforts have focused on development of ALDH1A1 selective inhibitors, the most deadly ovarian cancer subtype, high-grade serous (HGSOC), exhibits elevated expression of ALDH1A3. Herein, we report continued development of pan-ALDH1A inhibitors to assess whether broad spectrum ALDH1A inhibition is an effective adjunct to chemotherapy in this critical tumor subtype. Optimization of the CM39 scaffold, aided by metabolite ID and several new ALDH1A1 crystal structures, led to improved biochemical potencies, improved cellular ALDH inhibition in HGSOC cell lines, and substantial improvements in microsomal stability culminating in orally bioavailable compounds. We demonstrate that two compounds 68 and 69 are able to synergize with chemotherapy in a resistant cell line and patient-derived HGSOC tumor spheroids, indicating their suitability for future in vivo proof of concept experiments.
Graphical Abstract
1. Introduction
Approximately 14,000 deaths will occur in the United States as the result of ovarian cancer in 2020, making it the 5th most deadly cancer in U.S. females.1 Although new targeted therapies for ovarian cancer have recently been approved, the benefits they provide are marginal over the long-standing regimen of surgical tumor debulking in combination with platinum- and taxane-based chemotherapy.2-7 Poly-ADP Ribose inhibitors (PARPi) have recently proven to improve outcomes in the patients with BRCA mutations,8 but the majority of ovarian cancer patients do not have BRCA mutations and the standard of care for these patients remains chemotherapy.9 While many of these patients can obtain a complete clinical response with combined surgery and chemotherapy, the majority develop chemotherapy resistant disease due to residual cells spared by treatment. The ability to enhance the activity of chemotherapy to eliminate residual chemotherapy resistant cells could greatly improve the outcomes of patients with ovarian cancer.10
One potential clinical target to overcome chemotherapy resistance is the aldehyde dehydrogenase 1A family of enzymes (ALDH1A). ALDH1A enzymes can directly metabolize and inactivate chemotherapeutics such as cyclophosphamide.11 They are also indirectly linked with resistance to other chemotherapies such as platinums and taxanes.12-15 ALDH expression is even linked with resistance to therapeutics such as tyrosine kinase inhibitors.16
A potential explanation for ALDH1A enzymes and chemotherapy resistance may relate to their increased expression in cancer stem-like cells (CSCs). The presence of ALDH activity, as determined by the ALDEFLUOR assay, is a well-established means of identifying CSCs in ovarian and other cancers.17-21 We and others have postulated that ALDH plays a functional role in CSC biology and demonstrated that small molecule inhibitors of ALDH may offer an avenue to target CSCs and improve patient outcomes when combined with chemotherapy.12, 22-24 The three ALDH1A isoforms, ALDH1A1 (1A1), ALDH1A2 (1A2), and ALDH1A3 (1A3), exhibit a high degree of sequence homology (>70%), and are of particular interest in targeting CSCs. 1A1 and 1A3 are linked to stem-like characteristics in cancers arising from a variety of tissue types and all three have been implicated in chemoresistance.12, 20-31 High-grade serous ovarian cancer, the histologic subtype responsible for 70-80% of ovarian cancer deaths, exhibits a prominent elevation in 1A3 expression and more modest elevation of 1A1.32-33
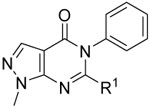
ALDH inhibitors were comprehensively reviewed in 2012 by Koppaka et al34 and more recently by Dinavahi et al.35 Notably, a concerted medicinal chemistry effort to develop ALDH1A1 selective inhibitors by Maloney and coworkers culminated in the discovery of 1a (NCT-506, Figure 1) and several analogs.36-37 This orally bioavailable compound potently inhibits ALDH in the 1A1 expressing OV-90 ovarian cancer cell line (ALDEFLUOR IC50 = 161 nM) and reverses taxane resistance in a SKOV-3 resistant cell line. Very recently, a number of moderately potent and very selective inhibitors of ALDH1A3 based on the natural product diadzin, such as compound 1b, have also been disclosed.38-39 Similar analogs have shown efficacy in a glioblastoma mouse model.40
Figure 1.
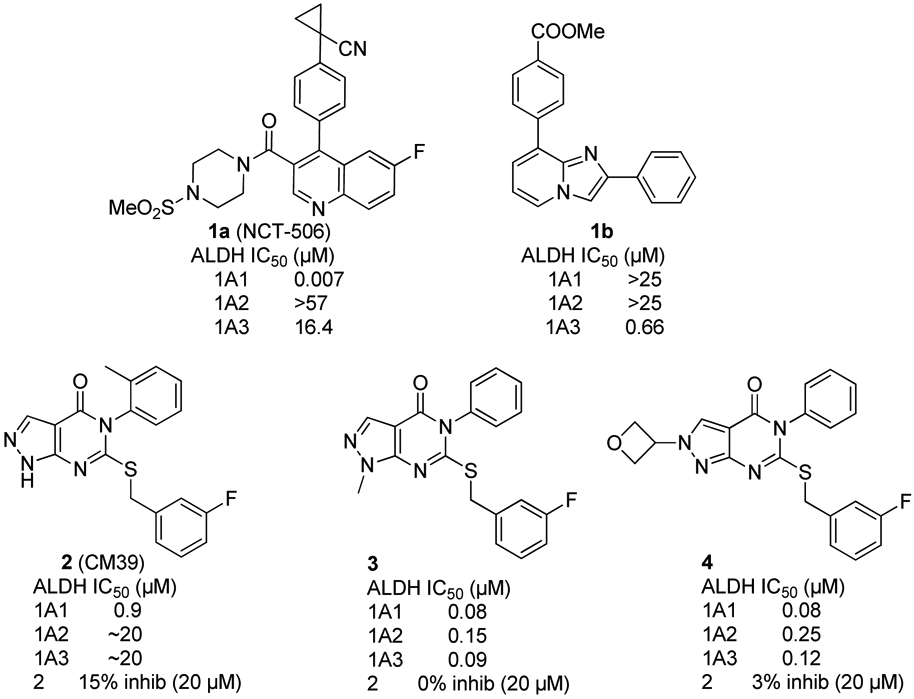
Previously developed ALDH1A inhibitors.
The overarching goal of our research has been to elucidate the SAR of ALDH1A isoform selectivity and the role of individual ALDH1A isoforms in chemoresistance; however, in light of the implication of multiple ALDH1A isoforms in ovarian cancer and the elevation of 1A3 in high-grade serous tumors, we have recently sought to develop more broad-spectrum ALDH1A inhibitors to facilitate the study of ALDH1A biology in a wider array of cell lines. In our previous publication, we disclosed development of first-in-class pan-ALDH1A inhibitors 3 and 4 with selectivity against ALDH2, beginning with HTS screening compound CM39 (2) (Figure 1).22 In this report we disclose our efforts toward the development of viable in vivo probes by significantly improving ALDEFLUOR inhibition, aqueous solubility and metabolic stability. To monitor these parameters, we assessed biochemical inhibition of the three ALDH1A isoforms and ALDEFLUOR inhibition in the high-grade serous ovarian cancer cell line PEO1, which expresses high levels of ALDH1A3.12 We felt that selectivity against ostensibly unrelated ALDH isoforms was desirable to provide unambiguous insight into the effects of ALDH1A inhibition. To monitor selectivity, we also tested compounds against the most homologous ALDH isoform not included in the ALDH1A family, ALDH2. None inhibited ALDH2 by more than 20% at 20 μM. Solubility and metabolic stability were assessed for key compounds to monitor progress towards suitable compounds for in vivo studies.
2. Results and Discussion
2.1. Chemistry
Synthesis of de-aza analog 7 began with known compound 5, obtained in 3 steps from 2,4,6-trichloropyridine (Scheme 1).41 Vigorous heating was required to displace the chloride with 3-fluorobenzyl mercaptan, forming 6. Finally, Chan-Lam coupling with phenylboronic acid afforded 7. Pyrrole 11 was prepared in a similar manner following methylation and hydrolysis of commercially available 8. Synthesis of imidazole 14 began by generating 13 from 12 with LiHDMS and the electrophilic NH2 source O-(diphenylphosphinyl)hydroxylamine.42 Subsequently, thiourea formation, cyclization and displacement of 3-fluorobenzyl bromide in one pot afforded 14.
Scheme 1.
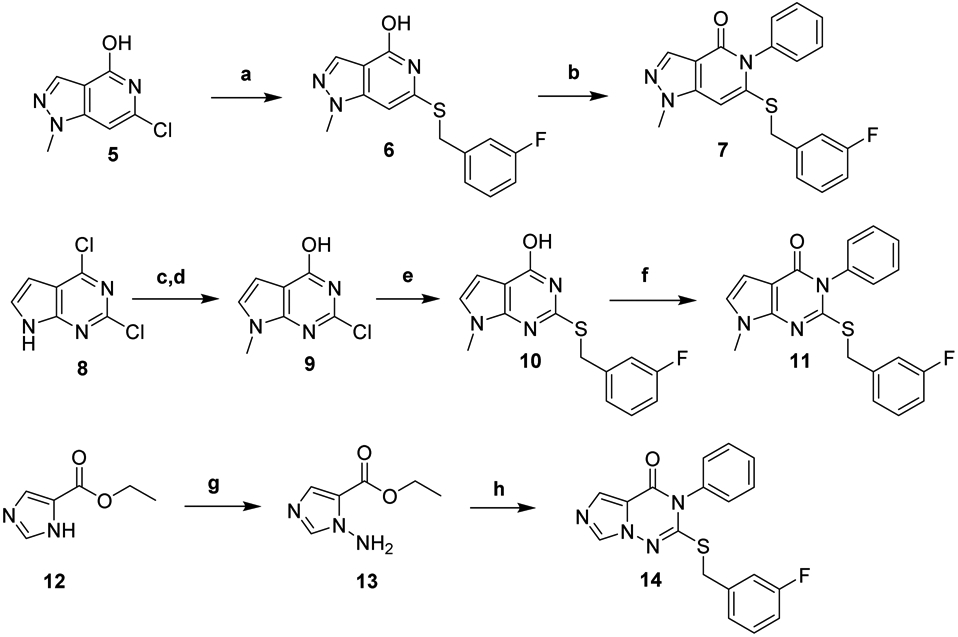
Synthesis of Bicyclic Heterocycle Analogsa
aReagents and Conditions: (a) 3-fluorobenzyl mercaptan, DIPEA, n-BuOH, 170 °C Microwave; (b) PhB(OH)2, Cu(OAc)2, pyridine, TEMPO, 3Å MS, air, DCE; (c) MeI, NaH, THF, 0 – 20 °C; (d) 10% aq. NaOH, Reflux; (e) 3-fluorbenzyl mercaptan, DIPEA, EtOH, 100 °C; (f) PhB(OH)2, Cu(OAc)2, pyridine, 3Å MS, air, DCM (g) O-(diphenylphosphinyl)hydroxylamine, LiHDMS, DMF, −10 – 20 °C; (h) PhNCS, NaH, 3-fluorobenzyl bromide, DMF, 0 − 20 °C.
Synthesis of analogs 18-19, and 32 (Scheme 2), began with formation of an unsymmetrical thiourea by addition of the commercially pyrazole 15 to phenyl isothiocyanate in the presence of NaH, which cyclized to form 16. Methylation and mCPBA oxidation of the sulfur afforded methyl sulfone 17 which was displaced with the appropriate amine or alcohol under basic conditions to afford 18-19, and 32. Synthesis of reversed linker analogs 21-24 began with nBuLi-mediated aminolysis of ester 15 with aniline to generate the corresponding phenyl amide followed by treatment with chloroacetyl chloride in a melt of chloroacetic acid to affordcompound 20. The chloride was displaced with the appropriate phenol, thiophenol, or aniline at moderate temperatures to provide analogs 21-23. Alkylation of amine 23 with methyl iodide afforded the tertiary aniline 24. Alkenyl analog 25 was generated by treating 20 with P(OEt)3 for a subsequent Horner-Wadsworth-Emmons olefination with 3-fluorobenzaldehyde. Pd-C catalyzed hydrogenation of the alkene afforded 26.
Scheme 2.
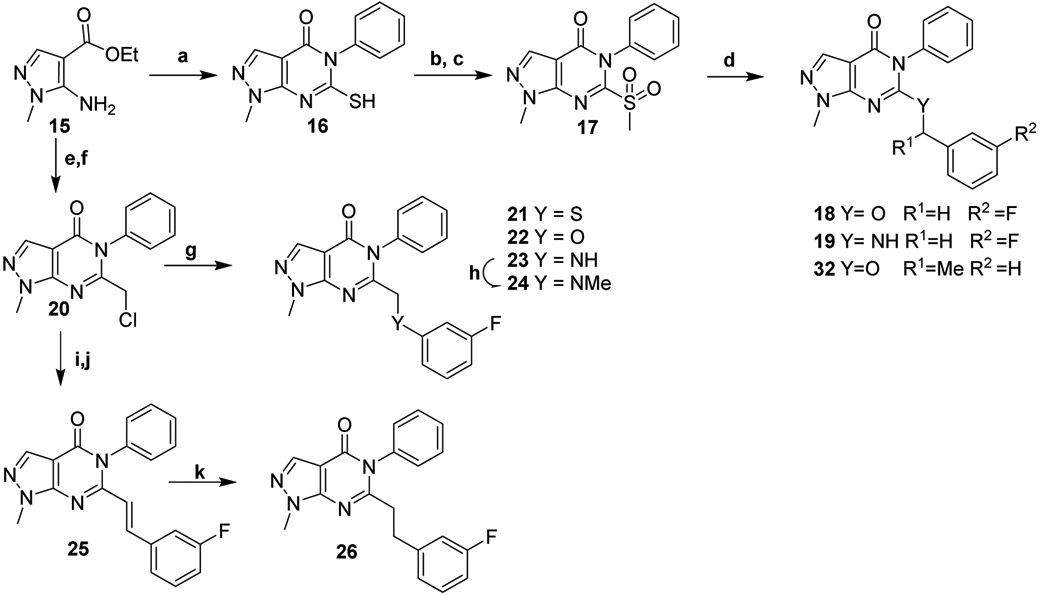
Synthesis of modified linker analogsa
aReagents and Conditions: (a) PhNCS, NaH, DMF, 0-50 °C; (b) K2CO3, MeI, DMF; (c) mCPBA, DCM; (d) NaH, 3-fluorobenzylamine or 3-fluorobenzyl alcohol or α-methyl benzyl alcohol, DMF, 0-20 °C; (e) nBuLi, aniline, THF, −78 °C to RT; (f) chloroacetic acid, chloroacetyl chloride, 80-120 °C; (g) 3-fluorophenol or 3-fluorothiophenol or 3-fluoroaniline, K2CO3, DMF, 50 or 70 °C; (h) MeI, K2CO3, DMF, 50 °C; (i) P(OEt)3, DMF, 150 °C; (j) NaH, 3-fluorobenzaldehyde, DMF, 0-20 °C; (k) Pd-C, H2, MeOH.
Branched alkyl analogs 27-28, 30-31, and 42-49 were generated by displacing the appropriate halide or mesylate with thiol 16 in the presence of K2CO3 (Scheme 3). In the case of phenol 46 removal of the TBS group was accomplished with HF-Pyridine. The enantiomers of 28 were obtained by first activating (R)- or (S)-1-phenylethanol with the Vilsmeier reagent. Subsequent displacement by thiol 16 with complete inversion of stereochemistry afforded enantiomers (R)-28 and (S)-28.43 Mitsunobu conditions provided the products in inferior yield and enantiopurity. The gem-dimethyl analog 29 was formed by treating thiol 16 with α-methylstyrene under acidic conditions.
Scheme 3.
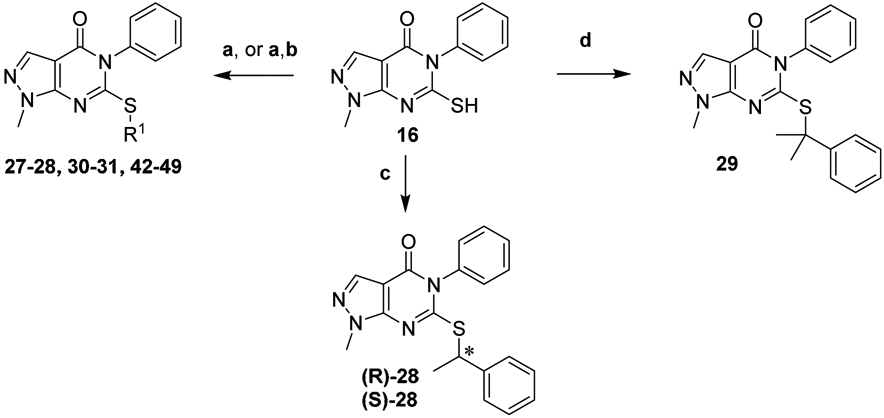
Synthesis of branched alkyl analogsa
aReagents and Conditions: (a) X-R1 or Ms-R1, K2CO3, DMF; (b) HF-Pyridine, THF; (c) (R)- or (S)-1-phenylethanol, Vilsmeier reagent, TEA, THF, 0-20 °C; (d) α-methylstyrene, TFA, DCM, 0-20 °C.
Synthesis of the conformationally restricted analogs began with Chan-Lam coupling of 5 with phenylboronic acid followed by displacement of the chloride with Na2S to generate intermediate 34 (Scheme 4). Synthesis of the 5-membered ring analog proceeded by alkylating the thiol with ethyl 2-bromo-2-phenyl acetate then hydrolyzing the ester to afford acid 35. Generating the acid chloride resulted in a spontaneous intramolecular Friedel-Crafts acylation to provide 36. To generate the 6-membered ring analogs, a survey of 1,4-conjugate addition conditions employing cinnamate esters did not afford any desired adducts. We postulate that under basic conditions the carbanion intermediate formed following addition of the thiol undergoes a retro-Michael elimination to regenerate starting materials faster than it can be protonated. Gratifyingly, treatment with 3-bromo-3-phenylpropionic acid44 efficiently alkylates thiol 34 in the presence of NaHCO3 to afford acid 37. Upon generating the acid chloride by treating 37 with oxalyl chloride, we found heat was required to promote cyclization to the 6-membered ring 38. Unfortunately, the Vilsmeier reagent formed from residual DMF in the starting material resulted in chlorination of the ketone. Alternatively, generating the mixed sulfonic anhydride by treatment with Ms2O in DCE at 85 °C promoted the cyclization to 38 without any undesired side reaction.45 Treatment of the resulting ketone with NaBH4 afforded 39 as an inconsequential, inseparable mix of diastereomers. We first attempted deoxygenation of the alcohol with chlorodiphenylsilane and InCl3 but obtained the unexpected elimination product 40.46 Barton-McCombie deoxygenation resulted in the desired saturated linker analog 41 as determined by mass spectrometry, however the product was too insoluble in common solvents to fully characterize or test in our assays.
Scheme 4.
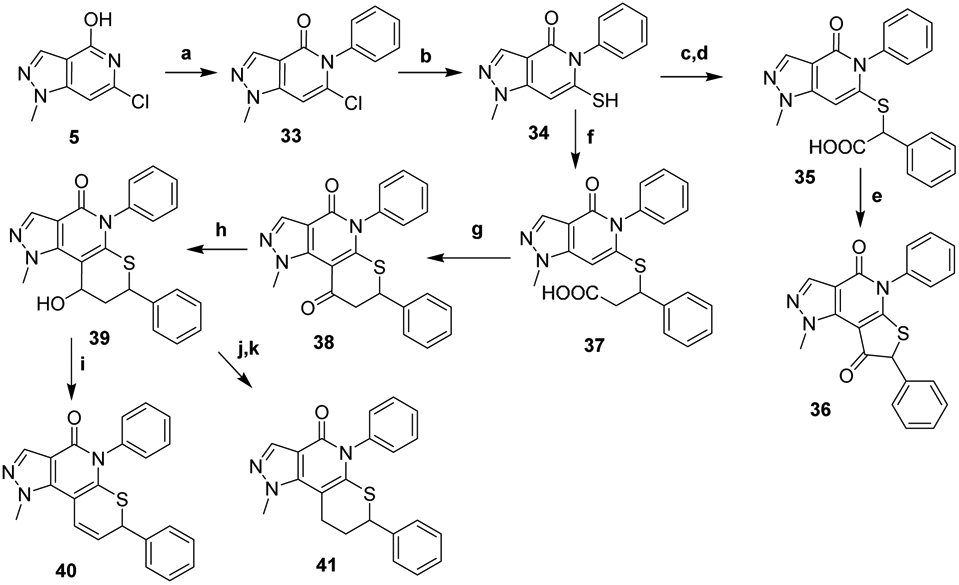
Synthesis of conformationally restricted analogsa
aReagents and Conditions: (a) PhB(OH)2, Cu(OAc)2, pyridine, 3Å MS, O2, DCE; (b) Na2S, DMF, 140 °C; (c) ethyl-2-bromo-2-phenylacetate, K2CO3, DMF; (d) Aq. LiOH, EtOH, THF; (e) (COCl)2, cat. DMF, DCM; (f) 3-bromo-3-phenylpropanoic acid, NaHCO3, DMF, 50 °C; (g) methanesulfonic anhydride, DCE, 85 °C; (h) NaBH4, EtOH; (i) chlorodiphenyl silane, InCl3, DCM; (j) CS2, NaH, MeI, THF, 0 °C; (k) tributyltin hydride, AIBN, toluene, reflux.
Alcohols 51 and 53 were generated from isothiocyanate 50 by addition of the requisite anilines then cyclization and alkylation of the resulting thiol under basic conditions in two pots as we have previously described (Scheme 5).22 Treatment of the alcohols with MeI under basic conditions afforded methyl ether analogs 52 and 54. 51 was converted to amine 55 upon mesylation, displacement with sodium azide, and Staudinger reduction. Dess-Martin oxidation of 51 and reductive amination with methyl or dimethylamine afforded 57-58. Synthesis of intermediate 59 and nitrile and alkynyl analogs 64-65 was accomplished starting with 50 according to our one-pot protocol.22 Deprotection of 59 yielded 61, which was bis-methylated to provide 63. Alkylation of the carbamate 59 with MeI afforded the mono-methylated analog 62 upon deprotection.
Scheme 5.
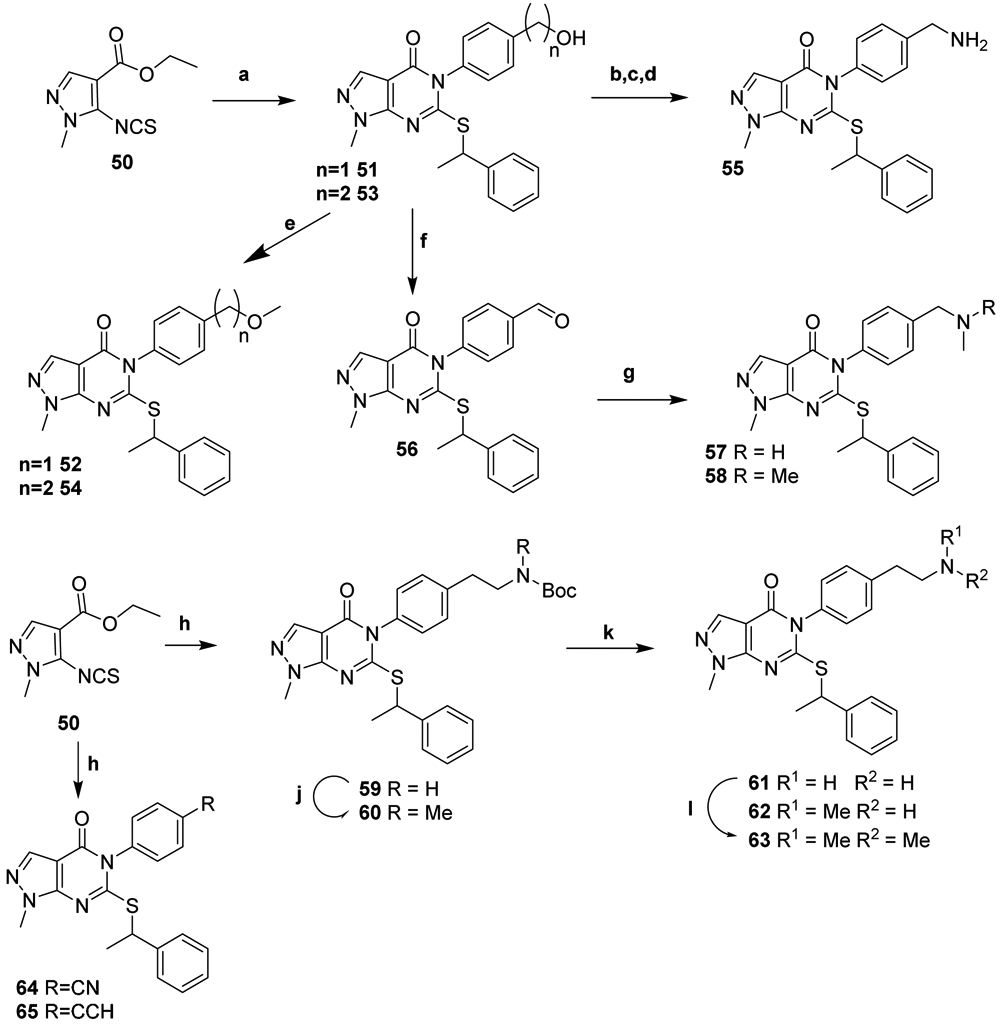
Synthesis of para-substituted N-phenyl analogsa
aReagents and Conditions: (a) (i) 4-aminobenzyl alcohol or 2-(4-aminophenyl)ethanol, NaH, DMF, 0-20 °C; (ii) 1-bromoethylbenzene, K2CO3, DMF, RT; (b) Ms-Cl, DIPEA, DCM, 0-20 °C; (c) NaN3, DMF; (d) PS-PPh3, H2O, THF; (e) MeI, NaH, DMF 0-20 °C; (f) Dess-Martin periodinane, DCM; (g) titanium isopropoxide, MeNH2 or Me2NH in EtOH, NaBH4, MeOH; (h) tert-butyl (4-aminophenethyl)carbamate, 4-amino benzonitrile or 4-ethynylaniline; 1-bromoethylbenzene, NaH, DMF, 0-20 °C; (j) MeI, NaH, DMF, 0-20 °C; (k) TFA, DCM;(l) Aq. formaldehyde, DIPEA, HOAc, sodium triacetoxyborohydride, DCE.
As shown in Scheme 6, synthesis of oxetanyl analogs 67-70 was accomplished by sequential one-pot alkylation of 66 at the sulfur and pyrazole, as we have previously described.22 Synthesis of methyl-oxetane analog 74 similarly began with the one-pot sequential alkylation of 66. Fortuitously, in contrast to other electrophiles which alkylated both positions of the pyrazole with a slight preference for the 1 position, diethyl 2-bromo-2-methylmalonate alkylated exclusively at the 2 position. The challenging reduction of the malonate ester 71 to diol 72 was completed by in situ generation of BH3 in dimethoxyethane from NaBH4 and Br2.47 71 was unaffected by fresh, commercially-obtained BH3-THF or 9-BBN at temperatures up to 70 °C, while reduction with NaBH4, LiBH4, and L-Selectride resulted primarily in the decarboxylated mono-alcohol 75. Reduction with aluminum-based reagents LiAlH4, or DIBAL-H resulted in rapid decomposition, even at −78 °C. With 72 in hand, tosylation and cyclization afforded oxetane 74.
Scheme 6.
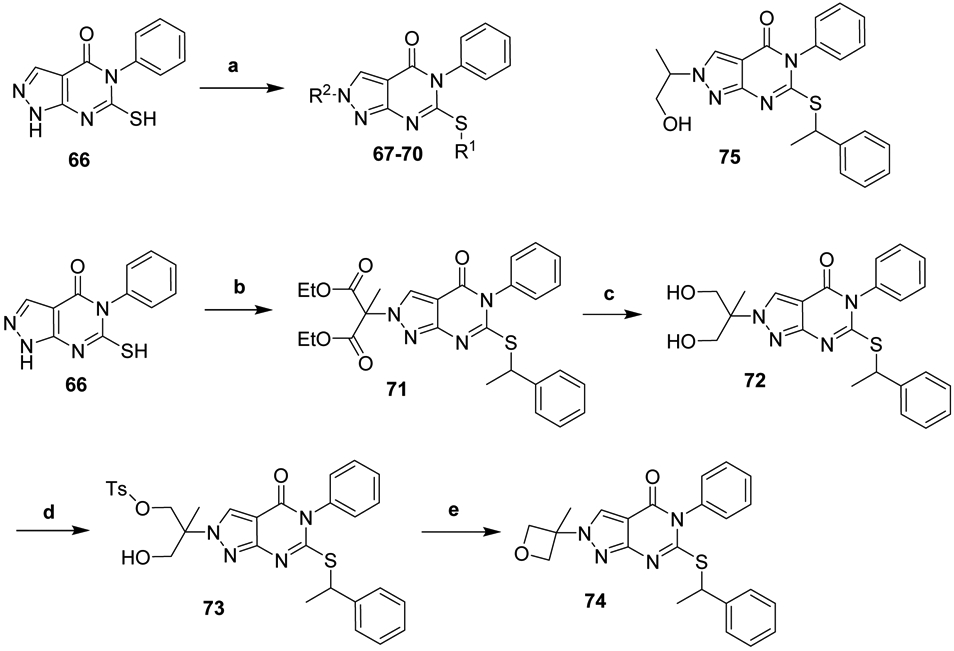
Synthesis of N-2 substituted analogsa
aReagents and Conditions: (a) R1-Br, R2-Br or R2-OMs, K2CO3, DMF, 20-80 °C; (b) 1-bromoethylbenzene, diethyl 2-bromo-2-methylmalonate, K2CO3, DMF, 20-50 °C; (c) NaBH4, Br2, dimethoxyethane, −20 °C to RT; (d) nBuLi, Ts-Cl, THF, −78 °C to RT; (e) nBuLi, THF, 0-80 °C.
2.2. Structure Activity Relationships
2.2.1. Modification of the heterocyclic core
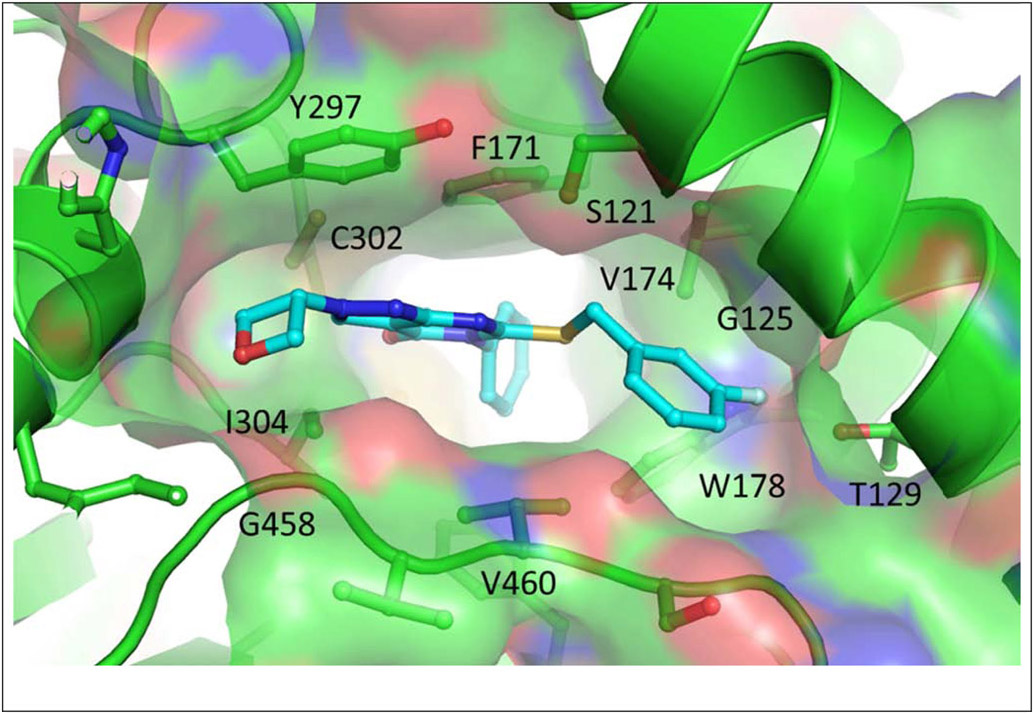
As shown in Table 1, compound 3, disclosed in our previous publication,22 potently inhibits ALDH1A1 but has poor stability to incubation with mouse liver microsomes (MLM), extremely low aqueous solubility and no significant inhibition of ALDEFLUOR activity at 1 μM in an ALDH1A3 expressing cell line. We speculated that either poor cell permeability or solubility might explain the lack of ALDEFLUOR activity for this compound. To determine if lowering the topological polar surface area (TPSA) could improve the ALEDFLUOR activity by increasing cell permeability. We synthesized a number of alternative heterocyclic cores. De-aza analog 7 (Table 1) was 3-fold less potent against 1A1 than 3 and unimproved in the ALDEFLUOR assay, but unexpectedly, given the similar cLogP, was substantially more soluble. The good physical properties of 7 seem to rule out insolubility as the sole explanation for the lack of ALDEFLUOR activity. The modest loss of ALDH potency was surprising given that the replaced NH is apparently solvent exposed in the 4-ALDH1A1 crystal structure previously published by us (Figure 2).22 According to that structure, the edge (1-position) of the pyrazole and the adjacent NH are facing towards solvent but the face is positioned to participate in an offset-parallel π-π interaction with Y297 3.5 Å away. Pyrrole and imidazole analogs 11 and 14 lost additional ALDH activity. The steep SAR for modifications to the pyrazole itself contrasts with the relatively flat SAR for pyrazole substituents we disclosed previously, perhaps indicating that the electronic properties of the pyrazole are optimal for the π-π interaction noted above (Figure 2).22
Table 1.
Characterization of alternative heterobicyclic core analogs.
| No. | Structure | ALDH IC50a (μM) | ADME Characterization | PEO1 Cell ALDEFLUORf (% inhib. @ 1 μM) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| 1A1 | 1A2 | 1A3 | MLMb (t1/2, min) |
Aq. Solc (μM) |
cLogPd | TPSAe (Å) |
|||
| 322 | 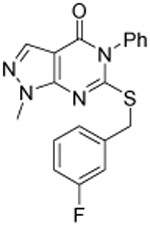 |
0.08 ±0.01 | 0.15 ±0.01 | 0.09 ±0.01 | 8 | <0.7 | 4.3 | 78 | 3 ±1 |
| 7 | 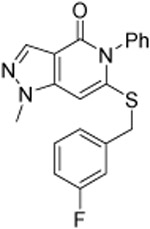 |
0.240 ±0.007 | 0.24 ±0.02 | 0.14 ±0.02 | 9 | 178 | 4.4 | 65 | 3 ±1 |
| 11 | 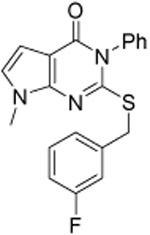 |
0.93 ±0.05 | 0.81 ±0.04 | 0.6 ±0.1 | 23 | 5.2 | 65 | ||
| 14 | 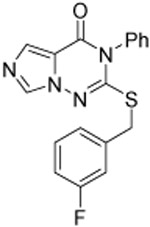 |
2.2 ±0.2 | 1.60 ±0.04 | 0.50 ±0.02 | 47 | 3.4 | 78 | ||
Values are expressed as mean ± SEM (n=3).
Half-life in mouse liver micorosomes.
Thermodynamic aqueous solubility (performed by Analiza Inc. using quantitative nitrogen detection (www.analiza.com)).
Calculated log P.
Topological polar surface area.
Mean ± SD as determined by flow cytometry (N=3).
Figure 2.
Structure of 4 bound to ALDH1A1 (PDB ID: 6DUM)22
2.2.2. Modification of the thioether linker
Due to the expected metabolic lability and lipophilicity of the thioether, we reasoned that finding an alternative linker could lead to substantial improvements in both metabolic stability and solubility (Table 2). Direct replacement of the sulfur with oxygen (18) or nitrogen (19) led to a substantial loss of potency across the ALDH1A family. The 4-ALDH1A1 structure (Figure 2) indicates the four closest contacts of the sulfur to be the lipophilic side chains of F171, V174, W178, and V460, which may explain the strong preference for a lipophilic group at this position.
Table 2.
Optimization of the thioether linker.
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| ALDH IC50a (μM) or % Control at 20 μM |
ADME Characterization | PEO1 Cell ALDEFLUORe (% inhib. @ 1 μM) |
||||||
| No. | R1 | 1A1 | 1A2 | 1A3 | MLMb t1/2 (min) |
Aq. Solc (μM) |
cLogPd | |
| 322 | 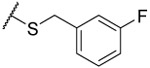 |
0.08 ±0.01 | 0.15 ±0.01 | 0.09 ±0.01 | 8 | <0.7 | 4.3 | 3 ±1 |
| 18 | 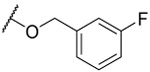 |
0.775 ±0.031 | 3.7 ±0.64 | 1.7 ±0.1 | 3.6 | |||
| 19 | 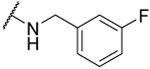 |
69% | 73% | 58% | 3.0 | |||
| 21 | 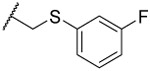 |
0.7 ±0.2 | 71% | 86% | 3.3 | |||
| 22 | 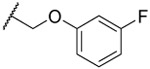 |
8.2 ±0.7 | 1.77 ±0.09 | 0.78 ± 0.09 | 194 | 2.8 | ||
| 23 | 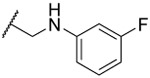 |
0.57 ±0.03 | 2.3 ±0.1 | 1.05 ±0.08 | 64 | 2.5 | ||
| 24 | 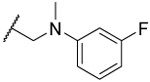 |
88% | 72% | 74% | 3.1 | |||
| 25 | 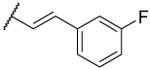 |
0.27 ±0.06 | 0.44 ±0.03 | 79% | 4 | 3.7 | ||
| 26 | 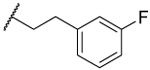 |
1.0 ±0.1 | 41% | 2.5 ±0.2 | 126 | 3.6 | ||
| 27 | 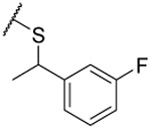 |
0.151 ±0.003 | 0.11 ±0.01 | 0.128 ±0.005 | 8 | 4.6 | ||
| 28 | 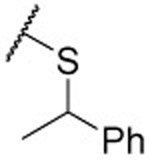 |
0.13 ±0.03 | 0.11 ±0.02 | 0.073 ±0.005 | 8 | 8 | 4.5 | 47 ±5 |
| (R)-28 | 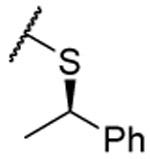 |
0.07 ±0.04 | 0.04 ±0.01 | 0.034 ±0.001 | 8 | 4.5 | 71 ±5 | |
| (S)-28 | 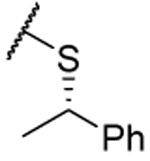 |
1.41 ±0.09 | 1.48 ±0.07 | 1.64 ±0.07 | 4.5 | 2 ±3 | ||
| 29 | 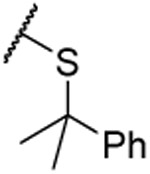 |
1.9 ±0.3 | 2.2 ±0.3 | 1.39 ±0.09 | 4.7 | |||
| 30 | 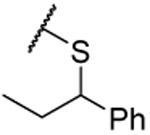 |
0.150 ±0.003 | 0.179 ±0.011 | 0.119 ±0.001 | 5.0 | |||
| 31 | 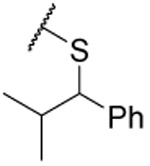 |
0.38 ±0.07 | 0.3 ±0.1 | 0.13 ±0.04 | 5.4 | |||
| 32 | 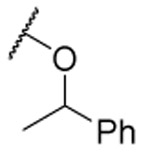 |
2.4 ±0.6 | 42% f | 0.6 ±0.2 | 3.9 | |||
Values are expressed as mean ± SEM (n=3).
Half-life in mouse liver micorosomes.
Thermodynamic aqueous solubility (performed by Analiza Inc. using quantitative nitrogen detection (www.analiza.com)).
Calculated log P.
Mean ± SD as determined by flow cytometry (N=3).
% Control at 5 μM.
We next endeavored to generate analogs with the position of the methylene and heteroatom reversed, in order to situate the heteroatom nearer the polar S121 sidechain in the polymorph used for crystallography (N121 in wild type ALDH1A1) (Figure 2). Consistent with their lower cLogP, oxygen and nitrogen analogs 22 and 23 had markedly improved aqueous solubility; however, these changes weakened activity against ALDH1A. Interestingly, oxygen analog 22 was modestly selective for 1A2 and 1A3 over 1A1. Methylation of the nitrogen (24) diminished the already weak activity of 23. Reversing the orientation of the thioether (21) results in greater predicted partial charge on the methylene due to the electron withdrawing nature of the pyrimidinone and as a result, significantly reduced cLogP vs. 3. Unfortunately, potentially as a result of repulsion between the polarized methylene and the lipophilic side chains mentioned above, this change was poorly tolerated by all of the ALDH1A isoforms.48 Taken together, these results suggest that the lipophilicity of the thioether contributes significantly to the ALDH1A potency in this series.
We next hypothesized that an ethyl linker might be the most suitable replacement for the thioether. Compared to nitrogen or oxygen, a methylene more closely mimics the electronegativity, lipophilicity, and bond angle of sulfur.49 On the other hand, we did not expect the highly planar alkenyl analog to be tolerated given the non-planar conformation of the thioether in Figure 2. Surprisingly, the ethyl analog (26) was unremarkable while the alkene (25) proved to be the most potent thioether replacement tested in terms of 1A1 and 1A2 potency, but was a poor inhibitor of 1A3. Despite their similar cLogP, 25 was much less soluble than 26, perhaps owing to its high degree of planarity which is often associated with undesirable physiochemical properties.50
Since we found the thioether linker to be critical to ALDH1A activity, we next turned to alkyl branching as a strategy to 1) improve solubility by reducing planarity and 2) sterically block metabolism of the thioether. Encouragingly, the racemic methyl-branched analogs 27 and 28 retained the potent pan-ALDH1A inhibition, and 28 improved ALDEFLUOR inhibition to 47% at 1 μM. The alkyl branching strategy did not improve metabolic stability as we predicted, but des-fluoro analog 28 was modestly more soluble than 3. As the fluoro substituent offered no advantage in biochemical potency or metabolic stability, we pursued des-fluoro analogs due to the greater abundance of starting materials and slight reduction in lipophilicity. We synthesized the two enantiomers of 28 and observed a 20:1 eudismic ratio in favor of the R-enantiomer (R)-28 over (S)-28. Consistent with the trend in biochemical potency, (R)-28 inhibited ALDEFLUOR by 71% at 1 μM while (S)-28 was inactive. We did not observe any change between the racemate and active enantiomer in MLM stability. Gem-dimethyl analog 29, which likely presents a methyl in the same apparently unfavorable orientation as (S)-28, was unsurprisingly not well tolerated by ALDH.
When we obtained a crystal structure by soaking ALDH1A1 crystals with racemic 28, we found that only (R)-28 was bound (Figure 3). The active conformation of the thioether was unchanged from that of 6DUM, and the newly installed CH3 projected toward a small cleft bounded by S121, V174 and G125. Attempts to fill this cleft by extending to ethyl (30) or isopropyl (31) substituents resulted in a loss of potency against 1A1/1A2. ALDH1A3 was more tolerant of the isopropyl substituent, rendering 31 very modestly 1A3 selective. Revisiting the sulfur to oxygen switch with the optimized linker of 28 resulted in a significant loss of potency across all three 1A isoforms (32) as previously observed with 18 vs 3.
Figure 3.
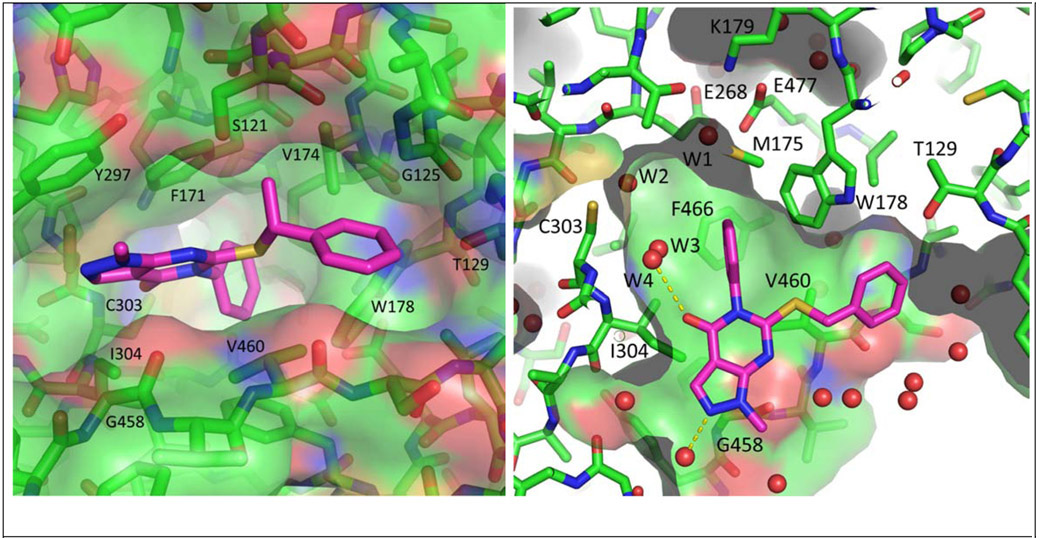
Structure of (R)-28 Bound to ALDH1A1 (PDB ID: 7JWS)
Based on our examination of the (R)-28 crystal structure (Figure 3), we envisioned restricting the thioether into the active conformation by forming a 5- or 6-membered ring joining the benzylic carbon of the thioether to the 7-position of the heterocyclic core. Our motivations to pursue conformational restriction were 1) to improve potency through reduction in the ΔS of binding and 2) to reduce metabolism through elimination of induced fit in metabolic enzymes.51 As shown in Table 3, the more rigidified analogs 36 and 40 favored binding to 1A2 and 1A3 with 3-to-5-fold selectivity over 1A1. Conversely, the more flexible 38 was slightly 1A1 selective. Only 36, the most potent 1A2/1A3 inhibitor from the series, inhibited ALDEFLUOR by >50% at 1 μM. Due to the loss of ALDEFLUOR potency and increased synthetic complexity, we did not pursue this conformationally restricted motif further. However, we did perform computational modeling to attempt to rationalize the interesting 5-fold selectivity for 1A2 and 1A3 by rigid cyclopentenone 36 (Supp. Figs. S-1 and S-2). Docking of the R-enantiomer into each of the enzyme active sites predicted a strong steric clash between the ring carbonyl and S121 of 1A1 that is not present in the other isoforms. Docking of the S-enantiomer predicted a flipped binding mode that resulted in a different steric clash between S121 and the 2-phenyl group that is absent in 1A2 and 1A3.
Table 3.
Characterization of conformationally restricted analogs.
| CMPD No. |
Structure | ALDH IC50a (μM) or % Control at 20 μM |
ADME Characterization | PEO1 Cell ALDEFLUORe (% inhib. @ 1 μM) |
||||
|---|---|---|---|---|---|---|---|---|
| 1A1 | 1A2 | 1A3 | MLMb t1/2 (min) |
Aq. Solc (μM) |
cLogPd | |||
| 36 | 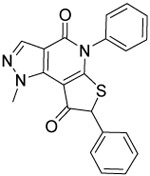 |
1.0 ±0.1 | 0.17 ±0.02 | 0.15 ±0.05 | 5 | 38 | 3.7 | 54 ±3 |
| 38 | 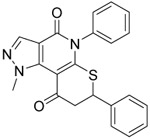 |
0.206 ±0.06 | 0.4 ±0.1 | 0.69 ±0.08 | 8 | 3.9 | 2 ±2 | |
| 40 | 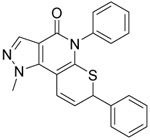 |
0.8 ±0.1 | 0.27 ±0.01 | 0.24 ±0.01 | 4.2 | 11 ±1 | ||
Values are expressed as mean ± SEM (n=3).
Half-life in mouse liver micorosomes.
Thermodynamic aqueous solubility (performed by Analiza Inc. using quantitative nitrogen detection (www.analiza.com)).
Calculated log P.
Mean ± SD as determined by flow cytometry (N=3).
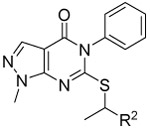
2.2.3. Optimization of the benzyl pendant
We then turned our attention to the optimization of the α-methylbenzyl pendant of 28 (Table 4). The crystal structure indicated the opportunity to engage the side chains of T129 and W178 in polar interactions (Figure 3) and potentially improve the lipophilic efficiency52 of our compounds. Gratifyingly, 2- and 3-pyridyl analogs (42, 43) retained potent biochemical inhibition of the ALDH1A family, improved ALDEFLUOR inhibition (≥ 95% at 1 μM), and improved the lipophilic ligand efficiency (LLE) substantially. Consistent with a reduced cLogP, 42 was more than 2-fold more soluble and 6-fold more stable in the MLM assay than 28. While 43 was exceptionally soluble, it did not possess the improved metabolic stability of 42, perhaps because the less-hindered 3-pyridyl is more susceptible to N-Oxidation. The 5-pyrimidinyl analog 44 retained strong 1A1 and 1A3 inhibition but was 4-fold less effective against 1A2 and only moderately effective in the ALDEFLUOR assay at 1 μM. Driven by a substantial reduction in cLogP, this compound has the best lipophilic efficiency, with respect to 1A1, of any compound in this series to date, but the diminution of ALDEFLUOR activity suggested that its TPSA was getting too high for optimal cell permeability. Based on the structure in Figure 3, the 4-pyridyl (45) was not expected to be in a position to make productive polar interactions and indeed showed diminished activity across the ALDH1A family. We also attempted to engage W178 with phenol analogs. Unfortunately, the 2-phenol analog rapidly reverted to thiol 16 in aqueous PBS and could not be tested. 3-phenol analog 46 was comparably potent against the 1A family as the 3-pyridyl analog 43, but was less soluble, less stable in the MLM assay, and a less effective ALDEFLUOR inhibitor. The 2- and 3-OMe analogs 47 and 48 were less active against 1A1, rendering them somewhat 1A2-1A3 selective. Conversely, 4-OMe analog 49 lost substantial activity against 1A2 and 1A3 rendering it 1A1 selective, consistent with what we observed with the 4-pyridyl analog 45.
Table 4.
Characterization of benzyl pendant modifications.
 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| CMPD No. |
R2 | ALDH IC50a (μM) or % Control at 5 μM |
ADME Characterization | PEO1 Cell ALDEFLUORf (% inhib. @ 1 μM) |
|||||
| 1A1 | 1A2 | 1A3 | MLMb (t1/2, min) |
Aq. Solc (μM) |
cLogPd | LLEe | |||
| 28 | Phenyl | 0.13 ±0.03 | 0.11 ±0.02 | 0.073 ±0.005 | 8 | 8 | 4.5 | 2.5 | 47 ±5 |
| 42 | 2-Pyridyl | 0.242 ±0.02 | 0.063 ±0.06 | 0.089 ±0.07 | 50 | 20 | 3.4 | 3.2 | 97 ±1 |
| 43 | 3-Pyridyl | 0.139 ±0.001 | 0.089 ±0.005 | 0.095 ±0.016 | 7 | 209 | 3.3 | 3.6 | 95 ±2 |
| 44 | 3,5-Pyrimidinyl | 0.094 ±0.007 | 0.38 ±0.08 | 0.10 ±0.03 | 8 | 264 | 2.6 | 4.4 | 39 ±0.6 |
| 45 | 4-Pyridyl | 0.451 ±0.031 | 68% | 45% | 3.3 | 3.0 | |||
| 46 | 3-OHPhenyl | 0.130 ±0.004 | 0.100 ±0.002 | 0.091 ±0.001 | 4 | 21 | 4.2 | 2.7 | 61 ±5 |
| 47 | 2-OMe Phenyl | 0.445 ±0.021 | 0.136 ±0.009 | 0.112 ±0.011 | 4.3 | 2.1 | |||
| 48 | 3-OMe Phenyl | 0.451 ±0.031 | 0.108 ±0.008 | 0.174 ±0.015 | 6 | < 2 | 4.3 | 2.0 | |
| 49 | 4-OMe Phenyl | 0.352 ±0.024 | 41% | 39% | 4.3 | 2.2 | |||
Values are expressed as mean ± SEM (n=3).
Half-life in mouse liver micorosomes.
Thermodynamic aqueous solubility (performed by Analiza Inc. using quantitative nitrogen detection (www.analiza.com)).
Calculated log P.
Lipophilic Ligand Efficiency.
Mean ± SD as determined by flow cytometry (N=3).
Assessment of a crystal structure of 2-pyridine 42 bound to 1A1 indicates a very similar binding mode compared to that of (R)-28 (Figure 4, left panel). The pyridyl nitrogen resides within 3.4 Å of the W178 N-H and suggests that there may indeed be a hydrogen bonding interaction. The crystal structure of phenol 46 (Figure 4, right panel) similarly indicates that this compound is able to engage W178 and T129, as well as the main chain carbonyl of G125. Despite the apparent successful engagement of polar residues as we had predicted, neither of these analogs are more potent at inhibiting 1A1 than simple phenyl 28. One possible explanation is that the strength of the new hydrogen bond(s) is not sufficient to overcome the expected desolvation penalty incurred upon binding.
Figure 4.
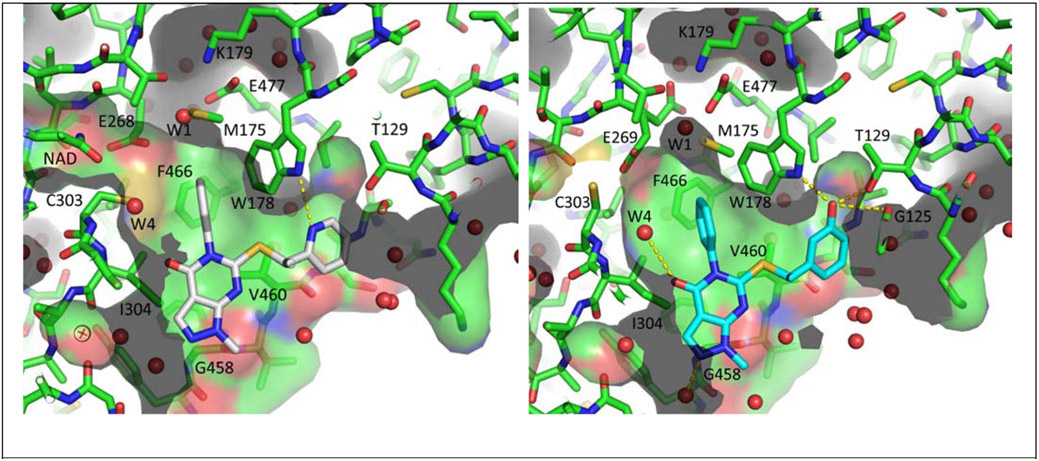
Structures of pyridine 42 (left, white) and phenol 46 (right, blue) bound to ALDH1A1 (PDBs: 7JWU and 7JWT
2.2.4. Exploring polar N-phenyl substituents
The (R)-28 crystal structure also indicated the proximity of a bound water and E477 to the distal carbon of the N-phenyl substituent (Figure 3, right panel). Although none of our attempts to engage these features (Table 5) improved the potency relative to proteo analog 28, comparison of alcohols 51 and 53 to their corresponding methyl ethers, 52 and 54, indicates an apparent preference by ALDH1A for hydrogen bond donors in this region. The most potent compound in the series, ethyl alcohol 53, was a 1A1-1A2 selective inhibitor. Examination of a 51-1A1 crystal structure (Figure 5, left panel) indicates that the methylene of the benzyl alcohol substituent is situated very close to M175, W178 and F466. The bound water and E477 move slightly to accommodate the alcohol and a new ordered water (W3) appears between the alcohol and C303. Interestingly, the structure of 53 bound to 1A1 (Figure 5, right panel) shows that the new water W3 is replaced by the alcohol OH, resulting in an unusual “tied back” conformation of the ethyl alcohol substituent.
Table 5.
Characterization of p-phenyl substituted analogs.
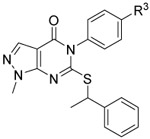 |
||||
|---|---|---|---|---|
| ALDH IC50a (μM) or % Control at 5 μMb | ||||
| No. | R3 | 1A1 | 1A2 | 1A3 |
| 51 | −CH2OH | 0.6 ±0.2 | 0.60 ±0.05 | 1.0 ±0.5 |
| 52 | −CH2OMe | 65% | 61% | 8 ±8 |
| 53 | −(CH2)2-OH | 0.36 ±0.09 | 0.216 ±0.003 | 3.0 ±0.1 |
| 54 | −(CH2)2-OMe | 27% | 54% | 34% |
| 55 | −CH2NH2 | 47% | 64% | 56% |
| 57 | −CH2NHMe | 58% | 70% | 57% |
| 58 | −CH2NMe2 | 47% | 86% | 53% |
| 61 | −(CH2)2NH2 | 39% | 45% | 50% |
| 62 | −(CH2)2NHMe | 89% | 94% | 80% |
| 63 | −(CH2)2NMe2 | 100% | 91% | 61% |
| 64 | −CN | 55%c | 3.1 ± 0.2 | 0.200 ±0.009 |
| 65 | −CCH | NPd | 0.8 ±0.1 | 0.52 ±0.05 |
Values are expressed as mean ± SEM (n=3).
Mean (n=3).
% Control at 20 μM.
No inhibition at 5 μM.
Figure 5.

Structures of alcohols 51 (orange) and 53 (salmon) bound to ALDH1A1 (PDBs: 7JWV and 7JWW)
Appending basic amines in an attempt to form a salt bridge with E477 was not tolerated across the 1A family (55-63). We again postulate that the desolvation penalty for the charged species is too great to be offset by a non-optimal polar interaction, and that the ability of E477 to interact with basic amines is likely diminished by the neighboring K179 (Figure 5). Serendipitously, nitrile 64, an intermediate in an abandoned synthetic route to 55, is to our knowledge the most potent 1A3-selective inhibitor reported to date. We performed docking studies using a 1A2 crystal structure (PDB: 6B5H), a 1A3 homology model,22 and a 1A3 crystal structure (PDB: 5FHZ)53 to generate one possible hypothesis for the observed selectivity (Supp. Fig. S-3A): that the nitrile can penetrate deeper into 1A3 and possibly engage in a semi-covalent interaction with the active site cysteine. Unfortunately 64 was rapidly metabolized (MLM t1/2 = 1 min) and was not suitable as an in vivo probe. Terminal alkynes possess similar linear geometry as nitriles, but are not sufficiently electrophilic to participate in semi-covalent interactions with cysteine. In support of our binding hypothesis, the alkyne 65 was 2-fold less potent against 1A3 than the nitrile, consistent with loss of reversible cysteine engagement. 65 was also greater than 3-fold more potent against 1A2. The apparent increase in potency of the alkyne against 1A2 vs the nitrile may actually just reflect the significantly higher desolvation penalty the nitrile has to pay to bind deep in the active site. The dramatic loss of activity of both 64 and 65 vs 1A1 is more difficult to rationalize. One hypothesis is that the I304 at the mouth of the pocket in 1A1 is larger than the corresponding T315 and T321 in 1A2 and 1A3, and cannot accommodate the N-phenyl ring when bearing rigid para substituents.
2.2.5. Combination of optimal thiol and pyrazole substituents
As shown in Table 6, we combined our optimized thiol substituents with the previously optimized 2-oxetanyl substituents on the pyrazole disclosed in our previous publication.22 Compound 4 from our previous work has been included in Table 6 for comparison. Homologated oxetanyl analog 67 exhibited potent pan-ALDH1A inhibition, as well as improved solubility and ALDEFLUOR activity relative to both 28 and 4. Oxetanyl analog 68 maintained comparable ALDH1A activity, more than doubled the MLM half-life of 67 and 4, and more potently inhibited ALDEFLUOR. Unfortunately, the improved MLM stability did not correlate well with in vivo exposure (Table 7).
Table 6.
Characterization of optimized compounds.
| CMPD No. |
ALDH IC50a (μM) | ADME Characterization | PEO1 Cell ALDEFLUORf (% inhib. @ 1 μM) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| 1A1 | 1A2 | 1A3 | MLMb t1/2 (min) |
Aq. Solc (μM) |
cLogPd | LLEe | |||
| 28 |  |
0.13 ±0.03 | 0.11 ±0.02 | 0.073 ±0.005 | 8 | 8 | 4.5 | 2.5 | 47 ±5 |
| 422 |  |
0.08 ±0.01 | 0.25 ±0.04 | 0.12 ±0.02 | 23 | 5 | 4.7 | 2.4 | 50 |
| 67 | 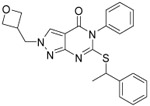 |
0.10 ±0.04 | 0.08 ±0.02 | 0.060 ±0.01 | 19 | 77 | 4.8 | 2.2 | 87 ±3 |
| 68 | 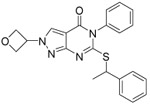 |
0.109 ±0.004 | 0.065 ±0.006 | 0.109 ±0.004 | 47 | 26 | 4.8 | 2.2 | 98 ±0.3 |
| 69 | 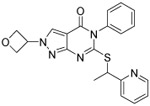 |
0.068 ±0.02 | 0.021 ±0.01 | 0.048 ±0.013 | >60 | 60 | 3.7 | 3.5 | 99 ±0.5 |
| 70 | 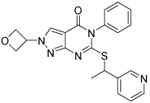 |
0.118 ±0.008 | 0.058 ±0.001 | 0.25 ±0.02 | 2.7 | 189 | 3.6 | 3.3 | 99 ±0.3 |
| 74 | 0.093 ±0.02 | 0.112 ±0.012 | 0.050 ±0.003 | >60 | 19 | 5.1 | 1.9 | 90 ±3 | |
Values are expressed as mean ± SEM (n=3).
Half-life in mouse liver micorosomes.
Thermodynamic aqueous solubility (performed by Analiza Inc. using quantitative nitrogen detection (www.analiza.com)).
Calculated log P.
Lipophilic Ligand Efficiency.
Mean ± SD as determined by flow cytometry (N=3).
Table 7.
Pharmacokinetic characterization of selected compounds.
| CMPD No. |
ADME Properties | Pharmacokinetic Parametersb | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 10 mg/kg i.p. | 20 mg/kg p.o. | ||||||||
| cLogP | Aq. Sola (μM) |
MLM t1/2 (min) |
% PPB @ 10 μM |
AUC0-7hobs (hr·μM) |
t1/2c (h) |
CMax (μM) |
AUC0-7hobs (hr·μM) |
CMax (μM) |
|
| 28 | 4.5 | 8 | 8 | 4.9 | 1.8 | 2.3 | 3.9 | 0.8 | |
| (R)-28 | 4.5 | 8 | 0.7 | 0.5 | 0.3 | ||||
| 68 | 4.8 | 26 | 47 | 97 | 19.5 | 2.1 | 7.9 | ||
| 42 | 3.4 | 20 | 50 | 7.9 | 1.2 | 3.9 | 17.4 | 7.3 | |
| 69 | 3.7 | 60 | >60 | 68 | 3.5 | 0.6 | 3.3 | 6.2 | 3.9 |
| 74 | 5.1 | 19 | >60 | 98 | 23 | 1.6 | 7.0 | 23.3 | 7.0 |
Thermodynamic solubility analysis was performed by Analiza Inc. using quantitative nitrogen detection. (www.analiza.com)
Results expressed as Mean (N=3) following a single administration as specified
Estimated using 2 and 4 hour timepoints only.
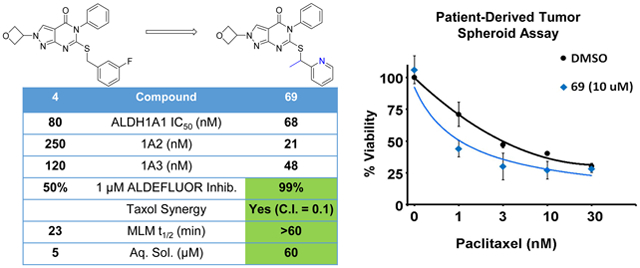
To guide design of more metabolically stable analogs, we performed an in vivo metabolite identification on 68, which revealed the oxetane and thioether to be the two primary sites of metabolism (Supp. Fig. S-5). Returning to the successful 2-pyridyl substituent as a means to reduce electron density at the metabolically labile benzylic carbon, we synthesized 2-pyridyl analog 69, which indeed exhibited excellent MLM stability and good solubility. Relative to the corresponding N-methyl analog 42 (Table 4), 69 improved inhibition across the 1A family, as well as ALDEFLUOR activity. In contrast, 3-pyridyl analog 70 did not gain significant potency compared to 42 and remained much less stable to MLM than the corresponding 2-pyridine 69. We also tried blocking the metabolically labile oxetane CH with a methyl group (74) which improved the MLM half-life from 47 min to over 60 min while maintaining ALDH1A potency and ALDEFLUOR inhibition.
To confirm the relationship between our IC50 values and inhibition constants (Ki), as well as to examine the mode of inhibition for compound 69, we performed steady-state kinetic analysis by varying the concentration of 69 as a function of varied aldehyde substrate, at fixed, saturating concentrations of coenzyme. The experiments gave an average Ki value of 38.1 ± 11.5 nM, which is within a factor of two relative to our observed IC50 value (68 nM, Table 6), thereby confirming the robustness of our SAR assay design. Fits to the tight-binding inhibition equations demonstrated best fits to either the noncompetitive (one experiment) or the uncompetitive (two experiments) models (Supp. Fig. S-7). The mode of inhibition is most consistent with the inhibitors binding to either a dead-end enzyme:NADH complex or an off-pathway enzyme conformation. Our structures with bound analogs of this series (Figures 2, 3, 4, and 5) are consistent with binding to enzyme species other than the catalytically poised enzyme:NAD+ complex. This mode of inhibition is similar to that obtained for CM26 and CM53 which have similar modes of binding to ALDH1A1 across the substrate binding pocket and produce noncompetitive modes of inhibition.54
2.3. In vivo pharmacokinetic analyses
To prioritize compounds for upcoming in vivo studies, we assessed the exposure of several compounds following i.p. and p.o. administration in mice. As shown in Table 7, compound 28 was only briefly able to surpass 1 μM following i.p. administration and had poor oral bioavailability. Surprisingly, active enantiomer (R)-28 achieved even worse exposure following i.p. administration despite having identical stability in the MLM assay. The more soluble 68 achieved 4-fold higher exposure than 28 following i.p. administration. Pyridyl 42 only had modestly higher exposure than 28 with i.p. administration, but achieved 4-fold higher exposure orally, likely due to protonation in the stomach. Given the excellent solubility and MLM stability of 69, we were surprised that the exposure for this compound was so poor. 74, which also had excellent MLM stability, achieved the highest exposure by both routes of administration. Noticing the correlation between higher cLogP and greater exposure, we hypothesized that a greater free drug fraction might be facilitating greater hepatic clearance.55 Indeed, based on PPB we calculate that the free drug fraction was 10-fold higher for the less lipophilic 69 than for 68 or 74. In fact, with regard to unbound drug levels, 69 had the highest i.p. exposure (1.1 hr*μM AUC0-7hobs) compared to 68 or 74 (0.6 and 0.5 hr*μM AUC0-7hobs, respectively). Importantly, the compounds 42 and 74 demonstrate comparable total exposure following oral administration at 20 mg/kg to that reported for 1a at 10 mg/kg (14.6 hr*μM AUC0-∞).
2.4. Comparison of ALDEFLUOR potency of selected compounds in ALDH1A1 and 1A3 high cell lines
As mentioned above, we hypothesized that our pan-inhibitors would be effective in a wider array of ovarian cancer cell lines. As shown in Figure 6, we found that compound 1a (NCT-506) is a very potent inhibitor of ALDEFLUOR in the OV90 cell line which expresses exclusively ALDH1A122 but is inactive in the PEO1 cell line which expresses predominantly ALDH1A3.12 Conversely, our ALDH1A pan inhibitors (R)-28, 68 and 69 were effective ALDEFLUOR inhibitors in both cell lines. Consistent with its selectivity for ALDH1A3, 64 was only active in PEO1 cells.
Figure 6.
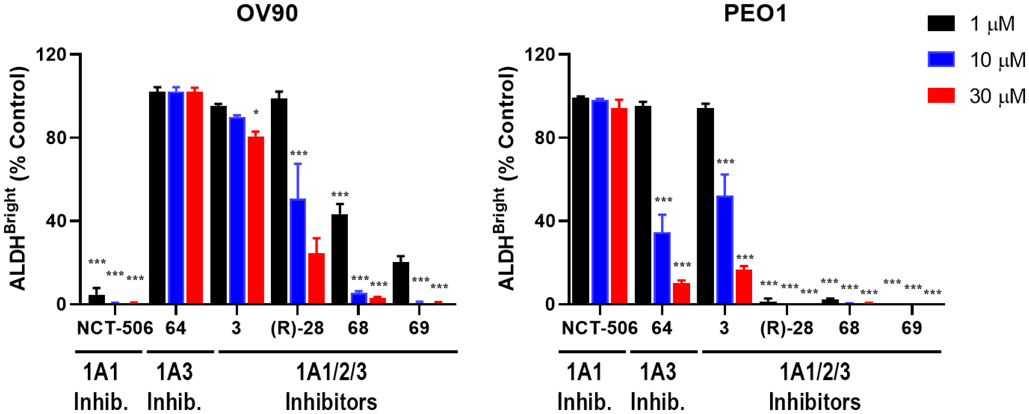
Comparison of selected ALDH inhibitors in ALDH1A1 and 1A3 high cell lines. ALDEFLUOR assay results from ALDH1A1-expressing OV90 and ALDH1A3-expressing PEO1 cells treated with the indicated inhibitors at the concentrations shown. Bars represent average percent ALDHbright compared to vehicle treated controls. (N=3) *p≤0.05, ***p≤0.0001.
2.5. Assessment of synergy between Taxol and selected ALDH inhibitors in taxane resistant SKOV-3 cells
Given the reported synergy for 1a and analogs with Taxol in resistant ALDH1A1 high SKOV-3 cells (SKOV3Trip2)27, 36, we next assessed our compounds under similar conditions (Figure 7). Because 1A1 is dominant in this cell line, we chose to compare 1A1-selective 1a with the three most potent ALDH1A pan-inhibitors from the Aldefluor study in Figure 6 ((R)-28, 68 and 69) to assess if there was any difference in activity between the two selectivity profiles. As single agents, two of our pan inhibitors (R)-28 and 69 were less toxic than 1a (EC50s of 86 and 148 μM respectively), while one of them (68) was somewhat more toxic (Figure 7). Given that the single agent toxicity of our compounds only occurred at high concentrations and was not correlated with ALDEFLUOR activity (Supp. Fig. S-6), we suspect this could be due to an off-target effect. All three of our compounds demonstrated excellent synergy, with a combination index (CI) as low as 0.01, and achieved synergy at modestly lower concentrations than 1a.
Figure 7.
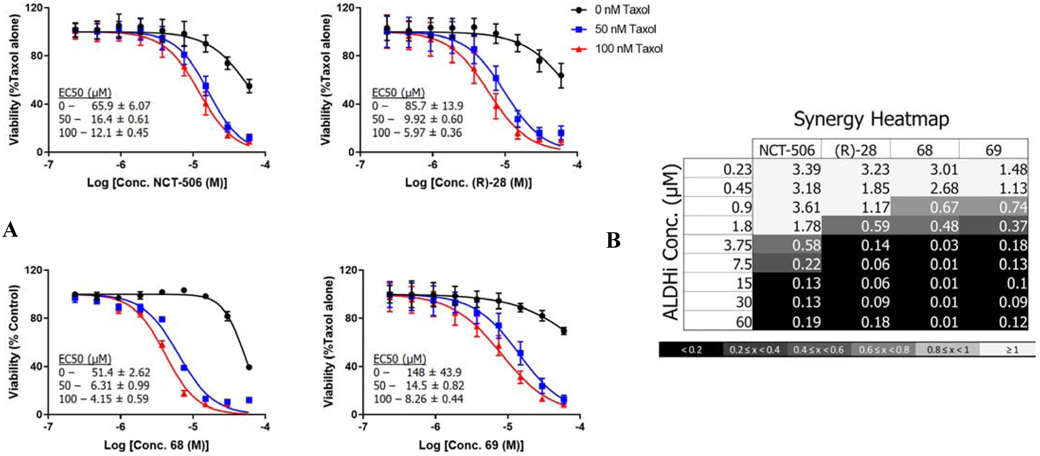
ALDH inhibitors reverse Taxol resistance in SKOV3 cells. A) Viability curves for Taxol resistant SKOV-3 cells treated with increasing concentrations of the indicated ALDH inhibitor alone or in combination with Taxol at the concentrations shown. Curves were generated in Prism 7 (GraphPad) and depict the average and SD from 3 experiments. B) Representative heatmaps of combination indices. Combination indices for the specified drug combinations were calculated using CompuSyn software (http://www.combosyn.com). (CI < 1 indicates synergy).
2.6. Demonstration of synergy with 3D patient-derived spheroids
Lastly, we tested the efficacy of our two most synergistic compounds using a stringent 3D tumor spheroid assay with primary chemotherapy resistant patient derived cells (Figure 8). High-grade serous ovarian cancer cells from two different patients were grown in poly-hema low adherence plates and rotated to increase cellular aggregation to increase growth and survival prior to treatment. Once spheroids were established, we initiated treatment with a titration of Taxol and the two most synergistic ALDH1A pan-inhibitors (68 and 69) from the taxane resistant cell line study in Figure 7. Both cell lines showed similar results, and thus the results are presented as a pooled analysis across the two cell lines. As noted above, both compounds showed synergy with Taxol with 69, which had no single agent toxicity, demonstrating the greatest synergy (CI 0.12-0.3).
Figure 8.
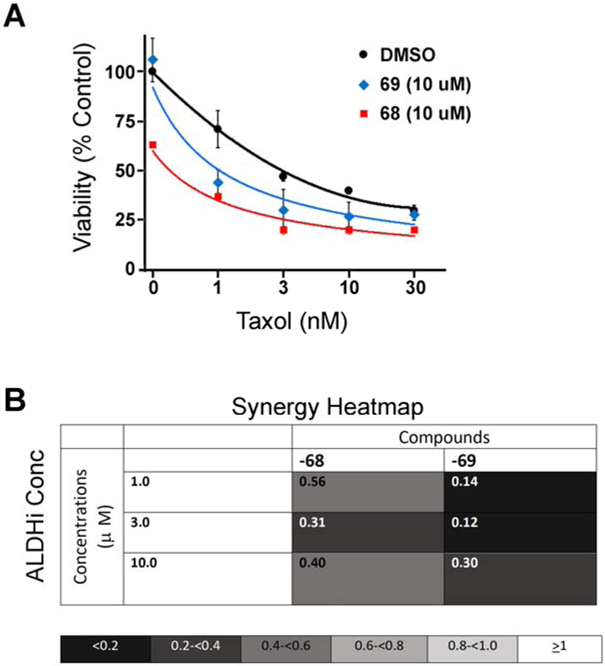
ALDH inhibitors reverse Taxol resistance in patient-derived tumor spheroids. A) Viability curves for cells treated with increasing concentrations of Taxol in the presence of the indicated ALDH inhibitor at a fixed concentration of 10 μM. Curves were generated in Prism 7 (GraphPad) and depict the average and SD from 3 experiments. B) Representative heatmaps of combination indices. Combination indices for the specified drug combinations were calculated using CompuSyn software (http://www.combosyn.com). (CI < 1 indicates synergy).
3. Conclusions
Encouraged by our early success in optimizing the CM39 series, we set out to further develop the series for ALDEFLUOR potency and pharmacokinetics. While replacement of the metabolically labile and lipophilic thioether linker was not successful, disrupting planarity with alkyl branching resulted in improved solubility for 28 relative to 3. Synthesis of the two enantiomers (R)-28 and (S)-28 revealed that the R-enantiomer (R)-28 possessed 20-fold greater ALDH activity and commensurately greater ALDEFLUOR inhibition. Inactive enantiomer (S)-28 was potently cytotoxic as a single agent (data not shown), indicating that the single agent cytotoxicity possessed by some analogs in the series is likely off-target. Efforts to alter the pyrazolopyrimidinone core to reduce tPSA or conformationally restrict the thioether afforded active, but unimproved compounds. Exploring polar substituents to engage W179 and T129 was more fruitful. While 2- and 3-pyridyl substituents did not result in increased enzymatic 1A1 potency relative to phenyl, they improved solubility, lipophilic efficiency, and ALDEFLUOR potency. Crystal structures of the 2-pyridyl and 3-hydroxyl analogs 42 and 46 indicate likely engagement with W179 in 1A1, but a desolvation penalty may counteract this favorable interaction. The 2-pyridyl analog 42 also improved metabolic stability 6-fold relative to phenyl analog 28. Pyrimidinyl analog 44 demonstrated that further increases in polarity in this region are tolerated by the enzyme, but detrimental to cell activity as measured by ALDEFLUOR, perhaps due to reduced cell permeability brought about by higher tPSA.
While attempts to engage polar residues deep in the active site were not successful at improving potency, they did result in compounds with interesting isoform selectivity. To our knowledge, nitrile analog 64 is the most potent ALDH1A3-selective inhibitor to date. Interestingly, a comparison of the predicted binding mode for 64 in ALDH1A3 with the bound conformation of GA1138, an analog of a second chemically distinct series of 1A3-selective inhibitors represented by 1b (Figure 1), reveals close proximity of the two ligands but very little actual overlap (Supp. Fig. S-3B), suggesting that hybrid structures of these two series could be designed in a fragment-like strategy with even greater potency.
Finally, combining the optimal linker and pyridyl substituents with the N-2 oxetane resulted in compounds with excellent MLM stability and ALDEFLUOR inhibition. Steric blocking of the oxetane, which was identified as a site of in vivo metabolism, also resulted in improved MLM stability. While our attempts thus far to improve in vivo exposure have been complicated by a presumed increase in hepatic extraction due to lower plasma protein binding, nevertheless multiple compounds were able to exceed the concentration at which they inhibited ALDEFLUOR by more than 50% in vitro following i.p. and oral administration.
Importantly, we were able to establish that our compounds were effective at inhibiting ALDEFLUOR in cell lines expressing either 1A1 or 1A3. In contrast, and as expected, the 1A1 selective inhibitor 1a (NCT-506) was not effective against the 1A3 expressing high-grade serous PEO1 line. Inhibition of both isoforms may be important for an effective therapeutic intervention as high-grade serous ovarian tumors tend to express higher levels of 1A3 than healthy tissue. Similarly, ALDH1A3 is upregulated in breast cancer and believed to be a primary driver of stemness56 and additional ALDH1A family members can be induced in therapy-resistant clones.57 Finally, we demonstrated that our compounds reversed chemoresistance in the SKOV3-TR line and synergized with Taxol in patient-derived tumor spheroids. Studies assessing the ability of our compounds to reverse taxane resistance in vivo are underway and will be reported in due time.
4. Experimental Section
4.1. Protein Purification and Enzymatic Assays
Human ALDH1A1, ALDH1A2, ALDH1A3, and ALDH2 were prepared and purified as previously described. 58-62 Inhibition of ALDH activity by compounds and IC50 curves were determined by measuring the formation of NAD(P)H spectrophotometrically at 340 nm (molar extinction coefficient of 6200 M−1 cm−1) on the Beckman DU-640 and Spectramax 340PC spectrophotometers using purified recombinant enzyme. Reaction components for ALDH1A and ALDH2 assays consisted of 100-200 nM enzyme, 200 μM NAD+, 100 μM propionaldehyde, and 1% DMSO in 25 mM BES buffer, pH 7.5. All assays were performed at 25 °C and were initiated by addition of substrate after a 2 min incubation period. IC50 curves were collected for compounds which substantially inhibited ALDH1A activity at 20 μM compound. Data were fit to the four parameter EC50 equation using SigmaPlot (v12) and the values represent the mean/SEM of three independent experiments (each n=3). The mode of inhibition for 69 was determined via steady-state kinetics by co-varying inhibitor and substrate concentrations at fixed concentration of NAD+ (1 mM; KM = 50 μM). All reactions contained 150 nM ALDH1A1 and 2% DMSO in 30 mM sodium BES, pH 7.5 at 25 °C. The reactions contained 5-80 μM propionaldehyde (KM = 10 μM). All data were fit to competitive, noncompetitive, uncompetitive, and mixed inhibition models using the tight binding inhibition equations in SigmaPlot (StatSys v13)(Supp. Fig. S-7). The appropriate model was selected through analysis of goodness-of-fit and the residuals of those fits. The values represent the average of three independent experiments for five concentrations of inhibitor and five concentrations of substrate.
4.2. ALDEFLUOR Assay
OV90 and PEO1 cells were used as ALDH1A1 and ALDH 1A3 high expression cell lines, respectively. Cells were grown and ALDEFLUOR (STEMCELL Technologies) assays performed as previously described.22 Briefly, cells were cultured to 70-80%, trypsinized, washed with PBS, and then re-suspended in ALDEFLUOR buffer. ALDEFLUOR reagent was added, cells were rapidly mixed and then equally distributed into tubes containing inhibitor, DEAB control, or vehicle. After a 30 minute incubation at 37 °C, cells were washed with ALDEFLUOR buffer and maintained on ice until flow-cytometric analysis. Gating was based on DEAB (inhibitor control- set to <1%) and vehicle control treated cells (positive control). The percent of ALDH inhibition was set as percentage of ALDEFLUOR positive cells for a particular sample and the percentage of ALDEFLUOR positive cells vs. vehicle treated control. The Two-way ANOVA with Tukeys multiple comparison test (Prism GraphPad) was used to determine statistical significance between samples treated with compound or vehicle.
4.3. Crystal Structure Determination.
All structures of ALDH1A1 with compounds were determined using crystals of the naturally occurring polymorphic variant, N121S. The crystals were grown by equilibrating 4-8 mg/mL ALDH1A1 N121S against 100 mM sodium BisTris, pH 6.2-6.5, 6-11% PEG3350, 200 mM NaCl, and 5-10 mM YbCl3. The complexes between ALDH1A1 N121S and compounds (R)-28, 42, 46, 51 and 53 were prepared by co-crystallizing in the presence of 500 uM compound (1% v/v DMSO final). For compound 42, 1 mM NAD was added to the solution for 6 hr prior to freezing. Cryoprotection for flash-freezing utilized 20% (v/v) ethylene glycol in ligand solution. Diffraction data for all complexes with ALDH1A1 N121S were collected at Beamline 19-ID operated by the Structural Biology Consortium at the Advance Photon Source (APS), Argonne National Laboratory. Diffraction data were indexed, integrated, and scaled using HKL3000.63 The CCP4 program suite was used for molecular replacement and refinement.64 The Coot molecular graphic application was used for model building.65 The TLSMD (translation/libration/screw motion determination) server was used to determine dynamic properties of ALDH1A1 N121S.66-67 Data collection and refinement statistics are compiled in Table S-1.
4.4. Cell Viability and Synergy Assays
PEO1 and OV90 cells were seeded in 96-well plates at 4000 cells/well (PEO1) or 2000 cells/well (OV90) as previously described.22 After 24 h, inhibitor or vehicle was added, Taxol was added, and then cells were incubated at 37 °C for 72 h. Viability was assessed using Cell-Titer Glo 2.0 (Promega) per manufacturer’s instructions. Viability was normalized to vehicle treated controls and graphed in Prism 7. Data are displayed as mean ± SD. Synergy was assessed using Chou-Talalay method and the CompuSyn program.68
4.4.1. Patient Derived Spheroid Assay
Two primary patient derived ovarian cancer cell lines were used (each derived from patients with high-grade serous stage IIIC ovarian cancer) obtained from IRB approved protocol, and collected from consented patients) and processed as previously described 69-70. 6,000 Patient cells were suspended in low-adherence plates in serum-free medium (SFM) supplemented with 5 ng/ml FGF, 5 ng/ml EGF, B27, 1X insulin-transferrin-selenium supplement, 1X nonessential amino acids, antibiotics and antimycotics. Cells were rotated for 30 minutes after plating to encourage spheroid formation 71. 24 hours after plating, treatment with the indicated compound and doses was initiated. Cells were retreated every 72 hours and total cell counts evaluated using CellTiter-Glo 3D viability assay (Promega).
4.5. Metabolic Stability in Mouse Liver Microsomes
The metabolic stability was assessed using CD-1 mouse liver microsomes (MLM). 1 μM of each compound was incubated with 0.5 mg/mL microsomes and 1.7 mM cofactor β-NADPH in 0.1 M phosphate buffer (pH = 7.4) containing 3.3 mM MgCl2 at 37 °C. The DMSO concentration was less than 0.1% in the final incubation system. At 0, 5, 10, 15, 30, 45, and 60 min of incubation, 40 μL of reaction mixture were taken out, and the reaction is quenched by adding 3-fold excess of cold acetonitrile containing 100 ng/mL of internal standard for quantification. The collected fractions were centrifuged at 15000 rpm for 10 min to collect the supernatant for LC–MS/ MS analysis, from which the amount of compound remaining was determined. The natural log of the amount of compound remaining was plotted against time to determine the disappearance rate and the half-life of tested compounds.
4.6. Pharmacokinetic Studies in Mice
All animal experiments in this study were approved by the University of Michigan Committee on Use and Care of Animals and Unit for Laboratory Animal Medicine (ULAM). The abbreviated pharmacokinetics of compounds were determined in female CD-1 mice following intraperitoneal (ip) injection of 10 mg/kg respectively. Compounds were dissolved in the vehicle containing 20% DMSO, 50% PEG-400, and 30% PBS. Four blood samples (50 μL) were collected over 7 h (at 0.5 h, 2 h, 4 h, and 7 h), centrifuged at 3500 rpm for 10 min, and plasma was frozen at −80°C for later analysis. Plasma concentrations of the compounds were determined by the LC–MS/MS method developed and validated for this study. The LC–MS/MS method consisted of a Shimadzu HPLC system, and chromatographic separation of tested compound which was achieved using a Waters Xbridge-C18 column (5 cm × 2.1 mm, 3.5 μm). An AB Sciex QTrap 4500 mass spectrometer equipped with an electrospray ionization source (ABI-Sciex, Toronto, Canada) in the positive-ion multiple reaction monitoring (MRM) mode was used for detection. All pharmacokinetic parameters were calculated by noncompartmental methods using WinNonlin, version 3.2 (Pharsight Corporation, Mountain View, CA, USA).
4.6.1. Identification of In vivo Metabolites of 68 from Mouse Plasma.
Female CD-1 mice were used to collect plasma samples for in vivo metabolite identification. Blank plasma was collected from one mouse without dosing drugs. Another mouse was dosed with target compound by IV (15 mg/kg), then the plasma samples were collected in 1 h after dosing. Both blank plasma and plasma with drug and metabolite were mixed with 3 times acetonitrile for protein precipitation and compound extraction. After vortex and centrifuge, the top extraction solutions were injected onto LCMS system for metabolite identification. The general approach for metabolite identification using AB Sciex QTrap 5500 mass spectrometer involves the following steps: 1. Obtain a product ion spectrum of the parent compound to establish fragmentation. 2. Interpret the spectrum to identify major fragment ion and possible neutral loss. 3. Collect spectra of samples using both established precursor ion scan and neutral loss scan, EMS scan of both control and samples were acquired also. 4. Run product ion scans and MRM scans for all possible metabolite identified from step 3 plus expected metabolite. 5. Interpret the spectrum of the metabolites and determine the structure with their logical fragmentation pattern.
4.7. Molecular Modeling.
Molecular docking studies were performed using GOLD (Genetic Optimization for Ligand Docking, The Cambridge Crystallographic Data Centre, Cambridge, UK) 72-73 against 1A1 (PDB: 7JWS), 1A2 (PDB: 6B5H)74 crystal structures and 1A3 homology model, previously reported by us.22 Prior to docking, ten different conformations were generated for each ligand using Omega (OpenEye Scientific, Santa Fe, NM), a systematic, knowledge-based conformer generator.75. GOLD uses a genetic algorithm to explore the conformational space of a compound inside the binding site of a protein.72-73 Docking studies were performed using the standard default settings with 100 genetic algorithms (GA) runs on each molecule.
4.8. Chemistry
4.8.1. General
All reagents were used in the condition received from commercial sources. Flash chromatographic purifications were performed using a Teledyne ISCO Combiflash RF with Redisep Gold RF silica columns. 1H NMR and 13C NMR were taken in CDCl3 or DMSO-d6 at room temperature on Varian Inova 400 or 500 MHz instruments. Reported chemical shifts are expressed in parts per million (ppm) on the δ scale from an internal standard of tetramethylsilane (0 ppm). Mass spectrometry data were obtained on either an Agilent TOF or Agilent Q-TOF. An Agilent 1100 series HPLC with an Agilent Zorbax Eclipse Plus-C18 column was used to determine purity of biologically tested compounds. All tested compounds were determined to be >95% pure using a 6 min gradient of 10-90% acetonitrile in water followed by a 2 min hold at 90% acetonitrile with detection at 254 nm. Enantiomeric excesses were determined by chiral analytical HPLC using a Daicel Chiralcel OD column 4.6X250mm. Optical rotations were determined by Robertson Microlit Laboratories (http://www.robertson-microlit.com) according to USP <781S> protocol.
NCT-506 used as a comparator in biological studies was prepared as described by Yang et al.36
4.8.2. 6-((3-Fluorobenzyl)thio)-1-methyl-1H-pyrazolo[4,3-c]pyridin-4(5H)-one (6).
A microwave tube was charged with 5 (300 mg, 1.6 mmol), DIPEA (1.1 ml, 6.5 mmol), (3-fluorophenyl)methanethiol (0.4 ml, 3.3 mmol), and 3 mL n-butanol. The headspace was purged with Ar and the tube was capped and heated to 170 °C in a microwave synthesizer for 48 h. Upon cooling a white precipitate formed which was collected by filtration and washed with water and hexanes to afford the titled compound (300 mg, 1.04 mmol, 64% yield).1H NMR (500 MHz, DMSO-d6) δ 11.37 (br. s., 1H), 7.95 (s, 1H), 7.31 - 7.41 (m, 1H), 7.16 - 7.25 (m, 2H), 7.09 (t, J = 8.56 Hz, 1H), 6.67 (s, 1H), 4.38 (s, 2H), 3.88 (s, 3H)
4.8.3. 6-((3-Fluorobenzyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[4,3-c]pyridin-4(5H)-one (7).
A flask was charged with 2 mL DCE, Pyridine (28 μL, 0.346 mmol), TEMPO (30 mg, 0.190 mmol), 6 (50 mg, 0.173 mmol), Cu(II)OAc hydrate (7 mg, 0.035 mmol), phenylboronic acid (42 mg, 0.346 mmol), and 3Å molecular sieves and stirred under air for 24 h at RT. The mixture was filtered with celite and the filter pad washed with DCM. The filtrate was washed with 1N HCl and brine, dried over sodium sulfate, and concentrated. The crude mixture was purified by flash (Elutes at 60% EtOAc in Hexanes) affording the titled compound as a white solid (32 mg, 0.088 mmol, 51% yield). MS (ESI): m/z 366.1066 [M+H]+ 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H), 7.45 - 7.55 (m, 3H), 7.23 - 7.31 (m, 3H), 7.04 (d, J = 7.43 Hz, 1H), 6.94 - 7.01 (m, 2H), 6.20 (s, 1H), 3.99 (s, 2H), 3.91 (s, 3H), 13C NMR (101 MHz, CDCl3) δ 162.8, 159.0, 144.9, 143.2, 137.6, 136.9, 130.3, 129.6, 129.3, 124.6, 115.9, 115.0, 111.5, 90.8, 38.5, 35.8. HPLC Purity: 95%
4.8.4. 2-chloro-7-methyl-3H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (9).
To a flask charged with 10 mL THF and 60 wt.% NaH (117 mg, 2.93 mmol) under N2 at 0 °C was added 8 (500 mg, 2.66 mmol) dissolved in 10 mL THF by syringe. The flask was stirred at 0 °C for an hour at which point MeI (183 μl, 2.93 mmol) was added by syringe. The flask was allowed to gradually warm to RT overnight and the next day the solvent was removed and the residue was treated with 20mL 2N NaOH and refluxed overnight. The product was precipitated by addition of conc. HCl, filtered, and washed with water and hexane to afford the titled compound as a white solid (415 mg, 2.26 mmol, 85% yield).1H NMR (400 MHz, DMSO-d6) δ 12.83 (br. s., 1H), 7.10 (d, J = 2.74 Hz, 1H), 6.45 (d, J = 3.13 Hz, 1H), 3.66 (s, 3H).
4.8.5. 2-((3-Fluorobenzyl)thio)-7-methyl-3H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (10).
A pressure tube was charged with 9 (100 mg, 0.545 mmol), DIPEA (0.190 ml, 1.089 mmol), (3-fluorophenyl)methanethiol (0.101 ml, 0.817 mmol) and 1mL abs. EtOH. The headspace was purged with Ar and the tube was heated to 100 °C overnight. Upon cooling a white precipitate formed which was collected by filtration and washed with an additional 1mL ice cold EtOH to afford the titled compound as a white solid (110 mg, 0.380 mmol, 70 % yield). 1H NMR (400 MHz, DMSO-d6) δ 12.12 (s, 1H), 7.41 - 7.24 (m, 3H), 7.06 (ddt, J = 9.5, 7.5, 2.1 Hz, 1H), 6.97 (d, J = 3.4 Hz, 1H), 6.35 (d, J = 3.3 Hz, 1H), 4.45 (s, 2H), 3.70 (s, 3H).
4.8.6. 2-((3-Fluorobenzyl)thio)-7-methyl-3-phenyl-3H-pyrrolo[2,3-d]pyrimidin-4(7H)-one (11).
A flask was charged with 10 (50 mg, 0.173 mmol), phenylboronic acid (23 mg, 0.190 mmol), Cu(II)OAc hydrate (52mg, 0.259 mmol), pyridine (28μl, 0.346 mmol), 3Å molecular sieves and 2mL DCM and stirred at RT under air for 5 days. The mixture was filtered through celite and the pad washed with additional DCM. The filtrate was washed with 1N HCl and brine before drying over sodium sulfate and concentrating. The crude residue was taken up in hot EtOH and the titled compound recrystallized as a white solid upon cooling (25 mg, 0.068 mmol, 40 % yield). MS (ESI): m/z 366.1073 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.66 - 7.41 (m, 3H), 7.33 - 7.22 (m, 3H), 7.18 - 7.08 (m, 2H), 6.94 (td, J = 8.7, 2.5 Hz, 1H), 6.80 - 6.71 (m, 1H), 6.67 - 6.61 (m, 1H), 4.34 (s, 2H), 3.80 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.67 (d, J = 245.7 Hz), 162.41, 159.18, 155.59, 146.87, 139.27, 136.39, 129.93 (d, J = 8.2 Hz), 129.70, 129.55, 129.49, 124.71 (d, J = 2.9 Hz), 123.06, 116.20, 114.35 (d, J = 21.1 Hz), 104.59, 102.96, 36.60 (d, J = 2.0 Hz), 31.52.
4.8.7. 2-((3-fluorobenzyl)thio)-3-phenylimidazo[5,1-f][1,2,4]triazin-4(3H)-one (14).
13 (12 mg, 0.077 mmol), prepared as previously described,76 was dissolved in dry DMF to which isothiocyanatobenzene (10 μl, 0.085 mmol) was added. The reaction was stirred under N2 overnight. The next day, 60 wt.% NaH (6 mg, 0.155 mmol) was added and the mixture was stirred for 2 h, at which point the flask was chilled to 0 °C and 3-fluorobenzyl bromide (10 μl, 0.085 mmol) was added. The mixture was stirred overnight. The reaction was diluted with brine and extracted with ethyl acetate. The organic portion was dried over sodium sulfate and the solvent removed. The crude product was purified by flash (Eluting 50-70% EtOAc in Hexanes) yielding the titled compound as a colorless residue (14 mg, 0.040 mmol, 51% yield). MS (ESI): m/z 353.0870 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.11 (s, 1H), 7.91 (s, 1H), 7.59 - 7.47 (m, 3H), 7.33 - 7.25 (m, 3H), 7.12 (d, J = 7.7 Hz, 1H), 7.05 (d, J = 9.4 Hz, 1H), 6.96 (t, J = 8.8 Hz, 1H), 4.26 (s, 2H). 13C NMR (126 MHz, CDCl3) δ 162.71 (d, J = 246.7 Hz), 153.51, 153.22, 137.36 (d, J = 7.6 Hz), 133.72, 133.52, 130.48, 130.20 (d, J = 8.4 Hz), 129.93, 129.74, 129.49, 124.95 (d, J = 3.2 Hz), 118.02, 116.24 (d, J = 22.2 Hz), 114.95 (d, J = 21.0 Hz), 36.59. HPLC Purity 98%
4.8.8. 6-mercapto-l-methyl-5-phenyl-lH-pyrazolo[3.4-dlpyrimidin-4(5H)-one (16).
To a solution of ethyl 5-amino-l -methyl- lH-pyrazole-4-carboxylate (15) (1 g, 5.91 mmol) in 15 mL DMF at 0°C was slowly added 60 wt.% NaH (0.591 g, 14.78 mmol) against a counterflow of nitrogen. The reaction was stirred for l hr at 0°C at which point phenyl isothiocyanate (0.706 mL, 5.91 mmol) was added and the reaction was warmed to 50°C and stirred overnight. The reaction was quenched by adding a 5 mL of sat. aq. ammonium chloride then the solvents were distilled off under high vacuum. The residue obtained was washed 3x with hexanes to remove the mineral oil then taken up in DCM and filtered to remove salts. After the DCM was evaporated the crude was triturated from hot EtOAc yielding the titled compound as a white solid (600mg, 2.323 mmol, 39% yield) 1H NMR (400 MHz, DMSO-d6) δ 8.23 (br. s., 1H), 7.65 (s, 1H), 7.33 (t, J= 7.60 Hz, 2H), 7.22 (t, J = 7.40 Hz, 1H), 6.96 (d, J= 8.22 Hz, 2H), 3.71 (s, 3H).
4.8.9. 1-Methyl-6-(methylsulfonyl)-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (17).
MeI (116 μl, 1.858 mmol) was added to a solution of 15 (prepared as previously described22) (400 mg, 1.549 mmol) and K2CO3 (321 mg, 2.323 mmol) in 5mL DMF. The mixture was stirred for 2 h at which point it was diluted with water, cooled to 0°C and stirred for 30 min. The resulting white precipitate was filtered and taken up in 5mL DCM to which mCPBA (896 mg, 3.64 mmol) was added. The mixture was stirred overnight at RT then 5mL Aq. Sodium Thiosulfate was added at which point the mixture was stirred an additional 1 h. The mixture was diluted with more DCM and the aqueous portion was discarded. The aqueous portion was washed with sat. aq. bicarbonate followed by brine and dried over sodium sulfate and concentrated. The reaction was redissolved in 5mL DCM, filtered and the filtrate concentrated. The residue was taken up in hot ethanol and slowly cooled to 0 °C to afford the titled compound as a white solid (360 mg, 1.183 mmol, 76 % yield).
4.8.10. 6-((3-Fluorobenzyl)oxy)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (18).
To a dry flask at 0 °C under N2 charged with 60 wt.% NaH in mineral oil (16.43 mg, 0.411 mmol) was added 1 mL of DMF and (3-fluorophenyl)methanol (0.038 mL, 0.353 mmol) by syringe. The mixture was stirred for 10 minutes at which point solid 17 (100 mg, 0.329 mmol) was added quickly. The mixture was stirred overnight and gradually warmed to RT. The following day the mixture was diluted with EtOAc and washed with water. The water was back extracted with a small portion of EtOAc and the combined organics were washed 3x with brine before drying over sodium sulfate and concentrating. The crude was purified by flash (0-100% EtOAc in Hex) yielding the titled compound as a white solid (10 mg, 0.029 mmol, 9 % yield). MS (ESI): m/z 351.1252 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.00 (s, 1H), 7.59 - 7.43 (m, 3H), 7.32 - 7.24 (m, 1H), 7.22 (d, J = 7.5 Hz, 2H), 7.04 - 6.90 (m, 2H), 6.84 (d, J = 9.6 Hz, 1H), 5.41 (s, 2H), 3.94 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.73 (d, J = 246.7 Hz), 157.90, 155.24, 150.75, 137.55 (d, J = 7.6 Hz), 135.64, 134.86, 130.08 (d, J = 8.3 Hz), 129.38, 128.95, 128.35, 122.58 (d, J = 3.0 Hz), 115.15 (d, J = 21.1 Hz), 114.12 (d, J = 22.3 Hz), 102.06, 69.24, 33.90. HPLC Purity: 97%
4.8.11. 6-((3-Fluorobenzyl)amino)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (19).
To a solution of (3-fluorophenyl)methanamine (0.112 ml, 0.986 mmol) in 5mL DMF at 0 °C was added 60 wt.% NaH in mineral oil (33 mg, 0.821 mmol). After 5 min and 17 (200 mg, 0.657 mmol) was added. The mixture turned immediately yellow and was stirred at 0 °C for 1 h at which point it was allowed to warm to RT. After 1 h the reaction was quenched with sat. aq. NH4Cl and diluted with water and extracted 2x with ethyl acetate. The combined organics were washed with brine 3x and dried over sodium sulfate before concentrating to yield a yellow solid. The crude solid was recrystallized from hot ethanol to afford the titled compound as a white crystalline solid upon cooling (29 mg, 0.083 mmol, 13 % yield). MS (ESI): m/z 350.1410 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.96 - 7.90 (m, 1H), 7.64 - 7.56 (m, 2H), 7.56 - 7.50 (m, 1H), 7.34 - 7.27 (m, 3H), 7.01 (d, J = 7.6 Hz, 1H), 6.98 - 6.92 (m, 2H), 4.67 - 4.54 (m, 3H), 3.85 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 162.93 (d, J = 246.7 Hz), 158.16, 152.49, 152.47, 140.69 (d, J = 6.9 Hz), 135.47, 134.63, 130.74, 130.20 (d, J = 8.1 Hz), 130.05, 129.15, 122.77 (d, J = 2.8 Hz), 114.48 (d, J = 21.1 Hz), 114.27 (d, J = 21.9 Hz), 100.24, 45.20 (d, J = 1.9 Hz), 33.57. HPLC Purity: 99%
4.8.12. 6-(chloromethyl)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (20).
A flask under N2 charged with chloroacetic acid (12.76 g, 135 mmol) and 15 (1.46 g, 6.75 mmol) was heated at 80 °C until a homogenous melt was obtained. Chloroacetyl chloride (1.35 mL, 16.88 mmol) was added dropwise and the mixture was stirred at 80 °C for 1 h. The flask was then fitted with a reflux condenser and heated to 120 °C overnight. The next day the flask was removed from the oil bath and immediately poured into 100 mL water. The mixture was stirred vigorously until cool. The titled compound, which precipitated, was collected by filtration as a white solid (1.22 g, 4.44 mmol, 66 % yield). 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.48 - 7.64 (m, 3H), 7.28 - 7.37 (m, 2H), 4.23 (s, 2H), 4.05 (s, 3H).
4.8.13. 6-(((3-Fluorophenyl)thio)methyl)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (21).
20 (60 mg, 0.218 mmol), K2CO3 (45 mg, 0.328 mmol), 3-fluorobenzenethiol (25 μL, 0.306 mmol) and 1 mL DMF were stirred overnight at 50 °C. The product was precipitated by the addition of water and collected by filtration. The filtrate was recrystallized from EtOH yielding the titled compound as a white crystalline solid (24 mg, 0.066 mmol, 30% yield). MS (ESI): m/z 367.1025 [M+H]+ 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.49 - 7.60 (m, 3H), 7.28 - 7.34 (m, 2H), 7.15 - 7.24 (m, 2H), 7.08 (d, J = 7.83 Hz, 1H), 6.92 (dt, J = 1.96, 8.41 Hz, 1H), 3.95 (s, 3H), 3.86 (s, 2H) 13C NMR (101 MHz, CDCl3) δ 164.1, 163.9, 161.4, 158.2, 155.9, 136.3, 135.3, 130.1, 129.9, 129.7, 128.9, 125.9, 117.2, 114.1, 104.3, 38.7, 34.1 HPLC Purity: 97%.
4.8.14. 6-((3-Fluorophenoxy)methyl)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (22).
To a dry flask under Ar charged with K2CO3 (55 mg, 0.400 mmol) and 20 (55 mg, 0.200 mmol) was added 1 mL DMF followed by 3-fluorophenol (27 μL, 0.300 mmol). The vial was flushed with argon, sealed, and heated to 70 °C for 3 h at which point the reaction was complete by HPLC. The mixture was diluted with 20 mL water and extracted 2x with EtOAc. The combined organics were washed 3x with brine and dried over sodium sulfate. Removal of solvent yielded a colorless oil which was subjected to flash (eluted at 40% EA/Hex) yielding the titled compound as a white crystalline solid (58 mg, 0.166 mmol, 83 % yield). MS (ESI): m/z 351.1255 [M+H]+ 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.43 - 7.55 (m, 3H), 7.28 (d, J = 7.04 Hz, 2H), 7.12 - 7.21 (m, 1H), 6.66 (dt, J = 1.57, 8.22 Hz, 1H), 6.52 - 6.58 (m, 1H), 6.46 - 6.52 (m, 1H), 4.71 (s, 2H), 4.02 (s, 3H) 13C NMR (101 MHz, CDCl3) δ 163.3, 158.7, 158.0, 153.9, 150.5, 135.6, 135.4, 130.3, 129.8, 129.7, 128.5, 110.3, 108.7, 104.7, 102.6, 68.3, 34.3 HPLC Purity: 98%.
4.8.15. 6-(((3-Fluorophenyl)amino)methyl)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (23).
A flask was charged with 1 mL DMF, K2CO3 (151 mg, 1.092 mmol), KI (18 mg, 0.109 mmol), 20 (150 mg, 0.546 mmol) and was briefly vacuum degassed and put under nitrogen at which point 3-fluoroaniline (63 μL, 0.655 mmol) was added by syringe. The flask was heated at 50 °C for 24 h. Water was added to precipitate the product which was collected by filtration yielding the titled compound as a light pink powder (177 mg, 0.507 mmol, 93 % yield). MS (ESI): m/z 350.1415 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.07 (s, 1H), 7.54 - 7.65 (m, 3H), 7.26 (d, J = 6.36 Hz, 2H), 7.07 (q, J = 7.83 Hz, 1H), 6.41 (t, J = 8.07 Hz, 1H), 6.29 (d, J = 7.83 Hz, 1H), 6.18 (d, J = 11.25 Hz, 1H), 5.00 (br. s., 1H), 4.05 (s, 3H), 3.88 (d, J = 5.20 Hz, 2H) 13C NMR (126 MHz, CDCl3) δ 163.9, 157.9, 156.2, 150.3, 148.4, 135.6, 135.4, 130.4, 130.3, 130.1, 128.3, 109.0, 104.8, 104.3, 99.9, 46.8, 34.3 HPLC Purity: 95%.
4.8.16. 6-(((3-Fluorophenyl)(methyl)amino)methyl)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (24).
A flask containing K2CO3 (25 mg, 0.179 mmol), MeI (9 μl, 0.150 mmol), 23 (50 mg, 0.143 mmol), and 1mL DMF was heated to 50 °C and stirred overnight. The next day the mixture was diluted with water and extracted 2x with EtOAc. The combined organic portion was washed 3x with brine, dried over sodium sulfate, and concentrated. The crude mixture was purified by flash chromatography (EtOAc in Hex) and the product obtained was further purified by recrystallization from ethanol yielding the titled compound as a white solid (10 mg, 0.028 mmol, 19% yield). MS (ESI): m/z 364.1568 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.05 (s, 1H), 7.59 (t, J = 7.5 Hz, 2H), 7.53 (t, J = 7.4 Hz, 1H), 7.28 (d, J = 8.1 Hz, 2H), 7.10 (q, J = 7.9 Hz, 1H), 6.41 (t, J = 8.3 Hz, 1H), 6.29 (dd, J = 16.8, 10.6 Hz, 2H), 4.16 (s, 2H), 3.87 (s, 3H), 2.99 (s, 3H).13C NMR (126 MHz, CDCl3) δ 163.94 (d, J = 242.4 Hz), 158.37, 156.25, 150.80, 150.59 (d, J = 10.6 Hz), 136.07, 135.24, 130.30, 130.07 (d, J = 10.3 Hz), 129.73, 128.15, 107.78 (d, J = 2.3 Hz), 104.16, 103.66 (d, J = 21.5 Hz), 99.46 (d, J = 26.3 Hz), 55.73, 39.56, 33.91. HPLC Purity: 96%.
4.8.17. (E)-6-(3-fluorostyryl)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (25).
20 (250 mg, 0.910 mmol) and triethyl phosphite (318 μl, 1.820 mmol) were dissolved in 2mL DMF and heated to 150 °C for 3 hours. The mixture was concentrated under vacuum and the crude residue was purified by flash chromatography (EA in Hex) yielding diethyl ((1-methyl-4-oxo-5-phenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)methyl)phosphonate as a yellow solid (250 mg, 0.664 mmol, 73.0 % yield). A dry flask charged with 60% NaH in mineral oil (11 mg, 0.279 mmol) and diethyl ((1-methyl-4-oxo-5-phenyl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-6-yl)methyl)phosphonate (100 mg, 0.266 mmol) was cooled to 0 °C and DMF was added by syringe. After stirring for 10 min 3-fluorobenzaldehyde (31 μL, 0.292 mmol) was added by syringe and the flask was allowed to warm to RT. After 1 h the reaction was complete by HPLC. Water was added to precipitate product which was collected by filtration, washed with additional water and hexanes, and then recrystallized from hot ethanol yielding the titled compound as a yellow crystalline solid (50 mg, 0.144 mmol, 54% yield). MS (ESI): m/z 347.1305 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.91 (d, J = 15.65 Hz, 2H), 7.53 - 7.64 (m, 3H), 7.22 - 7.34 (m, 3H), 7.09 (d, J = 7.83 Hz, 1H), 7.02 (dt, J = 2.45, 8.31 Hz, 1H), 6.96 (d, J = 9.78 Hz, 1H), 6.34 (d, J = 15.16 Hz, 2H), 4.10 (s, 3H) 13C NMR (126 MHz, CDCl3) δ 162.92 (d, J = 247.3 Hz), 158.17, 154.11, 151.05, 139.17, 137.23 (d, J = 7.5 Hz), 136.73, 135.40, 130.38 (d, J = 8.4 Hz), 129.96, 129.54, 128.88, 123.80 (d, J = 2.6 Hz), 120.96, 116.75 (d, J = 21.5 Hz), 114.04 (d, J = 21.8 Hz), 104.23, 34.17. HPLC Purity: 97%.
4.8.18. 6-(3-Fluorophenethyl)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (26).
A solution of 25 (40 mg, 0.115 mmol) and 10% Pd-C (37 mg, 0.035 mmol) in 5mL methanol was degassed under vacuum and the flask backfilled with an H2 balloon. The mixture was stirred vigorously for 2 hours then filtered through Celite and concentrated yielding the titled compound as a white solid (27 mg, 0.078 mmol, 67 % yield). MS (ESI): m/z 349.1461 [M+H]+ 1H NMR (400 MHz, CDCl3) δ 8.05 (s, 1H), 7.44 - 7.62 (m, 3H), 7.10 - 7.23 (m, 3H), 6.86 (t, J = 8.41 Hz, 1H), 6.81 (d, J = 7.83 Hz, 1H), 6.74 (d, J = 10.17 Hz, 1H), 4.04 (s, 3H), 3.04 (t, J = 7.83 Hz, 2H), 2.68 (t, J = 7.83 Hz, 2H) 13C NMR (101 MHz, CDCl3) δ 162.7, 158.9, 158.3, 150.8, 142.8, 136.9, 135.1, 130.0, 129.8, 129.4, 128.4, 123.9, 115.3, 113.2, 104.0, 37.2, 34.1, 32.3. HPLC Purity: 95%.
4.8.19. General Procedure A.
S-Alkylation of 16: To a solution of 16 (50 mg, 0.194 mmol, 1eq.), K2CO3 (2 eq.), in 1 mL DMF under Ar was added the appropriate alkyl halide (1.1 eq.). The reaction was stirred at RT overnight. The next day the reaction was diluted with 10 mL H2O and extracted 2x with EtOAc. The combined organics were washed 3x with brine, dried over sodium sulfate, and concentrated. The crude residue was taken up in Ethanol at reflux then allowed to cool to RT and rest for several hours at which point the titled compound was obtained by filtration as a crystalline solid.
4.8.20. 6-((1-(3-fluorophenyl)ethyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (27).
General Procedure A. White solid. (106 mg, 0.279 mmol, 68% yield). MS (ESI): m/z 381.1182 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.60 - 7.45 (m, 3H), 7.32 - 7.23 (m, 2H), 7.19 (dd, J = 4.8, 2.2 Hz, 1H), 7.17 (d, J = 8.3 Hz, 1H), 7.10 (dt, J = 10.0, 2.1 Hz, 1H), 6.93 (td, J = 8.4, 2.5 Hz, 1H), 4.99 (q, J = 7.2 Hz, 1H), 3.98 (s, 3H), 1.67 (d, J = 7.0 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 162.69 (d, J = 246.1 Hz), 161.07, 157.70, 150.94, 144.96 (d, J = 7.4 Hz), 135.45 (d, J = 23.9 Hz), 135.38, 130.02, 129.96, 129.73 (d, J = 9.7 Hz), 129.37 (d, J = 2.6 Hz), 123.07 (d, J = 2.9 Hz), 114.59 (d, J = 19.2 Hz), 114.42 (d, J = 18.5 Hz), 102.92, 46.26, 34.10, 21.78.HPLC Purity: 99%
4.8.21. 1-methyl-5-phenyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (28).
General Procedure A. White Solid (183 mg, 0.505 mmol, 65% yield) MS (ESI): m/z 363.1278 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.64 - 7.45 (m, 3H), 7.42 - 7.35 (m, 2H), 7.34 - 7.22 (m, 4H), 7.18 (d, J = 6.9 Hz, 1H), 5.02 (q, J = 7.2 Hz, 1H), 4.00 (s, 3H), 1.70 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.50, 157.82, 151.05, 142.12, 135.64, 135.36, 129.93, 129.72, 129.65, 129.41, 129.38, 128.53, 127.56, 127.52, 102.88, 46.89, 34.07, 21.97. HPLC Purity: 98%
4.8.22. (R)-1-methyl-5-phenyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one ((R)-28).
To a flask under N2 charged with 3mL DCM and DMF (0.096 ml, 1.239 mmol) was added oxalyl chloride (0.102 ml, 1.161 mmol) dropwise by syringe. The mixture was stirred for 15 minutes at which point the solvent was removed and residual acid removed with an additional DCM chase. The solid obtained was suspended in 3mL THF and cooled to 0 °C and (S)-1-phenylethanol (0.140 ml, 1.161 mmol) was added and stirred for ~1 minute until a homogenous solution was obtained. TEA (0.324 ml, 2.323 mmol) and 16 (200 mg, 0.774 mmol) were added. The mixture was stirred for 2h at which point the reaction was done by HPLC. The reaction was quenched with water and extracted 2x with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, and concentrated. The crude solid obtained was taken up in hot ethanol and the titled compound crystallized as a yellow solid upon cooling (156 mg, 0.430 mmol, 56% yield). HPLC Purity: 96%, >95% EE as determined by Chiral HPLC (15% IPA in Hexanes, 1 mL/min, RT = 19 min). NMR and MS data were verified to be identical to the racemate. Optical Rotation: (1.02, CHCl3)
4.8.23. (S)-1-methyl-5-phenyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one ((S)-28).
Prepared in the same manner as (R)-28 employing (R)-1-phenylethanol. (80 mg, 0.221 mmol, 29% yield) HPLC Purity: 98%, >95% EE as determined by Chiral HPLC (15% IPA in Hexanes, 1 mL/min, RT = 16 min). NMR and MS data were verified to be identical to the racemate. Optical Rotation: (1.05, CHCl3)
4.8.24. 1-methyl-5-phenyl-6-((2-phenylpropan-2-yl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (29).
To a slurry of 16 (75 mg, 0.290 mmol) and prop-1-en-2-ylbenzene (0.038 ml, 0.290 mmol) in DCM at 0 °C was added TFA (0.224 ml, 2.90 mmol) dropwise. The resulting homogenous solution was stirred for 30 min at 0 °C and 30 min at RT. The reaction was diluted with DCM and washed with sat. aq. NaHCO3. The organic portion was dried over sodium sulfate and concentrated. The crude product was purified by flash (eluting around 40-50% EtOAc in Hex) yielding a white solid. (65 mg, 0.173 mmol, 60% yield) MS (ESI): m/z 377.1434 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.91 (s, 1H), 7.62 - 7.50 (m, 5H), 7.31 (t, J = 7.6 Hz, 2H), 7.25 - 7.14 (m, 3H), 3.71 (s, 3H), 1.89 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 160.48, 157.80, 150.72, 145.95, 135.76, 135.19, 129.82, 129.65, 129.43, 127.91, 126.59, 126.28, 102.90, 54.87, 33.99, 29.71. HPLC Purity: 95%
4.8.25. 1-methyl-5-phenyl-6-((1-phenylpropyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (30).
General Procedure A. White solid. (110 mg, 0.292 mmol, 75% yield) MS (ESI): m/z 377.1321 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.58 - 7.46 (m, 3H), 7.37 - 7.32 (m, 2H), 7.30 (t, J = 7.7 Hz, 2H), 7.28 - 7.22 (m, 2H), 7.17 (d, J = 7.2 Hz, 1H), 4.77 (dd, J = 9.1, 6.1 Hz, 1H), 4.00 (s, 3H), 2.16 - 2.02 (m, 1H), 1.97 - 1.83 (m, 1H), 0.92 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.60, 157.83, 151.02, 140.91, 135.72, 135.34, 129.90, 129.70, 129.63, 129.42, 129.36, 128.40, 128.08, 127.46, 102.86, 53.65, 34.03, 29.25, 12.29. HPLC Purity: 98%
4.8.26. 1-methyl-6-((2-methyl-1-phenylpropyl)thio)-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (31).
Prepared from (1-bromo-2-methylpropyl)benzene77 according to general procedure A. White solid (29 mg, 0.074 mmol, 28% yield) MS (ESI): m/z 413.1418 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 1.1 Hz, 1H), 7.60 - 7.46 (m, 3H), 7.35 - 7.26 (m, 5H), 7.25 - 7.20 (m, 1H), 7.16 (d, J = 7.8 Hz, 1H), 4.74 (d, J = 7.8 Hz, 1H), 3.96 (s, 3H), 2.14 (dtt, J = 7.2, 6.8 Hz, 1H), 1.04 (d, J = 6.7 Hz, 3H), 0.85 (d, J = 6.7 Hz, 3H). 11C NMR (126 MHz, CDCl3) δ 161.57, 157.86, 150.95, 140.89, 135.87, 135.29, 129.88, 129.70, 129.64, 129.43, 129.37, 128.61, 127.98, 127.13, 102.81, 59.22, 34.04, 33.39, 21.07, 20.57. HPLC Purity: 99%
4.8.27. 1-methyl-5-phenyl-6-(1-phenylethoxy)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (32).
60% NaH in mineral oil (8 mg, 0.197 mmol) was added to a solution of 1-phenylethanol (0.026 ml, 0.214 mmol) in 1 mL DMF at 0 °C. The mixture was stirred for 10 min at which point 17 (50 mg, 0.164 mmol) was added and the mixture was allowed to warm to room temperature and stirred overnight. The mixture was diluted with water and extracted 2x with EtOAc. The combined organics were washed 3x with brine, dried over sodium sulfate, and concentrated. The crude residue was purified by flash (EtOAc and Hexanes) yielding the titled compound as a white solid (5 mg, 0.014 mmol, 9 % yield). MS (ESI): m/z 347.1504 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.97 (s, 1H), 7.60 - 7.44 (m, 3H), 7.28 (dd, J = 10.4, 7.1 Hz, 4H), 7.14 (d, J = 7.0 Hz, 3H), 6.20 (q, J = 6.6 Hz, 1H), 3.90 (s, 3H), 1.50 (d, J = 6.6 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 158.07, 154.93, 151.02, 141.03, 135.55, 135.21, 129.23, 128.72, 128.48, 128.44, 128.39, 128.02, 125.70, 109.99, 101.91, 33.77, 22.59. HPLC Purity: 96%
4.8.28. 6-chloro-1-methyl-5-phenyl-1H-pyrazolo[4,3-c]pyridin-4(5H)-one (33).
A dry flask was charged with 25mL DCE, 3Å molecular sieves, phenylboronic acid (1.966 g, 16.12 mmol), 5 (1.48 g, 8.06 mmol), copper(II) acetate hydrate (0.322 g, 1.612 mmol), and pyridine (1.304 ml, 16.12 mmol). The flask was vacuum purged and backfilled with an O2 balloon and stirred vigorously for 3 d. The crude reaction mixture was filtered with Celite and the filter pad washed with DCM. The filtrate was washed with sat. aq. NH4Cl and dried over sodium sulfate. Following concentration, the crude residue was washed with 10mL diethyl ether yielding an orange solid which was taken forward without further purification (1.05 g, 4.04 mmol, 50% yield).
4.8.29. 6-mercapto-1-methyl-5-phenyl-1H-pyrazolo[4,3-c]pyridin-4(5H)-one (34).
A flask containing 33 (1.05 g, 4.04 mmol), Na2S (400 mg, 4.4 mmol) and 20mL DMF was rigorously deoxygenated under vacuum, backfilled with N2 and heated to 140 °C for 12 h. The reaction was diluted with 1N HCl and EtOAc forming an emulsion. The entire mixture was filtered, affording the titled compound as a white solid (620 mg, 2.410 mmol, 60% yield). 1H NMR (400 MHz, DMSO-d6) δ 7.97 (s, 1H), 7.55 – 7.42 (m, 3H), 7.25 (d, J = 7.3 Hz, 2H), 6.89 (s, 1H), 3.87 (s, 3H).
4.8.30. 2-((1-Methyl-4-oxo-5-phenyl-4,5-dihydro-1H-pyrazolo[4,3-c]pyridin-6-yl)thio)-2-phenylacetic acid (35).
To a suspension of 34 (180 mg, 0.700 mmol) and K2CO3 (135 mg, 0.979 mmol) in 3mL DMF was added ethyl 2-bromo-2-phenylacetate (147 μl, 0.839 mmol). The mixture was stirred at RT overnight. The next day the mixture was diluted with water and extracted 2x with EtOAc. The combined organics were washed 3x with brine, dried over sodium sulfate, and concentrated. The crude mixture was purified by flash (Eluting at 50% EtOAc in Hexanes) to afford the titled compound as a white solid (213 mg, 0.508 mmol, 73% yield). 1H NMR (500 MHz, CDCl3) δ 8.10 (s, 1H), 7.56 - 7.46 (m, 3H), 7.34 - 7.23 (m, 7H), 6.47 (s, 1H), 4.69 (s, 1H), 4.23 - 4.02 (m, 2H), 3.93 (s, 3H), 1.18 (t, J = 7.1 Hz, 3H).
A slurry of ethyl 2-((1-methyl-4-oxo-5-phenyl-4,5-dihydro-1H-pyrazolo[4,3-c]pyridin-6-yl)thio)-2- phenylacetate (213 mg, 0.508 mmol) in 2mL 1N LiOH and 3mL THF was stirred for 2 hours at which point the reaction was complete by HPLC. Following removal of THF, the mixture was acidified with 1N HCl and extracted 3x with EtOAc. The combined organics were washed with brine, dried over sodium sulfate, and concentrated to afford the titled compound as a white solid (202 mg, 0.516 mmol, 102 % yield).
4.8.31. 1-Methyl-5,7-diphenyl-1H-pyrazolo[3,4-d]thieno[2,3-b]pyridine-4,8(5H,7H)-dione (36).
35 (138 mg, 0.353 mmol) was dissolved in 5mL DCM and cooled to 0 °C under nitrogen at which point (COCl)2 (0.037 ml, 0.423 mmol) and DMF (3 μl, 0.035 mmol) were added sequentially. The mixture was stirred for 10 minutes at 0 °C and then allowed to warm to room temperature and left overnight. The next day the solvent was removed and the residue was purified by flash (EA in Hex) to yield (90 mg, 0.241 mmol, 68% yield) as an oily white solid. MS (ESI): m/z 374.0956 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.17 (s, 1H), 7.63 - 7.53 (m, 3H), 7.44 - 7.34 (m, 5H), 7.34 - 7.28 (m, 2H), 4.97 (s, 1H), 4.48 (s, 3H).13C NMR (126 MHz, CDCl3) δ 192.45, 171.74, 158.04, 138.91, 138.16, 136.11, 134.16, 130.45, 130.00, 129.20, 128.86, 128.53, 128.34, 110.79, 101.00, 58.53, 41.37. HPLC Purity: 96%.
4.8.32. 3-((1-Methyl-4-oxo-5-phenyl-4,5-dihydro-1H-pyrazolo[4,3-c]pyridin-6-yl)thio)-3-phenylpropanoic acid (37).
Cinnamic acid (535 mg, 3.61 mmol) was heated to 70 °C in a sealed tube with 1mL 33% HBr/HOAc overnight. The next day the reaction was diluted with DCM and washed with water and brine, dried over sodium sulfate, and concentrated affording a white solid. The solid dissolved in 10mL DMF to which 34 (620 mg, 2.410 mmol) and sodium bicarbonate (304 mg, 3.61 mmol) were added. After 2 h the thiol was fully dissolved and the reaction was complete by HPLC and the reaction was neutralized with a couple drops of conc. HCl. The DMF was removed in vacuo and the crude product was purified by flash (5-10% MeOH in DCM) yielding the titled compound as a yellow oil. (670 mg, 1.652 mmol, 69% yield). 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 0.9 Hz, 1H), 7.52 – 7.40 (m, 3H), 7.32 – 7.19 (m, 5H), 7.13 (d, J = 7.0 Hz, 2H), 6.53 (s, 1H), 4.48 (t, J = 7.2 Hz, 1H), 3.86 (s, 3H), 2.97 – 2.83 (m, 2H).
4.8.33. 1-Methyl-5,7-diphenyl-7,8-dihydropyrazolo[3,4-d]thiopyrano[2,3-b]pyridine-4,9(1H,5H)-dione (38).
To a dry pressure tube was added a suspension of 37 (630 mg, 1.554 mmol) in 10mL DCE. Methanesulfonic anhydride (541 mg, 3.11 mmol) was added and the headspace was flushed with N2 and the tube sealed. The tube was heated to 70 °C for 2 h. The next day the reaction was diluted with DCM and washed with saturated Sodium Bicarbonate and brine before drying over sodium sulfate and concentrating. The crude brown oil was purified by flash (Elutes around 50% EA/Hex) yielding the product as a white-yellow foam (250 mg, 0.645 mmol, 42% yield). MS (ESI): m/z 388.1114 [M+H]+ 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1H), 7.57 - 7.46 (m, 3H), 7.40 - 7.30 (m, 5H), 7.29 - 7.21 (m, 3H), 4.66 (dd, J = 12.6, 4.5 Hz, 1H), 4.19 (s, 3H), 3.31 - 3.11 (m, 2H).13C NMR (101 MHz, CDCl3) δ 189.64, 159.55, 157.89, 141.28, 137.06, 136.73, 135.76, 130.17, 129.87, 129.77, 129.20, 129.15 (d, J = 1.1 Hz), 129.08, 127.79, 112.19, 105.21, 46.86, 45.66, 42.30. HPLC Purity: 99%
4.8.34. 9-hydroxy-1-methyl-5,7-diphenyl-5,7,8,9-tetrahydropyrazolo[3,4-d]thiopyrano[2,3-b]pyridin-4(1H)-one (39).
NaBH4 (5.86 mg, 0.155 mmol), and 38 (50 mg, 0.129 mmol) were added to 1mL abs. EtOH at 0 °C. The mixture was stirred for 30 minutes at the same temperature then quenched with saturated aqueous NH4Cl. The mixture was concentrated and taken up in EtOAc, washed with water and brine, dried over sodium sulfate, and concentrated to afford the titled compound (mixture of diastereomers) as a white solid (42 mg, 0.108 mmol, 84% yield).
4.8.35. 1-Methyl-5,7-diphenyl-5,7-dihydropyrazolo[3,4-d]thiopyrano[2,3-b]pyridin-4(1H)-one (40).
To a solution of 39 (24 mg, 0.062 mmol) and InCl3 (1.363 mg, 6.16 μmol) in 1mL DCM was added chlorodiphenylsilane (0.022 mL, 0.123 mmol). The mixture was stirred for 2 hours at room temperature at which point the mixture was concentrated and purified by flash (Eluting at 70% EA/Hex) to yield the titled compound as a yellow solid (14 mg, 0.038 mmol, 61% yield). MS (ESI): m/z 372.1162 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.12 (s, 1H), 7.53 - 7.47 (m, 1H), 7.47 - 7.40 (m, 2H), 7.38 - 7.34 (m, 2H), 7.34 - 7.27 (m, 4H), 7.13 (dt, J = 7.7, 2.3 Hz, 1H), 7.03 (dd, J = 9.8, 1.7 Hz, 1H), 5.81 (dd, J = 9.8, 4.8 Hz, 1H), 4.87 (dd, J = 4.8, 1.7 Hz, 1H), 4.21 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 158.29, 143.28, 141.58, 138.56, 138.02, 136.92, 129.44, 129.40, 129.39, 129.23, 128.79, 128.75, 128.40, 128.09, 123.47, 118.72, 112.30, 101.99, 45.63, 39.57. HPLC Purity: 99%
4.8.36. General Procedure B. Benzylic bromination.
The appropriate ethylpyridine (1.067 ml, 9.33 mmol), AIBN (0.153 g, 0.933 mmol), and NBS (1.744 g, 9.80 mmol) were refluxed in 20mL of CCl4 for 1 h. The reaction was cooled and filtered and the solid was taken up in DCM and filtered again. The combined filtrate was washed with saturated aqueous sodium bicarbonate and brine before drying over sodium sulfate and concentrating. The 1-bromoethyl pyridines were obtained as an orange oil and used without further purification.
4.8.36. 1-Methyl-5-phenyl-6-((1-(pyridin-2-yl)ethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (42).
2-ethylpyridine brominated by General Procedure B was used according to General Procedure A. White solid (56 mg, 0.154 mmol, 39.8 % yield) MS (ESI): m/z 364.1225 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.55 (d, J = 4.8 Hz, 1H), 8.00 (s, 1H), 7.64 (t, J = 7.7 Hz, 1H), 7.57 – 7.45 (m, 3H), 7.40 (d, J = 7.8 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 7.22 (d, J = 6.5 Hz, 1H), 7.16 (t, J = 6.2 Hz, 1H), 5.14 (q, J = 7.2 Hz, 1H), 3.96 (s, 3H), 1.73 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.52, 161.07, 157.78, 150.96, 149.52, 136.66, 135.53, 135.37, 129.96, 129.75, 129.38, 122.34, 121.76, 102.83, 48.47, 34.00, 20.95. HPLC Purity: 99%.
4.8.37. 1-Methyl-5-phenyl-6-((1-(pyridin-3-yl)ethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (43).
3-ethylpyridine brominated by General Procedure B was used according to General Procedure A. White solid (90 mg, 0.248 mmol, 64.0 % yield) MS (ESI): m/z 364.1225 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.72 (d, J = 2.2 Hz, 1H), 8.49 (d, J = 4.8 Hz, 1H), 7.99 (s, 1H), 7.70 (d, J = 7.9 Hz, 1H), 7.59 – 7.47 (m, 3H), 7.29 – 7.21 (m, 2H), 7.18 (d, J = 7.1 Hz, 1H), 5.01 (q, J = 7.3 Hz, 1H), 3.99 (s, 3H), 1.68 (d, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 160.67, 157.61, 150.85, 149.37, 148.81, 138.32, 135.47, 135.37, 134.47, 130.10, 129.79, 129.72, 129.37, 129.35, 123.49, 102.96, 44.04, 34.20, 21.59. HPLC Purity: 99%
4.8.38. 1-methyl-5-phenyl-6-((1-(pyrimidin-5-yl)ethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (44).
A dry vial was charged with polystyrene bound triphenylphosphine (Sigma Aldrich, 750mg, 3mmol/g loading), I2 (501 mg, 1.97 mmol), and 4mL THF and stirred for 15 minutes. Imidazole (154 mg, 2.26 mmol) was added and allowed to dissolve before adding 1-(pyrimidin-5-yl)ethanol78 (140mg, 1.128 mmol) dissolved in minimal THF. The mixture was agitated on an orbital shaker overnight. The reaction was filtered and the resin washed with additional DCM. The filtrate was washed with sat. aq. Na2S2O3, saturated aq. NaHCO3 and brine before drying over sodium sulfate and concentrating to afford 5-(1-iodoethyl)pyrimidine as an orange waxy solid which was used immediately without further purification due to rapid discoloration. 5-(1-iodoethyl)pyrimidine was used according to General Protocol A. White solid (41 mg, 0.113 mmol, 38.7 % yield). MS (ESI): m/z 387.1010 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 9.10 (s, 1H), 8.82 (s, 2H), 7.99 (s, 1H), 7.61 - 7.49 (m, 3H), 7.30 - 7.23 (m, 1H), 7.21 - 7.15 (m, 1H), 4.94 (q, J = 7.6 Hz, 1H), 3.98 (s, 3H), 1.68 (d, J = 7.4 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 160.07, 157.69, 157.41, 155.97, 150.67, 136.63, 135.42, 135.25, 130.29, 129.91, 129.80, 129.34, 103.07, 41.63, 34.31, 21.05. HPLC Purity: 96%.
4.8.39. 1-methyl-5-phenyl-6-((1-(pyridin-4-yl)ethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (45).
4-ethylpyridine brominated by General Procedure B was used according to General Procedure A. White solid (31 mg, 0.085 mmol, 29.4 % yield). MS (ESI): m/z 364.1229 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.55 (d, J = 4.9 Hz, 2H), 7.99 (s, 1H), 7.61 - 7.48 (m, 3H), 7.36 - 7.30 (m, 2H), 7.29 - 7.22 (m, 1H), 7.23 - 7.15 (m, 1H), 4.95 (q, J = 7.3 Hz, 1H), 3.95 (s, 3H), 1.64 (d, J = 7.3 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 160.55, 157.58, 151.66, 150.79, 150.04, 135.43, 135.41, 130.15, 129.79 (d, J = 9.4 Hz), 129.35 (d, J = 3.2 Hz), 122.37, 102.95, 45.48, 34.10, 21.20. HPLC Purity: 99%
4.8.40. 6-((1-(3-hydroxyphenyl)ethyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (46).
3-Ethylphenol (1 g, 8.19 mmol), TBS-Cl (1.357 g, 9.00 mmol), and imidazole (1.226 g, 18.01 mmol) were stirred overnight in 10 mL DMF. Diluted with 25 mL water and extracted with 25mL EtOAc. The organic portion was dried over Na2S2O3 and the solvent was removed. The crude oil was filtered through a pad of silica using ~5% EA in Hex yielding tert-butyl(3-ethylphenoxy)dimethylsilane (1.8 g, 7.61 mmol, 93 % yield) as a colorless oil which was used without further purification.
A suspension of tert-butyl(3-ethylphenoxy) dimethylsilane (1.8 g, 7.61 mmol), benzoyl peroxide (0.277 g, 1.142 mmol), and NBS (1.355 g, 7.61 mmol) in 15mL CCl4 was refluxed for 3 h. The mixture was cooled to RT and filtered. The filtrate was diluted with DCM and washed with water and brine before drying over sodium sulfate. The mixture was concentrated and the resulting residue was filtered through a pad of silica eluting with hexanes yielding (3-(1-bromoethyl)phenoxy)(tert-butyl)dimethylsilane as a colorless oil (1.2 g, 3.81 mmol, 50.0 % yield). 1H NMR (500 MHz, CDCl3) δ 7.19 (t, J = 7.9 Hz, 1H), 7.01 (d, J = 7.6 Hz, 1H), 6.92 (d, J = 2.1 Hz, 1H), 6.75 (dd, J = 8.1, 2.4 Hz, 1H), 5.15 (q, J = 6.9 Hz, 1H), 2.02 (d, J = 6.9 Hz, 3H), 1.00 - 0.95 (s, 9H), 0.21 (s, 6H).
(3-(1-bromoethyl)phenoxy)(tert-butyl)dimethylsilane was used in General Protocol A to afford 6-((1-(3-((tert-butyldimethylsilyl)oxy)phenyl)ethyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one as a yellow oil. (195 mg, 0.396 mmol, 82 % yield).
To an HDPE vial was added 6-((1-(3-((tert-butyldimethylsilyl)oxy)phenyl)ethyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (195 mg, 0.40 mmol) dissolved in 3mL THF. 70% HF in pyridine (250 μl, 10.2 mmol) was added and the vial was sealed under N2 and left overnight. The next morning the reaction was diluted with 15mL water and extracted with EtOAc. The organic portion was washed with sat. aq. NaHCO3 and brine then dried over sodium sulfate and concentrated yielding the titled compound as a white solid (123 mg, 0.325 mmol, 82 % yield). MS (ESI): m/z 379.1218 [M+H]+ 1H NMR (500 MHz, DMSO-d6) δ 9.43 (s, 1H), 8.04 (s, 1H), 7.59 - 7.53 (m, 1H), 7.53 - 7.48 (m, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.33 - 7.26 (m, 1H), 7.10 (t, J = 7.8 Hz, 1H), 6.83 (d, J = 7.7 Hz, 1H), 6.78 (s, 1H), 6.63 (dd, J = 8.1, 2.4 Hz, 1H), 4.94 (q, J = 7.0 Hz, 1H), 3.97 (s, 3H), 1.64 (d, J = 7.0 Hz, 3H).13C NMR (126 MHz, DMSO-d6) δ 161.31, 157.80, 157.30, 150.87, 143.59, 136.14, 135.10, 130.26, 130.07, 129.98, 129.95, 118.52, 114.93, 114.74, 102.80, 46.84, 34.34, 22.09. HPLC Purity: 98%
4.8.41. 6-((1-(2-methoxyphenyl)ethyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (47).
1-(1-bromoethyl)-2-methoxybenzene generated according to General Protocol B was employed in General Protocol A. White solid. (60 mg, 0.153 mmol, 52.7 % yield) MS (ESI): m/z 393.1383 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.00 (s, 1H), 7.61 - 7.43 (m, 3H), 7.33 (d, J = 7.6 Hz, 1H), 7.30 - 7.25 (m, 1H), 7.25 - 7.18 (m, 2H), 6.90 (t, J = 7.7 Hz, 1H), 6.87 (d, J = 8.0 Hz, 1H), 5.45 (q, J = 7.2 Hz, 1H), 3.98 (s, 3H), 3.85 (s, 3H), 1.69 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 162.17, 157.95, 156.54, 151.18, 135.77, 135.34, 129.94, 129.81, 129.62 (d, J = 4.8 Hz), 129.40 (d, J = 2.2 Hz), 128.62, 127.98, 120.63, 110.74, 102.76, 55.52, 41.03, 33.78, 21.14. HPLC Purity: 98%
4.8.42. 6-((1-(3-Methoxyphenyl)ethyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (48).
1-(1-bromoethyl)-3-methoxybenzene generated according to General Protocol B was employed in General Protocol A. White solid. (29 mg, 0.074 mmol, 25.4 % yield). MS (ESI): m/z 393.1383 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.00 (s, 1H), 7.60 - 7.43 (m, 3H), 7.27 (d, J = 6.7 Hz, 1H), 7.23 (t, J = 8.1 Hz, 1H), 7.21 - 7.17 (m, 1H), 6.97 (d, J = 7.6 Hz, 1H), 6.95 - 6.90 (m, 1H), 6.78 (d, J = 8.4 Hz, 1H), 4.99 (q, J = 7.2 Hz, 1H), 4.00 (s, 3H), 3.79 (s, 3H), 1.70 (d, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.49, 159.62, 157.82, 151.04, 143.66, 135.62, 135.36, 129.93, 129.68 (d, J = 9.5 Hz), 129.53, 129.38 (d, J = 5.0 Hz), 119.84, 113.81, 112.35, 102.88, 55.22, 46.85, 34.08, 21.91. HPLC Purity: 98%
4.8.43. 6-((1-(4-Methoxyphenyl)ethyl)thio)-1-methyl-5-phenyl-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (49).
1-(1-bromoethyl)-4-methoxybenzene generated according to General Protocol B was employed in General Protocol A. White solid. (38 mg, 0.097 mmol, 33.3 % yield) MS (ESI): m/z 393.1381 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.01 (s, 1H), 7.61 - 7.40 (m, 3H), 7.30 (d, J = 8.4 Hz, 2H), 7.28 - 7.24 (m, 1H), 7.18 (d, J = 6.6 Hz, 1H), 6.83 (d, J = 8.4 Hz, 2H), 5.00 (q, J = 7.1 Hz, 1H), 4.02 (s, 3H), 3.78 (s, 3H), 1.71 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.69, 158.95, 157.85, 151.09, 135.65, 135.37, 133.82, 129.88, 129.65 (d, J = 9.4 Hz), 129.37 (d, J = 5.7 Hz), 128.66, 113.87, 102.87, 55.25, 46.44, 34.06, 21.98. HPLC Purity: 96%
4.8.44. 5-(4-(Hydroxymethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (51).
5022 (250 mg, 1.183 mmol), and (4-aminophenyl)methanol (146 mg, 1.183 mmol) were stirred in 2mL DMF for 1 h. The flask was cooled in an ice bath and 60 wt.% NaH (99 mg, 2.485 mmol) was added. The mixture was stirred for 30 min at 0 °C and 1 h at RT. The reaction was quenched with a few drops of Sat. NH4Cl and diluted with water and ethyl acetate (Aq. Layer pH=7). The organic layer was discarded and the aqueous layer was acidified with 1N HCl and extracted 2x with EtOAc. The combined organics were washed 3x with brine, dried over sodium sulfate and concentrated. The crude sulfide was dissolved in 2mL DMF to which K2CO3 (164 mg, 1.183 mmol) and (1-bromoethyl)benzene (0.161 ml, 1.183 mmol) were added. The mixture was stirred for 2 h and diluted with water and extracted 2x with EtOAc. The combined organics were washed 3x with brine and dried over sodium sulfate and concentrated. The crude was purified by flash (eluting 50% EtOAc in Hexanes) yielding the product as a white solid. (100 mg, 0.255 mmol, 21.53 % yield) MS (ESI): m/z 393.1380 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.95 (d, J = 3.0 Hz, 1H), 7.46 (dd, J = 8.1, 2.0 Hz, 1H), 7.41 (dd, J = 8.1, 1.9 Hz, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.32 - 7.25 (m, 3H), 7.22 (dd, J = 7.5, 1.9 Hz, 2H), 7.13 (dd, J = 8.1, 2.2 Hz, 1H), 5.00 (q, J = 7.1 Hz, 1H), 4.65 (d, J = 3.3 Hz, 2H), 3.97 (d, J = 1.7 Hz, 3H), 3.38 (d, J = 24.4 Hz, 1H), 1.69 (dd, J = 7.2, 1.3 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 161.70, 158.05, 151.06, 143.53, 142.09, 135.26, 134.33, 129.25, 128.54, 127.88 (d, J = 7.7 Hz), 127.57, 127.52, 102.79, 64.08, 46.94, 34.06, 21.97. HPLC Purity: 96%
4.8.45. 5-(4-(methoxymethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (52).
To a solution of 51 (133 mg, 0.339 mmol) in 3mL dry DMF at 0 °C was added 60 wt.% NaH (13.55 mg, 0.339 mmol). MeI (42.4 μl, 0.678 mmol) was added immediately and the mixture was stirred for 10 min then allowed to warm to RT and stirred for 1 h. The reaction was diluted with water and extracted 2x with EtOAc. The combined organics were washed 3x with brine and dried over sodium sulfate. The crude product was purified by flash (eluting around 50% EtOAc in Hexanes) to yield the titled compound as a colorless oil which solidified over several days (45 mg, 0.111 mmol, 32.7 % yield). MS (ESI): m/z 407.1533 [M+H]+ 1H NMR (500 MHz, CDCl3 ) δ 7.96 (s, 1H), 7.50 (dd, J = 8.1, 1.9 Hz, 1H), 7.45 (dd, J = 8.1, 1.9 Hz, 1H), 7.41 - 7.35 (m, 2H), 7.33 - 7.27 (m, 2H), 7.27 - 7.21 (m, 2H), 7.17 (dd, J = 8.1, 2.3 Hz, 1H), 5.01 (q, J = 7.2 Hz, 1H), 4.53 (s, 2H), 3.99 (s, 3H), 3.45 (s, 3H), 1.70 (d, J = 7.2 Hz, 3H).13C NMR (126 MHz, CDCl3 ) δ 161.56, 157.82, 151.03, 142.13, 140.38, 135.30, 134.80, 129.36, 129.34, 128.57, 128.52, 128.50, 127.55, 127.51, 102.83, 73.99, 58.55, 46.89, 34.06, 21.98. HPLC Purity: 96%
4.8.46. 5-(4-(2-hydroxyethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (53).
50 (200 mg, 0.947 mmol) and 2-(4-aminophenyl)ethanol (130 mg, 0.947 mmol) were stirred in 5 mL DMF for 2 h at which point the reaction was cooled to 0 °C and 60 wt.% NaH (76 mg, 1.894 mmol) was added. The mixture was stirred at the same temperature for 30 min then allowed to warm to RT and stirred for 2 h. The mixture was diluted with 1N HCl and extracted 2x with EtOAc. The combined organics were washed with brine and dried over sodium sulfate before concentrating and suspending the solid in 2.5 mL DMF and adding K2CO3 (196 mg, 1.420 mmol) and (1-bromoethyl)benzene (0.129 ml, 0.947 mmol). The mixture was stirred at RT overnight. The following day the mixture was diluted with water and extracted 2x with EtOAc. The combined organics were washed 3x with brine and dried over sodium sulfate. The crude solid was purified by flash (Eluting at 50% EtOAc in Hexanes) to afford the titled compound as a white foam (125 mg, 0.308 mmol, 32.5 % yield). MS (ESI): m/z 407.1536 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.42 - 7.36 (m, 3H), 7.37 - 7.27 (m, 3H), 7.27 - 7.21 (m, 1H), 7.19 (dd, J = 8.0, 2.3 Hz, 1H), 7.11 (dd, J = 8.0, 2.3 Hz, 1H), 5.02 (q, J = 7.2 Hz, 1H), 3.99 (s, 3H), 3.90 (q, J = 5.7, 5.2 Hz, 2H), 2.93 (t, J = 6.6 Hz, 2H), 1.71 (d, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.60, 157.91, 151.04, 142.07, 140.80, 135.34, 133.82, 130.39, 130.34, 129.36, 129.33, 128.52, 127.56, 127.53, 102.84, 63.21, 46.91, 38.96, 34.05, 21.92. HPLC Purity 97%
4.8.47. 5-(4-(2-methoxyethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (54).
Prepared from 53 in a similar manner to 52. Colorless oil. (54 mg, 0.128 mmol, 49.7 % yield) MS (ESI): m/z 421.1689 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.96 (s, 1H), 7.43 - 7.36 (m, 3H), 7.36 - 7.27 (m, 3H), 7.27 - 7.21 (m, 1H), 7.18 (dd, J = 8.1, 2.3 Hz, 1H), 7.10 (dd, J = 8.1, 2.3 Hz, 1H), 5.01 (q, J = 7.1 Hz, 1H), 3.99 (s, 3H), 3.66 (t, J = 7.0 Hz, 2H), 3.38 (s, 3H), 2.96 (t, J = 7.0 Hz, 2H), 1.71 (d, J = 7.2 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 161.68, 157.85, 151.03, 142.16, 141.10, 135.30, 135.28, 133.64, 130.15 (d, J = 7.6 Hz), 129.18 (d, J = 4.5 Hz), 128.51, 127.53, 102.85, 73.06, 58.70, 46.87, 35.94, 34.04, 21.95. HPLC Purity: 97%
4.8.48. 5-(4-(aminomethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one hydrochloride (55).
DIPEA (200 μl, 1.147 mmol), and Ms-Cl (74.5 μl, 0.955 mmol) were added sequentially by syringe to a solution of 51 (300 mg, 0.764 mmol) in 1.5mL DCM at −10 °C. The mixture was stirred for 10 min then taken out of the cooling bath and stirred for 3 h. The reaction was diluted with DCM and washed with water and brine before drying the organic portion over sodium sulfate and concentrating to yield 4-(1-methyl-4-oxo-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)benzyl methanesulfonate as a colorless oil (347 mg, 0.737 mmol, 96 % yield).
A degassed solution of NaN3 (24.87 mg, 0.383 mmol) and 4-(1-methyl-4-oxo-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)benzyl methanesulfonate (150 mg, 0.319 mmol) was stirred in 2mL DMF at 60 °C overnight. The next day the mixture was diluted with water, and extracted 2x into EtOAc. The combined organics were washed 3x with brine and dried over sodium sulfate. The crude was purified by flash (eluting around 60% EA/Hex) to yield 5-(4-(azidomethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one as a white foam (62 mg, 0.149 mmol, 46.6 % yield). 1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 8.5 Hz, 1H), 7.38 (d, J = 7.6 Hz, 2H), 7.34 - 7.27 (m, 3H), 7.22 - 7.17 (m, 1H), 5.02 (q, J = 7.2 Hz, 1H), 4.47 (s, 2H), 4.00 (s, 3H), 1.71 (d, J = 7.2 Hz, 3H).
5-(4-(azidomethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (62 mg, 0.149 mmol) was taken up in 3mL THF to which polystyrene bound triphenylphosphine (Sigma Aldrich, 3mmol/g, .3mmol) and a few drops of water was added. The mixture was agitated on an orbit shaker for 2 days at which point 1 mL of water was added. The mixture was filtered after an hour and the resin washed with DCM. The filtrate was concentrated and purified by flash (5% MeOH/94% DCM/1% TEA) to yield a yellow oil. The product was additionally purified by C18 flash (30%-50% ACN in Water, 0.1% TFA) to yield the TFA salt as an oil. Residue was taken up in Ethyl Acetate and washed with sat. sodium carbonate. The organic portion was concentrated, taken up in EtOH, treated with a few drops Conc. HCl and then concentrated again to yield the tiled compound as a yellow residue (7 mg, 0.016 mmol, 11.01 % yield). MS (ESI): m/z 392.1542 [M+H]+ 1H NMR (500 MHz, Methanol-d4) δ 7.96 (s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 7.9 Hz, 1H), 7.40 - 7.32 (m, 3H), 7.29 (t, J = 7.4 Hz, 2H), 7.22 (t, J = 7.3 Hz, 1H), 5.07 (q, J = 6.9 Hz, 1H), 4.21 (s, 2H), 3.99 (s, 3H), 1.71 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, Methanol-d4) δ 161.48, 158.37, 151.06, 141.70, 136.38, 135.21, 134.48, 130.19 (d, J = 4.2 Hz), 130.05 (d, J = 3.7 Hz), 128.23, 127.32, 127.18, 102.33, 46.97, 42.44, 33.01, 20.76. HPLC Purity 98%
4.8.49. 4-(1-Methyl-4-oxo-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)benzaldehyde (56).
A solution of 51 (500 mg, 1.274 mmol) and Dess-Martin periodinane (648 mg, 1.53 mmol) was stirred in 10mL DCM overnight. The next day, 1mL sat. aq. sodium bicarbonate was added and the mixture vigorously stirred for 1 h then filtered. The organic portion of the filtrated was washed with water and brine then dried over sodium sulfate and concentrated to afford the titled compound as a white solid. (300 mg, 0.768 mmol, 60.3 % yield). 1H NMR (500 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.06 (dd, J = 8.1, 1.9 Hz, 1H), 8.04 (s, 1H), 8.02 (dd, J = 8.1, 2.0 Hz, 1H), 7.66 (dd, J = 8.1, 2.1 Hz, 1H), 7.56 (dd, J = 8.1, 2.1 Hz, 1H), 7.45 – 7.39 (m, 2H), 7.30 (dd, J = 8.4, 6.8 Hz, 2H), 7.25 – 7.13 (m, 1H), 5.06 (q, J = 7.1 Hz, 1H), 3.96 (s, 3H), 1.66 (d, J = 7.1 Hz, 3H).
4.8.50. 1-methyl-5-(4-((methylamino)methyl)phenyl)-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin- 4(5H)-one (57).
To a flask charged with 1mL MeOH and 56 (50 mg, 0.128 mmol) was added 33% methylamine in EtOH (0.032 ml, 0.256 mmol) and titanium isopropoxide (0.038 ml, 0.128 mmol) by syringe. The starting material gradually went into solution, and then a white precipitate formed over the course of an hour. At this point NaBH4 (7.27 mg, 0.192 mmol) was added and the mixture was stirred for 15 minutes at which point the reaction was once again homogenous and the starting material was consumed by HPLC. The mixture was diluted with water and extracted 3x with ethyl acetate. The combined organic portion was washed with brine and dried over sodium sulfate. Upon concentrating, 45 mg of an oily residue was obtained which was taken up in 1mL Et2O and a few drops of hexane was added until the mixture became slightly cloudy. Upon cooling in the freezer, a precipitate formed which was collected by filtration yielding the titled compound as a white solid (9 mg, 0.022 mmol, 17.33 % yield). (ESI): m/z 406.1693 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.49 (dd, J = 8.1, 2.0 Hz, 1H), 7.44 (dd, J = 8.1, 1.9 Hz, 1H), 7.38 (d, J = 7.1 Hz, 2H), 7.30 (t, J = 7.5 Hz, 2H), 7.25 - 7.18 (m, 2H), 7.13 (dd, J = 8.1, 2.3 Hz, 1H), 5.01 (q, J = 7.2 Hz, 1H), 3.99 (s, 3H), 3.83 (s, 2H), 2.51 (s, 3H), 1.83 (br. s, 1H), 1.70 (d, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.62, 157.85, 151.04, 142.13 (d, J = 2.2 Hz), 135.34, 134.27, 129.33, 129.28, 129.26, 128.50, 127.53, 127.51, 102.86, 55.55, 46.87, 36.21, 34.04, 21.95. HPLC Purity 96%
4.8.51. 5-(4-((dimethylamino)methyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (58).
Prepared in a similar manner to 57 using 5.6M dimethylamine in EtOH. (52 mg, 0.124 mmol, 48.4 % yield) (ESI): m/z 420.1849 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.98 (s, 1H), 7.47 (dd, J = 8.1, 1.9 Hz, 1H), 7.42 (dd, J = 8.1, 2.0 Hz, 1H), 7.40 - 7.34 (m, 2H), 7.33 - 7.27 (m, 2H), 7.25 - 7.18 (m, 2H), 7.12 (dd, J = 8.1, 2.3 Hz, 1H), 5.01 (q, J = 7.1 Hz, 1H), 3.99 (s, 3H), 3.49 (s, 2H), 2.29 (s, 6H), 1.70 (d, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 161.67, 157.84, 151.06, 142.20, 141.17, 135.34, 134.31, 130.10, 130.02, 129.19, 129.17, 128.51, 127.53, 127.51, 102.89, 63.89, 46.87, 45.59, 34.05, 21.99. HPLC Purity: 96%
4.8.52. tert-butyl 4-(1-methyl-4-oxo-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-5(4H)- yl)phenethylcarbamate (59).
Tert-butyl 4-aminophenethylcarbamate (prepared as previously described79, 290 mg, 1.227 mmol) was added to a solution of 50 (259 mg, 1.227 mmol) in 5mL DMF. After stirring for 2 h the mixture was cooled to 0 °C and 60 wt.% NaH (54.0 mg, 1.350 mmol) was added. The reaction was stirred at the same temperature for 15 min then allowed to warm to RT and stirred for 2 h. The flask was cooled to 0 °C and (1-bromoethyl)benzene (184 μl, 1.350 mmol) was added by syringe. The flask was warmed to RT and stirred for 1 h at which point the solvent was removed in vacuo. The residue was taken up in EtOAc and washed with water and brine before drying over sodium sulfate and concentrating. The crude residue was purified by flash (Eluting at 60% EtOAc in hexanes) then further purified by taking up the solid in 30mL hot EtOH, adding 5mL water, and allowing the mixture to cool to RT affording the titled compound as a white crystalline solid (330 mg, 0.653 mmol, 53.2 % yield). 1H NMR (500 MHz, CDCl3) δ 8.00 (s, 1H), 7.39 (d, J = 7.6 Hz, 2H), 7.37 - 7.28 (m, 3H), 7.25 - 7.22 (m, 1H), 7.19 (dd, J = 8.2, 2.2 Hz, 1H), 7.11 (dd, J = 8.1, 2.3 Hz, 1H), 5.02 (q, J = 7.2 Hz, 1H), 4.62 (s, 1H), 4.00 (s, 3H), 3.49 - 3.29 (m, 2H), 2.88 (t, J = 6.9 Hz, 2H), 1.71 (d, J = 7.2 Hz, 3H), 1.45 (s, 9H).
4.8.53. tert-butyl methyl(4-(1-methyl-4-oxo-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-5(4H)- yl)phenethyl)carbamate (60).
To a solution of 59 (48 mg, 0.095 mmol) in 1 mL DMF at 0 °C was added 60 wt.% NaH (4.94 mg, 0.123 mmol). The mixture was stirred under N2 at the same temperature for 30 min at which point MeI (7.12 μl, 0.114 mmol) was added by syringe. The mixture was stirred for an hour at RT then quenched with a few drops of sat. Aq. NH4Cl and concentrated in vacuo. The residue obtained was taken up in water and extracted 3x with EtOAc. The combined organics were washed 3x with brine and dried over sodium sulfate. The crude residue was purified by flash (Eluting around 50% EA in Hex) to yield the titled compound as a white foam (35 mg, 0.067 mmol, 70.9 % yield). 1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.42 - 7.14 (m, 8H), 7.10 (d, J = 7.8 Hz, 1H), 5.00 (q, J = 7.1 Hz, 1H), 3.98 (s, 3H), 3.47 (br.s, 2H), 2.98 - 2.72 (m, 4H), 1.70 (d, J = 7.1 Hz, 3H), 1.45 (s, 9H).
4.8.54. 5-(4-(2-Aminoethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one hydrochloride (61).
59 (105 mg, 0.208 mmol) was dissolved in 1 mL DCM to which 1 mL TFA was added. The mixture was stirred overnight then concentrated in vacuo. The residue was taken up in 1:3 conc. HCl and EtOH and evaporated 3 times to exchange TFA salt for HCl to afford the titled compound as an off-white solid (70 mg, 0.158 mmol, 76 % yield). (ESI): m/z 406.1695 [M+H]+ 1H NMR (500 MHz, DMSO-d6) δ 8.01 (s, 1H), 7.91 (br. s, 3H), 7.44 - 7.39 (m, 3H), 7.39 - 7.27 (m, 4H), 7.26 - 7.19 (m, 2H), 5.03 (q, J = 7.1 Hz, 1H), 3.95 (s, 3H), 3.11 (p, J = 5.3 Hz, 2H), 2.96 - 2.88 (m, 2H), 1.66 (d, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 161.26, 157.34, 150.84, 142.33, 139.55, 135.10, 134.70, 130.19, 130.12, 128.96, 128.06, 127.94, 102.77, 46.86, 39.92, 34.34, 33.16, 21.95. HPLC Purity: 98%.
4.8.55. 1-methyl-5-(4-(2-(methylamino)ethyl)phenyl)-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one hydrochloride (62).
60 (120 mg, 0.231 mmol) was dissolved in 1mL DCM and 1mL TFA. The mixture was stirred for 2 h, at which point the starting material was consumed by TLC. The reaction was concentrated and the residue taken up in sat. aq. sodium bicarbonate and extracted 3x into EtOAc. The combined organics were washed with brine, and dried over sodium sulfate. A few drops of 4N HCl in dioxane were added and the mixture was concentrated to yield the titled compound as a white solid (105 mg, 0.230 mmol, 100 % yield). (ESI): m/z 420.1853 [M+H]+ 1H NMR (500 MHz, DMSO-d6) δ 9.09 (s, 2H), 8.01 (s, 1H), 7.45 - 7.40 (m, 3H), 7.37 (dd, J = 8.1, 2.0 Hz, 1H), 7.35 - 7.27 (m, 3H), 7.26 - 7.20 (m, 2H), 5.02 (q, J = 7.1 Hz, 1H), 3.95 (s, 3H), 3.17 (dq, J = 10.9, 6.3, 5.9 Hz, 2H), 3.02 (dd, J = 10.0, 6.5 Hz, 2H), 2.56 (t, J = 5.3 Hz, 3H), 1.65 (d, J = 7.1 Hz, 3H).13C NMR (126 MHz, DMSO-d6) δ 161.27, 157.32, 150.84, 142.37, 139.50, 135.09, 134.67, 130.20 (d, J = 6.5 Hz), 130.15, 128.95, 128.06, 127.92, 102.77, 49.02, 46.86, 34.35, 32.76, 31.48, 21.97. HPLC Purity: 98%.
4.8.56. 5-(4-(2-(dimethylamino)ethyl)phenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (63).
61 (62 mg, 0.140 mmol) was dissolved in 2 mL DCE and treated with DIPEA (0.025 ml, 0.140 mmol), stirred for 5 min, then 37% aq. formaldehyde (0.031 ml, 0.421 mmol) and acetic acid (0.024 ml, 0.421 mmol) were added sequentially. The mixture was stirred for 10 min at which point sodium triacetoxyborohydride (89 mg, 0.421 mmol) was added and the mixture was stirred overnight. The next day the mixture was diluted with 10% aq. Na2CO3 and extracted with EtOAc. The organic portion was washed with brine, dried over sodium sulfate, and concentrated to yield the pure product as an off white foam (59 mg, 0.136 mmol, 97 % yield). (ESI): m/z 434.2009 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.37 (ddd, J = 15.8, 8.3, 1.8 Hz, 3H), 7.33 - 7.28 (m, 3H), 7.25 (d, J = 8.9 Hz, 1H), 7.16 (dd, J = 8.1, 2.3 Hz, 1H), 7.08 (dd, J = 8.1, 2.3 Hz, 1H), 5.01 (q, J = 7.1 Hz, 1H), 3.99 (s, 3H), 2.89 - 2.80 (m, 2H), 2.65 - 2.52 (m, 2H), 2.32 (s, 6H), 1.71 (d, J = 7.2 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 161.69, 157.86, 151.04, 142.47, 142.14, 135.35, 133.42, 129.98, 129.90, 129.18, 129.15, 128.50, 127.53, 127.52, 102.87, 61.08, 46.86, 45.47, 34.17, 34.03, 21.93. HPLC Purity: 98%
4.8.57. 4-(1-methyl-4-oxo-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)benzonitrile (64).
Prepared from 4-aminobenzonitrile and 50 in a similar manner to 59 affording the titled compound as a white solid (50 mg, 0.129 mmol, 15.76 % yield). (ESI): m/z 388.1225 [M+H]+ 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.83 (ddd, J = 8.2, 2.0, 0.6 Hz, 1H), 7.79 (ddd, J = 8.1, 2.0, 0.6 Hz, 1H), 7.42 (ddd, J = 8.1, 2.2, 0.6 Hz, 1H), 7.39 - 7.35 (m, 2H), 7.35 - 7.29 (m, 3H), 7.27 (d, J = 7.7 Hz, 1H), 5.04 (q, J = 7.2 Hz, 1H), 4.01 (s, 3H), 1.73 (d, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 160.20, 157.34, 150.88, 141.55, 139.74, 135.44 (d, J = 2.8 Hz), 133.55 (d, J = 6.9 Hz), 130.69, 128.65, 127.83, 127.44, 117.81, 114.08, 102.61, 47.28 (d, J = 8.1 Hz), 34.22, 21.94. HPLC Purity: 99%
4.8.58. 5-(4-ethynylphenyl)-1-methyl-6-((1-phenylethyl)thio)-1H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (65).
Prepared from 4-ethynylaniline and 50 in a similar manner to 59 affording the titled compound as a yellow solid. (117 mg, 0.303 mmol, 32.0 % yield) (ESI): m/z 387.1276 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 7.99 (s, 1H), 7.64 (dd, J = 8.2, 1.8 Hz, 1H), 7.60 (dd, J = 8.2, 1.9 Hz, 1H), 7.38 (d, J = 7.1 Hz, 2H), 7.31 (t, J = 7.5 Hz, 2H), 7.28 - 7.22 (m, 2H), 7.15 (dd, J = 8.2, 2.2 Hz, 1H), 5.02 (q, J = 7.1 Hz, 1H), 4.00 (s, 3H), 3.17 (s, 1H), 1.71 (d, J = 7.0 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 161.08, 157.62, 150.99, 141.93, 135.83, 135.39, 133.42 (d, J = 9.6 Hz), 129.51 (d, J = 2.9 Hz), 128.57, 127.64, 127.46, 124.07, 102.77, 82.53, 79.01, 47.01, 34.09, 21.97. HPLC Purity: 98%
4.8.59. 2-(oxetan-3-ylmethyl)-5-phenyl-6-((1-phenylethyl)thio)-2H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (67).
6622 (100 mg, 0.409 mmol), K2CO3 (141 mg, 1.023 mmol), and (1-bromoethyl)benzene (55.9 μl, 0.409 mmol) were stirred in 2mL DMF at room temperature for 1 h. Oxetan-3-ylmethyl methanesulfonate (prepared as previously described80, 68.0 mg, 0.409 mmol) was added by syringe and the mixture was heated to 60 °C overnight. The next morning the mixture was diluted with water and extracted 2x with EtOAc. The combined organic portions were washed 3x with brine, dried over sodium sulfate, and concentrated. The crude residue was purified by flash (Eluting at 60% EtOAc in Hex) yielding the titled compound as a colorless oil (26 mg, 0.062 mmol, 15.18 % yield). MS (ESI): m/z 419.1537 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.06 (s, 1H), 7.56 - 7.40 (m, 3H), 7.35 (d, J = 7.6 Hz, 2H), 7.31 - 7.25 (m, 3H), 7.24 - 7.14 (m, 2H), 5.18 (q, J = 7.0 Hz, 1H), 4.87 (t, J = 7.2 Hz, 2H), 4.57 (d, J = 7.5 Hz, 2H), 4.54 (q, J = 5.7 Hz, 2H), 3.66 (h, J = 6.9 Hz, 1H), 1.75 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 160.53, 158.69, 158.62, 141.62, 135.57, 129.82, 129.62, 129.59, 129.56, 129.51, 128.62, 128.54, 127.70, 127.48, 105.00, 74.46, 55.80, 46.52, 35.38, 22.10. HPLC Purity: 99%
4.8.60. 2-(oxetan-3-yl)-5-phenyl-6-((1-phenylethyl)thio)-2H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (68).
Prepared in a similar manner to 67 employing 3-bromooxetane. White Solid. (14 mg, 0.035 mmol, 8.45 % yield) MS (ESI): m/z 405.1381 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.19 (s, 1H), 7.55 - 7.42 (m, 3H), 7.36 (d, J = 7.6 Hz, 2H), 7.31 - 7.26 (m, 3H), 7.25 - 7.16 (m, 2H), 5.57 (p, J = 6.9 Hz, 1H), 5.26 - 5.18 (m, 3H), 5.09 (t, J = 7.3 Hz, 2H), 1.77 (d, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 160.97, 158.63, 141.50, 135.49, 129.87, 129.61, 129.54, 129.53, 128.57, 127.96, 127.70, 127.52, 105.33, 76.86, 76.82, 56.78, 46.58, 22.15. HPLC Purity: 98%
4.8.61. 2-(oxetan-3-yl)-5-phenyl-6-((1-(pyridin-2-yl)ethyl)thio)-2H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (69).
A dry flask was charged with 6622 (250 mg, 1.023 mmol), K2CO3 (424 mg, 3.07 mmol), 2-(1-bromoethyl)pyridine (190 mg, 1.023 mmol), and 3mL DMF. The mixture was stirred for 1 h at RT at which point 3-bromooxetane (0.127 ml, 1.535 mmol) was added before heating the mixture to 80 °C. After 48 hours the mixture was diluted with water and extracted 2x with EtOAc, The combined organic portion was washed 3x with brine and dried over sodium sulfate. The mixture was purified by flash (60% EtOAc in Hex) yielding the mostly pure product as the later peak. The product was recrystallized from ethanol to obtain the titled compound as a white solid (45 mg, 0.111 mmol, 10.84 % yield). MS (ESI): m/z 406.1340 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.51 (d, J = 4.3 Hz, 1H), 8.19 (s, 1H), 7.60 (td, J = 7.7, 1.9 Hz, 1H), 7.54 - 7.42 (m, 3H), 7.40 (d, J = 7.8 Hz, 1H), 7.28 (dt, J = 7.7, 1.8 Hz, 1H), 7.19 (dt, J = 7.2, 2.0 Hz, 1H), 7.12 (ddd, J = 7.6, 4.9, 1.2 Hz, 1H), 5.65 - 5.47 (m, 1H), 5.33 (q, J = 6.9 Hz, 1H), 5.21 (q, J = 6.2 Hz, 2H), 5.08 (td, J = 7.4, 1.5 Hz, 2H), 1.78 (d, J = 7.0 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 161.14, 160.59, 158.64, 158.58, 149.59, 136.65, 135.36, 129.89, 129.67, 129.64, 129.60, 129.54, 128.00, 122.44, 122.31, 105.30, 76.87, 76.83, 56.77, 48.08, 21.49. HPLC Purity 97%
4.8.62. 2-(oxetan-3-yl)-5-phenyl-6-((1-(pyridin-3-yl)ethyl)thio)-2H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (70).
Prepared in a similar manner to 69. White solid. (100 mg, 0.247 mmol, 12.0 % yield) MS (ESI): m/z 406.1335 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.68 (d, J = 2.3 Hz, 1H), 8.47 (dd, J = 4.8, 1.6 Hz, 1H), 8.19 (s, 1H), 7.69 (dt, J = 8.0, 2.0 Hz, 1H), 7.56 - 7.45 (m, 3H), 7.30 - 7.24 (m, 1H), 7.22 (dd, J = 7.9, 4.8 Hz, 1H), 7.18 (dt, J = 8.2, 1.8 Hz, 1H), 5.65 - 5.46 (m, 1H), 5.29 - 5.13 (m, 3H), 5.09 (td, J = 7.5, 1.3 Hz, 2H), 1.74 (d, J = 7.1 Hz, 3H). NOE at 5.6 when irradiating at 8.19. 13C NMR (126 MHz, CDCl3) δ 160.13, 158.50, 158.40, 149.30, 148.71, 137.73, 135.33, 135.07, 130.04, 129.68, 129.60, 129.58, 129.52, 127.97, 123.33, 105.34, 76.81, 56.82, 43.75, 21.54. HPLC Purity: 99%.
4.8.63. Diethyl 2-methyl-2-(4-oxo-5-phenyl-6-((1-phenylethyl)thio)-4,5-dihydro-2H-pyrazolo[3,4-d]pyrimidin-2-yl)malonate (71).
To a solution of 6622 (1g, 4.09 mmol) and K2CO3 (1.414 g, 10.23 mmol) in 25mL DMF under N2 was added (1-bromoethyl)benzene (0.557 ml, 4.09 mmol) by syringe. The mixture was stirred at RT for 1 h. Diethyl 2-bromo-2-methylmalonate (0.857 ml, 4.50 mmol) was added by syringe and the mixture was heated to 50 °C overnight at which point the reaction was complete by HPLC. The mixture was diluted with water and extracted 3x with EtOAc. The combined organics were washed 3x with brine and dried over sodium sulfate. The crude oil was purified by flash (Gradient from 25-30% EtOAc in hexanes) to yield the titled compound as a clear colorless oil.(1.76 g, 3.38 mmol, 83 % yield) 1H NMR (400 MHz, CDCl3) δ 8.41 (d, J = 1.1 Hz, 1H), 7.56 - 7.40 (m, 3H), 7.33 (d, J = 7.3 Hz, 2H), 7.30 - 7.14 (m, 6H), 5.21 (q, J = 6.9 Hz, 1H), 4.40 - 4.20 (m, 4H), 2.26 (d, J = 1.1 Hz, 3H), 1.78 - 1.69 (m, 3H), 1.45 - 1.05 (m, 6H).
4.8.64. 2-(1,3-dihydroxy-2-methylpropan-2-yl)-5-phenyl-6-((1-phenylethyl)thio)-2H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (72).
NOTE: REACTION PRODUCES POTENT THIOL SMELL. A dry flask under N2 charged with NaBH4 (0.481 g, 12.71 mmol) and 20 mL anhydrous dimethoxyethane was cooled in an ice bath. Bromine (0.305 ml, 5.93 mmol) was dissolved in 2 mL dimethoxyethane and added to the mixture dropwise by syringe (exothermic!). After complete addition, the reaction was removed from the cooling bath and stirred for 30 minutes at which point the solution was colorless with white NaBr precipitate. The flask was cooled in an ice/brine bath and 71 (1.47g, 2.82 mmol) dissolved in 10 mL dimethoxyethane was added by syringe. The mixture was kept in the bath for 1 h then allowed to warm to room temperature and stirred for 24 h. The reaction mixture was added dropwise to 200 mL ACN then 5mL of water was added slowly followed by 5 mL 1N HCl. The mixture was allowed to sit for 1 hour at which point EtOAc and 10 mL water were added and two layers formed. Sodium sulfate was added to saturate the aqueous solution and the organic portion decanted off. The aqueous portion was extracted again with EtOAc. The combined organic portions were dried over sodium sulfate and concentrated. The residue was taken up in DCM and dried with additional sodium sulfate and then loaded onto silica and purified by flash (80% EtOAc in Hexanes) to afford the titled compound as a white solid (303 mg, 0.694 mmol, 24.6 % yield). 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 0.6 Hz, 1H), 7.56 - 7.42 (m, 2H), 7.35 (d, J = 7.3 Hz, 3H), 7.32 - 7.25 (m, 3H), 7.25 - 7.14 (m, 2H), 5.26 - 5.08 (m, 1H), 4.09 - 3.60 (m, 4H), 1.76 (d, J = 7.0 Hz, 3H), 1.60 (s, 3H).
4.8.65. 3-hydroxy-2-methyl-2-(4-oxo-5-phenyl-6-((1-phenylethyl)thio)-4,5-dihydro-2H-pyrazolo[3,4-d]pyrimidin-2-yl)propyl 4-methylbenzenesulfonate (73).
A flask under N2 charged with 72 (303 mg, 0.694 mmol) in 6 mL THF was cooled to −78 °C. nBuLi (278 μl, 0.694 mmol) 2.5M in hexanes was added and the mixture was stirred for 1 h. TsCl (132 mg, 0.694 mmol) in 1mL THF was added and the reaction was stirred for 30 min at −78 °C and then allowed to warm to RT. The reaction was quenched with sat. aq. NH4Cl, diluted with water and extracted 2x with EtOAc. The combined organics were washed with brine and dried over sodium sulfate and concentrated. The crude mixture was purified by flash (Eluting at 50% EtOAc in Hexanes) to afford the titled compound as a white foam (140 mg, 0.237 mmol, 34.1 % yield). 1H NMR (400 MHz, CDCl3) δ 8.19 (d, J = 5.3 Hz, 1H), 7.68 (t, J = 8.6 Hz, 2H), 7.59 - 7.38 (m, 3H), 7.38 - 7.06 (m, 9H), 5.12 (qd, J = 7.0, 2.3 Hz, 1H), 4.51 (dd, J = 10.3, 4.4 Hz, 1H), 4.37 (dd, J = 10.2, 1.9 Hz, 1H), 3.95 (dd, J = 6.6, 2.9 Hz, 2H), 3.71 (t, J = 6.5 Hz, 1H), 2.41 (d, J = 17.6 Hz, 3H), 1.73 (t, J = 7.8 Hz, 3H), 1.64 (s, 3H).
4.8.66. 2-(3-methyloxetan-3-yl)-5-phenyl-6-((1-phenylethyl)thio)-2H-pyrazolo[3,4-d]pyrimidin-4(5H)-one (74).
To a dry pressure tube charged with 73 (140 mg, 0.237 mmol) in 2 mL at 0 °C was added nBuLi (2.5M in Hexanes, 0.114 ml, 0.284 mmol) in THF. The mixture was stirred for 20 min then heated to 65 °C overnight. The next morning the reaction was not complete by HPLC so the flask was cooled to 0 °C and another portion of nBuLi (2.5M in Hexanes, .05 ml, 0.125 mmol) was added. The mixture was heated to 80 °C for 4 hours at which point it was complete by HPLC and was quenched by adding a few drops of water then concentrated. The residue was taken up in water and ethyl acetate. The organic portion was washed with brine and dried over sodium sulfate and concentrated. The crude residue obtained was purified by flash (Eluting at 45% EtOAc in Hex). The white solid obtained was further recrystallized from hot ethanol to yield the titled compound as white needles (40 mg, 0.096 mmol, 40.3 % yield). MS (ESI): m/z 406.1340 [M+H]+ 1H NMR (500 MHz, CDCl3) δ 8.17 (s, 1H), 7.56 - 7.41 (m, 3H), 7.35 (d, J = 7.6 Hz, 2H), 7.31 - 7.25 (m, 3H), 7.25 - 7.15 (m, 2H), 5.29 (t, J = 5.9 Hz, 2H), 5.23 (q, J = 6.9 Hz, 1H), 4.72 (d, J = 6.5 Hz, 2H), 2.04 (s, 3H), 1.77 (d, J = 7.0 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 160.84, 158.77, 158.36, 141.44, 135.54, 129.84, 129.63, 129.60, 129.56, 129.52, 128.57, 127.70, 127.51, 126.17, 105.23, 81.35 (d, J = 5.0 Hz), 62.37, 46.51, 24.44, 22.18. NOE observed for oxetane and methyl protons at δ 5.29 and 2.04 respectively when irradiating pyrazole proton at δ 8.17. HPLC Purity 99%.
Supplementary Material
Figure S-1: Docking of cyclopentenone (R)-36 to 1A1, 1A2 and 1A3
Figure S-2: Docking of cyclopentenone (S)-36 to 1A1, 1A2 and 1A3
Figure S-3A: Docking of nitrile 64 to 1A2 and 1A3
Figure S-3B: Overlay of nitrile 64 and distinct chemotype GA11 in 1A3
Figure S-4: Electron density maps for new structures bound to ALDH1A1
Figure S-5: Summary of metabolite identification study on 68
Figure S-6. ALDEFLUOR inhibition by (R)-28 and 68 in taxane resistant SKOV3 cells
Figure S-7: Representative fit of the steady-state inhibition data to the tight-binding uncompetitive inhibition equation.
Table S-1: Data collection and refinement statistics for enzyme-compound complexes Representative NMR spectra for compounds 28, 36, 42, 53, 64, 68, 69 and 74
Highlights.
Aldehyde Dehydrogenase 1A inhibition has potential in chemotherapy-resistant cancer
Broad spectrum ALDH1A inhibitors are most effective in multiple cell types
ALDH1A inhibitors synergize with taxol chemotherapy in patient tumor spheroids
New ALDH1A inhibitors have improved pharmacokinetic properties
Acknowledgement
The authors gratefully acknowledge support by the National Cancer Institute of the National Institutes of Health through R01 CA214567-01 (SDL and RJB). Structural results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Society, A. C. Cancer Facts and Figures. https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html.
- 2.Ozols RF; Bundy BN; Greer BE; Fowler JM; Clarke-Pearson D; Burger RA; Mannel RS; DeGeest K; Hartenbach EM; Baergen R, Phase III Trial of Carboplatin and Paclitaxel Compared With Cisplatin and Paclitaxel in Patients With Optimally Resected Stage III Ovarian Cancer: A Gynecologic Oncology Group Study. Journal of Clinical Oncology 2003, 21 (17), 3194–3200. [DOI] [PubMed] [Google Scholar]
- 3.Thigpen T; duBois A; McAlpine J; DiSaia P; Fujiwara K; Hoskins W; Kristensen G; Mannel R; Markman M; Pfisterer J; Quinn M; Reed N; Swart AM; Berek J; Colombo N; Freyer G; Gallardo D; Plante M; Poveda A; Rubinstein L; Bacon M; Kitchener H; Stuart GC, First-line Therapy in Ovarian Cancer Trials. Int J Gynecol Cancer 2011, 21 (4), 756–762. [DOI] [PubMed] [Google Scholar]
- 4.Rossi L; Verrico M; Zaccarelli E; Papa A; Colonna M; Strudel M; Vici P; Bianco V; Tomao F, Bevacizumab in ovarian cancer: A critical review of phase III studies. Oncotarget 2017, 8 (7), 12389–12405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLachlan J; George A; Banerjee S, The current status of PARP inhibitors in ovarian cancer. Tumori 2016, 102 (5), 433–440. [DOI] [PubMed] [Google Scholar]
- 6.Bitler BG; Watson ZL; Wheeler LJ; Behbakht K, PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecologic oncology 2017, 147 (3), 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma NJCA, PARP Inhibitors for Cancer Therapy. Humana Press, Cham: 2015. [Google Scholar]
- 8.Poveda A; Floquet A; Ledermann JA; Asher R; Penson RT; Oza AM; Korach J; Huzarski T; Pignata S; Friedlander M; Baldoni A; Park-Simon T-W; Sonke GS; Lisyanskaya AS; Kim J-H; Filho EA; Vergote I; Rowe P; Pujade-Lauraine E, Final overall survival (OS) results from SOLO2/ENGOT-ov21: A phase III trial assessing maintenance olaparib in patients (pts) with platinum-sensitive, relapsed ovarian cancer and a BRCA mutation. Journal of Clinical Oncology 2020, 38 (15_suppl), 6002–6002. [Google Scholar]
- 9.Cancer Genome Atlas Research, N., Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474 (7353), 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cannistra SA, Cancer of the ovary. N Engl J Med 2004, 351 (24), 2519–29. [DOI] [PubMed] [Google Scholar]
- 11.Magni M; Shammah S; Schiro R; Mellado W; Dalla-Favera R; Gianni AM, Induction of cyclophosphamide-resistance by aldehyde-dehydrogenase gene transfer. Blood 1996, 87 (3), 1097–103. [PubMed] [Google Scholar]
- 12.Chefetz I; Grimley E; Yang K; Hong L; Vinogradova EV; Suciu R; Kovalenko I; Karnak D; Morgan CA; Chtcherbinine M; Buchman C; Huddle B; Barraza S; Morgan M; Bernstein KA; Yoon E; Lombard DB; Bild A; Mehta G; Romero I; Chiang CY; Landen C; Cravatt B; Hurley TD; Larsen SD; Buckanovich RJ, A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell reports 2019, 26 (11), 3061–3075.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awad O; Yustein JT; Shah P; Gul N; Katuri V; O'Neill A; Kong Y; Brown ML; Toretsky JA; Loeb DM, High ALDH activity identifies chemotherapy-resistant Ewing's sarcoma stem cells that retain sensitivity to EWS-FLI1 inhibition. PLoS One 2010, 5 (11), e13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dylla SJ; Beviglia L; Park IK; Chartier C; Raval J; Ngan L; Pickell K; Aguilar J; Lazetic S; Smith-Berdan S; Clarke MF; Hoey T; Lewicki J; Gurney AL, Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 2008, 3 (6), e2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croker AK; Allan AL, Inhibition of aldehyde dehydrogenase (ALDH) activity reduces chemotherapy and radiation resistance of stem-like ALDHhiCD44(+) human breast cancer cells. Breast Cancer Res Treat 2012, 133 (1), 75–87. [DOI] [PubMed] [Google Scholar]
- 16.Huang CP; Tsai MF; Chang TH; Tang WC; Chen SY; Lai HH; Lin TY; Yang JC; Yang PC; Shih JY; Lin SB, ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett 2013, 328 (1), 144–51. [DOI] [PubMed] [Google Scholar]
- 17.Silva IA; Bai S; McLean K; Yang K; Griffith K; Thomas D; Ginestier C; Johnston C; Kueck A; Reynolds RK; Wicha MS; Buckanovich RJ, Aldehyde Dehydrogenase in Combination with CD133 Defines Angiogenic Ovarian Cancer Stem Cells that Portend Poor Patient Survival. Cancer Res 2011, 71 (11), 3991–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma S; Chan KW; Lee TK; Tang KH; Wo JY; Zheng BJ; Guan XY, Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Molecular cancer research : MCR 2008, 6 (7), 1146–53. [DOI] [PubMed] [Google Scholar]
- 19.Kryczek I; Liu S; Roh M; Vatan L; Szeliga W; Wei S; Banerjee M; Mao Y; Kotarski J; Wicha MS; Liu R; Zou W, Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. International journal of cancer 2012,130 (1), 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo Y; Dallaglio K; Chen Y; Robinson WA; Robinson SE; McCarter MD; Wang J; Gonzalez R; Thompson DC; Norris DA; Roop DR; Vasiliou V; Fujita M, ALDH1A Isozymes are Markers of Human Melanoma Stem Cells and Potential Therapeutic Targets. Stem Cells 2012, 30 (10), 2100–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z; Xiang Y; Xiang L; Xiao Y; Li F; Hao P, ALDH Maintains the Stemness of Lung Adenoma Stem Cells by Suppressing the Notch/CDK2/CCNE Pathway. PLoS One 2014, 9 (3), e92669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huddle BC; Grimley E; Buchman CD; Chtcherbinine M; Debnath B; Mehta P; Yang K; Morgan CA; Li S; Felton J; Sun D; Mehta G; Neamati N; Buckanovich RJ; Hurley TD; Larsen SD, Structure-Based Optimization of a Novel Class of Aldehyde Dehydrogenase 1A (ALDH1A) Subfamily-Selective Inhibitors as Potential Adjuncts to Ovarian Cancer Chemotherapy. Journal of Medicinal Chemistry 2018, 61 (19), 8754–8773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Condello S; Morgan CA; Nagdas S; Cao L; Turek J; Hurley TD; Matei D, Beta-Catenin-Regulated ALDH1A1 is a Target in Ovarian Cancer Spheroids. Oncogene 2015, 34 (18), 2297–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duong HQ; Hwang JS; Kim HJ; Kang HJ; Seong YS; Bae I, Aldehyde Dehydrogenase 1A1 Confers Intrinsic and Acquired Resistance to Gemcitabine in Human Pancreatic Adenocarcinoma MIA PaCa-2 Cells. Int J Oncol 2012, 41 (3), 855–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomita H; Tanaka K; Tanaka T; Hara A, Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7 (10), 11018–11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang-Jie D; Jiao C; Yu-Feng G; Xiu-Wu B; Shi-Cang Y, ALDH1A3, a metabolic target for cancer diagnosis and therapy. International journal of cancer 2016, 139 (5), 965–975. [DOI] [PubMed] [Google Scholar]
- 27.Landen CN Jr.; Goodman B; Katre AA; Steg AD; Nick AM; Stone RL; Miller LD; Mejia PV; Jennings NB; Gershenson DM; Bast RC Jr.; Coleman RL; Lopez-Berestein G; Sood AK, Targeting Aldehyde Dehydrogenase Cancer Stem Cells in Ovarian Cancer. Mol Cancer Ther 2010, 9 (12), 3186–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moreb JS; Baker HV; Chang LJ; Amaya M; Lopez MC; Ostmark B; Chou W, ALDH Isozymes Downregulation Affects Cell Growth, Cell Motility and Gene Expression in Lung Cancer Cells. Mol Cancer 2008, 7, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokoyama Y; Zhu H; Lee JH; Kossenkov AV; Wu SY; Wickramasinghe JM; Yin X; Palozola KC; Gardini A; Showe LC; Zaret KS; Liu Q; Speicher D; Conejo-Garcia JR; Bradner JE; Zhang Z; Sood AK; Ordog T; Bitler BG; Zhang R, BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res 2016, 76 (21), 6320–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen MH; Weng JJ; Cheng CT; Wu RC; Huang SC; Wu CE; Chung YH; Liu CY; Chang MH; Chiang KC; Yeh TS; Su Y; Yeh CN, ALDH1A3, the Major Aldehyde Dehydrogenase Isoform in Human Cholangiocarcinoma Cells, Affects Prognosis and Gemcitabine Resistance in Cholangiocarcinoma Patients. Clin Cancer Res 2016, 22 (16), 4225–35. [DOI] [PubMed] [Google Scholar]
- 31.Cortes-Dericks L; Froment L; Boesch R; Schmid RA; Karoubi G, Cisplatin-Resistant Cells in Malignant Pleural Mesothelioma Cell Lines Show ALDH(high)CD44(+) Phenotype and Sphere-Forming Capacity. BMC cancer 2014, 14, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saw YT; Yang J; Ng SK; Liu S; Singh S; Singh M; Welch WR; Tsuda H; Fong WP; Thompson D; Vasiliou V; Berkowitz RS; Ng SW, Characterization of aldehyde dehydrogenase isozymes in ovarian cancer tissues and sphere cultures. BMC cancer 2012, 12, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowtell DD; Bohm S; Ahmed AA; Aspuria PJ; Bast RC Jr.; Beral V; Berek JS; Birrer MJ; Blagden S; Bookman MA; Brenton JD; Chiappinelli KB; Martins FC; Coukos G; Drapkin R; Edmondson R; Fotopoulou C; Gabra H; Galon J; Gourley C; Heong V; Huntsman DG; Iwanicki M; Karlan BY; Kaye A; Lengyel E; Levine DA; Lu KH; McNeish IA; Menon U; Narod SA; Nelson BH; Nephew KP; Pharoah P; Powell DJ Jr.; Ramos P; Romero IL; Scott CL; Sood AK; Stronach EA; Balkwill FR, Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nature reviews. Cancer 2015, 15 (11), 668–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koppaka V; Thompson DC; Chen Y; Ellermann M; Nicolaou KC; Juvonen RO; Petersen D; Deitrich RA; Hurley TD; Vasiliou V, Aldehyde Dehydrogenase Inhibitors: a Comprehensive Review of the Pharmacology, Mechanism of Action, Substrate Specificity, and Clinical Application. Pharmacological Reviews 2012, 64 (3), 520–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dinavahi SS; Bazewicz CG; Gowda R; Robertson GP, Aldehyde Dehydrogenase Inhibitors for Cancer Therapeutics. Trends Pharmacol Sci 2019, 40 (10), 774–789. [DOI] [PubMed] [Google Scholar]
- 36.Yang SM; Martinez NJ; Yasgar A; Danchik C; Johansson C; Wang Y; Baljinnyam B; Wang AQ; Xu X; Shah P; Cheff D; Wang XS; Roth J; Lal-Nag M; Dunford JE; Oppermann U; Vasiliou V; Simeonov A; Jadhav A; Maloney DJ, Discovery of Orally Bioavailable, Quinoline-Based Aldehyde Dehydrogenase 1A1 (ALDH1A1) Inhibitors with Potent Cellular Activity. J Med Chem 2018, 61 (11), 4883–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang SM; Yasgar A; Miller B; Lal-Nag M; Brimacombe K; Hu X; Sun H; Wang A; Xu X; Nguyen K; Oppermann U; Ferrer M; Vasiliou V; Simeonov A; Jadhav A; Maloney DJ, Discovery of NCT-501, a Potent and Selective Theophylline-Based Inhibitor of Aldehyde Dehydrogenase 1A1 (ALDH1A1). J Med Chem 2015, 58 (15), 5967–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quattrini L; Gelardi ELM; Coviello V; Sartini S; Ferraris DM; Mori M; Nakano I; Garavaglia S; La Motta C, Imidazo[1,2-a]pyridine Derivatives as Aldehyde Dehydrogenase Inhibitors: Novel Chemotypes to Target Glioblastoma Stem Cells. Journal of Medicinal Chemistry 2020, 63 (9), 4603–4616. [DOI] [PubMed] [Google Scholar]
- 39.Quattrini L; Gelardi ELM; Petrarolo G; Colombo G; Ferraris DM; Picarazzi F; Rizzi M; Garavaglia S; La Motta C, Progress in the Field of Aldehyde Dehydrogenase Inhibitors: Novel Imidazo[1,2-a]pyridines against the 1A Family. ACS Medicinal Chemistry Letters 2020, 11 (5), 963–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng P; Wang J; Waghmare I; Sartini S; Coviello V; Zhang Z; Kim S-H; Mohyeldin A; Pavlyukov MS; Minata M; Valentim CLL; Chhipa RR; Bhat KPL; Dasgupta B; La Motta C; Kango-Singh M; Nakano I, FOXD1-ALDH1A3 Signaling Is a Determinant for the Self-Renewal and Tumorigenicity of Mesenchymal Glioma Stem Cells. Cancer Research 2016, 76 (24), 7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kyungjin; LIU Jin-Jun; SCOTT Nathan Robert; YI Lin; ZAK Mark; ZHAO Guiling. PYRAZOLOPYRIMIDONE AND PYRAZOLOPYRIDONE INHIBITORS OF TANKYRASE. Patent: WO2013/182546 A1 2013.
- 42.Inhibitors Of PI3 Kinase. WO2011089400 (2011).
- 43.Yoshikazu K; Nobuya K; Teruaki M, A Convenient Method for the Preparation of Alkyl Aryl Sulfides from Alcohols and (Chloromethylene)dimethylammonium Chloride. Chemistry Letters 2005, 34 (12), 1612–1613. [Google Scholar]
- 44.Carceller E; Salas J; Merlos M; Giral M; Ferrando R; Escamilla I; Ramis J; García-Rafanell J; Forn J, Novel Azo Derivatives as Prodrugs of 5-Aminosalicylic Acid and Amino Derivatives with Potent Platelet Activating Factor Antagonist Activity. Journal of Medicinal Chemistry 2001, 44 (18), 3001–3013. [DOI] [PubMed] [Google Scholar]
- 45.Wilkinson MC, “Greener” Friedel–Crafts Acylations: A Metal- and Halogen-Free Methodology. Organic Letters 2011, 13 (9), 2232–2235. [DOI] [PubMed] [Google Scholar]
- 46.Yasuda M; Onishi Y; Ueba M; Miyai T; Baba A, Direct Reduction of Alcohols: Highly Chemoselective Reducing System for Secondary or Tertiary Alcohols Using Chlorodiphenylsilane with a Catalytic Amount of Indium Trichloride. The Journal of Organic Chemistry 2001, 66 (23), 7741–7744. [DOI] [PubMed] [Google Scholar]
- 47.Tudge M; Mashima H; Savarin C; Humphrey G; Davies I, Facile reduction of malonate derivatives using NaBH4/Br2: an efficient route to 1,3-diols. Tetrahedron Letters 2008, 49 (6), 1041–1044. [Google Scholar]
- 48.MarvinSketch Calculator Plugins were used for ADME predictions, Marvin 15.6.15, 2015, ChemAxon http://www.chemaxon.com (accessed: 06/05/2018).
- 49.Patani GA; LaVoie EJ, Bioisosterism: A Rational Approach in Drug Design. Chemical Reviews 1996,96 (8), 3147–3176. [DOI] [PubMed] [Google Scholar]
- 50.Lovering F; Bikker J; Humblet C, Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. Journal of Medicinal Chemistry 2009, 52 (21), 6752–6756. [DOI] [PubMed] [Google Scholar]
- 51.Fang Z; Song Y; Zhan P; Zhang Q; Liu X, Conformational restriction: an effective tactic in 'follow-on'-based drug discovery. Future medicinal chemistry 2014, 6 (8), 885–901. [DOI] [PubMed] [Google Scholar]
- 52.Freeman-Cook KD; Hoffman RL; Johnson TW, Lipophilic efficiency: the most important efficiency metric in medicinal chemistry. Future medicinal chemistry 2013, 5 (2), 113–115. [DOI] [PubMed] [Google Scholar]
- 53.Moretti A; Li J; Donini S; Sobol RW; Rizzi M; Garavaglia S, Crystal structure of human aldehyde dehydrogenase 1A3 complexed with NAD+ and retinoic acid. Scientific Reports 2016, 6 (1), 35710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morgan CA; Hurley TD, Characterization of two distinct structural classes of selective aldehyde dehydrogenase 1A1 inhibitors. J Med Chem 2015, 58 (4), 1964–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilkinson GR; Shand DG, A physiological approach to hepatic drug clearance. Clinical Pharmacology & Therapeutics 1975, 18 (4), 377–390. [DOI] [PubMed] [Google Scholar]
- 56.Marcato P; Dean CA; Pan D; Araslanova R; Gillis M; Joshi M; Helyer L; Pan L; Leidal A; Gujar S; Giacomantonio CA; Lee PW, Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells 2011, 29 (1), 32–45. [DOI] [PubMed] [Google Scholar]
- 57.Brady SW; McQuerry JA; Qiao Y; Piccolo SR; Shrestha G; Jenkins DF; Layer RM; Pedersen BS; Miller RH; Esch A; Selitsky SR; Parker JS; Anderson LA; Dalley BK; Factor RE; Reddy CB; Boltax JP; Li DY; Moos PJ; Gray JW; Heiser LM; Buys SS; Cohen AL; Johnson WE; Quinlan AR; Marth G; Werner TL; Bild AH, Combating subclonal evolution of resistant cancer phenotypes. Nat Commun 2017, 8 (1), 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morgan CA; Parajuli B; Buchman CD; Dria K; Hurley TD, N,N-Diethylaminobenzaldehyde (DEAB) as a Substrate and Mechanism-Based Inhibitor for Human ALDH Isoenzymes. Chemico-Biological Interactions 2015, 234 (0), 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hammen PK; Allali-Hassani A; Hallenga K; Hurley TD; Weiner H, Multiple Conformations of NAD and NADH when Bound to Human Cytosolic and Mitochondrial Aldehyde Dehydrogenase. Biochemistry 2002, 41 (22), 7156–7168. [DOI] [PubMed] [Google Scholar]
- 60.Parajuli B; Kimble-Hill AC; Khanna M; Ivanova Y; Meroueh S; Hurley TD, Discovery of Novel Regulators of Aldehyde Dehydrogenase Isoenzymes. Chem Biol Interact 2011, 191 (1-3), 153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parajuli B; Georgiadis TM; Fishel ML; Hurley TD, Development of Selective Inhibitors for Human Aldehyde Dehydrogenase 3A1 (ALDH3A1) for the Enhancement of Cyclophosphamide Cytotoxicity. Chembiochem 2014, 15 (5), 701–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buchman CD; Hurley TD, Inhibition of the Aldehyde Dehydrogenase 1/2 Family by Psoralen and Coumarin Derivatives. J Med Chem 2017, 60 (6), 2439–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Otwinowski Z; Minor W, Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol 1997, 276, 307–26. [DOI] [PubMed] [Google Scholar]
- 64.The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr D Biol Crystallogr 1994, 50 (Pt 5), 760–3. [DOI] [PubMed] [Google Scholar]
- 65.Emsley P; Cowtan K, Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr D Biol Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- 66.Painter J; Merritt EA, Optimal Description of a Protein Structure in Terms of Multiple Groups Undergoing TLS Motion. Acta Crystallogr D Biol Crystallogr 2006, 62 (Pt 4), 439–50. [DOI] [PubMed] [Google Scholar]
- 67.Painter J; Merritt EA, A Molecular Viewer for the Analysis of TLS Rigid-Body Motion in Macromolecules. Acta Crystallogr D Biol Crystallogr 2005, 61 (Pt 4), 465–71. [DOI] [PubMed] [Google Scholar]
- 68.Chou TC; Talalay P, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984, 22, 27–55. [DOI] [PubMed] [Google Scholar]
- 69.Ward Rashidi MR; Mehta P; Bregenzer M; Raghavan S; Fleck EM; Horst EN; Harissa Z; Ravikumar V; Brady S; Bild A; Rao A; Buckanovich RJ; Mehta G, Engineered 3D Model of Cancer Stem Cell Enrichment and Chemoresistance. Neoplasia (New York, N.Y.) 2019, 21 (8), 822–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raghavan S; Mehta P; Ward MR; Bregenzer ME; Fleck EMA; Tan L; McLean K; Buckanovich RJ; Mehta G, Personalized Medicine-Based Approach to Model Patterns of Chemoresistance and Tumor Recurrence Using Ovarian Cancer Stem Cell Spheroids. Clin Cancer Res 2017, 23 (22), 6934–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raghavan S; Mehta P; Horst EN; Ward MR; Rowley KR; Mehta G, Comparative analysis of tumor spheroid generation techniques for differential in vitro drug toxicity. Oncotarget 2016, 7 (13), 16948–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones G; Willett P; Glen RC, Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol 1995, 245 (1), 43–53. [DOI] [PubMed] [Google Scholar]
- 73.Jones G; Willett P; Glen RC; Leach AR; Taylor R, Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267 (3), 727–48. [DOI] [PubMed] [Google Scholar]
- 74.Chen Y; Zhu JY; Hong KH; Mikles DC; Georg GI; Goldstein AS; Amory JK; Schonbrunn E, Structural Basis of ALDH1A2 Inhibition by Irreversible and Reversible Small Molecule Inhibitors. ACS Chem Biol 2018, 13 (3), 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hawkins PC; Skillman AG; Warren GL; Ellingson BA; Stahl MT, Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 2010, 50 (4), 572–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heim-Riether A; Healy J, A Novel Method for the Synthesis of Imidazo[5,1-f][1,2,4]triazin-4(3H)-ones. The Journal of Organic Chemistry 2005, 70 (18), 7331–7337. [DOI] [PubMed] [Google Scholar]
- 77.Olah GA; Gupta BGB; Malhotra R; Narang SC, Chlorotrimethylsilane/lithium bromide and hexamethyldisilane/pyridinium bromide perbromide: effective and selective reagents for the conversion of alkyl (cycloalkyl and aralkyl) alcohols into bromides. The Journal of Organic Chemistry 1980, 45 (9), 1638–1639. [Google Scholar]
- 78.cGAS Antagonist Compounds. Patent: WO2017176812 A1 2017.
- 79.Piscitelli F; Ligresti A; La Regina G; Coluccia A; Morera L; Allarà M; Novellino E; Di Marzo V; Silvestri R, Indole-2-carboxamides as Allosteric Modulators of the Cannabinoid CB1 Receptor. Journal of Medicinal Chemistry 2012, 55 (11), 5627–5631. [DOI] [PubMed] [Google Scholar]
- 80.Kehler J; Rasmussen LK; Jessing M Triazolopyrazinones as PDE1 Inhibitors. Patent: WO2016055618 A1 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S-1: Docking of cyclopentenone (R)-36 to 1A1, 1A2 and 1A3
Figure S-2: Docking of cyclopentenone (S)-36 to 1A1, 1A2 and 1A3
Figure S-3A: Docking of nitrile 64 to 1A2 and 1A3
Figure S-3B: Overlay of nitrile 64 and distinct chemotype GA11 in 1A3
Figure S-4: Electron density maps for new structures bound to ALDH1A1
Figure S-5: Summary of metabolite identification study on 68
Figure S-6. ALDEFLUOR inhibition by (R)-28 and 68 in taxane resistant SKOV3 cells
Figure S-7: Representative fit of the steady-state inhibition data to the tight-binding uncompetitive inhibition equation.
Table S-1: Data collection and refinement statistics for enzyme-compound complexes Representative NMR spectra for compounds 28, 36, 42, 53, 64, 68, 69 and 74