

SUMMARY
The phosphorylation of G protein-coupled receptors (GPCRs) by GPCR kinases (GRKs) facilitates arrestin binding and receptor desensitization. While this process can be regulated by Ca2+-binding proteins such as calmodulin (CaM) and recoverin, the molecular mechanisms are poorly understood. Here, we report structural, computational and biochemical analysis of a CaM complex with GRK5, revealing how CaM shapes GRK5 response to calcium. The CaM N- and C-domains bind independently to two helical regions at the GRK5 N- and C-termini to inhibit GPCR phosphorylation, though only the C-domain interaction disrupts GRK5 membrane association thereby facilitating cytoplasmic translocation. The CaM N-domain strongly activates GRK5 via ordering of the amphipathic αN-helix of GRK5 and allosteric disruption of kinase-RH domain interaction for phosphorylation of cytoplasmic GRK5 substrates. These results provide a framework for understanding how two functional effects, GRK5 activation and localization, can cooperate under control of CaM for selective substrate targeting by GRK5.
Graphical Abstract

eToc Blurb
Komolov, Sulon et al. use structural, computational and functional approaches to characterize the mechanism of calmodulin-mediated activation of the G protein-coupled receptor kinase, GRK5. Additional findings reveal that GRK5 activation and localization cooperate under control of calmodulin for selective targeting of substrates at the plasma membrane and cytoplasm.
INTRODUCTION
G protein-coupled receptors (GPCRs) function to sense extracellular stimuli and activate intracellular signaling pathways to modulate various physiological processes (Lefkowitz, 2007). GPCR signaling is dynamically regulated with receptors undergoing rapid phosphorylation by GPCR kinases (GRKs) which facilitates β-arrestin binding to terminate G protein signaling and target receptors for endocytosis (Krupnick and Benovic, 1998). GRKs are AGC family kinases with 7 mammalian members across three sub-families: GRK1 (GRK1 and 7); GRK2 (GRK2 and 3); and GRK4 (GRK4, 5 and 6) (Komolov and Benovic, 2018). The GRK architecture includes a conserved kinase domain that is inserted into a regulator of G protein signaling homology (RH) domain that is bracketed by a short N-terminal α-helical domain (αN) and a variable C-terminal lipid-binding domain. Recent studies demonstrated high structural plasticity of GRKs (Komolov et al., 2017; Yao et al., 2017) with a transient electrostatic contact (“ionic lock”) between the RH and kinase domains being disrupted by GPCR binding. This promotes GRK domain dissociation, enhancing GRK-receptor complex stability and kinase activation (Komolov et al., 2017).
Given the essential role that GRKs play in GPCR regulation, it is not surprising that GRKs themselves are dynamically regulated (Gurevich et al., 2012). Ca2+-binding proteins like calmodulin (CaM) have an important role in this process and effectively inhibit GRK4 sub-family members with a particularly high affinity for GRK5 (Levay et al., 1998; Pronin et al., 1997). Although little is currently known about the structural organization of the CaM/GRK5 complex, two lipid-binding regions on the N- and C-termini of GRK5 have been implicated in CaM binding (GRK5 residues 20-39 and 546-562) (Levay et al., 1998) and a recent BioSAXS study supports a 1:1 binding model (Beyett et al., 2019). An alternative model of GRK regulation by calcium has been proposed for GRK1 where the neuronal calcium-sensor homolog of CaM, recoverin, binds to the GRK1 N-terminal α-helix (GRK1 residues 1-16) (Ames et al., 2006; Higgins et al., 2006), a region predicted to play an important role in GRK activity and GPCR binding (Huang et al., 2011).
While GRKs target GPCRs at the plasma membrane, GRK5 also phosphorylates multiple substrates in the cytoplasm and nucleus including HDAC5, p53, IkBα, nucleophosmin and α-synuclein (Arawaka et al., 2006; Chen et al., 2010b; Gold et al., 2013; So et al., 2012). Ca2+/CaM-mediated translocation of GRK5 from the plasma membrane to the cytoplasm/nucleus might contribute to targeting these substrates (Gold et al., 2013; Oda et al., 2018), though how CaM may spatially and temporally regulate GRK5 localization and substrate specificity is not well understood.
To delineate how GRKs are regulated by calcium, we set out to determine the molecular basis for GRK5 interaction with Ca2+/CaM. Through structural, computational and functional approaches, we uncover the stepwise mechanism of GRK5 activation by CaM and highlight key elements of GRK5 critical for CaM-mediated regulation. In addition, we demonstrate that GRK5 activation and localization cooperate under control of CaM for selective targeting of substrates at the plasma membrane and cytoplasm.
RESULTS AND DISCUSSION
Crystal Structure of a GRK5/CaM Complex
To better understand CaM regulation of GRK5, we solved a crystal structure of Ca2+/CaM bound to GRK5 at 2.0 Å resolution (Figure 1A and Table 1). The crystal structure contained one copy of the CaM/GRK5 complex in the asymmetric unit. The complex was also monomeric in solution with an estimated mass of ~81 kDa by size exclusion chromatography (SEC) (Figure S1A) and ~78 kDa by analytical ultracentrifugation (Figure S1B). A 1:1 complex was also suggested by native agarose electrophoresis (Figure S1C) and chemical cross-linking (Figure S1D). Taken together, our data strongly support a monomeric 1:1 complex.
Figure 1. High-resolution crystal structure reveals molecular details of GRK5/CaM interface.
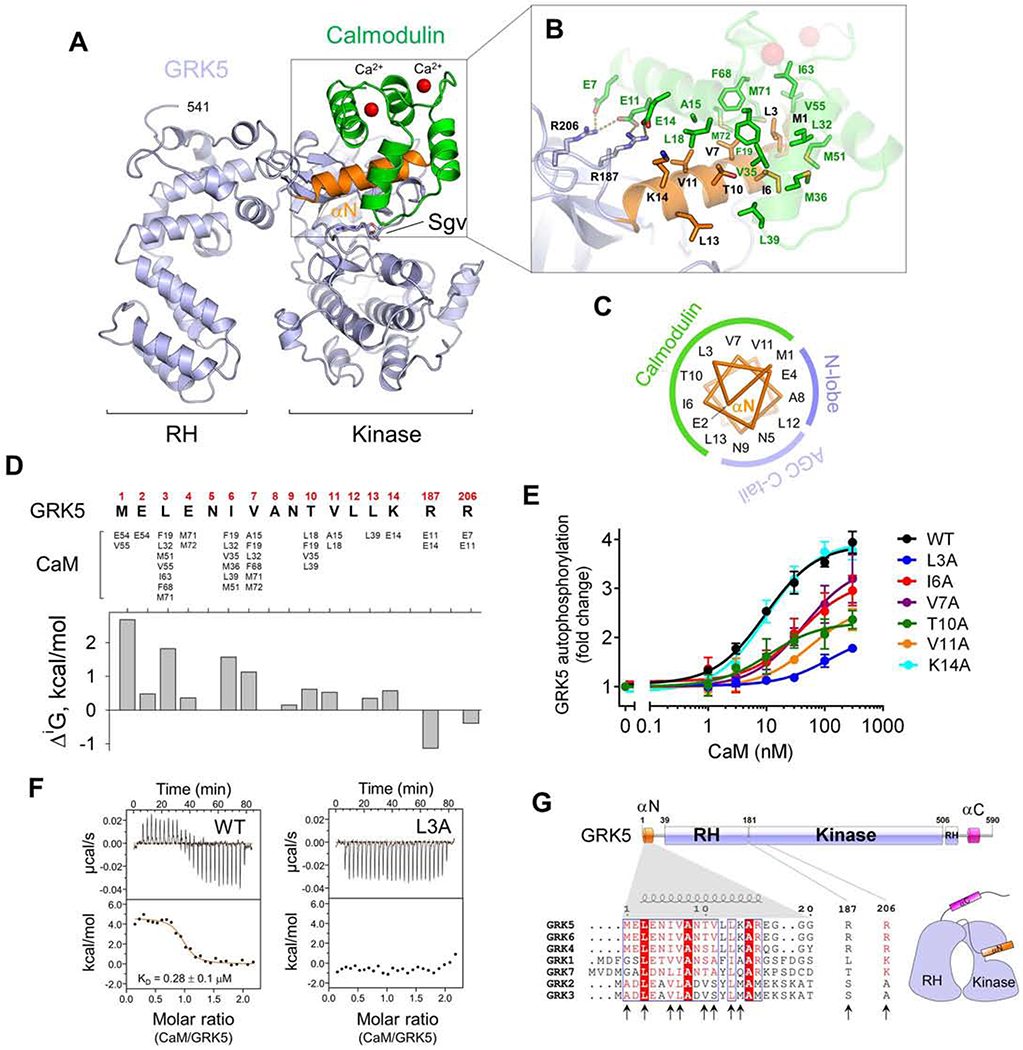
(A) Overall view of 2.0 Å crystal structure of GRK5/sangivamycin bound to Ca2+/CaM. GRK5 1542 (light blue) and CaM 4-73 (green) are resolved in the structure.
(B) Details of the GRK5-CaM interface.
(C) Helical-wheel analysis identifying a hydrophobic face of GRK5 αN in CaM binding while the opposing face forms intramolecular contacts with the GRK5 N-lobe and AGC kinase C-tail.
(D) Solvation energy (ΔiG) analysis identifies key residues that contribute to the GRK5-CaM interface.
(E) Key residues that form interactions with CaM in GRK5 αN were mutated to alanine and assessed for CaM-induced GRK5 autophosphorylation. Data are presented as mean ± SEM, n=3.
(F) Top, ITC raw heat of CaM binding to WT and L3A mutant GRK5. Bottom, binding isotherms. The fitted isotherm for CaM binding to GRK5 WT yielded n = 0.90 ± 0.06 (mol), KD = 278 ± 86 nM, ΔS = 46 ± 3 cal/mol·K, ΔH = 4.3 ± 0.6 kcal/mol.
(G) Domain architecture of GRK5 and sequence alignment of the GRK N-terminal regions plus residues R187/R206 lining the polar contact with CaM. Identical residues are boxed in red, and residues showing similarity are in red and grouped in a blue frame. Arrows indicate key residues in GRK5 that interact with CaM.
Table 1.
Crystallographic data collection and refinement statistics
| Data collection statistics | CaM/GRK5/sangivamycin |
|---|---|
| X-ray source | MicroMax-007 HF |
| X-ray detector | PILATUS3 R 200K |
| Wavelength (Å) | 1.54 |
| Space group | P212121 |
| Resolution (Å) | 50.00 - 1.96 (1.99 - 1.96) a |
| Unit cell dimensions (Å) | a = 69.875, b = 83.061, c = 137.521 |
| Angles (°) | α, β, γ = 90.0 |
| Solvent content (%) / VM (Å3/Da.) | 48.9 / 2.4 |
| Molecules per asymmetric unit | 1 |
| Total reflections | 1,999,159 (10,099) |
| Unique reflections | 51,717 (3,660) |
| Multiplicity | 3.9 (2.8) |
| Completeness (%) | 88.48 (63.68) |
| Mean I/sigma(I) | 14.03 (2.00) |
| Wilson B-factor (Å2) | 23.97 |
| Rmerge b | 0.1077 (0.5964) |
| Rmeas c | 0.1234 (0.7208) |
| Rpim d | 0.05851 (0.3934) |
| CC1/2 e | 0.994 (0.672) |
| Refinement statistics | |
| Resolution (Å) | 49.01 - 1.96 (2.028 - 1.96) |
| Number of Reflections | 51,637 (3,659) |
| Reflections used for Rfree f | 1998 (141) |
| CC* e | 0.998 (0.897) |
| Rwork | 0.1954 (0.3109) |
| Rfree f | 0.2296 (0.3266) |
| CCwork e | 0.956 (0.782) |
| CCfree e | 0.948 (0.769) |
| Number of non-hydrogen atoms | 5504 |
| - macromolecules | 4949 |
| - ligand | 35 |
| - water | 520 |
| Protein residues | 615 |
| Root mean square deviations from ideal geometry | |
| bond length (Å) | 0.008 |
| angles (°) | 1.21 |
| Ramachandran plot and MolProbity validation g | |
| Residues in favored region (%) | 96.87 |
| Residues in allowed region (%) | 3.13 |
| Residues as outliers (%) | 0.00 |
| Clashscore | 4.74 |
| Overall score | 1.43 |
| Average B-factor (Å2) | |
| Model (all atoms) | 35.47 |
| Protein | 35.36 |
| Ligand | 22.04 |
| Water | 37.44 |
| Number of TLS groups | 11 |
| PDB code | 6PJX |
Statistics for the highest-resolution shell are shown in parentheses.
Simple merging R factor for the multiple observations
Redundancy-independent merging R factor
Precision-indicating merging R factor
The CC1/2 is the correlation coefficient between two randomly selected half-datasets; CC* is a statistic metric for assessing the effective resolution limits of data and quality of unmerged data in the context of a refined model; CCwork and CCfree are the standard and cross-validated correlations of the observed intensities to the refined model-based intensities, for the work and test sets respectively.
Rfree value is calculated using the small subset of randomly selected reflections (test-set) that are set aside prior to refinement and not used in the refinement of the structural model.
From Chen et al. 2010a
A striking feature of the CaM/GRK5 structure is the GRK5 αN-helix at the core of the binding interface with CaM (orange in Figure 1A). While αN is typically disordered in GRK crystal structures, it folds as a 5-turn α-helix that packs near the kinase domain N-lobe in the CaM/GRK5 structure, next to the catalytic cleft. The interaction of αN with CaM is non-polar and engages a hydrophobic surface of the helix that forms a continuous interface with a large concave hydrophobic pocket of the CaM N-domain (Figure 1B). Polar contacts outside αN occur between R187 and R206 from the GRK5 N-lobe and CaM residues E7, E11 and E14. We did not observe densities for the CaM C-domain (residues 74-148) or the GRK5 C-terminal region (residues 542-590), although crystallization of full-length GRK5 and CaM was confirmed (not shown), suggesting that the missing regions are highly dynamic. The total buried surface area of the CaM/GRK5 interface is 742 Å2.
αN interacts with CaM on one side whereas the opposite side aligns against a pocket formed by the AGC kinase C-tail (residues 469-478), N-lobe (Y189, R190, Q204 and M211) and kinase domain hinge region (I265 and N267). Thus, αN is almost completely buried inside 2 cavities formed by CaM and the kinase domain as illustrated by a helical-wheel diagram (Figure 1C).
The conformation of GRK5 in our crystal structure demonstrates similarity with the crystal structure of GRK6 bound to sangivamycin, the only other structure of a GRK with αN fully resolved (Figure S1E) (Boguth et al., 2010). In the GRK6 structure, the N-terminus is also folded as an α-helix and packs near the kinase N-lobe. However, the GRK6 αN was ordered owing to crystal lattice contacts with a symmetry-related molecule of GRK6, raising the question of its physiological relevance. The CaM/GRK5 crystal structure validates helical formation of the N-terminus and also supports a physiological role of this conformation in Ca2+-dependent regulation of GRKs.
To analyze the crystallographic interface with CaM, the solvation energy (ΔiG) was calculated for relevant GRK5 residues (Figure 1D). A large ΔiG was indicated for R187 located on the N-lobe and αN residues M1, L3, I6, V7, T10, V11 and K14 that contact CaM. To assess the contribution of individual residues, GRK5 residues were mutated to alanine and CaM-stimulated autophosphorylation of GRK5 was used as a functional readout of CaM binding. Autophosphorylation was reduced in all mutants except K14A (Figure 1E), in agreement with the suggested role of these residues in CaM binding. The contribution of R187 and R206, which form polar contacts with CaM, could not be assessed since mutagenesis was detrimental to autophosphorylation. This is likely due to protein misfolding leading to a loss of catalytic function, as indicated by reduced solubility and autophosphorylation when expressed in cells (Figure S1F). Mutation of L3, which forms a core interaction with CaM in the crystal structure, demonstrated a particularly dramatic effect on CaM-stimulated GRK5 autophosphorylation as well as a loss of binding to CaM as detected by isothermal titration calorimetry (ITC) (Figure 1F).
All GRKs feature αN at the amino terminus, though not all GRK5 residues that contact CaM are well conserved (Figure 1G). While binding of GRK5 αN (residues 1-17) with the CaM N-domain (CaM N) resembles recoverin interaction with GRK1 αN (Ames et al., 2006; Higgins et al., 2006), this was not anticipated since prior studies found that a protein fragment containing residues 20-39 could bind CaM (Levay et al., 1998; Pronin et al., 1997). While this region can be classified as a CaM-binding domain based on physicochemical characteristics, its deletion in GRK5 ΔN35 mutant did not change GRK5 binding to CaM-Sepharose compared to the ΔN17 mutant (Figure 2A). Moreover, structural analysis showed that residues 20-39 maintain extensive intramolecular contacts with the RH domain as shown by the large buried surface area for many of these residues (Figure S1G), likely precluding a role in CaM binding.
Figure 2. Full-length model of GRK5-CaM complex.

(A) GRK5 binding to CaM-Sepharose was quantified by densitometry. Data are presented as mean ± SD, n=3. Cartoon of the CaM/GRK5 complex shows putative interaction between GRK5 C-terminus and CaM C (framed).
(B) GRK5 domain schematic and helical wheel of GRK5 region 548-559 that has a propensity to form an amphipathic α-helix (αC) (hydrophobic face in yellow). Arrows designate positions of GRK5 αC residues that have a critical role in CaM interaction.
(C) Full-length model of CaM/GRK5 complex. Modeled interaction of the CaM C/GRK5 αC interaction is framed.
(D) Validation of bipartite binding interface in CaM/GRK5 complex using HDX-MS. Blue and pink color-coding maps regions with HDX rate decrease and increase, respectively, in the complex as compared to individual proteins. Grey color-coding maps regions of either no change in HDX rate or not covered in the analysis (see also Figure S3).
See also Figures S2 and S3 and Movie S1.
Full-length Model of GRK5/CaM Interaction Features Bipartite CaM Coupling to GRK5
While electron density for the GRK5 C-terminus is missing in the CaM/GRK5 crystal structure, previous biochemical studies suggested that this region can bind CaM (Levay et al., 1998). Thus, we next examined GRK5 regions required for CaM binding using a direct binding assay on CaM-Sepharose. These studies revealed an ~90% decrease in GRK5 binding to CaM upon truncation of the C-terminal 49 residues (ΔC49) while truncation of αN (ΔN17) reduced CaM binding by only ~20% (Figure 2A). The contribution of the GRK5 C-terminus to CaM binding was further validated by binding on Strep-Tactin beads (Figure S2A) and by SEC where WT GRK5 and CaM migrated as a single homogeneous peak while GRK5 ΔC49 did not form a stable complex with CaM (Figure S2B). Additional evidence of bipartite CaM binding to GRK5 was obtained from limited proteolysis where CaM protects against trypsin cleavage of ~5 kDa C-terminal and ~2 kDa N-terminal fragments of GRK5 (Figure S2C and S2D). Thus, these data demonstrate that both GRK5 terminal regions contribute to CaM binding.
While GRK5 ΔC (ΔC49) has significantly reduced binding to CaM (Figure 2A), this was not observed in ITC binding studies, with isotherms for CaM binding to GRK5 ΔC (Figure S2E) and WT GRK5 (Figure 1F) being very similar. In contrast, N-terminal truncation (GRK5 ΔN and GRK5 ΔN/ΔC) caused a loss in the binding thermodynamics (Figure S2E), similar to the effect of L3A mutation (Figure 1F). We speculate that the heat generated in the ITC studies is likely due to the large conformational changes that result from CaM binding to the GRK5 N-terminus. Without effect on conformation, binding via the C-terminal site might not generate significant changes in enthalpy that would be detected by ITC.
We next sought to better define the CaM binding site within the GRK5 C-terminal region. Secondary structure analysis predicts that residues 548-559 possess a high propensity to adopt an amphipathic α-helix with basic residues on one side and hydrophobic residues on the other, typical for CaM-binding regions (Figure 2B). This helix can be classified as an IQ-like CaM binding motif based on the distance between the anchor residues (Figure S2F) (Yap et al., 2000). The circular dichroism spectrum of a GRK5 peptide containing residues 546-562 confirmed that it forms an α-helix in a hydrophobic environment (Figure S2G). Thus, the ability of this region to form an α-helix would likely be enhanced by binding to the hydrophobic pocket formed by CaM EF-hands.
Guided by biochemical and structural analysis, we built a full-length model of the CaM/GRK5 complex (Figure 2C) with CaM C-domain (CaM C) interaction with the GRK5 C-terminus modeled into the CaM/GRK5 crystal structure. The hydrophobic side of the GRK5 αC-helix (αC, residues 548-559) is comprised of non-polar residues L550, L551, L554 and F555 (LLLF motif) (Figure 2B) and was aligned in this model against a hydrophobic cavity formed by EF-3 and EF-4 of CaM C. The importance of the LLLF motif was shown using pull-down assays where alanine mutation (GRK5 4A) reduced GRK5 binding to CaM (Figures 2A and S2A), similar to the effect of C-terminus truncation (ΔC49). The final model of the CaM/GRK5 complex features the hydrophobic pockets of two CaM domains associated with GRK5 αN and αC separated by ~30 Å in an antiparallel arrangement.
To test our model and better understand in-solution dynamics of CaM binding to GRK5, we employed hydrogen-deuterium exchange mass spectrometry (HDX-MS) (Figures 2D and S3, Tables S2 and S3). Among regions with decreased rates of deuterium uptake (blue in Figure 2D), HDX analysis mapped the crystallographic interface of the N-terminal regions of GRK5 and CaM (lower box) as well as the modeled CaM C interaction with αC (upper box), further supporting the bipartite architecture of the CaM/GRK5 complex. In contrast, several regions in the RH and kinase domains of GRK5 had enhanced HDX rates in the complex (pink in Figure 2D), suggesting that they become more dynamic and/or solvent-exposed upon GRK5 interaction with CaM.
We also performed molecular dynamics (MD) simulations to evaluate the stability of protein contacts in the full-length model of CaM/GRK5 (Figure S2H). The N- and C-terminal interactions between CaM and GRK5 were stable and did not dissociate during simulations as indicated by the low RMSDs of the GRK5 αN/CaM N and GRK5 αC/CaM C regions. However, the GRK5 αC/CaM C connects to the complex core through flexible linkers and adopts a broader range of orientations than the GRK5 αN/CaM N region, which is stabilized on the kinase domain surface (Movie S1). Although CaM C remains bound to GRK5 αC in MD simulations, the observed dynamics of this region may be why this contact was not resolved in the crystal structure.
Insight into GRK5 Conformational Dynamics and Kinase Activation Induced by CaM Binding
Calcium stimulates proteins such as CaM and recoverin to regulate GPCR signaling via their ability to directly bind to GRKs and inhibit GRK-mediated phosphorylation of GPCRs (Komolov et al., 2009; Pronin et al., 1997). However, we also observed that CaM increased GRK5-mediated phosphorylation of α-synuclein (Figure S4A) and GRK5 autophosphorylation (Figure S4B), corroborating previous reports (Pronin et al., 1997, 2000). Such results contradict the classical view of CaM as an inhibitor of GRKs. To address this apparent dichotomy, we further characterized the structural consequences of CaM binding to GRK5.
The CaM/GRK5 co-crystal structure captures GRK5 in an active conformation. Structural alignment of GRK5 alone (PDB: 4TNB) and in complex with CaM reveals a 15° closure of the kinase active site upon CaM binding, yielding tighter packing of the N- and C-lobes for improved coordination of ATP (Figure 3A). Also, 22° rotation of the RH domain relative to the catalytic domain leads to disruption of the “ionic lock” that maintains this contact in the basal state (Komolov et al., 2017; Yao et al., 2017) and an ~5 Å further displacement of the RH domain from the catalytic domain (~10 Å total). In addition, the C-tail of the catalytic domain is completely ordered in the complex and occupies a new position near the catalytic domain N-lobe. Notably, formation of the AST loop (residues 468-473) facilitates inter-lobe and αN contacts, similar to AST loop packing in GRK6/sangivamycin (Figure S4C) (Boguth et al., 2010). Overall, GRK5 in complex with CaM displays features characteristic of the active state.
Figure 3. CaM binding facilitates GRK5 interdomain dynamics resulting in kinase activation.
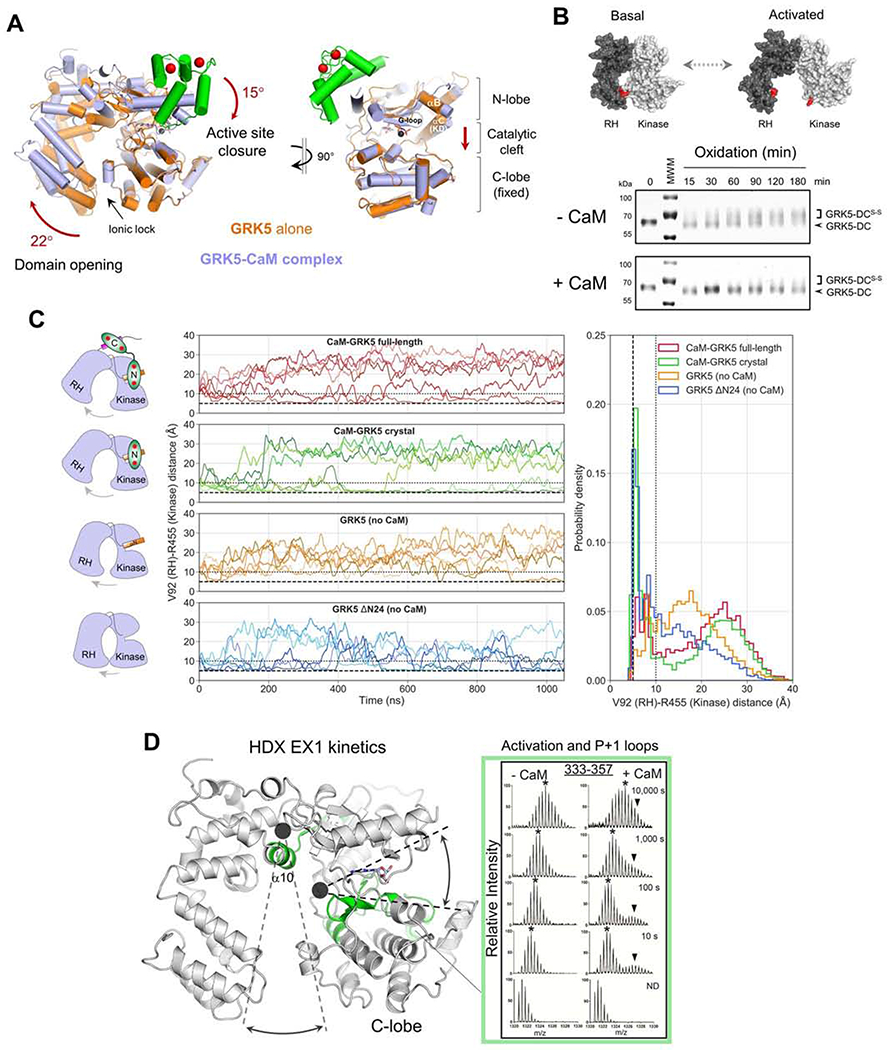
(A) Structural alignment of GRK5 with CaM (light blue, PDB: 6PJX) or GRK5 alone (orange, PDB: 4TNB). CaM is shown in green while calcium is red.
(B) Time course of interdomain disulfide bond formation in GRK5-DC. Positions of cross-linked (GRK5-DCS-S) and non-cross-linked (GRK5-DC) species are indicated.
(C) Effect of CaM association on GRK5 interdomain dynamics as revealed by MD simulations. RH domain opening is indicated as an increase in the interdomain distance between the α-carbons of V92 (RH domain) and R455 (kinase domain). Four different conditions were simulated (Table S1): (1) full-length model of the CaM/GRK5 complex (red), (2) crystallographic CaM/GRK5 complex (green), (3) crystallographic GRK5 without CaM (orange), and (4) crystallographic GRK5 without CaM and αN (blue). In all simulations, GRK5 starts from its conformation in the CaM/GRK5 crystal with an interdomain distance of ~10 Å (black dotted line). An interdomain distance of ~5 Å (black dashed line) is observed in the GRK5/sangivamycin crystal structure (PDB: 4TNB). A smoothed trace for each simulation is shown at left. On the right, the probability density of the V92-R455 distance is shown for each condition. Each histogram is based on six independent production simulations (1 μs each) with a snapshot frequency of 1 ns.
(D) GRK5 regions displaying mixed EX1/EX2 kinetics (green) in HDX-MS analysis of CaM/GRK5 complex. Mass spectra of non-deuterium (ND) and 10, 100, 1,000 and 10,000 s of deuterium exposure are shown for selected GRK5 peptide (333-357) revealing mixed EX1/EX2 kinetics. The single isotopic distributions (EX2 kinetics, marked by *) was indicated for mass spectrum of GRK5 alone (−CaM) while two isotopic distributions with mixed EX1/EX2 kinetics (▼ and *, respectively) were observed in GRK5 upon CaM binding (+CaM). Two hinges for GRK5 domain dynamics are shown as black dots with dashed lines mapping domain movement within the hinge regions.
Next, we sought to confirm the RH and kinase domain separation using a domain proximity assay that we developed to monitor GRK5 dynamics upon receptor docking (Komolov et al., 2017). A reduced electrophoretic mobility of a GRK5 double-cysteine mutant (GRK5-DC) under oxidative conditions indicates formation of a disulfide bond between two cysteines located at the contact site of the RH and kinase domains. In the basal state, the RH and kinase domains are bound via the ionic lock resulting in an upward shift of the GRK5-DC band, supporting domain cross-linking (GRK5-DCS-S) (Figure 3B). CaM effectively inhibits the upward shift of GRK5-DC supporting the dissociated domain arrangement in GRK5 observed in the crystal structure with CaM (Figure 3A).
To gain further insight into CaM effects on GRK5 interdomain dynamics, we employed MD simulations (Figure 3C and Table S1). Since RH domain opening allosterically regulates kinase active site closure, we measured interdomain distances at the GRK5 ionic lock site to follow RH domain opening and GRK5 activation. Throughout the simulations, the RH and kinase domains occasionally separated, swinging apart transiently between interdomain distances of 5 Å (basal compact conformation) and 30 Å (open elongated conformation). CaM binding to GRK5 (CaM-GRK5 full-length and crystal) substantially increased the average interdomain distance and the fraction of time GRK5 spends in an elongated conformation (Figure 3C, red and green traces; Movie S1). Modeling in the C-terminal contact (GRK5 αC/CaM C) showed no apparent impact on GRK5 interdomain dynamics suggesting that the interaction between GRK5 αN and CaM N might be essential for the observed dynamics. Indeed, removal of CaM and αN from the simulation diminished the sampling of the open elongated state (Figure 3C, blue traces). Notably, GRK5 in the absence of CaM can still sample an open conformation, albeit with average interdomain distances reduced to ~17 Å (Figure 3C, orange traces). Although GRK5 is not bound to CaM, the N-terminus still maintains an a-helical fold (Figure S4D, orange RMSD αN trace), which can support RH domain opening upon removal of CaM. Taken together, computational studies point out a key role of GRK5 αN in supporting the active conformation of GRK5.
The motion of GRK5 domains upon CaM binding centers around two main hinges for a swinging motion of the RH and kinase domains (Figure 3D, top black circle) and kinase active site closure (Figure 3D, right black circle). Peptides from these two hinge areas displayed unusual HDX mass spectra upon CaM binding featuring mixed EX1/EX2 kinetics (a bimodal isotopic distribution) (Figures 3D inset and S4E). The EX1 kinetics map protein regions that undergo substantial, cooperative conformational changes when the unfolding rate is much faster than the refolding rate and all of the amide hydrogens exchange with deuterium before refolding occurs (Engen et al., 2013). This was not observed in GRK5 alone which showed only EX2 kinetics (a single isotopic distribution). Mixed EX1/EX2 kinetics of peptide 499-515, comprising helix α10 and the AGC kinase “hydrophobic motif”, can be linked to the CaM-induced partial unwinding of the GRK5 region that facilitates the swinging motion of the RH and kinase domains (RH domain opening). A second area with mixed EX1/EX2 kinetics near the kinase domain hinge (peptides 297-318, 319-332 and 333-357) includes GRK5 regions that form the activation, catalytic and magnesium positioning loops of the kinase domain C-lobe. Structural perturbations within this area are expected to favor kinase active site closure and, in turn, GRK5 activation. Thus, HDX-MS analysis supports fast “relaxation” of both hinge areas, promoting global changes in GRK5 conformation and kinase activation in the CaM/GRK5 complex.
GRK5 Substrate Motif Profiling Reveals Preferred Amino Acid Sequence for Effective Phosphorylation
The functional role of the active conformation that GRK5 adopts in the crystal structure with CaM was supported by the enhanced rate of α-synuclein phosphorylation and GRK5 autophosphorylation observed in the presence of Ca2+/CaM (Figures S4A and S4B). To further investigate the functional relationship between CaM binding and phosphorylation of nonreceptor substrates by GRK5, we performed substrate motif profiling using a positional scanning peptide array (PSPA) screen (Miller and Turk, 2016). This screen helps to identify key specificity-determining residues enabling a protein kinase to target specific sites of phosphorylation. A library of 198 synthetic peptides was used to systematically substitute nine positions flanking a phosphorylation site (five residues N-terminal, −5 to −1, and four residues C-terminal, +1 to +4, from a Ser or Thr assigned as 0) with each of the 20 amino-acid residues as well as phosphothreonine (pThr) and phosphotyrosine (pTyr). The data are presented on a heat map to reveal positively (red) and negatively (blue) selected residues at each position in the presence (Figure 4A) or absence (Figure S5A) of Ca2+/CaM. CaM enhanced GRK5 activity on phosphorylation of arrayed peptides, albeit without a clear effect on specificity. This agrees with structural information showing no obvious structural rearrangements induced by CaM within the substrate docking surface on the GRK5 C-lobe. Acidic residues were primarily preferred at the +1 and +3 positions, aromatic residues at the −2 position, and pThr/pTyr and acidic residues at the −4 and +4 positions. GRK5 lacks selectivity for Arg at the −3 position, which is unusual for an AGC kinase (though not necessarily for a GRK) (Onorato et al., 1991). While no specific residue was clearly favored at any position, a consistent contribution of different classes of amino acids can be reflected in a GRK5 consensus motif shown in Figure 4B, some features of which are reflected in the phosphorylation site sequences of many known GRK5 substrates (Figure S5B).
Figure 4. GRK5 substrate specificity profiling and enzymatic analysis identify key elements critical for CaM-mediated activation and substrate targeting.
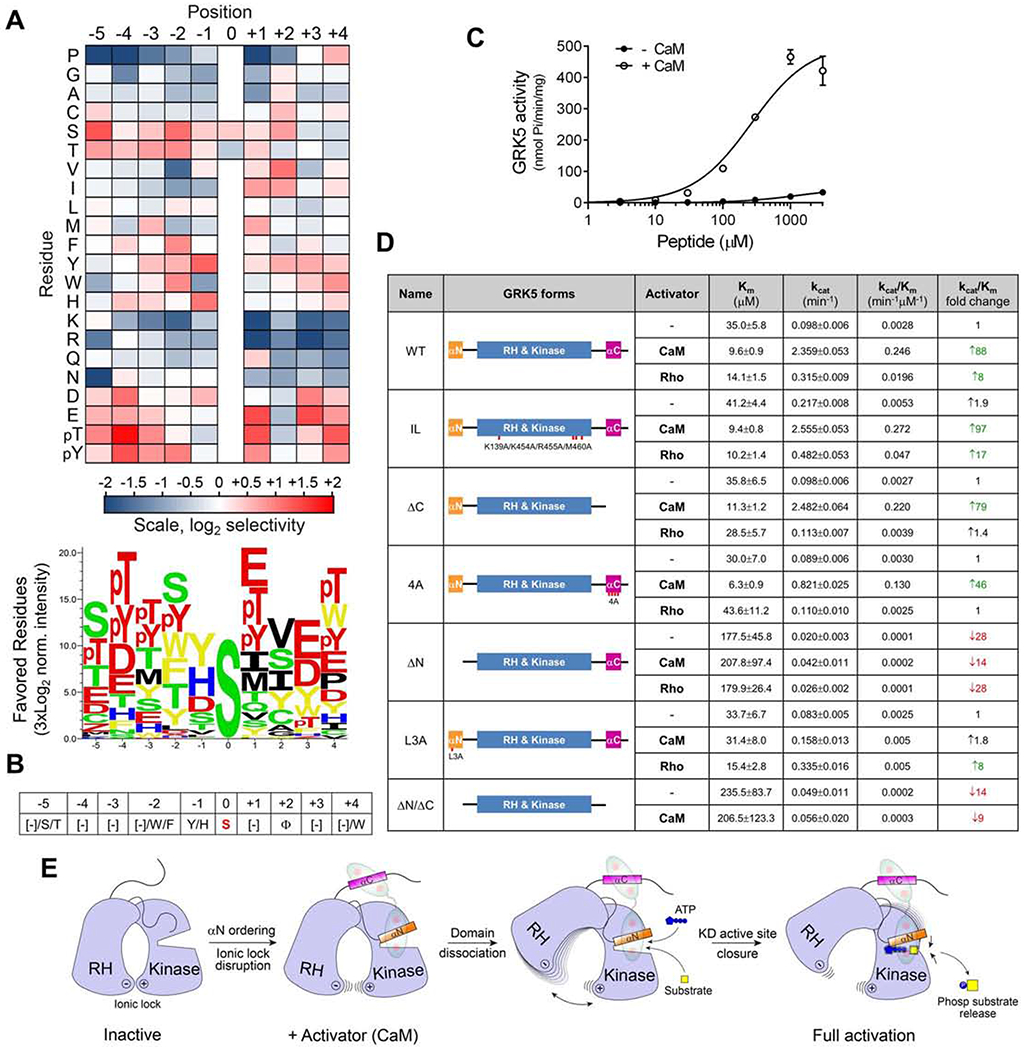
(A) Substrate specificity profiling of GRK5 bound to CaM (+CaM). Quantified PSPA spot intensities were normalized to an average value of 1 at each position within the peptide. Log2 transformed data are depicted as heat maps showing positively (red) and negatively (blue) selected residues by position. Data are the mean of two separate experiments. Sequence logo below the heat map shows positively selected residues from PSPA. See also Figure S5A.
(B) Consensus motif for GRK5 phosphorylation as revealed by PSPA analysis.
(C) Kinetic plots for AEMWYSEVEEARRR phosphorylation by GRK5 with or without CaM. Data are presented as mean ± SEM, n=3.
(D) Enzymatic parameters for WT or mutant GRK5-mediated phosphorylation of AEMWYSEVEEARRR at different ATP concentrations under basal (−), CaM-activated (CaM) or Rho-activated (Rho) conditions. The data are from three experiments and fitted to Michaelis-Menten kinetics. Data are presented as mean ± SD, n>3. See also Figure S5D.
(E) Cartoon for GRK5 activation by CaM illustrating possible steps in the progression of GRK5 from an initial inactive conformation to a fully-active state.
See also Figure S5.
Interestingly, surface charge distribution maps a basic character of the GRK5 catalytic cleft in contrast to the acidic active sites in PKA and PKB (Figure S5C). Different electrostatic characteristics of the catalytic sites agree with their distinct substrate preferences. In contrast to PKA and PKB, Arg and Lys were generally disfavored in GRK5, whereas negatively charged residues (including pThr and pTyr) or phospho-acceptor residues (Ser and Thr) were commonly preferred. This suggests that GRK5 may target a specific set of substrates, in part, due to specific substrate sequence preferences.
Another aspect of the preference of GRK5 for negatively charged residues is the presence of multiple phosphorylation sites at the C-terminus of GPCRs, which are often closely clustered and, therefore, the phosphorylation of one site might positively affect the phosphorylation of additional sites by GRKs. This could play a role in GRK-mediated phosphorylation of GPCRs and subsequent recruitment of arrestins, which require multiple phosphorylation sites for high affinity binding (Mendez et al., 2000; Vishnivetskiy et al., 2007).
Enzymatic Analysis Identifies Key Steps in GRK5 Activation by CaM
Based on the PSPA screen (Figure 4A), we designed the optimized peptide substrate AEMWYSEVEEA. GRK5 robustly phosphorylated this peptide in the presence of CaM with a Km = 265 ± 52 μM and Vmax = 509 ± 28 nmol Pi/min/mg (Figure 4C). We then measured the kcat and Km values for ATP of different GRK5 constructs to assess their catalytic properties in basal (no activator) and stimulated states (Figures 4D and S5D). Comparing the catalytic efficiency (kcat/Km) of WT GRK5 for ATP revealed an 88-fold increase in kcat/Km in the presence of CaM. This resulted from an ~3.6-fold decrease in Km and ~24-fold increase in kcat, suggesting that CaM promotes tighter binding of ATP and more efficient catalysis. We also evaluated an ionic lock mutant (GRK5 IL) that disrupts the RH/kinase domain contact and pre-activates GRK5 (Komolov et al., 2017). As expected, this mutant had an elevated basal activity (1.9-fold kcat/Km increase) but remained similarly responsive to CaM stimulation as WT (97-fold increase in kcat/Km). This suggests that the ionic lock disruption induced by CaM binding to GRK5 is likely stable in the complex.
We also assessed the contribution of each contact between CaM and GRK5 to kinase activation. Disruption of the C-terminal contact with CaM was evaluated using the GRK5 AC and 4A mutants and did not appreciably reduce CaM activation of GRK5 with a 79-fold and 46-fold increase in kcat/Km, respectively (Figure 4D). In striking contrast, GRK5 lacking the N-terminal CaM binding region (GRK5 ΔN) showed a dramatic loss of both basal and CaM-stimulated activity compared to GRK5 WT with a 28-fold and 14-fold decrease in kcat/Km, respectively (Figure 4D). This further supports the importance of the αN for GRK5 activity in both basal and CaM-stimulated states.
While an essential role for the N-terminus in GRK-mediated GPCR phosphorylation was previously demonstrated (Huang et al., 2009; Noble et al., 2003; Pao et al., 2009), we found that the N-terminus also plays a key role in the conformational dynamics observed when in complex with CaM. Though the N-terminus is disordered in the basal state, it folds as an α-helix via interaction with CaM and the kinase domain. This triggers a stepwise transition of GRK5 from an inactive to fully-active state (Figure 4E). αN integrates the kinase domain N-lobe, AGC C-tail and hinge regions via a series of intramolecular contacts. Consequently, as C-lobe contact with the N-lobe is strengthened, C-lobe connection to the RH domain (“ionic lock”) is weakened. Subsequent disruption of the structural constraints imposed by the ionic lock on GRK5 interdomain dynamics favors a fully-active enzyme conformation by facilitating RH domain dissociation and kinase active site closure. Thus, structural perturbations in GRK5 caused by αN folding help to remove the structural constraints maintaining the enzyme in an inactive state and thereby induce activation. The rapid conformational relaxation of two hinge regions also supports CaM-induced GRK5 structural dynamics (Figure 3D). Though the C-terminal contact between CaM and GRK5 seems dispensable for activation, it might enhance the N-terminal interaction through anchoring of CaM to GRK5. This notion is supported by the reduced efficacy of CaM to stimulate GRK5 C-terminal mutants (ΔC and 4A) (Figure 4D).
Structural Similarity between Mechanisms of GRK5 Activation by CaM and GPCRs
Previous studies have shown that GPCRs also activate GRKs (Chen et al., 1993; Palczewski et al., 1991). We found that light-activated rhodopsin (Rho) effectively enhances GRK5-mediated peptide phosphorylation (8-fold increase in kcat/Km for ATP), albeit with a potency that was 11-fold lower than CaM stimulation of GRK5 (Figure 4D). This lower potency of Rho to activate GRK5 was largely a result of reduced kcat measured for Rho activation. Thus, under these conditions, CaM is substantially more effective at stimulating GRK5 compared to Rho.
Unlike CaM, Rho was unable to stimulate the GRK5 C-terminal mutants (ΔC and 4A), most likely due to their membrane binding deficiency (Figures 4D and S5D). CaM and Rho also showed differences in activation of the GRK5 L3A mutant. While CaM failed to stimulate GRK5 L3A, Rho effectively activated GRK5 L3A (8-fold increase in kcat/Km) (Figure 4D). Thus, while L3 is essential for activation by CaM, it seems dispensable for activation by Rho. In contrast to the effect of L3A mutation, entire deletion of αN (ΔN) resulted in equal deficiency of Rho and CaM to stimulate GRK5 (Figure 4D). Interestingly, while the L3A mutation had no effect on Rho phosphorylation, the mutant exhibited a marked defect in phosphorylating the β2AR (Figure 5A). This might reflect different requirements for αN residues for GRK5 stimulation by different activators.
Figure 5. CaM and GPCRs can activate GRK5 via a similar mechanism of αN docking into hydrophobic pocket of activators.

(A) Rhodopsin (left panel) and β2AR (right panel) phosphorylation by GRK5 WT and αN mutants (L3A and ΔN). Data are presented as mean ± SEM, n=3.
(B) Hydrophobic surface representation of CaM in Ca2+-free and Ca2+-bound, and β2AR in inactive and active conformations. Calcium triggers hydrophobic pocket opening in CaM N that accommodates GRK5 αN (blue) whereas GPCR activation by agonist induces opening of the hydrophobic core utilized by α5 of Gαs (green).
(C) GRK5 N-terminal fragment can adopt a helical conformation. CD spectra of GRK5 N-terminal peptide 2-19 at the indicated trifluoroethanol (TFE) concentrations are shown.
(D) Equilibrium between inactive, basal and active forms of GRK5 can be shifted by αN stabilization.
To understand whether the functional deficiency of two activators to stimulate GRK5 ΔN originates from similar structural mechanisms controlling GRK5 activation, we compared the characteristics of the GRK5 αN docking site on both activators (Figure 5B). αN is hydrophobic which makes its association with the hydrophobic surface of the activators energetically favorable. Calcium triggers opening of the hydrophobic concave pocket in CaM N that accommodates GRK5 αN whereas GPCR activation by agonist induces opening of a hydrophobic groove near the cytoplasmic side of TM5/TM6 utilized by the α5-helix of a Gα-subunit (Rasmussen et al., 2011) and possibly by the αN-helix of a GRK (Figure 5B). Thus, a specific requirement for a hydrophobic environment for αN folding and kinase activation suggests structural parallels for the mechanisms of GRK5 activation by CaM and GPCRs.
Thus, our data support a common mechanism of GRK5 activation via engagement of hydrophobic concave surfaces of activators such as CaM and GPCRs to stabilize αN folding. However, we also noted that truncation of the GRK5 N-terminus results not only in severe defects in activation but also a large reduction in basal activity rendering the kinase nearly inactive (28-fold decrease in kcat/Km of ΔN, Figure 4D). This suggests that αN is also important for GRK5 activity even in the absence of activators. Interestingly, a GRK5 N-terminal peptide (residues 2-19) has 20% helical content in aqueous solution and can reach up to 100% helicity in the presence of trifluoroethanol (TFE), indicating that the GRK5 N-terminus has some helical character independent of the hydrophobic environment provided by activators (Figure 5C). Thus, if the basal activity of GRK5 (and likely other GRKs) is determined by this spontaneous αN formation, any factors that stabilize (like CaM and GPCRs) or destabilize αN could shift the equilibrium between the active and inactive state (Figure 5D).
Distinct Role of CaM Domains in GRK5 Activation, Membrane Binding and Substrate Targeting
Given the distinct stimulatory and inhibitory roles that CaM plays in phosphorylation of different GRK5 substrates, we next analyzed the functional response of GRK5 over a range of CaM concentrations. Increasing CaM inhibited GRK5-mediated phosphorylation of GPCRs (β2AR and rhodopsin) as well as phospholipid binding, while it activated GRK5 autophosphorylation and α-synuclein and peptide phosphorylation (Figure 6A). It is evident that similar concentrations of CaM control both the “on” and “off” effects on GRK5. This suggests that CaM might function as a switch to regulate GRK5 localization and activity in cells.
Figure 6. Distinct role of CaM domains in modulation of GRK5 function.

(A) GRK5 activity as a percent of basal activity (no CaM) for inhibitory effects and a percent of maximum activity (plus CaM) for activating effects (mean ± SD, n≥3). Data are presented as mean ± SD, n≥3.
(B) Surface representation of the full-length CaM/GRK5 model shown in Figure 2D. CaM binding (green) overlaps with the membrane-binding (blue dash) and receptor-binding (red dash) regions of GRK5.
(C) A cartoon of CaM and EF-hand mutants CaM 12 and CaM 34 with calcium shown as red dots.
(D) Peptide phosphorylation (top left), PC membrane binding (top right) and β2AR phosphorylation (bottom) by GRK5 with increasing concentrations of CaM, CaM 12 and CaM 34. GRK5 activity is expressed as the fold change of basal activity (no CaM). Data are presented as mean ± SEM, n=3.
See also Figure S6.
The dual effect of CaM is also reasonable from a structural perspective (Figure 6B). Prior reports found that GRK5 αC, engaged in CaM C interaction, contributes to both membrane binding (Thiyagarajan et al., 2004) and receptor interaction (Komolov et al., 2017). CaM binding largely overlaps with these regions suggesting that CaM C exerts steric hindrance on GRK5 binding to the membrane and receptor. CaM N also overlaps with receptor binding surfaces although it additionally stabilizes the active conformation of GRK5 via contact to αN. While CaM binding engages a large area on GRK5, the catalytic site remains accessible. This illustrates how activated GRK5 in complex with CaM can still target different substrates for phosphorylation despite inhibiting GPCR phosphorylation.
Next, we selectively mutated the individual CaM domains to verify their specific role. We prepared two CaM alanine mutants: one containing substitutions in the EF-1 and EF-2 hands (D22A, D58A) to abolish Ca2+ binding to the N-domain (CaM 12 mutant) and the other with substitutions in the EF-3 and EF-4 hands (D95A, D131A) to abolish Ca2+ binding to the C-domain (CaM 34 mutant) (Figure 6C) (Piazza et al., 2017). Impaired calcium coordination in the CaM EF-hand mutants was confirmed using a CaM mobility shift assay where the mutants had a reduced Ca2+-dependent shift compared to CaM WT (Figure S6A). Surface plasmon resonance (SPR) analysis also indicated a reduced binding capacity of the EF-hand mutants for GRK5 (Figure S6B). GRK5 bound to WT CaM with a KD = 5.9 ± 0.7 nM while binding to CaM with only a functional C-domain (CaM 12) or N-domain (CaM 34) had KDs of 31.4 ± 3.0 nM and 45.2 ± 4.1 nM, respectively. These results are consistent with disruption of one of the two GRK5 binding determinants for each mutant.
Selective knockout of each CaM domain revealed distinct functional effects on GRK5. Only the CaM mutant with a functional N-domain (CaM 34) was able to stimulate GRK5 phosphorylation of a peptide substrate (Figure 6D), GRK5 autophosphorylation (Figure S6C) and RH/kinase domain dissociation (Figure S6D), similar to the effect of CaM WT. This confirms the specific contribution of CaM N in enzyme activation. In contrast, CaM C (CaM 12) failed to activate GRK5, yet it was able to effectively inhibit GRK5 binding to phospholipids, a function not detected for CaM N (CaM 34) (Figure 6D). These data are also consistent with the phospholipid binding deficiency of the GRK5 C-terminal mutants (ΔC and 4A) observed in direct lipid binding studies (Figure S6E). Thus, we identified distinct roles for CaM domains with the C-domain specifically interfering with GRK5 membrane recruitment and the N-domain potently stimulating GRK5 enzymatic activity.
Both CaM mutants exhibited a similar potency to inhibit phosphorylation of the β2AR (Figure 6D) and rhodopsin (Figure S6C), although less effectively than CaM WT. This reveals a contribution of each domain in regulating GPCR phosphorylation. Interestingly, increasing concentrations of CaM WT and CaM 34 also induced an increase in GRK5 autophosphorylation which precisely follows the decrease in GPCR phosphorylation (Figure 6D, autoradiography of 32P incorporation). The similar effect was not observed for CaM 12 which inhibited β2AR phosphorylation without stimulation of GRK5 autophosphorylation. Given that CaM C targets GRK5 αC to inhibit GRK5 membrane binding while CaM N targets GRK5 αN to activate the kinase, the inhibitory mechanisms of CaM N- and C-domains on GPCR phosphorylation seem to be different and result from either disruption of membrane binding (CaM 12) or inhibition of GRK5 αN docking on the receptor (CaM 34). These results also imply that both CaM domains synergize within the full-length protein leading to more effective inhibition of GPCR phosphorylation.
CaM Serves as a Molecular Switch Linking Changes in Intracellular Calcium with Redistribution of GRK5 Activity in Cells
We next addressed whether CaM can serve as a molecular switch to redistribute GRK5 activity from one set of substrates to another in cells. To increase intracellular calcium levels and activate CaM, we treated HEK293 cells with the calcium ionophore A23187 or by stimulation of the endogenous Gq-coupled protease-activated receptor 1 (PAR1) via PAR1 activating peptide (PAR1-AP). A23187 (Figure S7A) and PAR1-AP (Figure S7B) stimulated sufficient calcium flux for CaM activation as confirmed by CaMKIIa autophosphorylation, a well-established target of Ca2+/CaM (Simon et al., 2015).
We next studied GRK5 localization and found that GRK5 translocated from the plasma membrane to the cytosol upon treatment with A23187 (Figure S7C) and PAR1-AP (Figure 7A). Translocation was not detected when extracellular calcium was chelated with EGTA or in the presence of the CaM inhibitor W7, suggesting that translocation is dependent on calcium and CaM. Cellular fractionation confirmed calcium-dependent GRK5 accumulation in the cytosol upon A23187 treatment (Figures S7D–F). In addition, a similar increase in cytosolic GRK5 was observed upon PAR1 stimulation (Figures 7B and S7G). Together, these findings further support that GRK5 translocation is driven by Ca2+/CaM activation, in agreement with a previous report (Gold et al., 2013).
Figure 7. CaM functions as a molecular switch linking changes in intracellular calcium with redistribution of GRK5 activity in cells.
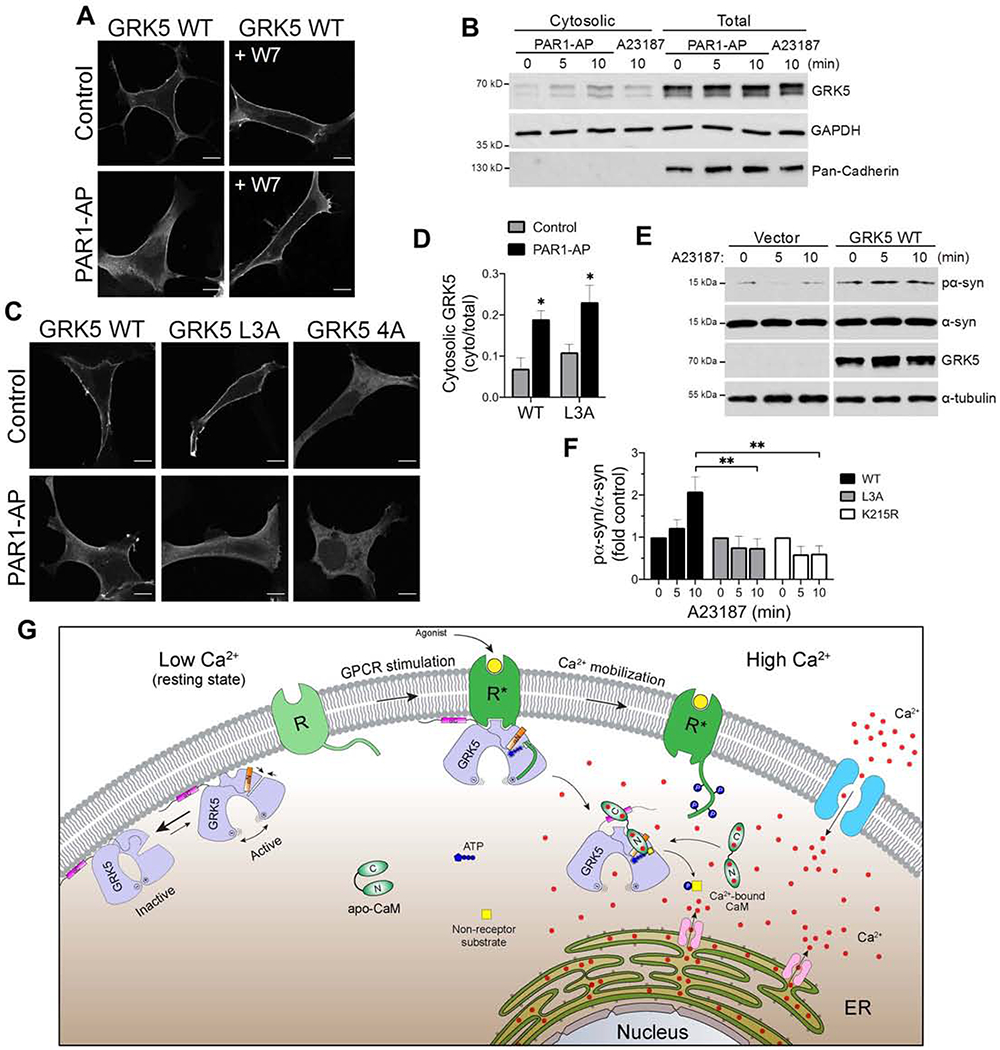
(A) Confocal microscopy of GRK5-GFP expressed in HEK293 cells preincubated without or with W7, followed by control or PAR1-AP treatment. Scale bar is 10 μm.
(B) Representative immunoblot of cytosolic and total GRK5 following 0 (control), 5 and 10 min of PAR1-AP treatment or 10 min of A23187 treatment. Cadherin and GAPDH serve as plasma membrane and cytosolic markers, respectively. See also Figure S7G.
(C) Confocal microscopy of GFP-tagged GRK5 WT, L3A or 4A expressed in HEK293 cells following control or PAR1-AP treatment. Scale bar is 10 μm.
(D) Quantification of cytosolic fraction of GRK5 WT and L3A mutant following 10 min of control or PAR1-AP treatment. Cytosolic GRK5 is represented as a fraction of total GRK5 (n=4, *p<0.05 compared to control). See also Figure S8C.
(E) Representative immunoblot of α-synuclein phosphorylation following 0 (DMSO), 5 and 10 min of A23187 treatment. Tubulin levels were assessed to verify equal protein loading. See also Figure S8D.
(F) Quantification of α-synuclein phosphorylation following 0 (DMSO), 5 and 10 min of A23187 treatment in HEK293 cells expressing GRK5 WT, L3A or K215R. Phosphorylation was normalized to total α-synuclein and presented as fold change over time 0 (n=3, **p<0.01). See also Figure S8E.
(G) Schematic for the structure-based mechanism of GRK5 regulation by CaM in the cell.
See also Figures S7 and S8.
We next utilized the GRK5 N-terminal mutant L3A and C-terminal mutant 4A to assess the contribution of each of the CaM binding sites to GRK5 translocation. Upon treatment with A23187 or PAR1-AP, GRK5 L3A was redistributed from the plasma membrane to the cytosol similar to GRK5 WT as shown by confocal microscopy (Figures 7C and S7C) and cellular fractionation (Figures 7D and S8A–C). This correlates with in vitro data showing effective recruitment of this mutant to lipid membranes in the absence of CaM (Figure S6E). In contrast, GRK5 4A did not respond to calcium activation, likely due to its cytosolic localization prior to treatment. This was expected, since GRK5 4A showed no binding to lipid membranes in vitro (Figure S6E). Thus, our cellular studies support the role of the CaM C in promoting cytosolic GRK5 accumulation by specifically targeting the GRK5 C-terminus.
The fact that GRK5 is strongly activated when bound to Ca2+/CaM suggests that CaM-mediated cytoplasmic accumulation of GRK5 might facilitate the phosphorylation of nonreceptor substrates. Indeed, we observed an increase in α-synuclein phosphorylation in cells overexpressing GRK5 WT upon stimulation by A23187 (Figures 7E and S8D), while no increase was observed with the GRK5 L3A or kinase-dead K215R mutants (Figures 7F and S8E). While the L3A mutation disrupts CaM activation of GRK5 (Figure 4D) and α-synuclein phosphorylation in vitro (Figure S4A), it does not alter GRK5 translocation to the cytoplasm (Figure 7D). Thus, this suggests that the increase in α-synuclein phosphorylation observed for GRK5 can be specifically attributed to GRK5 activation by CaM. Of note, GRK5 L3A retains a basal catalytic activity in complex with CaM (Figure 4D), yet this was not sufficient to promote α-synuclein phosphorylation (Figure 7F) upon L3A translocation to cytosol. Taken together, our findings support the role of CaM as a soluble activator that enables GRK5 phosphorylation of cytosolic substrates and illustrates how CaM serves as a switch to regulate GRK5 function in the cell. Further exploration is necessary to understand the extent to which CaM mediates GRK5 substrate phosphorylation in response to calcium activation and its role in various signaling pathways.
In summary, we performed a comprehensive analysis of CaM regulation of GRK5 using a number of biochemical, biophysical and cell biological approaches, and identified structural aspects of GRK5 activation by CaM that are relevant to GRK5 function in cells (Figure 7G). In the resting state, GRK5 is localized at the plasma membrane primarily via the C-terminus (magenta) and exists in dynamic equilibrium between inactive and active conformations. Activation is driven by ordering of the N-terminal α-helix (orange), which is not sustained in the absence of an activator such as a GPCR or CaM. Therefore, GRK5 is predominantly in an inactive state under basal conditions. Upon GPCR stimulation with agonist, GRK5 binds the receptor and this stabilizes formation of αN to favor the active conformation of GRK5 and promote receptor phosphorylation. The influx of extracellular Ca2+ or the release of Ca2+ from internal stores promotes CaM binding to GRK5. This effectively induces GRK5 cytosolic localization by binding to the C-terminal membrane anchoring helix (αC). CaM also stimulates GRK5 via stabilization of αN, RH domain opening and kinase domain closure, the hallmarks of GRK5 activation. This yields a catalytically competent state of the kinase for effective phosphorylation of soluble substrates in the cytosol. Thus, CaM acts as a strong activator of GRK5 cytosolic substrate phosphorylation while also inhibiting GPCR targeting. By simultaneously regulating multiple GRK5 functions, CaM serves as a molecular switch to modulate GRK5 subcellular localization and substrate targeting in response to calcium increases. Since increased intracellular calcium can be downstream of GPCR activation, CaM binding to GRK5 may potentiate G-protein signaling via relief of GRK5-mediated desensitization of GPCRs and mediate a positive feedback mechanism in GPCR activation.
Limitations
Our work provides mechanistic insight into CaM regulation of GRK5 activity and localization in the cell. While limited to GRK5, this can be the basis for future studies on the regulation of other members of the GRK family by calcium-binding proteins. Currently, there are a lack of known cytoplasmic and nuclear substrates for GRK5, limiting our ability to fully study how CaM regulates GRK5 substrate phosphorylation in a physiological setting. Therefore, a global proteomics approach would be useful to broadly assess how CaM regulates GRK5 interactions and phosphorylation. Finally, to extend our structural study of GRK5 dynamics to a more physiological situation, a FRET-based intramolecular biosensor would be a useful tool to capture GRK5 conformational changes in cells and study how such changes might differ between CaM vs GPCR binding. These lines of investigation should enable a better understanding of physiological and therapeutic factors that can modulate GRK5 activity.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey L. Benovic (jeffrey.benovic@jefferson.edu).
Materials Availability
All unique reagents generated in this study will be made available without restriction upon request.
Data and Code Availability
The coordinates for the crystal structure of Ca2+/CaM bound to GRK5 have been deposited in the Protein Data Bank under code PDB:6PJX.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
bacteria
E. coli BL21 Rosetta-2(DE3)pLysS (Novagen) was used for the expression of recombinant wild-type and mutant calmodulin. E. coli DH10Bac (Thermo Fisher Scientific) were used for production of recombinant bacmids.
Insect cells
The β2AR, wild-type and mutant GRK5 were expressed in Sf9 insect cells (Expression Systems) infected with recombinant baculovirus. Sf9 cells were maintained in Sf-900 II media (Gibco) containing 10% FBS and 10 μg/mL gentamicin (Gibco).
Human cells
Human embryonic kidney (HEK) 293T cells were obtained from American Type Culture Collection (ATCC). HEK293 cells were maintained in DMEM with L-glutamine (Corning), supplemented with 10% FBS (Corning), 22 mM HEPES (Corning), 10 U/mL penicillin and 10 μg/mL streptomycin (Corning) at 37°C, 5% CO2.
METHOD DETAILS
Plasmid generation
Point mutations were introduced into GRK5 and CaM using QuikChange II Site-Directed Mutagenesis Kit (Agilent) or Q5 site-directed mutagenesis (New England BioLabs) following the manufacturer instructions. GRK5 deletion mutants (ΔN, ΔN35, ΔC and ΔN/ΔC) were generated by polymerase chain reaction (PCR) using corresponding primers. To generate GRK5 GFP plasmids, PCR site-directed mutagenesis was first performed using pcDNA3.1 GRK5-strep, GRK5 L3A and GRK5 4A as templates to generate a 5’ HindIII restriction site, remove the stop codon (TAG) and generate a BamHI restriction site immediately after the GRK5 sequence. Following restriction digest with BamHI and HindIII, PCR products were ligated into the pEGFP vector. All mutations were verified by DNA sequencing.
Expression and purification of GRK5 from insect cells
High-titer baculoviruses were generated for human untagged wild-type GRK5 or C-terminally strep-tagged wild-type and mutant GRK5 using the Bac-to-Bac Baculovirus Expression System (Thermo Fisher) following the manufacturer’s instructions. For protein expression, cells were grown to a density of 2-3x106 cells/mL and infected at a MOI of 7-10 pfu/cell. Cells were harvested by centrifugation 46-48 hr post-infection and the pellet was washed once with phosphate-buffered saline (PBS) and resuspended in 150 mL buffer A (20 mM HEPES, pH 7.2, 250 mM NaCl, 0.02% Triton X-100, 5 mM ethylenediaminetetraacetic acid (EDTA), 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 3 mM benzamidine, 5 μg/mL leupeptin) per liter of cells. The cell suspension was flash frozen and stored at −80°C. Untagged GRK5 was purified as previously described (Komolov et al., 2015). Sf9 cells expressing strep-tagged GRK5 were thawed on ice and lysed by polytron at 25,000 rpm (2×30 s). The lysate was centrifuged at 42,000×g, 4°C for 30 min, followed by high-speed centrifugation at 285,000×g, 4°C for 1 hr. The supernatant was loaded on a 5 mL StrepTrap column (GE Healthcare) equilibrated in buffer B (20 mM HEPES, pH 7.2, 250 mM NaCl, 0.02% Triton X-100, 2 mM EDTA, 1 mM DTT). After washing the column with buffer B, bound GRK5 was eluted using 2.5 mM D-desthiobiotin in buffer B. GRK5 was further purified on a MonoS column (GE Healthcare), equilibrated in buffer C (20 mM HEPES, pH 7.2, 1 mM DTT, 200 mM NaCl), and eluted using a 50 mL gradient from 200 to 700 mM NaCl in 20 mM HEPES, pH 7.2, 1 mM DTT. Fractions containing GRK5 were combined and diluted to 200 mM NaCI with 20 mM HEPES, pH 7.2, 1 mM DTT. GRK5 was concentrated using a 30 kDa molecular weight cutoff centrifugal filter unit (Millipore) and the concentrated sample was centrifuged at 18,400×g, 4°C for 10 min to remove any protein aggregates. GRK5 protein concentration was determined by Bradford assay and diluted to the final concentration with buffer C. GRK5 preparations were ≥95% pure and were flash frozen and stored at −80°C. Strep-tagged GRK5 showed no change in catalytic activity compared to untagged GRK5, indicating that the tag does not alter GRK5 folding or function. Purified untagged GRK5 was used for crystallization of the CaM/GRK5 complex while all other assays were performed using strep-tagged GRK5 (unless otherwise stated).
Expression and purification of GRK5 from mammalian cells
HEK293 cells were seeded in 15 cm dishes and transfected at 70-80% confluence with 30 μg of strep-tagged wild-type or mutant GRK5 DNA using Lipofectamine 2000 (Thermo Fisher) reagent according to the manufacturer’s instructions. The media was exchanged 4 hr after transfection and the cells were harvested 72 hr post transfection, washed twice with ice cold PBS and stored at −80°C in 1 mL buffer A. All steps in the purification procedure were carried out at 4°C. Cells were thawed on ice, lysed using a polytron at 25,000 rpm (2×30 s), and the lysate clarified by centrifugation at 18,400×g for 10 min. To isolate GRK5, the lysate was incubated with 50 μL Strep-Tactin Sepharose beads (IBA Life Sciences) for 1 hr, agitating in a thermoshaker. The beads were then washed three times with 1 mL of wash buffer (20 mM HEPES, pH 7.2, 250 mM NaCl, 0.02% Triton X-100, 5 mM EDTA, 1 mM DTT). GRK5 was eluted twice with 75 μL and 50 μL of elution buffer (20 mM HEPES, pH 7.2, 200 mM NaCl, 1 mM DTT, 2.5 mM D-Desthiobiotin), agitating at 1050 rpm. Elution fractions were combined and GRK5 concentration was estimated by Western blot analysis, comparing samples to a standard curve of GRK5 and quantifying by densitometry (ImageJ). Samples were flash frozen and stored at −80°C.
Expression and purification of CaM
BL21 Rosetta-2(DE3)pLysS cells were transformed with a pET28b plasmid containing wild-type rat CaM DNA. A single colony grown on a LB agar plate with kanamycin was used to inoculate 5 mL of Luria broth (LB) medium, which was cultured overnight at 37°C, 250 rpm. The following day, the culture was diluted 1:250 in 1 L of LB containing chloramphenicol and kanamycin, and grown until OD600 of 0.6 was reached. CaM expression was then induced with 0.5 mM IPTG and cells were grown at 37°C for 3.5 hr, pelleted and stored at −80°C. All operations of CaM purification were carried out at 4°C. The pellet was thawed in 50 mL of lysis buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1 mM PMSF, 3 mM benzamidine, 10 μM leupeptin, 100 μg/mL lysozyme). Cells were sonicated for 2 min on ice (50% amplitude, 10 s on/off intervals), the lysate was centrifuged at 32,600×g for 20 min, and the CaCl2 concentration in the supernatant was adjusted to 5 mM and centrifuged at 285,000×g for 40 min. The supernatant was loaded onto a 15 mL column packed with Phenyl Sepharose resin (GE Healthcare) equilibrated in binding buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM CaCl2), washed with high-salt buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM CaCl2, 0.5 M NaCl) and eluted with EGTA buffer (50 mM Tris-HCl, pH 7.5, 2 mM EGTA). Fractions containing CaM were combined, loaded into 3.5 kDa molecular weight cutoff dialysis cassette (Millipore) and dialysed against 50 mM NH4HCO3. After dialysis, the protein was centrifuged at 32,600×g for 10 min and the concentration determined by absorbance at 276 nm using an extinction coefficient of 3006 cm−1M−1. The purified CaM was lyophilized and stored at −80°C. Two CaM EF-hand mutants having either alanine substitutions in EF1 and EF2 (D22A, D58A) (CaM 12 mutant) or in EF3 and EF4 (D95A, D131A) (CaM 34 mutant) were also expressed and purified as described above.
Expression and purification of GPCRs
Urea-treated rod outer segment (ROS) membranes containing rhodopsin were prepared from bovine retinas as described previously (Shichi and Somers, 1978). A truncated form of rhodopsin with 19 amino acid residues proteolytically removed from the carboxyl terminus (RhoΔC) was prepared using Endopeptidase Asp-N as previously described (Chen et al., 1993). The β2AR was expressed and isolated essentially as described earlier (Komolov et al., 2017).
In vitro-binding assays
Binding between CaM and GRK5 was evaluated using either a pull-down assay with CaM-Sepharose (GE Healthcare) or Strep-Tactin Sepharose (IBA Life Sciences). All operations were carried out at 4°C. For pull-down with CaM-Sepharose, 0.5 μM strep-tagged GRK5 was incubated with 15 μl CaM-Sepharose beads (75% v/v slurry in 20 mM Tris-HCl, pH 7.5) in 150 μl of binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% Tween 20, 1 mM DTT) containing either 2 mM CaCl2 (+Calcium) or 10 mM EGTA (−Calcium). For pull-down with Strep-Tactin Sepharose, 4 μM of a 1:1 mixture of CaM and strep-tagged GRK5 were incubated with 25 μl Strep-Tactin Sepharose beads (75% v/v slurry in 20 mM Tris-HCl, pH 7.5) in 100 μl of binding buffer containing either 2 mM CaCl2 (+Calcium) or 10 mM EGTA (−Calcium). After 1 hr incubation in a thermoshaker, the beads were washed three times with 1 ml of binding buffer and proteins were eluted with 50 μl of elution buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCI, 10 mM EGTA, 0.05% Tween 20). The amount of GRK5 bound to CaM-Sepharose or CaM bound to GRK5 immobilized on Strep-Tactin Sepharose were analyzed by SDS-PAGE and Coomassie blue staining. Protein bands were quantified by densitometry using Image Studio Lite (LI-COR Biosciences) and the amount of bound GRK5 or CaM was expressed as a percentage of the binding observed with GRK5 WT.
In vitro-kinase assays
A time course of GRK5-mediated phosphorylation of different substrates was assayed by incubating purified wild-type or mutant GRK5 (50 nM) with either ROS membranes (1.5 μM rhodopsin), β2AR (1.5 μM), α-synuclein (1.5 μM) or no substrate (GRK5 autophosphorylation) in reaction buffer (20 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 30 mM NaCl, 0.5 mM EDTA, 0.8 mM CaCl2, 100 μM [γ32P]ATP (1000-2000 cpm/pmol)) at 23°C with or without 0.5 μM CaM. For a titration of the effect of wild-type or mutant CaM on GRK5 phosphorylation, 200 nM GRK5 was incubated with either ROS membranes (4 μM rhodopsin), β2AR (4 μM), α-synuclein (4 μM) or 100 μM substrate peptide (AEMWYSEVEEARRR) in reaction buffer for 3 min at 23°C with the indicated concentrations of CaM (0-20 μM). To screen the effect of GRK5 αN-helix mutations on CaM interaction with GRK5, GRK5 WT or N-terminal mutants were initially expressed and partially purified from HEK293 cells, and then incubated at 10 nM concentration with the indicated concentrations of CaM (0.01-300 nM) in reaction buffer for 10 min at 23°C. Incubations were stopped with SDS sample buffer and samples were separated by SDS-PAGE. Gels were stained with Coomassie blue, dried, exposed to autoradiography film and 32P-labeled proteins were excised and counted to determine the pmol of phosphate transferred.
Michaelis-Menten kinetic analysis
Kinetic parameters of different GRK5 constructs were determined by phosphorylating the substrate peptide AEMWYSEVEEARRR in radiometric assay. The three C-terminal arginines were added to the peptide to improve its resolution in a gel. The peptide was synthesized by New England Peptide and reconstituted in 100 mM NH4HCO3. Km, kcat and catalytic efficiency Km/kcat for ATP were determined by incubating 50 μM peptide with either 500 nM GRK5 (basal activity) or 100 nM GRK5 (in the presence of activators) in 20 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 30 mM NaCl, 0.5 mM EDTA, 0.8 mM CaCl2 containing 2-200 μM [γ32P]ATP (1000-2000 cpm/pmol). Activators (CaM or RhoΔC) were added at a concentration of 0.5 μM and reactions proceeded for 5 min at 23°C. The kinetic parameters for peptide were determined by incubating 500 nM GRK5 and peptide (0.3-3000 μM) for 10 min at 23°C in 20 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 30 mM NaCl, 0.5 mM EDTA, 0.8 mM CaCl2, 25 mM NH4HCO3, 100 μM [γ32P] ATP (1000-2000 cpm/pmol) with or without 1 μM CaM. All incubations were stopped with SDS sample buffer and samples were electrophoresed on an 18% SDS-polyacrylamide gel. Gels were stained with Coomassie blue, dried, exposed to autoradiography film and 32P-labeled peptides were excised and counted to determine the pmol of phosphate transferred. Reaction velocities at the various ATP and peptide concentrations were fit to the Michaelis-Menten equation using GraphPad Prism.
Phospholipid-binding assay
Phospholipid vesicles were prepared by sonicating 40 mg of soybean phosphatidylcholine (PC) in 5 ml of assay buffer (20 mM Tris-HCl, pH 7.5, 10 mM CaCl2, 100 mM NaCl) on ice four times for 20 s. The ability of GRK5 to associate with phospholipids was analyzed by incubating 0.2 μM GRK5 in the presence or absence of indicated concentration of phospholipid vesicles (0-4 mg/ml PC) or indicated concentration of CaM at fixed concentration of PC (1 mg/ml) in 60 μl of assay buffer. After 15 min incubation at 15°C in a thermoshaker, the vesicles were pelleted at 200,000×g for 60 min (4°C), and the pellets were resuspended in 60 μl of assay buffer. The amount of GRK5 associated with the pelleted lipids was analyzed by SDS-PAGE and staining with Coomassie blue. GRK5 bands were quantified by densitometry using Image Studio Lite (LI-COR Biosciences), and the amount of GRK5 bound to phospholipids was expressed as a percentage of the total GRK5 after subtracting the amount of GRK5 pelleted in the absence of phospholipids.
Chemical cross-linking (zero-length cross-linking)
Cross-linking reactions were performed using a freshly prepared mixture of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC, Pierce)/N-hydroxysulfosuccinimide (sulfo-NHS, Pierce). Reactions containing a final concentration of 10 mM EDC/5 mM sulfo-NHS, 6 μM CaM and 3 μM GRK5 in cross-linking buffer (20 mM HEPES, pH 7.2, 200 mM NaCl, 5 mM CaCl2) were incubated for up to 180 min at 4°C. At the indicated time points, an aliquot was removed and the reaction quenched with 80 mM β-mercaptoethanol. Zero-length cross-linked samples were separated on 10% SDS-PAGE gels and stained with Coomassie Blue.
Native agarose gel electrophoresis
To monitor CaM and GRK5 complex formation, 7 μg of untagged GRK5 was incubated with the indicated amount of CaM and then run on a 0.8% horizontal agarose gel using TAMg buffer (40 mM Tris base, 20 mM acetic acid, pH 8.1, 1 mM MgSO4) supplemented with 1 mM CaCl2, essentially as previously described (Kim, 2011).
Limited trypsinolysis
To follow limited trypsinolysis of GRK5 alone or GRK5 plus CaM, 3 μM of untagged GRK5 with or without 6 μM CaM was incubated with 12 nM trypsin (250:1 ratio) in 20 mM Tris-HCl, pH 7.5, 115 mM NaCl and 2 mM CaCl2 for up to 90 min at 4°C. At the indicated time points, an aliquot was removed and the reaction was quenched with 5 mM PMSF and 10 mM EDTA. The proteolyzed GRK5 fragments were separated on 10% SDS-PAGE gels and stained with Coomassie Blue.
Ca2+-dependent gel mobility shift assay
To perform Ca2+-dependent gel mobility shift assays, 5 mM CaCl2 or 5 mM EGTA were added to 0.5 mg/ml CaM WT or EF-hand mutants CaM 12 or CaM 34 in buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl. The samples were incubated for 30 min at 4°C while rocking, followed by SDS-PAGE on 18% gel and Coomassie blue staining.
Analytical size-exclusion chromatography
Complex formation between GRK5 and CaM was tested by preparation of a 20 μM 1:1 complex between CaM and different GRK5 variants. The samples were run on a Superose 12 10/300 GL column (GE Healthcare) equilibrated with 20 mM HEPES, pH 7.2, 2 mM CaCl2, 200 mM NaCl and 1 mM DTT. Fractions containing GRK5, CaM or the complex were collected and analyzed by SDS-PAGE.
Analytical ultracentrifugation
Untagged GRK5 alone or a 1:1 complex with CaM isolated by SEC were diluted in 20 mM Hepes, pH 7.2, 200 mM NaCl, 1 mM DTT to a concentration of 5 μM and 6 μM, respectively. A sedimentation equilibrium analytical ultracentrifugation analysis was performed using a Beckman Coulter ProteomeLab XL-I/A analytical ultracentrifuge equipped with absorbance optics and an eight-hole An-50 Ti analytical rotor. The sample (100 μl) and reference buffer (120 μl) were loaded into six-sector centerpieces, sedimented to equilibrium at 18,000; 24,000; 30,000 and 36,000 rev min−1 at 4°C and absorption scans were collected at a fixed wavelength of 278 nm. The program SEDNTERP version 1.09 was used to correct the experimental s value to standard conditions at 20°C in water (s20,w) and to calculate the partial specific volume of each protein sample, solvent density and relative viscosity values along with the theoretical molecular mass (Laue et al, 1992). The resulting data were fitted to a “species analysis” model available in the program SEDPHAT (Rowe, 2005) and an estimated molecular mass was obtained. The fitted data were visualized and presented using program GUSSI (UT Southwestern).
Isothermal titration calorimetry (ITC)
Protein samples were prepared by dialyzing wild-type or mutant GRK5 and CaM into the same buffer (20 mM HEPES, pH 7.2, 200 mM NaCl, 10 mM CaCl2, 1 mM DTT). Following dialysis, samples were centrifuged for 5 min at 18,400×g. Before measurements, samples were degassed for 15 min at 4°C, stirring at 250 rpm. All titrations were performed using a NanoITC microcalorimeter (TA Instruments) with 195 μL sample cell volume. GRK5 or GRK5 mutants (25 μM) supplemented with 200 μM sangivamycin or dialysis buffer as a blank control were loaded into the sample cell and 175 μM CaM was titrated into the sample cell with 2 μL injections every 200 s for a total of 25 injections at 4°C. Peaks were baseline corrected, integrated and thermodynamic parameters were obtained using independent binding model in NanoAnalyze software (TA Instruments).
Circular dichroism
Circular dichroism spectra were measured using a Jasco J-810 spectropolarimeter equipped with a Peltier temperature control system. Peptide samples representing GRK5 regions 2-19 and 546-562 were diluted in PBS (3.2 mM Na2HPO4, 0.5 mM KH2PO4, pH 7.4, 135 mM NaCl) to 100 μM concentration with addition of various amounts of trifluoroethanol (GRK5 2-19) or methanol (GRK5 546-562). Measurements were conducted at 20°C using a quartz cell (Starna Cells, Inc.) with a 1-mm light path length. The apparent helical content was calculated from the mean residual ellipticity at 222 nm (Chen et al, 1972).
Surface plasmon resonance
GRK5 binding to CaM or the CaM EF-hand mutants was monitored using a Biacore 3000 (GE Healthcare). CaM was first immobilized by amine linkage to carboxy-activated dextran of a CM5 sensor chip. The dextran matrix was activated by injecting 200 μl of a fresh mixture of EDC/NHS (200 mM/50 mM) at a flow rate of 20 μl/min, followed by a 20-min injection at 2 μl/min of 50 μg/ml CaM diluted in 10 mM sodium acetate, pH 4.0. Any remaining reactive carboxy groups were deactivated using a 7-min pulse of 1 M ethanolamine hydrochloride, pH 8.5 at 25 μl/min. CaM/GRK5 binding was then analyzed at 20°C in a running buffer containing 10 mM Hepes, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.5 mM CaCl2, 0.05 mg/ml BSA, 0.005% Surfactant P20. GRK5 was diluted in the running buffer and injected in a three-fold concentration series (1, 3, 9, 27 and 81 nM) over the CaM surface at a flow rate of 50 μl/min with a contact time of 1 min and dissociation time of 2 min, followed by 1-min injection of 5 mM EGTA at the end of the cycle to regenerate CaM. Experimental data was double-referenced by subtracting a GRK5 response from a blank reference surface and a response from blank injection of running buffer.
Domain proximity assay
RH/kinase domain proximity in GRK5 at the ionic lock site was monitored by observation of the rate of interdomain disulfide cross-linking in a GRK5 double-cysteine mutant (GRK5-DC) under nonreducing conditions as described (Komolov et al., 2017). Briefly, GRK5-DC at 1.5 μM was incubated with 1.5 mM of freshly prepared K3Fe(CN)6 in 20 mM HEPES, pH 7.2, 200 mM NaCl and 1 mM CaCl2 at 15°C while rocking. At indicated time, an aliquot was withdrawn for SDS-PAGE gel analysis under nonreducing conditions. The effect of CaM binding on disulfide bond formation in GRK5-DC was evaluated in the presence of 4.5 μM CaM.
Positional scanning peptide array (PSPA)
The PSPA consisted of 198 peptide mixtures arrayed in a 1536 well plate. Peptides had the general sequence Y-A-x-x-x-x-x-S/T-x-x-x-x-G-K-K(biotin), where 8 of the 9 “x” positions were an equimolar mixture of 17 proteogenic amino acids excluding Ser, Thr and Cys, and S/T indicates an even mixture of Ser and Thr. In each well, a single “x” position was substituted with one of the 20 unmodified amino acids, phosphothreonine, or phosphotyrosine. In addition, two wells contained mixtures that fixed either Ser or Thr at the central position. Each well contained 50 μM peptide mixture, 50 μM [γ33P]ATP (37.5 μCi/mL) and GRK5 or GRK5/CaM complex (37.5 μg/mL) in 50 mM Tris, pH 7.5, 50 mM NaCl, 5 mM MgCl2, 0.2 mM CaCl2, 1 mM EDTA, 1 mM DTT, 0.1% Tween 20. Sealed plates were incubated at 30°C 2 hr, chilled on ice, unsealed, and 200 nL aliquots were transferred to streptavidin-coated membrane (Promega). Membranes were washed as described, dried, and exposed to a phosphor screen. Radiolabel incorporation was quantified by phosphor imager using QuantityOne software (BioRad), normalized to the total signal for all residues at a given position. Heat maps generated in Microsoft Excel show the average of two experiments.
Crystallization and structure determination
Lyophilized purified CaM was resuspended in gel filtration buffer (20 mM HEPES, pH 7.2, 200 mM NaCl, 2 mM CaCl2, 1 mM DTT) at a final concentration of 300 μM and centrifuged at 10,000×g for 10 min at 4°C. CaM and GRK5 were mixed at a 2.4:1 ratio and incubated at 4°C for 1 hr to facilitate complex formation. The sample was centrifuged for 10 min at 18,400×g at 4°C to remove any aggregates and then the complex was isolated on a Superdex 75 16/600 column (GE Healthcare) at 1 mL/min flow rate in gel filtration buffer. Fractions containing GRK5/CaM complex were combined and concentrated to approximately 35 mg/ml using a 10 kDa molecular weight cutoff centrifugal filter unit (Millipore), centrifuging at 1,000×g, 4°C. The concentrated sample was centrifuged for 10 min at 18,400×g, 4°C to remove possible protein aggregates. GRK5/CaM concentration was determined by Bradford assay and diluted to 30 mg/ml. 800 μM sangivamycin and 400 μM MgCl2 were added to the complex to stabilize GRK5 and samples were incubated at 4°C overnight while rocking.
The high-resolution diffraction quality crystals for GRK5/CaM complex were obtained using sitting drop vapor diffusion method in 96-well Swissci plate by mixing 0.4 μL protein at 25 mg/mL with an equal volume of crystallization solution containing 9:1 v/v ratio of the optimized condition (200 mM KCl, 50 mM Hepes, pH 7.5, 30% 5/4 v/v pentaerythritol propoxylate) and the additive (0.05% w/v L-Citrulline, 0.05% w/v Glycine, 0.05% w/v L-(−)-Threonine, 0.05% w/v L-(+)-Lysine, 0.05% w/v L-Alanine, 0.05% w/v L-Arginine, 0.05% w/v L-Asparagine monohydrate, 0.05% w/v L-Aspartic acid, 0.05% w/v L-Glutamic acid, 0.05% w/v L-Glutamine, 0.05% w/v L-Histidine, 0.05% w/v L-Isoleucine, 0.05% w/v L-Leucine, 0.05% w/v L-Methionine, 0.05% w/v L-Phenylalanine, 0.05% w/v L-Proline, 0.05% w/v L-Serine, 0.05% w/v L-Tryptophan, 0.05% w/v L-Tyrosine, 0.05% w/v L-Valine, 0.02 M HEPES sodium, pH 6). The crystals appeared within 12 weeks and grew over the course of 3-4 weeks before being harvested for diffraction quality screening and data collection. A number of medium to high-resolution diffraction datasets were collected using Rigaku MicroMaxx-007 HF diffractometer equipped with a Pilatus3 R 200K direct detector. All steps of data indexing, integration, and reduction were carried out using HKL3000. A 1.96 Å resolution crystal structure in an orthorhombic space group for GRK5/CaM complex was solved by molecular replacement with Phaser using kinase domain of GRK5 (PDB 4TNB) as the search model. A single copy of GRK5/CaM complex was located in the asymmetric unit with an estimated solvent content of 48.9%. The CaM, GRK5 N-terminal residues 1-65 and GRK5 AST loop region were located using omit maps and feature enhance maps in Phenix followed by iterative manual model building using Coot. The solutions were subjected to cycles of positional refinement and isotropic B-factor refinement using eleven TLS groups in phenix.refine (Afonine et al., 2012). The final model was refined to a Rwork/Rfree of 19.54/22.96 %. Final model validation was done using MolProbity (Chen et al., 2010a). Crystallographic data and refinement statistics are shown in Table 1. Ribbon diagrams and electrostatic surface representations were prepared using the program PyMOL (DeLano, 2010). PDBePISA server was used to analyze residues involved in complex interface, free energy of dissociation and interface total buried surface area calculations (Krissinel and Henrick, 2007). Positive ΔiG of a residue makes a negative contribution to the solvation energy gain of the interface, which corresponds to hydrophobic effect (Figure 1D).
Hydrogen-Deuterium Exchange/Mass Spectrometry (HDX-MS)
GRK5-CaM complex for HDX-MS study was prepared by mixing GRK5 and CaM at a 1:1 ratio to detect the conformational changes in CaM and at a 1:8 ratio to detect the conformational changes in GRK5. Briefly, the 150 μM apo-GRK5 was mixed with an equal volume of either 150 μM CaM (1:1) or 1200 μM CaM (1:8) in protein buffer containing 20 mM Hepes, pH 7.5, 200 mM NaCl, 10 mM CaCl2, 1 mM DTT, and incubated for 2 hr on ice to facilitate complex formation. For GRK5 or CaM alone, apo-GRK5 or CaM were used at 75 μM in protein buffer. Hydrogen/deuterium exchange was initiated by mixing 3 μl of protein sample with 27 μl D2O buffer (20 mM HEPES, pH 7.5, 200 mM NaCl, 10 mM CaCl2, 1 mM DTT in D2O) and incubated for 10, 100, 1,000 or 10,000 s on ice. At the indicated time points, the reaction was quenched with 30 μl ice-cold quench solution (100 mM Na2HPO4, 20 mM TCEP, pH 2.01) and then immediately frozen in dry ice. To minimize temperature fluctuation during the exchange reaction, all experiments were performed in a cold room maintained at 4°C. For non-deuterated samples, 3 μl sample was added to 27 μl H2O buffer (20 mM HEPES, pH 7.5, 200 mM NaCl, 10 mM CaCl2, 1 mM DTT in H2O) and mixed with 30 μl ice-cold quench solution. Three independent experiments were performed.
The samples were digested and analyzed by HDX-MS as described previously (Duc et al., 2015). Briefly, digestion was performed using an immobilized pepsin column (2.1 × 30 mm) (Life Technologies, Carlbad, CA, USA) at a flow rate of 100 μl/min with 0.05% formic acid in H2O at 10°C. Digested fragments were desalted and collected on a C18 VanGuard trap column (Waters, Milford, MA, USA) for 2 min and then analyzed by ultra-pressure liquid chromatography using an Acquity UPLC C18 column (1.7 μm, 1.0 × 100 mm) (Waters) at a flow rate of 40 μl/min with an acetonitrile gradient. To minimize back-exchange of deuterium to hydrogen, solvents were adjusted to pH 2.4 using 0.15% formic acid and the system including trap and UPLC columns were maintained at 0.5°C during analysis. The back-exchange was not corrected because all the data is comparison between two states. Mass spectra of peptide fragments were analyzed with a Xevo G2 Quadruple-Time of Flight (Q-TOF) mass spectrometer equipped with standard electrospray ionization (ESI) source in MSE mode. The mass spectra were acquired in the range of m/z 100-2000 Da for 10 min in positive ion mode.
Peptic peptides were identified from non-deuterated samples using Proteinlynx Global Server 2.4 software (Waters, Milford, MA, USA). The following parameters were applied: monoisotopic mass, nonspecific for the enzyme while allowing up to 1 missed cleavage, MS/MS ion searches, automatic fragment mass tolerance, and automatic peptide mass tolerance. Searches were conducted with the variable methionine oxidation modification and the peptides were filtered on a peptide score of no less than 6. The amount and percent deuterium exchange values for each peptide at variable time points were processed and determined by measuring the centroid of the isotopic distribution using DynamX 2.0 software (Waters). The detailed data for HDX-MS analysis is provided in Table S2 and S3.
Molecular dynamics simulations
Four different GRK5 systems were simulated via all-atom classical molecular dynamics (MD) in order to obtain a better understanding of the influence of CaM association on the conformation of GRK5 (Table S1): (1) the X-ray crystal structure of CaM/GRK5 (PDB: 6PJX), (2) our model of the full-length CaM/GRK5 complex, consisting of the X-ray crystal structure of CaM/GRK5 (PDB: 6PJX) with the modeled GRK5 αC/CaM C-domain region, (3) the X-ray crystal structure of CaM/GRK5 (PDB: 6PJX) without CaM, (4) the X-ray crystal structure of CaM/GRK5 (PDB: 6PJX) without CaM and the αN-helix of GRK5. The C-domain of CaM in Condition 2 was obtained from the Ca2+/CaM structure where it was resolved (PDB: 1K90). Since the C-terminus of GRK5 has not been experimentally resolved, the PEP-FOLD server (Thevenet et al., 2012) was employed to build a de novo structure of GRK5 residues 542-577 in Condition 2. MODELLER (version 9.15) (Eswar et al., 2006) was used to connect the C-domain of CaM and the GRK5 C-terminus to the X-ray crystal structure of the CaM-GRK5 complex. Modeled GRK5 residues 548-559 adopt an amphipathic α-helix (αC-helix). The hydrophobic side of the αC-helix comprising non-polar residues L550, L551, L554 and F555 (LLLF motif) was aligned against the CaM C-domain hydrophobic groove to build a second site for interaction between CaM and GRK5 within a 1:1 complex. Thus, the final model of the CaM/GRK5 complex features the two CaM domains displaying their hydrophobic pockets on the same side to reach both terminal GRK5 helices (αN and αC) oriented in an antiparallel arrangement at a distance of ~30 Å from each other. The CaM linker region connecting the two CaM domains in this model does not maintain a rigid α-helix and unfolds from M72 to D78, resulting in a moderate collapse of CaM.
Prime (Schrödinger) was used to model missing side chains and to add neutral acetyl and methylamide groups to cap protein termini, except for the C-domain of CaM in Condition 2 which has a carboxylated C-terminus. In all GRK5 simulations, we retained titratable residues in their dominant protonation state at pH 7. The structures were then inserted into a box of water using in-house simulation preparation software (Betz, 2018). Sodium and chloride ions were added to neutralize each system at a NaCl concentration of 150 mM. Water-box dimensions were chosen to maintain at least a 44 Å buffer between protein images in all dimensions. For all simulations, we used the CHARMM36m force field (Huang et al., 2017) for proteins and ions, and the TIP3P model (Jorgensen et al., 1983) for water molecules. We generated parameters for sangivamycin in the CHARMM General Force Field (CGenFF) (Vanommeslaeghe et al., 2009) using the ParamChem webserver, version 1.0.0 (paramchem.org).
Simulations were performed using the AMBER16 software (Case et al., 2016; Crowley et al., 2009). We performed all simulations using the Compute Unified Device Architecture (CUDA) version of Particle-Mesh Ewald Molecular Dynamics (PMEMD) in AMBER on one or two graphics processing units (GPUs) (Salomon-Ferrer et al., 2013). After minimization, systems were heated from 0 K to 100 K in the NVT ensemble over 12.5 ps and then from 100 K to 310 K in the NPT ensemble over 125 ps, using 10.0 kcal·mol−1·Å2 harmonic restraints applied to protein heavy atoms, Ca2+ ions and sangivamycin. Systems were then equilibrated at 310 K in the NPT ensemble at 1 bar with harmonic restraints on all protein heavy atoms, Ca2+ ions and sangivamycin, tapered off by 1.0 kcal·mol−1·Å2 starting at 5.0 kcal·mol−1·Å2 in a stepwise fashion every 2 ns for 10 ns and then by 0.1 kcal·mol−1·Å2 in a stepwise fashion every 2 ns for 20 ns. 50 ns of unrestrained MD was then performed as an additional step of equilibration for each simulation. Finally, production simulations were performed in the NPT ensemble at 310 K and 1 bar, using a Langevin thermostat for temperature coupling and an isotropic Monte Carlo barostat for pressure coupling. These simulations used a 4-fs time step with hydrogen mass repartitioning (Hopkins et al., 2015). Bond lengths to hydrogen atoms were constrained using SHAKE. Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were computed using the particle mesh Ewald method with an Ewald coefficient β of approximately 0.31 Å and a B-spline interpolation of order 4. The FFT grid size was chosen such that the width of a grid cell was approximately 1 Å. Trajectory snapshots were saved every 200 ps.
Stability of N- and C-terminal interactions in the CaM/GRK5 crystal structure or the full-length model of CaM/GRK5 complex was evaluated by measuring the backbone RMSD of GRK5 αN-helix (residues 4-17) and the N-domain of CaM (residues 7-75) for GRK5 αN/CaM N-domain region and the backbone RMSD of the αC-helix of GRK5 (residues 546-559) and the C-domain of CaM (residues 84-148) for GRK5 αC/CaM C-domain region. The RMSD of GRK5 αN/CaM N-domain is calculated with respect to the crystal structure whereas the RMSD of GRK5 αN/CaM N-domain was calculated with respect to the full-length model.
Analysis protocols for molecular dynamics simulations
The AmberTools16 CPPTRAJ package was used to reimage and center trajectories. Simulations were visualized and analyzed using Visual Molecular Dynamics (VMD) (Humphrey et al., 1996; Roe and Cheatham, 2013). Time traces from simulation were smoothed using a running average with a window size of 10 ns and visualized with the PyPlot package from Matplotlib. Pymol was used to generate Movie S1 (The PyMOL Molecular Graphics System).
HEK293 cell culture and plasmid transfection
HEK293 cells were seeded in a 6-well plate at 5-8x104 cells per well or in a 10 cm dish for 5070% confluency on the day of transfection. Cells were transfected with the indicated plasmid using Lipofectamine 2000 (Invitrogen), following the manufacturer’s instructions. To assess GRK5 salt bridge mutant solubility, cells in a 6-well plate were transfected with 1 μg GRK5 wild-type, R187A, R206A or R187A/R206A DNA. To assess α-synuclein phosphorylation, cells in a 6-well plate were co-transfected with 0.5 μg α-synuclein DNA and 0.1 μg GRK5 wild-type, L3A or K215R DNA or vector alone. For cell fractionation, microscopy and CaMKIIα autophosphorylation experiments with A23187 treatment, HEK293 cells were transfected in 10 cm dishes at ~70% confluency with 5 μg GRK5 wild-type or L3A DNA, 5 μg of GRK5-GFP wild-type, L3A or 4A DNA, or 8 μg CaMKIla DNA. The following day, 3x105 cells were seeded in 6 well plates. For cell fractionation and microscopy experiments with PAR1-AP treatment, HEK293 cells were transfected in a 10 cm dish with 0.5 μg GRK5 wild-type or L3A DNA or 0.5 μg of GRK5-GFP wild-type, L3A or 4A. The following day, 3x105 cells were seeded in 6 well plates. For CaMKIla autophosphorylation experiments with PAR1-AP treatment, cells in a 6-well plate were transfected with 0.5 μg CaMKIIa DNA.
Calcium activation
Forty-eight hours following transfection, culture media was aspirated and cells were treated with 2.5 μM of calcium ionophore A23187 (Sigma-Aldrich) or 100 μM PAR1-AP in cell culture media without FBS or antibiotics for the indicated times at 37°C. When indicated, EGTA (5 mM) was added to the treatment media or cells were pretreated with 10 μM W7 (20 μM W7 for CaMKIIα autophosphorylation experiments) in cell culture media without FBS or antibiotics for 1 hour at 37°C. For A23187 experiments, dimethyl sulfoxide (DMSO) treatment served as the vehicle control for the course of the experiment and represents the zero-time point. To assess α-synuclein phosphorylation, cells were pretreated with 250 nM okadaic acid (Sigma-Aldrich) in cell culture media without FBS or antibiotics for 10 min at 37°C prior to the addition of A23187.
Lysate preparation and immunoblotting
Cells were transferred on ice, washed once with ice-cold PBS and lysed with RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 DTT, 1 mM PMSF, 3 mM benzamidine, 10.5 μM leupeptin) containing PhosSTOP phosphatase inhibitor cocktail (Sigma-Aldrich). Cells were incubated with lysis buffer for 15 min on ice, scraped into 1.5 mL microcentrifuge tubes and centrifuged at 12,000xg for 10-30 min at 4°C to clarify the lysate. To assess α-synuclein and CaMKIIa phosphorylation, protein amount was determined by Bradford assay and samples were diluted to equal concentrations using RIPA buffer. The samples were electrophoresed on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membrane. To assess α-synuclein phosphorylation, samples were separated on 15% gels and transferred to polyvinylidene difluoride (PVDF) membranes at 250 mA for 1.5 h. PVDF and nitrocellulose membranes were blocked in 5% dry milk in PBS/Tween 20 for 30 min at room temperature. PBS with 0.1% Tween 20 was used for all applications except for α-synuclein assessment, which used 0.05% Tween 20. Membranes with transferred proteins were incubated overnight at 4°C with primary antibody diluted in 5% dry milk in PBS/Tween 20. The next day, blots were washed with PBS/Tween 20 and incubated with secondary antibody diluted in 5% dry milk in PBS/Tween 20 for 1 h at room temperature. Membranes were washed with PBS/Tween 20 and incubated with SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Scientific), following the manufacturer’s instructions. Blots were developed on X-ray film or imaged using the Amersham Imager 600 (GE Healthcare). Total levels of α-tubulin and α-synuclein were assessed after probing for GRK5 and phospho-α-synuclein, respectively. Membranes were stripped for 5 min with stripping buffer (20 mM Tris-HCl, pH 7.5, 30 mM NaCl, 0.1% Tween 20) and washed extensively with PBS/Tween 20. Membranes were blocked and probed as before, incubating in primary antibody for 1 h at room temperature. Otherwise, figures containing multiple blots were probed on separate gels with equivalent loading. X-ray films were scanned and images were quantified using ImageJ. Images obtained using the Amersham 600 Imager were quantified using the instrument software.
The following primary antibodies were used: GRK4-6 (1:1000-1:6000, 05-466: Millipore), α-synuclein (1:10,000, 4179: Cell Signaling), phospho-α-synuclein (Ser129) (1:1000, 01525191: Wako), pan CaMKII (1:1000, 3362S: Cell Signaling), CaMKIIα (1:3000, 11945: Cell Signaling), phospho-CaMKIIα (Thr286) (1:1000, 12716: Cell Signaling), α-tubulin (1:20000, T5168: Sigma-Aldrich), GAPDH (1:20000-1:80000, 5174: Cell Signaling), pan-cadherin (1:1000, 4068: Cell Signaling).
Cellular fractionation
To lyse cells in A23187 experiments for fractionation, cells were transferred on ice, washed with ice-cold PBS and lysed with 100 μL hypotonic lysis buffer (10 mM HEPES, pH 7.4, 1 mM EDTA, 2.5 mM MgCl2, 100 μM CaCl2, 1 mM DTT, 0.5 mM PMSF, 5 μg/ml leupeptin, 3 mM benzamidine, PhosSTOP phosphatase inhibitor cocktail) for 15 min. Lysates were scraped into 0.5 mL tubes fitted for pestles (REI) and manually homogenized with 100 strokes. Following homogenization, the NaCl concentration was adjusted to 100 mM. To lyse cells in PAR1-AP experiments for fractionation, cells were transferred on ice, washed with ice-cold PBS and lysed with 120 μL hypotonic lysis buffer (50 mM Tris, pH 8, 1 mM EDTA, 2.5 mM MgCl2, 1 mM CaCl2, 1 mM DTT, 0.5 mM PMSF, 5 μg/ml leupeptin, 3 mM benzamidine, PhosSTOP phosphatase inhibitor cocktail) for 15 min. Lysates were scraped into 0.5 mL tubes fitted for pestles (REI) and manually homogenized with 100 strokes. Samples were then centrifuged at 750xg for 5 min at 4°C and an aliquot was saved for total protein analysis. A fraction of the supernatant was transferred to a fresh 1.5 mL microcentrifuge tube and centrifuged at 150,000xg for 20 min, 4°C. The supernatant was saved as the cytosolic fraction. The samples were boiled in SDS sample buffer and proteins were separated by SDS-PAGE and analyzed by immunoblotting, as described above.
Confocal microscopy
HEK293 cells transiently expressing GRK5-GFP were seeded in a 6-well plate on coverslips. Following a 10 min treatment, cells were place on ice and washed with ice-cold PBS. To fix cells in A23187 experiments, cells were treated with 3.7% paraformaldehyde in PBS for 20 min at room temperature and washed twice with PBS. To fix cells in PAR1-AP experiments, cells were treated with 3.7% paraformaldehyde in PBS for 45 minutes on ice. Paraformaldehyde was aspirated and cells were incubated at −20°C with ice-cold 100% methanol for 5 min. Cells were washed three times with ice-cold PBS. Coverslips were mounted on glass slides with Prolong Diamond Antifade mounting media with DAPI (Invitrogen) and dried overnight. Cells were imaged on a Nikon A1R laser scanning confocal microscope using a Nikon Plan Apo 60x/1.40 oil objective and the NIS Elements AR software, version 5.11.02.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were produced using Prism 6.0 (GraphPad Software) or SigmaPlot 11.0 (Systat Software). All data are expressed as the mean ± standard deviation (SD) or standard error of the mean (SEM) where n indicates the number of biological replicates for each experiment. Statistical significance was determined for triplicate experiments by two-way ANOVA with a post-hoc Sidak’s correction for multiple comparisons (Figure 6F, S7F, S8B and S8D), one-way ANOVA with a post-hoc Dunnett’s test for multiple comparisons (Figure S7G), or multiple t-tests (Figure 6D).
Supplementary Material
Movie S1. Structural dynamics of the full-length complex of CaM/GRK5 in an MD simulation, Related to Figures 2 and 3
This simulation trajectory of the full-length CaM/GRK5 complex shows that the active conformation of GRK5 (blue with the αN-helix in orange and sangivamycin highlighted in licorice) maintains a stable interaction with CaM (green with Ca2+ ions in red). For clarity, water molecules and salt ions are not shown. The first frame shows the model of the CaM/GRK5 complex, from which the simulation was initiated. The remainder of the movie shows 50 ns of unrestrained MD (the last step in the equilibration protocol) followed by 900 ns of the production simulation. Simulation 2 of CaM-GRK5 (full length) was used to generate this movie (Table S1).
Table S3. HDX data for GRK5 and CaM, Related to Figure 2.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-GRK4-6 (clone A16/17) | Millipore | Cat# 05-466, RRID:AB_11213523 |
| Rabbit monoclonal anti-α-synuclein (clone D37A6) | Cell Signaling Technology | Cat# 4179, RRID:AB_1904156 |
| Mouse monoclonal anti-phospho-α-synuclein (Ser129) | Wako | Cat# 015-25191, RRID:AB_2537218 |
| Rabbit monoclonal anti-CaMKIIα (clone D10C11) | Cell Signaling Technology | Cat# 11945, RRID:AB_2797775 |
| Rabbit polyclonal anti-CaMKII (pan) | Cell Signaling Technology | Cat# 3362 RRID:AB_2067938 |
| Rabbit monoclonal anti-phospho-CaMKIIIα (Thr286) (clone D21E4) | Cell Signaling Technology | Cat# 12716, RRID:AB_2713889 |
| Mouse monoclonal anti-α-tubulin | Sigma-Aldrich | Cat# T5168, RRID:AB_477579 |
| Rabbit monoclonal anti-GAPDH (clone D16H11) | Cell Signaling Technology | Cat# 5174, RRID:AB_10622025 |
| Rabbit polyclonal anti-pan-cadherin | Cell Signaling Technology | Cat# 4068, RRID:AB_2158565 |
| Bacterial and Virus Strains | ||
| E. coli cells BL21 Rosetta-2(DE3)pLysS | Novagen | Cat# 71400 |
| E. coli DH10Bac | Thermo Fisher Scientific | Cat# 10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lysozyme | Sigma-Aldrich | Cat# L6876 |
| Sangivamycin | Sigma-Aldrich | Cat# S5895 |
| Protease Inhibitors | Roche | Cat# 4693159001 |
| ATP, [γ-32P] | PerkinElmer | Cat# BLU002Z001MC |
| Gel Filtration Markers Kit for Protein Molecular Weights 12,000-200,000 Da | Sigma-Aldrich | Cat# MWGF200-1KT |
| Bovine trypsin | Pronin et al., 2000, Sigma-Aldrich | Cat# T1426 |
| Human α-Synuclein | Sigma-Aldrich | Cat# S7820 |
| D-desthiobiotin | IBA Life Sciences | Cat# 2-1000-002 |
| Benzamidine | Sigma-Aldrich | Cat# 6506 |
| Leupeptin | Thermo Fisher Scientific | Cat# BP2662-100 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| FBS | Thermo Fisher Scientific | Cat# 35-010-CV |
| Sf-900 II media | Thermo Fisher Scientific | Cat# 10902088 |
| Gentamicin | Thermo Fisher Scientific | Cat# 15750060 |
| DMEM with L-glutamine | Corning | Cat# 10-017-CV |
| PhosSTOP phosphatase inhibitor cocktail | Sigma-Aldrich | Cat#4906837001 |
| ProLong Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36966 |
| Pseudomonas fragi endopeptidase Asp-N | Sigma-Aldrich | Cat# P3303 |
| L-α-Phosphatidylcholine | Sigma-Aldrich | Cat# P5638 |
| 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) | Pierce | Cat# 22980 |
| N-hydroxysulfosuccinimide (sulfo-NHS) | Pierce | Cat# 24510 |
| StrepTactin XT | IBA Life Sciences | Cat# 2-4370-000 |
| Ethanolamine hydrochloride | GE Healthcare | Cat# BR-1008-38 |
| Potassium hexacyanoferrate (III) (K3Fe(CN)6) | Sigma-Aldrich | Cat# 244023 |
| A23187 | Sigma-Aldrich | Cat# C7522 |
| Okadaic acid | Sigma-Aldrich | Cat# 49609 |
| GRK5 Peptide Substrate | New England Peptide | AEMWYSEVEEARRR |
| PAR1-AP | Peptide 2.0 | SFLLRN |
| Kinase Substrates Library | Anaspec | Cat# AS-62017-1 |
| Cellfectin II | Thermo Fisher Scientific | Cat# 10362100 |
| Penicillin-Streptomycin | Corning | Cat# 30-002-Cl |
| W-7, Hydrochloride | Sigma-Aldrich | Cat# 681629 |
| Amine Coupling Kit | GE Healthcare | Cat# BR100050 |
| Critical Commercial Assays | ||
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | Cat# 200523 |
| Deposited Data | ||
| CaM-GRK5 atomic coordinates | This paper | PDB: 6PJX |
| Experimental Models: Cell Lines | ||
| Insect: Sf9 cells | Expression Systems | Cat# 94-001S |
| Human: HEK293 cells | ATCC | N/A |
| Recombinant DNA | ||
| pET28b-wtCaM | This study | N/A |
| pET28b-mutCaM D22A/D58A | This study | N/A |
| pET28b-mutCaM D95A/D131A | This study | N/A |
| pVL1392-wtβ2AR | Komolov et al., 2017 | N/A |
| pVL1392-wtGRK5 | Komolov et al., 2017 | N/A |
| pVL1392-mutGRK5 D91C/K454C | Komolov et al., 2017 | N/A |
| pFastBac-mutGRK5 K139A/K454A/R455A /M460A | Komolov et al., 2017 | N/A |
| pFastBac-mutGRK5 18-590 (ΔN17) | This study | N/A |
| pFastBac-mutGRK5 36-590 (ΔN35) | This study | N/A |
| pFastBac-mutGRK5 1-541 (ΔC) | This study | N/A |
| pFastBac-mutGRK5 18-541 (ΔN/ΔC) | This study | N/A |
| pFastBac-mutGRK5 L3A | This study | N/A |
| pFastBac-mutGRK5 L550A, L551A, L554A and F555A (4A) | This study | N/A |
| pcDNA3.1-Human GRK5-strep | This paper | N/A |
| pcDNA3.1-Human GRK5-strep L3A | This paper | N/A |
| pcDNA3.1-Human GRK5-strep I6A | This paper | N/A |
| pcDNA3.1-Human GRK5-strep V7A | This paper | N/A |
| pcDNA3.1-Human GRK5-strep T10A | This paper | N/A |
| pcDNA3.1-Human GRK5-strep V11A | This paper | N/A |
| pcDNA3.1-Human GRK5-strep K14A | This paper | N/A |
| pcDNA3.1-Human GRK5 K215R | This paper | N/A |
| pcDNA3.1-Human GRK5 | This paper | N/A |
| pEGFP-Human GRK5 | This paper | N/A |
| pEGFP-Human GRK5 L3A | This paper | N/A |
| pEGFP-Human GRK5 4A | This paper | N/A |
| pRSV-CaMKII | Addgene | Cat#45064 |
| Software and Algorithms | ||
| Prism 6.0 | Graphpad | http://graphpad.com |
| PyMol 1.5 | Schrodinger | http://pymol.org |
| SigmaPlot 11 | Systat Software | https://systatsoftware.com/products/sigmaplot |
| Phenix 1.15 | (Liebschner et al., 2019) | http://www.phenix-online.org/ |
| Phaser 2.7.14 | (McCoy et al., 2007) | N/A |
| Coot 0.8.9.1 | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| HKL-3000 | (Minor et al., 2006) | https://hkl-xray.com/hkl-3000 |
| MolProbity | (Williams et al., 2018) | http://molprobity.biochem.duke.edu/ |
| DynDom server | (Lee et al., 2003) | http://dyndom.cmp.uea.ac.uk/dyndom |
| Proteinlynx Global Server 2.4 | Waters | HDX-MS Toolbox |
| DynamX 2.0 | Waters | HDX-MS Toolbox |
| MODELLER 9.15 | Shen and Sali, 2006 | http://salilab.org/modeller |
| Prime | Schrodinger | http://schrodinger.com/prime |
| CHARMM36m force field | Huang et al., 2017 | http://mackerell.umaryland.edu/charmm_ff.shtml |
| CHARMM General Force Field | Vanommeslaeghe et al., 2010 | http://cgenff.paramchem.org |
| ParamChem server 1.0 | N/A | http://paramchem.org |
| Dabble | Betz, 2018 | https://doi.org/10.5281/zenodo.836914 |
| PMEMD | AMBER16 | http://ambermd.org |
| SHAKE | AmberTools16 | http://ambermd.org |
| VMD | Humphrey et al., 1996 | www.ks.uiuc.edu/research/vmd |
| CPPTRAJ | AmberTools16 | http://ambermd.org |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij |
| ImageStudio Lite | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite |
| SEDNTERP 1.09 | Laue, T. M., Shah, B., Ridgeway, T. M. & Pelletier, 1992 | N/A |
| SEDPHAT | Rowe, 2005 | https://sedfitsedphat.nibib.nih.gov |
| GUSSI | UT Southwestern | N/A |
| NanoAnalyze | TA Instruments | N/A |
| BIAevaluation 4.1 | GE Healthcare | https://www.biacore.com/lifesciences/service/downloads/software_licenses/biaevaluation/ |
| PEP-FOLD server | Thevenet et al., 2012 | https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD |
| NIS Elements AR software 5.11.02 | Nikon Instruments | https://www.microscope.healthcare.nikon.com |
| Fiji (ImageJ) | Schindelin et al, 2012 | https://imagej.net/Fiji/Downloads |
| Seq2Logo 2.0 | Thomsen and Nielson et al, 2012 | http://www.cbs.dtu.dk/biotools/Seq2Logo/ |
HIGHLIGHTS.
CaM and GRK5 form a bipartite interface within a 1:1 complex
CaM triggers large conformational changes in GRK5
CaM activates GRK5-mediated phosphorylation of non-receptor substrates
The mechanism of GRK5 activation by CaM and GPCRs is largely conserved
ACKNOWLEDGEMENTS
We thank Drs. Yang Du and Brian Kobilka for providing purified β2AR, Dr. Philip Wedegaertner for the GRK5 4A construct, Dr. Ji-Fang Zhang for the CaM construct and Dr. Joao P.G.L.M. Rodrigues for help with generating the movie, and the Wistar Proteomics Facility for mass spectrometry analysis. This research was supported by NIH awards R35GM122541 and R01HL142310 (to J.L.B), R01GM104047 (to B.E.T), R01GM127359 (to R.O.D.) and the National Research Foundation of Korea funded by the Korean government (NFR-2019R1A5A2027340 and NRF-2018R1A2B6001554) (to K.Y.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames JB, Levay K, Wingard JN, Lusin JD, and Slepak VZ (2006). Structural basis for calcium-induced inhibition of rhodopsin kinase by recoverin. J. Biol. Chem 281, 37237–37245. [DOI] [PubMed] [Google Scholar]
- Arawaka S, Wada M, Goto S, Karube H, Sakamoto M, Ren C-H, Koyama S, Nagasawa H, Kimura H, Kawanami T, et al. (2006). The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson’s disease. J. Neurosci 26, 9227–9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz RM (2018). Dabble. 10.5281/zenodo.836914. [DOI] [Google Scholar]
- Beyett TS, Fraley AE, Labudde E, Patra D, Coleman RC, Eguchi A, Glukhova A, Chen Q, Williams RM, Koch WJ, et al. (2019). Perturbation of the interactions of calmodulin with GRK5 using a natural product chemical probe. Proc. Natl. Acad. Sci 201818547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguth CA, Singh P, Huang C, and Tesmer JJG (2010). Molecular basis for activation of G protein-coupled receptor kinases. EMBO J. 29, 3249–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case DA, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, et al. (2016). AMBER 2016 (University of California, San Francisco: ). [Google Scholar]
- Chen CY, Dion SB, Kim CM, and Benovic JL (1993). β-Adrenergic receptor kinase. Agonist-dependent receptor binding promotes kinase activation. J. Biol. Chem 268, 7825–7831. [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010a). MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhu H, Yuan M, Fu J, Zhou Y, and Ma L (2010b). G-protein-coupled receptor kinase 5 phosphorylates p53 and inhibits DNA damage-induced apoptosis. J. Biol. Chem 285, 12823–12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley MF, Williamson MJ, and Walker RC (2009). CHAMBER: Comprehensive support for CHARMM force fields within the AMBER software. Int. J. Quantum Chem 109, 3767–3772. [Google Scholar]
- Duc NM, Du Y, Thorsen TS, Lee SY, Zhang C, Kato H, Kobilka BK, and Chung KY (2015). Effective application of bicelles for conformational analysis of G protein-coupled receptors by hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom 26, 808–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen JR, Wales TE, Chen S, Marzluff EM, Hassell KM, Weis DD, and Smithgall TE (2013). Partial cooperative unfolding in proteins as observed by hydrogen exchange mass spectrometry. Int. Rev. Phys. Chem 32, 96–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen M-Y, Pieper U, and Sali A (2006). Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinforma Chapter 5, Unit-5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold JI, Martini JS, Hullmann J, Gao E, Chuprun JK, Lee L, Tilley DG, Rabinowitz JE, Bossuyt J, Bers DM, et al. (2013). Nuclear translocation of cardiac G protein-Coupled Receptor kinase 5 downstream of select Gq-activating hypertrophic ligands is a calmodulin-dependent process. PLoS One 8, e57324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Tesmer JJG, Mushegian A, and Gurevich VV (2012). G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther 133, 40–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins MK, Oprian DD, and Schertler GFX (2006). Recoverin binds exclusively to an amphipathic peptide at the N terminus of rhodopsin kinase, inhibiting rhodopsin phosphorylation without affecting catalytic activity of the kinase. J. Biol. Chem 281, 19426–19432. [DOI] [PubMed] [Google Scholar]
- Hopkins CW, Le Grand S, Walker RC, and Roitberg AE (2015). Long-Time-Step Molecular Dynamics through Hydrogen Mass Repartitioning. J. Chem. Theory Comput 11, 1864–1874. [DOI] [PubMed] [Google Scholar]
- Huang C-C, Orban T, Jastrzebska B, Palczewski K, and Tesmer JJG (2011). Activation of G protein-coupled receptor kinase 1 involves interactions between its N-terminal region and its kinase domain. Biochemistry 50, 1940–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Yoshino-Koh K, and Tesmer JJG (2009). A surface of the kinase domain critical for the allosteric activation of G protein-coupled receptor kinases. J. Biol. Chem 284, 17206–17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, Grubmuller H, and MacKerell AD (2017). CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph 14, 33-38, 27–28. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 79, 926–935. [Google Scholar]
- Kim R (2011). Native agarose gel electrophoresis of multiprotein complexes. Cold Spring Harb. Protoc 6, 884–887. [DOI] [PubMed] [Google Scholar]
- Komolov KE, and Benovic JL (2018). G protein-coupled receptor kinases: Past, present and future. Cell Signal 41, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komolov KE, Senin II, Kovaleva NA, Christoph MP, Churumova VA, Grigoriev II, Akhtar M, Philippov PP, and Koch K-W (2009). Mechanism of rhodopsin kinase regulation by recoverin. J. Neurochem 110, 72–79. [DOI] [PubMed] [Google Scholar]
- Komolov KE, Bhardwaj A, and Benovic JL (2015). Atomic Structure of GRK5 Reveals Distinct Structural Features Novel for G Protein-coupled Receptor Kinases. J. Biol. Chem 290, 20629–20647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komolov KE, Du Y, Duc NM, Betz RM, Rodrigues JPGLM, Leib RD, Patra D, Skiniotis G, Adams CM, Dror RO, et al. (2017). Structural and Functional Analysis of a β2-Adrenergic Receptor Complex with GRK5. Cell 169, 407–421. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Krupnick JG, and Benovic JL (1998). The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol 38, 289–319. [DOI] [PubMed] [Google Scholar]
- Laue TM, Shah B, Ridgeway TM & Pelletier SL (1992). Theory of computer aided interpretation of sedimentation data. (Royal Society of Chemistry, Cambridge, England: ), pp. 90–125. [Google Scholar]
- Lefkowitz RJ (2007). Seven transmembrane receptors: something old, something new. Acta Physiol. (Oxf). 190, 9–19. [DOI] [PubMed] [Google Scholar]
- Levay K, Satpaev DK, Pronin AN, Benovic JL, and Slepak VZ (1998). Localization of the sites for Ca2+-binding proteins on G protein-coupled receptor kinases. Biochemistry 37, 13650–13659. [DOI] [PubMed] [Google Scholar]
- Mendez A, Burns ME, Roca A, Lem J, Wu LW, Simon MI, Baylor DA, and Chen J (2000). Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron 28, 153–164. [DOI] [PubMed] [Google Scholar]
- Miller CJ, and Turk BE (2016). Rapid Identification of Protein Kinase Phosphorylation Site Motifs Using Combinatorial Peptide Libraries In Methods in Molecular Biology, Zegzouti H, and Goueli SA, eds. (New York, NY: Springer New York; ), pp. 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble B, Kallal LA, Pausch MH, and Benovic JL (2003). Development of a yeast bioassay to characterize G protein-coupled receptor kinases. Identification of an NH2-terminal region essential for receptor phosphorylation. J. Biol. Chem 278, 47466–47476. [DOI] [PubMed] [Google Scholar]
- Oda T, Yamamoto T, Kato T, Uchinoumi H, Fukui G, Hamada Y, Nanno T, Ishiguchi H, Nakamura Y, Okamoto Y, et al. (2018). Nuclear translocation of calmodulin in pathological cardiac hypertrophy originates from ryanodine receptor bound calmodulin. J. Mol. Cell. Cardiol 125, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorato JJ, Palczewski K, Regan JW, Caron MG, Lefkowitz RJ, and Benovic JL (1991). Role of acidic amino acids in peptide substrates of the beta-adrenergic receptor kinase and rhodopsin kinase. Biochemistry 30, 5118–5125. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Buczytko J, Kaplan MW, Polans AS, and Crabb JW (1991). Mechanism of rhodopsin kinase activation. J. Biol. Chem 266, 12949–12955. [PubMed] [Google Scholar]
- Pao CS, Barker B, and Benovic JL (2009). Role of the amino terminus of G protein-coupled receptor kinase 2 in receptor phosphorylation. Biochemistry 48, 7325–7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza M, Taiakina V, Dieckmann T, and Guillemette JG (2017). Structural Consequences of Calmodulin EF Hand Mutations. Biochemistry 56, 944–956. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Satpaev DK, Slepak VZ, and Benovic JL (1997). Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J. Biol. Chem 272, 18273–18280. [DOI] [PubMed] [Google Scholar]
- Pronin AN, Morris AJ, Surguchov A, and Benovic JL (2000). Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem 275, 26515–26522. [DOI] [PubMed] [Google Scholar]
- Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, and Cheatham TE (2013). PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- Rowe A (2005). Weak Interactions: Optimal Algorithms for Their Study in the AUC In Analytical Ultracentrifugation, Scott DJ, Harding SE, and Rowe AJ, eds. (Cambridge: Royal Society of Chemistry; ), pp. 484–500. [Google Scholar]
- Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, and Walker RC (2013). Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput 9, 3878–3888. [DOI] [PubMed] [Google Scholar]
- Schrodinger L (2010). The PyMOL Molecular Graphics System v.1.3r1. [Google Scholar]
- Shichi H, and Somers RL (1978). Light-dependent phosphorylation of rhodopsin. Purification and properties of rhodopsin kinase. J. Biol. Chem 253, 7040–7046. [PubMed] [Google Scholar]
- Simon B, Huart A-S, and Wilmanns M (2015). Molecular mechanisms of protein kinase regulation by calcium/calmodulin. Bioorg. Med. Chem 23, 2749–2760. [DOI] [PubMed] [Google Scholar]
- So CH, Michal AM, Mashayekhi R, and Benovic JL (2012). G Protein-coupled Receptor Kinase 5 Phosphorylates Nucleophosmin and Regulates Cell Sensitivity to Polo-like Kinase 1 Inhibition. J. Biol. Chem 287, 17088–17099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, and Tuffery P (2012). PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 40, W288–W293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiyagarajan MM, Stracquatanio RP, Pronin AN, Evanko DS, Benovic JL, and Wedegaertner PB (2004). A predicted amphipathic helix mediates plasma membrane localization of GRK5. J. Biol. Chem 279, 17989–17995. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. (2009). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem NA-NA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Raman D, Wei J, Kennedy MJ, Hurley JB, and Gurevich VV (2007). Regulation of arrestin binding by rhodopsin phosphorylation level. J. Biol. Chem 282, 32075–32083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X-Q, Cato MC, Labudde E, Beyett TS, Tesmer JJG, and Grant BJ (2017). Navigating the conformational landscape of G protein-coupled receptor kinases during allosteric activation. J. Biol. Chem 292, jbc.M117.807461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap KL, Kim J, Truong K, Sherman M, Yuan T, and Ikura M (2000). Calmodulin target database. J. Struct. Funct. Genomics 1, 8–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Structural dynamics of the full-length complex of CaM/GRK5 in an MD simulation, Related to Figures 2 and 3
This simulation trajectory of the full-length CaM/GRK5 complex shows that the active conformation of GRK5 (blue with the αN-helix in orange and sangivamycin highlighted in licorice) maintains a stable interaction with CaM (green with Ca2+ ions in red). For clarity, water molecules and salt ions are not shown. The first frame shows the model of the CaM/GRK5 complex, from which the simulation was initiated. The remainder of the movie shows 50 ns of unrestrained MD (the last step in the equilibration protocol) followed by 900 ns of the production simulation. Simulation 2 of CaM-GRK5 (full length) was used to generate this movie (Table S1).
Table S3. HDX data for GRK5 and CaM, Related to Figure 2.
Data Availability Statement
The coordinates for the crystal structure of Ca2+/CaM bound to GRK5 have been deposited in the Protein Data Bank under code PDB:6PJX.