

Abstract
Intellectual disability (ID) corresponds to several neurodevelopmental disorders of heterogeneous origin in which cognitive deficits are commonly associated with abnormalities of dendrites and dendritic spines. These histological changes in the brain serve as a proxy for underlying deficits in neuronal network connectivity, mostly a result of genetic factors. Historically, chromosomal abnormalities have been reported by conventional karyotyping, targeted fluorescence in situ hybridization (FISH), and chromosomal microarray analysis. More recently, cytogenomic mapping, whole-exome sequencing, and bioinformatic mining has led to the identification of novel candidate genes, including genes involved in neuritogenesis, dendrite maintenance, and synaptic plasticity. Greater understanding of the roles of these putative ID genes and their functional interactions might boost investigations into determining the plausible link between cellular and behavioral alterations as well as the mechanisms contributing to the cognitive impairment observed in ID. Genetic data combined with histological abnormalities, clinical presentation, and transgenic animal models provide support for the primacy of dysregulation in dendrite structure and function as the basis for the cognitive deficits observed in ID. In this review, we highlight the importance of dendrite pathophysiology in the etiologies of four prototypical ID syndromes, namely Down Syndrome (DS), Rett Syndrome (RTT), Digeorge Syndrome (DGS) and Fragile X Syndrome (FXS). Clinical characteristics of ID have also been reported in individuals with deletions in the long arm of chromosome 10 (the q26.2/q26.3), a region containing the gene for the collapsin response mediator protein 3 (CRMP3), also known as dihydropyrimidinase-related protein-4 (DRP-4, DPYSL4), which is involved in dendritogenesis. Following a discussion of clinical and genetic findings in these syndromes and their preclinical animal models, we lionize CRMP3/DPYSL4 as a novel candidate gene for ID that may be ripe for therapeutic intervention.
Keywords: Intellectual disability, dendrite dysgenesis, transgenic mice, CRMP3/DPYSL4, chromosome 10 (q26) deletion
Etiology of intellectual disability (ID)
Intellectual Disability is defined in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), by three primary criteria: 1) onset before age 18; 2) an intelligence quotient (IQ) below 70; 3) impairment of general mental ability that impacts adaptive functioning in three domains including conceptual, social, and practical skills. Neurodevelopmental abnormalities with cognitive and communication deficits are widely evident in children diagnosed with ID. Roughly two-thirds of ID patients present with a mild-to-moderate level of impairment and one-third develop a severe-to-profound level of impairment. In addition, these patients are at risk for developing various comorbidities, such as sensory impairments, convulsive disorders, behavioral disturbances, and psychiatric illnesses. Development of the human brain results from highly complex and coordinated activity involving genetic and environmental processes. This varied network of interactions suggests that the underlying causative factors leading to ID are heterogenous in nature. Numerous studies have provided evidence that ID is not a single disease, but rather a common hallmark of a collection of heterogeneous disorders [96]. The pathogenesis of these developmental disorders can best be viewed through the lens of altered intracellular signaling cascades, which arise from environmental stimuli or genetic factors, and culminate in the development of a pathological phenotype. The most common environmental factors leading to ID include exposure to neurotoxic compounds (alcohol, lead, mercury, drugs), malnutrition during pregnancy, and peri/post-natal asphyxia. These factors play a major role in contributing to the prevalence of mild forms of ID [44, 101]. In contrast to environmental factors, genetic causes can include a single genetic defect, such as deletion or mutation of a single gene (SYN1, POLR3B, SLC5A2; SYNGAP1, DYRK1A, SCN2). In addition, chromosomal deletion, aneusomies, inversion, and structural abnormalities may provoke more severe forms of ID [23, 72]. Commonly occurring forms include, but are not limited to, DS, RTT, DGS, and FXS. Laboratory findings and clinical data suggest that aberrant activity of metabolic, transcriptional, and signaling pathways are the most common molecular factors involved in the development of the various forms of ID [16, 224]. Neuropathologically, alterations in dendritic tree morphology have been reported in human neurological and psychiatric diseases and are particularly prevalent in those disorders associated with ID [51, 137].
Dendrite physiology: from specification to dysgenesis in ID
Neurons are characterized by a highly polarized cellular morphology, composed of an axon and dendrites, which are critical for the formation of neuronal circuits. These processes are the essential structural elements that allow information to be transmitted and processed in the nervous system. Dendrites are highly branched structures with characteristic arborization and morphology that can be distinguished after 5 days in a morphogenetic model of cultured hippocampal neurons (stage 4)[55]. The specific pattern of a dendritic tree can often be associated with a neuronal cell type, such as Purkinje or cortical neurons, and their physical structure reflects the complexity of their computational task. During development, initial wiring of the nervous system depends on precise refinement and remodeling of dendritic arbors through extension, addition, elongation, retraction, and pruning of individual dendrites [122]. These processes are highly dynamic, and dendritic structure is intimately influenced by local synaptic activity. Additionally, dendrites are crucial for the spatial and temporal integration of upstream information within neuronal circuits. The appearance of dendritic retraction, together with synaptic disassembly prior to soma/axon loss, correlates with the development of substantial functional deficits and is associated with early signs of pathology in a number of neurodegenerative disorders [58, 114].Various intrinsic signaling proteins mediate temporal and spatial control of dendritic arborization, which ensures dendritic growth is correctly generated and maintained over a lifetime [127, 166]. The roles of these molecules are functionally intertwined and highly regulated, which allows for tight control over dendritic extension and arborization. The plethora of molecular regulators of dendritic structure include neuromodulators and neurotransmitters, such as 5-hydoxytryptamine (5-HT) and gamma aminobutyric acid (GABA), as well as cell surface receptors, including neuropilin/plexin, Notch, Trks, and EpH receptors [75, 178, 205]. Ligands for these receptors, like the neurotrophic factors ciliary neurotrophic factor (CNTF), insulin-like growth factor (IGF), nerve growth factor (NGF), and brain-derived neurotrophic factor (BDNF) [29, 169], or the semaphorins, are fundamental to regulation of dendritic structure. Regulators also include cell adhesion molecules L1CAM, Cadherin-catenin adhesion complex, SAX7, and MNR-1[205]. Signaling molecules, such as CRMPs [172], Calmodulin Kinase 2b (CaMKIIb), and CRP1, as well as calcium signaling proteins and transcription factors NF-kB, CuX1-2, Neurogenin-2, CREB, and CREST [1, 37, 52] have critical roles. Cytoskeletal proteins are the final effectors supporting the morphology of dendrites, and synaptic scaffolding proteins, PSD95 and Cypin [2, 135, 157], as well as microtubule and tubulin polymerization regulators, APC, EB3, XMAP215, TACC3, and MAP1, are involved in regulation of dendritic structure.
Most dendrites have spines that can be classified into four morphological categories including, stubby, thin, filopodial, and mushroom. These structures serve as the main postsynaptic contact sites of glutamatergic excitatory synapses in the CNS [32]. Cytoskeletal elements within dendritic spines are primarily composed of cortactin, ARP2/3 complex, PSD 95/93, cofilin, actin filaments, and the regulatory Rho GTPases Rac, Rho, and Cdc42 [123]. These postsynaptic dendritic protrusions are important regulators of physiological Ca2+ gradients and serve to amplify local levels of calcium, which mediate activation of downstream calcium-dependent signaling pathways that are essential for synaptic plasticity [158]. Despite the small size of spines, a multitude of molecules have been implicated in their development and remodeling.
It is therefore tempting to assume that various etiological changes, such as environmental and genetic factors, will impact dendrite and spine structure and influence cognitive function [26, 153]. This is particularly true when considering neuronal mechanisms underlying ID, where defects in neuronal structure, network connectivity, and plasticity are critical features associated with common clinical presentations. In line with this concept of structural abnormality, alteration of the dendritic arbor is a consistent feature exhibited by patients with ID across multiple clinical presentations, as assessed on post-mortem neural tissues, with significant alterations of several important CNS areas, including the hippocampus, dentate gyrus, and cerebral cortex [180, 208]. Golgi staining of pyramidal neurons in the hippocampus and cerebral cortex in patients with DS, RTT, DGS and FXS, revealed a reduction in dendritic branching, arborization, and abnormalities in the morphology and density of dendritic spines compared to normal controls [22, 43]. This finding is highly suggestive of altered neuronal connectivity in these syndromes. Pathological spine loss can be caused by altered expression of various signaling proteins, such as Rho-GEF (KALRN), Rho-GAP (OPHN1), Cdc42-GEF (ARHGEF9), and Rac-GAP (OCRL1) [39, 161]. Moreover, SAP102 (DLG3; XLID) and PSD95 (DLG4) are associated with anchoring NMDA receptors as well as other major PSD proteins, such as HOMER, CaMKII, SynGAP, DLG-associated protein (DLGAP/GKAP) and SHANKs. Remarkably, a high number of de novo mutations and genomic aberrations have been identified in SHANK3, SHANK2, SYNGAP1, and GKAP in patients with ID, suggesting that alterations in glutamatergic neurons contribute, at least in part, to the cognitive impairment. Indeed in ID, dendritic abnormalities appear to correlate, to some extent, with the cognitive impairment profile of the patient [141].
Phenotypic features of patients with prototypic ID syndromes
ID of genetic origin, including DS, RTT, DGS and FXS, present with specific clinical and histopathological features and, although abnormalities can extend to many organs, we limited this review to those involving the nervous system.
DS.
A hallmark observation in neonatal DS patients is the presence of hypotonia, which is responsible for delayed motor development. Functionally, the IQ in these patients decreases over the course of the first decade of life and reaches a plateau that continues into adulthood with deficits in working memory/spatial skills, and reading/language [9]. Studies have reported microcephaly, reduced hippocampal and cerebral cortex size, and reduced cortical thickness. Neuropathological findings include abnormal neuronal differentiation, abnormal cortical lamination, decreased dendritic branching, reduced spine density, and altered dendritic arborization and spine morphology [192]. Aberrant electrophysiological properties of CNS neurons have also been reported. Patients also develop neuropathology closely resembling that of Alzheimer’s disease (AD) by the age of 50-60 years [214].
RTT.
RTT is almost exclusively found in female patients, with signs of the disorder often starting to emerge only after 6–18 months of life [40, 71], although they can appear ealier in the congenital variant. The onset of mental decline coincides with the emergence of motor abnormalities characterized by apraxia and often an ataxic gait that may develop into a Parkinsonian phenotype [94]. Patients exhibit partial or complete loss of spoken language, as well as a lack of hand movements. Many children with RTT display reduced brain volume, which is consistent with a smaller head circumference. This phenotype arises from the relatively small somatic volume of neurons associated with dense packing of these smaller cells in the cerebral cortex, cerebellum, and hippocampus, and reduced dendritic arborization. Neuropathologically, reduced dendritic morphological complexity is present throughout life. Apical dendrites from neurons of the hippocampus, prefrontal, motor, and middle temporal cortical areas are often asymmetric in structure and significantly reduced in number with some segments completely devoid of spines [11, 152]. In addition, phosphorylation of MAPs, which acts to stabilize microtubules, is decreased in postmortem tissues from these patients.
DGS.
DGS is a frequent cause of ID, with an estimated prevalence of approximately 1/3000 – 1/4000 live births [139]. Patients often exhibit complex and variable phenotypes, including developmental delay, hearing loss, as well as anomalies of the eye, craniofacial structures, and musculoskeletal systems. Approximately one-third of patients with DGS display mild to moderate ID, often associated with psychiatric disorders, such as schizophrenia and attention-deficit/hyperactivity disorder (ADHD) [206]. A global reduction in brain volume is observed in these patients, with widespread reductions in frontal, temporal, parietal, and occipital lobes, as well as atrophy of the cerebellum and hippocampus [92]. Post-mortem studies have revealed abnormalities in neuronal migration, organization, and connectivity [184]. Functional MRI reported patterns of hypoconnectivity in large-scale neuronal networks, which are necessary for higher order cognitive functions [110].
FXS.
FXS patients displaying an ID phenotype are found at a rate of approximately 1:7000 in males and 1:11000 in females [143]. They experience learning deficits, delayed language acquisition, impaired motor skills, and repetitive behavior. In addition, they exhibit a range of psychiatric symptoms including anxiety, poor social interaction, hyper-arousal/sensitivity, disrupted sleep, ADHD, and autism spectrum symptoms [130]. Male FXS patients demonstrate severe disability, while female patients are often less affected due to mosaicism resulting from X-chromosome inactivation, which randomly occurs early in embryogenesis [116]. During adulthood, FXS patients may be at increased risk of parkinsonism, and in some cases, dementia [187, 203]. These patients display impaired neurite extension with global failure of normal spine maturation and pruning during development that persists throughout adulthood.
Genetic basis of specific ID syndromes
Next-generation sequencing (NGS), using either whole exome or genome panels, as well as chromosomal microarray analysis (CMA), have emerged as powerful new tools for identification of genomic abnormalities associated with a wide range of ID syndromes. Genomic assays include single nucleotide polymorphism (SNP) arrays and comparative genomic hybridization (CGH) arrays, both of which are useful for detection of genomic copy number variants (CNV), such as microdeletions and microduplications. Together with karyotyping, these techniques have revealed that the frequency of disease-causing CNVs is highest (20%-25%) in children with moderate to severe ID and is accompanied by malformations/dysmorphic features and specific genetic mutations.
DS.
The phenotypic variability present in patients with this syndrome is due to the presence of three copies of functional genes on HSA21 loci. HSA21 is the smallest human chromosome with a density of 15 genes per megabase (Mb) and remains among the poorest chromosomes in terms of functional DNA elements per Mb. However, HSA21 shows significant enrichment for encoded proteins involved in the regulation and development of cytoskeletal structures [8]. These cytoskeletal proteins are known to be involved in neuronal development and neurological disorders, particularly in Alzheimer’s neuropathology. At least five genes located on chromosome 21 may affect neuronal development, including SIM2, DYRK1A, GART, PCP4, DSCAM [198]. The transmembrane protein DSCAM contains an intracellular domain that interacts with a number of known kinases, including PAK1, FAK, FYN, JNK, and p38 [125], as well as PSD95, and the MAGI scaffolding proteins. Importantly, overexpression of DSCAM can contribute to the intellectual disability phenotype of DS patients [182]. In this context, the reduced dendritic complexity observed in cortical and hippocampal neurons of DS patients is consistent with the reduced dendritic arborization observed following DSCAM overexpression in mouse hippocampal neurons [5, 64]. Alternatively, data from studies comparing post-mortem transcriptomes obtained from 58 ID patients with euploid controls showed that some of the dysregulated protein coding genes do not map to HSA21. Interestingly, amyloid precursor protein (APP) is encoded on chromosome 21 at location 21q21.3, and trisomy of this protein may explain the increased frequency of dementia in DS patients.
RTT.
Three genes, MECP2, CDKL5 and FOXG1, have been reported to cause RTT. Interestingly, expression of the well-known transcriptional regulator MeCP2 (Methyl-CpG-binding protein 2) correlates with neuronal differentiation and postnatal development of the CNS, which suggests a role in neuronal maintenance and activity [61, 71, 89]. MeCP2 has two known isoforms, MeCP2-e1 and MeCP2-e2, which result from differential splicing of exons 1 and 2, respectively. The existence of these isoforms contributes to the diverse functions associated with MECP2 expression, and mutations in exon 1 have been observed in RTT [3, 42, 197]. However, in a majority of cases, patients diagnosed with typical RTT have mutations in MECP2-e1/e2, which could disrupt synaptic proliferation and pruning during the restricted developmental window that coincides with peak synapse formation in the cerebral cortex between 7–18 months of age. RTT with early epilepsy, and the congenital variant of RTT, are mainly due to variations in the CDKL5 and FOXG1 genes [24, 103, 156], respectively. Additionally, because the coding region for MeCP2 is located on the X-chromosome, mutations in MECP2 contribute to the characteristic features of RTT. Since males only have one copy of the X-chromosome, mutations in males are generally incompatible with life and as such, RTT affects mainly females, although it has been reported that, in rare cases, male patients can survive until adulthood. Recently, integrative analysis of omics data has identified new genes and new signaling pathways that could be affected in RTT. Specifically, Sp1 and REST, known cis-regulatory elements for MeCP2, in addition to SIN3A and SMC3, which are parts of the MeCP2-HDAC protein complex [126], have been identified using this strategy [6, 20, 115], Other functions attributed to MeCP2 include histone deacetylase-independent repression, maintenance of DNA methylation, histone methylation, assembly of secondary chromatin structure independently of methyl CpG DNA binding capability, and interactions with RNA splicing factors. Furthermore, the expression of MeF2C, which is also known to cause ID and psychomotor impairment, is inhibited by MeCP2 together with the HDAC complex [126].
DGS.
The 22q11.2 microdeletion associated with DGS is one of the most common genetic microdeletion syndromes in humans. However, the relative importance of individual genes on chromosome 22 that contribute to cognitive and behavioral problems in patients with DGS has yet to be elucidated. Most of the coding region of chromosome 22 (~90%) is 3 Mb and contains approximately 60 genes, while the 1.5 Mb deletion (~8%) contains 28 genes critical for DGS [56, 149]. The decrease in gene dosage resulting from the deletion disrupts mitochondrial activity and limits neurite growth. Recent evidence suggests that ZDDHC8, DGCR8, and DGCT8 can compromise neuronal circuits in the hippocampus [138]. RANPB1, TRMT2A, HIRA, CDC45L, and UFD1L are additional genetic candidates that potentially contribute to DGS by altering cortical development. In addition, TBX1 is implicated in several clinical features including the formation of the craniofacial region, thymus, parathyroid gland, and the aortic arch, which are organs and structures affected in the 22q11.2 deletion. Furthermore, in the hippocampus, RTNR4 influences dendritic spine morphology and limits activity-dependent synaptic strengthening [30]. Finally, COMT, PRODH, and PIK4CA, have been investigated in the context of schizophrenia as the 22q11.2 deletion was the CNV the most significantly associated with the disease. DGS is one of the rare examples in humans of a cytogenetic abnormality occurring in conjunction with a psychiatric disease. Phenotypic pathology in a subset of DGS patients has also been associated with mutations on the non-deleted allele, leading to unmasking of additional conditions [140]. This suggests that variability in the deletion size and breakpoint locations may play an important role in various DGS phenotypes.
FXS.
Approximately 1100 genes have been annotated on the X chromosome, of which at least 800 are protein-coding, and approximately 696 are expressed in the brain. There are two clinical presentations associated with FXS. The first is a neurodevelopmental disorder that manifests following full mutation of the FMR1 gene, which is located on the q27.3 locus of the X chromosome in FXS patients. The second, known as Fragile X-associated tremor/ataxia syndrome (FXTAS), is a neurodegenerative disease experienced during aging of premutation carriers with the FMR1 gene. The molecular mechanism underlying silencing of the FMR1 gene arises from expansions in the length of the FMR1-5’untranslated region (UTR). Traditional FXS patients carry a full-length mutation, which is composed of >200 CGG repeats [83, 143]. FXS and FXTAS patients express differing levels of CGG repeats, which alter the translation of the FMR protein (FMRP), with deficiency observed in FXS and increases found in FXTAS. FMRP is an RNA-binding protein that controls the translation of a large array of mRNAs, which are critical for dendritic arborization and spine structure [54, 165]. In addition, FMRP modulates local protein synthesis in dendrites and optimizes the activity of several signaling cascades through interactions with Rac1, Arc, PSD95, MAP1B, CaMKII, calbindin, and cadherins, which are essential for spine morphogenesis [14, 77]. Furthermore, FMRP induces age-dependent impairments in synaptic plasticity, specifically in LTP function.
Evidence suggests that loss of function (LOF) associated with a subset of genes may give rise to a large variety of brain disorder phenotypes. For example, ID and schizophrenic patients, which also display dendritic abnormalities, have overlapping LOF mutations in POGZ, SCN2A and SYNGAP1 genes. Additionally, ITSN, which encodes intersectin -a specific guanine nucleotide exchange factor for Cdc42- has been implicated in both AD and DS. Mutations of these genes produce the dystrophic dendritic spine phenotype present in various forms of ID. This suggests that different categories of proteins that are altered in ID may operate in the same molecular and cellular processes, which points toward the next challenge of identifying a common molecular basis among various ID syndromes.
Animal models of ID
The interpretation of genetic causality associated with ID is not straightforward, as patients may have comorbid and unknown genomic abnormalities. Extensive efforts have been undertaken to generate mouse models with the aim of unravelling the underlying physiopathology of ID. Genetically null mouse models cannot completely recapitulate the complexity of human disease [18] and there are examples of non-concordant, and even paradoxical, relationships between human and murine findings. However, animal models frequently allow the identification of a single genetic target, such as through rescue of phenotypic abnormalities by experimental gene transfer, which can contribute to deciphering the detailed genetic processes underlying disease pathogenesis [65, 66].
DS mouse models.
Because DS is a contiguous genetic syndrome that spans the entire 35 Mb of the long arm of HSA21 [215], there is no simple or complete trisomy mouse model of DS. In the past, several DS mouse models (Ts65Dn, Ts1Cje, Tc(Hsa21)1TybEmcf, Dp(10/16/17)1Yey) [46, 87] have been generated, with each displaying trisomy of a different subset of HSA21 genes or their orthologs. Trisomy of multiple genes, including Dyrk1A, Synj1, and Sim2 have been found to cause learning and memory defects in Ts65Dn mice, which suggests the possibility that overexpression of these genes may be associated with learning disability in patients with DS [10, 175]. In addition, several Cre/loxP and transchromosomic transgenic mice bearing the entire, or a sub-segment of the HSA21 region is sufficient to cause a DS phenotype, including altered dendritic structure, abnormal LTP, and cognitive deficits [199, 217]. Importantly, overexpression of specific genes like Dyrk1a revealed that DYRK1A is implicated in changes to GABAergic- and glutamatergic-related protein expression. Changes in the expression profiles of these proteins are important because they are critically involved in cognitive function [68, 76, 175]. A meta-analysis of data obtained from proteomic and transcriptomic approaches identified several signaling pathways which are perturbed in DS mouse models [79] and DS patients [59, 154]: specifically, a subset of genes on HSA21 was shown to be altered (SOD1, APP, DONSON, TIAM1, COL6A2, ITSN1 and BACE2), as well as the BDNF-dependent pathway, which is involved in neuronal growth, differentiation, and survival. Finally, expression of CRMP3, which regulates dendrites, is decreased in the hippocampus of 141G6 and Ts65Dn transgenic DS mouse models [204].
RTT mouse models.
In mice, knock-down of the X-linked Mecp2 gene recapitulates many hallmark symptoms of RTT, such as decreased numbers and regional loss of dendritic spines in the frontal cortex. Also, loss of Mecp2 in Mecp2tm1.1Bird [80, 133] and Mecp2tm1.1Jae [28] transgenic mice results in abnormal dendritic spine morphology, decreased pyramidal dendritic arbor complexity, spine density [15, 25] as well as altered glial cell structure and function. MeCP2 present in astrocytes and microglia may indirectly regulate dendrites and synapses [108]. These mice also display higher spine turnover, with a net bias favoring spine elimination, reflecting the persistence of an immature mental state [106]. At four weeks of age, null mice begin to develop uncoordinated gait, hypoactivity, tremor, irregular breathing, and hindlimb clasping reminiscent of the hand stereotypies seen in RTT patients. As in humans, these mice exhibit abnormalities in neuronal metabolism [70], increased oxidative stress [42], increased serum cholesterol and triglycerides [21, 115], and abnormal mitochondrial structure with these phenotypes progressing in severity until death [189]. In an attempt to reverse the RTT phenotype, delayed expression of Mecp2 after the onset of neurological symptoms drastically decreased ataxia severity, hindlimb clasping, tremor, and irregular breathing, while also extending the lifespan of the animals. Additionally, overexpression of Mecp2 in Wild Type (WT) mice induced dendritic overgrowth, increasing the number of higher order branches of apical dendritic arbors in layer V pyramidal neurons. These findings suggest that lack of MeCP2 alone does not produce irreversible neuronal damage, which highlights the potential benefits of a therapeutic intervention targeting MECP2 in patients with RTT [207].
DGS mouse models.
The close homology between human chromosome 22 and portions of mouse chromosome 16 permits causal modeling of CNVs using targeted murine mutagenesis [128, 142]. The Df(16)A+/− mouse model, which carries a hemizygous 1.3 Mb chromosomal deficiency (Df(16)A+/−) syntenic to the 1.5 Mb human 22q11.2 deletion and encompassing 27 genes, has been generated using chromosomal engineering techniques [132]. Behavioral characterization of Df(16)A+/− mice has revealed specific deficits in sensorimotor gating, working memory, and fear conditioning. Furthermore, several studies of Df(16)A+/− mice have shown modest alterations in corticogenesis, relatively widespread deficits in dendritic complexity and spine density, as well as region-specific alterations in short- and long-term synaptic plasticity. Df(16)A+/− mice also display transcriptomic alterations in the prefrontal cortex and hippocampus [63, 162]. Furthermore, diminished synchrony between the hippocampus and medial prefrontal cortex during a memory retrieval task is indicative of long-range functional connectivity impairments resulting from 22q11.2 deletions. Additionally, Df(16)A+/− mutant mice lacking Comt show impaired emotional reactivity and increased aggressive behaviors [107]. The Df(16)A+/− mutant is among several transgenic lines that model the 22q11.2 microdeletion (LgDel, Df1, Dgcr8), and these mice have demonstrated alterations in brain microRNA biogenesis. Dgcr8 haploinsufficiency may contribute to these alterations through down-regulation of a specific subset of microRNA. Specifically, miR-185 was the top-scoring down-regulated microRNA in the prefrontal cortex and hippocampus, regions which are also implicated in the pathology of schizophrenia. Furthermore, this reduction in miR-185 expression contributed to deficits in dendrite and spine development in hippocampal neurons [212].
FXS mouse models.
To examine the function of FMRP in FXS, mouse models have been generated with either complete or conditional ablation of Fmr1 [78, 113]. These Fmr1−/− mice exhibit impairment of neurogenesis in the dentate gyrus, defects in neurite extension, deficits in various learning and memory tasks, as well as dysfunctional synaptic plasticity (LTP and LTD) in several brain structures. Collectively, this evidence suggests that the cognitive impairment observed in FXS is likely due to synaptic dysfunction [19]. Selectively increasing Htt expression, a long-non-coding RNA, using a modified CRISPR/Cas9 (dCas9–VP64–SAM) approach, rescued dendritic maturation deficits observed in Fmr1−/− neurons [129]. Fmr1−/− mice display only mild phenotypic characteristics, which are also seen in patients with FXS. This suggests a compensatory mechanism, likely through activity of FXR2P, which has a high degree of homology with FMRP. Additionally, Fmr1−/− Fxr2−/− double KO mice have demonstrated hyperactivity, increased anxiety, reduced PPI, impaired LTP, as well as altered spatial associative learning and memory. Together, these findings support the conclusion of a functional relationship between FXR2P and FMRP. Additionally, these double KO mice exhibited significant reductions in dendritic complexity, total dendrite length, and number of branching points that underscores how the interaction between FMRP and FXR can potentially contribute to the development of FXS phenotypes [194]. Furthermore, CGG knock-in mice, which serve as models for FXTAS, have been generated to study dendritic dystrophy [82].
Emerging mechanism: chromosome 10 (q26) deletion, characteristic phenotypes, and CRMP3/DPYSL4 as a novel candidate gene for ID
Previous work using animal models, as well as Golgi staining of post-mortem tissues obtained from patients with ID-associated syndromes, has indicated the presence of defects in dendritic growth, which points toward a need to elucidate the molecular mechanisms regulating this process [196]. Converging evidence arising from these experimental and clinical pathophysiological investigations has shown that dendritic malformation and synaptic dysfunction are frequent and prominent findings in ID. Our efforts directed toward uncovering new molecules responsible for dendritogenesis identified CRMP3 as a positive intracellular signaling regulator of dendrites and their spines in hippocampal neurons [68, 169, 174]. We found that CRMP3 has a profound influence on neurite initiation and dendritic complexity in vertebrate hippocampal neurons (Fig.1). Previous observations identified CRMP3 expression in axonal growth cones and the distal tip of dendrites in mammalian neurons [95]. Additionally, our work has revealed the involvement of CRMP3 in the development of dendritic spines (Fig.2) [172]. In a murine model with constitutive loss of CRMP3 expression, Crmp3−/− mice develop altered LTP and pre-pulse inhibition (PPI) [169, 173, 174], which suggests its involvement in neuronal plasticity and behaviors typically associated with psychiatric illness. Importantly, over-expression of CRMP3 induces dendrite outgrowth through increased Ca2+ influx (Fig.2), which can be inhibited using Crmp3 siRNA. In vitro, Crmp3−/− neurons, or neurons transfected with a dominant-negative truncated Crmp3 (Crmp3ΔC), displayed prototypical “fried-egg” morphology with complete absence of neurite extensions [172]. Additionally, over-expression of CRMP3 prevented prion-induced dendritic dystrophy in cultured neurons. Our observations suggest a distinct, but critical, contribution of CRMP3 activity in dendrite morphogenesis and maintenance [171, 172]. Work performed by other groups has found that CRMP3 interacts with the cytoskeletal proteins tubulin and actin. These studies have also identified that CRMP3 mediates Sema3A/Plexin-A/Nrp1 signaling and interacts with NrCAM, which is linked to Sema3F activity [147, 219]. Furthermore, CRMP3 expression is decreased in brain samples of AD patients, as well as the 3xTg-AD mouse model [60, 98], which both display dendritic dystrophy. It has been suggested that CRMP3 may be critically involved in learning and memory, since it is part of a group of long-lived synaptic proteins [86]. Indeed, treatment with Rosiglitazone, a peroxisome proliferator-activated receptor γ (PPARγ) agonist, increased CRMP3 expression in the hippocampus and improved cognition in the Tg2576 AD mouse model [51]. In contrast, in a mouse model of phenylketonuria, treatment with titanium dioxide nanoparticles (TiO2NPs) decreased the length of hippocampal dendrites, reduced CRMP3 expression, and impaired behavioral measures of memory [90, 93, 97]. From these findings, it is tempting to hypothesize that CRMP3 may participate in dendritic remodeling and arborization, which are critical for learning and memory processing in vivo.
Figure 1. Expression of CRMP3/DPYSL4 during nervous system development and role in cultured hippocampal neurons.

In situ hybridization of CRMP3 in sagittal sections of embryonic and adult mouse brain. (a) At E12.5, transcripts were detected in dorsal root ganglia (drg) and the central nervous system (CNS). Different regions of the rhombencephalon (Rhomb), mesencephalon (Mesen), and diencephalon (Dien) were stained and are denoted with white arrows. The black arrows denoted the CRMP3 expression in the ventricular neuroepithelia and the cortex (Cx). Staining was also observed in cerebellum, which is marked with unfilled arrows. No staining was observed in the choroid plexus (cp). (b). In sections obtained from adult mice, CRMP3 mRNA was detectable mainly in the cerebellar granular layer, the inferior olive, and the dentate gyrus. (c). Summary of In Situ Hybridization with an antisense riboprobe: highly positive signal (+++); weak signal (+). Representative Crmp3−/−cells with no visible processes (d; e) and Crmp3+/− cell with neurites (f; g) using β-galactosidase activity (d; f) or immunostaining with MAP2 antibodies (e; g). Transfected with Fl-CRMP3, neurons are characterized by active transport of the protein, increased formation of lamellipodia, and increased dendritic arborization. Representative CRMP3-over-expressing neuron (h; ov-crmp3) immunostained for Flag (red) and the dendritic marker MAP2 (green). Overlay of image from transfected neurons are in yellow (h, right). Yellow arrows show control (h) or LacZ− (d) cells. White arrows show neurites (f, g).
Figure 2. A proposed model for the regulation of dendrite morphology by CRMP3/DPYSL4 activity.

We propose a model involving CRMP3/DPYSL4 activity underlying neurite initiation and dendrite outgrowth following Ca2+ influx. Combinatorial stimulation of CRMP3/DPYSL4 gene expression, or activity by intrinsic and extrinsic factors (1). The localization and distribution of FL-CRMP3-positive puncta in dendrites suggest that CRMP3 could be associated with vesicles or large carrier protein complexes and is then actively transported to dendrites (2). Structural and biochemical studies support the notion that CRMP3 activity might be regulated by phosphorylation or other post-translational modifications (3–4) induced by neurotrophic factors or guidance cues (7). Activated CRMP3/DPYSL4 binds to protein partners and serves as an adaptor in a variety of signaling pathways including Ca2+ influx (CaV; 5). Our genetic studies have shown that CRMP3/DPYSL4 contributes to dendritic arborization, dendritic spine genesis (6) and neurite initiation (8). These effects might involve lamellipodia formation, cell migration (9), dendritic arborization, spine density and structure. Collectively, these functional clusters may affect cognition, learning and memory (10).
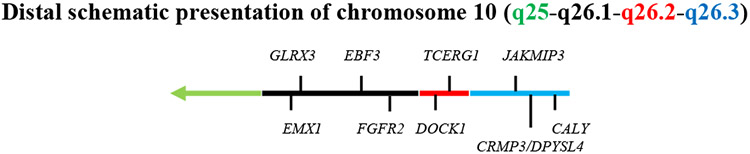
Approaches combining next-generation sequencing, cytogenomic mapping, and DECIPHER/IPA/GRAIL bioinformatics analyses have identified new chromosomic rearrangement, such as deletion or ring chromosome phenotypes. These studies have also identified chromosomal imbalance, along with new gene networks [84, 102, 223], and highlighted CRMP3/DPYSL4 as a new candidate gene for ID in data obtained from 1,354 patients. Additionally, these genetic/proteomic approaches have demonstrated interactions with CRMP2, DYNC11-1, HDGFRP3, and NrCAM, which is an integral component of the Sema3F receptor complex [49, 219]. Furthermore, recent studies using AGH have also detected CRMP3/DPYSL4 deletion in children with idiopathic ID [68]. Previously, we mapped the Crmp3/Dpysl4 gene onto chromosome 7 in mice [170] where it is linked to Mgmt, IGF2 and Drd2. However, in humans, we found that CRMP3/DPYSL4 is located on the distal region of chromosome 10 (10q.26) (Fig.3) [91]. Human chromosome 10 has 1.31 x 108 nucleotides, containing approximately 1,300 genes, of which 816 are protein coding and 430 are pseudogenes [48]. Most of these genes have both strands overlapping coding genes, with 67 that are likely antisense transcripts. Using the published draft of the complete human genome (International Human Genome Sequencing Consortium, 2001) [53], it is possible to correlate regions of chromosomal deletion or imbalance with corresponding gene sequences, and thus identify genes potentially responsible for clinical phenotypes (Table 1). Previous cytogenetic studies have shown that the 10q25-10qter deletion syndrome is clinically heterogeneous with common clinical features including ADHD [36], ID, craniofacial anomalies, growth failure, and congenital heart defects [117, 211]. Because the size and location of the deletions are variable, it remains unclear if the variability and severity of the phenotype correlates with specific genetic segments or critical genomic coding regions. However, the presence of a clearly recognizable phenotype, despite deletion size variability, has led to the hypothesis that deletion of a critical region could produce the characteristic clinical picture. Studies using AGH have shown that patients with 10q25.2-10qter deletion, including the CRMP3/DPYSL4 gene, have a recognizable ID phenotype [99] (Table 2). No other known genes in this region are clear candidates for the dendritic anomalies and characteristic phenotype observed in the patients harboring this deletion. Table 1 lists genes of interest, either up or downstream of CRMP3/DPYSL4, and Table 2 reports selected genes on chromosome 10q terminal involved in brain disorders. These characteristic phenotypes are found in patients with Ring Chromosome 10, which is a disorder that occurs following breakage and rejoining of both chromosome arms, as well as patients with a linear subterminal deletion of chromosome 10. Patients with deletion of the CRMP3/DPYSL4 gene (10q26.3) always present with an associated ID phenotype. Interestingly, a subset of patients without CRMP3/DPYSL4 gene deletion do not present with ID syndromes (Table 2). Collectively, these observations demonstrate an intriguing correlation, suggesting that a critical region containing the CRMP3/DPYSL4 gene could contribute to the characteristic ID phenotypes seen in the 10q26 deletion. A role for CRMP3 in dendrite dystrophy in ID would be in agreement with its altered expression in AD patients, as well as in AD/PKU mouse models, which are characterized by dendrite degeneration.
Figure 3. Schematic representation of gene distribution on chromosome 10.

Mapping the q26 terminal area (a) and the genes involved in brain disorders (b).
Table 1.
Schematic presentation of the 10q25.2-q26.3 region and gene positioning from top to bottom, with clinical features.
| Genes of interest on Chromosome 10 (q26.1-26.3) | ||
|---|---|---|
| Genes | Functions | References |
| PAX2 | Chronic kidney diseases | Deng et al; 2019. Ref.50 |
| EMX2 | Transcription Factor | Cecchi et al; 2002. Ref.31 |
| GLRX3 | Vesicoureteric reflux | Darlow et al; 2017. Ref.41 |
| EBF3 | Facial dysmorphisms; ID candidate | Lopes et al; 2017. Ref.131 |
| MGMT | Glioblastomas biomarkers | Jovcevska; 2019. Ref.105 |
| MDR1 | Amplified in glioblastomas | Suzuki et al; 2004. Ref. 193 |
| ADAM12 | Cancer; arthritis; cardiac hypertrophy | Ray et al; 2011. Ref.177 |
| DOCK1 | Cranio facial dysmorphism; ID candidate | Yatsenko et al; 2009. Ref.221 |
| TXNL2 | Breast, colorectal cancers | Qu et al; 2011. Ref.167 |
| FGFR2 | Microcephaly; ID candidate | Sangu et al; 2016. Ref.185 |
| PTPRE | Obesity-induced disease development | Kogelman et al; 2016 Ref.112 |
| MK167 | Cancer risk factor | Joensuu et al; 2004 Ref.104 |
| TCERG1 | Apoptotic agent; spliceosome | Montes et al; 2015 Ref.148 |
| BNIP3 | Mitophagy; cancer | Panigrahi et al; 2019 Ref.159 |
| JAKMIP3 | Candidate for COPD | Mostafaei et al; 2018 Ref.150 |
| DPYSL4/CRMP3 | Dendrite maintenance; AD, ID candidate | Quach et al; 2020 Ref.174 |
| LRRC27 | Intra-ocular pressure; glaucoma | Woo Kim et al; 2020 Ref.216 |
| PWWP2B | Histone modifications regulation | Zhang et al; 2018 Ref.225 |
| NKX6-2 | Hypomyelination; spastic ataxia | Chelban et al; 2020 Ref.27 |
| GPR123 | Candidate gene for asthma | Tremblay et al; 2008 Ref. 200 |
| VENTX | Putative tumor suppressor | Gao et al; 2016 Ref.67 |
| MIR202 | Reduced pancreatic tumor | Mody et al; 2017 Ref.146 |
| ADAM8 | Neuro-inflammation; asthma | Knolle et al; 2009 Ref.111 |
| TUBGCP2 | Neuronal migration defect | Mitani et al; 2019 Ref.145 |
| CALY | Attention deficit hyperactivity disorders | Plaisancie et al; 2014 Ref.163 |
| PRAP | Developmental regulation during pregnancy | Duan et al; 1997 Ref. 57 |
| ECHS1 | Deafness; epilepsy; optic atrophy | Haack et al; 2015 Ref.81 |
| MTG1 | Cardiac hypertrophy; mitochondrial function | Xu et al; 2019 Ref.219 |
| SPRN | Influence in TSE susceptibility | Lampo et al; 2007 Ref.118 |
| CYP2E1 | Alcohol abuse/immune interactions | Ratna et al; 2017 Ref.176 |
| FRG2B | Neuro-endocrine tumors | Snezhkina et al; 2019 Ref.190 |
| UTF | Germ cell lineage development | Kasowitz et al; 2017 Ref.109 |
| PAOX | Over-expressed in myelofibrosis | Salati et al; 2016 Ref.183 |
| SYCE | Non-obstructive azoospermia | Maor-Sagie et al; 2015 Ref.134 |
| PPP2R2 | Mitochondrial fragmentation; autophagy | Fang et al; 2013 Ref. 62 |
| DRD1IP | Attention deficit/hyperactivity disorders | Laurin et al; 2007 Ref.121 |
Table 2.
Identification of commonly expressed genes surrounding CRMP3/DPYSL4: delineating the critical region for intellectual disability.
 | ||||
|---|---|---|---|---|
|
CRMP3/ DPYSL4 |
Deletion interval | ID | Number
of Patients |
References |
| Yes | 10q26.2-10q26.3 | Yes | 4 patients | Plaisancie et al; 2014. Ref 163 |
| Yes | 10q26.2-10q26.3 | Yes | 1 patient | Courtens et al; 2006. Ref 36 |
| Yes | 10q25.2-10q26.3 | Yes | 13 patients | Irving et al; 2003. Ref 99 |
| Yes | 10q26-10qter | Yes | 1 patient | Turleau et al; 1979. Ref 120 |
| Yes | 10q26.13-10qter | Yes | Patient N# 2 | Yatsenko et al; 2009. Ref 221 |
| No | 10q26.2 | No | Patient N#3 | Yatsenko et al; 2009. Ref 221 |
| No | 10q26.2 | No | 1 patient | Laudier et al; 2016. . Ref 120 |
| No | 10q23.2-10q23.3 | No | Patients N#1, 2, 3 | Delnatte et al; 2006. Ref 47 |
| No | 10q21-10q23 | No | 12 patients | Bowles et al; 1996. Ref 17 |
| No | 10q22.3-10q24.1 | No | 1 patient | Alimi et al; 2015. Ref 4 |
| No | 10q22.1-10q22.3 | No | 1 patient | Tzschach et al; 2006. Ref 202 |
| No | 10q26.2 | No | Meta-analysis | Darlow et al; 2017. Ref 41 |
| No | 10q24.2-10q24.32 | No | 1 patient | Hoefele et al; 2012 . Ref 89 |
| No | 10q24-q25 | No | 3 patients | Sanyanusin et al; 1995. Ref 186 |
An unholy trinity: reflections on the molecular crosstalk between ID, AD, and IQ
Numerous neurodevelopmental disorders that present with comorbid ID and neurodegenerative pathologies (e.g. AD) display similar patterns of altered dendrite and spine structures in the cerebral cortex and hippocampus [73, 191]. However, candidate gene targets, as well as the molecular signaling pathways underlying dendrite and spine dysgenesis are potentially distinct among these disorders. While neurodegenerative diseases are typically considered age-associated disorders where the accumulation and aggregation of pathogenic proteins leads to aberrant regulation of gene expression within dendrites and spines, in neurodevelopmental diseases with associated ID, alterations to dendrites and spines arise from genetic mutations that affect protein expression and gene transcription during the maturation of these structures [88].
Drawing on data from our laboratory, in combination with findings from animal models of various brain disorders, post-mortem tissue studies, and reviews of the current literature describing phenotypes in AD patients and ID patients with chromosome 10 q26.3 terminal deletion, we hypothesize that impaired expression of CRMP3/DPYSL4 may contribute to phenotypes in both ID and AD. Additionally, profiles of dendritic exuberance, positively correlating with performance in learning and memory-related tasks, have been previously posited as a proof of concept regarding this hypothesis [13, 35]. Ultimately, this raises questions of whether this gene and its protein product may also influence some aspects of human intelligence. Although a strict and comprehensive definition of human intelligence has been elusive due to the presence of different types, including sense of reasoning, creativity, and novel problem-solving ability, it is most commonly measured as an “IQ score” using the Wechsler Adult Intelligent Scale (WAIS) [100, 213]. Sequencing the human genome has identified ~30,000 genes, suggesting that the complex functional phenotype of an individual is not only directly related to the number of coding genes, but is also reliant upon genomic and proteomic networks. These networks are regulated through epigenetic factors, in addition to non-linear crosstalk mechanisms, leading to complex interactomes that produce divergent outputs from a small number of input genes [85, 124]. Consequently, it is important to highlight interactions between different levels of molecular regulatory elements and how this could contribute to the functional diversity observed during different periods over the span of a human lifetime. Although there is still much debate regarding the effects of various genes associated with high IQ scores, there are several observations that can be made. First, high IQ scores are linked to larger, more complex dendritic arbors and dendritic spine morphologies in human pyramidal neurons [33, 74, 164] (Fig.4). Second, Sema3F, which is important in the formation and refinement of neuronal networks [26, 45, 147, 180, 181], is associated with intelligence [34] and involved in cognitive decline. Furthermore, in a meta-analysis of 87,740 individuals, the Sema3F signaling pathway was significantly enriched in subjects with high IQ score [222] whereas, a large-scale GWAS meta-analysis involving 269,867 individuals listed CRMP3/DPYSL4 as a candidate gene correlated with high IQ [188]. Molecules like CRMP3, critical for dendrite development, may represent a common final pathway in ID from various etiologies. Additionally, the similar alterations of dendrites and spines found in patients with AD and ID, as well as related animal models, highlights the parallels between these disease states. While these observations are compelling, more comprehensive investigations and further analysis of the vast amounts of genetic information currently available will be required in order to effectively promote a candidate ID-AD-IQ gene to a validated target [210].
Figure 4. Dendritic tree and spine morphology changes associated with intellectual ability.

The model depicts a pyramidal neuron displaying high dendritic spine density, which is correlated with higher levels of intelligence. Lower scores on the IQ assessment, < 70, are associated with lower dendritic arbor complexity as well as decreased spine density in numerous brain regions. These morphological characteristics are highly correlated with scores on the IQ assessment and likely underlie the functional outcomes associated with ID.
Looking forward, it is easy to image CRMP3 as a potential target for gene therapy for a variety of brain disorders associated with these common structural features since CRMP3 induces dendritic outgrowth through increased Ca2+ and is essential for maintenance of dendritic spine structural integrity. Future studies regarding the role of CRMP3 in dendritic spine pathology associated with ID are warranted to better characterize this protein in human disease states along with the therapeutic potential offered by understanding its function.
ACKNOWLEDGEMENTS
We would like to acknowledge all of those whose work has contributed to the knowledge reviewed here as well as the work of those not mentioned. This work was supported in part by National Institute of Health awards R01NS09877202 & R01DA04285202 to R.K.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCES
- 1.Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A (2004) Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303:197–202. 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- 2.Akum BF, Chen M, Gunderson SI, Riefler GM, Scerri-Hansen MM, Firestein BL (2004) Cypin regulates dendrite patterning in hippocampal neurons by promoting microtubule assembly. Nat Neurosci 7:145–52. 10.1038/nn1179. [DOI] [PubMed] [Google Scholar]
- 3.Aldosary M, Al-Bakheet A, Al-Dhalaan H, Almass R, Alsagob M, Al-Younes B, AlQuait L, Mustafa OM, Bulbul M, Rahbeeni Z, Alfadhel M, Chedrawi A, Al-Hassnan Z, AlDosari M, Al-Zaidan H, Al-Muhaizea MA, AlSayed MD, Salih MA, AlShammari M, Faiyaz-Ul-Haque M, Chishti MA, Al-Harazi O, Al-Odaib A, Kaya N, Colak D (2020) Rett Syndrome, a Neurodevelopmental Disorder, Whole-Transcriptome, and Mitochondrial Genome Multiomics Analyses Identify Novel Variations and Disease Pathways. OMICS 24:160–171. 10.1089/omi.2019.0192. [DOI] [PubMed] [Google Scholar]
- 4.Alimi A, Lauren A, Weeth-Feinstein LA, Stettner A , Caldera F, Weiss JM (2015) Overlap of Juvenile Polyposis Syndrome and Cowden Syndrome Due to De Novo Chromosome 10 Deletion Involving BMPR1A and PTEN: Implications for Treatment [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alves-Sampaio A, Troca-Marín JA, Montesinos ML (2010) NMDA-mediated regulation of DSCAM dendritic local translation is lost in a mouse model of Down's syndrome. J Neurosci. 30:13537–48. 10.1523/JNEUROSCI.3457-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpGbinding protein 2. Nat Genet 23:185–8. 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 7.Antonarakis SE (2001) Chromosome 21: from sequence to applications. Curr Opin Genet Dev 11:241–6. 10.1016/s0959-437x(00)00185-4. [DOI] [PubMed] [Google Scholar]
- 8.Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S (2004) Chromosome 21 and down syndrome: from genomics to pathophysiology. Nat Rev Genet 5:725–38. 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- 9.Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, Sherman SL, Reeves RH (2020) Down syndrome. Nat Rev Dis Primers 6:9 10.1038/s41572-019-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arbones ML, Thomazeau A, Nakano-Kobayashi A, Hagiwara M, Delabar JM (2019) DYRK1A and cognition: A lifelong relationship. Pharmacol Ther 194:199–221. 10.1016/j.pharmthera.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong DD (2005) Neuropathology of Rett syndrome. J Child Neurol 20:747–53. 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- 12.Bar-Nur O, Caspi I, Benvenisty N (2012) Molecular Analysis of FMR1 Reactivation in fragile-X Induced Pluripotent Stem Cells and Their Neuronal Derivatives J Mol Cell Biol 4:180–3. 10.1093/jmcb/mjs007. [DOI] [PubMed] [Google Scholar]
- 13.Baudry M, Zhu G, Liu Y, Wang Y, Briz V, Bi X (2015) Multiple cellular cascades participate in long-term potentiation and in hippocampus-dependent learning. Brain Res 1621:73–81. 10.1016/j.brainres.2014.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayés A, Collins MO, Croning MD, van de Lagemaat LN, Choudhary JS, Grant SG (2012) Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS One 7:e46683 10.1371/journal.pone.0046683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U (2009) Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol 514:240–58. 10.1002/cne.22009. [DOI] [PubMed] [Google Scholar]
- 16.Borrie SC, Brems H, Legius E, Bagni C (2017) Cognitive dysfunctions in intellectual disabilities: the contributions of the Ras-MAPK and PI3K-AKT-mTOR pathways. Annu Rev Genomics Hum Genet 18:115–142. 10.1146/annurev-genom-091416-035332. [DOI] [PubMed] [Google Scholar]
- 17.Bowles KR, Gajarski R, Porter P, Goytia V, Bachinski L, Roberts R, Pignatelli R, Towbin JA (1996) Gene mapping of familial autosomal dominant dilated cardiomyopathy to chromosome 10q21-23. J Clin Invest 98:1355–60. 10.1172/JCI118922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Branchi I, Bichler Z, Berger-Sweeney J, Ricceri L (2003) Animal models of mental retardation: from gene to cognitive function. Neurosci Biobehav Rev 27:141–53. 10.1016/s0149-7634(03)00016-2. [DOI] [PubMed] [Google Scholar]
- 19.Brennan FX, Albeck DS, Paylor R (2006) Fmr1 knockout mice are impaired in a leverpress escape/avoidance task. Genes Brain Behav 5:467–71. 10.1111/j.1601-183X.2005.00183.x. [DOI] [PubMed] [Google Scholar]
- 20.Brindisi M, Saraswati AP, Brogi S, Gemma S, Butini S, Campiani G (2020) Old but gold: Tracking the new guise of histone deacetylase 6 (HDAC6) enzyme as a biomarker and therapeutic target in rare diseases. J Med Chem 63:23–39. 10.1021/acs.jmedchem.9b00924. [DOI] [PubMed] [Google Scholar]
- 21.Buchovecky CM, Turley SD, Brown HM, Kyle SM, McDonald JG, Liu B, Pieper AA, Huang W, Katz DM, Russell DW, Shendure J, Justice MJ (2013) A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat Genet 45:1013–20. 10.1038/ng.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardozo PL, de Lima IBQ, Maciel EMA, Silva NC, Dobransky T, Ribeiro FM (2019) Synaptic elimination in neurological disorders. Curr Neuropharmacol 17:1071–1095. 10.2174/1570159X17666190603170511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carvill GL, Mefford HC (2013) Microdeletion syndromes. Curr Opin Genet Dev 23:232–9. 10.1016/j.gde.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 24.Chahrour M, Zoghbi HY (2007) The story of Rett syndrome: from clinic to neurobiology. Neuron 56:422–37. 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Chapleau CA, Boggio EM, Calfa G, Percy AK, Giustetto M, Pozzo-Miller L (2012) Hippocampal CA1 pyramidal neurons of Mecp2 mutant mice show a dendritic spine phenotype only in the presymptomatic stage. Neural Plast 2012:976164 10.1155/2012/976164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chédotal A, Richards LJ (2010) Wiring the Brain: The Biology of Neuronal Guidance. Cold Spring Harb Perspect Biol. 2(6): a001917 10.1101/cshperspect.a001917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chelban V, Kaya N, Alkuraya F, Houlden H (2018) In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. NKX6-2-Related Disorder. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–202. [PubMed] [Google Scholar]
- 28.Chen RZ, Akbarian S, Tudor M, Jaenisch R (2001) Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27:327–31. 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 29.Chen XQ, Sawa M, Mobley WC (2018) Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic Biol Med 114:52–61. 10.1016/j.freeradbiomed.2017.10.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH, Xu W, Qiu Z (2014) MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell 28:547–60. 10.1016/j.devcel.2014.01.032. [DOI] [PubMed] [Google Scholar]
- 31.Cecchi C (2002) Emx2: a gene responsible for cortical development, regionalization and area specification. Gene 291:1–9. 10.1016/s0378-1119(02)00623-6. [DOI] [PubMed] [Google Scholar]
- 32.Chidambaram SB, Rathipriya AG, Bolla SR, Bhat A, Ray B, Mahalakshmi AM, Manivasagam T, Thenmozhi AJ, Essa MM, Guillemin GJ, Chandra R, Sakharkar MK (2019) Dendritic spines: Revisiting the physiological role. Prog Neuropsychopharmacol Biol Psychiatry. 92:161–193. 10.1016/j.pnpbp.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Choi YY, Shamosh NA, Cho SH, DeYoung CG, Lee MJ, Lee JM, Kim SI, Cho ZH, Kim K, Gray JR, Lee KH (2008) Multiple bases of human intelligence revealed by cortical thickness and neural activation. J Neurosci 28:10323–9. 10.1523/JNEUROSCI.3259-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coleman JRI, Bryois J, Gaspar HA, et al. (2019) Biological annotation of genetic loci associated with intelligence in a meta-analysis of 87,740 individuals. Mol Psychiatry 24:182–197. 10.1038/s41380-018-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conrad CD, Ortiz JB, Judd JM (2017) Chronic stress and hippocampal dendritic complexity: Methodological and functional considerations. Physiol Behav 178:66–81. 10.1016/j.physbeh.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 36.Courtens W, Wuyts W, Rooms L, Pera SB, Wauters J (2006) A subterminal deletion of the long arm of chromosome 10: a clinical report and review. Am J Med Genet A 140: 402–9. 10.1002/ajmg.a.31053. [DOI] [PubMed] [Google Scholar]
- 37.Crampton SJ, O'Keeffe GW (2013) NK-κB: emerging roles in hippocampal development and function Int J Biochem Cell Biol 45:1821–4. 10.1016/j.biocel.2013.05.037. PMID: 23764620 [DOI] [PubMed] [Google Scholar]
- 38.Crawley JN (2007) Mouse behavioral assays relevant to the symptoms of autism. Brain Pathol 17:448–59. 10.1111/j.1750-3639.2007.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crocker-Buque A, Currie SP, Luz LL, Grant SG, Duffy KR, Kind PC, Daw MI (2016) Altered thalamocortical development in the SAP102 knockout model of intellectual disability. Hum Mol Genet 25:4052–4061. 10.1093/hmg/ddw244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cutri-French C, Armstrong D, Saby J, Gorman C, Lane J, Fu C, Peters SU, Percy A, Neul JL, Marsh ED (2020) Comparison of core features in four developmental encephalopathies in the Rett natural history study. Rett and Rett related disorders natural history study. Ann Neurol 10.1002/ana.25797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darlow JM, Darlay R, Dobson MG, Stewart A, Charoen P, Southgate J, Baker SC, Xu Y, Hunziker M, Lambert HJ, Green AJ, Santibanez-Koref M, Sayer JA, Goodship THJ, Puri P, Woolf AS, Kenda RB, Barton DE, Cordell HJ (2017) Genome-wide linkage and association study implicates the 10q26 region as a major genetic contributor to primary nonsyndromic vesicoureteric reflux. Sci Rep 7:14595 10.1038/s41598-017-15062-9.PMID: 29097723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Felice C, Ciccoli L, Leoncini S, Signorini C, Rossi M, Vannuccini L, Gianni Guazzi G, Latini G, Comporti M, Valacchi G, Hayek J (2009) Systemic oxidative stress in classic Rett syndrome. Free Radic Biol Med 47:4408 10.1016/j.freeradbiomed.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 43.De Rubeis S, Fernández E, Buzzi A, Di Marino D, Bagni C (2012) Molecular and cellular aspects of mental retardation in the Fragile X syndrome: from gene mutations to spine dysmorphogenesis. Adv Exp Med Biol 970:517–51. 10.1007/978-3-7091-0932-8_23. [DOI] [PubMed] [Google Scholar]
- 44.De Toma I, Manubens-Gil L, Ossowski S, Dierssen M (2016) Where environment meets cognition: A focus on two developmental intellectual disability disorders. Neural Plast. 2016:4235898 10.1155/2016/4235898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degano AL, Pasterkamp J, Ronnett GV (2012) MeCP2 deficiency disrupts axonal guidance, fasciculation, and targeting by altering Semaphorin 3F function. Mol Cell Neurosci 42: 243–254. 10.1016/j.mcn.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delabar JM, Aflalo-Rattenbac R, Créau N (2006) Developmental defects in trisomy 21 and mouse models. Scientific World Journal 6:1945–64. 10.1100/tsw.2006.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delnatte C, Sanlaville D, Mougenot JF, Vermeesch JR, Houdayer C, Blois MC, Genevieve D, Goulet O, Fryns JP, Jaubert F, Vekemans M, Lyonnet S, Romana S, Eng C, Stoppa-Lyonnet D (2006) Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet 78:1066–74. 10.1086/504301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deloukas P, Earthrowl ME, Grafham DV , Rubenfield M, French L, Steward CA, Sims SK (2004) The DNA sequence and comparative analysis of human chromosome 10. Nature 429:375–81. 10.1038/nature02462. [DOI] [PubMed] [Google Scholar]
- 49.Demyanenko GP, Mohan V, Zhang X, Brennaman LH, Dharbal KE, Tran TS, Manis PB, Maness PF (2014) Neural cell adhesion molecule NrCAM regulates Semaphorin 3Finduced dendritic spine remodeling. J Neurosci 34:11274–87. 10.1523/JNEUROSCI.1774-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng H, Zhang Y, Xiao H, Yao Y, Liu X, Su B, Zhang H, Xu K, Wang S, Wang F, Ding J (2019) Diverse phenotypes in children with PAX2-related disorder. Mol Genet Genomic Med 7:e701 10.1002/mgg3.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denner LA, Rodriguez-Rivera J, Haidacher SJ, Jahrling JB, Carmical JR, Hernandez CM, Zhao Y, Sadygov RG, Starkey JM, Spratt H, Luxon BA, Wood TG, Dineley KT (2012) Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. J Neurosci 32:16725–35a. 10.1523/JNEUROSCI.2153-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dijkhuizen PA, Ghosh A (2005) Regulation of dendritic growth by calcium and neurotrophin signaling. Prog Brain Res 147:17–27. 10.1016/S0079-6123(04)47002-2 [DOI] [PubMed] [Google Scholar]
- 53.Disease Genes and Chromosomes: Disease maps of the human chromosome 10. In Genetic Testing; 5(1), 2001. [DOI] [PubMed] [Google Scholar]
- 54.Dockendorff TC, Labrador M (2019)The Fragile X Protein and Genome Function. Mol Neurobiol 56:711–721. 10.1007/s12035-018-1122-9. [DOI] [PubMed] [Google Scholar]
- 55.Dotti CG, Sullivan CA, Banker GA (1988) The establishment of polarity by hippocampal neurons in culture. J Neurosci 8:1454–68. 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Du Q, de la Morena MT, van Oers NSC (2020) The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front Genet 10:1365 10.3389/fgene.2019.01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duan WR, Parmer TG, Albarracin CT, Zhong L, Gibori G (1997) PRAP, a prolactin receptor associated protein: its gene expression and regulation in the corpus luteum. Endocrinology 138:3216–21. 10.1210/endo.138.8.5336. [DOI] [PubMed] [Google Scholar]
- 58.Emoto K (2011) Dendrite remodeling in development and disease. Dev Growth Differ. 53:277–86. 10.1111/j.1440-169X.2010.01242.x. [DOI] [PubMed] [Google Scholar]
- 59.Engidawork E, Lubec G (2003) Molecular changes in fetal Down syndrome brain. J Neurochem 84:895–904. 10.1046/j.1471-4159.2003.01614.x. [DOI] [PubMed] [Google Scholar]
- 60.Expression Atlas: https://www.ebi.ac.uk/gxa/home.124. [Google Scholar]
- 61.Fagiolini M, Patrizi A, LeBlanc J, Jin LW, Maezawa I, Sinnett S, Gray SJ, Molholm S, Foxe JJ, Johnston MV, Naidu S, Blue M, Hossain A, Kadam S, Zhao X, Chang Q, Zhou Z, Zoghbi H (2020) Intellectual and developmental disabilities research centers: A multidisciplinary approach to understand the pathogenesis of Methyl-CpG binding protein 2-related disorders. Neuroscience 445:190–206. 10.1016/j.neuroscience.2020.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fang K, Li H, Hsieh C, Li D, Song D, Cheng W, Guo Z (2013) Differential autophagic cell death under stress with ectopic cytoplasmic and mitochondrial-specific PPP2R2B in human neuroblastoma cells. Apoptosis 18:627–38. 10.1007/s10495-013-0809-7. [DOI] [PubMed] [Google Scholar]
- 63.Fénelon K, Xu B, Lai CS, Mukai J, Markx S, Stark KL, Hsu PK, Gan WB, Fischbach GD, MacDermott AB, Karayiorgou M, Gogos JA (2013) The pattern of cortical dysfunction in a mouse model of a schizophrenia-related microdeletion. J Neurosci 33:14825–39. 10.1523/JNEUROSCI.1611-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferrer I, Gullotta F (1990) Down's syndrome and Alzheimer's disease: Dendritic spine counts in the hippocampus. Acta Neuropathol 79:680–5. 10.1007/BF00294247. [DOI] [PubMed] [Google Scholar]
- 65.Filosa S, Pecorelli A, D'Esposito M, Valacchi G, Hajek J (2015) Exploring the possible link between MeCP2 and oxidative stress in Rett syndrome. Free Radic Biol Med 88(Pt A):81–90. 10.1016/j.freeradbiomed.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 66.Foote M, Arque G, Berman RF, Santos M (2016) Fragile X-associated tremor/ataxia syndrome (FXTAS) motor dysfunction modeled in mice. Cerebellum 15:611–22. 10.1007/s12311-016-0797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao H, Wu B, Le Y, Zhu Z (2016) Homeobox protein VentX induces p53-independent apoptosis in cancer cells. Oncotarget 7:39719–39729. 10.18632/oncotarget.9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Friedman J, Adam S, Arbour L, Armstrong L, Baross A, Birch P, Boerkoel C, Chan S, Chai D, Delaney AD, Flibotte S, Gibson WT, Langlois S, Lemyre E, Li HI, MacLeod P, Mathers J, Michaud JL, McGillivray BC, Patel MS, Qian H, Rouleau GA, Van Allen MI, Yong SL, Zahir FR, Eydoux P, Marra MA (2009) Detection of pathogenic copy number variants in children with idiopathic intellectual disability using 500 K SNP array genomic hybridization. BMC Genomics 10:526 10.1186/1471-2164-10-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gianakopoulos PJ, Zhang Y, Pencea N, OrlicMilacic M, Mittal K, Windpassinger C, White S-J, Kroisel PM, Chow EWC, Saunders CJ, Minassian BA, Vincent JB (2012) Mutations in MECP2 exon 1 in classical Rett patients disrupt MECP2_e1 transcription, but not transcription of MECP2_e2. Am J Med Genet Part B 159B:210–216. 10.1002/ajmg.b.32015. [DOI] [PubMed] [Google Scholar]
- 70.Goffin D, Zhou ZJ (2012) The neural circuit basis of Rett syndrome. Front Biol (Beijing). 7:428–435. 10.1007/s11515-012-1248-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gold WA, Krishnarajy R, Ellaway C, Christodoulou J (2018) Rett syndrome: A genetic update and clinical review focusing on comorbidities. ACS Chem Neurosci 9:167–176. 10.1021/acschemneuro.7b00346. [DOI] [PubMed] [Google Scholar]
- 72.Goldenberg P (2018) An update on common chromosome microdeletion and microduplication syndromes. Pediatr Ann 47(5):e198–e203. 10.3928/19382359-20180419-01. [DOI] [PubMed] [Google Scholar]
- 73.Gomez W, Morales R, Maracaja-Coutinho V, Parra V, Nassif M (2020) Down syndrome and Alzheimer's disease: common molecular traits beyond the amyloid precursor protein. Aging 12: 1011–1033. 10.18632/aging.102677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goriounova NA, Djai B, Heyer D, Wilbers R, Verhoog MB, Giugliano M, Verbist C, Obermayer J, Kerkhofs A, Smeding H, Maaike Verberne M, Idema S, Baayen JC, Pieneman AW, de Kock CP, Klein M, Mansvelder HD (2018) Large and fast human pyramidal neurons associate with intelligence. Elife. 7:e41714 10.7554/eLife.41714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goshima Y, Yamashita N, Nakamura F, Sasaki Y (2016) Regulation of dendritic development by semaphorin 3A through novel intracellular remote signaling. Cell Adh Migr 10:627–640. 10.1080/19336918.2016.1210758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grau C, Arató K, Fernández-Fernández JM, Valderrama A, Sindreu C, Fillat C, Ferrer I, de la Luna S, Altafaj X (2014) DYRK1A-mediated phosphorylation of GluN2A at Ser(1048) regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front Cell Neurosci 8:331 10.3389/fncel.2014.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grossman AW, Elisseou NM, McKinney BC, Greenough WT (2006) Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res 1084: 158–64. 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- 78.Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL, Eisch AJ, Oostra BA, Nelson DL, Jin P, Zhao X (2011) Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med 17:559–65. 10.1038/nm.2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gupta M, Dhanasekaran AR, Gardiner KJ (2016) Mouse models of Down syndrome: gene content and consequences. Mamm Genome 27:538–555. 10.1007/s00335-016-9661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 27:322–6. 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 81.Haack TB, et al. (2015) Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann Clin Transl Neurol 2:492–509. 10.1002/acn3.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hagerman P (2013) Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol 126:1–19. 10.1007/s00401-013-1138-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hagerman RJ, Protic D, Rajaratnam A, Salcedo-Arellano MJ, Aydin EY, Schneider A (2018) Fragile X-associated neuropsychiatric disorders (FXAND). Front Psychiatry 9:564 10.3389/fpsyt.2018.00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Harripaul R, Noor A, Ayub M, Vincent JB (2017) The use of next-generation sequencing for research and diagnostics for intellectual disability. Cold Spring Harb Perspect Med 7(3): a026864 10.1101/cshperspect.a026864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hawe JS, Theis FJ, Heinig M (2019) Inferring interaction networks from multi-omics data. Front Genet 10:535 10.3389/fgene.2019.00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heo S, Diering GH, Na CH, Nirujogi RS, Bachman JL, Pandey A, Huganir RL (2018) Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc Natl Acad Sci USA 115: E3827–E3836. 10.1073/pnas.1720956115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herault Y, Delabar JM, Fisher EMC, Tybulewicz VLJ, Yu E, Brault V (2017) Rodent models in Down syndrome research: impact and future opportunities. Dis Model Mech 10:1165–1186. 10.1242/dmm.029728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Herms J, Dorostkar MM (2016) Dendritic spine pathology in neurodegenerative diseases.Annu Rev Pathol 11:221–50. 10.1146/annurev-pathol-012615-044216. [DOI] [PubMed] [Google Scholar]
- 89.Hoefele J, Gabert M, Heinrich U, Benz K, Rompel O, Rost I, Klein HG, Kunstmann E (2012) A novel interstitial deletion of 10q24.2q24.32 in a patient with renal coloboma syndrome. Eur J Med Genet 55:211–5. 10.1016/j.ejmg.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 90.Hong F, Ze Y, Zhou Y, Hong J, Yu X, Sheng L, Wang F (2017) Nanoparticulate TiO2 - mediated inhibition of the Wnt signaling pathway causes dendritic development disorder in cultured rat hippocampal neurons. J Biomed Mater Res A 105:2139–2149. 10.1002/jbma.36073. [DOI] [PubMed] [Google Scholar]
- 91.Honnorat J, Byk T, Kusters I, Aguera M, Ricard D, Rogemond V, Quach T, Aunis D, Sobel A, Mattei MG, Kolattukudy P, Belin MF, Antoine JC (1999) Ulip/CRMP proteins are recognized by autoantibodies in paraneoplastic neurological syndromes. Eur J Neurosci 11:4226–32. 10.1046/j.1460-9568.1999.00864.x. [DOI] [PubMed] [Google Scholar]
- 92.Hopkins SE, Chadehumbe M, Blaine Crowley T, Zackai EH, Bilaniuk LT, McDonald-McGinn DM (2018) Neurologic challenges in 22q11.2 deletion syndrome. Am J Med Genet A 176:2140–2145. 10.1002/ajmg.a.38614. [DOI] [PubMed] [Google Scholar]
- 93.Hu R, Gong X, Duan Y, Li N, Che Y, Cui Y, Zhou M, Liu C, Wang H, Hong F (2010) Neurotoxicological effects and the impairment of spatial recognition memory in mice caused by exposure to TiO2 nanoparticles. Biomaterials 31:8043–50. 10.1016/j.biomaterials.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 94.Humphreys P, Barrowman N (2016) The incidence and evolution of Parkinsonian rigidity in Rett syndrome: a pilot study. Can J Neurol Sci 43:567–73. 10.1017/cjn.2016.8. [DOI] [PubMed] [Google Scholar]
- 95.Igarashi M (2014) Proteomic identification of the molecular basis of mammalian CNS growth cones. Neurosci Res 88:1–15. 10.1016/j.neures.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 96.Ilyas M, Mir A, Efthymiou S, Houlden HF (2020) The genetics of intellectual disability: advancing technology and gene editing.1000 Res 9:F1000 Faculty Rev-22. eCollection 2020 10.12688/f1000research.16315.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Imperlini E, Orrù S, Corbo C, Daniele A, Salvatore F(2014) Altered brain protein expression profiles are associated with molecular neurological dysfunction in the PKU mouse model. J. Neurochem 129:1002–12. 10.1111/jnc.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Iqbal J, Zhang K, Jin N, Zhao Y, Liu Q, Ni J, Shen L (2018) Effect of sodium selenate on hippocampal proteome of 3×Tg-AD mice-exploring the antioxidant dogma of Selenium against Alzheimer's disease. ACS Chem Neurosci 9:1637–1651. 10.1021/acschemneuro.8b00034. [DOI] [PubMed] [Google Scholar]
- 99.Irving M, Hanson H, Turnpenny P, Brewer C, Ogilvie CM, Davies A, Berg J (2003) Deletion of the distal long arm of chromosome 10; is there a characteristic phenotype? A report of 15 de novo and familial cases. Am J Med Genet A 1 23A:153–63. 10.1002/ajmg.a.20220. [DOI] [PubMed] [Google Scholar]
- 100.Iverson GL (2001) Interpreting change on the WAIS-III/WMS-III in clinical samples. Arch Clin Neuropsychol 16:183–91. PMID: 14590186. [PubMed] [Google Scholar]
- 101.Iwase S, Bérubé NG, Zhou Z, Kasri NN, Battaglioli E, Scandaglia M, Barco A (2017) Epigenetic etiology of intellectual disability. J Neurosci 37:10773–10782. 10.1523/JNEUROSCI.1840-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jackson M, Marks L, May GHW, Wilson JB (2018) The genetic basis of disease. Essays Biochem 62: 643–723. 10.1042/EBC20170053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jdila MB, Triki CC, Ghorbel R, Bouchalla W, Ncir SB, Kamoun F, Fakhfakh F (2020) Unusual double mutation in MECP2 and CDKL5 genes in Rett-like syndrome: Correlation with phenotype and genes expression. Clin Chim Acta 508:287–294. 10.1016/j.cca.2020.05.037. [DOI] [PubMed] [Google Scholar]
- 104.Joensuu H, Lehtimäk T, Holli K, Elomaa L, Turpeenniemi-Hujanen T, Kataja V, Anttila A, Lundin M, Isola J, Lundin J (2004) Risk for Distant Recurrence of Breast Cancer Detected by Mammography Screening or Other Methods. JAMA 292:1064–73. 10.1001/jama.292.9.1064. [DOI] [PubMed] [Google Scholar]
- 105.Jovčevska I, Bosn J (2019) Genetic secrets of long-term glioblastoma survivors. Basic Med Sci 19:116–124. 10.17305/bjbms.2018.3717.Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH (2005) Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis 19:18–27. 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 107.Jurata LW, Gallagher P, Lemire AL, Charles V, Brockman JA, Illingworth EL, Altar CA (2006) Altered expression of hippocampal dentate granule neuron genes in a mouse model of human 22q11 deletion syndrome. Schizophr Res 88:251–9. 10.1016/j.schres.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 108.Kahanovitch U, Patterson KC, Hernandez R, Olsen ML (2019) Glial Dysfunction in MeCP2 Deficiency Models: Implications for Rett Syndrome. Int J Mol Sci 20:3813 10.3390/ijms20153813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kasowitz SD, Luo M, Ma J, Leu A, Wang J (2017) Embryonic lethality and defective male germ cell development in mice lacking UTF1. Sci Rep 7:17259 10.1038/s41598-017-17482-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kiehl TR, Chow EW, Mikulis DJ, George SR, Bassett AS (2009) Neuropathologic features in adults with 22q11.2 deletion syndrome. Cereb Cortex 19:153–64. 10.1093/cercor/bhn066. [DOI] [PubMed] [Google Scholar]
- 111.Knolle MD, Owen CA (2009) ADAM8: a new therapeutic target for asthma. Expert Opin Ther Targets 13:523–40. 10.1517/14728220902889788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kogelman LJA , Fu J , Franke L , Greve JW , Hofker M , Rensen SS , Kadarmideen HN (2016) Inter-Tissue Gene Co-Expression Networks Between Metabolically Healthy and Unhealthy Obese Individuals. PLoS One 11:e0167519 10.1371/journal.pone.0167519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kooy RF, D'Hooge R, Reyniers E, Bakker CE, Nagels G, De Boulle K, Storm K, Clincke G, De Deyn PP, Oostra BA, Willems PJ (1996) Transgenic mouse model for the fragile X syndrome. Am J Med Genet 64:241–5. . [DOI] [PubMed] [Google Scholar]
- 114.Kulkarni VA, Firestein BL (2012) The dendritic tree and brain disorders. Mol Cell Neurosci 50:10–20. 10.1016/j.mcn.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 115.Kyle S M, Vashi N, Justice MJ (2018) Rett Syndrome: A neurological disorder with metabolic components. Open Biol 8:170216 10.1098/rsob.170216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.La Fata G, Gärtner A, Domínguez-Iturza N, Dresselaers T, Dawitz J, Poorthuis RB, Averna M, Himmelreich U, Meredith RM, Achsel T, Dotti CG, Bagni C (2014) FMRP regulates multipolar to bipolar transition affecting neuronal migration and cortical circuitry. Nat Neurosci 17:1693–700. 10.1038/nn.3870. [DOI] [PubMed] [Google Scholar]
- 117.Lacaria M, Srour M, Michaud JL, Doja A, Miller E, Schwartzentruber J, Goldsmith C, Majewski J; FORGE Canada Consortium, Boycott KM (2017) Expansion of the clinical phenotype of the distal 10q26.3 deletion syndrome to include ataxia and hyperemia of the hands and feet. Am J Med Genet A 173:1611–1619. 10.1002/ajmg.a.38231. [DOI] [PubMed] [Google Scholar]
- 118.Lampo E, Van Poucke M, Hugot K, Hayes H, Van Zeveren A, Peelman LJ (2007) Characterization of the genomic region containing the Shadow of Prion Protein (SPRN) gene in sheep. BMC Genomics 8:138 10.1186/1471-2164-8-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Landi S , Putignano E, Boggio EM, Giustetto M, Pizzorusso T, Ratto GM (2011) The Short-Time Structural Plasticity of Dendritic Spines Is Altered in a Model of Rett Syndrome. Sci Rep 1:45 10.1038/srep00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Laudier B, Epiais T, Pâris A, Menuet A, Briault S, Ozsancak C, Perche O (2016) Molecular and clinical analyses with neuropsychological assessment of a case of del (10)(q26.2qter) without intellectual disability: Genomic and transcriptomic combined approach and review of the literature. Am J Med Genet A 170:1806–12. 10.1002/ajmg.a.37677. [DOI] [PubMed] [Google Scholar]
- 121.Laurin N, Misener VL, Wigg KG, Anderson B, Cate-Carter T, Tannock R, Humphries T, Lovett MW, Barr CL (2007) Association of the dopamine receptor D1 gene, DRD1, with inattention symptoms in families selected for reading problems. Mol Psychiatry 12:776–85. 10.1038/sj.mp.4001972. [DOI] [PubMed] [Google Scholar]
- 122.Lefebvre JL, Sanes JR, Kay JN (2015) Development of dendritic form and function. Annu Rev Cell Dev Biol 31:741–77. 10.1146/annurev-cellbio-100913-013020. [DOI] [PubMed] [Google Scholar]
- 123.Lei W, Omotade OF, Myers KR, Zheng JQ (2016) Actin cytoskeleton in dendritic spine development and plasticity. Curr Opin Neurobiol 39:86–92. 10.1016/j.conb.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li Y, McGrail DJ, Latysheva N, Yi S, Babu MM, Sahni N (2020) Pathway perturbations in signaling networks: Linking genotype to phenotype.Semin Cell Dev Biol 99:3–11. 10.1016/j.semcdb.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li W, Aurandt J, Jürgensen C, Yi Rao, Guan K (2006) FAK and Src kinases are required for Netrin-Induced tyrosine phosphorylation of UNC5. J Cell Sci 119:47–55. 10.1242/jcs.02697. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 126.Liao W (2019) Psychomotor dysfunction in Rett Syndrome: Insights into the neurochemical and circuit roots. Dev Neurobiol 79:51–59. 10.1002/dneu.22651. [DOI] [PubMed] [Google Scholar]
- 127.Libersat F, Duch C (2004) Mechanisms of dendritic maturation Mol Neurobiol 29:303–20. 10.1385/MN:29:3:303. [DOI] [PubMed] [Google Scholar]
- 128.Lindsay EA, Botta A, Jurecic V, Carattini-Rivera S, Cheah YC, Rosenblatt HM, Bradley A, Baldini A (1999) Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 401:379–83. 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- 129.Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, Hnisz D, Li CH, Yuan B, Xu C, Li Y (2018) Rescue of Fragile X Syndrome neurons by DNA methylation editing of the FMR1 gene. Cell 172:979–992. 10.1016/j.cell.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Loesch D, Hagerman R (2012) Unstable mutations in the FMR1 gene and the phenotypes. Adv Exp Med Biol 769:78–114. 10.1007/978-1-4614-5434-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lopes F, Soares G, GonÇalves-Rocha M, Pinto-Basto J, Maciel P (2017) Whole gene deletion of EBF3 supporting haploinsufficiency of this gene as a mechanism of neurodevelopmental disease. Front Genet 8:143 10.3389/fgene.2017.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lund J, Roe B, Chen F, Budarf M, Galili N, Riblet R, Miller RD, Emanuel BS, Reeves RH (1999) Sequence-ready physical map of the mouse chromosome 16 region with conserved synteny to the human velocardiofacial syndrome region on 22q11.2. Mamm Genome. 10:438–43. 10.1007/s003359901019. [DOI] [PubMed] [Google Scholar]
- 133.Lyst MJ, Bird A (2015) Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 16:261–75. 10.1038/nrg3897. [DOI] [PubMed] [Google Scholar]
- 134.Maor-Sagie E, Cinnamon Y, Yaacov B, Shaag A, Goldsmidt H, Zenvirt S, Neri Laufer, Richler C, Frumkin A (2015) Deleterious mutation in SYCE1 is associated with non-obstructive azoospermia. J Assist Reprod Gene 32:887–91. 10.1007/s10815-015-0445-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mardones MD, Jorquera PV, Herrera-Soto A, Ampuero E, Bustos FJ, van Zundert B, Varela-Nallar L (2019) PSD95 regulates morphological development of adult-born granule neurons in the mouse hippocampus. J Chem Neuroanat 98:117–123. 10.1016/j.jchemneu.2019.04.009. [DOI] [PubMed] [Google Scholar]
- 136.Marin-Padilla M (1972) Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res 44:625–9. 10.1016/0006-8993(72)90324-1 [DOI] [PubMed] [Google Scholar]
- 137.Martínez-Cerdeño V (2017) Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev Neurobiol 77: 393–404. 10.1002/dneu.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Cheindel C, Tugimoto TJ, Wu Y, Lieberman JA, Lamantia AS (2008) Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci 39:439–51. 10.1016/j.mcn.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McDonald-McGinn DM, Sullivan KE (2011) Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome /velocardiofacial syndrome). Medicine (Baltimore) 90:1–18. 10.1097/MD.0b013e3182060469. [DOI] [PubMed] [Google Scholar]
- 140.McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS (2015) 22q11.2 deletion syndrome. Nat Rev Dis Primers 1:15071 10.1038/nrdp.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Medvedev NI, Popov VI, Rodriguez Arellano JJ, Dallérac G, Davies HA, Gabbott PL, Laroche S, Kraev IV, Doyère V, Stewart MG (2010) The N-methyl-D-aspartate receptor antoganist CPP alters synapse abd spine structure and impairs long-term potentiation and long-term depression induced morphological plasticity in dentate gyrus of the awake rat. Neuroscience 165:1170–81. 10.1016/j.neuroscience.2009.11.047. [DOI] [PubMed] [Google Scholar]
- 142.Merscher S , Funke B, Epstein JA , Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, Jore BS, Lopez M, Pandita RK, Lia M, Carrion D D, Xu H H, Schorle H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucherlapati R (2001) TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell 104:619–29. 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 143.Mila M, Alvarez-Mora MI, Madrigal I, Rodriguez-Revenga L (2018) Fragile X syndrome: An overview and update of the FMR1 gene. Clin Genet 93:197–205. 10.1111/cge.13075. [DOI] [PubMed] [Google Scholar]
- 144.Mir YR, Kuchay RAH (2019) Advances in identification of genes involved in autosomal recessive intellectual disability: a brief review. J Med Genet 56:567–573. 10.1136/jmedgenet-2018-105821. [DOI] [PubMed] [Google Scholar]
- 145.Mitani T, et al. (2019) Bi-allelic pathogenic variants in TUBGCP2 cause microcephaly and lissencephaly spectrum disorders Am J Hum Genet 105:1005–1015. 10.1016/j.ajhg.2019.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mody HR, Hung SW, Pathak RK, Griffin J, Cruz-Monserrate Z, Govindarajan R (2017) miR-202 diminishes TGFβ receptors and attenuates TGFβ1 -induced EMT in pancreatic cancer. Mol Cancer Res 15:1029–1039. 10.1158/1541-7786.MCR-16-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mohan V, Sullivan CS, Guo J, Wade SD, Majumder S, Agarwal A, Anton ES, Temple BS, Maness PF (2019) Temporal regulation of dendritic spines through NrCAMSemaphorin3F receptor signaling in developing cortical pyramidal neurons. Cereb Cortex 29:963–977. 10.1093/cercor/bhy004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Montes M, Cloutier A, Sánchez-Hernández N, Michelle L, Lemieux B, Blanchette M, Cruz-Monserrate Z, Govindarajan R, Chabot B, Suñé C (2012) TCERG1 regulates alternative splicing of the Bcl-x gene by modulating the rate of RNA polymerase II transcription. Mol Cell Biol 32:751–762. 10.1128/MCB.06255-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morrow BE, McDonald-McGinn DM, Emanuel BS, Vermeesch JR, Scambler PJ (2018) Molecular genetics of 22q11.2 deletion syndrome. Am J Med Genet A 176:2070–2081. 10.1002/ajmg.a.40504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mostafaei S, Kazemnejad A, Azimzadeh Jamalkandi S Amirhashchi S, Donnelly SC, Armstrong ME, Doroudian M (2018) Identification of novel genes in human airway Epithelial Cells associated with Chronic Obstructive Pulmonary Disease (COPD) using Machine-Based Learning Algorithms. Sci Rep. 8:15775 10.1038/s41598-018-33986-8(JAKMIP3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mullen SA, Carvill GL, Bellows S, Bayly MA, Trucks H, Lal D, Sander T, Berkovic SF, Dibbens LM, Scheffer IE, Mefford HC (2013) Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology 81:1507–14. 10.1212/WNL.0b013e3182a95829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nerli E, Roggero OM, Baj G, Tongiorgi E (2020). In vitro modeling of dendritic atrophy in Rett syndrome: determinants for phenotypic drug screening in neurodevelopmental disorders. Sci. Rep 10:2491 10.1038/s41598-020-5968-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Nishiyama J (2019) Plasticity of dendritic spines: Molecular function and dysfunction in neurodevelopmental disorders. Psychiatry Clin Neurosci 73:541–550. 10.1111/pcn.12899. [DOI] [PubMed] [Google Scholar]
- 154.Nosheny RL, Belichenko PV, Busse BL, Weissmiller AM, Dang V, Das D, Fahimi A, Salehi A, Smith SJ, Mobley WC (2015) Increased cortical synaptic activation of TrkB and downstream signaling markers in a mouse model of Down Syndrome. Neurobiol Dis 77:173–90. 10.1016/j.nbd.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.O'Kusky J, Ye P (2012) Neurodevelopmental effects of insulin-like growth factor signaling. Front Neuroendocrinol. 33:230–51. 10.1016/j.yfrne.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Operto FF, Mazza R, Pastorino GMG, Verrotti A, Coppola G (2019) Epilepsy and genetic in Rett syndrome: A review. Brain Behav 9:e01250 10.1002/brb3.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ori-McKenney KM, McKenney RJ, Huang HH, Li T, Meltzer S, Jan LY, Vale RD, Wiita AP, Jan YN (2016) Phosphorylation of β-tubulin by the Down syndrome kinase, minibrain/DYRK1a, regulates microtubule dynamics and dendrite morphogenesis. Neuron 90:551–63. 10.1016/j.neuron.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Padamsey Z, Foster WJ, Emptage NJ (2019) Intracellular Ca2+ release and synaptic plasticity: A tale of many stores. Neuroscientist 25:208–226. 10.1177/1073858418785334. [DOI] [PubMed] [Google Scholar]
- 159.Panigrahi DP, Praharaj PP, Bhol CS, Mahapatra KK, Patra S, Behera BP, Mishra SR, Bhutia SK (2020) The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol 66:45–58. 10.1016/j.semcancer.2019.07.015. [DOI] [PubMed] [Google Scholar]
- 160.Penzes P, Cahill ME, Jones KA, Srivastava DP (2008) Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol 18:405–13. 10.1016/j.tcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 161.Petralia RS, Sans N, Wang YX, Wenthold RJ (2005) Ontogeny of postsynaptic density proteins at glutamatergic synapses. Mol Cell Neurosci 29:436–52. 10.1016/j.mcn.2005.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Piskorowski RA, Nasrallah K, Diamantopoulou A, Mukai J, Hassan SI, Siegelbaum SA, Gogos JA, Chevaleyre V (2016) Age-dependent specific changes in area CA2 of the hippocampus and social memory deficit in a mouse model of the 22q11.2 deletion syndrome. Neuron 89:163–76. 10.1016/j.neuron.2015.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Plaisancié J, Bouneau L, Cances C, Garnier C, Benesteau J, Leonard S, Bourrouillou G, Calvas P, Vigouroux A, Julia S, Bieth E (2014) Distal 10q monosomy: new evidence for a neurobehavioral condition? Eur J Med Genet 57:47–53. 10.1016/j.ejmg.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 164.Plomin R, von Stumm S (2018) The new genetics of intelligence. Nat Rev Genet 19:148–159. 10.1038/nrg.2017.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Prieto M, Folci A, Martin S (2019) Post-translational modifications of the Fragile X Mental Retardation Protein in neuronal function and dysfunction. Mol Psychiatry 10.1038/s41380-019-0629-4. [DOI] [PubMed] [Google Scholar]
- 166.Prigge CL, Kay JN (2018) Dendrite morphogenesis from birth to adulthood. Curr Opin Neurobiol 53:139–145. 10.1016/j.conb.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Qu Y, et al. (2011) Thioredoxin-like 2 regulates human cancer cell growth and metastasis via redox homeostasis and NF-κB signaling. J Clin Invest 121:212–25. 10.1172/JCI43144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Quach TT, Auvergnon N, Khanna R, Belin MF, Kolattukudy PE, Honnorat J, Duchemin AM (2018) Opposing morphogenetic defects on dendrites and mossy fibers of dentate granular neurons in CRMP3-deficient mice. Brain Sci 8:196 10.3390/brainsci.8110196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Quach TT, Massicotte G, Belin MF, Honnorat J, Glasper ER, Devries AC, Jakeman LB, Baudry M, Duchemin AM, Kolattukudy PE (2008) CRMP3 is required for hippocampal CA1 dendritic organization and plasticity. FASEB J 22:401–9. 10.1096/fj.07-9012com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Quach TT, Mosinger B Jr, Ricard D, Copeland NG, Gilbert DJ, Jenkins NA, Stankoff B, Honnorat J, Belin MF, Kolattukudy P (2000) Collapsin response mediator protein-3/unc-3 3-like protein-4 gene: organization, chromosomal mapping and expression in the developing mouse brain. Gene 242:175–82. 10.1016/s0378-1119(99)00528-4. [DOI] [PubMed] [Google Scholar]
- 171.Quach TT, Wang Y, Khanna R, Chounlamountri N, Auvergnon N, Honnorat J, Duchemin AM (2011) Effect of CRMP3 expression on dystrophic dendrites of hippocampal neurons. Mol Psychiatry 16:689–91. 10.1038/mp.2011.6. [DOI] [PubMed] [Google Scholar]
- 172.Quach TT, Wilson SM, Rogemond V, Chounlamountri N, Kolattukudy PE, Martinez S, Khanna M, Belin MF, Khanna R, Honnorat J, Duchemin AM (2013) Mapping CRMP3 domains involved in dendrite morphogenesis and voltage-gated calcium channel regulation. J Cell Sci 126:4262–73. 10.1242/jcs.131409. [DOI] [PubMed] [Google Scholar]
- 173.Quach TT, Glasper ER, Devries AC, Honnorat J, Kolattukudy PE, Duchemin AM (2008) Altered prepulse inhibition in mice with dendrite abnormalities of hippocampal neurons. Mol Psychiatry 13:656–8. 10.1038/mp.2008.27. [DOI] [PubMed] [Google Scholar]
- 174.Quach TT, Moutal A, Khanna R, Deems NP, Duchemin AM, Barrientos RM (2020) Collapsin response mediator proteins: Novel targets for Alzheimer's disease. J Alzheimers Dis 10.3233/JAD-200721. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rachidi M, Lopes C (2010) Molecular and cellular mechanisms elucidating neurocognitive basis of functional impairments associated with intellectual disability in Down syndrome. Am J Intellect Dev Disabil 115:83–112. 10.1352/1944-7558-115.2.83. [DOI] [PubMed] [Google Scholar]
- 176.Ratna A, Mandrekar P (2017) Alcohol and cancer: mechanisms and therapies. Biomolecules 7:61 10.3390/biom7030061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ray BK, Dhar S, Shakya A, Ray A (2011) Z-DNA-forming silencer in the first exon regulates human ADAM-12 gene expression Proc Natl Acad Sci USA 108:103–8. 10.1073/pnas.1008831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361:1545–64. 10.1098/rstb.2006.1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Meredith Rhiannon M., Mansvelder Huibert D. (2010) STDP and mental retardation: Dysregulation of dendritic excitability in Fragile X syndrome. Front Synaptic Neurosci 2:10 10.3389/fnsyn.2010.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Riccomagno MM, Kolodkin AL (2015) Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol 31: 779–805. 10.1146/annurev-cellbio-100913-013038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sahay A, Molliver ME, Ginty DD, Kolodkin AL (2003) Semaphorin 3F is critical for development of limbic system circuitry and is required in neurons for selective CNS axon guidance events. J Neurosci 23:6671–6680. 10.1523/JNEUROSCI.23-17-06671.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Saito Y, Oka A, Mizuguchi M, Motonaga K, Mori Y, Becker LE, Arima K, Miyauchi J, Takashima S (2000). The developmental and aging changes of Down's syndrome cell adhesion molecule expression in normal and Down's syndrome brains. Acta Neuropathol 100:654–64. 10.1007/s004010000230. [DOI] [PubMed] [Google Scholar]
- 183.Salati S, et al. (2016) Integrative analysis of copy number and gene expression data suggests novel pathogenetic mechanisms in primary myelofibrosis. Int J Cancer 138:1657–69. 10.1002/ijc.29920. [DOI] [PubMed] [Google Scholar]
- 184.Sanders AF, Hobbs DA, Stephenson DD Jr, Laird RD, Beaton EA (2017) Working memory impairments in chromosome 22q11.2 deletion syndrome: The roles of anxiety and stress physiology. J Autism Dev Disord 47:992–1005. 10.1007/s10803-016-3011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sangu N, Okamoto N, Shimojima K, Ondo Y, Nishikawa M, Yamamoto T (2016) A de novo microdeletion in a patient with inner ear abnormalities suggests that the 10q26.13 region contains the responsible gene. Hum Genome Var 3:16008 10.1038/hgv.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Saré RM, Figueroa C, Lemons A, Loutaev I, Beebe Smith C (2019) Comparative behavioral phenotypes of Fmr1 KO, Fxr2 Het, and Fmr1 KO/Fxr2 Het mice. Brain Sci. 9:13 10.3390/brainsci9010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sauna-Aho O, Bjelogrlic-Laakso N, Siren A, Arvio M (2018) Signs indicating dementia in Down, Williams and Fragile X syndromes. Mol Genet Genomic Med 6:855–860. 10.1002/mgg3.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Savage JE, et al. (2018) Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 51:404–413. 10.1038/s41588-018-0311-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shulyakova N, Andreazza AC, Mills LR, Eubanks JH (2017) Mitochondrial dysfunction in the pathogenesis of Rett syndrome: Implications for mitochondriatargeted therapies. Front Cell Neurosci 11:58 10.3389/fncel.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Snezhkina AV, Lukyanova EN, Fedorova MS, Kalinin DV, N V Melnikova NV, O A Stepanov OA, Kiseleva MV, Kaprin AD, Pudova EA, Kudryavtseva AV (2019) Novel genes associated with the development of carotid paragangliomas. Mol Biol (Mosk) 53:613–626. 10.1134/S0026898419040141. [DOI] [PubMed] [Google Scholar]
- 191.Sokol DK, Maloney B, Long JM, Ray B, Lahiri DK (2011) Autism, Alzheimer disease, and fragile X: APP, FMRP, and mGluR5 are molecular links. Neurology 76: 1344–1352. 10.1212/WNL.0b013e3182166dc7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Suetsugu M, Mehraein P (1980) Spine distribution along the apical dendrites of the pyramidal neurons in Down's syndrome. A quantitative Golgi study. Acta Neuropathol 50:207–10. 10.1007/BF00688755. [DOI] [PubMed] [Google Scholar]
- 193.Suzuki T, Maruno M, Wada K, Kagawa N, Fujimoto Y, Hashimoto N, Izumoto S, Yoshimine T (2004) Genetic analysis of human glioblastomas using a genomic microarray system. Brain Tumor Pathol 21:27–34. 10.1007/BF02482174. [DOI] [PubMed] [Google Scholar]
- 194.Spencer CM, Serysheva E, Yuva-Paylor LA, Oostra BA, Nelson DL, Paylor R (2006) Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins. Hum Mol Genet 15:1984–1994. 10.1093/hmg/ddl121. [DOI] [PubMed] [Google Scholar]
- 195.Szczałuba K, Demkow U (2017) Array comparative genomic hybridization and genomic sequencing in the diagnostics of the causes of congenital anomalies. J Appl Genet 58: 185–198. 10.1007/s13353-016-0376-z. [DOI] [PubMed] [Google Scholar]
- 196.Takashima S, Ieshima A, Nakamura H, Becker LE (1989) Dendrites, dementia and the Down syndrome brain. Dev 11:131–3. 10.1016/s0387-7604(89)80082-8. [DOI] [PubMed] [Google Scholar]
- 197.Takeguchi R, Takahashi S, Kuroda M, Tanaka R, Suzuki N, Tomonoh Y, Ihara Y, Sugiyama N, Itoh M (2020) MeCP2_e2 partially compensates for lack of MeCP2_e1: A male case of Rett syndrome. Mol Genet Genomic Med 8:e1088. 10.1002/mgg3.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Haydar Tarik F., Reeves RH (2012) Trisomy and early brain development. Trends Neurosci 35: 81–91. 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Trazzi S, Fuchs C, Valli E, Perini G, Bartesaghi R, Ciani E (2013) The amyloid precursor protein (APP) triplicated gene impairs neuronal precursor differentiation and neurite development through two different domains in the Ts65Dn mouse model for Down syndrome..J Biol Chem 288:20817–29. 10.1074/jbc.M113.451088. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 200.Tremblay K, Lemire M, Potvin C, Tremblay A, Hunninghake GM, Raby BA, Hudson TJ, Perez-Iratxeta C, Andrade-Navarro MA, Laprise C (2008) Genes to Diseases (G2D) computational method to identify asthma candidate genes. PLoS One 3:e2907 10.1371/journal.pone.0002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Turleau C, de Grouchy J, Ponsot G, Bouygues D (1979) Monosomy 10qter. Hum Genet 47:233–7. 10.1007/BF00321014. [DOI] [PubMed] [Google Scholar]
- 202.Tzschach A, Krause-Plonka I, Menzel C, Knoblauch A, Toennies H, Hoeltzenbein M, Radke M, Ropers HH, Kalscheuer V (2006) Molecular cytogenetic analysis of a de novo interstitial chromosome 10q22 deletion. Am J Med Genet A 140:1108–10. 10.1002/ajmg.a.31226..PMID: 16619204 [DOI] [PubMed] [Google Scholar]
- 203.Utari A, Adams E, Berry-Kravis E, Chavez A, Scaggs F, Ngotran L, Boyd A, Hessl D, Gane LW, Tassone F, Tartaglia N, Leehey MA, Hagerman RJ (2010) Aging in fragile X syndrome. J Neurodev Disord 2:70–76. 10.1007/s11689-010-90472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Vacano GN, Gibson DS, Turjoman AA, Gawryluk JW, Geiger JD, Duncan M, Patterson D (2018) Proteomic analysis of six- and twelve-month hippocampus and cerebellum in a murine Down syndrome model. Neurobiol Aging 63:96–109. 10.1016/j.neurobiolaging.2017.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Valnegri P, Puram SV, Bonni A (2015) Regulation of dendrite morphogenesis by extrinsic cues. Trends Neurosci 38:439–47. 10.1016/j.tins.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Van L, Boot E, Bassett AS (2017) Update on the 22q11.2 deletion syndrome and its relevance to schizophrenia. Curr Opin Psychiatry 30:191–196. 10.1097/YCO.0000000000000324. [DOI] [PubMed] [Google Scholar]
- 207.Vashi N, Justice MJ (2019) Treating Rett syndrome: From mouse models to human therapies. Mamm Genome. 30:90–110. 10.1007/s00335-019-09793-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Martínez-Cerdeño Verónica (2017) Dendrite and spine modifications in autism and related neurodevelopmental disorders in patients and animal models. Dev Neurobiol 77: 393–404. 10.1002/dneu.22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Villard L (2007) MECP2 mutations in males. J Med Genet 44:417–423. 10.1136/jmg.2007.049452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Vissers LE, Gilissen C, Veltman JA (2016). Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17:9–18. 10.1038/nrg3999. [DOI] [PubMed] [Google Scholar]
- 211.Waggoner DJ, Chow CK, Dowton SB, Watson MS (1999) Partial monosomy of distal 10q: Three new cases and a review. Am J Med Genet 86:1–5. PMID: 10440820. [PubMed] [Google Scholar]
- 212.Wesseling H, Xu B, Want EJ, Holmes E, Guest PC, Karayiorgou M, Gogos JA, Bahn S (2017) System-based proteomic and metabonmic analysis of the Df (16)A(+/−) mouse identifies potential miR-185 targets and molecular pathway alterations. Mol Psychiatry 22:384–395. 10.1038/mp.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wicherts JM (2016) The importance of measurement invariance in neurocognitive ability testing. Clin Neuropsychol 30:1006–16. 10.1080/13854046.2016.1205136. [DOI] [PubMed] [Google Scholar]
- 214.Wilcock DM (2012) Neuroinflammation in the aging down syndrome brain; lessons from Alzheimer's disease. Curr Gerontol Geriatr Res 170276 10.1155/2012/170276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wiseman FK, Alford KA, Tybulewicz VL, Fisher EM (2009) Down syndrome--recent progress and future prospects. Hum Mol Genet 18(R1):R75–83. 10.1093/hmg/ddp010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Woo Kim Y, et al. (2020) Exploring the novel susceptibility gene variants for primary open-angle glaucoma in East Asian cohorts: The GLAU-GENDISK study. Sci Rep 10:221 10.1038/s41598-019-57066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Xing Z, Li Y, Pao A, Bennett AS, Tycko B, Mobley WC, Yu YE (2016) Mouse-based genetic modeling and analysis of Down syndrome. Br Med Bull 120:111–122. 10.1093/bmb/ldw040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Xu D, Zhao Y, Weng X, Lu Y, Li W, Tang K, Chen W, Liu Z, Xinrui Qi, Jialing Zheng J, Fassett J, Zhang Y, Xu Y(2019) Novel role of mitochondrial GTPases1 in pathological cardiac hypertrophy. J Mol Cell Cardiol 128:105–116. 10.1016/j.yjmcc.2019.01.025. [DOI] [PubMed] [Google Scholar]
- 219.Xu F, Li L, Schulz VP, Gallagher PG, Xiang B, Zhao H, Li P (2014) Cytogenomic mapping and bioinformatic mining reveal interacting brain expressed genes for intellectual disability. Mol Cytogenet 7:4 10.1186/1755-8166-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yamagata M, Sanes JR (2010) Synaptic localization and function of sidekick recognition molecules require MAGI scaffolding proteins. J Neurosci 30:3579–88. 10.1523/JNEUROSCI.6319-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yatsenko SA, Kruer MC, Bader PI, Corzo D, Schuette J, Keegan CE, Nowakowska B, Peacock S, Cai WW, Peiffer DA, Gunderson KL, Ou Z, Chinault AC, Cheung SW (2009) Identification of critical regions for clinical features of distal 10q deletion syndrome. Clin Genet 76:54–62. 10.1111/j.1399-0004.2008.01115.x. [DOI] [PubMed] [Google Scholar]
- 222.Zabaneh D, Krapohl E , Gaspar HA , Curtis C, Lee SH , Patel H, Newhouse S, Wu HM , Simpson MA, Putallaz M , Lubinski D, Plomin R, Breen G (2018) A genome-wide association study for extremely high intelligence. Mol Psychiatry 23:1226–1232. 10.1038/mp.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Zahir FR, Mwenifumbo JC, Chun HJE, Lim EL, Van Karnebeek CDM, Couse M, Mungall KL, Lee L, Makela N, Armstrong L, Boerkoel CF, Langlois SL, McGillivray BM, Jones SJM, Friedman JM, Marra MA (2017) Comprehensive whole genome sequence analyses yields novel genetic and structural insights for Intellectual Disability. BMC Genomics 18: 403 10.1186/s12864-017-3671-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Zamboni V, Jones R, Umbach A, Ammoni A, Passafaro M, Hirsch E, Merlo GR (2018) Rho GTPases in intellectual disability: From genetics to therapeutic opportunities. Int J Mol Sci 19:1821 10.3390/ijms19061821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Zhang T, Wei G, Millard CJ, Fischer R, Konietzny R, Kessler BM, Schwabe JWR, Brockdorff N. (2018) A variant NuRD complex containing PWWP2A/B excludes MBD2/3 to regulate transcription at active genes. Nat Commun. 9:3798 10.1038/s41467-018-06235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]