

Abstract
T cell exhaustion represents one of the most pervasive strategies tumors employ to circumvent the immune system. While repetitive, cognate T cell receptor signaling is recognized as the primary driving force behind this phenomenon, it remains unknown what other forces drive T cell exhaustion in the tumor microenvironment (TME). Here, we show that activation of the self-ligand SLAMF7 immune receptor on T cells induced STAT1 and STAT3 phosphorylation, expression of multiple inhibitory receptors, and transcription factors associated with T cell exhaustion. Analysis of The Cancer Genome Atlas revealed that SLAMF7 transcript levels were strongly correlated with various inhibitory receptors, and that high SLAMF7 expression was indicative of poor survival in clear cell renal cell carcinoma (ccRCC). Targeted reanalysis of a CyTOF dataset which profiled the TME in 73 ccRCC patients, revealed cell-type specific SLAMF7 expression patterns, strong correlations between exhausted T cells and SLAMF7+ tumor-associated macrophages (TAMs), and a unique subset of SLAMF7highCD38high TAMs. These SLAMF7highCD38high TAMs showed the strongest correlations with exhausted T cells and were an independent prognostic factor in ccRCC. Confirmatory ex vivo co-culture studies validated that SLAMF7-SLAMF7 interactions between murine TAMs and CD8+ T cells induces expression of multiple inhibitory receptors. Finally, mice lacking SLAMF7 show restricted growth of B16-F10 tumors and CD8+ T cells from these mice express less PD-1 and TOX, and exhibited an impaired ability to progress through the exhaustion developmental trajectory to terminal exhaustion. These findings suggest that SLAMF7 might play an important role in modulating T cell function in the TME.
Keywords: SLAMF7, CRACC, TOX, T cell exhaustion, renal carcinoma
Introduction:
The biological process of T cell exhaustion (or dysfunction), resulting from chronic T cell stimulation and activation has emerged as one of the most important topics in immunology and cancer biology (1–4). T cells undergoing persistent T cell receptor (TCR) stimulation, as occurs in the tumor microenvironment (TME), begin a process whereby they assume a cellular state less amenable to proliferation, cytokine production, and cytotoxicity (1, 3, 4). Conversion of infiltrating T cells to an exhaustion phenotype is a well-known hallmark tumors use to evade the host immune response and continue unchecked growth (5–7).
These exhausted or dysfunctional T cells are characterized at the cellular level by epigenetic remodeling (3, 8, 9) and expression of a number of inhibitory receptors, also known as checkpoint receptors, including: PD-1, CTLA-4, LAG3, Tim3, TIGIT, VISTA, CD38, 2B4 (10), and others (1, 2). Through both known and unknown mechanisms, these inhibitory receptors directly inhibit regulatory and effector T cell functions and are responsible for the characteristic loss of effector function observed in exhausted T cells (3). Antibodies targeting these inhibitory receptors are showing great promise in generating responses in cancer patients, however responses vary by tumor type and the majority of patients do not respond effectively to checkpoint blockade of PD-1, PD-L1, or CTLA-4 (11), and a combination of PD-1 and CTLA-4 blockade results in substantial risk of serious adverse events (12, 13). Consequently, there is a need to understand why some patients do not respond to checkpoint blockade, what additional signals besides repetitive TCR signaling induce inhibitory receptors on T cells in the TME, and if T cell exhaustion can be prevented from occurring in the first place (3).
Renal cell carcinoma (RCC) is the most common cancer of the genitourinary system, with over 400,000 new cases and over 175,000 deaths each year, and continues to increase in prevalence (14). The clear cell renal cell carcinoma (ccRCC) subtype is the most common form of RCC, and while a heterogeneous disease, it has a unique TME making it an attractive target for immunotherapy (14, 15). In contrast to most tumors where the TME is dominated by M-2 macrophages, the ccRCC TME is composed primarily of T cells, with a large portion consisting of CD8+ T cells (15, 16). However, the infiltrate of cytotoxic T lymphocytes does not portend a favorable prognosis due to extensive levels of T cell exhaustion (17–19). ccRCC is notable in that, of all the cancers in The Cancer Genome Atlas (TCGA), immune evasion occurs in ccRCC almost entirely via T cell exhaustion (19). This has paved the way for trials of various checkpoint inhibitors in ccRCC, with the most recent findings of the CheckMate 214 trial establishing combination ipilimumab (anti-CTLA-4) and nivolumab (anti-PD-1) as first-line treatment for intermediate or poor-risk ccRCC patients (14, 20). However, this regimen lacks effectiveness in all ccRCC patients (11) and can cause significant adverse effects (12, 13, 20), signifying the need to better understand T cell exhaustion mechanisms in ccRCC.
The SLAMF7 (CRACC, CD319, CS-1) receptor is a member of the signaling lymphocytic activation molecules (SLAM) family of receptors (21), with expression restricted to hematopoietic cells, and is present at differing frequencies on various types of immune cells (21–23). SLAMF7, and other SLAM family members (except 2B4), are unique in that they function as homotypic receptors, which once activated, recruit various SH2 domain-containing proteins to their cytoplasmic immunoreceptor tyrosine-based switch motifs (ITSMs) (21, 24). In doing so, SLAMF7 is able to modulate a host of immune cell-specific functions across various immune cell types (21, 23–25). SLAM family receptors are increasingly being linked to T cell exhaustion, with expression of SLAMF6 recently discovered as a marker to identify progenitor exhausted CD8+ T cells (26), and both SLAMF6 and SLAMF4 (2B4) being shown to function as inhibitory receptors on CD8+ T cells (10, 27). Notably, SLAMF7 has also recently been linked to a CD8+ T cell subset enriched in melanoma patients who fail to respond to checkpoint blockade (28), and expressed on certain memory-precursor and effector CD8+ T cells which respond indirectly to checkpoint blockade (29). Here, we set out to define the role of SLAMF7 on T cell regulation in the context of T cell exhaustion. We uncovered a novel role for SLAMF7 in regulating the expression of various T cell inhibitory receptors, exhaustion-promoting transcription factors, and STAT1/3 phosphorylation in a TCR-independent manner. Also, we find that SLAMF7 self-ligation between TAMs and T cells is sufficient to induce the upregulation of these inhibitory receptors. Our data identify SLAMF7 as a novel regulator of T cell inhibitory programs, with potential clinical implications and therapeutic opportunities.
Materials and Methods:
Reagents used.
The following human antibodies were used: CD3-FITC (HIT3a), CD4-V450 (RPA-T4), CD8-Alexa700 (HIT8a), SLAMF7-PerCp-eFluor710 (162), LAG-3-APC-eFluor780 (T47-530), TIM-3-PE-Cy7 (F38-2E2), EZH2-PE (11/EZH2), PD-1-Alexa488 (EH12.2H7), FoxP3-BV421 (206D), YY1-Alexa594 (H-10), Blimp-1–DyLight650 (3H2-E8), CTLA4-FITC (14D3), IL-2-APC (MQ1-17H12), TNFα-Alexa488 (Mab11), IFNγ-PE (4S.B3), STAT1 (pY701)-Alexa647, STAT2 (p690)-PE, STAT3 (p704)-Alexa647, STAT5 (p694)-Alexa647, STAT6 (p641)-Alexa647, P38 MAPK (pT180/pY182)-Alexa647, ERK1/2 (pT202/pY204)-Alexa647, ZAP70 (p319/p352)-PE, and SLAMF7 (162.1) (used for cross-linking). Mouse antibodies used include: CD3-APC (145-2C11), CD38-BV510 (90/CD38), CD8a-Alexa700 (53-6.7), LAG3-PE (C9B7W), Tim3-APC-Fire750 (B8.2C12), PD-1-PerCp-eFluor710 (J43), 2B4-FITC (eBio244F4), SLAMF7-BV421 (4G2), CD11b-BV570 (M1/70), CD19-PerCp-Cy5.5 (1D3), Ly6G-BV711 (1A8), Ly6C-BV421 (HK1.4), MHC-II-BV785 (M5/114.15.2), CD206-Alexa647 (C06862), CD45-Alexa532 (30-F11), NK1.1-PE-Cy7 (PK136), SLAMF7-APC (4G2), CD11c-PE-CF594 (HL3), CCR2-BV750 (475301), LAG3-BV785 (C9B7W), CD3-BUV737 (17A2), TOX-eFluor660 (TXRX10), SLAMF6-BUV395 (13G3), CD69-PE-Cy7 (H1.2F3), and CD4-eFluor450 (RM4-5). All antibodies were purchased from BioLegend, BD Biosciences, or ThermoFisher. Recombinant human IL-2 (Peprotech) was used at 300 IU/mL. Recombinant murine IL-2 (R&D Systems) was used at 300 IU/mL. CD3/28 Dynabeads (ThermoFisher) were added at a 1:1 cell:bead ratio.
Blood sample collection and human T cell isolation.
Fresh PBMCs were obtained from buffy coats and processed as previously described (30). CD3+ T cells were isolated from PBMCs via negative selection using Dynabeads Untouched Human T Cells Kit (ThermoFisher) per manufacturer’s instructions.
In vitro human T cell culture and stimulation.
Isolated, fresh, primary human CD3+ T cells were plated at 2x105 cells/well in 96-well plates. Cells were cultured in complete RPMI (RPMI 1640, 10% FBS, 1X penicillin, streptomycin, and fungizone). For cross-linking experiments, 8 μg/ml anti-SLAMF7 (162.1) mAb was added to a sterile, high-binding 96-well cell culture plate overnight at 4 °C. Wells were washed with PBS three times before addition of cells. Cells were cultured for indicated times at 37 °C and 5% CO2. For 6 day time points, cells were cultured in the presence of stimuli for 3 days before being moved into a fresh SLAMF7 mAb cross-linked plate with fresh media and rhIL-2. For re-stimulation experiment, cells were washed and all stimuli removed after 6 days of culture. Cells were re-plated in fresh complete media with 300 IU/mL IL-2 and CD3/28 Dynabeads at a 0.2:1 bead:cell ratio, and cultured for an additional 3 days. GolgiPlug was added for the last 4 hrs of culture and intracellular staining of cytokines was performed with BD CytoFix/Perm kit per manufacturer’s instructions.
Phosphoflow experiments.
Isolated, primary human CD3+ T cells were rested for 24hrs in 40 IU/mL rhIL-2 in complete RPMI. Cells (300,000 per well) were stimulated in 96-well plates as indicated for various time points at 37 °C and 5% CO2 before being fixed in BD CytoFix/Perm buffer at 37 °C and 5% CO2. Permeabilization was performed with BD Phosflow Perm Buffer IV (0.5X) per manufacturer’s instructions. Cells were stained with anti-CD4, anti-CD8, and a mAb against a single phosphorylated protein at the same time. Samples were run on a Cytek Aurora spectral cytometer.
Animal procedures and generation of SLAMF7−/− mice
All mice used in experiments were bred in house. SLAMF7−/− genotype was verified by PCR. 8-12 week old WT (C57BL/6J) or SLAMF7−/− (C57BL/6J background) were used for all experiments. SLAMF7−/− mice were generated at MSU Transgenic and Genome Editing Facility (MSU-TGEF) using CRISPR-Cas9 with gRNAs targeting introns between exon 2/3 and exon 5/6 of SLAMF7. Genomic deletion of this region of SLAMF7 completely abrogates SLAMF7 expression on all cells. A single founder mouse with homozygous deletions in both introns was bred to a WT mouse, and the F1 progeny were bred together to establish a stable line of SLAMF7−/− mice.
B16 tumor experiments and murine immune cell isolations.
Investigators were blinded to mouse genotypes when measuring tumors. Either 7x105 B16 tumor cells (for experiments involving isolation of TAMs) or 4x105 B16 tumor cells (for tumor growth experiments) were injected into the hind flank of WT and SLAMF7−/− mice. For TAM isolation experiments tumors were harvested 25 days later and manually dissociated into a single-cell suspension. For tumor growth and TIL phenotyping experiments tumors were harvested at completion of tumor measurements. Cell suspension was pelleted and resuspended in RPMI with 0.5 mg/mL Collagenase IV (Milipore-Sigma) and 1,000 IU/mL DNaseI (Millipore-Sigma) under constant, gentle agitation at 37 °C for 1 hr. Digestion was stopped with EDTA and cell suspension was filtered through a 40μm cell strainer. Cells were then subject to a Ficoll-Paque gradient centrifugation step to enrich for immune cells. Cells were washed three times with complete RPMI before immune cell isolation procedures. TAMs were isolated from dissociated B16 tumors using a mouse CD11b positive selection kit (StemCell) per manufacturer’s instructions. CD8+ T cells were isolated from excised B16 tumors and splenocytes using a CD8a positive selection kit (Miltenyi Biotec) per manufacturer’s instructions. Murine immune cells were either stained directly for spectral cytometry analysis or plated for co-culture experiments. For TAM-CD8 T cell co-culture experiments, 5x104 TAMs and 1.5x105 CD8+ T cells (1:3 ratio) were plated in a 96-well cell culture plate in complete RPMI supplemented with an additional 1% PSF and 50 IU/mL rmIL-2. Cells were cultured for 6 days at 37 °C and 5% CO2 with a media change at 3 days. For murine in vitro CD3/28 stimulation experiment, mouse total splenocytes were cultured for 6 days in complete RPMI and 50 IU/mL rmIL-2 with or without murine CD3/28 Dynabeads at a 1:1 cell:bead ratio. BMDMs were generated as previously described (31).
Spectral cytometry.
Cells were prepared and stained as previously described (23). Transcription factor and cytokine staining was performed with the Transcription Factor Buffer Set (BD Biosciences) per manufacturer’s instructions. For IL-2 intracellular staining, GolgiPlug (BD Biosciences) was added for the final 4.5 hrs of cell culture. Viability staining was performed with either Zombie NIR Fixable Viability Kit (BioLegend) or LIVE/DEAD Aqua fixable viability dye (ThermoFisher), and was included in all experiments. Fc receptors were blocked in all samples during staining with human Fc block (BD biosciences) or mouse Fc block (BD biosciences). Samples were acquired on a 5-laser Cytek Aurora Spectral Cytometer. Spectra were unmixed using SpectroFlo software and data was analyzed using FlowJo version 10.6.1 (Tree Star). SPICE plots were generated using Pestle version 2.0 and SPICE version 6.0 (35). For (Fig. 2e), tSNE dimensionality reduction was performed in FlowJo on a concatenated FCS file containing Mock and SLAMF7 CL samples, in duplicate, from two healthy donors with the following tSNE parameters: iterations=7000, perplexity=70. FlowSOM clustering was performed in FlowJo with the number of meta-clusters set to 10. Clusters with very few cells and high similarity to existing clusters were manually merged with those clusters to obtain a final consensus of 4 CD8 T cell clusters and 2 CD4 T cell clusters. Merged FlowSOM clusters were color-coded and overlaid onto the tSNE map of all T cells. The same approach was repeated for (Fig. 3e).
FIGURE 2. SLAMF7 activation induces expression of inhibitory receptors and exhaustion promoting transcription factors in CD8+ and CD4+ T cells.
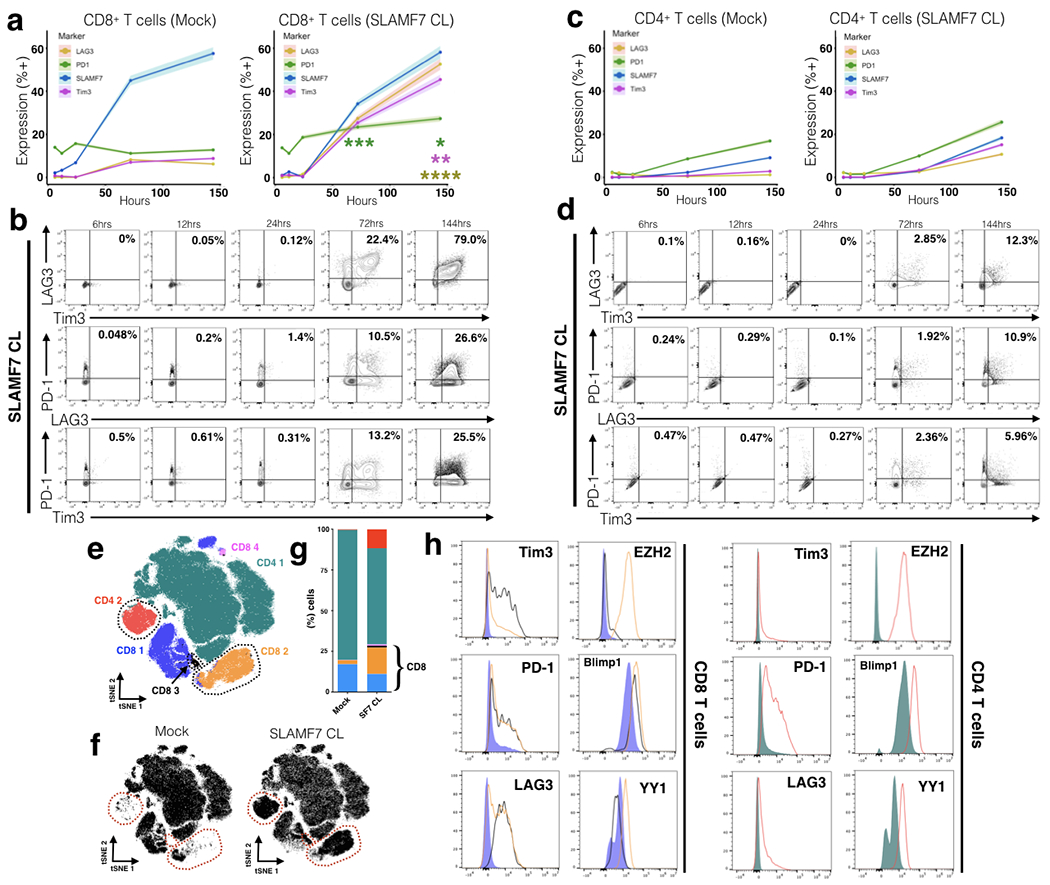
(a, b) Time course of exhaustion marker expression on CD8+ T cells in the presence or absence of in vitro SLAMF7 activation. SLAMF7 was activated by receptor cross-linking with an anti-SLAMF7 mAb. Exhaustion marker expression is shown as mean of two replicates of 2-4 healthy donors with the upper and lower 5% bounds of data shaded in (a) or as biaxial plots (b). Asterisks indicate significant differences in marker expression between Mock and SLAMF7 CL conditions at various time points and are colored by marker. (c, d) Time course of exhaustion marker expression on SLAMF7 activated CD4+ T cells shown in the same manner as (a, b). (a-d) Representative of (n=5) independent experiments with (n=11) total healthy donors. (e) FlowSOM clustering on Mock and SLAMF7 activated T cells reveals unique clusters of T cells following SLAMF7 activation. Individual cells (n=200,000) are depicted on a tSNE plot and colored according to FlowSOM clusters. Clusters only present, or greatly enriched, during SLAMF7 activation are outlined. (f) tSNE maps of T cells separated by condition with clusters only present during SLAMF7 CL outlined. (g) Cluster composition of Mock and SLAMF7 CL conditions. (h) Comparison of exhaustion markers and T cell exhaustion-linked transcription factors between various FlowSOM clusters. Histograms are colored corresponding to clusters from (e). Results in (e-h) are representative of 4 independent experiments showing similar results with a total of (n=8) healthy donors. Pooled results from (n=2) healthy donors displayed in (e-h). Groups in (i) compared using a paired student’s t-test. *p<0.05. Conditions in (a) compared by fitting a mixed model and with Sidak’s multiple comparison test. Mock results from (a) and (c) are duplicated from Figure 1.
Figure 3. SLAMF7-activated T cells are distinct from terminally exhausted T cells and are capable of producing cytokines upon re-stimulation.
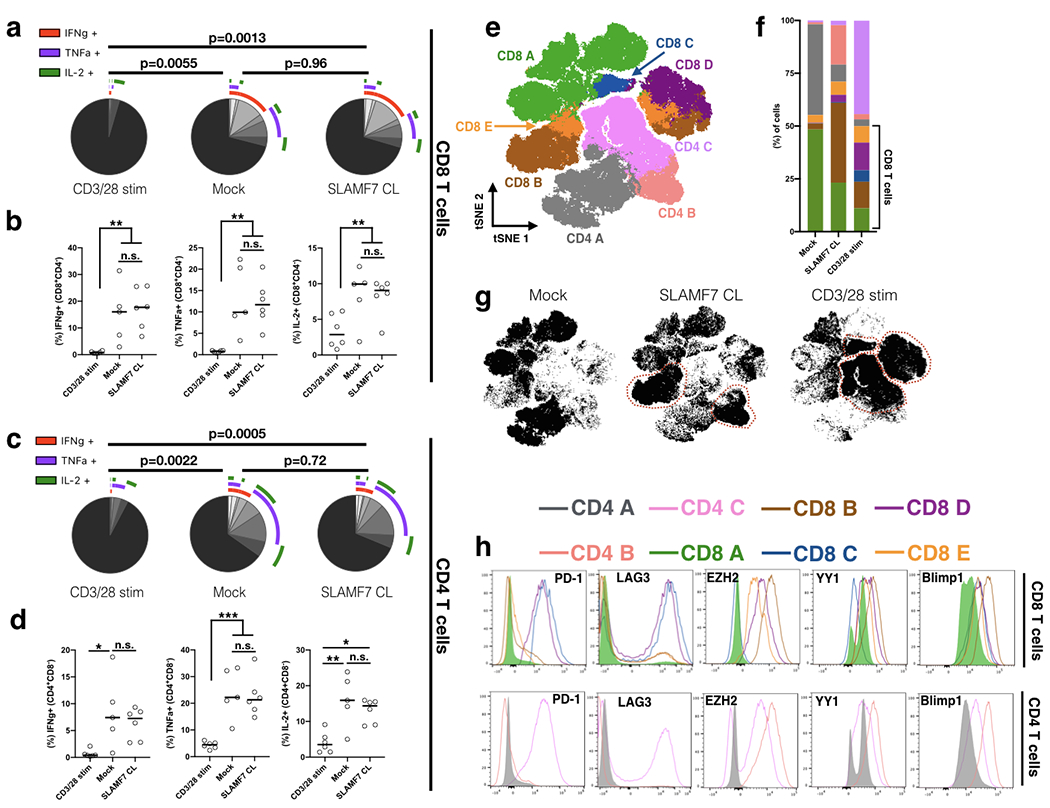
(a, b) Primary human T cells were stimulated for 6 days as described previously and re-stimulated on the seventh day with CD3/28 Dynabeads at a 0.2:1 bead:cell ratio for an additional 3 days. Expression of pro-inflammatory cytokines by CD8+ T cells was measured by intracellular staining and depicted as SPICE plots in (a) and dot plots in (b).
(c, d) Pro-inflammatory cytokine expression of CD4+ T cells.
(e) tSNE map of primary human T cells stimulated for 6 days as described previously with Mock, SLAMF7 CL, or CD3/28 stimulation. Shown are 400,000 cells pooled from 5 healthy donors and colored by FlowSOM clusters.
(f) Comparison of various FlowSOM clusters by stimulation condition.
(g) tSNE maps separated by condition with clusters enriched in each stimulation condition outlined.
(h) Comparison of exhaustion markers and T cell exhaustion-linked transcription factors between various FlowSOM clusters. Histograms are colored corresponding to clusters from (e).
(a-d) Representative of a single experiment with a total of 5 healthy donors.
SPICE plots in (a, c) representative of all samples combined and compared using Permutation test and results in (b, d) compared by fitting a mixed model and with Tukey’s multiple comparison test.
(e-h) is representative of a single experiment with 5 healthy donors.
*p<0.05, **p<0.01; ***p<0.001
IL-2 ELISA.
Primary human CD3+ T cells were plated at 2x105 cells/well in a 96-well plate with indicated stimuli. Exogenous rhIL-2 was not added. Supernatant was collected at indicated time points and IL-2 concentration was assessed by sandwich ELISA (BioLegend).
TCGA data analysis.
FPKM RNA-seq expression values for genes of interest and survival data were obtained from The Cancer Genome Atlas TCGA-TARGET-GTex dataset using Xena (xena.ucsc.edu). Gene expression values were normalized to account for differences in total immune cells by dividing by PTPRC (CD45) and plotted using matplotlib in Python 3.7. Kaplan-Meier curves were generated using the lifelines package. Survival metrics used in each Kaplan-Meier plot were chosen based on previously published work (32). TIDE analysis was performed on the TIDE webpage (http://tide.dfci.harvard.edu/query/) by querying the SLAMF7 gene. Figures were taken directly from the resulting TIDE analysis.
ccRCC CyTOF data re-analysis.
The entire ccRCC CyTOF dataset from Chevrier et al., 2017 (15) was downloaded from the Bodenmiller Lab webpage (https://cytobank.org/bodenmillerlab/index.html). All immune cell subsets were identified based on predetermined Phenograph-assigned cluster IDs present in the raw FCS files and were colored in figures in the same manner as in Chevrier et al., 2017 (15). SLAMF7 expression on various immune cell types was manually gated based on a priori knowledge of expected SLAMF7 expression patterns and it was determined that a gate set at “1” was most appropriate and accurate for all immune cell subsets. This threshold was used consistently for SLAMF7 gating on all immune cell types. While this threshold is lower than what some investigators use for CyTOF analysis, to our knowledge, SLAMF7 has never been assessed or manually gated on CyTOF. Furthermore, while SLAMF7 is expressed at a very high antigen density on certain immune cells (ie. DCs and plasma cells), it is also present on many other immune cell types, albeit at a lower antigen density. For this reason we utilized manual gating to allow us to accurately assess marker expression, whereas some other techniques can make appreciating cells with a low density of SLAMF7 receptor difficult. Samples from healthy controls and rcc #17 (too few cells) were excluded.
Statistics.
Violin plots were generated using the ggplot2 package in R. Heatmap correlation matrix was generated using the corrplot package in R. FlowJo v10.6.1 software was used for all spectral cytometry analysis and high-dimensional analyses. Pie charts were made in Prism v7 (GraphPad). All t-tests were paired and performed in GraphPad Prism v7. Log-rank test was used to analyze all Kaplan-Meier plots. 2-way ANOVA was used to compare tumor growth curves. Details on statistical analyses used for TIDE analysis can be found in the original publication (19). SPICE plots were compared using the Permutation test included in the software. Groups in (Fig. 2a and Fig. 3b, d) compared by fitting to a mixed model before performing multiple comparison test. This was accomplished in Prism v8 and performed due to the presence of missing values due to random chance. No mice or human samples were censored from any analysis for any reason (except the CyTOF samples listed above).
Study approval.
All animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee.
Results
SLAMF7 is co-expressed with multiple exhaustion markers on activated T cells.
To examine associations between SLAMF7 and various inhibitory receptors on T cells we used a chronic, polyclonal, in vitro T cell activation assay. CD3+ human T cells were cultured with only rhIL-2 (Mock) or rhIL-2 + CD3/CD28 Dynabeads and exhaustion markers were assessed over a period of six days using spectral cytometry (Supplementary Fig. 1a–c). We observed time-dependent increases in expression of LAG3, PD-1, and Tim3 on CD3/28 activated CD8+ and CD4+ T cells (Fig. 1a and b). SLAMF7 expression was minimal on both CD8+ (1.12%) and CD4+ (0%) T cells initially, but increased over time with CD3/28 stimulation, more in CD8+ than CD4+ T cells, and interestingly, was robustly induced in a time-dependent manner in the absence of CD3/28 activation on CD8+ T cells (Fig. 1a, b). To assess co-expression patterns of SLAMF7 with LAG3, Tim3, PD-1, and CTLA-4 we utilized SPICE (33) and observed that SLAMF7 is highly co-expressed in cells expressing any combination of exhaustion markers, and is consistently present on CD8+ T cells expressing multiple exhaustion markers (Fig. 1c). This effect was also observed on CD4+ T cells, but less pronounced, as these cells do not up-regulate SLAMF7 expression to the levels seen on CD8+ T cells (Fig. 1d). Additionally, we repeated this assay with mouse splenocytes and observed the same pattern of co-expression (Supplementary Fig. 1d–h).
FIGURE 1. SLAMF7 is co-expressed with multiple exhaustion markers on CD3/CD28 stimulated human T cells.
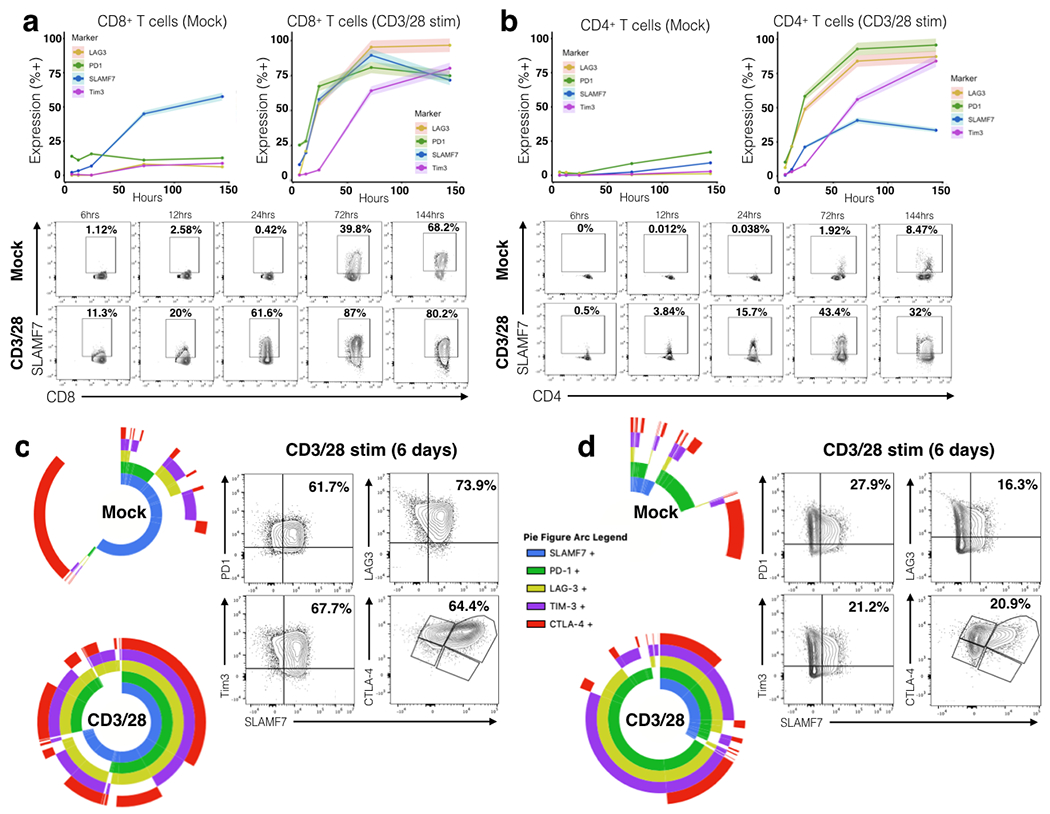
(a, b) Time course of SLAMF7 and exhaustion marker expression on Mock or CD3/CD28 in vitro stimulated primary human CD8+ T cells (a) and CD4+ T cells (b). Top, exhaustion marker expression is shown as mean of two replicates of 2-4 healthy donors with the upper and lower 5% bounds of data shaded in. Bottom, representative plots of SLAMF7 expression on Mock and CD3/28 stimulated cells over time. (c, d) Left, SPICE plots showing co-expression of SLAMF7 with exhaustion markers on Mock and CD3/28 stimulated CD8+ T cells (c) and CD4+ T cells (d). SPICE plots in (c) and (d) each represent two replicate samples from two healthy donors. Right, representative biaxial plots of SLAMF7 co-expression on CD3/28 stimulated T cells after 6 days. Data is representative of 4 total healthy donors from two independent experiments.
Activation of SLAMF7 alone on T cells induces inhibitory receptors and exhaustion-associated transcription factors.
Since we identified that SLAMF7 is co-expressed with multiple exhaustion markers, we decided to examine if inducing SLAMF7 signaling in T cells impacts the expression level of T cell inhibitory receptors. We utilized a similar in vitro T cell stimulation assay as before, but instead activated the SLAMF7 receptor on T cells with an agonistic antibody via cross-linking, as previously described (23). We found that activating the SLAMF7 receptor on human T cells induced expression of PD-1, Tim3, and LAG3 in a time-dependent manner on CD8+ T cells (Fig. 2a, b), but to a lesser extent on CD4+ T cells (Fig. 2c, d). We also noted considerable numbers of T cells co-expressing multiple exhaustion markers by three and six days (Fig. 2b, d). This finding was not observed when cells were cultured in the absence of IL-2 (data not shown).
To gain a more in-depth understanding of the effects SLAMF7 activation had on T cell exhaustion we employed unbiased, neural-network-based, computational clustering using FlowSOM (34), and dimensionality reduction using tSNE, which revealed six distinct T cell clusters in Mock and SLAMF7 activated samples following six days of stimulation (Fig. 2e). We identified two CD4+ T cell clusters (CD4 1-2) and four CD8+ T cell clusters (CD8 1-4) (Fig. 2e). Importantly, the CD4 2, CD8 2, and CD8 3 clusters were virtually absent in Mock stimulation and only appeared in SLAMF7 activated samples (Fig. 2f, g). Comparison of these clusters with clusters present during Mock stimulation revealed an inhibitory receptor and transcription factor (TF) expression profile characteristic of exhausted/dysfunctional T cells (35–38) (Fig. 2h and Supplementary Fig. 2a, d, e). Specifically the CD4 2 cluster had high expression of PD-1 and the epigenetic-reprogramming transcription factors YY1, Blimp1, and EZH2 (Fig. 2h). The two SLAMF7 cross-linking-induced CD8+ T cell clusters shared an exhaustion phenotype, but differed in expression of various markers such as EZH2, Tim3, and YY1 (Fig. 2h). Together, this suggests that SLAMF7 activation on human T cells is able to induce expression of inhibitory receptors and transcription factors linked to control of T cell exhaustion, independent of TCR signaling.
Due to the fact that regulatory T cells (Tregs) are known to express high levels of certain exhaustion markers (39, 40) we wanted to rule out the possibility that SLAMF7 activation was merely inducing CD4+ and CD8+ Tregs. FoxP3 staining showed minor, but consistent increases in FoxP3+ CD4+ T cells (Supplemental Fig. 2b) and CD8+ T cells (Supplementary Fig. 2c) following SLAMF7 activation, but these few cells did not account for the far greater percentage of cells expressing multiple exhaustion markers.
In vitro SLAMF7-activated T cells are distinct from terminally exhausted T cells and are capable of producing cytokines upon re-stimulation.
While the T cells induced following SLAMF7 activation have the appearance of exhausted T cells, we wished to confirm this by performing a functional assay. Primary human T cells were stimulated as described above for six days and then re-stimulated with fresh CD3/28 Dynabeads for an additional three days. We then assessed the ability of these cells to produce pro-inflammatory cytokines following re-stimulation. We found that T cells first stimulated for six days with CD3/28 Dynabeads were unable to produce IL-2, TNFα, and IFNγ upon re-stimulation consistent with their inhibitory marker profile, identifying them as bona fide exhausted T cells (Fig. 3a–d). In contrast to these cells, cells receiving Mock stimulation (only IL-2) first were able to produce significant quantities of all three cytokines (Fig. 3a–d). Interestingly though, we found that cells first stimulated with SLAMF7 CL for six days were still able to produce cytokines equivalent to Mock samples (Fig. 3a–d). This indicates that while in vitro SLAMF7-activated T cells upregulate exhaustion markers, they still retain the ability to produce pro-inflammatory cytokines suggesting they have not reached terminal exhaustion yet.
We next compared the inhibitory receptor and transcription factor profile of SLAMF7-activated T cells to terminally exhausted T cells (CD3/28 stimulation) in an unbiased manner. FlowSOM clustering of Mock, SLAMF7 CL, and CD3/28 stimulated T cells (all stimulated for six days) revealed three CD4+ T cell clusters and five CD8+ T cell clusters (Fig. 3e). Each of the CD4 T cell clusters was strongly enriched in one of the stimulation conditions with Mock being primarily composed of CD4 A, SLAMF7 CL composed mostly of CD4 B, and CD3/28 stimulation composed primarily of CD4 C (Fig. 3f, g). Similarly, Mock samples primarily contained the CD8 A cluster, while SLAMF7 CL samples contained mostly the CD8 B cluster, and CD3/28 stimulation containing mostly CD8 C and CD8 D clusters (Fig. 3f, g). Comparison of inhibitory receptors and TF’s in these clusters revealed important distinctions between SLAMF7-activated T cell clusters and terminally exhausted T cell clusters. We found that exhausted T cell clusters (CD8 C, CD8 D, and CD4 C) have high expression of PD-1 and LAG-3, along with lower expression of the transcription factors YY1, EZH2, and Blimp1 (Fig. 3h). SLAMF7 CL samples still co-expressed multiple inhibitory receptors, but at a low level than exhausted T cells, along with much stronger expression of all three transcription factors (Fig. 3h). Together, this reveals that although in vitro SLAMF7-activated T cells resemble exhausted T cells, they are not functionally (terminally) exhausted, yet still have an increased susceptibility to multiple inhibitory signals due to their inhibitory receptor profile. As they show high expression of TF’s involved in T cell exhaustion, in vitro SLAMF7-activated T cells may be progressing along the path to terminal exhaustion through a pathway independent from TCR activation.
SLAMF7 signaling in CD8+ T cells induces STAT1 and STAT3 phosphorylation.
To mechanistically understand how SLAMF7 signaling in CD8+ T cells modulates the expression levels of multiple inhibitory receptors and their associated transcription factors, we performed phosphoflow on human T cells utilizing a panel containing common intermediary signaling proteins implicated in T cell responses (41). We found that SLAMF7 activation induced a progressive increase in STAT3 phosphorylation over time and a burst of STAT1 phosphorylation at 90 minutes post-activation, without affecting numerous other known T cell signaling intermediaries (Fig. 4a–h). Additionally, we observed that SLAMF7 activation induced CD69 expression and minimally altered CD44 expression on CD8+ T cells (Fig. 4i, j); an important observation as CD69 has been previously shown to drive inhibitory receptor expression on T cells (42). SLAMF7 activation on CD8+ T cells also shifted the subset distribution away from naive and terminal differentiated effector memory re-expressing CD45RA cells (EMRA) towards effector memory cells (Supplemental Fig. 2f). Finally, as high levels of IL-2 can drive both T cell inhibitory receptor expression (43, 44) and Treg formation (43, 45), we assessed IL-2 production and found that SLAMF7 activation does not stimulate IL-2 production or secretion from T cells (Fig. 4k, l). Together, these findings tie STAT1 and STAT3 phosphorylation to SLAMF7 activation in CD8+ T cells, suggesting that these factors might be involved in SLAMF7-dependent T cell signaling.
Figure 4. Alterations in T cell signaling pathways following SLAMF7 activation.
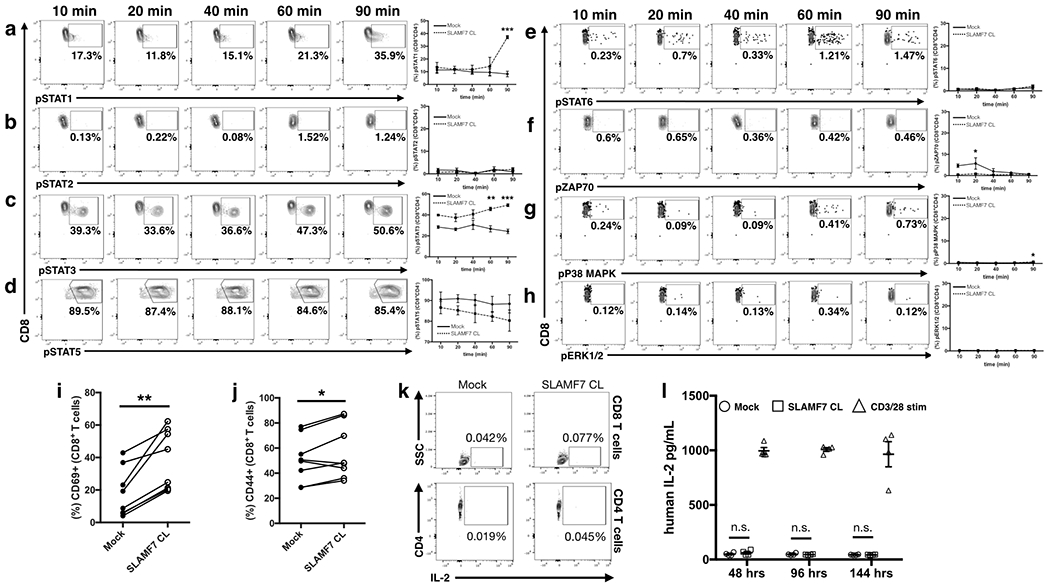
(a-h) Time course of isolated primary human CD8+ T cells stimulated in vitro by SLAMF7 cross-linking (n=2 donors). Representative biaxial plots are shown for only SLAMF7 CL conditions. Surface expression of CD69 (i) and CD44 (j) was assessed on (n=8) donors following 6 days of in vitro stimulation. (k) Biaxial plots showing lack of IL-2 expression in CD8+ (top) and CD4+ primary human T cells following 3 days of in vitro stimulation (n=2), representative of two independent experiments. (l) Secretion of IL-2 by in vitro stimulated primary human CD3+ T cells was assessed over time by ELISA (n=4). No exogenous IL-2 was added to cultures and results are representative of two independent experiments. (a-h) is representative of two independent experiments with a total of 5 healthy donors and was analyzed by two-way ANOVA with Sidak’s multiple comparison test. (i, j) contains pooled samples from 2 independent experiments and was analyzed with a paired students t-test. Two-way ANOVA with Tukey’s multiple comparison test used for (l)
SLAMF7 expression is correlated with inhibitory receptor expression in clear cell renal cell carcinoma patients and is linked to poor survival.
To begin to identify if the SLAMF7 receptor functions to modulate T cell responses in cancer patients in vivo, we assessed co-expression of SLAMF7 mRNA with mRNA from various exhaustion markers in patient tumor samples. Using TCGA RNA-seq data, we first assessed all cancers to see which had the highest levels of CD8+ T cells and SLAMF7 expression (Supplementary Fig. 2g, h). We focused our further analyses on clear cell renal cell carcinoma (ccRCC) due to its high levels of CD8+ T cells, large sample size, and the knowledge that immune dysfunction in ccRCC occurs primarily via T cell exhaustion (19). Correlation analyses between SLAMF7 with CTLA-4, LAG3, and PD-1 revealed very strong co-expression in ccRCC (Fig. 5a). Co-expression was weaker with Tim3 (Fig. 5a), an effect possibly due to the expression of Tim3 on various myeloid cell subsets (46). Importantly, stratifying ccRCC patients into the highest and lowest SLAMF7 expressing quartiles revealed reduced disease-specific survival in patients with high expression of SLAMF7 (Fig. 5b).
FIGURE 5. SLAMF7 correlates with exhaustion markers and is linked to poor survival in ccRCC.
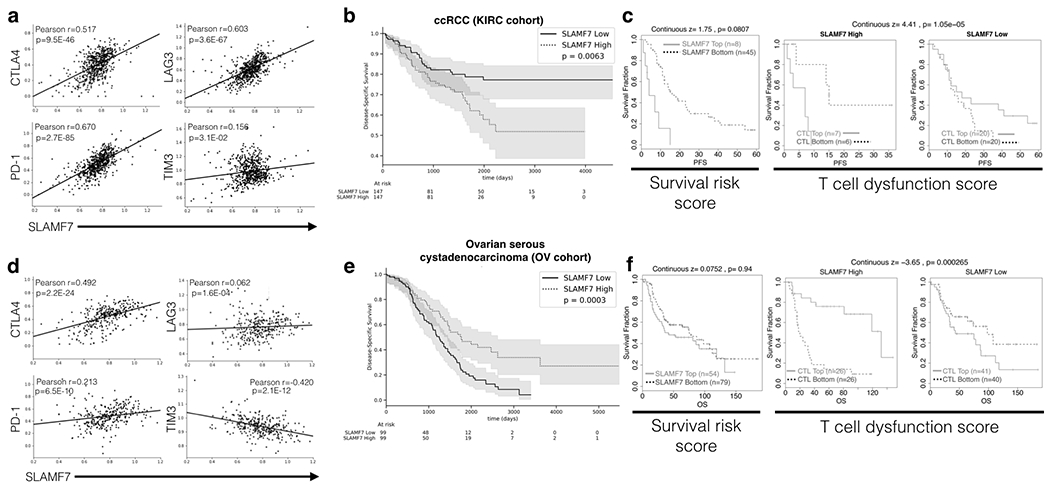
(a) TCGA data was analyzed to examine co-expression of SLAMF7 with PD-1, CTLA-4, TIM-3, and LAG-3 at the mRNA level in ccRCC. Data was normalized to CD45 (PTPRC) to control for differential levels of immune cells. Pearson R and p values are displayed in each figure. (b) Disease specific survival in ccRCC patients from the KIRC cohort (n=603) in the top and bottom SLAMF7 expression quartiles. (c) SLAMF7-specific TIDE analysis of ccRCC patients from the E-MTAB-3267 cohort (n=53). Left, progression free survival of patients with high versus low SLAMF7 mRNA expression, with continuous z-score and associated p value signifying gene-associated death risk from a CoxPH model displayed. Right and middle, SLAMF7 expression effects on progression free survival with patients from the E-MTAB-3267 cohort stratified by CTL levels. Right-most plot displays patients with low SLAMF7 mRNA expression and middle plot shows patients with high SLAMF7 mRNA expression. Kaplan-Meier plots show survival differences based on TIDE-determined CTL levels in each patient. Continuous z-score and associated p value is displayed. (d) SLAMF7 associations with exhaustion markers from patients in the ovarian serious cystadenocarcinoma (OV) cohort from TCGA (n=426) displayed as in (a). (e) Kaplan-Meier plot for patients in the OV cohort stratified by highest and lowest SLAMF7 expression quartiles as in (b). (f) TIDE analysis of ovarian cancer patients from the GSE139@PRECOG cohort (n=203) performed as in (c). Overall survival is used in (f) as opposed to progression free survival used in (c). Log-rank test was used for Kaplan-Meier plots in (b) and (e) and shaded regions represent 95% confidence interval.
To further determine if SLAMF7 expression in ccRCC was linked to T cell exhaustion and dysfunction, we used the Tumor Immune Dysfunction and Exclusion (TIDE) tool (19). TIDE analysis of a separate, smaller cohort of ccRCC patients confirmed impaired survival in patients with high SLAMF7 expression (Fig. 5c). It also showed that high levels of cytotoxic T lymphocytes (CTLs) are moderately protective, except when SLAMF7 expression is high, in which case there is reduced survival (Fig. 5c). Analysis of other cancers in the TCGA revealed this phenomenon was not conserved (Supplemental Fig. 2i–l). In cancers where immune dysfunction is not primarily occurring via T cell exhaustion, such as ovarian cancer (Fig. 5d–f) and squamous cell lung carcinoma (Supplementary Fig. 2k) (19), this trend disappears. Together, these results suggest that high SLAMF7 mRNA expression is linked to poor survival, only in cancers dominated by T cell exhaustion.
SLAMF7 expression patterns in human ccRCC tumors, associations with exhausted T cells, and links to patient survival.
While the correlations and effects on patient survival noted using TCGA data are informative, it does not address immune cell-specific expression patterns of SLAMF7 in the ccRCC TME. To address this we re-analyzed a previously published CyTOF dataset of ccRCC patient tumors, which contained expression data for SLAMF7 (15). As predicted by CIBERSORT analysis of bulk RNA-seq data (Supplementary Fig. 2g), the ccRCC TME is composed primarily of T cells, followed by myeloid cells (Fig. 6a) (15). The SLAMF7+ compartment largely mimics that seen in the periphery, with major contributions from plasma cells and NK cells (Fig. 6a). Looking at SLAMF7 expression on major immune cell types, as well as various predetermined TAM subsets (15), we observed considerable heterogeneity in SLAMF7 expression on numerous immune cell types across patients (Fig. 6b and Supplementary Fig. 3a). In particular, NK cells, plasmacytoid dendritic cells (pDCs), and various TAM subsets show the greatest heterogeneity (Fig. 6b).
FIGURE 6. SLAMF7 expression in the ccRCC immune niche and associations with T cell phenotypes and patient survival.
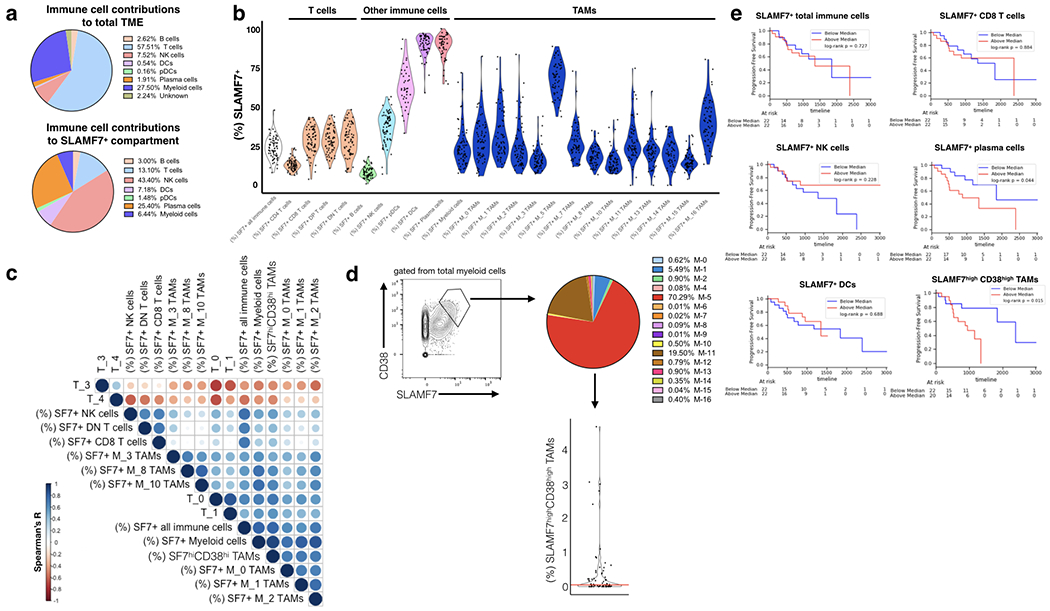
(a) Top, relative contributions of various immune cell subsets in the ccRCC TME as assessed by CyTOF. Percentages are calculated from a concatenated sample of (n=73) ccRCC tumor samples using Phenograph-assigned clusters previously determine by Chevrier et al., 2017. Bottom, SLAMF7+ cells were gated out from total immune cells and contributions of various Phenograph clustered immune cell subsets was determined. (b) SLAMF7 expression across various immune cell types in the ccRCC TME. Phenograph-assigned clusters were used for immune subset identification with TAMs being further subdivided based on TAM-specific subsets identified by Chevrier et al., 2017. Each dot represents an individual patient tumor sample. (c) Correlation matrix heatmap of SLAMF7 expression on various immune cell types and frequencies of various T cell subsets. T cell frequencies are calculated as the frequency of each T cell subset out of the total T cell compartment of each patient. T cell subsets are identified by Phenograph in Chevrier et al., 2017. Spearman correlation is displayed as a circle scaled by color and size based on the magnitude of the R value. Only the top 10 positive and negative correlations are displayed. (d) Left, gating of SLAMF7highCD38high TAMs from total myeloid cells. Right, breakdown of SLAMF7highCD38high TAMs by Phenograph-determined TAM subsets. Results are from a concatenated sample containing all 73 ccRCC tumor samples. Bottom, frequency of SLAMF7highCD38high TAMs per total immune cells per patient. Red line indicates median (0.052). (e) Kaplan-Meier plots of progression free survival, with ccRCC patients stratified by high or low numbers of various immune cell subsets expressing SLAMF7. For all plots the relative frequency of SLAMF7+ cells from each immune subset out of the total immune compartment is considered. Log-rank test is used to compare groups in (e).
To begin to identify which SLAMF7 expressing cell types in the TME might be responsible for activating SLAMF7 on T cells and inducing exhaustion, we performed a correlation analysis of various SLAMF7+ immune cells to all of the predetermined T cell subsets (15) (Fig. 6c). We found strikingly strong positive correlations between subsets of exhausted T cells (T-0, T-1, T-7, T-16, and T-18) (15) and SLAMF7+: total immune cells, total myeloid cells, M-0 TAMs, M-1 TAMs, M-2 TAMs, M-3 TAMs, and M-10 TAMs (Fig. 6c and supplementary table I). Importantly the “exhausted” T cell subsets were labeled as such based on high expression of inhibitory receptors and were never functionally characterized (15). We also noted relatively strong positive correlations of CD4+ Tregs (T-6) (15) to SLAMF7+ total immune cells (Spearman r=0.3) and SLAMF7+ total myeloid cells (Spearman r=0.38) (Fig. 6c). Furthermore, we observed strong negative correlations between the above listed SLAMF7+ TAM subsets and subsets of effector memory T cells (T-3 and T-4) (15) (Fig. 6c and supplementary table 1). It is recognized that TAMs are responsible for driving tumor infiltrating leukocytes (TILs) towards exhaustion phenotypes (47). Our results here suggest that SLAMF7-SLAMF7 interactions between TAMs and TILs may be a novel mechanism through which tumors are able to create an inhospitable immune niche, by the induction of T cell exhaustion, Treg formation, and shifts away from beneficial effector memory T cells (48).
Previous analysis of this data set concluded that high CD38 expression marks pathogenic TAMs (15). We discovered a population of SLAMF7highCD38high TAMs present at varying levels between patients (Fig. 6d). Hypothesizing that this may be a pathogenic TAM subset linked to T cell exhaustion, we correlated the percentage of this cell population per total immune cells in the TME of each patient to T cell subsets in our correlation matrix. Levels of this unique TAM subset showed the strongest positive correlations with the above listed exhausted T cell subsets and Tregs (Fig. 6c and supplementary table I). Further analysis of this TAM subset revealed it is composed primarily of M-5 and M-11 TAMS (Fig. 6d), both previously linked to T cell exhaustion and poor survival in this cohort (15). To identify which SLAMF7 expressing immune cell types were linked to poor survival we performed a series of Kaplan-Meier analyses by splitting patients up based on high or low frequencies of various SLAMF7+ cell types. We found that high levels of SLAMF7+ total immune cells, CD8+ T cells, DCs, and NK cells were not linked to poor survival (Fig. 6e). Patients with high levels of SLAMF7+ plasma cells (Fig. 6e) had poor survival, however, as nearly all plasma cells express SLAMF7, further analysis showed this effect was merely due to differences in numbers of plasma cells (Supplementary Fig. 3b). Unsurprisingly, we also noted that patients with high levels of Tregs (T-6) had markedly worse survival (Supplementary Fig. 3c). Most interestingly, we found that stratifying patients by high levels of the SLAMF7highCD38high TAM subset was the most effective parameter for identifying patients with poor survival (Fig. 6e). This effect was not just due to this population containing high levels of M-5 TAMs (previously linked to T cell exhaustion (15)), as patients with high levels of M-5 TAMs showed no decrease in survival (Supplementary Fig. 3d). Finally, patients in the high and low SLAMF7highCD38high TAM groups were otherwise balanced in available demographic and clinical parameters (Supplementary Fig. 3e). Together, these findings suggest that SLAMF7highCD38high TAMs may be driving T cell dysfunction and immune evasion in the ccRCC TME.
SLAMF7 expression in the TME propels CD8+ T cells towards terminal exhaustion and impacts tumor growth.
In order to validate our findings from human samples in vivo, we generated SLAMF7−/− mice and assessed growth of B16-F10 tumors. We found tumor growth to be significantly attenuated in SLAMF7−/− mice compared to WT mice (Fig. 7a). We also observed high co-expression of SLAMF7 with various inhibitory receptors on CD8+ TILs from WT mice (Fig. 7b, c). In comparing WT to SLAMF7−/− TILs, we observed significantly fewer CD8+ T cells expressing multiple exhaustion markers (Fig. 7d) and individual markers (Fig. 7e) in SLAMF7−/− mice. To validate if SLAMF7 was modulating levels of bona fide exhausted T cells we compared expression of the exhaustion-defining TF, TOX (49–51). SLAMF7−/− CD8+ intra-tumoral T cells expressed significantly fewer TOX+ cells (Fig. 7f) and PD-1+/TOX+ cells (Fig. 7g). Taking a more granular approach, we used the markers SLAMF6 (Ly108) and CD69 to track where CD8+ TILs from WT and SLAMF7−/− mice were on the exhaustion developmental trajectory, as recently described (52). We found that CD8+ T cells from WT mice tended to be at either end of the exhaustion spectrum (Texprog1 and Texterm) (Fig. 7h), while SLAMF7−/− CD8+ T cells occupied the intermediate stages more often (Texprog2 and Texint) with significantly less terminally exhausted cells (Fig. 7h). We also observed SLAMF7 expression to be highest on the two progenitor stages (Texprog1 and Texprog2) (Fig. 7i), which together with the above findings, suggests that SLAMF7 signaling may help to propel CD8+ T cells through the exhaustion developmental pathway to terminal exhaustion.
FIGURE 7. SLAMF7 expression in the TME propels CD8+ T cells towards terminal exhaustion and impacts tumor growth.
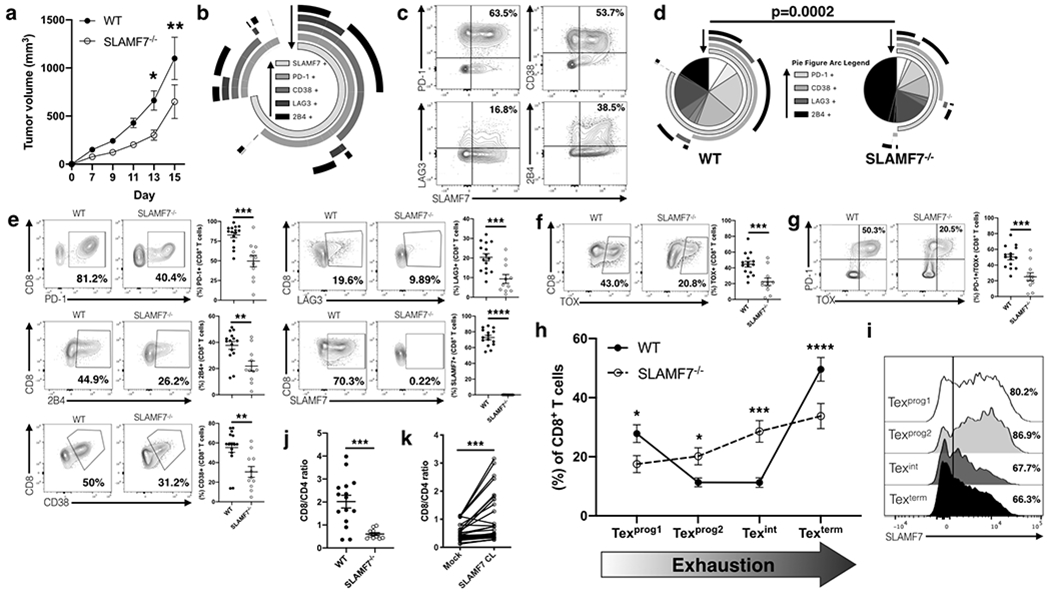
(a) B16-F10 tumor growth in WT and SLAMF7−/− mice. (b) SPICE plot of inhibitory receptor expression on intra-tumoral CD8+ T cells from WT mice; plot shows response from all samples combined. Arrows assist in identifying which arc each marker corresponds too. (c) Biaxial representative plots of SLAMF7 co-expression with various inhibitory receptors on intra-tumoral CD8+ T cells from WT mice. (d) SPICE plots comparing exhaustion marker expression on intra-tumoral CD8+ T cells from WT and SLAMF7−/− mice. Plots are aggregate from all samples and compared with a Permutation test. Arrows assist in identifying which arc each marker corresponds too. (e) Comparison of individual inhibitory receptors from intra-tumoral CD8+ T cells showing both representative biaxial plots and responses of individual mice. (f) Expression of TOX in intra-tumoral CD8+ T cells from WT and SLAMF7−/− mice. (g) Co-expression of TOX and PD-1 from intra-tumoral CD8+ T cells from WT and SLAMF7−/− mice. (h) Frequency of CD8+ intra-tumoral T cells across the four populations that make up the exhaustion developmental trajectory (Beltra et al., 2020). Populations are defined as: Texprog1 (SLAMF6+CD69+), Texprog2 (SLAMF6+CD69−), Texint (SLAMF6−CD69−), Texterm (SLAMF6−CD69+). (i) SLAMF7 expression across exhausted CD8+ T cell subsets. (j, k) Ratio of CD8+ to CD4+ T cells from B16-F10 melanomas of WT and SLAMF7−/− mice (j), or PBMCs of healthy human individuals with either mock or SLAMF7 CL stimulation (k). Results from (a) are pooled from two independent experiments using n=14-15 mice per genotype, composed of equal numbers of males and females, and compared with 2-way ANOVA and Sidak’s multiple comparison test. Results from (b-i) are pooled results from two independent experiments with n=15 WT and n=11 SLAMF7−/− mice composed of equal numbers of males and females and compared with unpaired two-tailed students t-test. Results in (k) are pooled from seven independent experiments with n=32 and compared with paired two-tailed students t-test. All data are represented as mean ± SEM. **p<0.01; ***p<0.001; ****p<0.0001.
Interestingly, we also noticed a significantly decreased CD8/CD4 T cell ratio in TILs from SLAMF7−/− mice (Fig. 7j). Returning to our in vitro human data, we observed a corresponding reverse trend in the presence of SLAMF7 activation (Fig. 7k). Thus, while SLAMF7 appears to drive T cell exhaustion, it may also be playing a role in CD8+ T cell survival or ability to maintain tissue residency.
Expression of SLAMF7 on TAMs drives T cells to express inhibitory receptors.
To determine if SLAMF7-SLAMF7 self-ligation between TAMs and tumor-infiltrating T cells induces T cell inhibitory receptor expression, we developed an ex vivo TAM-T cell co-culture model (Fig. 8a). B16-F10 tumors were grown in WT and SLAMF7−/− mice, TAMs were isolated from established tumors at day 25, and co-cultured with CD8+ T cells from splenocytes of non-tumor bearing mice (Fig. 8a). We observed that T cells co-cultured with SLAMF7+ TAMs expressed multiple inhibitory receptors (Fig. 8b, c). Notably, T cells cultured with SLAMF7−/− TAMs had significantly fewer cells positive for various exhaustion markers, with dramatic decreases in T cells co-expressing multiple markers appearing similar to CD8+ T cells cultured in the absence of TAMs (Fig. 8c). Importantly, this finding is specific to TAMs, as CD8+ T cells co-cultured with bone marrow-derived macrophages (BMDMs) from WT or SLAMF7−/− mice do not show alterations in inhibitory receptor expression (Supplemental Fig. 3f, g). Additionally, comparison of TAMs from WT and SLAMF7−/− mice reveled that they have similar expression of numerous markers, except for SLAMF7 (Fig. 8d). This finding suggests that direct SLAMF7-SLAMF7 interactions between TAMs and CD8+ T cells are sufficient to induce expression of multiple inhibitory receptors on T cells.
FIGURE 8. SLAMF7 expression on TAMs drives T cell exhaustion.
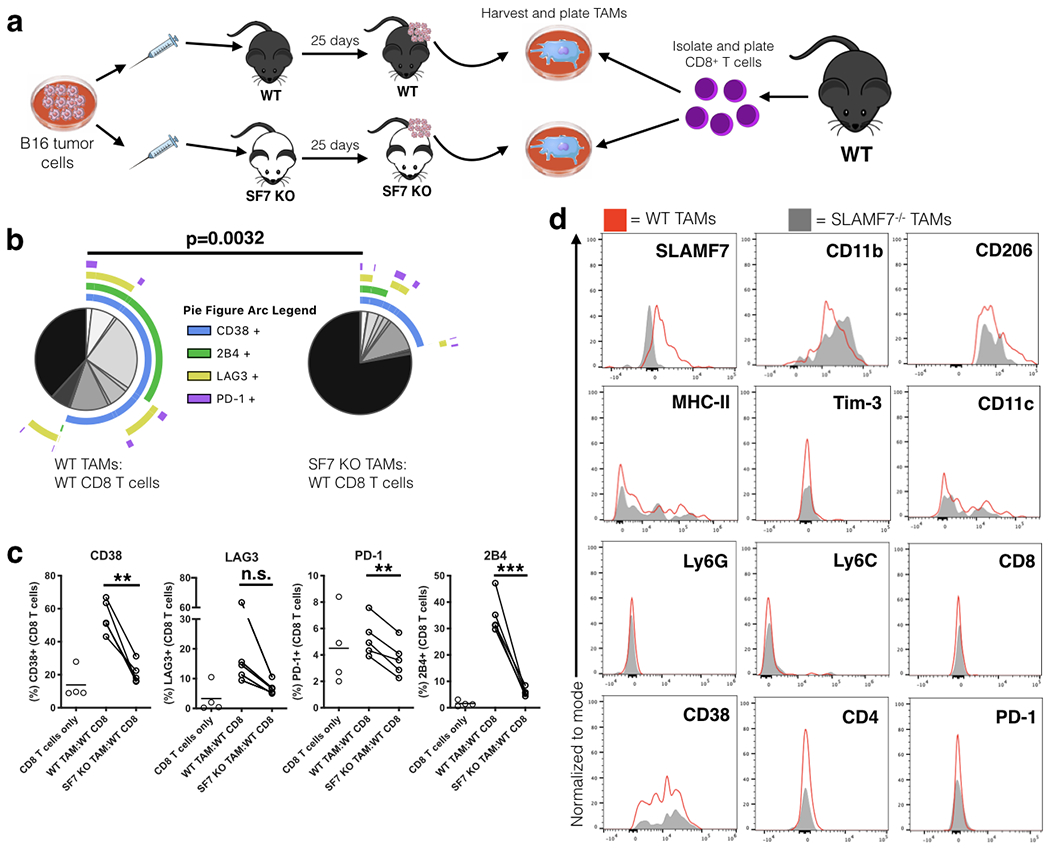
(a) Experimental approach used for ex vivo T cell:TAM co-culture. (b) SPICE plots of CD8+ T cells co-cultured with TAMs for 6 days (n=5); plots show response from all samples combined. (c) Changes in expression of exhaustion markers on CD8+ T cells from co-culture experiment (n=5). Groups containing CD8+ T cells and TAMs compared using a paired student’s t-test. (d) Comparison of various markers on TAMs from B16-F10 tumors grown in WT and SLAMF7−/− mice. Results from (b-d) are representative of a single experiment using TAMs pooled from (n=4) mice per genotype and CD8+ T cells from splenocytes of (n=5) WT, non-tumor bearing mice. n.s., not significant; **p<0.01; ***p<0.001.
Discussion:
The impressive response rates and instances of complete remission following checkpoint inhibitor therapy have catapulted immunotherapy and T cell exhaustion to the forefront of cancer research. While therapies targeting PD-1 (and PD-L1) and CTLA-4 have proven efficacious, there are still a great many patients who do not respond or develop resistance to checkpoint blockade, suggesting the need for a greater understanding of the biological processes regulating T cell dysfunction. To this end, a host of additional inhibitory checkpoint receptors have been identified and found to be expressed on exhausted T cells (2), with blocking antibodies against these receptors now in phase I, II, and III clinical trials for various cancers (2). However, this approach of targeting additional T cell inhibitory receptors does not address the root problem, which is: what are the TCR-independent mechanisms responsible for inducing inhibitory receptor expression in the TME in the first place (3). Here, we have begun to answer this question in ccRCC and provide early evidence that SLAMF7-SLAMF7 self-ligation between CD8+ T cells and SLAMF7+ TAMs may be driving T cell inhibitory receptor expression and exhaustion.
Based on the temporal expression patterns we observe, it would seem that SLAMF7 is actually one of the first receptors expressed on activated CD8+ T cells following any sort of pro-inflammatory stimuli. This observation has been noted at the single-cell level (29) and lends credence to the idea that SLAMF7 may be one of the earliest counter-measures T cells use to rein in over-activation. It accomplishes this by providing an early TCR-independent signal to begin up-regulating expression of multiple inhibitory receptors immediately upon initial activation or entrance into an inflammatory microenvironment. It also appears to reprogram these cells by inducing numerous transcription factors known to positively modulate T cell exhaustion programs. Notably, while SLAMF7 signaling on T cells does not impair their ability to produce cytokines, it does lend them highly susceptible to signaling from multiple inhibitory receptors, which would still result in the induction of non-functional T cells in the TME where these signals are abundant. This is an important subtlety to the biology of SLAMF7 signaling on T cells, and suggests that interruption of this signaling could result in effective anti-tumor responses by T cells.
Already we have taken the first steps at understanding the underlying mechanism by finding that SLAMF7 activation on CD8+ T cells induces STAT1 and STAT3 phosphorylation. Phosphorylated STAT3 in intra-tumoral T cells has been shown to induce PD-1 expression (53), inhibit effector T cells (53), and promote expansion of Tregs (53); findings that we also observe following SLAMF7 activation on T cells. Furthermore, there is evidence that EZH2 is able to induce phosphorylation of STAT3 (54) and that CD38 is able modulate EZH2 activity (55), suggesting SLAMF7 signaling in T cells may be inducing expression of inhibitory receptors via a CD38-STAT3-EZH2 axis.
Contrary to STAT3, the role of STAT1 in modulating CD8+ T cell signaling and exhaustion marker expression is less well understood. There is evidence showing the potential for pSTAT1 to induce expression of PD-1 in T cells and other immune cells (56). Further confirming that SLAMF7-mediated STAT1 phosphorylation is implicated in driving T cell exhaustion is the finding that Texterm CD8+ T cells are enriched with open chromatin regions bound by STAT1 (52), and we find that in the absence of SLAMF7 there are fewer of these cells. Additionally, SLAMF7-mediated increases in the expression of the exhaustion-controlling transcription factors YY1 (38), Blimp1 (35, 36), and EZH2 (37, 38), are likely potential mechanisms regulating T cell exhaustion. The choice to study these transcription factors specifically, was made not only because they are linked to T cell exhaustion, but because both Blimp1 and YY1 have been found to control SLAMF7 expression (57, 58), which implies there may be reciprocal regulation involved. Interestingly, Texterm CD8+ T cells are enriched in open chromatin regions bound by Blimp-1, we find that SLAMF7 activation induces Blimp-1, and in the absence of SLAMF7, there are fewer Texterm cells, implicating another potential mechanism behind SLAMF7-specific effects on T cells.
The restricted B16-F10 tumor growth in SLAMF7−/− mice can potentially be explained by the CD8+ T cell population shifts we see along the exhaustion development spectrum. Texint cells (which we find significantly more of in the tumors of SLAMF7−/− mice) have been found to have high cytotoxic capacity and lower expression of inhibitory receptors compared to the other subsets recently described (52). Having more of these cells is likely to provide a beneficial anti-tumor response. How exactly SLAMF7 signaling drives cells away from Texint towards Texterm, and if SLAMF7 mediates changes in TOX expression remain to be discovered.
Our discovery that high expression of both CD38 and SLAMF7 on TAMs marks a unique subset strongly linked to T cell exhaustion and poor survival, helps to mechanistically explain why a pervious study found M-5 TAMs to be so strongly associated with exhausted CD8+ T cells (15). M-5 TAMs have the highest expression of SLAMF7 of any of the other TAM subsets that were identified, and the SLAMF7highCD38high TAM subset is composed of 70% M-5 TAMs (15). It will be important for future studies to determine if this unique TAM subset is present in the TME of other cancers, and if so, does it also induce T cell inhibitory receptor expression. Along the same lines, the results from our ex vivo TAM-T cell co-culture model support this proposed mechanism, but do not provide conclusive, in vivo proof.
An important concept to consider as future studies are conducted to both better understand the physiology of SLAMF7 signaling and attempt to therapeutically modulate it, is the sensitivity of SLAMF7 to different methods of activation or inhibition. By targeting SLAMF7 with a ligand attached to a solid surface (such as that of a cell culture plate or a cell membrane) as we have done here and previously (23), the receptor becomes activated and down-stream signaling is initiated. However, adding soluble SLAMF7 ligands to the system might result in blocking of the receptor and achieve effects directly opposite to that of receptor activation (23). This makes the study of SLAMF7 signaling very dependent on the experimental methods used and may explain why other studies (59) reached different conclusions from this study.
Our analysis of the roles played by SLAMF7 on both T cells and tumor-associated macrophages, incorporating a combination of multi-parametric spectral cytometry and targeted analysis of existing datasets, provide early evidence implicating a novel immune evasion mechanism employed by tumors.
Supplementary Material
Key points:
SLAMF7 signaling on T cells reprograms them to an exhaustion-like state.
SLAMF7 expression is prognostic in a cancer dominated by T cell exhaustion; ccRCC.
SF7-SF7 interactions between TAMs and T cells drive inhibitory receptor expression.
Acknowledgments:
We thank the MSU Transgenic and Genome Editing Facility, Elena Demireva, and Huirong Xie for generating SLAMF7−/− mice. We appreciate assistance performing spectral cytometry provided by Matthew Bernard and the MSU South Campus Flow Cytometry Core Facility. Thanks to Austen N. Grooms for assistance with IL-2 ELISA experiments.
Financial support: P.O is supported by the John A. Penner Endowed Research Fellowship. Y.A.A. was supported by the National Institutes of Health Grant 1R21AI122808-01. A.A. is supported by the Osteopathic Heritage Foundation.
Abbreviations:
- SLAMF7
SLAM family member 7
- SF7
SLAMF7
- TME
Tumor microenvironment
- ccRCC
clear cell renal cell carcinoma
- TCGA
The Cancer Genome Atlas
- TAM
tumor-associated macrophage
- CyTOF
Cytometry by time of flight
- CL
Cross-linking
- tSNE
t-distributed stochastic neighbor embedding
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
References
- 1.Wherry EJ, and Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews LP, Yano H, and Vignali DAA. 2019. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol 20: 1425–1434. [DOI] [PubMed] [Google Scholar]
- 3.Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, Lynn RC, Philip M, Rao A, Restifo NP, Schietinger A, Schumacher TN, Schwartzberg PL, Sharpe AH, Speiser DE, Wherry EJ, Youngblood BA, and Zehn D. 2019. Defining ‘T cell exhaustion’. Nat Rev Immunol 19: 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Philip M, and Schietinger A. 2019. Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Curr Opin Immunol 58: 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanahan D, and Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 6.Pauken KE, and Wherry EJ. 2015. Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36: 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thommen DS, and Schumacher TN. 2018. T Cell Dysfunction in Cancer. Cancer cell 33: 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, Tonnerre P, Chung RT, Tully DC, Allen TM, Frahm N, Lauer GM, Wherry EJ, Yosef N, and Haining WN. 2016. The epigenetic landscape of T cell exhaustion. Science 354: 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, and Schietinger A. 2017. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545: 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto T, Price DA, Casazza JP, Ferrari G, Nason M, Chattopadhyay PK, Roederer M, Gostick E, Katsikis PD, Douek DC, Haubrich R, Petrovas C, and Koup RA. 2011. Surface expression patterns of negative regulatory molecules identify determinants of virus-specific CD8+ T-cell exhaustion in HIV infection. Blood 117: 4805–4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haslam A, and Prasad V. 2019. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA network open 2: e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Soldatos TG, Dimitrakopoulou-Strauss A, Larribere L, Hassel JC, and Sachpekidis C. 2018. Retrospective Side Effect Profiling of the Metastatic Melanoma Combination Therapy Ipilimumab-Nivolumab Using Adverse Event Data. Diagnostics 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou S, Khanal S, and Zhang H. 2019. Risk of immune-related adverse events associated with ipilimumab-plus-nivolumab and nivolumab therapy in cancer patients. Therapeutics and clinical risk management 15: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kotecha RR, Motzer RJ, and Voss MH. 2019. Towards individualized therapy for metastatic renal cell carcinoma. Nat Rev Clin Oncol 16: 621–633. [DOI] [PubMed] [Google Scholar]
- 15.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, Ries CH, Ailles L, Jewett MAS, Moch H, van den Broek M, Beisel C, Stadler MB, Gedye C, Reis B, Pe’er D, and Bodenmiller B. 2017. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 169: 736–749 e718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senbabaoglu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, Miao D, Ostrovnaya I, Drill E, Luna A, Weinhold N, Lee W, Manley BJ, Khalil DN, Kaffenberger SD, Chen Y, Danilova L, Voss MH, Coleman JA, Russo P, Reuter VE, Chan TA, Cheng EH, Scheinberg DA, Li MO, Choueiri TK, Hsieh JJ, Sander C, and Hakimi AA. 2016. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome biology 17: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M, Crozet L, Ouakrim H, Goc J, Cazes A, Flejou JF, Gibault L, Verkarre V, Regnard JF, Pages ON, Oudard S, Mlecnik B, Sautes-Fridman C, Fridman WH, and Damotte D. 2013. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res 19: 4079–4091. [DOI] [PubMed] [Google Scholar]
- 18.Giraldo NA, Becht E, Pages F, Skliris G, Verkarre V, Vano Y, Mejean A, Saint-Aubert N, Lacroix L, Natario I, Lupo A, Alifano M, Damotte D, Cazes A, Triebel F, Freeman GJ, Dieu-Nosjean MC, Oudard S, Fridman WH, and Sautes-Fridman C. 2015. Orchestration and Prognostic Significance of Immune Checkpoints in the Microenvironment of Primary and Metastatic Renal Cell Cancer. Clin Cancer Res 21: 3031–3040. [DOI] [PubMed] [Google Scholar]
- 19.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, Liu J, Freeman GJ, Brown MA, Wucherpfennig KW, and Liu XS. 2018. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24: 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthelemy P, Porta C, George S, Powles T, Donskov F, Neiman V, Kollmannsberger CK, Salman P, Gurney H, Hawkins R, Ravaud A, Grimm MO, Bracarda S, Barrios CH, Tomita Y, Castellano D, Rini BI, Chen AC, Mekan S, McHenry MB, Wind-Rotolo M, Doan J, Sharma P, Hammers HJ, Escudier B, and CheckMate I. 2018. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 378: 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cannons JL, Tangye SG, and Schwartzberg PL. 2011. SLAM family receptors and SAP adaptors in immunity. Annu Rev Immunol 29: 665–705. [DOI] [PubMed] [Google Scholar]
- 22.O’Connell P, Amalfitano A, and Aldhamen YA. 2019. SLAM Family Receptor Signaling in Viral Infections: HIV and Beyond. Vaccines (Basel) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Connell P, Pepelyayeva Y, Blake MK, Hyslop S, Crawford RB, Rizzo MD, Pereira-Hicks C, Godbehere S, Dale L, Gulick P, Kaminski NE, Amalfitano A, and Aldhamen YA. 2019. SLAMF7 Is a Critical Negative Regulator of IFN-alpha-Mediated CXCL10 Production in Chronic HIV Infection. Journal of immunology 202: 228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruz-Munoz ME, Dong Z, Shi X, Zhang S, and Veillette A. 2009. Influence of CRACC, a SLAM family receptor coupled to the adaptor EAT-2, on natural killer cell function. Nat Immunol 10: 297–305. [DOI] [PubMed] [Google Scholar]
- 25.Guo H, Cruz-Munoz ME, Wu N, Robbins M, and Veillette A. 2015. Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol Cell Biol 35: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, Manos M, Gjini E, Kuchroo JR, Ishizuka JJ, Collier JL, Griffin GK, Maleri S, Comstock DE, Weiss SA, Brown FD, Panda A, Zimmer MD, Manguso RT, Hodi FS, Rodig SJ, Sharpe AH, and Haining WN. 2019. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20: 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yigit B, Wang N, Ten Hacken E, Chen SS, Bhan AK, Suarez-Fueyo A, Katsuyama E, Tsokos GC, Chiorazzi N, Wu CJ, Burger JA, Herzog RW, Engel P, and Terhorst C. 2019. SLAMF6 as a Regulator of Exhausted CD8(+) T Cells in Cancer. Cancer Immunol Res 7: 1485–1496. [DOI] [PubMed] [Google Scholar]
- 28.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, Freeman SS, Reuben A, Hoover PJ, Villani AC, Ivanova E, Portell A, Lizotte PH, Aref AR, Eliane JP, Hammond MR, Vitzthum H, Blackmon SM, Li B, Gopalakrishnan V, Reddy SM, Cooper ZA, Paweletz CP, Barbie DA, Stemmer-Rachamimov A, Flaherty KT, Wargo JA, Boland GM, Sullivan RJ, Getz G, and Hacohen N. 2018. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175: 998–1013 e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, Kuchroo VK, Regev A, and Anderson AC. 2019. Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 50: 181–194 e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Connell P, Zheng YH, Amalfitano A, Aldhamen YA 2019. In vitro Infection of Primary Human Monocytes with HIV-1. bio-protocol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aldhamen YA, Appledorn DM, Seregin SS, Liu CJ, Schuldt NJ, Godbehere S, and Amalfitano A. 2011. Expression of the SLAM family of receptors adapter EAT-2 as a novel strategy for enhancing beneficial immune responses to vaccine antigens. Journal of immunology 186: 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V, Cancer N Genome Atlas Research, and Hu H. 2018. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173: 400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roederer M, Nozzi JL, and Nason MC. 2011. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry. Part A : the journal of the International Society for Analytical Cytology 79: 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y. 2015. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry. Part A : the journal of the International Society for Analytical Cytology 87: 636–645. [DOI] [PubMed] [Google Scholar]
- 35.Shin H, Blackburn SD, Intlekofer AM, Kao C, Angelosanto JM, Reiner SL, and Wherry EJ. 2009. A role for the transcriptional repressor Blimp-1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 31: 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shankar EM, Che KF, Messmer D, Lifson JD, and Larsson M. 2011. Expression of a broad array of negative costimulatory molecules and Blimp-1 in T cells following priming by HIV-1 pulsed dendritic cells. Molecular medicine 17: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He S, Liu Y, Meng L, Sun H, Wang Y, Ji Y, Purushe J, Chen P, Li C, Madzo J, Issa JP, Soboloff J, Reshef R, Moore B, Gattinoni L, and Zhang Y. 2017. Ezh2 phosphorylation state determines its capacity to maintain CD8(+) T memory precursors for antitumor immunity. Nat Commun 8: 2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balkhi MY, Wittmann G, Xiong F, and Junghans RP. 2018. YY1 Upregulates Checkpoint Receptors and Downregulates Type I Cytokines in Exhausted, Chronically Stimulated Human T Cells. iScience 2: 105–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ, and Anderson AC. 2013. TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology 2: e23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, Morikawa H, Kawazoe A, Kinoshita T, Shitara K, Sakaguchi S, and Nishikawa H. 2019. PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A 116: 9999–10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perez OD, Mitchell D, Campos R, Gao GJ, Li L, and Nolan GP. 2005. Multiparameter analysis of intracellular phosphoepitopes in immunophenotyped cell populations by flow cytometry. Curr Protoc Cytom Chapter 6: Unit 6 20. [DOI] [PubMed] [Google Scholar]
- 42.Mita Y, Kimura MY, Hayashizaki K, Koyama-Nasu R, Ito T, Motohashi S, Okamoto Y, and Nakayama T. 2018. Crucial role of CD69 in anti-tumor immunity through regulating the exhaustion of tumor-infiltrating T cells. Int Immunol 30: 559–567. [DOI] [PubMed] [Google Scholar]
- 43.Hofer T, Krichevsky O, and Altan-Bonnet G. 2012. Competition for IL-2 between Regulatory and Effector T Cells to Chisel Immune Responses. Front Immunol 3: 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beltra JC, Bourbonnais S, Bedard N, Charpentier T, Boulange M, Michaud E, Boufaied I, Bruneau J, Shoukry NH, Lamarre A, and Decaluwe H. 2016. IL2Rbeta-dependent signals drive terminal exhaustion and suppress memory development during chronic viral infection. Proc Natl Acad Sci U S A 113: E5444–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, and Ritz J. 2006. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 108: 1571–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banerjee H, and Kane LP. 2018. Immune regulation by Tim-3. F1000Res 7: 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dannenmann SR, Thielicke J, Stockli M, Matter C, von Boehmer L, Cecconi V, Hermanns T, Hefermehl L, Schraml P, Moch H, Knuth A, and van den Broek M. 2013. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunology 2: e23562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chapuis AG, Thompson JA, Margolin KA, Rodmyre R, Lai IP, Dowdy K, Farrar EA, Bhatia S, Sabath DE, Cao J, Li Y, and Yee C. 2012. Transferred melanoma-specific CD8+ T cells persist, mediate tumor regression, and acquire central memory phenotype. Proc Natl Acad Sci U S A 109: 4592–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, Utzschneider DT, von Hoesslin M, Cullen JG, Fan Y, Eisenberg V, Wohlleber D, Steiger K, Merkler D, Delorenzi M, Knolle PA, Cohen CJ, Thimme R, Youngblood B, and Zehn D. 2019. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571: 265–269. [DOI] [PubMed] [Google Scholar]
- 50.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, Werner MT, Huang AC, Alexander KA, Wu JE, Attanasio J, Yan P, George SM, Bengsch B, Staupe RP, Donahue G, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Kaye J, Berger SL, and Wherry EJ. 2019. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 571: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, Trivedi P, Menocal L, Appleby H, Camara S, Zamarin D, Walther T, Snyder A, Femia MR, Comen EA, Wen HY, Hellmann MD, Anandasabapathy N, Liu Y, Altorki NK, Lauer P, Levy O, Glickman MS, Kaye J, Betel D, Philip M, and Schietinger A. 2019. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571: 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, Yan P, Nzingha K, Xu W, Amaravadi RK, Xu X, Karakousis GC, Mitchell TC, Schuchter LM, Huang AC, and Wherry EJ. 2020. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52: 825–841 e828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnson DE, O’Keefe RA, and Grandis JR. 2018. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol 15: 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I, Lee C, Joo KM, Rich JN, Nam DH, and Lee J. 2013. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer cell 23: 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Katsuyama E, Suarez-Fueyo A, Bradley SJ, Mizui M, Marin AV, Mulki L, Krishfield S, Malavasi F, Yoon J, Sui SJH, Kyttaris VC, and Tsokos GC. 2020. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep 30: 112–123 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bally AP, Austin JW, and Boss JM. 2016. Genetic and Epigenetic Regulation of PD-1 Expression. Journal of immunology 196: 2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dongre P, Mathew S, Akopova I, Gryczynski I, and Mathew P. 2013. YY1 and a unique DNA repeat element regulates the transcription of mouse CS1 (CD319, SLAMF7) gene. Mol Immunol 54: 254–263. [DOI] [PubMed] [Google Scholar]
- 58.Kim JR, Mathew SO, and Mathew PA. 2016. Blimp-1/PRDM1 regulates the transcription of human CS1 (SLAMF7) gene in NK and B cells. Immunobiology 221: 31–39. [DOI] [PubMed] [Google Scholar]
- 59.Comte D, Karampetsou MP, Yoshida N, Kis-Toth K, Kyttaris VC, and Tsokos GC. 2017. Signaling Lymphocytic Activation Molecule Family Member 7 Engagement Restores Defective Effector CD8+ T Cell Function in Systemic Lupus Erythematosus. Arthritis & rheumatology 69: 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.