

SUMMARY
Deubiquitinating enzymes (DUBs) catalyze the removal of ubiquitin, thereby reversing the activity of E3 ubiquitin ligases and are central to the control of protein abundance and function. Despite the growing interest in DUBs as therapeutic targets, cellular functions for DUBs remain largely unknown and technical challenges often preclude the identification of DUB substrates in a comprehensive manner. Here, we demonstrate that treatment with potent DUB inhibitors coupled to mass spectrometry-based proteomics can identify DUB substrates at a proteome-wide scale. We applied this approach to USP7, a DUB widely investigated as a therapeutic target and identified many known substrates and additional targets. We demonstrate that USP7 substrates are enriched for DNA repair enzymes and E3 ubiquitin ligases. This work provides not only a comprehensive annotation of USP7 substrates, but a general protocol widely applicable to other DUBs, which is critical for translational development of DUB targeted agents.
In Brief
Bushman et al. profile commonly used inhibitors of deubiquitinating enzymes to assess their biochemical selectivity and cellular activity. USP7 is used to demonstrate proof-of-concept for using potent and selective inhibitors of deubiquitinating enzymes to identify their substrates by mass spectrometry.
Graphical Abstract
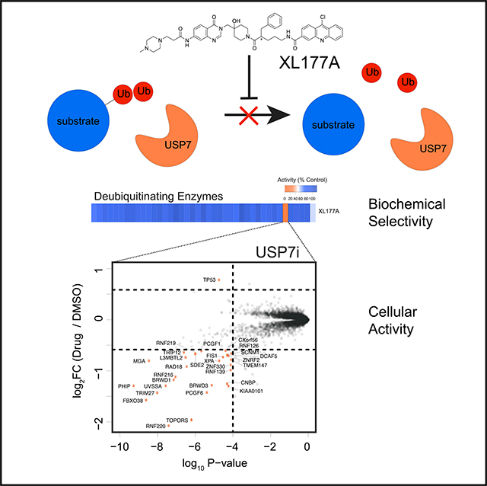
INTRODUCTION
The ubiquitin-proteasome system (UPS) is one of the central mechanisms used by eukaryotic cells to maintain protein homeostasis. In the UPS, ubiquitin is covalently attached to proteins through the activity of a three-enzyme cascade consisting of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3) (Deshaies and Joazeiro, 2009). Substrate specificity of the UPS is largely conferred by E3 ligases, of which more than 600 are encoded by the human genome. The removal of ubiquitin chains is catalyzed by DUBs, a family of more than 100 enzymes that in metazoans belong to 7 distinct families, 6 of which are cysteine proteases while the seventh are metalloproteases (Harrigan et al., 2018). DUBs have been linked to a myriad of cellular processes, such as DNA damage repair, epigenetic regulation, oncogenesis, and cell growth and homeostasis (Jackson and Durocher, 2013; Pinto-Fernandez and Kessler, 2016). The relatively limited number of enzymes with respect to the abundance of ubiquitin ligases and substrates suggests that each DUB likely interfaces with a large set of substrates.
The enzymatic addition and removal of ubiquitin chains from proteins is a primary mechanism by which cells regulate the post-translational abundance of proteins. Accordingly, modulating the function of UPS enzymes is a viable route to affect protein abundance and downstream function. However, this approach relies on mapping the relationships between protein substrates and the enzymes that add and remove ubiquitin. Many tools and methodologies have been developed for the study of endogenous ubiquitination sites by mass spectrometry (MS), including tandem ubiquitin binding entities and anti-diglycine antibodies, which have been successfully used to identify substrates of specific E3 ligases and DUBs (Altun et al., 2011; Hjerpe et al., 2009; Thompson et al., 2014; Udeshi et al., 2013). Previous efforts to systematically map the interacting partners of DUBs in yeast and human cells have provided important insights into possible substrates (Kouranti et al., 2010; Sowa et al., 2009). However, many substrates evade detection due to the transient and weak nature of binding to DUBs. MS has been used to study the functional deletion of DUBs and identify substrates in yeast (Fang et al., 2016; Poulsen et al., 2012), but systematic identification of human DUB substrates remains an understudied area of research. Recent development of highly selective DUB inhibitors provides the opportunity to interrogate individual DUB function in a minimally invasive cellular context. Here, we describe a method for identifying DUB substrates with highly selective DUB inhibitors coupled to proteomics. As demonstrated for USP7, this approach enables comprehensive mapping of DUB substrates in unmodified cells and confirms existing biology while also unraveling targets and functions for USP7.
RESULTS
In Vitro Biochemical Selectivity of USP7 Inhibitors
It is widely appreciated that that tandem mass tag (TMT)-based quantitative proteomics (Figure S1A) is an effective tool to interrogate ubiquitin-mediated changes to protein abundance in response to small-molecule treatment (An et al., 2017; Donovan et al., 2018; Kronke et al., 2014). Pre-requisites for such experiments include potent and selective small-molecule probes. To optimize the proteomics-based identification of DUB substrates and ensure controlled experimental conditions, we settled on non-covalent and covalent pairs of USP7 inhibitors (XL188 and XL177A) for which enantiomeric control compounds (XL203C and XL177B) with significantly reduced activity were available (Figure S1B) (Lamberto et al., 2017; Schauer et al., 2020). The enantiomer pair XL188 and XL203C inhibit USP7 in a purified enzyme biochemical assay with half maximal inhibitory concentration (IC50) values of 90 nM and 7 μM, respectively, while XL77A and XL177B exhibit IC50 values of 0.34 and 340 nM, respectively. Importantly, the small-molecule inhibitors used are highly selective for a single DUB. XL188 and XL177A were tested against a panel of 41 purified DUBs for inhibition in an in vitro ubiquitin cleavage assay (Figure 1A). XL188 (tested at 10 μM) and XL177A (tested at 1 μM) exhibited 100% inhibition of USP7 while little to no inhibition was detected for any of the other 40 DUBs representing the breadth of the entire DUB family (Clague et al., 2019). The covalent inhibitor XL177A was further shown to be highly selective for USP7 relative to native DUBs as well as across the broader proteome using chemoproteomic assays (Schauer et al., 2020). Taken together, the potency and selectivity data demonstrate the suitability of these enantiomeric pairs to profile substrates of USP7 using MS-based proteomics.
Figure 1. Differential Proteomics Analysis of MM.1S Cells Treated with Selective USP7 Inhibitors.

(A) In vitro biochemical selectivity of XL series USP7 inhibitors. Compounds XL188, XL203C, and XL177A were added to in vitro deubiquitination assays at concentrations of 10, 10, and 1 μM, respectively.
(B) Scatterplot representation of protein abundance changes after USP7 inhibitor treatment. Orange points represent proteins that pass the threshold of >1.5-fold change and p < 0.0001. p values and log2FC values were calculated using a moderated t test as implemented in limma. See Data S1 in the Supplemental Information for matching tables.
(C) Rank-order representation of protein abundance changes after USP7 inhibition. Proteins were ranked in ascending order using p value-adjusted log2 fold change as a score (Pi score = −log10(p value) × log2FC). Thresholds same as in (B).
Proteomics Approach Identify DUB Substrates
To benchmark the use of quantitative proteomics for the identification of novel DUB substrates, we adapted previously described experimental and analysis workflows to identify proteins destabilized following inhibition of USP7 with multiple inhibitors. We treated MM.1S myeloma cells with 10 μM non-covalent USP7 inhibitors, XL188 and XL203C, 1 μM covalent USP7 inhibitors, XL177A and XL177B, or 0.1% DMSO control (Figure S1A). Treatments were limited to 6 h to reduce secondary effects common to prolonged drug exposure. Protein abundances were quantified using a TMT MS strategy previously optimized for the interrogation of drug-induced changes to protein abundance (Donovan et al., 2018; McAlister et al., 2014). Of ~ 9,000 quantified protein groups, less than 30 proteins exceeded cutoffs defined as 1.5-fold change in protein abundance and significance of p value < 0.0001 (Figure 1B). The focal changes to the cellular proteome were consistent with the high selectivity of the USP7 inhibitors and short drug treatment expected to limit downstream transcriptional effects. Several previously reported substrates were destabilized following USP7 inhibition, including TRIM27, RNF220, FBXO38, UVSSA, and RAD18. Consistent with previous reports, the sole protein observed to be significantly stabilized after USP7 inhibition was p53. In accordance with the role of USP7 in de-ubiquitinating MDM2 and thereby destabilizing p53, treatment with XL188 or XL177A has been shown to result in loss of MDM2 and accumulation of p53 (Lamberto et al., 2017; Schauer et al., 2020). In addition to known substrates, several other proteins are destabilized by USP7 inhibition: TOPORS, RNF216, DCAF12, BRWD1, GTF2B, RLIM, CCDC71L, EAPP, C12orf45, OTUD5, SDE2, RNF219, HLTF, and SCNM1. Since previously validated substrates were identified in this experiment, our data strongly suggests that these proteins are likely bona fide substrates of USP7 as well.
Time Course of Covalent and Non-covalent USP7 Inhibition
Modulation of USP7 activity can be accomplished in multiple ways, including genetic ablation or transient overexpression of USP7. We reasoned that the long time points associated with these techniques would be prohibitive for identification of USP7 substrates due to confounding tertiary effects of disrupting these proteins. To assess this, we performed a time course using covalent and non-covalent USP7 inhibitors XL177A and XL188 for 2, 6, 12, or 24 h. Only nine proteins were differentially quantified after 2 h of covalent USP7 inhibition: MDM2, RNF220, RNF111, TOPORS, BRWD1, BRWD3, TCEAL4 were downregulated and TP53 was upregulated (Figure 2A). After 6 h of USP7 inhibition, the majority of disrupted proteins (21 out of 24) were downregulated, consistent with the role of USP7 is stabilizing its substrates. In contrast, the longer treatments had a higher proportion of upregulated proteins, 11/33 at 12 h and 49/101 at 24 h. Comparison of covalent XL177A and non-covalent XL188 revealed nearly identical protein abundance changes at all time points (Figures 2B and S2). The USP7 substrate MDM2 was destabilized after 2 h but recovered to baseline abundance by 6 h, which is in accordance with MDM2 as a target gene of p53 being transcriptionally upregulated.
Figure 2. Time Course of Protein Abundance Changes Following USP7 Inhibition.

(A) Scatterplot representation of protein abundance changes over time with XL177A (see Figure S2 for XL188). Thresholds and statistical test are the same as in Figure 1.
(B) Heatmap of log2FC protein abundance values for differentially quantified proteins following USP7 inhibition by XL177A or XL188 after either 2, 6, 12, or 24 h treatment. Proteins indicated in bold were differentially quantified after 2 h XL177A treatment. See Data S1 in the Supplemental Information for matching tables.
High-Confidence USP7 Substrate Candidates
The identification and validation of high-confidence hits is an outstanding problem for large proteomics datasets. We demonstrate that this problem can be overcome by use of orthogonal cell perturbations, in this case two distinct USP7 inhibitors (covalent versus reversible), with high-confidence substrates identified by multiple inhibitors targeting the same DUB and negative control compound exhibiting diminished yet similar effects. Proteome changes for each of the selective USP7 inhibitors were nearly identical, with strong and statistically significant correlations in protein abundance changes among XL177A, XL188, and XL203C (Figure S3A). The Pearson correlations among XL188 and its covalent and enantiomeric counterparts (XL177A and XL203C) are r = 0.85 and r = 0.67, respectively. There was a clear dose-like response between compound potency and changes in protein abundance, with the more potent enantiomers (XL177A, XL188) destabilizing known USP7 substrates more strongly than the less potent enantiomers (XL177B, XL203C). TRIM27, TOPORS, RNF220, FBXO38, RNF216, UVSSA, and BRWD1 were among the 10 top-ranked destabilized proteins for both non-covalent XL188 and covalent XL177A compounds. Strikingly, several of these proteins (TRIM27, RNF216, FBXO38, RNF220, and UVSSA) were also top-ranked hits for the XL203C control (Figure 1C), even though they did not meet our stringent thresholds (1.5-fold change, p < 0.0001). The low-level activity of XL203C thus provides an additional internal control and further supports the identified USP7 substrates. Combining the data from XL177A, XL188, and XL203C, we can derive a hit list of high-confidence USP7 substrates that rank in the top 1% of destabilized proteins for three USP7 inhibitors (Figure 3A). This set of proteins includes many previously described substrates and known USP7 interactors (Table S1), as well as several additional proteins for which we did not find USP7-related annotations. Several of these USP7 targets are more strongly destabilized than previously identified substrates, highlighting the advantage of unbiased proteomics in identifying DUB substrates from the complete context of the cellular proteome.
Figure 3. Evaluation of High-Confidence USP7 Candidate Substrates.

(A) Heatmap of Pi scores for top differentially quantified proteins and known USP7 targets (see Table S1) following USP7 inhibition.
(B) Heatmap comparison of USP7 inhibition against USP7 IP-MS data from the DUB Interactome database (see Figure S3A for Pearson correlation, Figure S3B for BioPlex database).
(C) STRING network analysis of proteins destabilized by XL177A and XL188 treatment (D) Annotation of USP7 targets with enriched Gene Ontology molecular function terms. Gene enrichment was calculated using Fisher’s exact test as implemented in Enrichr.
To further evaluate our list of high-confidence USP7 substrates, we examined existing literature and databases for evidence of interaction with USP7. The DUB Interactome database of immunoprecipitation (IP)-MS experiments contained 317 proteins identified by USP7 pull-down that were also present in our data (Sowa et al., 2009). Of these, 24 are considered high-confidence interacting proteins based on ComPASS IP-MS metrics (WDN > 1.0) and several were modestly destabilized by one or more of our USP7 inhibitors (Figure 3B). Consistent with the reported role of USP7 in stabilizing its binding partners, there was a modest anti-correlation between protein abundances following USP7 inhibition and USP7 IP-MS scores (Figure S3A). Several known USP7 targets were identified by both IP and protein abundance change following inhibition, including FBXO38, RNF220, DNMT1, TRIP12, and HLTF. USP7 substrates DDX24 and USP11 were similarly destabilized by USP7 inhibition but not detected by IP. There were 27 baits for USP7 from the BioPlex network (Huttlin et al., 2015), with PCGF1, PCGF6, and PHF8 also destabilized by covalent inhibition of USP7 by XL177A (Figure S3B). The STRING database was also examined for physical and functional interactions among proteins destabilized by the two more potent enantiomers and Enrichr was used to calculate Gene Ontology enrichment scores (Chen et al., 2013; Szklarczyk et al., 2019). As expected, proteins disrupted by these inhibitors were highly enriched for annotated connections to USP7 and several Gene Ontology terms (Figure 3C). Proteins, including p53, RNF168, TRIP12, UHRF1, UVSSA, and CLSPN demonstrate the well-established role of USP7 in DNA repair (Jackson and Durocher, 2013). Consistent with the role of USP7 in DUB-E3 ubiquitin “switches” (Kim and Sixma, 2017), including the USP7/TRIM27 or USP7/UHRF1/DNMT1 complexes, this network is also highly enriched for proteins with ubiquitin ligase activity (Figure 3D). Collectively, these data validate the consistent and focal changes in proteome abundance after treatment with XL177A and XL188, and provide strong support for the USP7 substrate candidates identified by these probes.
Increased Throughput Profiling of DUB Inhibitors
While multiplexed MS-based proteomics is a powerful approach to detect changes in protein abundance, throughput is limited to the number of channels available (currently 6–16plex for commercially available TMT reagents). To increase throughput for the routine profiling of small molecules targeting enzymes of the UPS, we evaluated a study design that would maximize the number of independent treatments in a single experiment. The empirical Bayes approach implemented in the limma software package has proven beneficial for analysis of microarray, RNA sequencing, and proteomics datasets by robust estimation of sample variance using a statistical shrinkage approach (Ritchie et al., 2015). We have previously observed the established benefits of using limma in the analysis of proteomics data with limited numbers of replicates (An et al., 2017; Donovan et al., 2018). We sought to determine empirically if data analyzed using a simple control (n = 3) versus drug (n = 1) design would be sufficient to screen for proteome changes, increasing throughput to 7–13 compounds per experiment. To test this, we applied this approach to previously published treatments with thalidomide and found that, although this experimental design is statistically underpowered, the interpretation of data remains comparable (Figure 4A). This increased throughput enables broader profiling of DUB inhibitors earlier in the development pipeline, and we sought to determine if experiments with less-optimized molecules could still be used to identify candidate DUB substrates and yield important information.
Figure 4. Differential Proteomics Analysis of Additional DUB Inhibitors.

(A) Comparison of singlicate (3× DMSO versus 1× Drug) and triplicate (3× DMSO versus 3× Drug) analysis of protein abundance changes after thalidomide treatment.
(B) Rank-order representation of differentially quantified proteins (as in Figure 1C) after singlicate cell treatments with various dual-selective DUB inhibitors. Thresholds same as Figures 1 and 2. See Data S1 and Data S2 in the Supplemental Information for matching tables and scatterplots.
(C) Heatmap comparison of proteins disrupted by DUB inhibition in two independent treatment doses or experiments.
While extensive annotation of USP7 targets was achieved with the highly potent and selective XL molecules, there are a number of less active and/or less selective DUB inhibitors that have not been extensively characterized. We collected and profiled inhibitors described in the literature or patents reported with either single or dual specificity: P22077 (USP7/USP47), GNE-6640 (USP7), ML323 (USP1), WH-9943–91 (USP1), AZ1 (USP25/USP28), SB1-B-55 (USP25/USP28), SB1-F-78 (USP30), and SB1-F-70 (UCHL1/USP30) (Altun et al., 2011; Buckmelter et al., 2017; Dexheimer et al., 2010; Guerin et al., 2017; Kategaya et al., 2017; Stockley et al., 2018; Wrigley et al., 2017). When tested in vitro at 10 μM for inhibition of a panel of deubiquitinating enzymes, compounds demonstrated varying levels of potency and off-target activity for other DUBs (Figure S4). Compared with the XL series of USP7 inhibitors, P22077 and GNE-6640 had inferior selectivity for USP7 with modest (40%–50%) off-target inhibition of several DUBs along with significantly weaker on-target inhibition of USP7. In contrast, the dual USP25/USP28 inhibitors were highly selective and showed no off-target activity. Intermediate selectivity was seen with USP1, USP30, and UCHL1 inhibitors, which demonstrated modest or high inhibition of one or more DUBs in addition to their reported targets. Taken together, these inhibitors represent the wide range of potency and selectivity profiles for currently available DUB inhibitors.
We treated MM.1S cells with concentrations estimated to be above the IC50 values for each compound (1, 10, or 15 μM) and conducted proteomics analysis 6 h post-treatment. Data were analyzed as described above for singlicate treatments, revealing proteins that change in abundance in response to small-molecule treatment (Figure 4B). While changes in protein abundance were not as large as with the XL compounds, it was still possible to clearly discern proteins disrupted above background signal. Comparing the two scaffolds targeting USP30 (SB1-F-78 and SB1-F-70), of which SB1-F-78 is significantly more selective in biochemical assays, we conclude that proteomics profiling can serve as an orthogonal selectivity assay. The inferior selectivity of SB1-F-70 compared with SB1-F-78 manifests in a large number of up- and downregulated proteins, compared with the more focal changes of SB1-F-78 treatment (Figure 4B). Many of the aggregate top-ranked differentially quantified proteins from each singlicate comparison were confirmed either in independent experiments or with a different dose, supporting the reproducibility of top hits identified in this screening approach (Figure 4C). While the interpretation with respect to specific substrates is difficult for these weaker/less selective compounds, the data allow some insight on the quality of a molecule with respect to on-target efficacy and selectivity. In the absence of highly selective inhibitors or well-characterized DUB biology, our data demonstrate that singlicate profiling can provide an unbiased first look at potential targets for these enzymes.
DISCUSSION
We describe a strategy to for the unbiased identification of DUB substrates that takes advantage of highly selective USP7 inhibitors. To ensure we could adequately judge our approach for identifying DUB substrates, we placed emphasis on (1) the use of multiple highly selective inhibitors with the same target, (2) short treatment times to minimize confounding secondary effects, and (3) study of a well-characterized DUB with known substrates as a proof-of-concept case report. USP7 satisfies these criteria given that it represents the best studied DUB, for which several substrates have previously been identified and validated. Moreover, USP7 is one of the few mammalian DUBs for which highly selective inhibitors are available, such as the XL series utilized here. We demonstrate that highly selective and potent compounds are necessary for substrate identification, as contrasted by the USP7 inhibitors GNE-6640 and P22077 (Altun et al., 2011; Kategaya et al., 2017). Interestingly, the enantiomeric control of the non-covalent inhibitor (XL203C) behaved similar to a low-dose treatment of XL188 with all hits observed in response to XL188 downregulated to a lesser degree (Figure 1C). This is a surprising finding, given the lack of proteome changes with similarly potent molecules, such as GNE-6640, and indicates that the combination of potent on-target activity and the absence of off-target activity (DUBs and other proteins) is necessary for successful substrate identification. As is seen with P22077 and GNE-6640, reduced on-target activity along with even modest off-target activity can preclude unambiguous identification of substrates. This is an important finding as it suggests that cellular profiling along with biochemical selectivity panels can help to prioritize chemotypes early in a drug discovery campaign for DUB inhibitors.
USP7 has been shown to be highly expressed and found in a large number of interaction networks (Sowa et al., 2009). The formation of stable protein complexes is likely a major regulatory mechanism for USP7. It has long been thought that auto-ubiquitination and degradation may be a mechanism for E3 ligases to turn off their own activity once they fulfill their signaling role. The fact that our results show that many USP7 substrates are E3 ligases provides strong support for such a concept. Considering that many USP7 binding partners are also E3 ligases, it is possible that USP7 could antagonize auto-ubiquitination inactivation of the E3 ligases. Indeed, our data suggest that the stability of many E3 ligase USP7-binding partners depends on USP7 DUB activity itself. The E3 ligases TRIM27, RNF220, and RAD18 have all been reported to form stable complexes with USP7 and are among the proteins most strongly destabilized by selective inhibition of USP7 (Figure 3A). The stability of DNA methyltransferase 1 (DNMT1) has been reported to be regulated by opposing USP7 DUB activity and UHRF1 E3 activity (Du et al., 2010). Our data show that inhibition of USP7 results in destabilization of both DNMT1 and UHRF1. USP7 seems to regulate the stability of both a downstream substrate and the cognate E3 ligase for its degradation. We have identified TOPORS, RNF216, DCAF12, RLIM, and HLTF as candidate USP7 substrates that are also E3 ligases. Further experimentation will be necessary to determine if USP7 and these proteins act together as a “ubiquitin switch” for downstream substrates.
In summary, we demonstrate that selective inhibitors and MS-based proteomics represent a powerful approach for the identification of functional DUB substrates in their endogenous context. We further demonstrate that, for profiling of selective DUB inhibitors or other compounds targeting the UPS, the statistical disadvantage of singlicate treatments is not prohibitive to screening changes to the proteome. The increase in throughput can facilitate broader use of proteomics earlier in inhibitor development, which we believe is broadly applicable in targeting the UPS.
SIGNIFICANCE
Deubiquitinating enzymes (DUBs) are critical components of the ubiquitin-proteasome system and regulate key biological pathways. A major limitation for understanding the role of DUBs in biology and DUB-directed drug discovery is the lack of methods to identify DUB substrates. Here, we demonstrate that DUB substrates can be identified by unbiased mass spectrometry-based proteomics using selective DUB inhibitors. These findings are important to better understand the complex biology of DUBs and to effectively guide the development of novel therapeutics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric S. Fischer (Eric_Fischer@dfci.harvard.edu).
Materials Availability
All unique/stable reagents generated in this study will be made available on request but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The mass spectrometry datasets generated during this study are available in the Proteomics Identification Database (PRIDE) archive under the project accessions PXD020873, PXD020881, PXD020882, and PXD020865. All other data presented are available in the main text and Supplemental Information.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
MM.1S cells were purchased from ATCC and handled according to ATCC instructions. MM.1S cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and maintained in a humidified 37°C / 5% CO2 incubator. Cell lines are authenticated using ATCC STR service and regularly tested to ensure they are free of mycoplasma contamination.
METHOD DETAILS
In Vitro DUB Selectivity Profiling
Compounds were screened for in vitro selectivity using the Ubiquigent DUBprofiler SPT system. Compounds were incubated with purified DUBs for 15 minutes before ubiquitin-rhodamine(110)-glycine or Ubiquitin-AMC substrate was added. Percent remaining DUB activity was calculated based on release of fluorescent rhodamine or 7-amino-4-methylcoumarin (AMC) from substrate relative to DMSO control.
Cell Treatments
MM.1S cells were counted using an automated cell counter (ThermoFisher Countess II FL) and cell viability was assessed by staining with 0.4% Trypan Blue solution to ensure >99% of cells were alive prior to treatment. The evening before treatment, 10 × 106 cells were seeded in 10ml of fresh culture media in upright T25 cell culture flasks. The next morning, cells were treated with 0.1% DMSO, 15 μM XL188, 15 μM XL203C, 1 μM XL177A, 1 μM XL177B, 1 μM P22077, 1 μM GNE-6640, 15 μM AZ1, 15 μM SB1-B-55, 10 or 15 μM SB1-F-78, 10 or 15 μM SB-1-F70, 10 μM ML323, or 10 or 15 μM WH-9943–91. After 2, 6, 12, or 24 hours, cells were harvested by centrifugation and washed three times with PBS. Cell pellets were either frozen in liquid nitrogen and stored at −80°C or used immediately prepared for mass spectrometry.
Sample Preparation for TMT LC-MS3 Mass Spectrometry
Cell pellets were lysed by resuspending in lysis buffer (8 M urea, 50 mM NaCl, 50 mM EPPS pH 8.5, 1× Roche protease inhibitor, and 1× Roche PhosphoStop) and passing through a 21 gauge × 1.25 inch needle 20 times. The homogenized sample was clarified by centrifugation at 20,000 × g for 10 min at 4°C. Protein concentration of the cell lysate was determined by micro-BCA assay (Pierce) according to the manufacturer protocol. Proteins were reduced and alkylated with 10 mM TCEP for 30 min, 15 mM iodoacetamide for 30 min, and 10 mM DTT for 15 min. For each sample, 100 or 200 μg of protein was precipitated using four volumes of methanol, one volume of chloroform, and three volumes of water. The mixture was vortexed and centrifuged at 14,000 × g for 5 min to separate the organic and aqueous phases and pellet the protein. The precipitated protein was washed with three volumes of methanol and allowed to air dry. Precipitated protein was resuspended in 4 M urea, 50 mM HEPES pH 7.4, followed by dilution to 1 M urea with 200 mM EPPS pH 8. Samples were digested with LysC (1:50 enzyme:protein) for 12 hours at room temperature, diluted to 0.5 M urea, and digested with trypsin (1:50 enzyme:protein) for 6 hours at 37°C. Digested peptide samples were diluted with anhydrous acetonitrile (ACN) to a final concentration of 30% v/v and labeled with tandem mass tag (TMT) reagents (Thermo Fisher Scientific) according to manufacturer’s instructions at a ratio of 1:4 peptide:TMT label for 1.5 hours at room temperature. The labeling reactions were quenched with 0.3% hydroxylamine for 15 min at room temperature. The sample channels were combined in equal amounts, desalted using C18 solid phase extraction cartridges (Waters) and analyzed by LC-MS for channel ratio comparison. Samples were then normalized and recombined using adjusted volumes calculated by the channel ratio comparison. Samples were dried down in a speed vacuum and resuspended in 1% formic acid, acidified to pH 2–3, and desalted with C18 SPE (Sep-Pak, Waters). Samples were then offline fractionated into 96 fractions by high pH reverse-phase HPLC (Agilent LC1260) through an Aeris XB-C18 column (Phenomenex) with mobile phase A (5% ACN, 10 mM NH4HCO3, pH 8.0) and mobile phase B (90% ACN, 10 mM NH4HCO3, pH 8.0) in LC-MS grade H2O. The resulting 96 fractions were then pooled in a non-contiguous manner into 24 fractions, desalted with SOLAμ SPE plates (Thermo Fisher Scientific), dried down by speed vacuum, and resuspended in 5% ACN / 5% formic acid. Each of the 24 fractions were used for subsequent mass spectrometry analysis.
TMT LC-MS3 Mass Spectrometry
Data were collected using an Orbitrap Fusion Lumos mass spectrometry (Thermo Fischer Scientific) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific) or an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific) coupled with an UltiMate 3000 RSLCnano System. Peptides were separated on a ES803a/ES803a.rev2 75 μm inner diameter microcapillary column (Thermo Fisher Scientific). Peptides were separated using a 3 hour gradient of 6–27% acetonitrile in 1.0% formic acid with a flow rate of 300 nL/min. Each analysis used a MS3-based TMT method as described previously (McAlister et al., 2014). The data were acquired using a mass range of m/z 350–1350, resolution 120,000, AGC target 1 × 106, maximum injection time 100 ms, dynamic exclusion of 90 s for the peptide measurements in the Orbitrap. Data-dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 1.8 × 104, and a maximum injection time of 120 ms. MS3 scans were acquired in the Orbitrap with a HCD collision energy set to 55%, AGC target set to 1.5 × 105, maximum injection time of 150 ms, resolution at 50,000, and with a maximum synchronous precursor selection (SPS) precursors set to 10.
QUANTIFICATION AND STATISTICAL ANALYSIS
LC-MS Data Analysis
RAW files produced by mass spectrometry were processed by Proteome Discoverer 2.2 (Thermo Fisher Scientific), which assembled and quantified proteins from peptides and controlled peptide and protein level false discovery rates. MS/MS spectra were searched against a Uniprot Homo sapiens database using both forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 20 ppm, fragment ion mass tolerance of 0.6 Da, static alkylation of cysteine (57.02146 Da), static TMT labeling of lysine residues and N-termini of peptides (229.16293 Da), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were quantified using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor-quality MS3 spectra were excluded from quantitation (summed signal-to-noise across 10 channels > 200 and precursor isolation specificity < 0.5). Reporter ion intensities were normalized and scaled using inhouse scripts and the R framework (R Core Team, 2019). Statistical analysis was carried out using the limma package within the R framework (Ritchie et al., 2015).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| Tandem Mass Tag (TMT) Reagents, 10plex, 11plex, and 16plex sets | Thermo Fisher Scientific | Cat# 90110 Cat# A34808 Cat# A44521 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| cOmplete, Mini Protease Inhibitor Cocktail | Sigma-Aldrich | Cat# 11836153001 |
| PhosSTOP Phosphatase Inhibitor Tablets | Sigma-Aldrich | Cat# 04906837001 |
| XL188 | Sara Buhrlage, Dana Farber Cancer Institute (DFCI) (Lamberto et al., 2017) | N/A |
| XL203C | Sara Buhrlage, DFCI (Lamberto et al., 2017) | N/A |
| XL177A | Sara Buhrlage, DFCI (Schauer et al., 2020) | N/A |
| XL177B | Sara Buhrlage, DFCI (Schauer et al., 2020) | N/A |
| P22077 | Sara Buhrlage, DFCI (Altun et al., 2011) | N/A |
| GNE-6640 | Sara Buhrlage, DFCI (Kategaya et al., 2017) | N/A |
| AZ1 | Sara Buhrlage, DFCI (Wrigley et al., 2017) | N/A |
| SB1-B-55 | Sara Buhrlage, DFCI (Guerin et al., 2017) | N/A |
| SB1-F-70 | Sara Buhrlage, DFCI (Stockley, Kemp and Madin, 2018) | N/A |
| SB1-F-78 | Sara Buhrlage, DFCI (Stockley, Kemp and Madin, 2018) | N/A |
| ML323 | Sara Buhrlage, DFCI (Dexheimer et al., 2010) | N/A |
| WH-9943-91 | Sara Buhrlage, DFCI (Buckmelter et al., 2017) | N/A |
| Deposited Data | ||
| wp-esf_038 | This paper | PXD020873 |
| wp-esf_039 | (Donovan et al., 2018) | PXD010428 |
| wp-esf_061 | This paper | PXD020881 |
| wp-esf_098 | This paper | PXD020882 |
| wp-esf_194 | This paper | PXD020865 |
| Experimental Models: Cell Lines | ||
| MM.1S (H. sapiens) | ATCC | CRL-2974; RRID: CVCL_8792 |
| Software and Algorithms | ||
| Proteome Discoverer 2.2 | Thermo Fisher Scientific | RRID: SCR_014477 |
| R Framework | (R Core Team, 2019) | http://www.R-project.org/ |
| Limma Package (R framework) | (Ritchie et al., 2015) | https://doi.org/doi:10.18129/B9.bioc.limma |
Highlights.
Commonly used DUB inhibitors show a wide range of selectivity and cellular activity
Cellular proteomics of USP7, USP47, USP25/28, USP30, UCHL1, and USP1 inhibitors
Use of multiple USP7 inhibitors to confirm known and new targets of USP7
Practical insights for profiling DUB inhibitors earlier in the development pipeline
ACKNOWLEDGMENTS
This work was supported by the following grants from the NIH: R01CA214608 and R01CA218278 (to E.S.F.). We thank Milka Kostic and Tao Wu for critical feedback on the manuscript. E.S.F. is a Damon Runyon-Rachleff Innovator in part supported by the Damon-Runyon Cancer Research Foundation DRR-50-18.
Footnotes
DECLARATION OF INTERESTS
E.S.F. is a founder, advisor, and equity holder in Civetta Therapeutics and Jengu Therapeutics. E.S.F. is an equity holder in C4 Therapeutics, and a consultant to Astellas, Novartis, Deerfield, and EcoR1 capital. The Fischer lab receives or has received research funding from Novartis Astellas, and Deerfield.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chembiol.2020.09.005.
REFERENCES
- Altun M, Kramer HB, Willems LI, McDermott JL, Leach CA, Goldenberg SJ, Kumar KG, Konietzny R, Fischer R, Kogan E, et al. (2011). Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol 18, 1401–1412. [DOI] [PubMed] [Google Scholar]
- An J, Ponthier CM, Sack R, Seebacher J, Stadler MB, Donovan KA, and Fischer ES (2017). pSILAC mass spectrometry reveals ZFP91 as IMiDdependent substrate of the CRL4(CRBN) ubiquitin ligase. Nat. Commun 8, 15398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmelter AJ, Ioannidis S, Follows BC, Gustafson G, Wang M, Caravella JA, Wang Z, Fritzen EL, and Lin J (2017). Purinones as Ubiquitin-Specific Protease 1 Inhibitors (U.S.A.: Forma Therapeutics, Inc.). [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Urbe S, and Komander D (2019). Breaking the chains: deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol 20, 338–352. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, and Joazeiro CA (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem 78, 399–434. [DOI] [PubMed] [Google Scholar]
- Dexheimer TS, Rosenthal AS, Liang Q, Chen J, Villamil MA, Kerns EH, Simeonov A, Jadhav A, Zhuang Z, and Maloney DJ (2010). Discovery of ML323 as a novel inhibitor of the USP1/UAF1 deubiquitinase complex. Probe Reports from the NIH Molecular Libraries Program (National Center for Biotechnology Information (US)). [PubMed] [Google Scholar]
- Donovan KA, An J, Nowak RP, Yuan JC, Fink EC, Berry BC, Ebert BL, and Fischer ES (2018). Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. eLife 7, 10.7554/eLife.38430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S, Kao HY, Xu Y, Willis J, Markowitz SD, et al. (2010). DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signaling 3, ra80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang NN, Zhu M, Rose A, Wu KP, and Mayor T (2016). Deubiquitinase activity is required for the proteasomal degradation of misfolded cytosolic proteins upon heat-stress. Nat. Commun 7, 12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerin DJ, Bair KW, Caravella JA, Ioannidis S, Lancia DR, Li H, Mischke S, NG PY, Richard D, Schiller SER, et al. (2017). Thienopyrazine carboxamides as ubiquitin-specific protease inhibitors, Patent WO/2017/139779, filed February 13, 2107, and published August 17, 2017.
- Harrigan JA, Jacq X, Martin NM, and Jackson SP (2018). Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat. Rev. Drug Discov 17, 57–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, and Rodriguez MS (2009). Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep 10, 1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. (2015). The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Durocher D (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807. [DOI] [PubMed] [Google Scholar]
- Kategaya L, Di Lello P, Rouge L, Pastor R, Clark KR, Drummond J, Kleinheinz T, Lin E, Upton JP, Prakash S, et al. (2017). USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 550, 534–538. [DOI] [PubMed] [Google Scholar]
- Kim RQ, and Sixma TK (2017). Regulation of USP7: a high incidence of E3 complexes. J. Mol. Biol 429, 3395–3408. [DOI] [PubMed] [Google Scholar]
- Kouranti I, McLean JR, Feoktistova A, Liang P, Johnson AE, Roberts-Galbraith RH, and Gould KL (2010). A global census of fission yeast deubiquitinating enzyme localization and interaction networks reveals distinct compartmentalization profiles and overlapping functions in endocytosis and polarity. PLoS Biol 8, 10.1371/journal.pbio.1000471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberto I, Liu X, Seo HS, Schauer NJ, Iacob RE, Hu W, Das D, Mikhailova T, Weisberg EL, Engen JR, et al. (2017). Structure-guided development of a potent and selective non-covalent active-site inhibitor of USP7. Cell Chem Biol 24, 1490–1500.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Fernandez A, and Kessler BM (2016). DUBbing cancer: deubiquitylating enzymes involved in epigenetics, DNA damage and the cell cycle as therapeutic targets. Front. Genet 7, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen JW, Madsen CT, Young C, Kelstrup CD, Grell HC, Henriksen P, Juhl-Jensen L, and Nielsen ML (2012). Comprehensive profiling of proteome changes upon sequential deletion of deubiquitylating enzymes. J. Proteomics 75, 3886–3897. [DOI] [PubMed] [Google Scholar]
- R Core Team (2019). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing). [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer NJ, Liu X, Magin RS, Doherty LM, Chan WC, Ficarro SB, Hu W, Roberts RM, Iacob RE, Stolte B, et al. (2020). Selective USP7 inhibition elicits cancer cell killing through a p53-dependent mechanism. Sci. Rep 10, 5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, and Harper JW (2009). Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley ML, Kemp MI, and Madin A (2018). Cyano-substituted Heterocycles with Activity as Inhibitors of USP30, Patent WO2018065768, filed October 4, 2017, and published April 12, 2018
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47, D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JW, Nagel J, Hoving S, Gerrits B, Bauer A, Thomas JR, Kirschner MW, Schirle M, and Luchansky SJ (2014). Quantitative Lys-Gly-Gly (diGly) proteomics coupled with inducible RNAi reveals ubiquitin-mediated proteolysis of DNA damage-inducible transcript 4 (DDIT4) by the E3 ligase HUWE1. J. Biol. Chem 289, 28942–28955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, and Carr SA (2013). Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell Proteomics 12, 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrigley JD, Gavory G, Simpson I, Preston M, Plant H, Bradley J, Goeppert AU, Rozycka E, Davies G, Walsh J, et al. (2017). Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem. Biol 12, 3113–3125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry datasets generated during this study are available in the Proteomics Identification Database (PRIDE) archive under the project accessions PXD020873, PXD020881, PXD020882, and PXD020865. All other data presented are available in the main text and Supplemental Information.