

Abstract
Clinical trial results have recently demonstrated that inhibiting inflammation by targeting the interleukin-1β pathway can offer a significant reduction in lung cancer incidence and mortality, highlighting a pressing and unmet need to understand the benefits of inflammation-focused lung cancer therapies at the genetic level. While numerous genome-wide association studies (GWAS) have explored the genetic etiology of lung cancer, there remains a large gap between the type of information that may be gleaned from an association study and the depth of understanding necessary to explain and drive translational findings. Thus, in this work we jointly model and integrate extensive multi-omics data sources, utilizing a total of 40 genome-wide functional annotations that augment previously published results from the International Lung Cancer Consortium (ILCCO) GWAS, to prioritize and characterize single nucleotide polymorphisms (SNPs) that increase risk of squamous cell lung cancer through the inflammatory and immune responses. Our work bridges the gap between correlative analysis and translational follow-up research, refining GWAS association measures in an interpretable and systematic manner. In particular, re-analysis of the ILCCO data highlights the impact of highly-associated SNPs from nuclear factor-κB signaling pathway genes as well as major histocompatibility complex mediated variation in immune responses. One consequence of prioritizing likely functional SNPs is the pruning of variants that might be selected for follow-up work by over an order of magnitude, from potentially tens of thousands to hundreds. The strategies we introduce provide informative and interpretable approaches for incorporating extensive genome-wide annotation data in analysis of genetic association studies.
Keywords: genome-wide annotation, integrative omics, lung cancer, major histocompatibility complex
1. Introduction
Genome-wide association studies (GWAS) have successfully identified many genetic loci associated with lung cancer, however it is often difficult to explain exactly how associated variants perturb biological processes and increase risk of disease (Edwards, Beesley, French, & Dunning, 2013; Tam et al., 2019). Such difficulties arise in part from a conventional focus on only a few data types - for instance, genotype or gene expression data - which may naturally limit our understanding of the processes involved in carcinogenesis. Recently, many quantitative tools such as fine-mapping have been applied to better prioritize single nucleotide polymorphisms (SNPs) implicated in lung cancer (Ferreiro-Iglesias et al., 2018), but these approaches are also generally applied to only a few types of data. Less effort has been focused on cogently integrating the dozens of multi-omic annotations that offer a range of distinct perspectives regarding the mechanistic roles of individual SNPs (ENCODE Project Consortium, 2012; Visscher et al., 2017) associated with lung cancer.
This paper integrates forty different genome-wide variant functional annotations in a generalized linear mixed model (GLMM) based approach to prioritize and characterize inflammatory- and immune-related risk SNPs identified by a large-scale GWAS of lung squamous cell carcinoma (SCC) recently conducted by the International Lung Cancer Consortium (ILCCO) (McKay et al., 2017). Functional annotations describe characteristics of SNPs in a manner that parallels the way covariates describe subjects in a traditional GWAS regression analysis; one example of an annotation is the distance (in bp) between a SNP and the nearest transcription start site. The 40 annotations we use can thus be thought of as 40 covariates describing each SNP. In contrast to association-based methods such as fine-mapping, our model utilizes genome-wide annotation data from a variety of modalities to predict the biological roles of a SNP. The multi-dimensional functional prediction recognizes that different types of variants can possess vastly different consequences and thus allows SNPs to be evaluated on a range of attributes. This crucial feature distinguishes the model from other aggregation algorithms, most of which produce a single composite score that may be difficult to interpret and can over- or under-weight certain traits based on factors such as the type of training data used (Huang, Gulko, & Siepel, 2017; Kircher et al., 2014; Rogers et al., 2018).
Our re-analysis of the previously published ILCCO GWAS data – which reported a table of over 21,000 highly-associated (i.e. p<10−5) SNPs - emphasizes the inflammatory response and related immune mechanisms, which have long been studied as risk factors for lung cancer (Takahashi, Ogata, Nishigaki, Broide, & Karin, 2010; Walser et al., 2008). We take this approach because of a pressing and unmet need to more clearly explain significant results from the recent CANTOS clinical trial. In this trial (P. M. Ridker et al., 2017), patients treated with 300 mg of canakinumab, an IL-1β inhibitor, demonstrated a large decrease in lung cancer incidence (hazard ratio=0.33, p<0.0001) and mortality (hazard ratio=0.23, p=0.0002) during median follow-up of 3.7 years, one of the first such successes when targeting IL-1β in randomized human trials for lung cancer. CANTOS was primarily designed to evaluate cardiovascular outcomes and also demonstrated a significant reduction in risk of recurrent cardiovascular events (Paul M Ridker et al., 2017). It is of great interest to understand the genetic underpinnings of inflammatory- and immune-based risk in SCC to, for example, provide a more direct view into the biological mechanisms underlying disease, suggest possible therapeutic targets, or guide more accurate risk screening policies, all goals difficult to achieve through association studies alone (Karczewski & Snyder, 2018). The main objective of our manuscript is to move toward these goals by identifying and characterizing translationally relevant SNPs that can be prioritized for functional follow-up studies. Accomplishing this objective can conserve many resources that would be needed to validate the tens of thousands of variants identified through GWAS alone, as prioritization can allow researchers to focus on a subset of GWAS variants that are likely to possess functional roles.
This synthesis of the multi-faceted variant functional annotation data highlights the roles of many SNPs in genes belonging to intricate regulatory networks that affect cytokine signaling cascades and the immune response. For example, we prioritize a number of regulatory SNPs in nuclear factor-κB (NF-κB) signaling pathway genes. We also uncover a number of variants in the HLA region that may perturb disease risk through disruption of standard antigen presenting processes. Comparisons with risk variants in other lung cancer subtypes suggest that the sets of genetic variants driving inflammation risk in these diseases differ from those highlighted in our analysis of SCC. Taken together, our results help explain the CANTOS findings at a genetic level, advance understanding of the inflammation and immune related systems driving lung cancer risk across different histologies, and demonstrate how to integrate varied sources of multi-omic variant annotation data in a coherent statistical framework.
2. Materials and Methods
2.1. Multi-dimensional annotation class estimation
To interrogate the mechanistic roles of individual variants, we utilized a Multi-dimensional Annotation Class Integrative Estimator (MACIE) (Li, 2020; Yung, 2016) that modeled a SNP’s annotation values using a generalized linear mixed model in which the annotation values were assumed to depend on a SNP’s membership in multiple latent binary classes. In the MACIE framework, function was defined as a composite of these unobserved classes, with each class designed to summarize the functionality described by a different set of annotations. This distinctive formulation of functionality as a set of multiple characteristics allowed for a more versatile and more interpretable model than was possible when considering only a single holistic score. Existing integrative methods that produce one-dimensional ratings often either sacrifice data to focus on a single attribute or sacrifice specificity to incorporate more annotations (Supplementary Figure S1).
The model-fitting procedure estimated latent variant functional classes in the GLMM setting, while accounting for correlations between multiple annotations using random effects, under previously specified models for this framework (Sammel, Ryan, & Legler, 1997). Estimation proceeded with the EM algorithm, and computation of MACIE occurred in two main stages. In the training stage, the MACIE model was constructed using functional scores (36 for noncoding and synonymous variants and 12 for nonsynonymous coding variants) from a properly selected training dataset. This step obtained the fitted model parameters. In the prediction stage, the fitted model parameters were applied to a new set of SNPs to calculate the probability that each SNP belonged to a specific functional category given the data (see Appendix A for technical details). The functionality of a noncoding or synonymous coding SNP was defined with a regulatory class and an evolutionarily conserved function class. The membership of a SNP in each class was denoted by a binary indicator, and as such, the MACIE score was a composite measure corresponding to the probability that a SNP belonged to each of the 2 × 2 = 4 possible classes. Marginal probabilities for either the regulatory or conserved classes could also be calculated by summing the two probabilities corresponding to that class.
Functional annotation scores were selected and partitioned into three groups (regulatory function, evolutionarily conserved function, damaging protein function) based on previous experience (Yung, 2016) with modeling the annotations most likely to add novel and useful information in predicting functional roles. Different scores and different partitions could additionally be used for different phenotypes; such a step would require the model to be retrained. All precomputed scores for the specific MACIE model used in this analysis are available online (see Data Availability Statement).
All 40 scores used in the model were downloaded from the EIGEN (Ionita-Laza, McCallum, Xu, & Buxbaum, 2016) and CADD (Kircher et al., 2014) databases. Missing values were imputed as described in the original databases. As with EIGEN (Ionita-Laza et al., 2016), we retrieved each variant’s functional class from the CADD database and grouped “Regulatory”, “Intronic”, “Downstream”, “Upstream”, “Noncoding change”, “3prime UTR”, “5prime UTR”, “Intergenic”, and “Synonymous” SNPs all in the same noncoding and synonymous variants group. The MACIE training dataset for these variants consisted of 10% of variants randomly selected from the 1000 Genomes Project data set (excluding those in dbNSFP) that were located within 500 bp upstream of a gene start site.
For each variant, the conserved class integrated eight evolutionary conservation scores downloaded from the EIGEN database: GERP_NR, GERP_RS, PhyloPri, PhyloPla, PhyloVer, PhastPri, PhastPla, and PhastVer (names given as they appear in EIGEN). These scores corresponded to outputs from the GERP++, phyloP, and phastCons algorithms and were previously described (Davydov et al., 2010; Ionita-Laza et al., 2016; Pollard, Hubisz, Rosenbloom, & Siepel, 2010; Siepel et al., 2005). The transcription regulatory class integrated 28 functional scores from the CADD database (Kircher et al., 2014), including GC, CpG, ENCODE histone modification marks (EncH3K27Ac, EncH3K4Me1, EncH3K4Me3), ENCODE open chromatin marks (EncExp, EncOCCombPVal, EncOCDNasePVal, EncOCFairePVal, EncOCpolIIPVal, EncOCctcfPVal, EncOCmycPVal, EncOCDNaseSig, EncOCFaireSig, EncOCpolIISig, EncOCctcfSig, EncOCmycSig), ENCODE transcription factor binding sites data (TFBS, TFBSPeaks, TFBSPeaksMax), ChromHMM states collapsed into 5 groups (cHmmTSS, cHmmTx, cHmmEnh, cHmmZnf, cHmmRepr), bStatistic, minDistTSS, and minDistTSE (all names as they appear in the CADD database). The MACIE GLMM for noncoding and synonymous SNPs was then fit by jointly integrating all 36 functional scores in two classes.
For the nonsynonymous model, the training set used 10% of variants in the dbNSFP database (Liu, Wu, Li, & Boerwinkle, 2016), excluding sex chromosomes. The same eight conservation scores as above were used to evaluate an evolutionarily conserved class, and then four protein function scores (SIFT (Ng & Henikoff, 2003), PolyPhenDiv (Adzhubei et al., 2010), PolyPhenVar, and Mutation Assessor (Reva, Antipin, & Sander, 2011)) were extracted from the EIGEN database and used to predict a second class assessing the damaging function of coding substitutions. The MACIE marginal probabilities were then calculated using the same procedures described for noncoding variants.
2.2. Gene-based association analysis
To focus the analysis on inflammatory- and immune-related processes, we first performed gene-level inference on the largest existing lung cancer GWAS dataset. While MACIE results were calculated for all ILCCO SNPs, a key feature of the model was interpretability of the predicted classes, and so to most clearly demonstrate this advantage we concentrated discussion on those SNPs located near inflammatory- and immune-related genes. SNPs that were not located near genes but still predicted to belong to MACIE functional classes were interesting in their own right, although we left their exploration for future work.
Collection and preparation of the complete ILCCO OncoArray dataset was described previously (McKay et al., 2017), and from this overall compendium we restricted our initial gene-based association analysis to a subset of 7,426 squamous cell carcinoma cases and 55,630 controls, all of European descent. We mapped (Aken et al., 2016; Chang et al., 2015) individual SNP test statistics to genes if they fell inside or within 5 kb of the gene.
Gene-based inference was conducted with the Generalized Berk-Jones (GBJ) statistic (Sun & Lin, 2019). GBJ was derived from the Berk-Jones statistic, which demonstrates certain asymptotic optimality properties in set-based testing situations, and GBJ was previously shown to provide more power than comparable methods such as the Sequence Kernel Association Test (Wu et al., 2011) over a variety of commonly-occurring testing situations.
All summary statistics used were derived from SNPs with a minor allele frequency greater than 1% in the 1000 Genomes European cohort, thus the normality assumption of GBJ was likely to be satisfied, given the very large sample size. For very large genes comprising more than 1,000 SNPs, we pruned the set to remove SNPs that were correlated at r2>0.5 (Chang et al., 2015). Each different histology was tested with the ILCCO summary statistics corresponding to that histology.
In searching for functional SNPs conferring risk of SCC, we also limited our main analysis to variants possessing a marginal association of p<5 × 10−6 or uncommon and rare variants (minor allele frequency less than 5%) demonstrating p<5 × 10−4. The common threshold of 5 × 10−8 used for genome-wide significance was not employed both because this was not a hypothesis testing study and because highly functional SNPs may demonstrate only modest association due to the limitations of marginal models. A less stringent measure was used for uncommon and rare variants because they are widely believed to demonstrate more functionality.
2.3. Fine-mapping of HLA
Many variants in the HLA region showed both highly significant p-values and high MACIE predictions, and the density of strongly associated SNPs was much larger compared to other portions of the genome. Thus, we first performed additional imputation and fine-mapping to discover independent association signals amidst the strong long-range LD of this region. The original genotypes were imputed using 1000 Genomes Project data (1000 Genomes Project Consortium, 2015), but larger and richer datasets have been made available since then. We utilized the SNP2HLA (Jia et al., 2013) software to reimpute data from the original OncoArray chip with a reference dataset from the Type I Diabetes Genetics Consortium (Rich & Concannon, 2015). We then followed previous lung cancer association studies (Ferreiro-Iglesias et al., 2018) by taking a forward stepwise regression approach with all original ILCCO SNPs as well as newly imputed SNPs to help determine the strongest independent signals in the highly polymorphic region.
3. Results
3.1. SCC-associated genes
Gene-based inference uncovered 243 genes (Supplementary Table S1) significantly associated with lung cancer risk at the Bonferroni-corrected level of p=1.96 × 10−6 (Figure 1a–c), with a number of the most highly associated genes mapping to regions of the genome that were validated in previous lung cancer studies (IREB2, CHRNA5, ADAMTS7, and others) (Bosse & Amos, 2018; Timofeeva et al., 2012). Of the 243 significant genes, 187 (77%) fell inside or within 2 Mb of the HLA complex (Supplementary Figure S2). Notably, many significant genes comprised important components of NF-κB signaling pathways. Examples included TNF, CHUK, NFKBIL1, and TRIM38. Multiple major histocompatibility complex (MHC) class I and class II genes were implicated as well, including HLA-A, HLA-DQA1, and HLA-DQB1. These inflammation and immune-related loci guided our search for functional variants.
Figure 1.
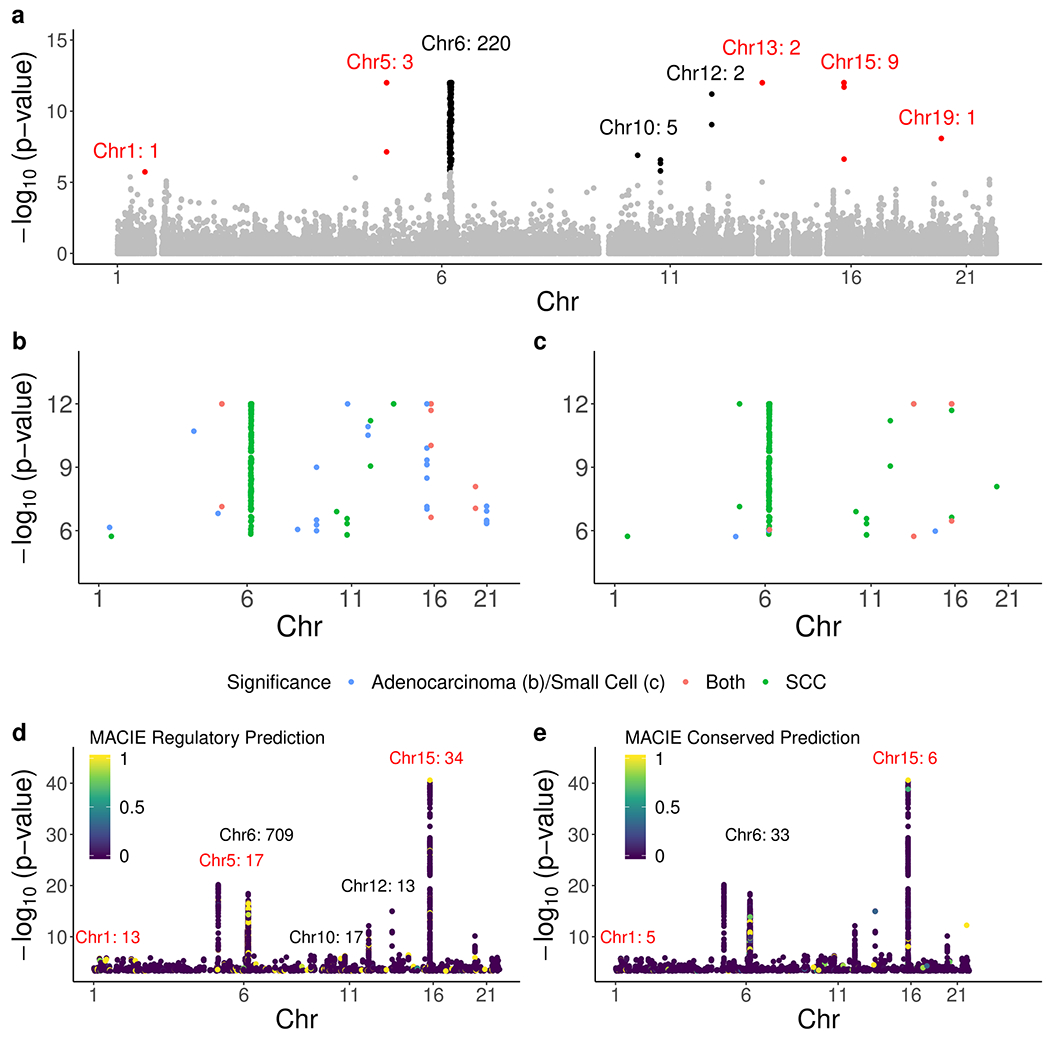
Gene-level Manhattan plots for association with lung cancer and SNP-level Manhattan plots marked with MACIE predictions. a, Gene-level Manhattan plot for lung squamous cell carcinoma. Each point represents one gene. All genes passing the Bonferroni-corrected threshold are colored black (even chromosomes) or red (odd chromosomes), with non-significant genes shown in grey. Labeled numbers note count of significant genes on each chromosome (see Supplementary Table S1 for list of all significant genes). The majority of significant SCC genes fall on chromosome 6. b-c, Gene-level Manhattan plot comparing significant squamous cell carcinoma genes against significant genes in (b) adenocarcinoma and (c) small cell carcinoma. Color corresponds to whether the gene is significant in just one disease or both. Only genes passing the Bonferroni-corrected significance threshold are displayed. d-e, SNP-level Manhattan plot for association with lung cancer where the points are additionally colored according to the MACIE prediction of inclusion in the (d) regulatory class and (e) conserved class. Labeled numbers note count of significant genes on top six chromosomes with most SNPs passing the marginal association threshold (see Materials and Methods) and possessing a MACIE regulatory prediction greater than 0.9 as well as top three chromosomes with most SNPs passing the marginal association threshold and possessing a MACIE conserved prediction greater than 0.9.
3.2. Model-highlighted variants
In total, we found 868 SNPs meeting the marginal association threshold (see Materials and Methods) that were predicted to belong to the regulatory class with probability greater than 0.9 (Figure 1d), and we found 65 SNPs meeting the marginal association threshold that were predicted to belong to the evolutionarily conserved class with probability greater than 0.9 (Figure 1e). While some SNPs highlighted by the model possessed extremely significant p-values, the range of marginal association was also quite large (Supplementary Table S2–S5). SNPs with MACIE predictions close to one generally possessed many elevated functional scores (Figure 2–3), while variants with low predictions were often characterized by less remarkable annotation values. In the following, we highlight some of the strongest model predictions to show how results can supplement previous literature and existing work in a novel and efficient manner.
Figure 2.
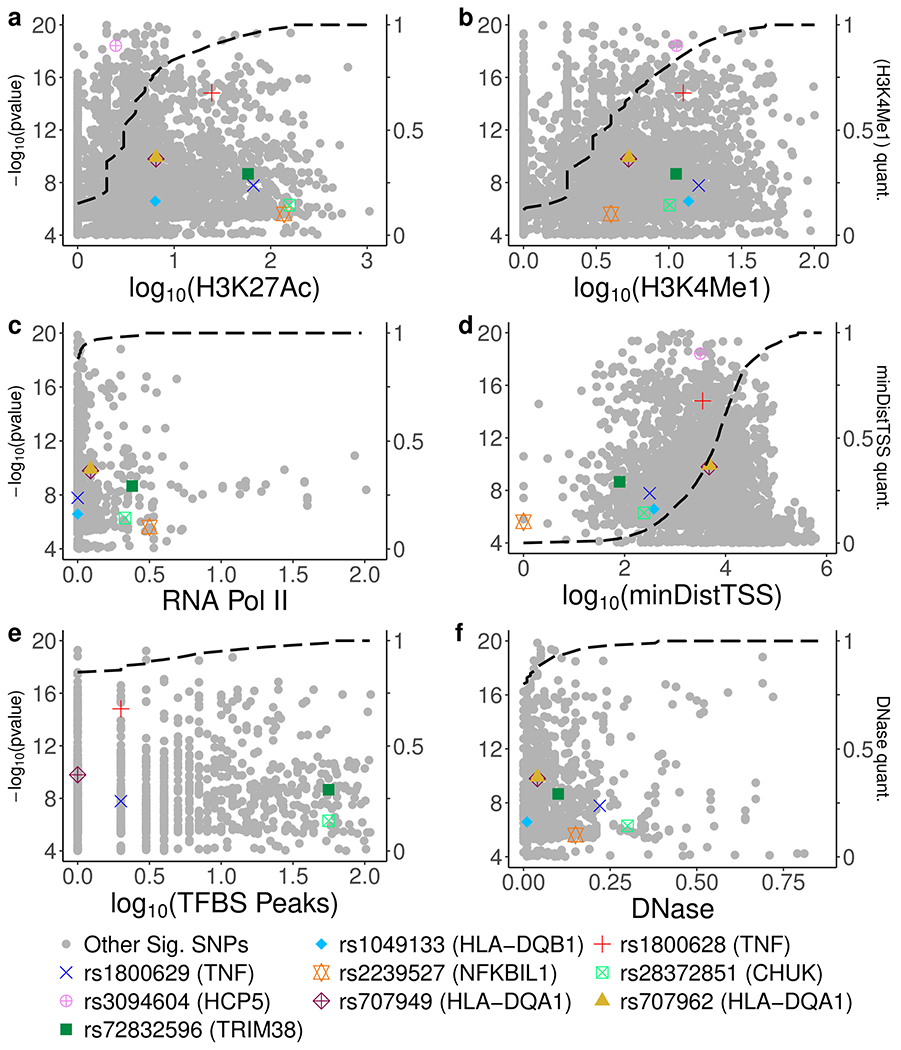
Highly-weighted annotation scores used to calculate MACIE probability of regulatory class in noncoding SNPs. a-f, Highly-weighted (see Supplementary Figure S1 for weights) regulatory annotation scores for SNPs associated with the inflammatory and immune responses, including (a) logarithm of peak H3K27Ac signal, (b) logarithm of peak H3K4Me1 signal, (c) peak RNA polymerase II signal, (d) logarithm of distance to closest transcription start site, (e) logarithm of number of ChIP transcription factor binding site peaks across all cell types and tissues, and (f) peak DNase I signal, all in ENCODE data. Dashed black line denotes an empirical cumulative distribution function for the measure on the x-axis, i.e. the (x,y) position of a point on the line illustrates that a variant scoring x on the given annotation has a value that is greater than y percent (where y is measured according the secondary y-axis on the right) of all variants passing the threshold for marginal association (see Materials and Methods) with SCC (SNPs not discussed are shaded in gray). If a line drawn straight up from the x-axis through a SNP intersects the black line at a value near 1, then that SNP possesses an annotation value greater than most other SNPs demonstrating evidence of association with SCC. The x-axis shows at least the second through ninety-eighth percentile for all annotations. All SNPs mentioned in main text are plotted for completeness, including missense SNPs that are not evaluated using regulatory annotations. Discussed gene is given in parentheses next to each SNP, although a single SNP may be proximal to multiple genes. Not all SNPs possess a value for all annotations; missing values are not plotted but are imputed as described in the original data sources (see Materials and Methods). While SNPs predicted by MACIE as belonging to the regulatory class do not always show the lowest p-values or most extreme scores, they possess an annotation profile containing multiple notable measures that cumulatively contribute to a significant MACIE prediction. SNPs with low regulatory predictions may demonstrate one or a few large scores, but their overall annotation profile for this class consists of mostly unremarkable values.
Figure 3.

Highly-weighted annotation scores used to calculate MACIE probability of evolutionarily conserved class. a-f, Highly-weighted (see Supplementary Figure S1 for weights) conservation annotation scores for SNPs associated with the immune and inflammatory responses, including (a) GERP++ rejected substitution score, (b) phastCons primate score, (c), phastCons placental mammal score, (d) phyloP placental mammal score, (e) phastCons vertebrate score, and (f) phyloP vertebrate score, all plotted against marginal P-value for association with SCC. Dashed black line denotes an empirical cumulative distribution function for the measure on the x-axis. The x-axis shows at least the second through ninety-eighth percentile for all annotations. While SNPs predicted by MACIE as belonging to the conserved class do not always show the lowest p-values or most extreme scores, they possess an annotation profile containing multiple elevated measures that cumulatively contribute to a significant MACIE prediction.
3.3. Variants near genes involved in NF-κB signaling
The NF-κB family possesses a number of different roles in the immune response and other biological processes (Zhang, Lenardo, & Baltimore, 2017), and regulation of NF-κB has been implicated in a wide variety of inflammation-based diseases, including many cancers (Hoesel & Schmid, 2013; Lawrence, 2009; Pikarsky et al., 2004). In their inactive state, NF-κB proteins are bound to the inhibitors of NF-κB (IκB) family (Dolcet, Llobet, Pallares, & Matias-Guiu, 2005). Activation can occur through the IκB kinase (IKK) complex (Karin & Greten, 2005).
TNF superfamily activation is an inducer of NF-κB signaling (Aggarwal, 2003) and is also one of the pathways that can be blocked by canakinumab-induced inhibition of IL-1β (Taniguchi & Karin, 2018). Members of the family including TNF, LTA, and LTB were three neighboring genes highly associated with SCC (all with gene-level p<1 × 10−12 for association with SCC), highlighting a link that has been observed in other inflammatory phenotypes as well (Dretzke et al., 2011; Van Schouwenburg, Rispens, & Wolbink, 2013). Two of the most significant SNPs in the region were rs1800629 and rs1800628, found less than 1 kb upstream and downstream of the TNF transcription start and end sites, respectively.
Based on data from 28 different measures of epigenetic activity, the regulatory class prediction for rs1800629 was greater than 99% (Table 1). This estimate was driven by many elevated annotation scores including, for example, ENCODE experiments demonstrating evidence of open chromatin (Figure 2f). The variant rs1800629 was previously cataloged in curated sources such as ClinVar (Landrum et al., 2014), has been referred to in the literature as TNF-308A, and has seen its adenine substitution repeatedly associated with increased expression of TNF (Karimi, Goldie, Cruickshank, Moses, & Abraham, 2009; Mira et al., 1999). Consistent with the CANTOS hypothesis that inflammation contributes to lung cancer risk, the adenine minor allele was risk-conferring for SCC (OR=1.16, p=1.66 × 10−8), indicating the possibility that rs1800629 was linked with SCC because it increased TNF expression and thus also inflammatory activity.
Table 1.
MACIE model predictions for highlighted SNPs with connections to inflammatory and immune processes.
| Noncoding and Synonymous Variants Model | Nonsynonymous Coding Variants Model | |||||||
|---|---|---|---|---|---|---|---|---|
| SNP | MACIE01a | MACIE10 | MACIE00 | MACIE11 | MACIE01 | MACIE10 | MACIE00 | MACIE11 |
| rs72832596 | 0.99* | 7.1 × 10−30 | 4.5 × 10−25 | 3.4 × 10−5 | - | - | - | - |
| rs2239527 | 5.8 × 10−4 | 1.8 × 10−12 | 2.2 × 10−15 | 0.99* | - | - | - | - |
| rs3094604 | 1.3 × 10−11 | 9.2 × 10−7 | 0.99* | 2.6 × 10−17 | - | - | - | - |
| rs1800629 | 0.99* | 4.4 × 10−14 | 1.1 × 10−11 | 8.6 × 10−3 | - | - | - | - |
| rs1800628 | 2.7 × 10−10 | 2.9 × 10−7 | 0.99* | 1.7 × 10−16 | - | - | - | - |
| rs1049133 | 1.5 × 10−4 | 0.93 | 0.07 | 4.6 × 10−3 | - | - | - | - |
| rs28372851 | 0.99* | 1.9 × 10−10 | 1.5 × 10−6 | 1.8 × 10−4 | - | - | - | - |
| rs707949 | - | - | - | - | 2.2 × 10−3 | 0.62 | 0.14 | 0.23 |
| rs707962 | - | - | - | - | 0.01 | 0.09 | 0.88 | 0.02 |
MACIE01, probability of regulatory class only (noncoding and synonymous variants model) or probability of damaging protein class only (nonsynonymous coding variants model); MACIE10, probability of evolutionarily conserved class only; MACIE00, probability of neither class; MACIE11, probability of both evolutionarily conserved class and regulatory class (Noncoding and Synonymous Variants Model) or probability of both evolutionarily conserved class and damaging protein class (Nonsynonymous Coding Variants Model). See Materials and Methods for more details on models and classes.
Probabilities in MACIE classes that would be normally rounded to 1 are written as 0.99 and denoted with an asterisk to reduce confusion, as the sum of all four classes for a single SNP must sum to 1.
Although rs1800628 (OR=1.28, p=1.54 × 10−15) demonstrated a level of marginal association that was multiple orders of magnitude more significant than its upstream counterpart rs1800629, the MACIE prediction was less than 0.01 for both classes (Table 1) due to a lack of regulatory and evolutionary conservation evidence (Figure 2–3) among the available data. The disparity in p-values was possibly due to the limitations of marginal regressions, for example, rs1800628 may have fallen in strong linkage disequilibrium with another important SNP. rs1800628 may also simply have possessed roles that were not probed by the existing classes as defined in the model.
As a subunit of the IKK complex, CHUK (gene-level p=1.60 × 10−6 for association with SCC) was another key member of the NF-κB signaling cascade (Hacker & Karin, 2006) determined to be significantly associated with lung cancer risk. Although no SNPs in CHUK reached the standard genome-wide significance level of 5 × 10−8, the combination of 26 SNPs with p-values less than 5 × 10−6 pushed the gene-level association between CHUK and SCC to significance. Integrated annotation analysis (Supplementary Figure S3) suggested the signal originated in part from rs28372851 (OR=1.25, p=5.18 × 10−7), a variant located approximately 2 kb upstream of the transcription start site. This SNP was previously predicted (Fishilevich et al., 2017) to lie in an enhancer region of CHUK, and the combined data strongly estimated that rs28372851 belonged to the regulatory class, with a prediction score greater than 0.99 (probability conserved class less than 0.1%). The MACIE regulatory prediction was driven by multiple epigenetic features including high histone modification signal peaks (Supplementary Figure S3b–c). Increased expression of CHUK can further activate NF-κB (Hacker & Karin, 2006).
3.4. Variants near inhibitors of NF-κB signaling
The CANTOS trial illustrated that an IL-1β inhibitor could provide significant therapeutic benefits in lung cancer, so it followed that searching for genetic variation mimicking the function of canakinumab might also uncover SNPs with key protective roles. One such example was identified in TRIM38 (gene-level p=1.26 × 10−12), which was previously observed to inhibit cytokines, including TNF and IL-1β, that activate NF-κB (Hu & Shu, 2017). TRIM38 reached gene-level significance in association with SCC due in part to 15 genome-wide significant SNPs, although many of them fell in high LD (Supplementary Figure S4a). In particular, the SNP rs72832596 (OR=1.21, p=2.02 × 10−9) possessed many elevated annotation values (Supplementary Figure S4b–c) that contributed to a model prediction of greater than 0.99 for inclusion in the class of regulatory SNPs (probability conserved class less than 0.1%, Supplementary Figure S4d–e). Further validation was provided by the GTEx (GTEx Consortium et al., 2013) project, which showed rs72832596 to be an eQTL of TRIM38 in whole blood, as the minor allele was associated with decreased expression of TRIM38. While GTEx discoveries suffer from the same drawbacks as other association results, rely on small sample sizes, and are not available for all SNPs with annotation data, positive eQTL findings do offer some evidence of regulatory function. The minor allele also showed an effect direction (Supplementary Figure S4f) consistent with a positive correlation between inflammation and disease risk. As immune cells may comprise a significant proportion of whole blood, the expression-increasing allele at rs72832596 could possibly be interpreted as a pseudo-dose of canakinumab increasing the level of cytokine inhibition, thus reducing the amount of signaling by IL-1β and associated proteins and leading to a decrease in inflammation.
A final highly significant gene in SCC with connections to cytokine regulatory networks was NFKBIL1 (gene-level p=3.04 × 10−12), which has been shown to demonstrate IκB-like functions (Chiba, Matsuzaka, et al., 2011; Hayden & Ghosh, 2012) and has been linked with, among other inflammation-related phenotypes, rheumatoid arthritis (Chiba, Miyashita, et al., 2011; Okamoto et al., 2003). The integrated annotation data highlighted rs2239527 (OR=1.10, p=2.02 × 10−6), a variant approximately 5 kb upstream (Supplementary Figure S5a) of NFKBIL1 that showed exceptional conservation across primates, mammals, and vertebrates with, for example, large phastCons, phyloP, and GERP++ scores. These scores and others led the model to predict that rs2239527 belonged to both the regulatory and conserved classes with probability approaching 1 (Supplementary Figure S5b–c). Results from multiple other alternative variant scoring systems also highlighted this variant (Supplementary Figure S5d–f).
3.5. HLA risk variants
Although the CANTOS trial demonstrated the therapeutic benefit of targeting cytokine signaling processes, annotation data also implicated other features of inflammatory and immune responses in conferring lung cancer risk. In particular, we previously noted that many of the most significant SNPs and genes associated with SCC fell in the HLA region. More specifically, variants in HLA genes prioritized by the MACIE model (Figure 4a–b) demonstrated a variety of links to the dysregulated immune response that has been observed in lung cancers (Carbone, Gandara, Antonia, Zielinski, & Paz-Ares, 2015; Gandhi et al., 2018; Palucka & Banchereau, 2012).
Figure 4.

MHC region MACIE scores as well as annotation scores and comprehensive variant scores for HLA-DQA1 SNPs. a-b, MACIE regulatory (a) and conserved class (b) scores plotted against p-value for MHC SNPs. SNPs are colored according to their score for the complementary class, i.e. when the regulatory score is on the y-axis, the color corresponds to the conservation score, and when the conservation score is on the y-axis, the color corresponds to either the damaging protein function score (for nonsynonymous SNPs) or regulatory score (other SNPs). A very small p-value is not necessarily indicative of a high MACIE prediction. c-e, Individual SNP annotation scores and comprehensive ratings for significant SNPs in HLA-DQA1 including (c) GERP++ rejected substitutions score, (d) phastCons vertebrate score, (e) CADD PHRED comprehensive score (higher indicates more deleterious), and (f) SIFT protein score (lower indicates more deleterious) plotted against P -value. We only show those SNPs passing the marginal association threshold with SCC (see Materials and Methods). Black dashed line shows the empirical distribution of these measures (using secondary y-axis on right side of plots) for all SNPs across the genome meeting this significance threshold. Variants are colored as described in EIGEN.
Re-imputation and fine-mapping in the HLA selected two independent association signals amidst the strong long-range LD of the HLA (Supplementary Table S6). The association and prediction models converged for HLA-DQB1 variant rs1049133 (unconditional OR=1.18, p=3.45 × 10−8), which ranked as the second strongest independent association signal in the entire HLA and was predicted by the available annotation data to belong to the conserved class with probability 93.4%. HLA-DQB1 (gene-level p=4.10 × 10−10) was significantly associated with SCC risk, and the protein is a receptor found on antigen-presenting cells. In contrast, the most significant SNP in the reimputation, rs3094604, did not show an annotation profile indicating that it belonged to one of the model classes (Table 1). Thus, this example again succinctly demonstrated the added information generated by modeling annotation data. HLA-DQB1 was also notable for its status as the gene with the second largest number of SNPs both passing the marginal association threshold and possessing a MACIE regulatory prediction of greater than 0.9, with 35 such SNPs. HLA-DQA1 (gene-level p<1 × 10−12) held the most such SNPs with 86, and no other MHC-region gene held more than 11.
Additionally, many of the model-prioritized SNPs located outside HLA-DQB1 and HLA-DQA1 still showed strong links to these genes. Across all chromosomes there were only 868 SNPs passing the marginal association threshold and possessing a MACIE regulatory prediction of greater than 0.9, with 613 (71%) falling in the HLA region. Of these 613, 21% were eQTLs of HLA-DQA1 in one or both of blood or lung tissue according to GTEx, and 29% were eQTLs of HLA-DQB1 in one or both of blood or lung tissue according to GTEx (Supplementary Table S7), for a total of 562 eQTL findings across both tissues. Of the 562 SNP-expression pairs, over 99% of the effect directions were oriented such that the expression-lowering allele corresponded to the same allele associated with a marginal increase in SCC risk. The abundance of highly associated and model-prioritized SNPs that were previously validated as HLA eQTLs in blood and lung tissue suggested that many SNPs may contribute to SCC risk by modulating expression of MHC genes.
Many of the significantly associated SNPs in the HLA region were nonsynonymous coding SNPs, and for these variants we calculated a damaging protein substitution class instead of the regulatory class (see Materials and Methods). In general, highly linked nonsynonymous variants tended to demonstrate more variation in conservation class predictions. For example, in HLA-DQA1 the missense variants rs707949 (OR=1.17, p=1.61 × 10−10) and rs707962 (OR=1.18, p=1.23 × 10−10), demonstrated correlation falling just below 1. However (Figure 4c–f), rs707949 earned a higher evolutionarily conserved class prediction of 0.85 compared to 0.12 for rs707962. As with all previous predictions, results should be interpreted with caution as some functionality of nonsynonymous SNPs – for example, their potential to influence the folding of HLA-DQA1 - are roles not directly covered by existing classes in the MACIE model and demonstrate the potential for MACIE modeling of additional attributes once more annotations are available to predict other categories.
3.6. Significantly associated genes in other histologies
Previous reports have described the genetic bases of different lung cancer histologies as highly dissimilar (Wang et al., 2015); thus, important variants may differ and therapeutic strategies such as those used in CANTOS may see efficacy vary by subtype. To investigate whether the variants highlighted above may also contribute to risk of other lung cancer subtypes, we reperformed the initial step of our investigation by conducting separate gene-level analyses with the ILCCO adenocarcinoma and small cell lung cancer cases. Consistent with previous reports, we found that the top genes associated with SCC showed little overlap with the other two histologies. Compared to the 243 genes significant at the Bonferroni-corrected significance level in SCC, only 39 genes reached significance in adenocarcinoma (Fig 1b), even with a larger number of adenocarcinoma cases (11,270 compared to 7,426 for SCC). In particular, the inflammation-related genes TNF, CHUK, TRIM38, and NFKBIL1 did not rank among the top 2,000 most significant genes, and no HLA genes passed the Bonferroni-corrected significance level. This difference could partly be attributable to the increased mutation burden associated with squamous cell carcinoma of the lung. Small cell lung cancer showed even fewer significant genes (Figure 1c) with only 12 passing the Bonferroni threshold, although this low number was likely also impacted by the reduced sample size of 2,170 small cell lung cancer cases. Marginal association strength for previously mentioned SNPs also varied across histology (Supplementary Table S8).
As we previously detailed, association results are limited in scope and interpretation. However, by demonstrating the differences in significant genes between SCC and other histologies, we showed that a similar integrated analysis of adenocarcinoma or small cell lung cancer would likely identify many distinct variants.
4. Discussion
Genetic studies of lung cancer have often focused on extracting information from association analyses using only a few types of data. Yet, for such studies to further disentangle the disease’s complex genetic etiology and provide more translational value, it is becoming increasingly important to integrate and utilize the full scope of available omics information, recorded from a wide range of experimental modalities (Freedman et al., 2011; Karczewski & Snyder, 2018). We attempted to advance this objective and refine results from the recent CANTOS clinical trial by harnessing a variety of diverse genomic datasets to elucidate how genetic variants identified through GWAS may confer lung SCC risk through inflammation and immune networks.
Our analysis identified hundreds of SNPs that demonstrate evidence of association with SCC and possessed a greater than 90% chance of inclusion in at least one of three classes: regulatory, evolutionarily conserved, and protein damaging. These variants represented a filtration of the tens of thousands of highly associated SNPs documented in the largest existing lung cancer GWAS, which, for example, reported a table of over 21,000 highly associated SNPs. Our results demonstrated that the most significant variants at GWAS risk loci may not necessarily be functional SNPs (Supplementary Table 9), and hence it may not be desirable to simply select the most significant variants for follow-up studies. By leveraging functional annotation information, we showed that it is possible to select a much smaller subset of significantly associated SNPs for follow-up. Such work can reduce the number of variants prioritized for additional studies by over an order of magnitude and can translate to large cost and effort savings. In addition, our work markedly contributed to knowledge about the functional mechanisms at each identified risk locus, providing further quantitative and qualitative insight about how individual variants possess roles in the major biological themes leading to lung cancer risk. We next summarize some of these broader findings regarding risk factors for lung cancer.
In further analyzing SNPs selected by the integrative annotation analysis, we found indications that the NF-κB inflammation signaling pathway and HLA-mediated immune responses were key mechanisms in SCC disease progression. Specifically, multi-omics data from dozens of diverse sources demonstrated that a number of annotation-prioritized SNPs were located near genes involved in regulation of NF-κB activation and antigen presentation. Although our study is not the first to link lung cancer with inflammation, NF-κB (Ben-Neriah & Karin, 2011; Taniguchi & Karin, 2018), or the HLA region (Ferreiro-Iglesias et al., 2018), to our knowledge, it is among the first attempts to integrate genome-wide functional annotation data with large-scale lung cancer GWAS findings in pinpointing relevant variants and explaining the specific roles of these substitutions.
The abundance of variants highlighted in this report reinforces the view that lung cancer boasts a highly complex genetic etiology, with large networks of SNPs possessing non-trivial amounts of important behavior. Thus, as a complement to the traditional GWAS individual SNP association analysis, the novel approach outlined in this paper – identifying significant genes through set-based inference and leveraging rich external annotation data to assess the roles of significant SNPs in these genes – could likely be applied to search for genetic determinants of lung cancer risk in other biological systems as well. We emphasize that MACIE offers an unsupervised and highly interpretable tool allowing researchers to move beyond the conventional search for associations and instead determine the biological consequences of genetic variation. When applying the method to different traits, researchers should leverage disease-specific biological knowledge and should ensure that the classes and functional annotations used are relevant for the outcome of interest.
Our manuscript focused on squamous cell carcinoma because the vast majority of ILCCO SCC cases identified as current or past smokers; these subjects likely demonstrated a larger mutation burden and increased pulmonary inflammation due to tobacco smoke (Spitz et al., 2011), and therefore we expected inflammatory mechanisms to display a more direct connection with SCC than other lung cancer subtypes. Comparisons of SCC GWAS results with adenocarcinoma and small cell lung cancer revealed largely disparate profiles of genetically-induced inflammation risk. Hence our findings illustrated the importance of studying disease subtypes and considering differing underlying biological mechanisms when utilizing IL-1β inhibitors as a strategy for treating or preventing lung cancer.
The specific findings discussed in this manuscript offer many avenues for follow-up research. The profusion of functional SNPs indicates that more precise interventions tailored to each patient’s individual genome may be more successful than wide-ranging therapies aimed at the general population. Integrative investigations of other processes critical to lung cancer progression will be essential as well. Further experimental validation will also be necessary to continue refining the performance of data-driven predictions and provide additional evidence linking specific variants to disease.
Supplementary Material
Acknowledgements
We thank the International Lung Cancer Consortium for use of their data and Dr. Song Gao of Sun Yat-sen University Cancer Center for helpful conversations. We would also like to thank the reviewers for their helpful comments that greatly improved the paper.
Funding: National Institutes of Health: R35-CA197449, R01-HL113338, P42-ES016454, U01CA209414, U19CA203654, and T32-ES007142. Christopher I. Amos is a research scholar of the Cancer Prevention Institute of Texas and is partially supported by RR170048.
Appendix A: Fitting the MACIE Model
Suppose for a noncoding or synonymous SNP i and annotation class j (j = 1,2) we observe a set of Kj different functional scores. Let ci = (ci1,i2) denote the vector of unobserved latent functional binary class indicators, with ci1 a binary indicator for SNP i possessing regulatory function and ci2 a binary indicator for SNP i possessing evolutionarily conserved function. Then for j = 1,2 and k = 1, …, Kj, the functional scores yijk are modeled using a GLMM that assumes their means μijk = E(yijk) are linear functions of the latent binary functional status indicators cij and the random effects bijk. Specifically, we assume
where gjk is a canonical link function. We set bij = (bij1, ⋯, bijk)′ = Λjfij with fij ~ MVN(0, I) as a vector of length Pj < Lj to reduce computational complexity, with Λj acting as factor loadings. This step is reasonable because many functional annotations likely attempt to measure the same few underlying variables, and we assume there are Pj of these variables.
The expectation maximization algorithm is used to fit the model. The algorithm, expectation steps, and maximization steps are similar to those previously described (Sammel et al., 1997) for latent variable models with mixed discrete and continuous outcomes. Expectations that cannot be evaluated in closed form are taken with Gauss-Hermite quadrature, and score equations that cannot be solved in closed form are updated with a one-step Fisher scoring procedure (Sammel et al., 1997). Given the fitted model parameters and a new SNP i′ with corresponding annotation scores yi′1 and yi′2, the probability p(ci′ = z|yi′1, yi′2) is the MACIE score, where z = (z1, z2) ∈ {0,1} × {0,1}. The same model fitting procedure is used for nonsynonymous coding SNPs, except we utilize a different set of annotations and training data (see Material and Methods). Other sets of annotations may be used to probe different functional classes as well.
Footnotes
Data Availability Statement
Portions of the data used in this study were generated by the International Lung Cancer Consortium and are available at the database of Genotypes and Phenotypes (dbGaP) under accession phs001273.v1.p1 and phs000876.v1.p1. This is the same data that was published in McKay et al., 2017. Additional ILCCO data is available upon request from https://oncoarray.dartmouth.edu. The data are not publicly available due to privacy restrictions. Annotation data was downloaded as described in Methods. All MACIE code, scores, and annotation data used in this paper are available at www.github.com/ryanrsun/lungCancerMACIE.
References
- 1000 Genomes Project Consortium. (2015). A global reference for human genetic variation. Nature, 526(7571), 68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal BB (2003). Signalling pathways of the TNF superfamily: a double-edged sword. Nature Reviews: Immunology, 3(9), 745–756. doi: 10.1038/nri1184 [DOI] [PubMed] [Google Scholar]
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, … Searle SM (2016). The Ensembl gene annotation system. Database (Oxford), 2016. doi: 10.1093/database/baw093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Neriah Y, & Karin M (2011). Inflammation meets cancer, with NF-kappaB as the matchmaker. Nature Immunology, 12(8), 715–723. doi: 10.1038/ni.2060 [DOI] [PubMed] [Google Scholar]
- Bosse Y, & Amos CI (2018). A decade of GWAS results in lung cancer. Cancer Epidemiology, Biomarkers & Prevention, 27(4), 363–379. doi: 10.1158/1055-9965.Epi-16-0794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone DP, Gandara DR, Antonia SJ, Zielinski C, & Paz-Ares L (2015). Non–small-cell lung cancer: role of the immune system and potential for immunotherapy. Journal of Thoracic Oncology, 10(7), 974–984. doi: 10.1097/JTO.0000000000000551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, & Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. doi: 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Matsuzaka Y, Warita T, Sugoh T, Miyashita K, Tajima A, … Kimura M (2011). NFKBIL1 confers resistance to experimental autoimmune arthritis through the regulation of dendritic cell functions. Scandinavian Journal of Immunology, 73(5), 478–485. doi: 10.1111/j.1365-3083.2011.02524.x [DOI] [PubMed] [Google Scholar]
- Chiba T, Miyashita K, Sugoh T, Warita T, Inoko H, Kimura M, & Sato T (2011). IkappaBL, a novel member of the nuclear IkappaB family, inhibits inflammatory cytokine expression. FEBS Letters, 585(22), 3577–3581. doi: 10.1016/j.febslet.2011.10.024 [DOI] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, & Batzoglou S (2010). Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol, 6(12), e1001025. doi: 10.1371/journal.pcbi.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolcet X, Llobet D, Pallares J, & Matias-Guiu X (2005). NF-kB in development and progression of human cancer. Virchows Arch, 446(5), 475–482. doi: 10.1007/s00428-005-1264-9 [DOI] [PubMed] [Google Scholar]
- Dretzke J, Edlin R, Round J, Connock M, Hulme C, Czeczot J, … Meads C (2011). A systematic review and economic evaluation of the use of tumour necrosis factor-alpha (TNF-α) inhibitors, adalimumab and infliximab, for Crohn’s disease. Health Technology Assessment (Winchester, England), 15(6), 1. doi: 10.3310/hta15060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Beesley J, French JD, & Dunning AM (2013). Beyond GWASs: illuminating the dark road from association to function. American Journal of Human Genetics, 93(5), 779–797. doi: 10.1016/j.ajhg.2013.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature, 489(7414), 57–74. doi: 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreiro-Iglesias A, Lesseur C, McKay J, Hung RJ, Han Y, Zong X, … Li Y (2018). Fine mapping of MHC region in lung cancer highlights independent susceptibility loci by ethnicity. Nature Communications, 9(1), 1–12. doi: 10.1038/s41467-018-05890-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, … Cohen D (2017). GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford), 2017. doi: 10.1093/database/bax028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman ML, Monteiro AN, Gayther SA, Coetzee GA, Risch A, Plass C, … Mills IG (2011). Principles for the post-GWAS functional characterization of cancer risk loci. Nature Genetics, 43(6), 513–518. doi: 10.1038/ng.840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, … Powell SF (2018). Pembrolizumab plus chemotherapy in metastatic non–small-cell lung cancer. New England Journal of Medicine, 378(22), 2078–2092. doi: 10.1056/NEJMoa1801005 [DOI] [PubMed] [Google Scholar]
- GTEx Consortium, Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, … Young N (2013). The genotype-tissue expression (GTEx) project. Nature Genetics, 45(6), 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker H, & Karin M (2006). Regulation and function of IKK and IKK-related kinases. Sci STKE, 2006(357), re13. doi: 10.1126/stke.3572006re13 [DOI] [PubMed] [Google Scholar]
- Hayden MS, & Ghosh S (2012). NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes & Development, 26(3), 203–234. doi: 10.1101/gad.183434.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesel B, & Schmid JA (2013). The complexity of NF-κB signaling in inflammation and cancer. Molecular Cancer, 12(1), 86. doi: 10.1186/1476-4598-12-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M-M, & Shu H-B (2017). Multifaceted roles of TRIM38 in innate immune and inflammatory responses. Cellular & Molecular Immunology, 14(4), 331–338. doi: 10.1038/cmi.2016.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YF, Gulko B, & Siepel A (2017). Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nature Genetics, 49(4), 618–624. doi: 10.1038/ng.3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, McCallum K, Xu B, & Buxbaum JD (2016). A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nature Genetics, 48(2), 214–220. doi: 10.1038/ng.3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, … de Bakker PI (2013). Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One, 8(6), e64683. doi: 10.1371/journal.pone.0064683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, & Snyder MP (2018). Integrative omics for health and disease. Nature Reviews: Genetics, 19(5), 299–310. doi: 10.1038/nrg.2018.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M, Goldie LC, Cruickshank MN, Moses EK, & Abraham LJ (2009). A critical assessment of the factors affecting reporter gene assays for promoter SNP function: a reassessment of− 308 TNF polymorphism function using a novel integrated reporter system. European Journal of Human Genetics, 17(11), 1454–1462. doi: 10.1038/ejhg.2009.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, & Greten FR (2005). NF-kappaB: linking inflammation and immunity to cancer development and progression. Nature Reviews: Immunology, 5(10), 749–759. doi: 10.1038/nri1703 [DOI] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, & Maglott DR (2014). ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research, 42(Database issue), D980–985. doi: 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T (2009). The nuclear factor NF-κB pathway in inflammation. Cold Spring Harbor Perspectives in Biology, 1(6), a001651. doi: 10.1101/cshperspect.a001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yung G, Zhou H, Sun R, Li Z, Liu Y, Ionita-Laza I, Lin X (2020). A multi-dimensional integrative scoring framework for predicting functional regions in the human genome. Harvard University Department of Biostatistics Technical Report. [Google Scholar]
- Liu X, Wu C, Li C, & Boerwinkle E (2016). dbNSFP v3. 0: A one‐stop database of functional predictions and annotations for human nonsynonymous and splice‐site SNVs. Human Mutation, 37(3), 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, … Amos CI (2017). Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nature Genetics, 49(7), 1126–1132. doi: 10.1038/ng.3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira J-P, Cariou A, Grall F, Delclaux C, Losser M-R, Heshmati F, … Riché F (1999). Association of TNF2, a TNF-α promoter polymorphism, with septic shock susceptibility and mortality: a multicenter study. Journal of the American Medical Association, 282(6), 561–568. doi: 10.1001/jama.282.6.561 [DOI] [PubMed] [Google Scholar]
- Ng PC, & Henikoff S (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research, 31(13), 3812–3814. doi: 10.1093/nar/gkg509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Makino S, Yoshikawa Y, Takaki A, Nagatsuka Y, Ota M, … Inoko H (2003). Identification of I kappa BL as the second major histocompatibility complex-linked susceptibility locus for rheumatoid arthritis. American Journal of Human Genetics, 72(2), 303–312. doi: 10.1086/346067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palucka K, & Banchereau J (2012). Cancer immunotherapy via dendritic cells. Nature Reviews Cancer, 12(4), 265–277. doi: 10.1038/nrc3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, … Ben-Neriah Y (2004). NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature, 431(7007), 461–466. doi: 10.1038/nature02924 [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, & Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Research, 20(1), 110–121. doi: 10.1101/gr.097857.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, & Sander C (2011). Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Research, 39(17), e118. doi: 10.1093/nar/gkr407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich SS, & Concannon P (2015). Role of Type 1 Diabetes-Associated SNPs on Autoantibody Positivity in the Type 1 Diabetes Genetics Consortium: Overview. Diabetes Care, 38 Suppl 2, S1–3. doi: 10.2337/dcs15-2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, … Anker SD (2017). Antiinflammatory therapy with canakinumab for atherosclerotic disease. New England Journal of Medicine, 377(12), 1119–1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, & Glynn RJ (2017). Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet, 390(10105), 1833–1842. doi: 10.1016/s0140-6736(17)32247-x [DOI] [PubMed] [Google Scholar]
- Rogers MF, Shihab HA, Mort M, Cooper DN, Gaunt TR, & Campbell C (2018). FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics, 34(3), 511–513. doi: 10.1093/bioinformatics/btx536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammel MD, Ryan LM, & Legler JM (1997). Latent variable models for mixed discrete and continuous outcomes. Journal of the Royal Statistical Society: Series B (Statistical Methodology), 59(3), 667–678. [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, … Haussler D (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Research, 15(8), 1034–1050. doi: 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz MR, Gorlov IP, Amos CI, Dong Q, Chen W, Etzel CJ, … Zhang D (2011). Variants in inflammation genes are implicated in risk of lung cancer in never smokers exposed to second-hand smoke. Cancer Discovery, 1(5), 420–429. doi: 10.1158/2159-8290.CD-11-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, & Lin X (2019). Genetic Variant Set-Based Tests Using the Generalized Berk–Jones Statistic With Application to a Genome-Wide Association Study of Breast Cancer. Journal of the American Statistical Association, (ePub ahead of print), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Ogata H, Nishigaki R, Broide DH, & Karin M (2010). Tobacco smoke promotes lung tumorigenesis by triggering IKKβ-and JNK1-dependent inflammation. Cancer Cell, 17(1), 89–97. doi: 10.1016/j.ccr.2009.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam V, Patel N, Turcotte M, Bosse Y, Pare G, & Meyre D (2019). Benefits and limitations of genome-wide association studies. Nature Reviews: Genetics, 20(8), 467–484. doi: 10.1038/s41576-019-0127-1 [DOI] [PubMed] [Google Scholar]
- Taniguchi K, & Karin M (2018). NF-kappaB, inflammation, immunity and cancer: coming of age. Nature Reviews: Immunology, 18(5), 309–324. doi: 10.1038/nri.2017.142 [DOI] [PubMed] [Google Scholar]
- Timofeeva MN, Hung RJ, Rafnar T, Christiani DC, Field JK, Bickeboller H, … Landi MT (2012). Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Human Molecular Genetics, 21(22), 4980–4995. doi: 10.1093/hmg/dds334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schouwenburg PA, Rispens T, & Wolbink GJ (2013). Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nature Reviews Rheumatology, 9(3), 164. doi: 10.1038/nrrheum.2013.4 [DOI] [PubMed] [Google Scholar]
- Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, & Yang J (2017). 10 Years of GWAS Discovery: Biology, Function, and Translation. American Journal of Human Genetics, 101(1), 5–22. doi: 10.1016/j.ajhg.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walser T, Cui X, Yanagawa J, Lee JM, Heinrich E, Lee G, … Dubinett SM (2008). Smoking and lung cancer: the role of inflammation. Proceedings of the American Thoracic Society, 5(8), 811–815. doi: 10.1513/pats.200809-100TH [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wei Y, Gaborieau V, Shi J, Han Y, Timofeeva MN, … Houlston RS (2015). Deciphering associations for lung cancer risk through imputation and analysis of 12,316 cases and 16,831 controls. Eur J Hum Genet, 23(12), 1723–1728. doi: 10.1038/ejhg.2015.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, & Lin X (2011). Rare-variant association testing for sequencing data with the sequence kernel association test. American Journal of Human Genetics, 89(1), 82–93. doi: 10.1016/j.ajhg.2011.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung GYH (2016). Statistical methods for analyzing genetic sequencing association studies. (PhD thesis). Harvard University, Harvard University; Retrieved from https://dash.harvard.edu/bitstream/handle/1/33493313/YUNG-DISSERTATION-2016.pdf?sequence=4&isAllowed=y [Google Scholar]
- Zhang Q, Lenardo MJ, & Baltimore D (2017). 30 years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell, 168(1-2), 37–57. doi: 10.1016/j.cell.2016.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.