

Abstract
Although an abundance of drug candidates exists which are aimed at the remediation of central nervous system (CNS) disorders, the utility of some are severely limited by their inability to cross the blood brain barrier. Potential drug delivery systems such as the Angiopep family of peptides have shown modest potential; however, there is a need for novel drug delivery candidates that incorporate peptidomimetics to enhance the efficiency of transcytosis, specificity, and biocompatibility. Here, we report on the first in vitro cellular uptake and cytotoxicity study of a peptidomimetic, cationic peptide, L57. It binds to cluster 4 of the low-density lipoprotein receptor-related protein 1 (LRP1) receptor which is expressed in numerous cell types, such as brain endothelial cells. We used early-passage-number brain microvascular endothelial cells and astrocytes harvested from rat pup brains that highly express LRP1, to study the uptake of L57 versus Angiopep-7 (A7). Uptake of L57 and A7 showed a concentration-dependent increase, with L57 being taken up to a greater degree than A7 at the same concentration. Additionally, peptide uptake in LRP1-deficient PEA 10 cells had greatly reduced uptake. Furthermore, L57 demonstrated excellent cell viability versus A7, showing promise as a potential drug delivery vector for CNS therapeutics.
Keywords: Drug delivery system(s), Polymeric drug delivery system(s), Peptide transporter(s), Peptide(s), Blood-brain barrier (BBB), Endothelial, Cell culture, Lipoprotein(s), Receptor(s)
Introduction
The delivery of therapeutics into the brain for the remediation of specific central nervous system (CNS) disorders is impeded by their suboptimal blood-brain barrier (BBB) penetration. Macromolecules, such as proteins and nucleic acids, are prevented from paracellular and transcellular transport by specialized CNS barriers such as brain capillary endothelial cells of the BBB. This formidable challenge is one key reason why these disorders remain largely untreated, despite the ready availability of effective candidate drugs to treat them.1 As such, there is a growing need for effective modes of delivery to efficiently transport therapeutics across the BBB.
Over the last decade, there has been a heightened interest in neuro-pharmaceutics for CNS disorders, as reviewed in.2,3 Potential advantages of neuropharmaceuticals include high specificity, potency, minimal cross-reactivity, low immunogenicity and relative ease of modification to optimize their pharmacokinetic properties. Consequently, the pharmaceutical industry has made great strides in the development of peptide therapeutics, with over 70 therapeutic peptides on the market and more than 150 in clinical studies.3 Therapeutics targeted to the brain can be delivered through various routes of entry such as intravenous or subcutaneous injection, or intranasal administration.2,3 However, not all peptides cross the BBB.2 More recent approaches have incorporated the use of targeted nanoparticles to deliver therapeutic peptides to the brain.4 Small cationic peptides, in particular, have been used in a Trojan horse-like approach to remediate brain disorders such as using a SynB1 protegrin derivative to deliver benzylpenicillin across the BBB5 and treating ischemic brain damage using Tat-NR2B9c and pTat-PDZ1–2.6–8
Two well-characterized modes of peptide transport across the BBB are adsorptive-mediated transcytosis (AMT) and receptor-mediated transcytosis (RMT). Via the AMT mode, poly-lysine and poly-arginine peptides are translocated as efficiently as Tat peptides.9 Kamei et al.10 concluded that efficient transduction is achieved for a peptide with eight sequential arginines (RRRRRRRR, R8) in a mouse microvascular endothelial model based upon bEnd.3 cells. Using bEnd.3 cells in vitro and rat brains in vivo, they concluded that R8 was safe and effective for the non-covalent delivery of certain hydrophilic macromolecules such as the peptide drug insulin, which led us to select this peptide as a positive control for our studies in the main cells of interest, rat brain microvascular endothelial cells (BMVECs).
As for the RMT mode, the cationic peptides Angiopep-2 (A2, TFFYGGSRGKRNNFKTEEY)11,12 and its analogue Angiopep-7 (A7, TFFYGGSRGRRNNFRTEEY) cross the BBB in mouse models.12,13 A2 and A7 are peptides derived from the Kunitz domain of the low-density lipoprotein receptor-related protein 1 (LRP1) endocytic receptor12 which is expressed in many cell types including brain capillary endothelial cells and is enriched at the lipid rafts. LRP1 is also implicated in numerous biological processes such as transcytosis of macromolecules such as receptor-associated proteins and matrix proteins.14,15 In 2008, the uptake of ANG1005, an A2-Paclitaxel drug conjugate, was found to exhibit high brain accumulation and antitumor efficacy in both in vitro and in vivo animal models of brain and lung cancer, compared to the unconjugated Paclitaxel drug.16 This has shown promising results in Phase II clinical trials.17 Two other A2 conjugates, ANG1007 and ANG1009, using the anticancer drugs doxorubicin and etoposide, respectively, have also been developed.18 These drugs were effective in inducing cytotoxicity against human cancer cell lines such as NCI–H460 lung carcinoma. A non-covalent mode of delivery has also been developed which carries immunoglobulins to the brain using the K16ApoE synthetic peptide.19
The success of A2 peptide-drug conjugates in preclinical studies opened the door to the discovery of the first artificial LRP1 ligand, the cationic peptide L57, which specifically binds to cluster 4 of the LRP1 receptor. This peptide, developed by Sakamoto et al.,13 is less basic than the well-studied cell-penetrating peptide R8; thus, there may be a lower risk of non-specific binding. In their study, L57 showed significant BBB permeability and higher LRP1 binding affinity over both A2 and A7. Moreover, A7 was chosen as the reference peptide for their in vivo studies as it exhibited better BBB permeability than A2. This factored into the decision to use A7 in our study to compare it against L57. Importantly, Sakamoto et al. did not study the toxicity of the A7 and L57 peptides in vitro (in a suitable BBB model such as BMVECs) or in vivo. Furthermore, they did not provide an assessment of the levels of cellular uptake of these peptides in various cells in vitro - which was addressed in the present study.
In this study, we used primary rat BMVECs to visualize the intracellular accumulation and localization of the fluorescent L57, A7 and R8 peptides in vitro through fluorescence microscopy. The principal reason we chose these cells was because LRP1 receptors are found to be highly expressed in brain microvessels in mouse pups.20 Furthermore, BMVECs are the brain’s first structural barrier to the CNS; thus these cells provide a suitable in vitro model to compare uptake and potential cytotoxicity of peptide carriers. We quantified the uptake of various concentrations of these peptides and rein-forced the observations made by Sakamoto et al.13 that L57 peptides exhibited better cell uptake than A7, with our observations being made in BMVECs compared to their brain perfusion assay in mice. We also compared the cellular uptake of these peptides in two other cell types, LRP1-expressing brain astrocytes (another key component of the BBB) and PEA 10 cells (LRP1-deficient cells),21 with varying, but still encouraging, results. We also conducted cytotoxicity testing, which was previously unstudied. Our results showed that L57 has greater biocompatibility than both A7 and R8 peptides in BMVECs and PEA 10 cells. In summary, our results suggest that the L57 peptide could be a promising new carrier to enable more effective delivery of CNS therapeutics to the brain.
Materials and Methods
Materials
The fluorenylmethoxycarbonyl (Fmoc)-protected amino acids, Rink Amide AM resin, 1-hydroxybenzotriazole (HOBt), O-(benzo-triazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) and O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) were purchased from Aapptec (Louisville, KY, USA). N,N-diisopropylethylamine (DIPEA) was acquired from Sigma-Aldrich (St. Louis, MO, USA). Fluorescein (disodium salt) and diethyl ether were purchased from Fisher Scientific (Hampton, NH, USA). N,N-dimethylformamide (DMF), N, N′-dicyclohexylcarbodiimide (DCC), 99%, isopropyl alcohol (IPA), triisopropylsilane (TIPS), N-hydroxysuccinimide (NHS), 1, 2-thanedithiol (EDT) and trifluoroacetic acid, 99% was from MilliporeSigma (St. Louis, MO, USA). Anisole and thioanisole were purchased from TCI America (Boston, MA, USA). Dimethyl sulfoxide (DMSO, cell culture grade) was bought from MP Biomedical (Irvine, CA, USA).
Peptide Synthesis
L57 (FITC-Ahx-TWPKHFDKHTFYSILKLGKH-CONH2) was custom-made by Biomatik (Cambridge, ON, Canada). Angiopep-7 (A7, TFFYGGSRGRRNNFRTEEY-CONH2) and R8 (RRRRRRR-CONH2) were synthesized by Fmoc-based solid phase peptide synthesis methodology (Table 1). First, Rink amide AM resin was swollen with DMF. For Fmoc deprotection, 25% piperidine in DMF was added to the resin, under nitrogen bubbling for 20–25 min. The resin was washed with DMF (3×), IPA (2×) and DMF (3×). For each amino acid coupling, 2 equivalents (eq.) of HOBt, HBTU (2.5 eq.) and DIPEA (6 eq.) was added to 2.5 eq. of Fmoc-amino acid in DMF. Nitrogen was bubbled for 3–4 h and resin was washed with DMF (3×), IPA (2×) and DMF (3×). A7 was also coupled to two Beta-alanine spacer amino acids using the same strategy. Both A7 and R8 peptides were then fluorescently tagged with fluorescein at the C-terminal. This was done by first activating the fluorescein with NHS (1.3 eq. based on fluorescein), DCC (1.3 eq. based on fluorescein), HATU (1.5 eq. based on resin), and DIPEA (5 eq. based on fluorescein) in DMF for 3 h while stirring. The activated fluorescein-NHS ester was then transferred to the main vessel together with 3 eq. of DIPEA for another 3 h. Final fluorescently-tagged A7 (Fl-A7) was then washed with DCM (3×) and cleaved from the resin with a cocktail of TFA (98% v/v), TIPS (1% v/v) and distilled water (1% v/v). Cleavage of fluorescently-tagged R8 (Fl-R8) was carried out with a cocktail of TFA (90% v/v), thioanisole (5% v/v), EDT (3% v/v), and anisole (2% v/v). The cleaved mixture was concentrated under vacuum and precipitated dropwise in cold ether, centrifuged, frozen in water and then lyophilized. Peptide molecular weight was confirmed by matrixassisted laser desorption ionization time-of-flight (MALDI TOF) mass spectrometry with a Bruker UltrafleXtreme MALDI TOF/TOF mass spectrometer at Louisiana State University, Baton Rouge, USA (See Supplementary Figs. S1–S3).
Table 1.
Amino Acid Sequences of the Peptides Used in This Study.
| Peptides | Sequencesa,b |
|---|---|
| Fl-L57 | FITC-Ahx-TWPKHFDKHTFYSILKLGKH-CONH2 |
| Fl-Angiopep-7 | Fluorescein-AA-TFFYGGSRGRRNNFRTEEY-CONH2 |
| Fl-R8 | Fluorescein-RRRRRRRR-CONH2 |
All sequences contain the L-forms of the amino acids.
A in second sequence refers to beta-alanine spacer.
Animal Care and Harvesting of Primary rat BMVECs and Astrocytes
Pregnant Sprague Dawley dams were maintained in a 12-h day-light cycle and provided with food and water ad libitum. Rat pups remained with the dam until sacrificed by cervical disarticulation between post-natal day 1–3. All procedures were conducted according to a protocol approved by the Louisiana Tech University Animal Care and Use Committee.
Cells were isolated from the cortex of Sprague–Dawley pups (2–4 days) and cultured in F12 Nutrient Mixture containing 10% Fetal Bovine Serum, 10% Horse Serum, and other components (heparin, glutamine, NaHCO3), as previously described.22,23 The glial cells thus obtained are purified by treating them with 5.51 µM of Puromycin (MilliporeSigma, St. Louis, MO, USA) and cultured in rat endothelial cell growth medium with endothelial growth factors to obtain BMVECs alone.24,25 Rat brain astrocytes were obtained by sub culturing the glial cells in Ham’s F–12K medium containing 5% Horse serum and 5% FBS and washing off the overlaying microglial cells a few times before being subcultured.
Cells were plated at a density of 10,000 cells per well onto poly-l-lysine-coated 96-well black-walled cell culture plates (Corning Costar), and grown at 37 °C, in 5% CO2 in humidified incubators. Cells were grown for 24 h then used for cell uptake and cytotoxicity studies at 50%–70% confluency. Cells were tested at early passage numbers (not tested after passage 9) so that BBB model characteristics could be retained.26
Mouse embryonic fibroblasts (PEA 10) that are heterozygous for LRP receptor expression were purchased from ATCC (Manassas, VA, USA). These were used to study the effect of LRP deficiency on the uptake of these LRP1-targeted peptide ligands.21
Visualization of Peptide Uptake and Intracellular Distribution
Fl-A7 was initially dissolved in a minimal volume of DMSO, 20 µl, and then this solution was diluted in endothelial cell media to prepare working concentrations of 10, 30 and 100 µM. Fl-L57 and Fl-R8 were dissolved in distilled water and then in endothelial media. Fl-L57 was prepared at 3, 10 and 30 µM. Fl-R8, our positive control, was prepared at 30 µM concentration. Each concentration was tested in two separate passages of cells, in triplicate wells of 96-well plates. For each test, six wells containing cells with no peptide were used as negative controls. Of these, three wells with media were used as control wells for the Fl-L57, and three wells contained 0.5% DMSO in media (equivalent amount of DMSO as the 100 µM Fl-A7) control wells for the Fl-A7. For each peptide concentration, the media in the corresponding well was exchanged with the peptide media. After peptides were added to the respective wells, the plates were incubated for 4 h at 37 °C, in 5% CO2 in humidified incubators. After incubation, the cells were washed twice with warm phosphate buffered saline (PBS) and once with phenol red-free RPMI media and then fixed for 10 min with paraformaldehyde.27 After fixation, the cells were washed once with PBS, and then stored for imaging in PBS. Some cells were also stained with DAPI nuclear counterstain to visualize the individual cell nuclei.
Epifluorescence images of FITC-labeled peptides and DAPI were acquired at three random locations at 200× magnification in both fluorescence (FITC: 470 nm excitation, 530 nm emission; DAPI: 358 nm excitation, 461 nm emission) and phase contrast images using a Leica DMI 6000B inverted microscope with a DP71 color camera (Leica Microsystems, Wetzlar, Germany) and at consistent equipment settings and controls. A few time-lapse image sets of live cells were acquired to show dynamics of uptake. Images were saved as TIFF files for analysis.
Quantification of Fluorescence Intensity
Mean fluorescence intensity from FITC emission was used to compare levels of peptide uptake. Cells in TIFF images were analyzed by first setting the background fluorescence for each image as a threshold value using FIJI software (ImageJ)28,29 and then selecting regions of interest using random assignment, but taking care to avoid regions with undissolved fluorescent peptide aggregates. Finally, the corrected mean fluorescence intensity was calculated in relative fluorescence units (rfu) according to the formula:
Corrected mean fluorescence intensity (in rfu) = mean fluorescence of cell - mean fluorescence of background.
For each treatment, two experimental trials were conducted with triplicate wells, and the corrected mean fluorescence intensity was calculated for 20 cells in each well, for a total of 120 cells. For each well incubated with peptide, this value was normalized to the mean of the vehicle control wells to obtain the normalized fluorescence intensity (NFI). There was no significant difference between cells incubated in the vehicle with DMSO versus without it; thus, these results were grouped.
MTT Assay
The MTT assay, which measures cytotoxicity by comparing cellular metabolic activity, was performed as described previously by Vives et al.30 Cells were cultured in poly-l-lysine-coated 96-well black-walled cell culture plates in humidified incubators at 5% CO2 and 37 °C temperature for 24 h. The cells were then treated with varying concentrations of the Fl-L57, Fl-A7 or Fl-R8 peptides. Control wells for Fl-L57 and Fl-R8 received media with no peptide. Control wells for Fl-A7 received media with no peptide, but with DMSO equivalent to that in the highest concentration (100 µM) of Fl-A7. After 4 h, MTT (1.25 mg/ml in RPMI media) was added and incubated for 1.5 h at 37 °C. The resulting formazan product was dissolved in 90% isopropyl alcohol and absorbance was read at 570 nm. The absorbance values of peptide-treated wells were normalized to the mean absorbance of wells incubated with the respective vehicle.
Statistical Analysis
Statistical comparisons for the cellular uptake assay were performed using one-way ANOVA with Tukey’s test (comparisons between groups) for the fluorescence intensity data and Dunnett’s (compared to control) post hoc correction for the cytotoxicity assay. For comparisons between two groups, two-tailed t-tests were performed. All statistical tests were performed using GraphPad Prism version 8.3.0 for Windows (GraphPad Software, San Diego, California USA, www.graphpad.com). Results were considered statistically significant when p < 0.05. Values are represented as mean ± standard error of the mean (SEM) of n = 6 wells for each treatment condition.
Results
Internalization of FITC-Labeled Peptides
Cells incubated with Fl-L57 (Figs. 1A, 2a and 3a) displayed diffuse staining throughout the cells for all three cell types. Incubation of both types of primary brain cells with 100 µM Fl-A7 exhibited high uptake in (Figs. 1g and 2g) with very low uptake by PEA 10 cells (Fig. 3g). In BMVECs, the fluorescence was saturated when incubated with 30 µM Fl-L57; therefore, these results were excluded from analysis for this cell type. BMVECs showed the highest uptake efficiency for the Fl-L57 and Fl-A7 peptides, followed by astrocytes and then PEA 10 cells.
Fig. 1.
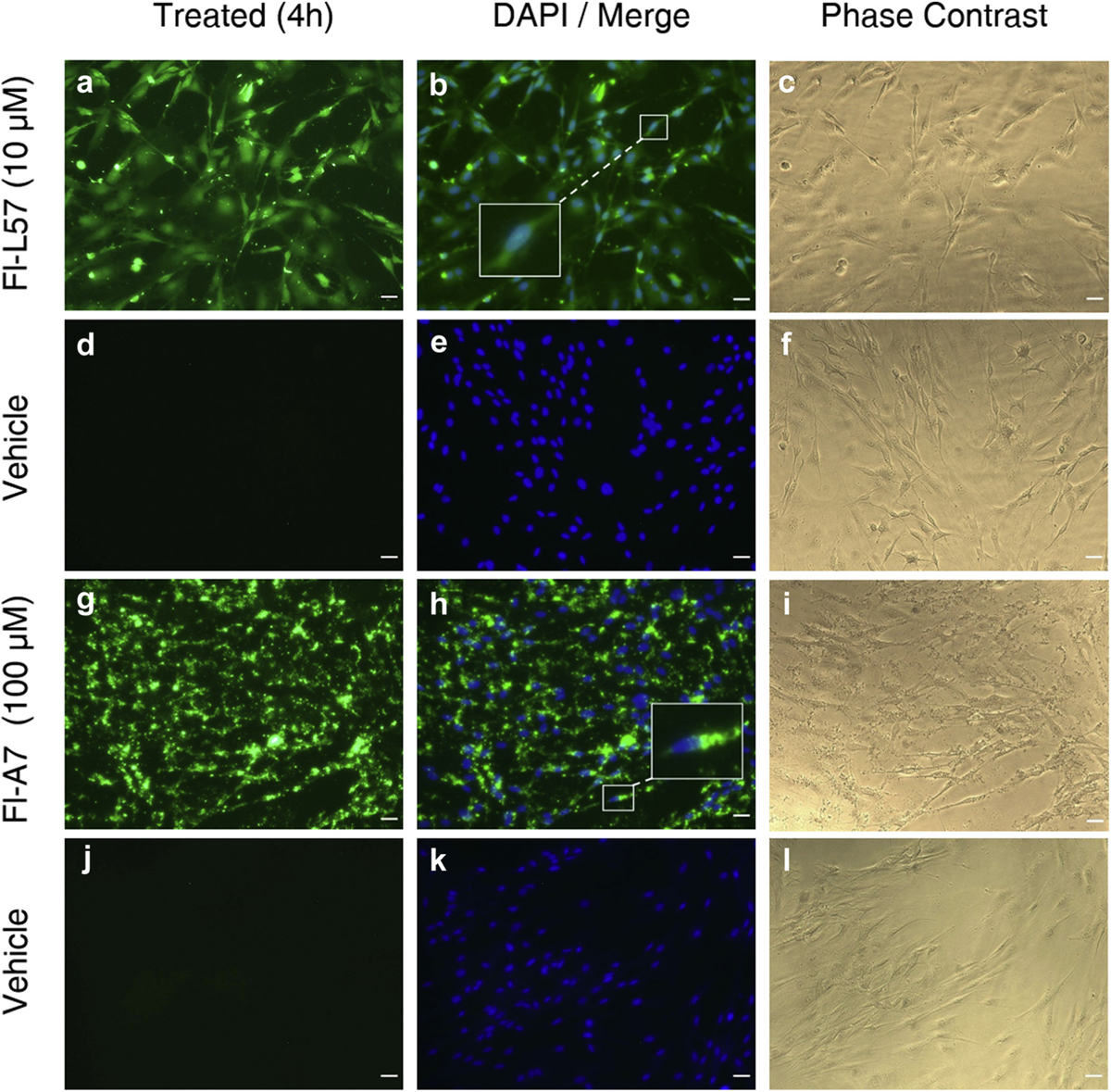
Uptake of fluorescein-labeled L57 (Fl-L57) and A7 peptides (Fl-A7) in cultured rat brain microvascular endothelial cells (BMVECs). Panels a–f: Representative image set of cells incubated with Fl-L57 (10 µM) (panels a–c) and vehicle (panels d–f) for 4 h. A. BMVECs after incubation with Fl-L57 (green). b. Image in A (green) was merged with image of DAPI-counterstained nuclei (blue). c. Phase contrast image of the same cells shown in a-b. d. BMVECs incubated with vehicle. e. Image in d was merged with image of DAPI-counterstained nuclei (blue). f. Phase contrast image of the same cells shown in d-e. Panels d–l: Representative image set of cells incubated with Fl-A7 (100 µM) (panels g–i) and vehicle (panels j–l) for 4 h. g. BMVECs after incubation with Fl-A7 (green). h. Image in g (green) was merged with image of DAPI-counterstained nuclei (blue). i. Phase contrast image of the same cells shown in g-h. j. BMVECs incubated with vehicle. k. Image in j was merged with image of DAPI-counterstained nuclei (blue). l. Phase contrast image of the same cells shown in j-k. Scale bar represents 30 µm.
Fig. 2.
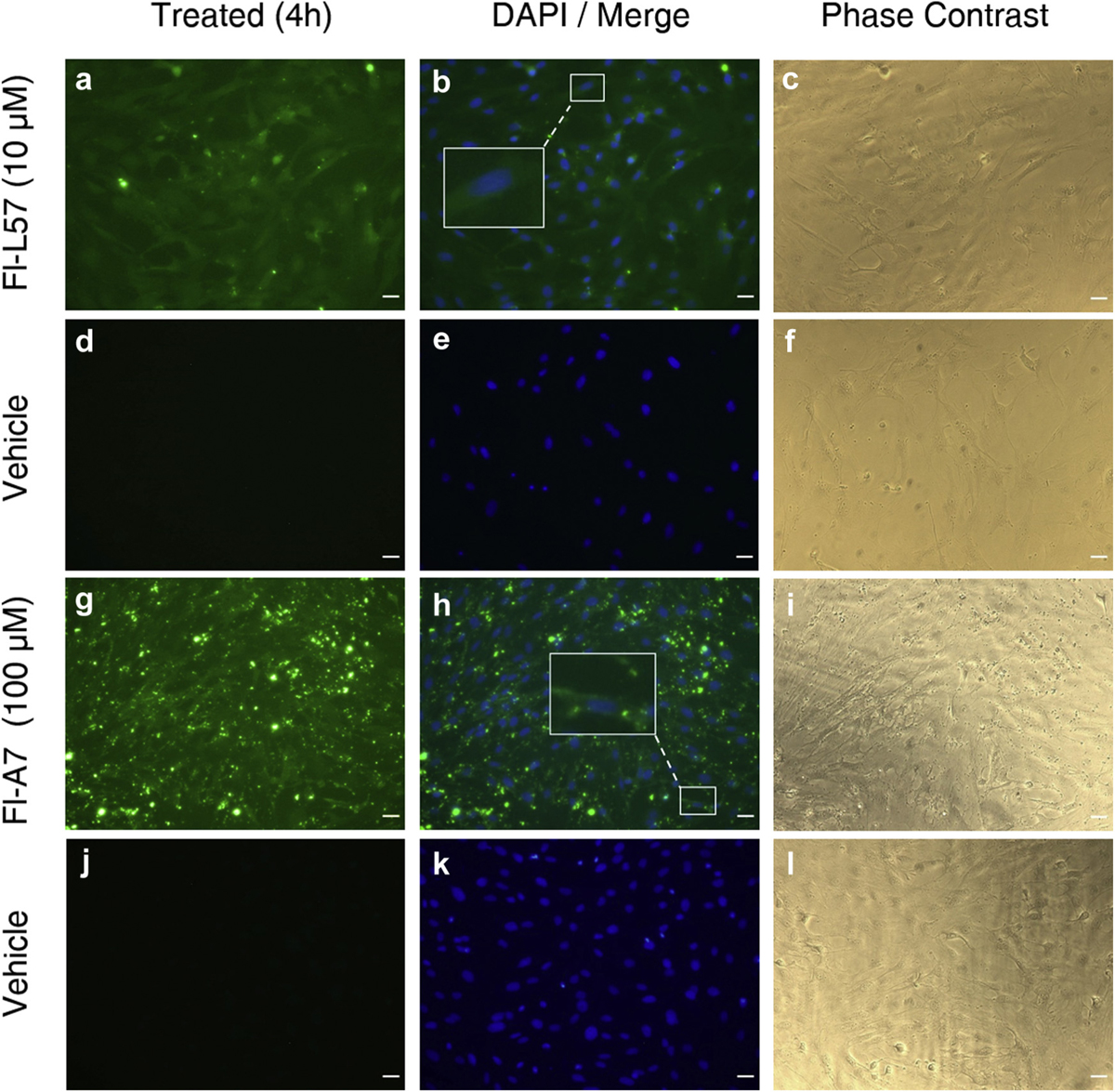
Uptake of fluorescein-labeled L57 (Fl-L57) and A7 peptides (Fl-A7) in cultured rat brain astrocytes. Panels a–f: Representative image set of cells incubated with Fl-L57 (10 µM) (panels a–c) and vehicle (panels d–f) for 4 h. a. Astrocytes after incubation with Fl-L57 (green). b. Image in a (green) was merged with image of DAPI-counterstained nuclei (blue). c. Phase contrast image of the same cells shown in a-b. d. Astrocytes incubated with vehicle. e. Image in d was merged with image of DAPI-counterstained nuclei (blue). f. Phase contrast image of the same cells shown in d-e. Panels g–l: Representative image set of cells incubated with Fl-A7 (100 µM) (panels g–l) and vehicle (panels j–l) for 4 h. g. Astrocytes after incubation with Fl-A7 (green). h. Image in g (green) was merged with image of DAPI-counterstained nuclei (blue). i. Phase contrast image of the same cells shown in g-h. j. Astrocytes incubated with vehicle. k. Image in j was merged with image of DAPI-counterstained nuclei (blue). l. Phase contrast image of the same cells shown in j-k. Scale bar represents 30 µm.
Fig. 3.
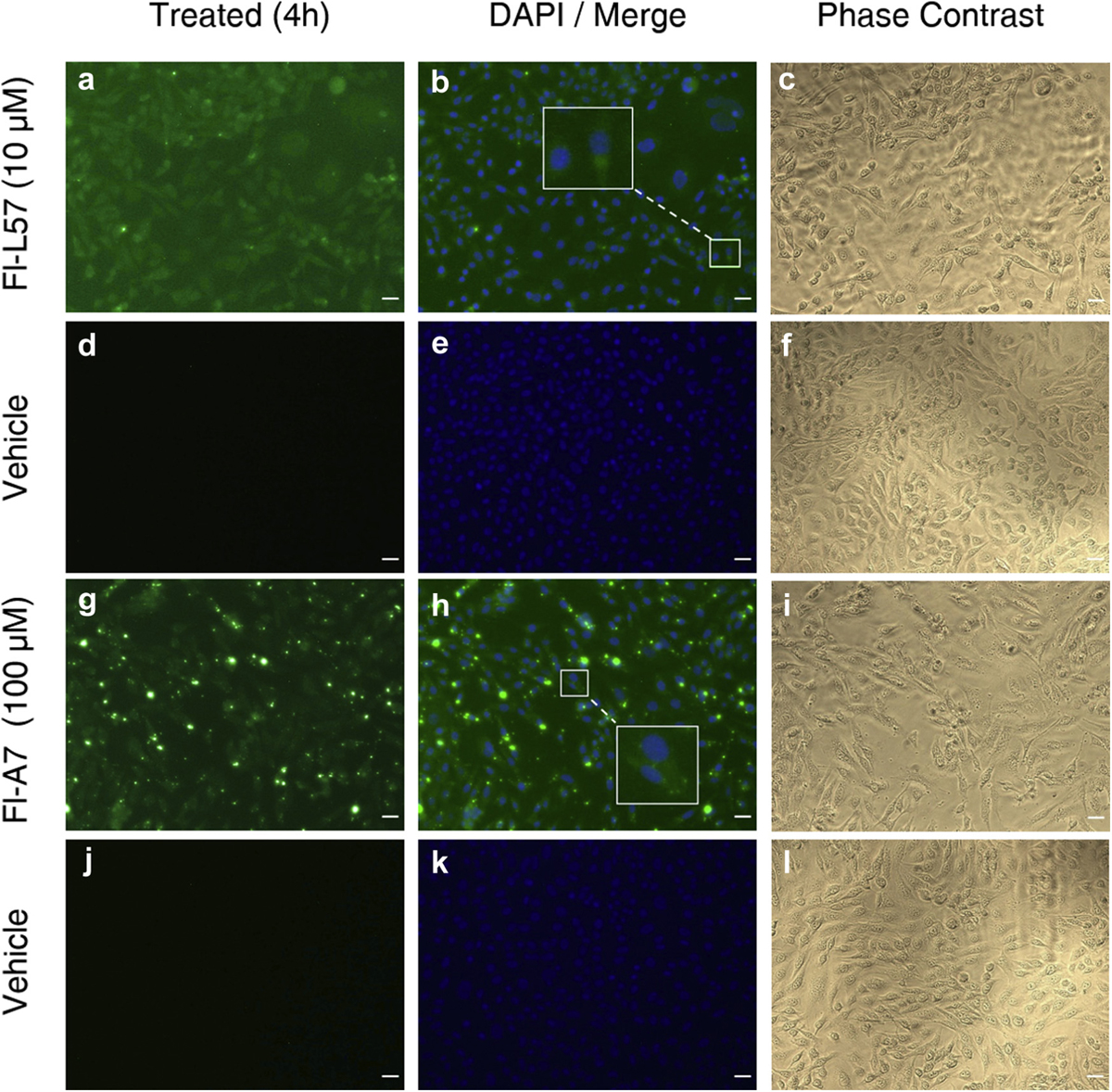
Uptake of fluorescein-labeled L57 (Fl-L57) and A7 peptides (Fl-A7) in cultured PEA 10 cells. Panels a–f: Representative image set of cells incubated with Fl-L57 (10 µM) (panels a–c) and vehicle without peptide (panels d–f) for 4 h. a. PEA 10 cells after incubation with Fl-L57 (green). b. Image in a (green) was merged with image of DAPI-counterstained nuclei (blue). c. Phase contrast image of the same cells shown in a-b. d. PEA 10 cells incubated with vehicle. e. Image in d was merged with image of DAPI-counterstained nuclei (blue). f. Phase contrast image of the same cells shown in d-f. Panels g–l: Representative image set of cells incubated with Fl-A7 (100 µM) (panels g–i) and vehicle (panels j–l) for 4 h. g. PEA 10 cells after incubation with Fl-A7 (green). h. Image in g (green) was merged with image of DAPI-counterstained nuclei (blue). i. Phase contrast image of the same cells shown in g-h. j. PEA 10 cells incubated with vehicle. K. Image in J was merged with image of DAPI-counterstained nuclei (blue). l. Phase contrast image of the same cells shown in j-k. Scale bar represents 30 µm.
Another remarkable observation, conducted in live BMVECs (Fig. 4), as well as in astrocytes (images not shown), was the rapid time course of Fl-L57 uptake. We observed cells taking up the peptide in real time with numerous cells exhibiting increases in fluorescence within the first minute of incubation. As seen in Fig. 4, some cells initially exhibited only perinuclear localization (Fig. 4a, ars) that redistributed over time throughout the cell (Fig. 4b, ars). This redistribution was observed as a gradual, and sometimes rapid, increase in fluorescence spreading radially throughout the cytoplasm of the cells (Supplementary Time-Lapse Videos, V1 and V2). However, this phenomenon was not observed in the PEA 10 cells.
Fig. 4.
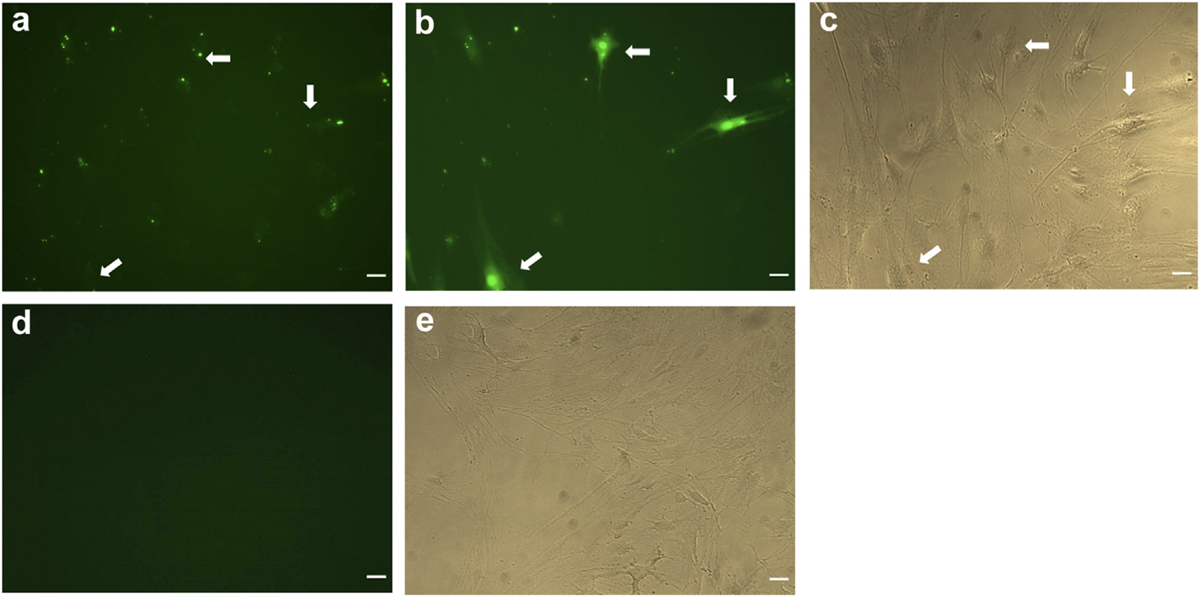
Time course images show uptake and distribution of Fl-L57 in BMVECs. Representative images of live cells incubated with Fl-L57 at 10 µM over the first 1 min of incubation (panels a–c) and vehicle (panels d–e). a. Image of cell culture well immediately after adding Fl-L57. b. Visualization of Fl-L57 in the same well at 1 min after exposure shows uptake by BMVECs. c. Phase contrast image of the same cells in a-b. Supplementary Time-Lapse Videos (V1 and V2) show the dynamics of peptide uptake in cells. d. BMVECs without peptide. e. Phase contrast image of the same cells shown in d. Scale bar represents 30 µm.
Quantification of Cellular Uptake of the Peptides
NFI was used to compare the relative levels of cellular uptake of Fl-L57 and Fl-A7. Fl-R8 also exhibited significant uptake and, therefore, was useful as a positive control in BMVECs. Internalization of Fl-A7 was concentration-dependent with 100 µM Fl-A7 producing significantly more uptake than 10 µM (p < 0.0001 for all cell types) and 30 µM Fl-A7 (p = 0.013 for BMVECs, p = 0.0002 for astrocytes and p = <0.0001 for PEA 10 cells, Fig. 5). Likewise, in the BMVECs and astrocytes, cellular uptake of Fl-L57 scaled with concentration. Incubation with 10 µM Fl-L57 resulted in an approximately two-fold higher NFI versus incubation with 3 µM of the peptide in all cell types (Fig. 5). Overall, PEA 10 cells had markedly lower uptake efficiency compared to BMVECs, and much lower uptake compared to astrocytes at higher concentrations (Fig. 5).
Fig. 5.
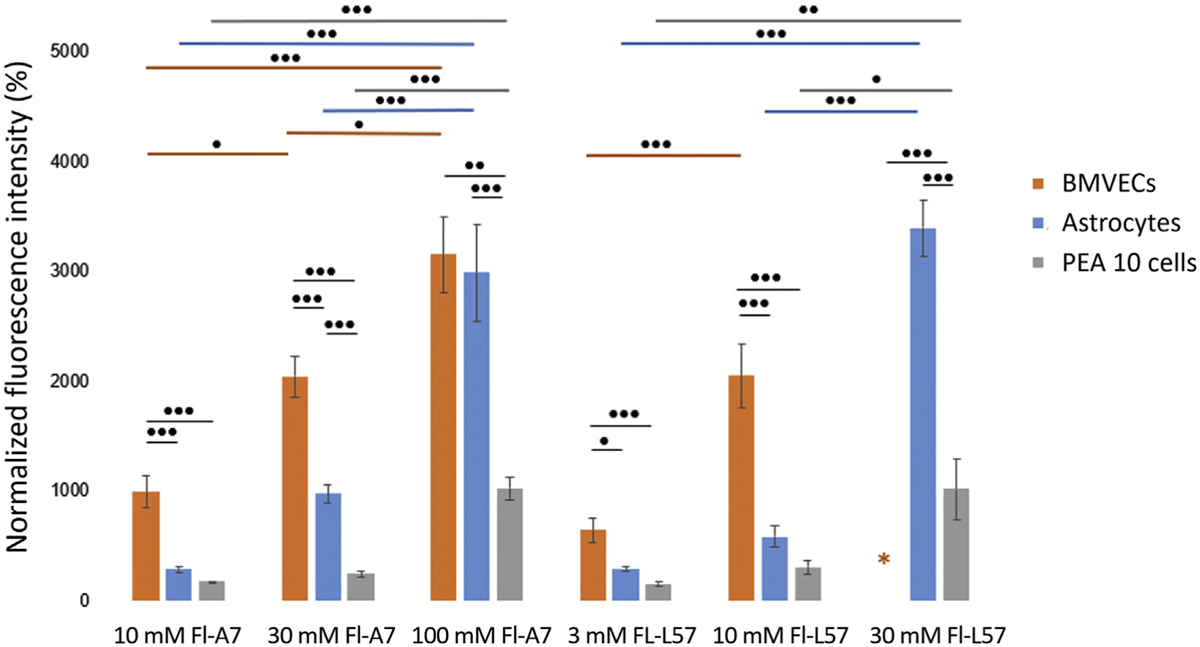
Peptide uptake is concentration-dependent across the three cell lines tested – BMVECs, astrocytes and PEA 10 cells. Normalized fluorescence intensity (NFI) was used to compare peptide uptake. Cells (10,000/well) were incubated with various concentrations of the three peptides Fl-A7, Fl-R8 and Fl-L57 for 4 h and then fixed. The fluorescence intensity of the peptides was quantified using ImageJ/FIJI software and then normalized to controls. Fl-R8 images were taken at a different exposure; therefore, comparison is not shown. Twenty cells were randomly selected per well (n = 6 wells), for a total of 120 cells per peptide concentration (error bars denote mean ± SEM). In BMVECs, the fluorescence was saturated when incubated with 30 µM Fl-L57; therefore, these results were excluded from analysis (orange asterisk). Statistical analysis was performed using one-way ANOVA with Tukey’s post hoc comparisons between all groups. For comparisons between two groups, two-tailed t-tests were performed. Horizontal colored bars represent comparisons of peptide concentrations within the same cell lines (color corresponds to cell type). Black bars represent comparisons of peptide concentrations between different cell lines. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine the difference in uptake efficiency between Fl-L57 and Fl-A7, we compared them at the same concentration (10 µM) across all the cell lines tested. Consistent with an in situ study,13 Fl-L57 demonstrated significantly better uptake overall. We observed significant differences between NFI of 10 µM Fl-L57 and 10 µM Fl-A7 when incubated with BMVECs (p = 0.0082) and astrocytes (p = 0.013) but not PEA 10 cells (p = 0.075). In addition, significant differences were seen between NFI of 30 µM Fl-L57 and 30 µM Fl-A7 when incubated with astrocytes (p < 0.0001) and with PEA 10 cells (p = 0.018).
Cytotoxicity of Peptides
The MTT assay indicated high cell viability in both BMVECs and PEA 10 cells when incubated with Fl-L57, and with Fl-R8 in BMVECs (Fig. 6a). In contrast, viability for BMVECs incubated with Fl-A7 significantly decreased in a concentration-dependent manner (10 µM, p = 0.015), 30 µM (p = 0.0001) and 100 µM Fl-A7 (p < 0.0001). A similar result was seen in PEA 10 cells (Fig. 6b), with the viability of Fl-L57 being comparable to the control condition at all concentrations, in contrast to lower viability for 30 µM Fl-A7 (p = 0.017) and 100 µM Fl-A7 (p = 0.0002).
Fig. 6.
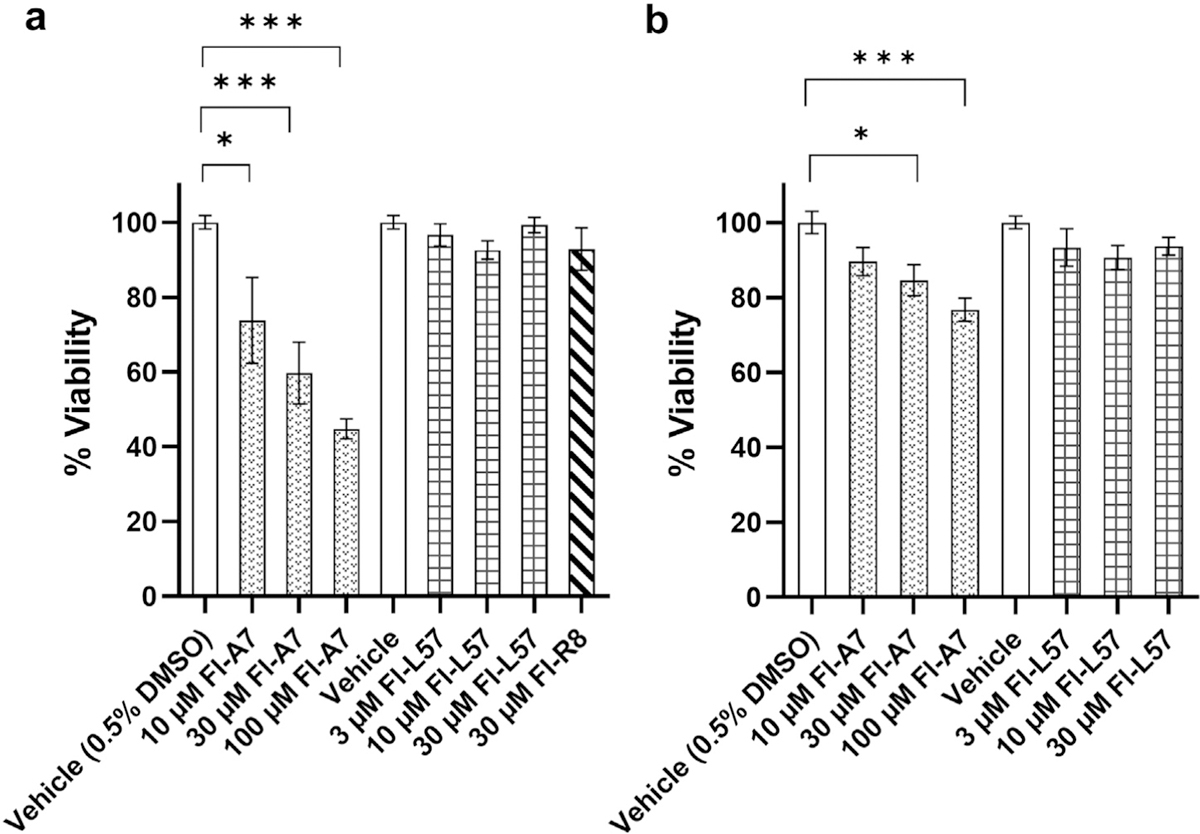
Viability of cells after incubation with Fl-A7 versus Fl-L57. BMVECs (a) and PEA 10 cells (b) were seeded at 10,000 cells per well and then incubated with Fl-A7, FL-L57, or Fl-R8 (in BMVECs only) peptides, or vehicle, for 4 h. An MTT assay was carried out to assess cellular metabolic activity. The y-axis represents % cell viability which is normalized to viability of vehicle-treated cells (n = 6 wells, error bars are mean ± SEM). For the Fl-A7 peptides, media with 0.5% DMSO concentration (corresponding to that in 100 µM Fl-A7) was used as the vehicle. Statistical analysis was performed using one-way ANOVA with Dunnett’s post hoc correction. A. Normalized percent viability in BMVECs (ANOVA F (7, 38) = 12.8, p < 0.0001); B. Normalized percent viability in PEA 10 cells using MTT assay (ANOVA F (6, 35) = 14.5, p = 0.0017). For pairwise comparisons, *p < 0.05, ***p < 0.001.
Discussion
The primary goals of this study were to assess the efficiency of uptake and cytotoxicity of the novel artificial LRP1-binding peptide L57 in primary BMVECs, and to compare this to previously-studied cationic peptides, A7 and R8, which are known to cross the BBB.10,13 Cellular uptake and cytotoxicity of Fl-L57 were evaluated in vitro for the first time in BMVECs, which are crucial for uptake of this peptide due to their high expression of the LRP1 receptor and location in the microvasculature of the brain. BMVECs are a suitable BBB model owing to their enhanced tight junction complexity and monolayer tightness, which demonstrates their ability to better recapitulate characteristics of the brain than animal-derived cell lines and immortalized human BMVECs.31 Astrocyte end feet cover 90–98% of the surface area of the brain microvasculature.2 Thus, astrocytes were also used to evaluate the uptake efficiency of these peptides. The BMVECs and astrocytes used in this study were not from cell lines, they were early passages from freshly harvested cells from rat pups; therefore, they very closely mimic BBB phenotype. Our study provides a foundation for more extensive cellular uptake studies in BBB models using this peptide. A key observation made in this study is the significantly higher uptake of Fl-L57 versus Fl-A7 at the same concentration (10 µM) in all three cell types tested, suggesting that when compared to other LRP1 binding peptides, we may see similar or improved drug delivery of CNS therapeutics complexed with L57.
The third cell type we employed was the genetically-modified PEA 10 cell line, which is heterozygous for the LRP1 gene. It has one wild-type and one disrupted allele, and it expresses approximately half of the wild-type level of LRP1.21 This cell line was selected to determine whether the LRP-1 receptor is required for endocytosis of Fl-L57 and Fl-A7. The greatly reduced uptake of Fl-L57 and Fl-A7 by LRP1-deficient PEA 10 cells compared to BMVECs and astrocytes that have high levels of LRP expression strongly suggests that these peptides are internalized via a LRP1-mediated mechanism.
The mode of internalization of L57 was through interaction with the cluster 4 (Ser3332–Asp3779) of LRP1 receptor, as studied by Sakamoto et al., using diversified peptide libraries displayed on T7 phages.13 The rationale for our choices of R8 and A7 were manifold. R8 was chosen as the positive control for comparing peptide internalization, due to its ability to efficiently internalize and even transcytose through the BBB when used both in vitro32 and in vivo.10 On the other hand, A7 is an LRP1-binding peptide like L57; although, it lacks sequence homology to L57. The two positive charges on the A7 peptide and LRP1 binding mechanism are mainly responsible for the transcytosis of A7 across the BBB.12,13 The L57 peptide is similarly cationic and also binds to the LRP1 receptor. Thus, it can be postulated that the same factors are responsible for the transcytosis of L57, in addition to the advantages of possibly having a reduced risk of non-specific binding and being stable in mouse plasma.13 Another group used A7 as a negative control due to its lack of in vivo transcytosis.12 However, Sakamoto concluded that A7 demonstrated “comparable or slightly better BBB permeability than Angiopep-2” when used as a reference peptide in their in situ brain perfusion study.13 Therefore, these conflicting results compelled us to use A7 in this preliminary study to gather further information on the utility of this peptide for delivery to the brain.
The MTT assay suggested excellent viability of cells incubated with Fl-L57, which further supports its potential for drug delivery studies in CNS diseases. The metabolic activities of the cells were normal for Fl-L57 and Fl-R8 (Fig. 5a) and suggests that these peptides would not have adverse effects on the metabolism of cells at the concentrations studied. In contrast, Fl-A7 induced cytotoxicity in a concentration-dependent manner in both cell types (Fig. 5a and b). This was unexpected because A7 has been studied before; however, its cytotoxicity had not been evaluated in non-cancerous cells prior to the present study. Given its greater internalization and the lack of cytotoxicity, it appears that L57 would be more suitable as a delivery vector than A7, particularly if higher concentrations of the peptide are required. Furthermore, our results suggest caution when using A7 for intravenous delivery of drugs.
Our real-time observation of spreading fluorescence seen in live BMVECs (Fig. 4 and Supplementary Videos V1 and V2) and astrocytes incubated with Fl-L57 warrants further study. Notably, this phenomenon was not observed for Fl-A7 or Fl-R8 in live cells. Future studies could provide valuable insight into the length of time needed for BMVECs and astrocytes to internalize the peptide and the intracellular route of internalized Fl-L57. The ability to monitor trafficking of Fl-L57 in these cell models will enable future studies of the peptide when conjugated or complexed with therapeutic compounds.
In summary, we tested the first artificial LRP1-binding peptide, L57, developed by Sakamoto et al. in suitable in vitro models, rat BMVECs and astrocytes, and discovered that this peptide has concentration-dependent uptake. Reduced uptake in LRP1-deficient PEA 10 cells strongly suggests that Fl-L57 and Fl-A7 uptake is mediated by LRP1-receptors. The Fl-L57 peptide also shows excellent cell viability, suggesting it could be an efficient and safe vector for CNS therapeutics. The unexpected, yet exciting real-time fluorescence phenomenon we observed for internalization of the L57 peptide can be used to gain insight into cellular uptake and trafficking of this peptide in vitro, whether alone or conjugated to potential therapeutics. Testing different modes of administration and uptake of a variety of therapeutics could be used to further evaluate L57’s pharmacokinetic properties. Further experiments are necessary to compare the BBB-penetrating ability of Fl-A7 vs. Fl-L57 at higher concentrations and determine the optimal dose of each peptide.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
Funding was provided by the Edmonson/Crump Professorship [Louisiana Board of Regents Support Fund Endowed Professorships] to TAM, a Louisiana Board of Regents Grant [LEQSF(2018–21)-RD-A-13] and a Louisiana Biomedical Research Network Grant [NIH 5P20GM103424 LSU, Subcontract No. PO-0000002131] to SP, and a Louisiana Tech University, College of Engineering and Science Graduate Scholarship to JPR.
Footnotes
Declarations of interest: none.
Appendix A. Supplementary Data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.xphs.2020.09.019.
References
- 1.Neuwelt E, Abbott NJ, Abrey L, et al. Strategies to advance translational research into brain barriers. Lancet Neurol 2008;7(1):84–96. [DOI] [PubMed] [Google Scholar]
- 2.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today 2007;12(1–2): 54–61. [DOI] [PubMed] [Google Scholar]
- 3.McGonigle P Peptide therapeutics for CNS indications. Biochem Pharmacol 2012;83(5):559–566. [DOI] [PubMed] [Google Scholar]
- 4.Papademetriou I, Vedula E, Charest J, Porter T. Effect of flow on targeting and penetration of angiopep-decorated nanoparticles in a microfluidic model blood-brain barrier. PLoS One 2018;13(10):e0205158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rousselle C, Clair P, Temsamani J, Scherrmann JM. Improved brain delivery of benzylpenicillin with a peptide-vector-mediated strategy. J Drug Target 2002;10(4):309–315. [DOI] [PubMed] [Google Scholar]
- 6.Aarts M, Liu Y, Liu L, et al. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 2002;298(5594):846–850. [DOI] [PubMed] [Google Scholar]
- 7.Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol Ther 2004;104(1):29–45. [DOI] [PubMed] [Google Scholar]
- 8.Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci 2004;27(2):85–131. [DOI] [PubMed] [Google Scholar]
- 9.Park J, Ryu J, Kim KA, et al. Mutational analysis of a human immunodeficiency virus type 1 Tat protein transduction domain which is required for delivery of an exogenous protein into mammalian cells. J Gen Virol 2002;83(Pt 5):1173–1181. [DOI] [PubMed] [Google Scholar]
- 10.Kamei N, Yamaoka A, Fukuyama Y, Itokazu R, Takeda-Morishita M. Non-covalent strategy with cell-penetrating peptides to facilitate the brain delivery of insulin through the blood-brain barrier. Biol Pharm Bull 2018;41(4):546–554. [DOI] [PubMed] [Google Scholar]
- 11.Demeule M, Currie JC, Bertrand Y, et al. Involvement of the low-density lipoprotein receptor-related protein in the transcytosis of the brain delivery vector angiopep-2. J Neurochem 2008;106(4):1534–1544. [DOI] [PubMed] [Google Scholar]
- 12.Bertrand Y, Currie JC, Demeule M, et al. Transport characteristics of a novel peptide platform for CNS therapeutics. J Cell Mol Med 2010;14(12):2827–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakamoto K, Shinohara T, Adachi Y, Asami T, Ohtaki T. A novel LRP1-binding peptide L57 that crosses the blood brain barrier. Biochem Biophys Rep 2017;12:135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JA, Casalini T, Brambilla D, Leroux JC. Presumed LRP1-targeting transport peptide delivers beta-secretase inhibitor to neurons in vitro with limited efficiency. Sci Rep 2016;6:34297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, Strickland DK. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev 2008;88(3):887–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regina A, Demeule M, Che C, et al. Antitumour activity of ANG1005, a conjugate between paclitaxel and the new brain delivery vector Angiopep-2. Br J Pharmacol 2008;155(2):185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumthekar P, Tang SC, Brenner AJ, et al. ANG1005, a brain penetrating peptide-drug conjugate, shows activity in patients with breast cancer with leptomeningeal carcinomatosis and recurrent brain metastases. Clin Cancer Res 2020;26:2789–2799. [DOI] [PubMed] [Google Scholar]
- 18.Che C, Yang G, Thiot C, et al. New Angiopep-modified doxorubicin (ANG1007) and etoposide (ANG1009) chemotherapeutics with increased brain penetration. J Med Chem 2010;53(7):2814–2824. [DOI] [PubMed] [Google Scholar]
- 19.Sarkar G, Curran GL, Mahlum E, et al. A carrier for non-covalent delivery of functional beta-galactosidase and antibodies against amyloid plaques and IgM to the brain. PLoS One 2011;6(12):e28881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000;106(12):1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Willnow TE, Herz J. Genetic deficiency in low density lipoprotein receptor-related protein confers cellular resistance to Pseudomonas exotoxin A. Evidence that this protein is required for uptake and degradation of multiple ligands. J Cell Sci 1994;107(3):719–726. [PubMed] [Google Scholar]
- 22.Scoggin JL, Tan C, Nguyen NH, et al. An enzyme-based electrochemical biosensor probe with sensitivity to detect astrocytic versus glioma uptake of glutamate in real time in vitro. Biosens Bioelectron 2019;126:751–757. [DOI] [PubMed] [Google Scholar]
- 23.Karekar N, Karan A, Khezerlou E, et al. Self-assembled metal-organic biohybrids (MOBs) using copper and silver for cell studies. Nanomaterials (Basel) 2019;9(9):1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saleh MY, Prajapati N, DeCoster MA, Lvov Y. Tagged halloysite nanotubes as a carrier for intercellular delivery in brain microvascular endothelium. Front Bioeng Biotechnol 2020;8:451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abbott NJ, Hughes CC, Revest PA, Greenwood J. Development and characterisation of a rat brain capillary endothelial culture: towards an in vitro blood-brain barrier. J Cell Sci 1992;103(Pt 1):23–37. [DOI] [PubMed] [Google Scholar]
- 26.Brown RC, Morris AP, O’Neil RG. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res 2007;1130(1):17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kosuge M, Takeuchi T, Nakase I, Jones AT, Futaki S. Cellular internalization and distribution of arginine-rich peptides as a function of extracellular peptide concentration, serum, and plasma membrane associated proteoglycans. Bioconjug Chem 2008;19(3):656–664. [DOI] [PubMed] [Google Scholar]
- 28.Wong KA, O’Bryan JP. Bimolecular fluorescence complementation. J Vis Exp 2011;15(50):e2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 1997;272(25):16010–16017. [DOI] [PubMed] [Google Scholar]
- 31.Steiner O, Coisne C, Engelhardt B, Lyck R. Comparison of immortalized bEnd5 and primary mouse brain microvascular endothelial cells as in vitro blood-brain barrier models for the study of T cell extravasation. J Cereb Blood Flow Metab 2011;31(1):315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Futaki S, Suzuki T, Ohashi W, et al. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem 2001;276(8):5836–5840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.