

Abstract
High-dimensional cytometry is a powerful technique for deciphering the immunopathological factors common to multiple individuals. However, rational comparisons of multiple batches of experiments performed on different occasions or at different sites are challenging due to batch effects. Here we describe the integration of multi-batch cytometry datasets (iMUBAC), a flexible, scalable, and robust computational framework for unsupervised cell-type identification across multiple batches of high-dimensional cytometry datasets, even without technical replicates. After overlaying cells from multiple healthy controls across batches, iMUBAC learns batch-specific cell-type classification boundaries and identifies aberrant immunophenotypes in patient samples from multiple batches in a unified manner. We illustrate unbiased and streamlined immunophenotyping using both public and in-house mass cytometry and spectral flow cytometry datasets. The method is available as the R package iMUBAC (https://github.com/casanova-lab/iMUBAC).
Keywords: immunophenotyping, high-dimensional cytometry, mass cytometry, spectral flow cytometry, batch correction, unsupervised clustering, machine learning
Introduction
High-dimensional cytometry — including mass cytometry (i.e., cytometry by time-of-flight, CyTOF) and spectral flow cytometry (e.g., Cytek Aurora) — facilitates the phenotyping of precious samples from patients for various immune cell subsets at single-cell resolution, which is of particular interest in human immunology. Ideally, a large number of samples should be processed simultaneously to ensure comparability. However, this is not always possible. For example, patients may be recruited across the globe, and over decades, and they may be longitudinally monitored in prospectively expanding cohorts for rare diseases, such as inborn errors of immunity (1). Similarly, some investigations may involve multiple pilot studies with small numbers of patients followed by larger-scale validation studies. In such situations, the integration of multiple batches of experiments processed on different occasions and at different sites is crucial. The simplest solution for multi-batch integration is to gate cell subsets with manual batch-to-batch adjustments. However, manual analyses of cytometry data are inherently subjective, knowledge-driven, and non-scalable for multiple batches of datasets. More objective, unbiased, and scalable methods are therefore desired.
Efforts have been made to facilitate high-dimensional data inspection and unsupervised cell-subset identification [e.g., viSNE (2), SPADE (3), FlowSOM (4, 5), CITRUS (6), and CellCNN (7)]. However, these automated approaches are themselves sensitive to the batch effects resulting from the separate processing of different experiments. Both single-dimensional [i.e., CytoNorm (8) and CytofBatchAdjust (9)] and multi-dimensional [i.e., CytofRUV (10)] signal intensity normalization methods have been described for reducing batch effects before unsupervised cell-type identification. However, the requirement for technical replicates shared across all batches, is a major practical obstacle inherent to these methods. For example, in a prospectively expanding cohort of patients with rare immunological disorders, as experiments accumulate, with the discovery and recruitment of new patients, it becomes increasingly difficult, if not almost impossible, to include identical technical replicate samples in all experiments. Moreover, batch correction on cells from patients is undesirable due to the inherent uncertainty about over- or undercorrection, particularly for patients expected to have markedly altered immunophenotypes. Thus, the computational approaches currently available remain inadequate for unbiased and high-resolution immunophenotyping on multiple batches of cytometry datasets without technical replicates.
Materials and Methods
Cells
Peripheral blood mononuclear cells (PBMCs) were isolated from heparinized venous blood samples by Ficoll-Hypaque density gradient centrifugation (GE Healthcare). Cells were cryopreserved in fetal calf serum supplemented with 10% dimethyl sulfoxide (DMSO) and stored at -150°C until use. Patients with various severe infectious diseases were included in this study. When blood samples from patients were collected at distant sites, the blood samples were transported to either the Paris or the New York branch of our laboratory overnight and processed. We accounted for the effect of blood sample transportation by collecting samples from healthy volunteers or healthy family members and transporting and processing them simultaneously with the transported samples (travel/family controls). Cells from healthy local volunteers collected and processed locally were also used (local controls). We included multiple local controls in each cytometry experiment.
Mass cytometry (CyTOF)
PBMCs from 38 healthy individuals (29 adults and nine children/adolescents aged one to 17 years), 31 patients with various forms of unusually severe infectious diseases and autoimmune diseases, and 16 travel/family controls for these patients were studied in seven different batches of experiments. Nine healthy adult individuals and one patient were studied twice in different batches as either technical or biological replicates. Freshly thawed PBMCs (1.0×106 cells per panel) were first stained with Fc block and then with a panel of metal-conjugated antibodies obtained from Fluidigm or by customized conjugation. The panel consisted of the following antibodies: anti-CD45–89Y, anti-CD57–113In, anti-CD11c-115In, anti-CD33–141Pr, anti-CD19–142Nd, anti-CD45RA-143Nd, anti-CD141–144Nd, anti-CD4–145Nd, anti-CD8–146Nd, anti-CD20–147Sm, anti-CD16–148Nd, anti-CD127–149Sm, anti-CD1c-150Nd, anti-CD123–151Eu, anti-CD66b-152Sm, anti-PD-1–153Eu, anti-CD86–154Sm, anti-CD27–155Gd, anti-CCR5–156Gd, anti-CD117–158Gd, anti-CD24–159Tb, anti-CD14–160Gd, anti-CD56–161Dy, anti-γδTCR-162Dy, anti-CRTH2–163Dy, anti-CLEC12A-164Dy, anti-CCR6–165Ho, anti-CD25–166Er, anti-CCR7–167Er, anti-CD3–168Er, anti-CX3CR1–169Tm, anti-CD38–170Er, anti-CD161–171Yb, anti-CD209–172Yb, anti-CXCR3–173Yb, anti-HLA-DR-174Yb, anti-CCR4–176Yb, and anti-CD11b-209Bi. Stained cells were washed, fixed, and permeabilized with barcode permeabilization buffer (Fluidigm) and barcoded with the Cell-ID 20-Plex Pd Barcoding Kit (Fluidigm). Samples were then washed, pooled into a single tube, and stored until acquisition. Dead cells and doublets were excluded by staining the cells with a rhodium-based dead cell exclusion intercalator (Rh103) before fixation and with cationic iridium nucleic acid intercalators (Ir191 and Ir193) after fixation. Cells were acquired on a Helios mass cytometer (Fluidigm). Data are available from FlowRepository (accession: FR-FCM-Z3YK, https://flowrepository.org/id/FR-FCM-Z3YK).
Spectral flow cytometry
Two experiments were performed on separate dates. We studied frozen PBMCs from 11 locally recruited healthy adult controls and three patients (two patients with homozygous loss-of-function mutations of FAS and one patient with a heterozygous gain-of-function STAT3 mutation). Freshly thawed PBMCs (2×106 cells for controls and 5×106 cells for the patient) were stained with the Zombie NIR Fixable Viability Kit (BioLegend, 1:2000) for 15 minutes on ice. Cells were then stained with the following panel on ice for 30 minutes: FcR Blocking Reagent (Miltenyi Biotec, 1:50), anti-CD3-BD Horizon V450 (BD Biosciences, 1:450), anti-CD4-BUV563 (BD Biosciences, 1:450), anti-CD8-BUV737 (BD Biosciences, 1:150), anti-CD14-BUV395 (BD Biosciences, 1:100), anti-CD16-PE-Dazzle (BioLegend, 1:150), anti-CD20-BV785 (BD Biosciences, 1:150), anti-CD56-BV605 (BioLegend, 1:50), anti-γδTCR-Alexa Fluor 647 (BioLegend, 1:50), anti-Vδ1-FITC (Miltenyi Biotec, 1:450), anti-Vδ2-APC-Fire750 (BioLegend, 1:1350), anti-Vα7.2-Alexa Fluor 700 (BioLegend, 1:50), MR1-BV421 (provided by the NIH Tetramer Core Facility, 1:200), anti-Vα24-Jα18-BV480 (BD Biosciences, 1:50), and anti-Vβ11-APC (Miltenyi Biotec, 1:150) antibodies. The cells were fixed on ice for 30 minutes and permeabilized with the True-Nuclear Transcription Factor Buffer Set (BioLegend). The cells were then stained for intracellular transcription factors overnight in the dark at 4°C with the following panel: anti-T-bet-PE-Cy7 (BioLegend, 1:1350), anti-GATA3-BV711 (BioLegend, 1:50), anti-RORγT-PE (BD Biosciences, 1:50), and anti-EOMES-PerCP-eFluor 710 (eBioscience, 1:50) antibodies. Cells were acquired with an Aurora cytometer (Cytek). Data were inspected with FlowJo, and manually gated live singlets were then exported as FCS files for subsequent iMUBAC analysis. Data are available from FlowRepository (accession: FR-FCM-Z3YL, https://flowrepository.org/id/FR-FCM-Z3YL).
As a technical validation, PBMCs from two locally recruited healthy adult controls and three patients (two patients with homozygous loss-of-function mutations of FAS and one patient with a homozygous gain-of-function STAT3 mutation) were also analyzed by conventional flow cytometry. Cells were stained with the following panel on ice for 20 minutes: FcR Blocking Reagent (Miltenyi Biotec, 1:50), anti-αβTCR-PE/Cy7 (BioLegend, 1:100), anti-CD3-BV421 (BioLegend, 1:100), anti-CD4-redFluor 710 (Tonbo Biosciences, 1:100), anti-CD8-PerCP/Cy7 (BioLegend, 1:100), anti-CD14-APC/Cy7 (BioLegend, 1:100), anti-CD16-APC (BioLegend, 1:100), anti-CD19-Super Bright 645 (eBioscience, 1:100), and anti-CD56-Alexa Fluor 488 (BD Biosciences, 1:100). Cells were then stained with 7-AAD (Tonbo Biosciences, 1:200) on ice for 10 minutes, and then acquired with a BD FACS Aria (BD Biosciences). Compensation was performed with single-stained PBMCs as controls. Data were analyzed with FlowJo.
Public CyTOF datasets
The CyTOF dataset used in the CytofRUV study (10) was downloaded from FlowRepository (accession: FR-FCM-Z2L2, https://flowrepository.org/id/FR-FCM-Z2L2). The dataset consists of PBMC samples from nine healthy controls. All samples are stained with a panel of antibodies targeting 19 surface and 12 intracellular proteins. Each sample was studied twice in two batches of experiments (i.e., technical replicates). Only samples from healthy controls were used in this study. In the benchmark analysis, the first healthy control (VBDR996) was used as an anchor sample for CytoNorm, CytofBatchAdjust, and CytofRUV.
The CyTOF dataset from the checkpoint blockade study (11) was downloaded from FlowRepository (accession: FR-FCM-ZYQR, https://flowrepository.org/id/FR-FCM-ZYQR). The dataset consists of PBMC samples from six healthy controls studied in four different batches of experiments. All samples are stained with a panel of antibodies targeting 33 surface and nine intracellular proteins. Only samples from healthy controls were used in this study. One control (GCND398064) was included as a technical replicate in all batches.
The CyTOF datasets from the PD-1 blockade study (12) were downloaded from FlowRepository (accession: FR-FCM-ZY34, https://flowrepository.org/id/FR-FCM-ZY34). The datasets consist of PBMC samples from 20 patients with stage IV melanoma before and about 12 weeks after treatment with either nivolumab or pembrolizumab, and 10 healthy donors at two corresponding time points. Patients are classified as responders or non-responders based on treatment outcomes for the first 15 weeks of treatment. The datasets consist of two batches (i.e., experiments performed on two different dates), designated as discovery and validation cohorts in the original study. The first batch contains five healthy donors, five responders, and five non-responders, whereas the second batch contains five healthy donors, six responders, and four non-responders. The samples are stained with three different antibody panels.
Computational analysis
All computational analyses were performed with a laptop PC equipped with quad-core CPUs and 64GB RAM, using the free versions of R (ver 4.0) (https://www.r-project.org/) (13) and RStudio Desktop (ver 1.3) (https://rstudio.com/products/rstudio/). The R package iMUBAC and its instructions, including example codes and datasets, are available from GitHub (https://github.com/casanova-lab/iMUBAC). Full scripts are available upon request.
Integration of multi-batch cytometry datasets (iMUBAC)
The iMUBAC workflow consists of four steps: i) preprocessing, ii) batch correction, iii) unsupervised clustering and cell-type annotation, and iv) batch-specific cell-type prediction.
Preprocessing.
Batch-specific preprocessing was performed as follows. First, CyTOF data files in the FCS format were imported into R with the ncdfFlow package. The ncdfFlow package can be used for the memory-efficient HDF5-based storage of cytometry data. The truncate_max_range option was disabled. For CyTOF data, the transformation option was also disabled, as the transformation implemented in the underlying read.FCS package is optimized for flow cytometry. Channel names were then organized. This step resolves batch-to-batch differences in the panel design such that identical markers measured with different channels (e.g., fluorochrome- or metal-conjugated antibodies) are aligned. For CyTOF data, we then excluded doublets and dead cells in a data-adaptive manner. In this step, all cells from all samples in a single batch of an experiment were pooled, such that identical gates were applied to all samples in a given batch. For DNA-based gating, the dnaGate function in the cydar package was used, and outliers on both the higher and lower sides (considered to be doublets and debris, respectively) were excluded. For event length and dead cell exclusion dye-based gating, the outlierGate function in the cydar package was used, and outliers on the higher side were excluded. In our in-house CyTOF datasets, the intercalator Rh103 was used to exclude dead cells, whereas the intercalators Ir191 and Ir193 were used to exclude doublets and debris. In the PD-1 blockade CyTOF datasets, the intercalator Pt198 was used to exclude dead cells, whereas the intercalators Ir191 and Ir193 were used to exclude doublets and debris. For our in-house pre-gated spectral flow cytometry datasets, the channel for the Zombie NIR Fixable Viability dye was used to exclude dead cells, and automated gating for doublets and DNA content was disabled. The expression values were then transformed. For CyTOF data, a hyperbolic arcsin transformation was applied, with a cofactor of five. For spectral flow cytometry data, Logicle transformation was applied, with parameters estimated in a data-adaptive manner with the estimateLogicle function implemented in the flowCore package. Finally, any event with zeros for all markers was discarded.
After the batch-specific preprocessing, the outputs were concatenated, with only markers common to all batches retained, to form a single SingleCellExperiment object in R.
Batch correction.
The goal of this step is to enable the system to learn batch-to-batch deviations due to technical effects but not biological variability. We, therefore, used only data from healthy controls for batch correction, excluding data for patients and travel/family controls. Data were first down-sampled to 200,000 cells per batch (unless otherwise stated) by taking approximately equal numbers of cells from each control, to reduce the computational burden. For the in-house spectral flow datasets, we used 500,000 cells per batch to ensure the robust identification of invariant natural killer T (iNKT) cells, an extremely rare innate-like T-cell subset. For the PD-1 blockade CyTOF Panel 3 (Myeloid Panel) dataset, we used 50,000 cells per batch, due to the low total cell counts in the dataset. We then batch-corrected expression values for all markers with Harmony (14), using the default parameters. Rather than performing principal component analysis (PCA), we used each marker directly as an input for batch correction. The effect of batch correction was assessed by manual inspection, with uniform manifold approximation and projection (UMAP) used for data visualization (15).
Unsupervised clustering and cell-type identification.
The goal of this step was to identify cell types in an unsupervised manner. We implemented two methods: i) FlowSOM-guided clustering and ii) UMAP-based dimension reduction followed by the shared nearest neighbor (SNN) graph-based clustering (16) as described below.
FlowSOM method.
This approach was inspired by the workflow described by Nowicka et al. (5). Briefly, batch-corrected expression values were subjected to unsupervised clustering with FlowSOM (4), using the FlowSOM package, followed by metaclustering with the ConsensusClusterPlus package. Euclidean distance was used for metaclustering. For the in-house spectral flow cytometry and CyTOF datasets, we generated 50 and 60 clusters, respectively, to improve the resolution of cell-type identification. For the PD-1 blockade CyTOF datasets, we generated 40 clusters. We manually determined the identity of these clusters from their locations in the UMAP plot and the heatmap summarizing the median expression levels of all markers for each cluster.
SNN graph method.
This approach was inspired by the workflow for single-cell RNA sequencing datasets implemented in the scran package. Batch-corrected expression values were first dimension-reduced via UMAP into 10 dimensions. These dimensions were then used to construct an SNN graph with the buildSNNGraph function in scran, using the default settings. Finally, the graph was divided into clusters with the Louvain algorithm implemented as the cluster_louvain function in the igraph package (17).
Batch-specific cell-type prediction.
The goal of this step is to allow the system to learn, automatically, the cell-type classification rules, or cell-type boundaries, in a batch-specific manner, and to propagate the boundaries to all the cells in a given batch, including cells from travel/family controls and patients. Importantly, we used non-batch-corrected expression values tied to cluster labels defined in the unsupervised clustering section. First, cells from the healthy controls used for unsupervised clustering were further downsampled, retaining a maximum of 100 cells per cluster from a given batch. This step reduces both the computational burden and the class imbalance problem during machine learning, as there are both highly abundant cell subsets (e.g., CD14+ monocytes) and rare cell subsets (e.g., plasmacytoid dendritic cells) among human PBMCs. We tested several conditions and found that cell-type classification rules can be learned successfully from 100 events per cell type. A classifier was then trained, using the caret package (18). We selected the extremely randomized trees (19) algorithm implemented in the extraTrees package. After centering and scaling the non-batch-corrected expression values, we performed five-repeat 10-fold cross-validations with internal upsampling to maximize the Kappa statistic. Hyperparameters were tuned for each batch; mtry was tuned from five to 15, whereas numRandomCuts was tuned from one to two, the ranges being empirically determined. The batch-specific classifier was then applied for all cells in a given batch, including the cells of patients and travel/family controls, to determine the best-matching clusters in a probabilistic manner. This approach assigns all cells of patients and travel/family controls into one of the clusters defined with the cells of healthy controls. It is, therefore, still possible that the cells of patients assigned to a particular cluster display differential expression of a certain subset of markers relative to the cells of controls in the same cluster. Cluster-wise differential expression (DE) analysis is required to characterize these DE markers comprehensively, but this topic is not dealt with further here.
Benchmarking
CytoNorm (8), CytofBatchAdjust (9), and CytofRUV (10) were downloaded from their GitHub repositories and used with default settings. Batch correction was performed with one technical replicate sample (replicated across all batches). For iMUBAC analysis, batch correction was performed without specifying technical replicates. Signals for all markers were simultaneously corrected. Earth mover’s distance (EMD) was calculated between batches for each of the markers, with the EMDomics package. When multiple healthy controls were included in a given batch, the EMD was calculated without distinguishing between individuals. Computation time was measured in R. Per-cluster batch representation was defined as the proportion of cells from a certain batch in a given cluster. All clusters defined through unsupervised clustering were subjected to the analysis.
Differential abundance analysis
The raw counts of cell subsets identified from the three panels were jointly tested for differential abundance (DA) with the quasi-likelihood F-test (QLF) framework of the edgeR package (20). When both pre- and post-treatment datasets were used, the DA between groups (i.e., responders and non-responders) was assessed, with adjustment for both treatment and batch effects. When only pretreatment datasets were used, the DA between groups was assessed with adjustment for batch effects only. The DA values for subsets with an absolute log2 fold-change of at least 0.5 and an adjusted P-value below 0.05 were considered statistically significant.
Results
We present iMUBAC (integration of multi-batch cytometry datasets), a flexible, robust, and scalable computational framework for rational interbatch comparisons through high-dimensional batch correction and unsupervised cell-type identification across multiple batches (Fig. 1). The workflow can be broken down into four steps. First, iMUBAC performs data-adaptive, automated preprocessing, including the exclusion of doublets and dead cells and ensuring inter-batch consistency in panel design. Second, iMUBAC batch-corrects cells from healthy local controls, but not from travel/family controls or patients, using Harmony (14) to reduce batch effects before clustering. The major advantage of Harmony is that it does not require technical replicates shared across batches. Third, iMUBAC performs unsupervised clustering with the batch-corrected expression values, by i) FlowSOM-guided clustering (4) or ii) dimension reduction by uniform manifold approximation and projection (UMAP) (15) followed by shared nearest-neighbor (SNN) graph-based clustering (16). If desired, the clusters can be further merged manually and identified to improve interpretability in subsequent analyses. Fourth, iMUBAC trains batch-specific classifiers through machine learning with non-corrected expression values. Here, the idea is to “back-propagate” cell-type annotations defined in the batch-corrected high-dimensional space into the non-corrected, batch-specific spaces in which the patients’ cells are embedded. The entire workflow is independent of patients (and of travel/family controls), for whom immunophenotypes need to be determined, thereby preventing over- or undercorrection.
Figure 1.

Integration of multi-batch cytometry datasets (iMUBAC). Multiple cytometry datasets can be integrated for rational inter-batch and inter-individual comparisons. Even experiments with heterogeneous designs (e.g., differences in the numbers of local controls and patients) or inconsistent panels without shared technical replicates can be integrated. Only cells from healthy local controls were used for batch correction. The batch-corrected expression values were then used for unsupervised clustering, followed by manual annotation if desired. The non-batch-corrected expression values, tied to cell-type annotations, were then used to train batch-specific cell-type classifiers. Finally, cell types were predicted for the rest of the cells, including the cells of patients and their travel/family controls.
We first benchmarked the batch-correction efficacy of iMUBAC against other algorithms, using two different public multi-batch CyTOF datasets for peripheral blood mononuclear cells (PBMCs) from multiple healthy volunteers, including technical replicate samples shared across all batches of experiments. The CytofRUV dataset reported by Trussart et al. (10) contains nine healthy controls analyzed in two batches of experiments. The first control (VBDR996) was used as an anchor for CytoNorm, CytofBatchAdjust, and CytofRUV. By contrast, Harmony-guided batch-correction in iMUBAC used 200,000 cells per batch randomly sampled from all the available healthy controls. The checkpoint blockade dataset reported by Wei et al. (11) contains 14 samples from six healthy controls studied in four batches of experiments, including one control (GCND398064) studied as technical replicates in all batches. The shared control was used as an anchor for CytoNorm, CytofBatchAdjust, and CytofRUV. By contrast, Harmony-guided batch-correction in iMUBAC used 1,000,000 cells per batch randomly sampled from all the available controls. We compared the earth mover’s distance (EMD) between batches for each marker before and after batch correction, as described in previous studies (8, 10). We aggregated all controls in each batch for EMD calculation. We therefore expect inter-individual variability to persist even after the removal of batch effects due to technical variability. The comparison showed that iMUBAC reduced EMD in both datasets as effectively as CytoNorm, and perhaps more effectively than CytofBatchAdjust (Fig. 2A), whereas CytofRUV gave the most effective reduction of EMD in both datasets.
Figure 2.

Benchmarking of the iMUBAC workflow. (A and B) The efficacy of batch correction in (A) mass cytometry (CyTOF) datasets with technical replicates, and (B) mass and spectral flow cytometry datasets lacking technical replicates. Only cells from healthy controls were used. The earth mover’s distance (EMD) was used as a metric of “dissimilarity” in marker expression patterns between batches of experiments. The EMD was calculated for each marker, across all batches of experiments, in a given dataset. Smaller EMDs indicate more effective removal of batch effects. The EMD distributions of all markers are shown. (C) The impact of batch correction on unsupervised clustering. We calculated the proportion of cells from a given batch in a given cluster as a metric of the evenness of the representation of each batch in each cluster. (D and E) The impact of the number of healthy controls per batch on (D) batch correction and (E) unsupervised clustering. The in-house CyTOF dataset reconstructed with the indicated number of randomly selected controls was analyzed with iMUBAC. In (A, B, and D), the horizontal bars represent the mean. In (C and E), the vertical dashed lines indicate the expected values based on the number of batches in the datasets.
We then evaluated the efficacy of batch correction by iMUBAC using datasets lacking shared technical replicates across batches. To this end, we used the following multi-batch cytometry datasets: in-house CyTOF, in-house spectral flow, and PD-1 blockade CyTOF datasets. The in-house CyTOF dataset contains 47 PBMC samples from 38 healthy local controls analyzed in seven batches of experiments with a panel consisting of 38 cell-surface markers. The in-house spectral flow cytometry dataset contains 11 PBMC samples from 11 healthy controls analyzed in two batches of experiments with a panel consisting of 14 cell-surface and four intracellular markers. The PD-1 blockade CyTOF dataset, reported by Krieg et al. (12), contains 10 PBMC samples from 10 healthy controls analyzed in two batches of experiments (i.e., discovery and validation cohorts) with three different antibody panels. These datasets cannot be processed by existing methods (i.e., CytoNorm, CytofBatchAdjust, or CytofRUV) because of the lack of replicates. Batch-correction was performed by iMUBAC, using cells randomly sampled from all controls (N=500,000 for the spectral flow dataset, N=50,000 for the PD-1 blockade Panel 3 CyTOF dataset, and N=200,000 for other datasets). We found that batch-correction decreased EMD in all datasets (Fig. 2B). We then evaluated the benefits of batch correction for subsequent unsupervised clustering, by calculating the proportion of cells from a given batch of experiments in a given cluster, as the “per-cluster batch representation”. FlowSOM defined 60, 50, 40, 30, and 40 clusters, using non-corrected or corrected expression values, for the in-house CyTOF, in-house spectral flow, and PD-1 blockade CyTOF Panel 1, 2, and 3 datasets, respectively. Batch correction improved the evenness of per-cluster batch representation in all datasets (Fig. 2C). Finally, we assessed the impact of the number of healthy controls per batch on the batch-correction performance of iMUBAC, using the in-house CyTOF dataset as a case study. We calculated both EMDs and per-cluster batch representations with the inclusion of a limited number of controls. We found that at least three controls per batch were required to achieve effective batch correction (Figs. 2D and E). Collectively, iMUBAC successfully integrates multi-batch CyTOF and spectral flow cytometry datasets, even without shared technical replicates, by overlaying cells from multiple healthy controls as anchors.
Scalability is also an issue when a large number of high-dimensional cytometry experiments need to be integrated. We thus evaluated the scalability of the iMUBAC workflow by assessing the computation time required. We used our in-house CyTOF dataset, which was the largest dataset tested here. We first evaluated the relationship between the number of batches and the computation time required for Harmony-guided batch correction (using ~200,000 cells per batch). We found that computation time increased linearly with the number of batches (Fig. 3A). It took ~2.5 hours to integrate all seven batches. We also assessed the time required for cell-type back-propagation by machine learning. Our in-house CyTOF dataset contains 95 PBMC samples from healthy local controls, patients with unusually severe infectious diseases and autoimmune diseases, and their travel/family controls (~18 million cells from 85 individuals in total), analyzed in seven batches of experiments. After batch correction with healthy local controls, FlowSOM defined 60 clusters. Batch-specific classifiers were trained on non-batch-corrected expression values, and the cluster labels were back-propagated to all cells, including cells from travel/family controls and patients. The classifier training phase took less than an hour per ~200,000 cells per batch, whereas the prediction phase was completed in most cases within 15 minutes per batch (Figs. 3B and 3C). Collectively, iMUBAC scales up linearly as the number of batches or cells increases.
Figure 3.

Linear scalability of the iMUBAC workflow. The computation time of each step of the iMUBAC workflow was measured with the in-house CyTOF dataset as a case study, in which peripheral blood mononuclear cells (PBMCs) from healthy local controls, travel/family controls, and patients with unusually severe infectious diseases and autoimmune diseases were studied in seven batches of experiments (N=95 samples, 18,344,759 cells in total) with a general immunophenotyping panel (38 surface markers). The results of three computations were compiled. (A) Batch correction via Harmony. (B) Batch-specific cell-type classifier training. (C) Batch-specific cell-type prediction. R, Kendall’s correlation coefficients. In (B), the error bars represent the SEM.
The successful identification of both abundant and rare leukocyte subsets is an absolute requirement for optimal immunophenotyping. We, thus, evaluated the accuracy and immunological interpretability of the iMUBAC workflow with our in-house CyTOF dataset. After batch correction with healthy local controls, FlowSOM defined 60 clusters, which we merged and identified manually (Fig. S1). We then calculated the percentages of the various subsets among live single leukocytes (CD45+CD66b− cells, excluding CD45−CD66b+ granulocytes). The cell-type frequencies of both technical (i.e., experiments performed on different dates with aliquots of identical biological materials) and biological (i.e., experiments performed on different dates with biological materials obtained from identical donors on different occasions) duplicates correlated well between the two batches tested (Fig. S2). We also evaluated the accuracy of automatic cell-type identification by iMUBAC, by manually gating the same CyTOF dataset in FlowJo. The manually determined frequencies of both abundant (e.g., αβ T cells) and rare (e.g., conventional dendritic cells) subsets were remarkably consistent with the frequencies determined by the iMUBAC workflow (independent of manual gating) across a wide range of percentages (from ~0.01% to ~70%), not only for local controls, but also for travel/family controls and patients (Fig. 4). Indeed, iMUBAC successfully identified both the reduction (e.g., NK cells) and expansion (e.g., B cells) of multiple cell subsets in samples from patients, demonstrating the robustness of cell-type identification regardless of abundance in a given sample. Collectively, iMUBAC rationally integrates multi-batch cytometry datasets to identify cell types consistent with state-of-the-art gating in both controls and patients.
Figure 4.
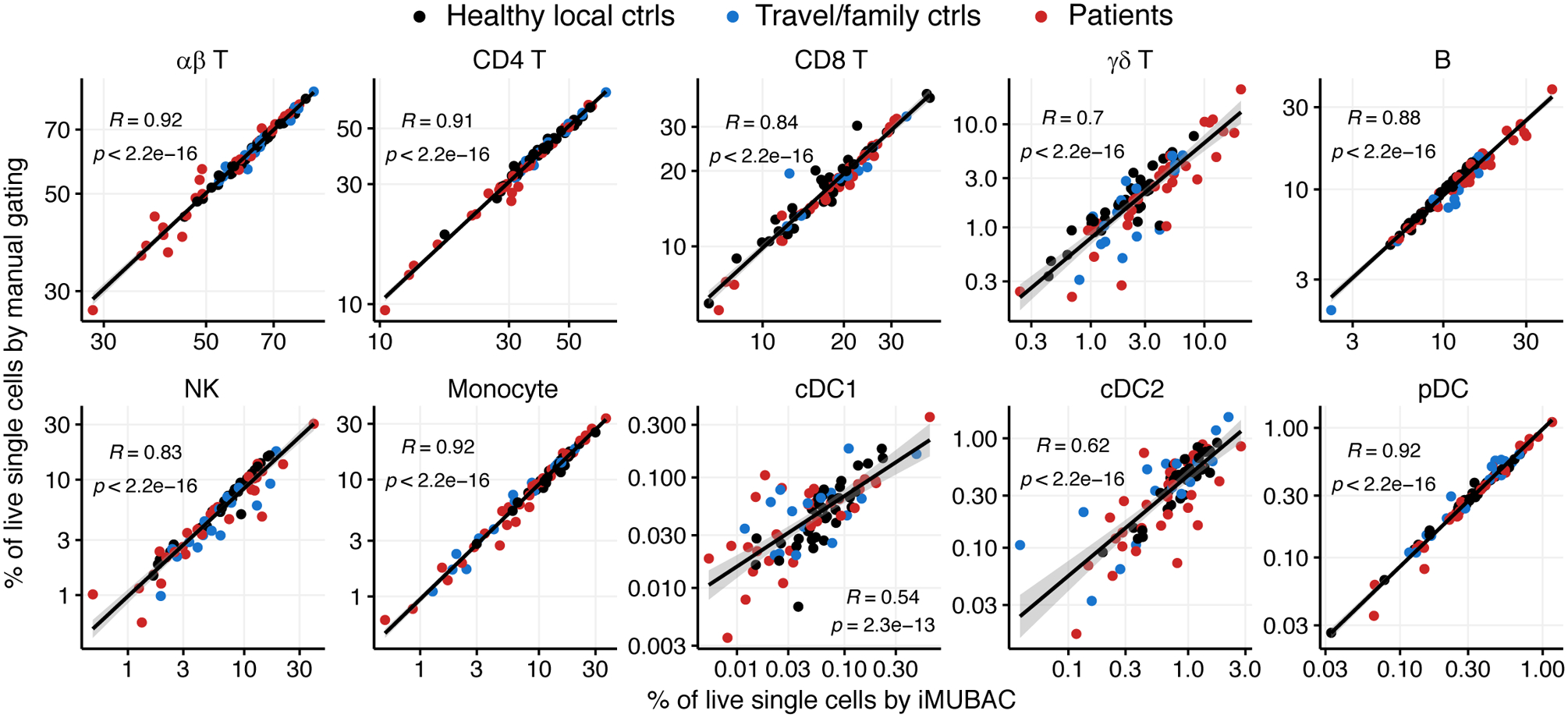
iMUBAC identifies immune cell subsets consistently with manual gating. The in-house CyTOF dataset was analyzed, in which PBMCs from healthy local controls, travel/family controls, and patients with unusually severe infectious diseases and autoimmune diseases were studied in seven batches of experiments (N=95 samples, 18,344,759 cells in total) with a general immunophenotyping panel (38 surface markers). We evaluated the accuracy of cell-type identification by iMUBAC, by also manually gating the same dataset in a batch-specific manner, and comparing the relative frequencies of various immune cell subsets among live single leukocytes (CD45+CD66b−, excluding CD45−CD66b+granulocytes). During the comparison, multiple clusters for a given cell subset (e.g., “NK CD16+”, “NK CD16dim” and “NK CD56bright” for NK cells) were merged before the calculation of frequencies. Representative results for 10 subsets are shown. NK, natural killer cells; cDC, conventional dendritic cells; pDC, plasmacytoid dendritic cells. R, Kendall’s correlation coefficients.
The identification, description, and quantification of disease-associated rare leukocyte subsets are crucial in many immunological studies. We, therefore, applied iMUBAC to our in-house multi-batch spectral flow cytometry dataset, which contains PBMCs from 11 healthy controls and three patients with two monogenic forms of autoimmunity: FAS deficiency (N=2) (21, 22) and STAT3 gain-of-function (N=1) (23–25). Both these disorders are associated with lymphoproliferation and high counts of CD4−CD8− double-negative αβ T (DN T) cells in the peripheral bloodstream. After the manual gating out of dead cells and doublets in FlowJo, iMUBAC batch corrected the cells (~500,000 per batch) from healthy controls and defined 50 clusters, which we then merged and identified manually (Figs. 5A, B, and S3). As expected, we observed an expansion of a cluster representing DN T cells in patients with FAS deficiency and, to a lesser extent, in those with STAT3 gain-of-function (Fig. 5C). Conventional flow cytometry with a different antibody panel validated this expansion of DN T cells (data not shown). iMUBAC also revealed a decrease in the levels of Vδ2 γδ T, CD8+ mucosal-associated invariant T (MAIT), and CD16+ natural killer (NK) cells in both groups, possibly reflecting a previously unappreciated level of immunopathological homogeneity between these two inborn errors of immunity (Fig. 5C). Thus, iMUBAC readily identifies both known and unappreciated immunophenotypes in patients with rare immunological disorders.
Figure 5.

iMUBAC identifies both known and previously unrecognized disease-associated immunophenotypes. (A-C) Analysis of the in-house spectral flow cytometry dataset for PBMCs from patients with monogenic forms of autoimmunity (N=3) and healthy controls (N=11) containing 11,202,354 cells in total. (A and B) UMAP visualization of the single-cell measurements of all markers with the following hyperparameters: n_neighbors=75 and min_dist=0.75. One million cells were selected at random for visualization. (A) Batch correction. (B) Cell-type identification through unsupervised clustering. (C) Selected immunophenotypes of patients with FAS deficiency and STAT3 gain-of-function mutations. (D-F) Analysis of the CyTOF datasets for PBMCs from patients with advanced melanoma before and after PD-1 blockade immunotherapy (N=20) and healthy controls (N=10) (12). The original study compared immunophenotypes between responders and non-responders and reported higher levels of CD14+CD16−HLA-DRhi monocytes among responders at baseline. Panel 1, T-cell phenotyping (1,092,973 cells). Panel 2, cytokines (1,730,805 cells). Panel 3, myeloid phenotyping (262,006 cells). (D) UMAP visualization of all markers (n_neighbors=25 and min_dist=0.4) showing the effect of batch correction. (E and F) Differential abundance (DA) analysis. All subsets from the three panels were simultaneously tested for DA with edgeR (20) after adjustment for treatment and batch effects. Percentages among live single leukocytes before PD-1 blockade are shown. (E) Expansion of the “CD14 Mono 2” cluster, corresponding to CD14+CD16−HLA-DRhiCD86hiPD-L1hi monocytes, in responders. (F) DA for subsets identified as statistically significant when only pretreatment datasets were used for DA testing. Estimated log2 fold-changes between responders and non-responders are also shown. In (C, E, and F), bars represent the mean and SEM.
Finally, we demonstrated the potential advantage of streamlined immunophenotyping on multi-batch cytometry datasets by applying iMUBAC to the PD-1 blockade CyTOF datasets previously reported by Krieg et al. (12). These datasets consist of PBMCs from 10 healthy controls and 20 patients with stage IV melanoma before and after PD-1 blockade immunotherapy (11 responders and nine non-responders) studied in two batches of experiments (i.e., the discovery and validation cohorts) with three different antibody panels. Healthy controls were used for batch correction, clustering with FlowSOM, and classifier training (Figs. 5D and S4). We tested the differential abundance (DA) between responders and non-responders, using the quasi-likelihood F-test (QLF) in edgeR (20), as previously applied to CyTOF analysis (26, 27). Eight DA subsets were identified, which remained statistically significant after adjustment for treatment status and batch effects. In particular, the “CD14 Mono 2” cluster from Panel 3, corresponding to CD14+CD16−HLA-DRhiCD86hiPD-L1hi monocytes, was expanded in responders, consistent with the findings of the original report (12) (Figs. 5E and S4A). Moreover, among the eight DA subsets, four T-cell subsets from Panel 1 were reproducibly identified even when only pretreatment datasets were used for the DA analysis (Figs. 5F and S4B). These subsets could potentially be used as biomarkers of a better prognosis, before the initiation of PD-1 blockade immunotherapy. Thus, iMUBAC can be used to streamline an exploratory immunophenotyping analysis in clinical pilot studies of common diseases.
Discussion
High-dimensional cytometry is a promising technique for human immunological studies, which are often characterized by the limited amounts of clinical samples available. A rational comparison of leukocyte subsets from patients with immunological conditions studied on different occasions would make it possible to identify the immunophenotypes unique to particular patients, and those common to these patients. Moreover, platforms such as FlowRepository and Cytobank facilitate the sharing of cytometry data, making it possible to foster discoveries through meta-analysis. However, the lack of appropriate technical replicates across experiments hinders batch correction, making integrative analyses challenging. We present iMUBAC, a technical replicate-independent batch correction and unsupervised clustering framework, available on GitHub as a readily installable R package (https://github.com/casanova-lab/iMUBAC), for the rational comparative immunophenotyping analysis of multiple batches of high-dimensional cytometry datasets. We show that iMUBAC is flexible in terms of study design (e.g., technical replicates), linearly scalable with increases in the number of batches and cells to be integrated, and robust with respect to the abundance of each of the immune cell subsets in a given sample. The combination of batch correction and unsupervised clustering provided by this package enables investigators to define leukocyte subsets comparatively across batches, eliminating the need to interpret “batch-specific” clusters originating from batch effects, or artifacts. Furthermore, we demonstrate that iMUBAC can successfully identify known disease condition-specific subsets in both CyTOF and spectral flow cytometry datasets. Given the increasing popularity of high-dimensional spectral flow cytometry, a unified workflow applicable to both CyTOF and spectral flow cytometry is of major pragmatic significance.
There are several caveats to the use of iMUBAC. First, we assume that both healthy controls and patients have identical sets of cell subsets. All samples must, therefore, be derived from the same type of biological material. For example, iMUBAC cannot simultaneously handle PBMC samples from controls and tumor-infiltrating cells from patients. However, we successfully show that the subset of DN T cells, a rare T-cell subset, in healthy controls, is expanded in patients with lymphoproliferative autoimmunity, suggesting that iMUBAC can handle samples from patients that are quantitatively different from control samples. Second, iMUBAC makes use of multiple healthy controls per batch, rather than a single shared technical replicate, enabling it to capture an “average” immunophenotype across controls. Given the inherent variability of human immunophenotypes, any single control may deviate from the average immunophenotype. As we showed, the use of multiple controls (at least three) mitigates this unpredictable variability. Third, the functionality of iMUBAC may be restricted by the computational resources available, although this package can operate on any conventional desktop PC or laptop computer. The computation time (of the order of hours) of iMUBAC is undoubtedly longer than that for the other tools currently available. When shared technical replicates are available, CytofRUV may be a better choice, as it outperforms iMUBAC in terms of both the speed and efficacy of batch correction. Nevertheless, we demonstrate here the feasibility of comparative immunophenotyping analysis on multi-batch cytometry experiments without relying on shared technical replicates. We envisage that our framework can be applied in many areas of immunological studies, thereby fostering discoveries that might otherwise be overlooked.
Supplementary Material
Key points.
iMUBAC integrates multi-batch cytometry datasets without shared technical replicates.
Streamlined immunophenotyping identifies disease-associated phenotypes across batches.
Acknowledgments
We would like to thank the patients, their relatives, and their physicians for participating in this study; Dominick Papandrea, Yelena Nemirovskaya, Mark Woollett, Lazaro Lorenzo-Diaz, and Cécile Patissier for administrative assistance; Tatiana Kochetkov for technical assistance; the members of the laboratory for helpful discussions. We thank the Flow Cytometry Resource Center at the Rockefeller University for technical support for flow cytometry. We also thank the Human Immune Monitoring Core at the Icahn School of Medicine at Mount Sinai for technical assistance with mass cytometry. We thank the National Institutes of Health (NIH) Tetramer Core Facility (NTCF) for providing the MR1 tetramer, which was developed jointly with Dr. James McCluskey, Dr. Jamie Rossjohn, and Dr. David Fairlie.
This study was supported in part by a grant from the St. Giles Foundation, The Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), Université de Paris, Howard Hughes Medical Institute, the National Institute of Allergy and Infectious Diseases/National Institutes of Health (R37AI095983 to J.-L.C., U19AI142737 to S.B.-D., and R01AI127372 and R01AI148963 to D.B.), the French Foundation for Medical Research (FRM) (EQU201903007798), the French National Research Agency under the “Investments for the Future” program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID), the GENMSMD project (ANR-16-CE17-0005-01 to J.B.), the SCOR Corporate Foundation for Science, and Fondation du Souffle (SRC2017 to J.B.). M.O. was supported by the David Rockefeller Graduate Program, the Funai Foundation for Information Technology (FFIT), the Honjo International Scholarship Foundation (HISF), and the New York Hideyo Noguchi Memorial Society, Inc. (HNMS). R.Y. was supported by the Immune Deficiency Foundation and Stony Wold-Herbert Fund. A.N.S. was supported by the European Commission (EC, Horizon 2020 Marie Skłodowska-Curie Individual Fellowship #789645), the Dutch Research Council (NWO, Rubicon Grant #019.171LW.015), and the European Molecular Biology Organization (EMBO, Long-Term Fellowship #ALTF 84-2017, non-stipendiary). J.R. was supported by INSERM PhD program (poste d’accueil INSERM).
References
- 1.Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, Franco JL, Holland SM, Klein C, Morio T, Ochs HD, Oksenhendler E, Picard C, Puck J, Torgerson TR, Casanova J-L, and Sullivan KE. 2020. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol 40: 24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amir ED, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D. 2013. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol 31: 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qiu P, Simonds EF, Bendall SC, Gibbs KD, V Bruggner R, Linderman MD, Sachs K, Nolan GP, and Plevritis SK. 2011. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol 29: 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y. 2015. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 87: 636–645. [DOI] [PubMed] [Google Scholar]
- 5.Nowicka M, Krieg C, Weber LM, Hartmann FJ, Guglietta S, Becher B, Levesque MP, and Robinson MD. 2017. CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. F1000Research 6: 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruggner RV, Bodenmiller B, Dill DL, Tibshirani RJ, and Nolan GP. 2014. Automated identification of stratifying signatures in cellular subpopulations. Proc. Natl. Acad. Sci. U. S. A 111: E2770–E2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arvaniti E, and Claassen M. 2017. Sensitive detection of rare disease-Associated cell subsets via representation learning. Nat. Commun 8: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Gassen S, Gaudilliere B, Angst MS, Saeys Y, and Aghaeepour N. 2020. CytoNorm: A Normalization Algorithm for Cytometry Data. Cytom. Part A 97: 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuyler RP, Jackson C, Garcia-Perez JE, Baxter RM, Ogolla S, Rochford R, Ghosh D, Rudra P, and Hsieh EWY. 2019. Minimizing Batch Effects in Mass Cytometry Data. Front. Immunol 10: 2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trussart M, Teh CE, Tan T, Leong L, Gray DH, and Speed TP. 2020. Removing unwanted variation with CytofRUV to integrate multiple CyTOF datasets. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei SC, Anang N-AAS, Sharma R, Andrews MC, Reuben A, Levine JH, Cogdill AP, Mancuso JJ, Wargo JA, Pe’er D, and Allison JP. 2019. Combination anti–CTLA-4 plus anti–PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci 116: 22699 LP – 22709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, Dummer R, Robinson MD, Levesque MP, and Becher B. 2018. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med 24: 144–153. [DOI] [PubMed] [Google Scholar]
- 13.R Core Team. 2018. R: A Language and Environment for Statistical Computing. .
- 14.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, ru Loh P, and Raychaudhuri S. 2019. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16: 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McInnes L, Healy J, and Melville J. 2018. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv . [Google Scholar]
- 16.Xu C, and Su Z. 2015. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 31: 1974–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csárdi G, and Nepusz T. 2006. The igraph software package for complex network research. InterJournal Complex Sy: 1695. [Google Scholar]
- 18.Kuhn M 2008. Building Predictive Models in R Using the caret Package. J. Stat. Softw 28: 1–26.27774042 [Google Scholar]
- 19.Geurts P, Ernst D, and Wehenkel L. 2006. Extremely randomized trees. Mach. Learn 63: 3–42. [Google Scholar]
- 20.Robinson MD, McCarthy DJ, and Smyth GK. 2009. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, Rieux-Laucat F, Siegel RM, Su HC, Teachey DT, and Rao VK. 2010. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood 116: e35–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magerus-Chatinet A, Stolzenberg M-C, Loffredo MS, Neven B, Schaffner C, Ducrot N, Arkwright PD, Bader-Meunier B, Barbot J, Blanche S, Casanova J-L, Debré M, Ferster A, Fieschi C, Florkin B, Galambrun C, Hermine O, Lambotte O, Solary E, Thomas C, Le Deist F, Picard C, Fischer A, and Rieux-Laucat F. 2009. FAS-L, IL-10, and double-negative CD4−CD8− TCR α/β+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood 113: 3027–3030. [DOI] [PubMed] [Google Scholar]
- 23.Haapaniemi EM, Kaustio M, Rajala HLM, van Adrichem AJ, Kainulainen L, Glumoff V, Doffinger R, Kuusanmäki H, Heiskanen-Kosma T, Trotta L, Chiang S, Kulmala P, Eldfors S, Katainen R, Siitonen S, Karjalainen-Lindsberg M-LM-L, Kovanen PE, Otonkoski T, Porkka K, Heiskanen K, Hänninen A, Bryceson YT, Uusitalo-Seppälä R, Saarela J, Seppänen M, Mustjoki S, Kere J, Kuusanmaki H, Heiskanen-Kosma T, Trotta L, Chiang S, Kulmala P, Eldfors S, Katainen R, Siitonen S, Karjalainen-Lindsberg M-LM-L, Kovanen PE, Otonkoski T, Porkka K, Heiskanen K, Hanninen A, Bryceson YT, Uusitalo-Seppala R, Saarela J, Seppanen M, Mustjoki S, and Kere J. 2015. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 125: 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen AA, Niemela JE, Lyons JJ, Engelhardt KR, Zhang Y, Topcagic N, Roberson EDOO, Matthews H, Verbsky JW, Dasu T, Vargas-Hernandez A, Varghese N, McClain KL, Karam LB, Nahmod K, Makedonas G, Mace EM, Sorte HS, Perminow GG, Koneti Rao V, O’Connell MP, Price S, Su HC, Butrick M, McElwee J, Hughes JD, Willet J, Swan D, Xu Y, Santibanez-Koref M, Slowik V, Dinwiddie DL, Ciaccio CE, Saunders CJ, Septer S, Kingsmore SF, White AJ, Cant AJ, Hambleton S, Cooper MA, Rao VK, O’Connell MP, Price S, Su HC, Butrick M, McElwee J, Hughes JD, Willet J, Swan D, Xu Y, Santibanez-Koref M, Slowik V, Dinwiddie DL, Ciaccio CE, Saunders CJ, Septer S, Kingsmore SF, White AJ, Cant AJ, Hambleton S, and Cooper MA. 2015. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 125: 591–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nabhani S, Schipp C, Miskin H, Levin C, Postovsky S, Dujovny T, Koren A, Harlev D, Bis AM, Auer F, Keller B, Warnatz K, Gombert M, Ginzel S, Borkhardt A, Stepensky P, and Fischer U. 2017. STAT3 gain-of-function mutations associated with autoimmune lymphoproliferative syndrome like disease deregulate lymphocyte apoptosis and can be targeted by BH3 mimetic compounds. Clin. Immunol 181: 32–42. [DOI] [PubMed] [Google Scholar]
- 26.Lun ATL, Richard AC, and Marioni JC. 2017. Testing for differential abundance in mass cytometry data. Nat. Methods 14: 707–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weber LM, Nowicka M, Soneson C, and Robinson MD. 2019. diffcyt: Differential discovery in high-dimensional cytometry via high-resolution clustering. Commun. Biol 2: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.